
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAl2O3 promoted mechanochemical nucleophilic aromatic substitution†
Luca
Vaghi
 *,
Eva
Palomba
and
Antonio
Papagni
*,
Eva
Palomba
and
Antonio
Papagni
Department of Materials Science, University of Milano-Bicocca, Via Roberto Cozzi 55, 20125 Milano, Italy. E-mail: luca.vaghi@unimib.it
First published on 9th May 2024
Abstract
An auxiliary mediated solventless mechanochemical methodology for the nucleophilic aromatic substitution of aryl fluorides by nitrogen nucleophiles without the aid of any base has been developed. The excellent affinity of Al2O3 for hydrogen fluoride is the key for this process. Thanks to the use of Al2O3 as a milling auxiliary yields of up to 99% were achieved, and the easily processable crude derived from the milling jar allowed a simple and fast work-up procedure, making this method very attractive.
Introduction
Nucleophilic aromatic substitution (SNAr) has played an important role in organic synthesis for a long time.1,2 Indeed, the possibility of introducing functional groups on aromatic substrates by replacing a leaving group (often halogen atoms, in particular fluorine) is successfully exploited in pharmaceutical chemistry and in industry,3–5 thanks to its simplicity, cheapness, and effectiveness, and the absence of traces of metal catalysts in the final products. However, these processes often require high temperature, long reaction time (up to days) and high-boiling polar aprotic solvents, such as dimethylformamide, which are difficult to eliminate and environmentally unfriendly.In recent years, mechanochemical synthetic methodologies have been constantly gaining importance as a valid alternative to conventional solution processes,6–17 and can address the problem of the use of toxic solvents and high temperature by exploiting mechanical energy furnished by the impacts inside the reactor (jar). In this context, the effectiveness of mechanochemistry applied to SNAr reactions has been recently demonstrated, providing a substantial advantage in the reaction kinetics compared to the classical procedures in solution.18 However, a common issue in mechanochemical procedures is the recovery of the crude from the jar. Indeed, especially when organic and inorganic reagents are involved together, a sticky dough remains at the end of the reaction and its recovery requires an huge amount of solvent, compromising the benefits of a solvent-free procedure.18,19 This issue can be addressed using a grinding auxiliary, i.e., an inert material to be added into the reactor together with the reagents, since this often allows the obtainment of a final solid dispersion that is easy to recover and process.20 Besides, the auxiliary can also be effective in switching the reaction selectivity towards the desired products,21,22 affecting also the yields depending on its peculiar features.23,24 The main drawbacks of grinding auxiliary addition are the dispersion of mechanical energy and the less close contact among the reactants resulting in worsening the reaction kinetic and yield. For these reasons, the individuation of auxiliaries that are able to promote selected transformations is particularly appealing.
Aiming to develop a grinding auxiliary assisted mechanochemical methodology for the SNAr of aromatic fluorides, we found impressive differences in reaction outputs depending on the nature of the auxiliary used.
Results and discussion
We selected 4-fluoronitrobenzene 1 and piperidine 2a as a model substrate and model nucleophile to set up the milling auxiliary mediated SNAr reaction (Table 1), as these reagents have already proved to be effective under the auxiliary free milling conditions.18 The more commonly used grinding auxiliary, i.e., silica (SiO2), was firstly tested. At a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 1/2a ratio results were not good both without and in the presence of K2CO3 as a base, affording 3a in only 3% and 4% yields, respectively (Table 1, entries 1 and 2). Yield increased to a still unsatisfactory 24% only using a large excess of 2a (Table 1, entry 4). However, the powdery crudes derived from these preliminary attempts were easily recovered, allowing also an easy and fast purification on a silica pad (Fig. S1 and S2†). Indeed, thanks to the significant difference in retention factors of 2a, 3a and 1, respectively, and the absence of by-products, the vacuum-aided dry column chromatography methodology25 with a limited quantity of selected eluent was able to afford pure 3a even if the yields were extremely low, a significant improvement if compared with the hard to process sticky dough observed in the case of the reported auxiliary-free procedure.18
:1 1/2a ratio results were not good both without and in the presence of K2CO3 as a base, affording 3a in only 3% and 4% yields, respectively (Table 1, entries 1 and 2). Yield increased to a still unsatisfactory 24% only using a large excess of 2a (Table 1, entry 4). However, the powdery crudes derived from these preliminary attempts were easily recovered, allowing also an easy and fast purification on a silica pad (Fig. S1 and S2†). Indeed, thanks to the significant difference in retention factors of 2a, 3a and 1, respectively, and the absence of by-products, the vacuum-aided dry column chromatography methodology25 with a limited quantity of selected eluent was able to afford pure 3a even if the yields were extremely low, a significant improvement if compared with the hard to process sticky dough observed in the case of the reported auxiliary-free procedure.18
|
|
|||||
|---|---|---|---|---|---|
| Entry |
1:2a |
Auxiliary | Base | Time (min) | 3a Isolated yield (%) |
| a Reactions were performed using a 50 mL grinding reactor (ZrO2) and 4 milling balls (1 cm diameter, ZrO2) at a shaking frequency of 15 Hz, on 2 mmol of 1, using 5 g of auxiliary and 2 equiv. of base (when employed). | |||||
| 1 | 1:1 |
SiO2 | — | 60 | 3 |
| 2 | 1:1 |
SiO2 | K2CO3 | 60 | 4 |
| 3 | 1:1.5 |
SiO2 | K2CO3 | 60 | 5 |
| 4 | 1:5 |
SiO2 | — | 60 | 24 |
| 5 | 1:1 |
TiO2 | — | 60 | 44 |
| 6 | 1:1 |
TiO2 | K2CO3 | 60 | 67 |
| 7 | 1:1 |
Al2O3 | — | 60 | 83 |
| 8 | 1:1 |
Al2O3 | K2CO3 | 60 | 83 |
| 9 | 1:1 |
Al2O3 | — | 30 | 88 |
| 10 | 1:1.25 |
Al2O3 | — | 30 | 91 |
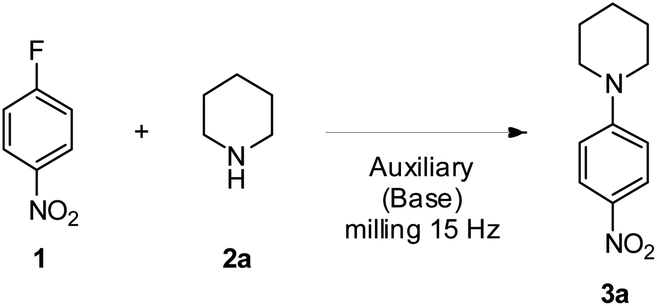
The use of a base in SNAr reactions is generally considered essential and credited to a strong catalytic effect, even though several reports raise doubts on this belief,26,27 and looking to a recent high throughput screening of SNAr the absence of such a catalytic effect is evident.28 This was confirmed in herein reported preliminary tries. It is our opinion that the role of the base is primary to intercept the HF produced during the SNAr reaction, which, in turn, can protonate the nucleophile making it unreactive. In fact, the use of two equivalents of nucleophile can provide the same results of base addition,29,30 and in particular cases, when the base could interfere with the non-bonding interactions required to achieve the transformation, the use of two equivalents of nucleophile is mandatory.31,32
Keeping in mind the concept of intercepting HF, our attention was drawn to costless materials showing also a good affinity for HF, to be used as milling auxiliaries in our procedures. Widely used metal oxides, i.e., titania (TiO2) and alumina (Al2O3), caught our attention; indeed, both have been reported to have an excellent affinity for HF. In the case of TiO2 this behaviour is mainly exploited as a functionalization strategy to improve the photoelectric and photocatalytic properties of titania-based devices and materials,33–36 whereas the alumina–fluorine affinity has been studied in catalysis,37–39 in biological systems,40 and in industrial applications to scavenge F− ions.41 Moreover, hydrogen fluoride adsorption on alumina has been investigated by computational methods by Teng and co-workers in 2011.42
First, TiO2 was tested with our experimental setup at a 1:1 1/2a ratio without a base for 60 min of milling, affording 3a in 44% yield (Table 1, entry 4). This result confirmed our suppositions on the HF sequestering ability of titania; under the same conditions with SiO2 as an auxiliary the yield was only 3% (Table 1, entry 1). Addition of K2CO3 as a base increased the yield to 67% (Table 1, entry 6), suggesting a cooperative HF sequestration; thus, TiO2 showed a good but non-optimal HF sequestering ability.
Next, Al2O3 was experimented with at a 1:1 1/2a ratio without a base for 60 min of milling, and under these conditions an astonishing 83% yield of pure 3a was achieved (Table 1, entry 7). Right after, we repeated the experiment in the presence of K2CO3 (Table 1, entry 8), and base addition had no improving effect achieving the same 83% yield. This confirmed the optimal Al2O3–HF affinity to be exploited in the auxiliary mediated mechanochemical SNAr. Further optimization, i.e., reduction of milling time to 30 min and a 1:1.25 1/2a ratio, allowed the isolation of 3a with a good yield of 91% (Table 1, entry 10).
With these optimized conditions for the Al2O3 mediated mechanochemical SNAr, we tested the procedure with different amines (2b–l, Table 2). The effect of milling time was re-evaluated with morpholine 2b (Table 2, entries 1, 2 and 3), and 60 min was found to be the best option since a longer reaction time resulted in a slight advantage in yield; consequently, this parameter was not modified in the subsequent experiments. Increasing the excess of 2b had a more positive impact on the 3b isolated yield, going from 60% to 70% to 96% for 1/2b ratios of 1:1, 1:1.5 and 1:2, respectively (Table 2, entries 3, 4 and 5). The same behaviour was observed for 3-pyrrolidinol 2c (Table 2, entries 6 and 7), 4-amino-1-butanol 2h (Table 2, entries 12 and 13) and 2-aminoethanol 2i (Table 2, entries 14 and 15), whereas for the more reactive piperidine 2a a 25% excess was sufficient to exceed 90% yield. The choice for less reactive nucleophiles should be at least a 1:1.5 substrate/amine ratio. As a matter of fact, an amine excess is a strategy often used in SNAr methodologies.43
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Nucleophile | 1:Nu |
Time (min) | Product | Isolated yield (%) |
| a Reactions were performed using a 50 mL grinding reactor (ZrO2) and 4 milling balls (1 cm diameter, ZrO2) at a shaking frequency of 15 Hz, on 2 mmol of 1 or 1b, using 5 g of Al2O3. b 2 equiv. of K2CO3 were added. | ||||||
| 1 | 1 | 2b | 1:1 |
30 | 3b | 55 |
| 2 | 1 | 2b | 1:1 |
60 | 3b | 60 |
| 3 | 1 | 2b | 1:1 |
120 | 3b | 62 |
| 4 | 1 | 2b | 1:1.5 |
60 | 3b | 70 |
| 5 | 1 | 2b | 1:2 |
60 | 3b | 96 |
| 6 | 1 | 2c | 1:1 |
60 | 3c | 48 |
| 7 | 1 | 2c | 1:2 |
60 | 3c | 80 |
| 8 | 1 | 2d | 1:2 |
60 | 3d | 32 |
| 9 | 1 | 2e | 1:1.5 |
60 | 3e | 3 |
| 10 | 1 | 2f | 1:1.5 |
60 | 3f | Traces |
| 11 | 1 | 2g | 1:1 |
60 | 3g | — |
| 12 | 1 | 2h | 1:1 |
60 | 3h | 29 |
| 13 | 1 | 2h | 1:2 |
60 | 3h | 52 |
| 14 | 1 | 2i | 1:1 |
60 | 3i | 17 |
| 15 | 1 | 2i | 1:2 |
60 | 3i | 62 |
| 16 | 1 | 2j | 1:2 |
60 | 3j | 4 |
| 17 | 1 | 2k | 1:1 |
60 | 3k | Traces |
| 18 | 1 | 2l | 1:1 |
60 | 3l | — |
| 19 | 1b | 2a | 1:1 |
60 | 3a | 15 |
| 20b | 1b | 2a | 1:1 |
60 | 3a | 4 |

Primary alkylamines were less prone to react with respect to cyclic secondary amines; indeed, 3d was achieved in a modest 32% yield (Table 2, entry 8). Driven by the hypothesis that the lower reactivity was caused by the volatility of butylamine 2d, we experimented with a much heavier alkyl amine, i.e., hexadecylamine 2e, yet the result was significantly worse, and product 3e was isolated in only 3% yield (Table 2, entry 9).
Non-cyclic secondary amines were ineffective; thus, 2f and 2k afforded only barely detectable traces of SNAr products (Table 2, entries 10 and 17), and N-ethylethanolamine 2j gave 3j in poor 4% yield (Table 2, entry 16).
It is evident that steric hindrance around the nucleophilic site plays a crucial role in SNAr type reactions; in fact, when hindered primary amine 2g (adamantylamine) was experimented, no traces of expected product 3g were detected (Table 2, entry 11). A particular discussion on this point must be reserved for cyclic amines. Even though they should be more hindered at the nitrogen with respect to linear primary amines, their particular conformation makes the nitrogen lone pair sensibly more prone to carry out a nucleophilic attack; indeed, their nucleophilicity is more pronounced.44,45
In the presence of an OH group on the amine nucleophile (i.e., 2c and 2h–k) N- vs. O-selectivity was complete, and no O-substitution products were observed. It is worth noticing that 2h and 2i afforded N-substitution products 3h and 3i in 52% and 62% yield, respectively (Table 2, entries 13 and 15); hence, their reactivity was unexpectedly much better with respect to butylamine 2d, whose yield was limited to 32% (Table 2, entry 8). Since the nucleophilic reactivity of 2h and 2i is lower than that of 2d,44 we hypothesized a non-bonding interaction between OH and Al2O3 which somehow favours the SNAr reaction.46 The low nucleophilicity of the aromatic nitrogen makes aniline 2l unreactive; indeed, no traces of (4-nitrophenyl)phenylamine 3l were detected (Table 2, entry 18). Yet, even in solution this kind of reaction is challenging, requiring very long time at high temperature and/or insanely high pressures.47,48
At last, we experimented under the same conditions with 1-chloro-4-nitrobenzene 1b as a substrate with the more reactive amine nucleophile, i.e., piperidine 2a. However, 3a was achieved in only 15% yield (Table 2, entry 19), and even in a worse 4% yield upon adding 2 equiv. of base (Table 2, entry 20). Of course, aryl chlorides are far less reactive than aryl fluorides in SNAr; accordingly, with a big increase of the milling time improvements could be observed, but long reaction times in mechanochemical procedures are not convenient; furthermore, it is reported that Cl− adsorption on Al2O3 is much less effective compared to that of F−;49 thus, we decided to not further investigate aryl chlorides with our methodology.
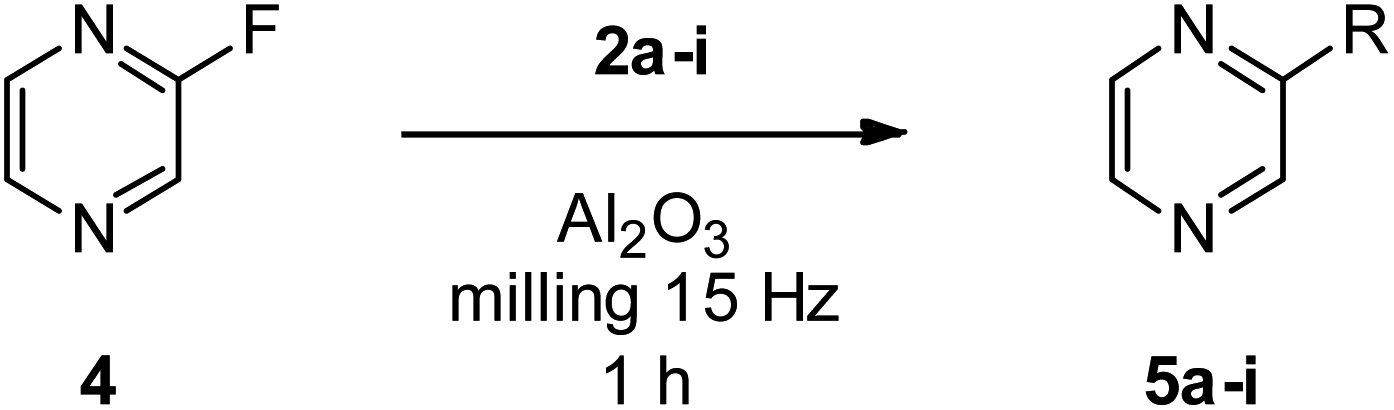
In order to explore the generality of the method, we used 2-fluoropyrazine 4 as a substrate employing the more efficient amine nucleophiles from previous experiments (Table 3). In this case too, the best results were achieved with cyclic secondary amines, i.e., piperidine 2a, morpholine 2b and 3-pyrrolidinol 2c; thus, we prepared pure 5a, 5b, and 5c in good yields of 84, 69 and 86%, respectively (Table 3, entries 2, 4 and 5). Good results were obtained also in the synthesis of 5h from 4-amino-1-butanol 2h and 5i from 2-aminoethanol 2i with a 63 and 61% isolated yield, respectively (Table 3, entries 7 and 8). Conversely, reaction with butylamine 2d afforded only barely detectable traces of expected product 5d. N- versus O-selectivity was complete even in these cases, and we did not observe any presence of O-substituted products.
|
|
||||
|---|---|---|---|---|
| Entry | Nucleophile |
4:Nu |
Product | Isolated yield (%) |
| a Reactions were performed using a 50 mL grinding reactor (ZrO2) and 4 milling balls (1 cm diameter, ZrO2) at a shaking frequency of 15 Hz for 1 h, on 2 mmol of 4, using 5 g of Al2O3 as an auxiliary. | ||||
| 1 | 2a | 1:1 |
5a | 73 |
| 2 | 2a | 1:1.25 |
5a | 84 |
| 3 | 2b | 1:1.5 |
5b | 55 |
| 4 | 2b | 1:2 |
5b | 69 |
| 5 | 2c | 1:2 |
5c | 86 |
| 6 | 2d | 1:2 |
5d | Traces |
| 7 | 2h | 1:2 |
5h | 63 |
| 8 | 2i | 1:2 |
5i | 61 |
Next, we tested our methodology with a selection of commercially available fluoroaryl and fluoroheteroaryl substrates in the reaction with piperidine 2a (Scheme 1). First, the effect of the nitro group position in fluoronitrobenzenes was investigated. It is well known that the electronic effect of the activating group in ortho and para positions with respect to the leaving group is far more pronounced than the steric effect in SNAr reactions;1 indeed, 1-(2-nitrophenyl)piperidine 6a was achieved in an excellent 98% yield; conversely, no traces of 1-(3-nitrophenyl)piperidine 7a were detected when 1-fluoro-3-nitrobenzene 7 was reacted under the same conditions. Subsequently, different substituted p-fluoronitrobenzenes (8–12, Scheme 1) were experimented with. Methyl substituted 8a was obtained in 95% yield, and the presence of an additional electron-withdrawing group, i.e., CF3 in 9 and CN in 10, allowed us to achieve 9a and the key intermediate for the synthesis of antimalarial and antibacterial antimetabolites 10a,50 in almost quantitative yields. Noteworthily, the presence of an NH2 group on substrate 11 did not prevent the preparation of compound 11a with a good isolated yield of 87%. The latter is a useful intermediate for the synthesis of ferroptosis inhibitors,51 AMP-activated protein kinase activators,52 fibroblast growth factor receptor inhibitors,53 and antitumor agents.54,55 Substrate 12 was recovered unreacted from the milling attempt due to the high steric hindrance caused by the two methyl groups ortho to fluorine.
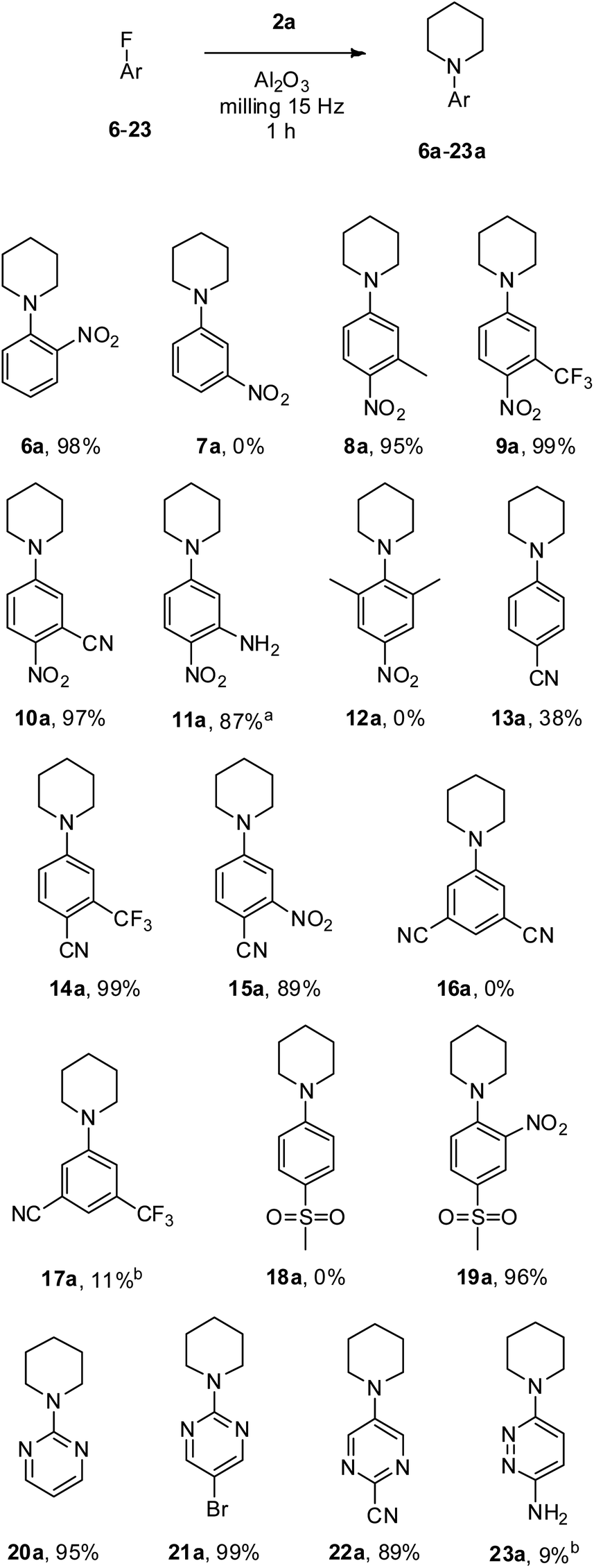 | ||
| Scheme 1 Scope of substrates in the Al2O3 promoted mechanochemical SNAr. Reagents and conditions: substrate (2 mmol), piperidine (3 mmol), Al2O3 (5 g), milling (15 Hz), 1 h. a 8 g of Al2O3 were used to avoid dough formation. b Determined by 1H NMR. | ||
At this point, we experimented with fluorobenzenes bearing less powerful activating groups, i.e., CN and SO2Me (13–19, Scheme 1).1 By reacting 4-fluorobenzonitrile 13 under our conditions 4-(1-piperidinyl)benzonitrile 13a was isolated in a modest 38%. However, the presence of an electron-withdrawing group ortho to the CN sensibly improved the outcome of the milling reaction; indeed, 14a and 15a were achieved in 99% and 89% isolated yield, respectively. It is worth noting that the quantitative yield obtained for the pharmacologically significant nonsteroidal androgen receptor antagonist 14a56 outperformed the yields reported to date in the literature, which were limited to 75%.56,57 As expected, when the CN activating groups are placed meta to the leaving fluorine the SNAr reaction was unfruitful, and no traces of product 16a were detected, while a useless 11% yield of 17a was identified by 1H NMR analysis of the eluted residue. When 1-fluoro-4-(methylsulfonyl)benzene 18 was employed the SNAr product 18a was not observed, while the simultaneous presence of SO2Me and NO2 groups in the activating positions enabled the synthesis of 19a in an excellent 96% isolated yield.
Finally, pyrimidinyl and pyridazyl fluorides were investigated. Pyrimidine substrates 20 and 21 in which the leaving fluorine is located in the favourable position 2 were successfully transformed into piperidine substituted 20a (an effective antioxidant for the coadjutant treatment of neurodegenerative diseases)58 and 21a with an excellent isolated yield of 95% and 99%, respectively. Pyrimidine compounds which bear the fluorine in the less favourable position 5 are not expected to react in SNAr, nevertheless, the presence of the CN activating group in the right position in 22 allowed the isolation of pure 22a in 89% yield. 3,6-Diaminopyridazines are key intermediates for the production of pharmaceutically important compounds;59 thus, 6-fluoro-3-pyridazinamine 23 was tested; however, 1H NMR analysis revealed a poor 9% yield of 23a. It is worth underlining that the powdery crude residues from all experiments were recovered and processed really easily, allowing the isolation of the desired product in pure form in a short time.
To prove the scalability of our methodology we tried a reaction with 10 mmol of aryl substrate. Obviously to maintain the same level of mechanical energy a jar with greater volume should be used, as increasing the quantity of the reagent and auxiliary results in muffling the impacts, and therefore the reaction output could be worse. Nevertheless, 6 (10 mmol), 2a (15 mmol) and Al2O3 (15 g) were charged in a 50 mL reactor (ZrO2) with 4 milling balls (1 cm diameter, ZrO2), and the mixture was shaken at 15 Hz for 1 h. After the usual fast purification by vacuum-aided chromatography on silica gel product 6a was achieved in pure form in a good yield of 91% (Fig. S3†).
Experimental
The detailed synthetic procedures, the characterization of all synthesized compounds and copies of NMR and IR spectra are available in the ESI.†Conclusions
In summary, we have described a new simple and convenient solventless mechanochemical procedure for the nucleophilic aromatic substitution of aryl fluorides. The HF scavenging properties of Al2O3 were the key factor, allowing the use of any base to be avoided. Using it as grinding auxiliary facilitated an easy and fast preparation of several amino substituted aryls and heteroaryls in good yields, greatly decreasing the quantity of organic solvent in the work-up step and avoiding the production of aqueous waste that is costly to treat. Cyclic secondary amines and properly activated substrates gave the best results, as expected for this kind of transformation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Giorgio Patriarca for the NMR analyses and Sara Peverali for the photo support.Notes and references
- J. F. Bunnett and R. E. Zahler, Aromatic Nucleophilic Substitution Reactions, Chem. Rev., 1951, 49, 273–412 CrossRef CAS.
- F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley-VCH Verlag, Weinheim, Germany, 2013 Search PubMed.
- J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Expanding the medicinal chemistry synthetic toolbox, Nat. Rev. Drug Discovery, 2018, 17, 709–727 CrossRef PubMed.
- D. G. Brown and J. Boström, Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS.
- S. D. Roughley and A. M. Jordan, The Medicinal Chemist's Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- E. Juaristi and C. G. Avila-Ortiz, Salient achievements in Synthetic Organic chemistry enabled by Mechanochemical Activation, Synthesis, 2023, 55, 2439–2459 CrossRef CAS.
- B. R. Naidu, T. Sruthi, R. Mitty and K. Venkateswarlu, Catalyst-free mechanochemistry as a versatile tool in synthetic chemistry: a review, Green Chem., 2023, 25, 6120–6148 RSC.
- N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Green metrics in mechanochemistry, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC.
- V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Advancing mechanochemical synthesis by combining milling with different energy sources, Nat. Rev. Chem, 2022, 7, 51–65 CrossRef PubMed.
- J. Andersen and J. Mack, Mechanochemistry and organic synthesis: from mystical to practical, Green Chem., 2018, 20, 1435–1443 RSC.
- J. L. Howard, Q. Cao and D. L. Browne, Mechanochemistry as an emerging tool for molecular synthesis: what can it offer?, Chem. Sci., 2018, 9, 3080–3094 RSC.
- T. Friščić, C. Mottillo and H. M. Titi, Mechanochemistry for Synthesis, Angew Chem. Int. Ed. Engl., 2020, 59, 1018–1029 CrossRef PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Mechanochemistry: opportunities for new and cleaner synthesis, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- C. Espro and D. Rodríguez-Padrón, Re-thinking organic synthesis: Mechanochemistry as a greener approach, Curr. Opin. Green Sustainable Chem., 2021, 30, 100478 CrossRef CAS.
- Y. S. Zholdassov, L. Yuan, S. R. Garcia, R. W. Kwok, A. Boscoboinik, D. J. Valles, M. Marianski, A. Martini, R. W. Carpick and A. B. Braunschweig, Acceleration of Diels-Alder reactions by mechanical distortion, Science, 2023, 380, 1053–1058 CrossRef CAS PubMed.
- T. Seo, K. Kubota and H. Ito, Mechanochemistry-directed ligand design: Development of a high-performance phosphine ligand for palladium-catalyzed mechanochemical organoboron cross-coupling, J. Am. Chem. Soc., 2023, 145, 6823–6837 CrossRef CAS PubMed.
- K. Kubota, T. Seo, K. Koide, Y. Hasegawa and H. Ito, Olefin-accelerated solid-state C–N cross-coupling reactions using mechanochemistry, Nat. Commun., 2019, 10, 111 CrossRef PubMed.
- J. M. Andersen and H. F. Starbuck, Rate and Yield Enhancements in Nucleophilic Aromatic Substitution Reactions via Mechanochemistry, J. Org. Chem., 2021, 86, 13983–13989 CrossRef CAS PubMed.
- T. Dalidovich, J. V. Nallaparaju, T. Shalima, R. Aav and D. G. Kananovich, Mechanochemical nucleophilic substitution of alcohols via isouronium intermediates, ChemSusChem, 2022, 15, e202102286 CrossRef CAS PubMed.
- I. Andreosso, A. Papagni and L. Vaghi, Mechanochemical oxidation of fluorinated anilines to symmetric azobenzenes, J. Fluorine Chem., 2018, 216, 124–127 CrossRef CAS.
- R. Thorwirth, F. Bernhardt, A. Stolle, B. Ondruschka and J. Asghari, Switchable Selectivity during Oxidation of Anilines in a Ball Mill, Chemistry, 2010, 16, 13236–13242 CrossRef CAS.
- G. Cravotto, K. Martina, L. Rinaldi, F. Baricco and L. Boffa, Highly Efficient Mechanochemical N-Arylation of Amino Alcohols and Diamines with Cu0 Powder, Synlett, 2015, 26, 2789–2794 CrossRef.
- R. Schmidt, A. Stolle and B. Ondruschka, Aromatic substitution in ball mills: formation of aryl chlorides and bromides using potassium peroxomonosulfate and NaX, Green Chem., 2012, 14, 1673–1679 RSC.
- T. Szuppa, A. Stolle, B. Ondruschka and W. Hopfe, Solvent-free dehydrogenation of c-terpinene in a ball mill: investigation of reaction parameters, Green Chem., 2010, 12, 1288–1294 RSC.
- D. Pedersen and C. Rosenbohm, Dry column vacuum chromatography, Synthesis, 2001, 16, 2431–2434 Search PubMed.
- A. J. Kirby and W. P. Jencks, Base catalysis of the reaction of secondary amines with p-nitrophenyl phosphate. Kinetic evidence for an addition intermediate in nucleophilic aromatic substitution1, J. Am. Chem. Soc., 1965, 87, 3217–3224 CrossRef CAS.
- C. F. Bernasconi, Kinetic behavior of short-lived anionic σ complexes, Acc. Chem. Res., 1978, 11, 147–152 CrossRef CAS.
- Z. Jaman, D. L. Logsdon, B. Szilágyi, T. J. P. Sobreira, D. Aremu, L. Avramova, R. G. Cooks and D. H. Thompson, High-throughput experimentation and continuous flow evaluation of nucleophilic aromatic substitution reactions, ACS Comb. Sci., 2020, 22, 184–196 CrossRef CAS PubMed.
- C. R. Johnson, B. Zhang, P. Fantauzzi, M. Hocker and K. M. Yager, Libraries of N-alkylaminoheterocycles from nucleophilic aromatic substitution with purification by solid supported liquid extraction, Tetrahedron, 1998, 54, 4097–4106 CrossRef CAS.
- A. J. Blacker, G. Moran-Malagon, L. Powell, W. Reynolds, R. Stones and M. R. Chapman, Development of an SNAr reaction: A practical and scalable strategy to sequester and remove HF, Org. Process Res. Dev., 2018, 22, 1086–1091 CrossRef CAS.
- L. Vaghi, M. Coletta, P. Coghi, I. Andreosso, L. Beverina, R. Ruffo and A. Papagni, Fluorine substituted non-symmetric phenazines: a new synthetic protocol from polyfluorinated azobenzenes, ARKIVOC, 2019, 2019, 340–351 Search PubMed.
- K. Liu, M. Brivio, T. Xiao, V. M. Norwood IV, Y. S. Kim, S. Jin, A. Papagni, L. Vaghi and R. W. Huigens III, Modular synthetic routes to fluorine-containing halogenated phenazine and acridine agents that induce rapid iron starvation in methicillin-resistant staphylococcus aureus biofilms, ACS Infect. Dis., 2022, 8, 280–295 Search PubMed.
- J. Tang, H. Quan and J. Ye, Photocatalytic properties and photoinduced hydrophilicity of surface-fluorinated TiO2, Chem. Mater., 2007, 19, 116–122 CrossRef CAS.
- M. Samadpour, P. P. Boix, S. Giménez, A. Iraji Zad, N. Taghavinia, I. Mora-Seró and J. Bisquert, Fluorine treatment of TiO2 for enhancing quantum dot sensitized solar cell performance, J. Phys. Chem. C Nanomater. Interfaces, 2011, 115, 14400–14407 CrossRef CAS.
- A. M. Czoska, S. Livraghi, M. Chiesa, E. Giamello, S. Agnoli, G. Granozzi, E. Finazzi, C. D. Valentin and G. Pacchioni, The nature of defects in fluorine-doped TiO2, J. Phys. Chem. C Nanomater. Interfaces, 2008, 112, 8951–8956 CrossRef CAS.
- K. Lv, B. Cheng, J. Yu and G. Liu, Fluorine ions-mediated morphology control of anatase TiO2 with enhanced photocatalytic activity, Phys. Chem. Chem. Phys., 2012, 14, 5349–5362 RSC.
- K. Maruoka and T. Ooi, The synthetic utility of the hypercoordination of boron and aluminum, Chemistry, 1999, 5, 829–833 CrossRef CAS.
- W. J. Smith III and J. S. Sawyer, A novel and selective method for the N-arylation of indoles mediated by KF/Al2O3, Tetrahedron Lett., 1996, 37, 299–302 CrossRef.
- F. Schneider and B. Ondruschka, Mechanochemical solid-state Suzuki reactions using an in situ generated base, ChemSusChem, 2008, 1, 622–625 CrossRef CAS PubMed.
- A. Lubkowska, B. Zyluk and D. Chlubek, Interactions between fluorine and aluminum, Fluoride, 2002, 35, 73–77 CAS.
- Z. Tian, W. Guo, Z. Zhang, Y. Lai, S. Ye and J. Li, Removal of fluorine ions from industrial zinc sulfate solution by a layered aluminum-based composite, Hydrometallurgy, 2017, 171, 222–227 CrossRef CAS.
- J.-L. Quan, B.-T. Teng, X.-D. Wen, Y. Zhao, R. Liu and M.-F. Luo, Hydrogen fluoride adsorption and reaction on the α-Al2O3(0001) surface: A density functional theory study, J. Chem. Phys., 2012, 136, 114701 CrossRef PubMed.
- Y.-J. Cherng, Efficient nucleophilic substitution reactions of pyrimidyl and pyrazyl halides with nucleophiles under focused microwave irradiation, Tetrahedron, 2002, 58, 887–890 CrossRef CAS.
- T. Kanzian, T. A. Nigst, A. Maier, S. Pichl and H. Mayr, Nucleophilic reactivities of primary and secondary amines in acetonitrile, Eur. J. Org. Chem., 2009, 2009, 6379–6385 CrossRef.
- W.-Z. Ge, B.-M. Wu and W.-Y. Huang, Nucleophilic substitution reaction in polyfluoroaromatics: I. Reactions with secondary amine nucleophiles, Acta Chim. Sin., 1985, 3, 349–355 CrossRef.
- S. Roy, G. Mpourmpakis, D.-Y. Hong, D. G. Vlachos, A. Bhan and R. J. Gorte, Mechanistic study of alcohol dehydration on γ-Al2O3, ACS Catal., 2012, 2, 1846–1853 CrossRef CAS.
- H. Suhr, N. Substitutionen and V. Umsetzung, von 4-Nitro-fluorbenzol mit primären Aminen, Adv. Cycloaddit., 1965, 687, 175–182 CAS.
- H. Kotsuki, S. Kobayashi, K. Matsumoto, H. Suenaga and H. Nishizawa, High pressure organic chemistry; XII.1A convenient synthesis of aromatic amines from activated aromatic fluorides, Synthesis , 1990, 1147–1148 CrossRef CAS.
- W. Szczepaniak and H. Kościelna, Specific adsorption of halogen anions on hydrous γ-Al2O3, Anal. Chim. Acta, 2002, 470, 263–276 CrossRef CAS.
- E. F. Elslager, J. Clarke, L. M. Werbel, D. F. Worth and J. Davoll, Folate antagonists. 3. 2,4-Diamino-6-(heterocyclic)quinazolines, a novel class of antimetabolites with potent antimalarial and antibacterial activity, J. Med. Chem., 1972, 15, 827–836 CrossRef CAS PubMed.
- Y. Fang, X. Chen, Q. Tan, H. Zhou, J. Xu and Q. Gu, Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: A new mechanism of action, ACS Cent. Sci., 2021, 7, 980–989 CrossRef CAS PubMed.
- J. Charton, S. Girault-Mizzi, M.-A. Debreu-Fontaine, F. Foufelle, I. Hainault, J.-G. Bizot-Espiard, D.-H. Caignard and C. Sergheraert, Synthesis and biological evaluation of benzimidazole derivatives as potent AMP-activated protein kinase activators, Bioorg. Med. Chem., 2006, 14, 4490–4518 CrossRef CAS PubMed.
- W. Yan, X. Wang, Y. Dai, B. Zhao, X. Yang, J. Fan, Y. Gao, F. Meng, Y. Wang, C. Luo, J. Ai, M. Geng and W. Duan, Discovery of 3-(5′-Substituted)-Benzimidazole-5-(1-(3,5-dichloropyridin-4-yl)ethoxy)-1H-indazoles as Potent Fibroblast Growth Factor Receptor Inhibitors: Design, Synthesis, and Biological Evaluation, J. Med. Chem., 2016, 59, 6690–6708 CrossRef CAS PubMed.
- V. A. Mamedov, N. A. Zhukova, A. D. Voloshina, V. V. Syakaev, T. N. Beschastnova, A. P. Lyubina, S. K. Amerhanova, A. I. Samigullina, A. T. Gubaidullin, D. N. Buzyurova, I. K. Rizvanov and O. G. Sinyashin, Synthesis of Morpholine-, Piperidine-, and N-Substituted Piperazine-Coupled 2-(Benzimidazol-2-yl)-3-arylquinoxalines as Novel Potent Antitumor Agents, ACS Pharmacol. Transl. Sci., 2022, 5, 945–962 CrossRef CAS PubMed.
- J. S. Kim, Q. Sun, C. Yu, A. Liu, L. F. Liu and E. J. LaVoie, Quantitative structure-activity relationships on 5-substituted terbenzimidazoles as topoisomerase I poisons and antitumor agents, Bioorg. Med. Chem., 1998, 6, 163–172 CrossRef CAS PubMed.
- I. Kinoyama, N. Taniguchi, T. Yoden, H. Koutoku, T. Furutani, M. Kudoh and M. Okada, Synthesis and pharmacological evaluation of novel arylpiperazine derivatives as nonsteroidal androgen receptor antagonists, Chem. Pharm. Bull., 2004, 52, 1330–1333 CrossRef CAS PubMed.
- R. P. Trump, J.-B. E. Blanc, E. L. Stewart, P. J. Brown, M. Caivano, D. W. Gray, W. J. Hoekstra, T. M. Willson, B. Han and P. Turnbull, Design and synthesis of an array of selective androgen receptor modulators, J. Comb. Chem., 2007, 9, 107–114 CrossRef CAS PubMed.
- H. Kawada and P. F. Kador, Orally bioavailable metal chelators and radical scavengers: Multifunctional antioxidants for the coadjutant treatment of neurodegenerative diseases, J. Med. Chem., 2015, 58, 8796–8805 CrossRef CAS PubMed.
- P. A. Bethel, B. Roberts and A. Bailey, Preparation of differentially substituted 3,6-diaminopyridazines under mild conditions, Tetrahedron Lett., 2014, 55, 5186–5190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization of all synthesized compounds and copies of NMR and IR spectra. See DOI: https://doi.org/10.1039/d4mr00039k |
| This journal is © The Royal Society of Chemistry 2024 |