
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal mechano-synthesis of 2-cyclopropyl-4-(4-fluorophenyl)quinoline-3-acrylaldehyde—a pivotal intermediate of pitavastatin†
Jingbo
Yu
 *ab,
Yanhua
Zhang
a,
Zehao
Zheng
a and
Weike
Su
*ab
*ab,
Yanhua
Zhang
a,
Zehao
Zheng
a and
Weike
Su
*ab
aLaboratory of Pharmaceutical Engineering of Zhejiang Province, Key Laboratory for Green Pharmaceutical Technologies and Related Equipment of Ministry of Education, Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou, 310014, P. R. China. E-mail: yjb@zjut.edu.cn
bHuzhou Key Laboratory of Mechanochemistry, Zhejiang Yangtze River Delta Biomedical Industry Technology Research Park, Deqing 313200, P. R. China
First published on 1st July 2024
Abstract
Pitavastatin (PTV), a potent cholesterol-lowering agent, holds considerable commercial appeal, driving chemists to fervently pursue its efficient and sustainable synthesis. Despite prolonged efforts over several decades, the quest for a simplified, more efficacious, and environmentally conscious manufacturing process for PTV remains a significant challenge. Our study introduces a three-step total mechano-synthesis, commencing with readily available 4-bromoquinoline, to produce the key intermediate (2-cyclopropyl-4-(4-fluorophenyl)quinoline-3-acrylaldehyde) of PTV. This methodology incorporates an extrusive Suzuki–Miyaura coupling, mechanochemical Minisci C–H alkylation, and extrusive oxidation Heck coupling, each thoroughly presented to display their scalability. Notably, we emphasize the extensive exploration of substrate versatility in Minisci reactions to access cyclopropane-bearing pharmaceutical compounds and natural products. This total mechano-synthesis route distinguishes itself through eco-friendly reaction conditions, exceptional stepwise efficiency, intuitive operability, and pronounced potential for large-scale implementation, paving the way for PTV's streamlined and sustainable manufacture.
Introduction
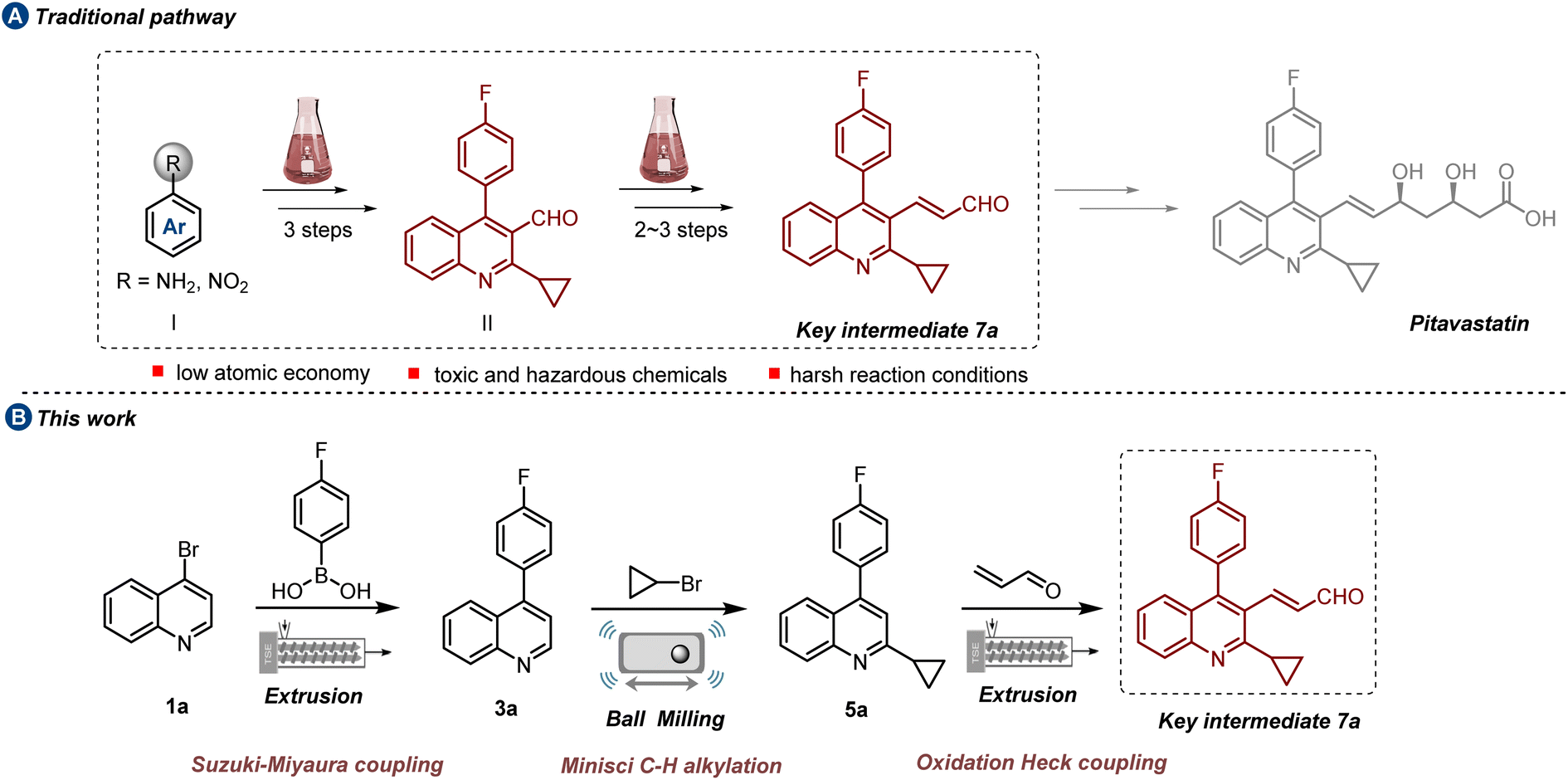
Pitavastatin (PTV), approved by the FDA in 2009, is commonly referred to as a “Superstatin” in the pharmaceutical industry due to its excellent therapeutic effects and small dosage.1 Acting as an HMG-CoA reductase inhibitor, its prominent efficacy is depicted by the increase of high-density lipoprotein cholesterol and the reduction of low-density lipoprotein cholesterol.2 Besides its lipid-lowering effects, PTV exhibits pleiotropic effects like anti-inflammatory, immunomodulatory, and antithrombotic activities.3 Over recent decades, the preparation of PTV drug has garnered persistent attention.4 Apart from heptanoate as the basic structure, a core quinoline ring assembled with fluorophenyl and cyclopropyl moieties provides improved pharmacokinetics and effective HMG-CoA inhibition at low doses. Central to accessing PTV is the synthesis of 2-cyclopropyl-4-(4-fluorophenyl)quinoline-3-acrylaldehyde, which serves as the key intermediate 7a (Scheme 1a). Typically, this intermediate is initially derived from polysubstituted quinoline formaldehyde II via a Wittig reaction,5 a well-established synthetic pathway for introducing the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds group, albeit challenged by the generation of significant amounts of phosphine oxide byproduct, leading to low atom economy efficiency. Besides, the synthetic methods for quinoline formaldehyde II always necessitate multiple functional group preinstallations6a–f and the use of toxic and hazardous chemicals such as PBr3,6d acyl chloride6e and concentrated sulfuric acid,6f which result in low overall efficiency and cause numerous environmental concerns. Therefore, the development of a novel and green synthetic route with competitive advantages is highly appealing.
C bonds group, albeit challenged by the generation of significant amounts of phosphine oxide byproduct, leading to low atom economy efficiency. Besides, the synthetic methods for quinoline formaldehyde II always necessitate multiple functional group preinstallations6a–f and the use of toxic and hazardous chemicals such as PBr3,6d acyl chloride6e and concentrated sulfuric acid,6f which result in low overall efficiency and cause numerous environmental concerns. Therefore, the development of a novel and green synthetic route with competitive advantages is highly appealing.
 | ||
| Scheme 1 Background and reaction design of this work. | ||
In recent years, research on essentially solvent-free methodologies for organic synthesis using mechanochemistry7 has gained popularity due to its potential to eliminate or significantly reduce the need for solvent, enhance reaction rates, and offers alternative selectivity. The application of mechanochemical techniques in the synthesis for drug molecule has proven to be a more environmentally friendly and sustainable approach compared to conventional synthesis pathways. Recent advancements in medicinal mechanochemistry have witnessed many successful cases,8 wherein active pharmaceutical ingredients (APIs) are synthesized entirely via mechanochemical methods. These include Phenytoin8d (antiepileptic drug), CEDMH8e/CADMH8f (antiseptic), Glibenclamide8g/Chlorpropamide8g (hypoglycemic drugs), Procainamide8h (anti-arrhythmia agent), Paracetamol8h (antipyretic analgesic), Axitinib8i (anticancer drug) and Pyrimethamine8j (anti osteoporosis drug), illustrating the breadth and potential of this greener methodology. Although most mechanochemical researches have been achieved by grinding in ball mills,9 the shift from small-scale reactions to continuous and scalable mechano-synthesis has attracted great attention of chemists very recently.10 In 2019, alongside flow chemistry, mechanochemical reactive extrusion, promising in sustainability and scalability, was recognized as one of the top 10 chemical innovations by IUPAC.11
As a prevalent form of reactive extrusion, twin-screw extruder (TSE) technology has been widely applied in the preparation of co-crystals, metal organic frameworks (MOFs) and supramolecular porous cages.12 However, in drug synthesis, there have been only a few sporadic studies.13 Building upon our previous works on total mechanosynthesis of pharmaceuticals and drug-like compounds,14 herein we turned our attention to the underexplored scalable mechanochemistry for the green synthesis of PTV.
Results and discussion
The synthetic route depicted in Scheme 1b was chosen for the initial evaluation. Following our proposed pathway, 4-bromoquinoline undergoes a mechanochemical Suzuki–Miyaura coupling reaction with 4-fluorophenylboric acid 2, resulting in the generation of 3a. This intermediate subsequently undergoes a one-pot Minisci C–H alkylation reaction with bromocyclopropane 4, leading to the formation of 5a.Ultimately, the pivotal intermediate 7a is obtained via a mechanochemical oxidative Heck reaction. Scalability experiments are carried out at each stage of the process to assess its industrial feasibility. Additionally, due to the essential role of the cyclopropane structure in pharmaceutical compounds, an extensive examination was conducted on the substrate scope of the Minisci reaction, specifically focusing on the direct alkylation of drug-relevant molecules and natural products.
Synthesis of 3a
The synthesis began with the coupling of commercially available 4-bromoquinoline 1a and fluorophenylboronic acid by classical palladium catalysis, using K3PO4 (3.0 equiv.) as a base and NaCl (0.5 mass equiv.) as the grinding auxiliary in a mixer mill (30 Hz, 30 min). Inspired by the works of Kubota and Ito,15 as well as Browne,16 which indicated that increasing overall collision energy by inputting of supplementary thermal energy can improve the efficiency of coupling reactions, a heat gun was employed to facilitate the reaction, giving the desired product 3a in 75–81% isolated yields (Table 1, entries 1 and 2). Due to the cost-effectiveness and lower toxicity of nickel catalysts compared to palladium catalysts,17 our focus then shifted to nickel-catalysed coupling reactions. Among the tested nickel catalysts, NiCl2(dppp) (10 mol%) provided the best yield of 83% (Table 1, entry 3 vs. entries 6 and 7); however, reducing the catalyst loading hindered the reaction. Notably, a decreased amount of 2 contributed positively to the transformation with the best yield of 93% obtained by prolonging the milling time to 60 min (Table 1, entry 4). Modifying either the type (Table 1, entries 8 and 9) or the quantity of the base (K3PO4) resulted in unfavourable outcomes (Table 1, entry 5 and Table S1†).|
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Base (equiv.) | 3a (%) |
| a Reaction conditions unless specified otherwise: 1a (0.4 mmol), 2 (0.6 mmol), catalyst, base and NaCl (0.5 mass equiv.) were placed in a 15 mL stainless-steel vessel with a stainless-steel ball (dMB = 10 mm), heated by heat-gun, milling in a mixer mill (RETSCH MM 400) for 30 min at 30 Hz. MM = mixer mill. b Isolated yields. c 2 (0.56 mmol). d 2 (0.64 mmol). e LAGs (H2O: 0.08 μL mg−1). f Milling for 60 min. | |||
| 1 | Pd(OAc)2 (10) | K3PO4 (3) | 81 |
| 2 | PdCl2(PPh3)2 (10) | K3PO4 (3) | 75 |
| 3 | NiCl2(dppp) (10/8/6) | K3PO4 (3) | 83/71/56 |
| 4 | NiCl 2 (dppp) (10) | K 3 PO 4 (3) | 86c/80d/93c,f |
| 5 | NiCl2(dppp) (10) | K3PO4 (2/4) | 76/65 |
| 6 | NiCl2(PPh3)2 (10) | K3PO4 (3) | 75 |
| 7 | NiCl2(PCy3)2 (10) | K3PO4 (3) | 82 |
| 8 | NiCl2(dppp) (10) | K2CO3/Na2CO3/Cs2CO3 (3) | 43/56/39 |
| 9 | NiCl2(dppp) (10) | NaHCO3/KF/CsF (3) | 65/43/70 |
| 10 | FeCl3(PPh3)3 (10) | K3PO4 (3) | 29 |
| 11 | FeCl3(dppe)3 (10) | K3PO4 (3) | 32 |
| 12 | FeCl2 (10) | K3PO4 (3) | 33 |
| 13 | FeCl3 (10) | K3PO4/K2CO3 (3) | 50/53(48c) |
| 14 | FeCl 3 (10/8/6) | K 2 CO 3 (3) | 80/65/48 |

Iron, as one of the most abundant metallic elements on earth, has the advantages of being nontoxic, cost-effective, and environmentally and biologically compatible, making it an ideal green catalyst. In the screening process for catalyst utilizing iron salts, it was observed that ligand-free iron catalysts outperformed the coordinated ones (Table 1, entries 10–13). However, a reduction in the amount of 2 led to a decline in the product yield (Table 1, entry 13, and Table S1†), which was contrasted to nickel catalysis. Considering the benefits of liquid assisted grinding technology (LAG) in mechanochemistry,18 where a small amount of liquid can manipulate reactivity, we also applied the LAG protocol in this study (see details in ESI†). The results showcased that the addition of H2O as a liquid additive (LAGs) along with K2CO3 as an alternative base led to a superior yield of up to 80% (Table 1, entry 14). While the yield of iron catalysts may not match that of nickel catalysts, its cost-effectiveness provides an advantage for industrial production. Consequently, further large-scale extrusion studies will investigate the potential industrial applications of both iron and nickel catalyst systems.
Next, we explored the extrusion conditions using a twin-screw extruder (SJZS-7A). A mixture of 1a (36 mmol, 7.50 g), 2 (50.4 mmol, 7.56 g), iron or nickel catalyst (10 mol%), K2CO3 or K3PO4 (3.0 equiv.), with or without NaCl and with or without of H2O (0.08 μL mg−1) was manually introduced into the extruder through the feeding port (with a feed rate of 2.63 g min−1). The extrusion process was performed at a screw rotation rate of 18 rpm and with three-stage heating temperatures (Fig. 1). Following an optimization study of this three-stage heating protocol, it was determined that the nickel catalytic reaction exhibited the best transformation at temperatures of 65 °C, 75 °C, 80 °C, while the iron catalytic reaction required higher temperatures (80 °C, 90 °C, 80 °C) to achieve optimal results. Subsequent experiments revealed that lower screw speeds of 18 and 15 rpm were more effective compared to 25 rpm. This improvement in efficiency at lower speeds can be attributed to the longer residence time [Ni cat. (from 11 min to 15 min); Fe cat. (from 13 min to 15 min)] of the reactants within the extruder, which facilitates enhanced conversion of the reactants. Ultimately, the highest Space-Time Yield (STY) values were determined for the Ni catalyst and Fe catalyst, respectively. The Ni catalyst achieved an STY of 2.78 × 103 kg per m3 per day (E-factor 5.55, see details in ESI†), while the Fe catalyst demonstrated an STY of 2.13 × 103 kg per m3 per day. It should be noted that approximately 18 g of extrudate (out of the total 40 g blend loaded) was processed before the system reached a steady state. Losses in continuous processes are a frequent occurrence, with their significance diminishing notably as material inputs scale up.19
 | ||
| Fig. 1 Extrusion optimization for Suzuki–Miyaura coupling.a a Condition A: 1a (36 mmol), 2 (50.4 mmol), NiCl2(dppp) (10 mol%) and K3PO4 (3.0 equiv.) were reacted in a twin-screw extruder (SJZS-7A). Screw speed = 18 rpm, feed rate: 2.63 g min−1, temperature as specified. STY = total product mass (kg)/(reactant volume (m3) × time (day). b Condition B: 1a (36 mmol), 2 (50.4 mmol), FeCl3 (10 mol%), K2CO3 (3.0 equiv.), LAGs (H2O: 0.08 μL mg−1) and NaCl (10.00 g) were reacted in a twin-screw extruder (SJZS-7A). Screw speed = 18 rpm, feed rate: 2.63 g min−1, temperature as specified. STY = total product mass (kg)/reactant volume (m3) × time (day). | ||
Synthesis of 5a
Our recent study demonstrated the generation of alkyl radicals through the mechanochemical interaction of alkyl halides with freshly prepared magnesium surfaces, enabling the Minisci-type alkylation of pyrimidine14c and pyridine8 derivatives. Consequently, we envisage employing this methodology for the synthesis of PTV, through the C–H alkylation reaction of p-fluorophenylquinoline 3a with bromocyclopropane 4. The investigation commenced by employing these two substrates, resulting in an 18% yield of 5a with the presence of 2 equiv. of magnesium along with Na2SO4 (1.0 g) as the grinding auxiliary (Table 2, entry 2). Control experiments confirmed the necessity of magnesium for the reaction (Table 2, entry 1), demonstrating the significant impact of its quantity on the product yield (Table 2, entry 2). Other grinding auxiliary gave inferior results (Table 2, entry 3) and the experiments indicated the generation of a certain amount of defluorinated by-product alongside the desired product. In an effort to reduce these by-products, potassium fluoride (KF) was introduced as a grinding auxiliary. However, contrary to expectations, KF not only failed to inhibit the formation of by-products but also decreased the yield of the desired product notably (Table 2, entries 3 and 4). Interestingly, optimizing the amount of grinding auxiliary Na2SO4 (0.9 g) led to a substantial increase in the product yield up to 64%, especially when the amount of bromocyclopropane 4 is reduced from 5 to 4 equivalents (Table 2, entries 5 and 6; Tables S5 and S6†), however further decrease its amount did not give any help for this transformation (Table S5,† entry 9). To further demonstrate the efficiency and synthetic utility of this methodology (Scheme 2), a large-scale experiment (8 mmol) was conducted involving the reaction of 3a and 4 in a 50 mL stainless-steel ball-milling jar equipped with two stainless-steel balls (dMB = 14 mm). This procedure successfully produced the desired compound 5a with a satisfactory yield (51%, E-factor 2.92/15.65, see details in ESI†). The cyclopropyl motif plays a crucial role in bioactive molecules and natural products.20 Research indicates that incorporating a cyclopropane fragment enhances drug properties such as lipid solubility, specificity, and resistance to metabolic degradation.21 Given the importance of the cyclopropyl motif in medicinal chemistry, this study further investigates the substrate scope and adaptability of heteroarenes as potential drug structural components under the optimal conditions (Fig. 2). A range of 4-arylquinolines with both electron-donating and electron-withdrawing substituents at different positions on the phenyl ring successfully underwent reactions with bromocyclopropane 4, giving the desired products 5a–5f in 48–51% yields. 4-Methylquinoline (3g) and 4-bromoquinoline (3h) also served as compatible substrates, albeit resulting in relatively lower yields. Encouragingly, the reaction scope extended to other valuable heteroarenes such as 4-methoxypyridine (4i), 4-tert-butylpyridine (4j), 4-methylpyrimidine (4k), benzothiazole (4l), and the challenging N-methyl-1H-indazole (4m).22 Notably, the potential of this method in the late-stage modification of drug-relevant molecules and natural products was demonstrated by utilizing caffeine, theophylline and a clioquinol derivative as substrates, resulting in products 5n–5p with synthetically acceptable yields.|
|
|||
|---|---|---|---|
| Entry | Mg chip (equiv.) | Grinding auxiliary (g) | 5a (%) |
| a Reaction conditions: 3a (0.2 mmol), 4 (1.0 mmol), Mg chip (3.0 equiv.) and Na2SO4 (1.0 g) were placed in a 15 mL stainless-steel vessel with a stainless-steel ball (dMB = 12 mm), milling in a mixer mill (RETSCH MM 400) for [3 × (30 min + 2 min break)] at 30 Hz. MM = mixer mill. b Isolated yields. c 4 (0.8 mmol), a stainless-steel ball (dMB = 14 mm). | |||
| 1 | — | Na2SO4 (1.0) | n.d. |
| 2 | 2.0/3.0/4.0 | Na2SO4 (1.0) | 18/43/37 |
| 3 | 3.0 | NaCl/silica gel/KF (1.0) | n.d./n.d./15 |
| 4 | 3.0 | KF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Na2SO4 = 3:7/2:8 (1.0) :Na2SO4 = 3:7/2:8 (1.0) |
28/32 |
| 5 | 3.0 | Na2SO4 (0.8/0.9) | 45/53 |
| 6 | 3.0 | Na 2 SO 4 (0.9) | 64 |

 | ||
| Scheme 2 Gram-scale for the Minisci C–H alkylation reaction.a a Reaction conditions: 3a (8 mmol), 4 (16 mmol), Mg chip (3.0 equiv.) and Na2SO4 (14 g) were placed in a 50 mL stainless-steel vessel with two stainless-steel balls (dMB = 14 mm), milling in a mixer mill (RETSCH MM 400) for [3 × (30 min + 2 min break)] at 30 Hz. MM = mixer mill. b Without grinding auxiliary. | ||
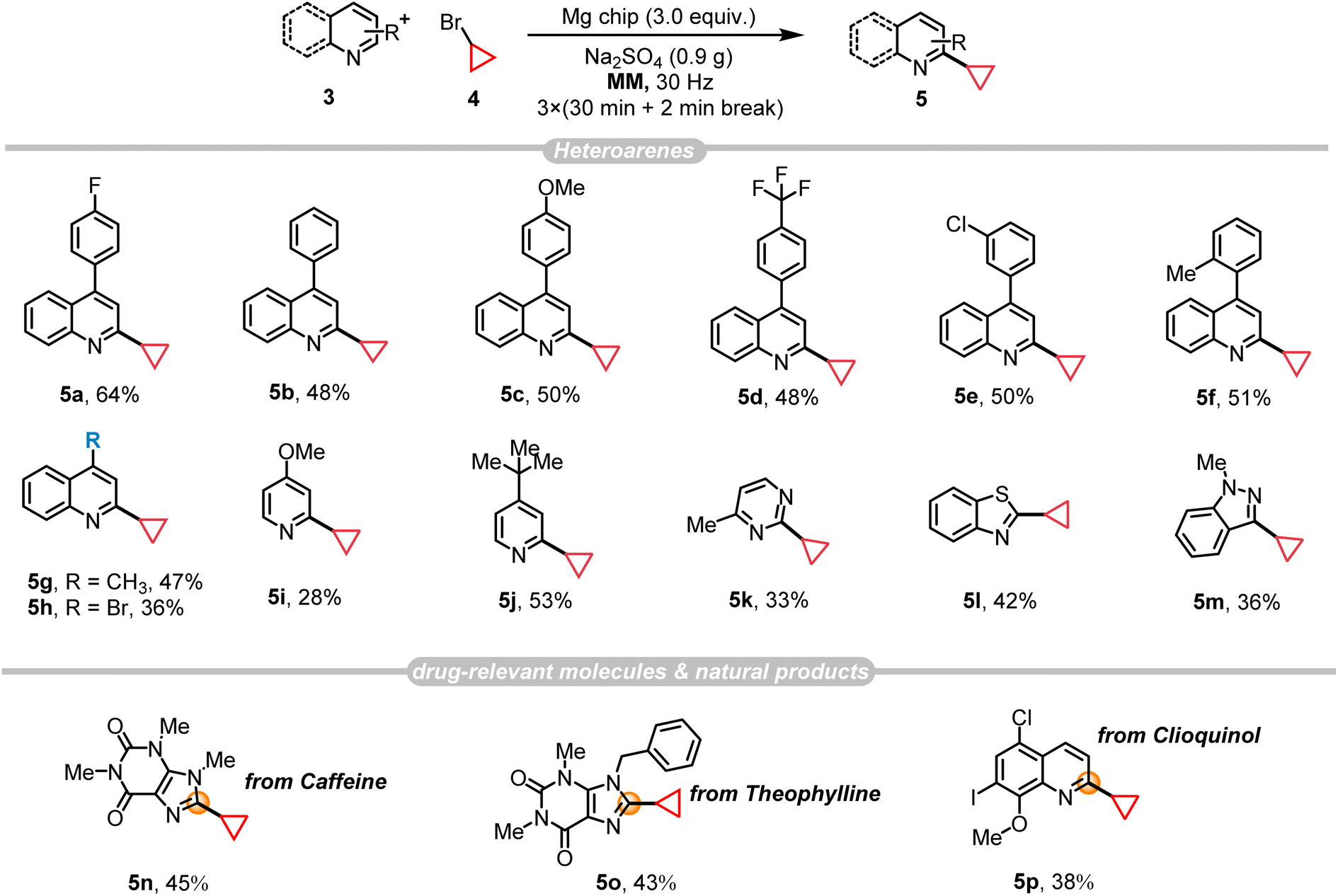 | ||
| Fig. 2 Substrate scope for Minisci C–H alkylation reaction.a a Reaction conditions: 3 (0.2 mmol), 4 (0.8 mmol), Mg chip (3.0 equiv.) and Na2SO4 (0.9 g) were placed in a 15 mL stainless-steel vessel with a stainless-steel ball (dMB = 14 mm), milling in a mixer mill (RETSCH MM 400) for [3 × (30 min + 2 min break)] at 30 Hz. MM = mixer mill. Isolated yield. | ||
Synthesis of the key intermediate 7a
The final step involves the conversion of 5a to 3-(2-cyclopropyl-4-(4-fluorophenyl)quinolin-3-yl)acrylaldehyde 7a, which is the key intermediate in the synthesis of PTV (Table 3, see details in Tables S9–S11†). Initially, this transformation was carried out using Pd(OAc)2 (10 mol%) as the catalyst, 1,10-phenanthroline (13 mol%) as the ligand, Ag2CO3 (1.0 equiv.) as the oxidant, DMF (0.12 μL mg−1) as LAGs, and Na2SO4 (0.5 g) as the grinding auxiliary in a mixer mill (RETSCH MM 400) equipped with a heat gun operating at 30 Hz for (2 × 30) min, offering the desired product in 39% yield (Table 3, entry 1). The investigation of alternative palladium catalysts (Pd[O2C(CH3)3]2, Pd(acac)2, and PdCl2(PPh3)2) or silver oxidants (Ag2CO3, Ag2O, CF3SO2OAg, AgBF4, AgOAc, Ag2CO3:Cu(OAc)2 = 1:1, and Ag2CO3:CuCO3 = 1:1) revealed a significant reduction in yield (Table 3, entries 2–4). Moreover, altering the catalyst loading (from 10% to 8 mol%) and stoichiometric amount of oxidant (from 1.0 equiv. to 1.5 equiv.) gave comparative results (Table 3, entry 5). Further experiments demonstrated that 1,10-phenanthroline as the ligand exhibited superior performance compared to other ligands (Table 3, entry 6). Switching the grinding auxiliary from Na2SO4 to silica gel/NaHCO3/Na2CO3, or changing the LAGs from DMF to MeCN/EtOAc/DMSO/hexane all led to diminished yields (Table 3, entries 7 and 8). The optimal quantities for these two additives were found to be 0.5 g and 0.12 μL mg−1, respectively (Table 3, entry 9 and Table S10†). Lastly, adjusting the substrate ratios to 1:1/1:2 did not improve the formation of the coupling product (Table 3, entry 10).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) |
| a Reaction conditions: 5a (0.4 mmol), 6 (0.6 mmol), Pd(OAc)2 (10 mol%), 1,10-phenanthroline (13 mol%), Ag2CO3 (1.0 equiv.) DMF (0.12 μL mg−1) and Na2SO4 (0.5 g) were placed in a stainless-steel vessel (15 mL) with a stainless-steel ball (dMB = 10 mm), milling in a mixer mill (RETSCH MM 400) at 30 Hz for (2 × 30) min. b Isolated yields. MM = mixer mill. n. d. = not detected. CEMTPP = ethoxycarbonylmethylene triphenylphosphorane. DPPE = 1,2-bis(diphenylphosphine)ethane. | ||
| 1 | None | 43 |
| 2 | Pd[O2C(CH3)3]2/Pd(acac)2/PdCl2(PPh3)2 instead of Pd(OAc)2 | 24/27/30 |
| 3 | Ag2CO3/Ag2O/CF3SO2OAg/AgBF4/AgOAc instead of Ag2CO3 | 23/12/18/n.d./n.d. |
| 4 | Ag2CO3:Cu(OAc)2 (1:1)/Ag2CO3:CuCO3 (1:1) instead of Ag2CO3 |
27/trace |
| 5 | 8 mol% instead of 10 mol% of Pd(OAc)2/1.5 eq. instead of 1.0 eq. of Ag2CO3 | 35/34 |
| 6 | CEMTPP/DPPE instead of 1,10-phenanthroline | 32/31 |
| 7 | Silica gel/NaHCO3/Na2CO3 instead of Na2SO4 | 32/n.d./trace |
| 8 | MeCN/EtOAc/DMSO/hexane instead of DMF | 23/18/25/22 |
| 9 | 0.10/0.08 μL mg−1 of DMF | 34/28 |
| 10 | 1.0 eq./2.0 eq. of 6 | 28/34 |
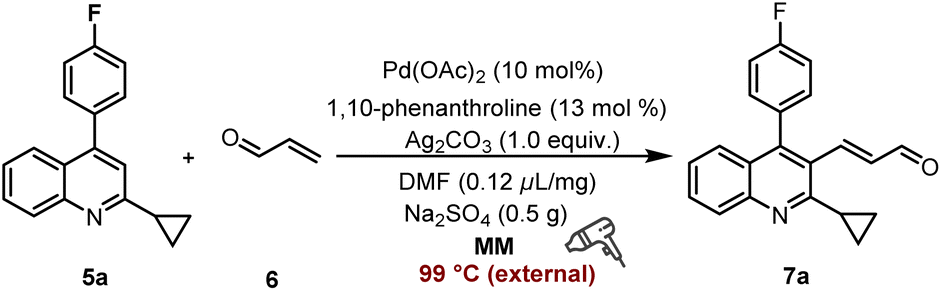
After establishing the optimal conditions through mixer milling tests, our study advanced to investigate the extrusion parameters suitable for large-scale reactions using a twin-screw extruder (SJZS-7A) (Fig. 3). Scaling our methodology up by 80-fold to a 32 mmol scale of quinoline starting material 5a involved manual mixing of all the reactants (5a, 16 mmol 6, and 32 mmol Ag3CO3), catalyst (3.2 mmol Pd(OAc)2/4.2 mmol 1,10-phenanthroline), and additives (20 g Na2SO4 and 0.12 μL mg−1 DMF) with a spatula, then feeding them into the extruder through a hopper over 15 minutes. The initial investigation was started by adjusting the three-stage heating temperatures to establish steady-state conditions ensuring the mass neither too wet nor too dry. In this context, our research revealed that the optimal temperature was determined to be 90 °C, 105 °C, and 80 °C, respectively (Table S12†). Subsequent analysis focused on the impact of reducing the screw speed from 25 to 18 rpm for mixtures processed at 90°C–105°C–80 °C, consequently increasing the residence time (from 15 min to 18 min) in the extruder barrel. Thereby, an optimal Space-Time Yield (STY) of 1.32 × 103 kg per m−3 per day was achieved (E-factor 5.96/11.27, see details in ESI†).
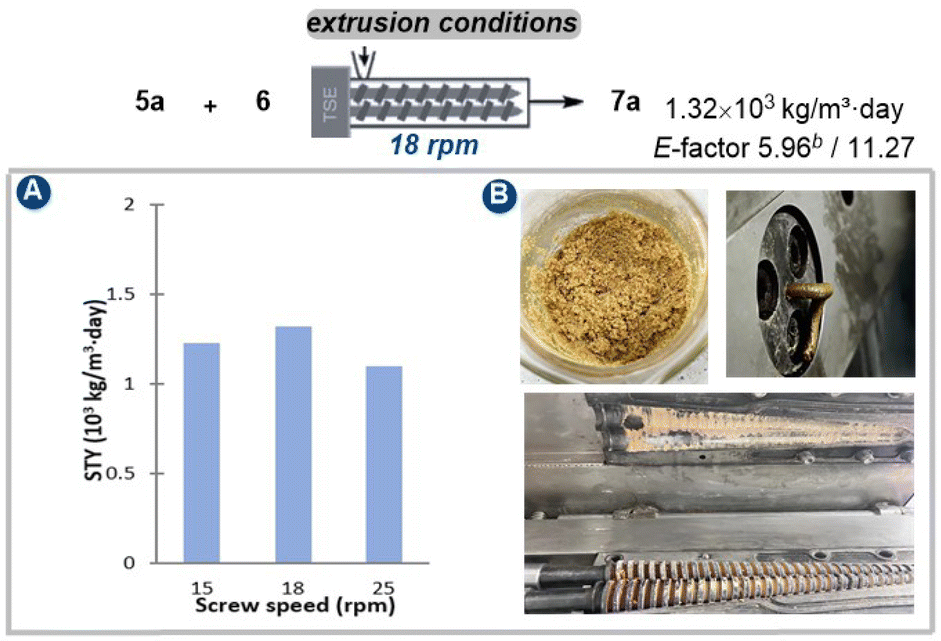 | ||
| Fig. 3 Extrusion optimization for oxidative Heck reaction conditions.a a Reaction conditions: 5a (32 mmol), 6a (16 mmol), Pd(OAc)2 (10 mol%), 1,10-phenanthroline (13 mol%), Ag2CO3 (1.0 equiv.), DME (0.12 μL mg−1) and Na2SO4 (20 g) were reacted in a twin-screw extruder (SJZS-7A). Screw speed = 18 rpm, feed rate: 2.63 g min−1, temperature as specified. STY = total product mass (kg)/reactant volume (m3) × time (day). | ||
A comprehensive comparison was then carried out between the total mechanosynthesis methodology and the conventional route for the preparation of the pivotal intermediate 7a of pitavastatin (Table 4, see details in Fig. S1–S4†). The findings unmistakably established that the total mechano-synthesis strategy introduced herein outperformed the traditional process in both time efficiency and procedural economy, necessitating the fewest steps. Furthermore, the environmental factor (E-factor) was meticulously computed for every individual step within the ESI.† These computations revealed that three stages within the total mechano-synthesis displayed relatively low E-factor values, ranging from 2.92 to 15.65.
| Method | Steps | Time (h) | Total yield (%) |
|---|---|---|---|
| a The yield derived from small-scale experimental studies, wherein the Suzuki–Miyaura cross-coupling reaction is catalyzed by utilizing nickel metal. | |||
| Mechanochemical method | 3 | 10.8 | 25.6a |
| Traditional method 1 | 7 | 51.0 | 17.0 |
| Traditional method 2 | 6 | 68.5 | 17.7 |
| Traditional method 3 | 6 | 202.0 | 15.5 |
Conclusion
In summary, we have successfully developed a novel, environmentally friendly, and industrially promising mechanochemical route for the green synthesis of the key intermediate 7a of pitavastatin (PTV), overcoming environmental issues associated with conventional methods. Following meticulous optimization, the synthesis was accomplished in a streamlined three-step processes (Suzuki–Miyaura coupling, Minisci C–H alkylation, and oxidation Heck coupling). Significantly, two of these steps were effectively upscaled utilizing continuous-flow extrusion technology, with the second step demonstrating its scalability at the gram-scale level. Moreover, given the substantial therapeutic significance of cyclopropyl moieties, we probed the versatility of the mechanochemical Minisci methodology, revealing its promising potential for the targeted cyclopropylation of active pharmaceutical compounds. The developed process underscores the untapped potential of mechanochemistry in drug synthesis, particularly when integrated into continuous flow systems that offer shortened production times, fewer procedural steps, solvent-free operations, and enhanced product yields. Consequently, this advancement contributes to a more environmentally sustainable and resource-efficient synthesis paradigm, aligning with contemporary imperatives for greener pharmaceutical synthesis.Author contributions
JB suggested the idea, YH and ZH performed the experiments and processed the data. WK and JB supervised the project. Writing – review, and editing were conducted by YH and JB. All authors have read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Zhejiang Provincial Natural Science Foundation of China (No. LY23B060005), the National Natural Science Foundation of China (No. 21978270), and National Key R&D Program of China 2021YFC2101000 for financial support.References
- A. Sahebkar, N. Kiaie, A. M. Gorabi, M. R. Mannarino, V. Bianconi, T. Jamialahmadi, M. Pirro and M. Banach, Prog. Lipid Res., 2021, 84, 101127 CrossRef CAS PubMed.
- A. Ramadan and A. Elnour, J. Pharm. BioAllied Sci., 2022, 14, 72–80 CrossRef CAS.
- K. Tajiri, N. Shimojo, S. Sakai, T. Machino-Ohtsuka, K. Imanaka-Yoshida, M. Hiroe, Y. Tsujimura, T. Kimura, A. Sato, Y. Yasutomi and K. Aonuma, Cardiovasc, Drugs Ther., 2013, 27(5), 413–424 CAS.
- (a) Y. N. Fuiikawa and M. N. Suzuki, EP Pat., 0304063, 1989; (b) Z. Časar, Curr. Org. Chem., 2010, 14, 816–845 CrossRef; (c) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- (a) Y. Ohara and M. Suzuki, World Pat., 2000005213A1, 2000; (b) Y. Ohara, M. Suzuki and Y. Yanagawa, EP Pat., 0520406A1, 1992.
- (a) Y. Wan and X. Hu, Org. Lett., 2022, 24, 5797–5801 CrossRef CAS PubMed; (b) C. Counceller, C. Eichman, B. Wray and J. Stambuli, Org. Lett., 2008, 10, 1021–1023 CrossRef CAS PubMed; (c) P. Li, L. Yu, X. Zhang and M. Shi, Org. Lett., 2018, 20, 4516–4520 CrossRef CAS PubMed; (d) Z. Chasar, D. Cituock and M. Jukic, CN Pat., 103119022B, 2016; (e) C. M. Counceller, C. C. Eichman, B. C. Wray and J. P. Stambuli, Org. Lett., 2008, 10, 1021–1023 CrossRef CAS PubMed; (f) Z. Li and S. Li, CN Pat., 104496898A, 2015.
- For selected reviews: (a) G.-W. Wang, Chem. Sci. Rev., 2013, 42, 7668–7700 RSC; (b) S.-E. Zhu, F. Li and G.-W. Wang, Chem. Sci. Rev., 2013, 42, 7535–7570 RSC; (c) J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed; (d) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (e) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed; (f) F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e202200362 CrossRef CAS PubMed; (g) V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Nat. Rev. Chem., 2023, 7, 51–65 CrossRef CAS PubMed; (h) J. Reynes, V. Isoni and F. García, Angew. Chem., Int. Ed., 2023, 62, e202300819 CrossRef CAS PubMed; (i) C. G. Avila-Ortiz and E. Juaristi, Molecules, 2020, 25, 3579 CrossRef CAS PubMed; (j) E. Juaristi and C. G. Ávila Ortiz, Synthesis, 2023, 55, 2439–2459 CrossRef CAS.
- For selected reviews: (a) X. Yang, C. Wu, W. Su and J. Yu, Eur. J. Org Chem., 2022, e202101440 CrossRef CAS; (b) F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e202200362 CrossRef CAS PubMed; (c) O. Bento, F. Luttringer, T. Mohy El Dine, N. Pétry, X. Bantreil and F. Lamaty, Eur. J. Org Chem., 2022, e202101516 CrossRef CAS ; for selected examples; (d) M. Pérez-Venegas and E. Juaristi, ACS Sustainable Chem. Eng., 2020, 8, 8881–8893 CrossRef; (e) L. Konnert, B. Reneaud, R. M. de Figueiredo, J.-M. Campagne, F. Lamaty, J. Martinez and E. Colacino, J. Org. Chem., 2014, 79, 10132–10142 CrossRef CAS PubMed; (f) L. Konnert, M. Dimassi, L. Gonnet, F. Lamaty, J. Martinez and E. Colacino, RSC Adv., 2016, 6, 36978–36986 RSC; (g) A. Porcheddu, F. Delogu, L. De Luca and E. Colacino, ACS Sustain. Chem. Eng., 2019, 7, 12044–12051 CAS; (h) D. Tan, V. Štrukil, C. Mottillo and T. Friščić, Chem. Commun., 2014, 50, 5248–5250 RSC; (i) T. Portada, D. Margetić and V. Štrukil, Molecules, 2018, 23, 3163 CrossRef PubMed; (j) J. Yu, Z. Hong, X. Yang, Y. Jiang, Z. Jiang and W. Su, Beilstein J. Org. Chem., 2018, 14, 786–795 CrossRef CAS PubMed; (k) C. Wu, T. Ying, H. Fan, C. Hu, W. Su and J. Yu, Org. Lett., 2023, 25, 2531–2536 CrossRef CAS PubMed.
- For selected examples: (a) A. Stolle, R. Schmidt and K. Jacob, Faraday Discuss., 2014, 170, 267C286 RSC; (b) J. L. Do, C. Mottillo, D. Tan, V. Strukil and T. Friscic, J. Am. Chem. Soc., 2015, 137, 2476–2479 CrossRef CAS PubMed; (c) V. S. Pfennig, R. C. Villella, J. Nikodemus and C. Bolm, Angew. Chem., Int. Ed., 2022, 61, e202116514 CrossRef CAS PubMed; (d) L. Li, C. Niu and G.-W. Wang, Chin. J. Chem., 2022, 40, 2539–2545 CrossRef CAS; (e) G. Shao, C. Niu, H.-W. Liu, H. Yang, J.-S. Chen, Y.-R. Yao, S. Yang and G.-W. Wang, Org. Lett., 2023, 25, 1229–1234 CrossRef CAS PubMed; (f) R. Takahashi, P. Gao, K. Kubota and H. Ito, Chem. Sci., 2023, 14, 499–505 RSC.
- (a) Q. Cao, J. L. Howard, D. E. Crawford, S. L. James and D. L. Browne, Green Chem., 2018, 20, 4443–4447 RSC; (b) K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed; (c) R. R. A. Bolt, J. A. Leitch, A. C. Jones, W. I. Nicholson and D. L. Browne, Chem. Soc. Rev., 2022, 51, 4243–4260 RSC; (d) R. R. A. Bolt, S. E. Raby-Buck, K. Ingram, J. A. Leitch and D. L. Browne, Angew. Chem., Int. Ed., 2022, 61, e202210508 CrossRef CAS PubMed; (e) Q. Cao, D. E. Crawford, C. Shi and S. L. James, Angew. Chem., Int. Ed., 2020, 59, 4478–4483 CrossRef CAS PubMed.
- F. Gomollón-Bel, Angew. Chem., Int. Ed., 2019, 41, 12–17 Search PubMed.
- (a) D. E. Crawford, Beilstein J. Org. Chem., 2017, 13, 65–75 CrossRef CAS PubMed; (b) D. E. Crawford and J. Casaban, Adv. Mater., 2016, 28, 5747–5754 CrossRef CAS PubMed; (c) D. Crawford, J. Casaban, R. Haydon, N. Giri, T. McNally and S. L. James, Chem. Sci., 2015, 6, 1645–1649 RSC.
- (a) E. Colacino, A. Porcheddu, C. Charnay and F. Delogu, React. Chem. Eng., 2019, 4, 1179–1188 RSC; (b) D. E. Crawford, A. Porcheddu, A. S. McCalmont, F. Delogu, S. L. James and E. Colacino, ACS Sustainable Chem. Eng., 2020, 8, 12230–12238 CrossRef CAS; (c) I. Sović, S. Lukin, E. Meštrović, I. Halasz, A. Porcheddu, F. Delogu, P. C. Ricci, F. Caron, T. Perilli, A. Dogan and E. Colacino, ACS Omega, 2020, 5, 28663–28672 CrossRef PubMed.
- (a) J. Yu, P. Ying, H. Wang, K. Xiang and W. Su, Adv. Synth. Catal., 2020, 362, 893–902 CrossRef CAS; (b) J. Yu, X. Yang, C. Wu and W. Su, J. Org. Chem., 2020, 85, 1009–1102 CrossRef CAS PubMed; (c) C. Wu, T. Ying, X. Yang, W. Su, A. V. Dushkin and J. Yu, Org. Lett., 2021, 23, 6423–6428 CrossRef CAS PubMed; (d) H. Shou, Z. He, G. Peng, W. Su and J. Yu, Org. Biomol. Chem., 2021, 19, 4507–4514 RSC; (e) X. Yang, H. Wang, Y. Zhang, W. Su and J. Yu, Green Chem., 2022, 24, 4557–4565 RSC; (f) K. Xiang, P. Ying, T. Ying, W. Su and J. Yu, Green Chem., 2023, 25, 2853–2862 RSC; (g) J. Yu, H. Chen, Z. Zhang, Y. Fang, T. Ying and W. Su, Green Chem., 2024, 26, 6570–6577 RSC.
- (a) Y. Gao, C. Feng, T. Seo, K. Kubota and H. Ito, Chem. Sci., 2022, 13, 430–438 RSC; (b) R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, S. Maeda, J. Jiang, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed; (c) R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, S. Maeda, J. Jiang, H. Takaya, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2022, 61, e202210508 CrossRef PubMed.
- (a) R. R. A. Bolt, S. E. Raby-Buck, K. Ingram, J. A. Leitch and D. L. Browne, Angew. Chem., Int. Ed., 2022, 61, e202210508 CrossRef CAS PubMed; (b) R. R. A. Bolt, S. E. Raby-Buck, K. Ingram, J. A. Leitch and D. L. Browne, Angew. Chem., 2022, 134, e202210508 CrossRef.
- B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed.
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- R. H. Hastings, M. M. Mokhtar, A. Ruggles, C. Schmidt, D. Bourdeau, M. C. Haibach, H. Geneste, J. Mack, S. S. Co and I. R. Speight, Org. Process Res. Dev., 2023, 27, 1667–1676 CrossRef CAS.
- (a) L.-Q. Sun, F. McPhee and P. M. Scola, J. Med. Chem., 2016, 59, 8042–8060 CrossRef CAS PubMed; (b) N. A. Meanwell, J. Med. Chem., 2016, 59, 7311–7351 CrossRef CAS PubMed.
- (a) L. A. Dawson, Bioorg. Med. Chem. Lett., 2009, 19, 837–840 CrossRef PubMed; (b) Y. Yoshida, Y. Naoe, T. Terauchi, F. Ozaki, T. Doko, A. Takemura, T. Tanaka, K. Sorimachi, C. T. Beuckmann, M. Suzuki, T. Ueno, S. Ozaki and M. Yonaga, J. Med. Chem., 2015, 58, 4648–4664 CrossRef CAS PubMed.
- S. Faarasse, S. El Kazzouli, F. Suzenet and G. Guillaumet, J. Org. Chem., 2018, 83, 12847–12854 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, optimization data, environmental factor (E-factor) calculation, characterization data and NMR spectra. See DOI: https://doi.org/10.1039/d4mr00036f |
| This journal is © The Royal Society of Chemistry 2024 |