
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base-mediated trimerization of enones under solvent-free and ball-milling conditions†
Gang
Shao
a,
Pinhua
Li
 ab,
Zheng-Chun
Yin
a,
Jun-Shen
Chen
a,
Xu-Ling
Xia
a and
Guan-Wu
Wang
*ac
ab,
Zheng-Chun
Yin
a,
Jun-Shen
Chen
a,
Xu-Ling
Xia
a and
Guan-Wu
Wang
*ac
aHefei National Research Center for Physical Sciences at Microscale and Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: gwang@ustc.edu.cn
bDepartment of Chemistry, Anhui Polytechnic University, Wuhu, Anhui 241000, P. R. China
cState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, Gansu 730000, P. R. China
First published on 29th January 2024
Abstract
An efficient mechanochemical trimerization of enones with KOtBu as the base and water as the proton source under solvent-free and ambient conditions has been developed. This protocol provides novel, simple, rapid and scalable access to 1,3,5-triaryl-2,4-acyl-cyclohexanols, which exist as chair conformations with all bulky substituents located at equatorial positions. In addition, the formed cyclohexanol derivatives can be further dehydrated to afford the corresponding cyclohexene derivatives with β,γ-unsaturation. By changing the type or amount of the employed base, another type of stereoisomer, where the 4-acyl group is situated at the axial position, can be favorably generated as the major product.
Mechanochemical organic transformations have drawn increasing attention over the past few decades.1 Apart from providing efficient and ambient organic processes in modern synthetic chemistry, mechanochemical reactions can even alter the chemical selectivity, providing unexpected products that cannot be generated by their solution-based counterparts.2
Over the past two decades, enones have been frequently applied in the area of mechanochemistry, such as Michael additions,3 aminohalogenations,4 Mn(OAc)3-mediated radical reactions5 and cascade reactions.6 Recently, the trimerization of enones was realized using N-heterocyclic carbene (NHC)/NaOtBu/O2 in ether for 5–96 h via an initial single-electron-transfer (SET) step, subsequent reaction with O2, radical coupling and homolytic O–O bond breaking to generate the key alkoxyl radical, followed by reactions with two other enones through multiple steps (Scheme 1a).7 This reaction required the combination of NHC and NaOtBu to generate radical intermediates from enones for further reactions. In continuation of our interest in mechanochemistry,8 we herein disclose our finding that enones can undergo mechanochemical solvent-free trimerization with KOtBu as the base and water as the proton source through a cascade process of the retro-aldol reaction of enones, followed by double Michael additions and a final intramolecular aldol reaction (Scheme 1b).
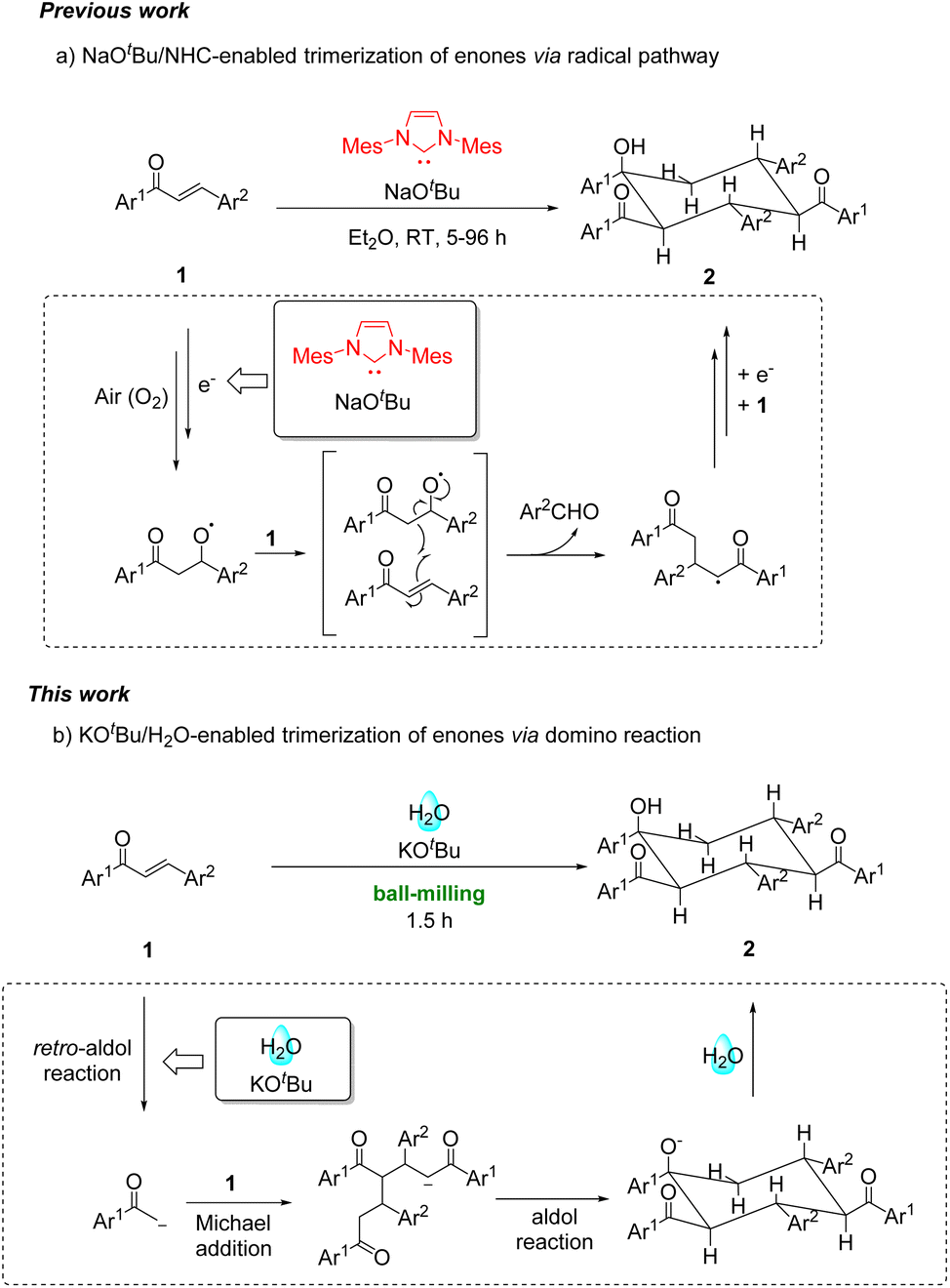 | ||
| Scheme 1 Strategies for the trimerization of enones. | ||
Chalcone 1a was chosen as the model substrate to determine the optimal reaction conditions. Initially, 0.20 mmol of 1a and 2 equiv. of KOtBu were added into a stainless steel jar (5 mL) together with 4 stainless steel balls (5 mm in diameter) under solvent-free and ambient conditions and mixed at a milling frequency of 1800 rounds per minute (rpm) in a Retsch MM 400 mixer mill at room temperature for 1.5 h. Unfortunately, 2,4-dibenzoyl-1,3,5-triphenylcyclohexanol (2a) could be observed only in a trace amount, together with its stereoisomer 2a′ in 34% yield (Table 1, entry 1). The stereochemistry of 2a was determined from its NMR spectra7 and unambiguously established by single-crystal X-ray crystallography (Fig. 1), while the stereochemistry of 2a′ was confirmed by comparison of its NMR data with those reported in the previous literature.9 The single-crystal structure of 2a clearly showed that all bulky phenyl and benzoyl groups were located at equatorial positions in the chair conformation. In contrast, most bulky substituents of 2a′ were situated at equatorial sites except for the axial 4-benzoyl group. To our satisfaction, the outcome was dramatically changed when 2 equiv. of H2O was added as the proton source, and 2a was obtained selectively and efficiently in 86% yield (Table 1, entry 2). These results indicated that H2O was crucial in promoting this reaction. With the increase in the amount of H2O to 3 equiv., the product yield was enhanced to 95%, yet further increasing the amount of H2O to 4 equiv. did not improve the efficiency of the reaction (Table 1, entries 3 and 4). In addition, no benefit to the yield of 2a could be achieved by increasing the amount of KOtBu from 2 equiv. to 3 equiv. (Table 1, entry 5). Reducing the amount of KOtBu to 1 equiv. gave 2a in only 21% yield, yet its stereoisomer 2a′ could be favorably obtained in 58% yield (Table 1, entry 6). Reducing the reaction time from 1.5 h to 1 h resulted in a lower product yield, while increasing the reaction time to 2 h led to a similar yield (Table 1, entries 7 and 8 vs. entry 3). Product 2a could also be efficiently obtained by replacing KOtBu with KOH or NaOH in the presence of 18-crown-6 or 15-crown-5, respectively (Table 1, entries 9 and 10). Thus, the combination of alkali hydroxide with a crown ether was an alternative way to obtain 2a efficiently under mechanochemical conditions. However, when KOH or NaOH was used alone, 2a′ became the major product, and 2a was the minor product (Table 1, entries 11 and 12). These results showed that stronger bases (KOtBu) under mechanochemical conditions resulted in more efficient formation of product 2a, while relatively weaker bases (KOH or NaOH alone) favored the generation of its stereoisomer 2a′. In the solid state, the different tightness of the arrangement between the cations and anions of bases might lead to different effective sizes, resulting in different reactivities of the anions. The utilization of a double-component solid base system9 consisting of NaOH and K2CO3 in a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 led to a similar result to that of using NaOH only, and no reaction occurred when K2CO3 was employed alone (Table 1, entries 13 and 14). Other inorganic and organic bases, such as Cs2CO3, KOAc and Et3N, were also examined. Nevertheless, all these bases were ineffective in this reaction (Table 1, entries 15–17). The above experimental results showed that the type and amount of the employed base exhibited strong influences on the reaction efficiency, stereoselectivity and product distribution. The exact reason remains to be clarified. To compare the present solvent-free reaction with its solution-based counterpart, the reaction of 1a (0.20 mmol) with KOtBu (0.40 mmol) and H2O (0.60 mmol) performed in 1.5 mL of diethyl ether (Et2O) at 25 °C for 48 h provided 2a (22%), 1,3,5-tribenzoyl-2,4,6-triphenylcyclohexane7 (3a, 27%) and other unidentified byproducts, showing much lower selectivity (Table 1, entry 18). Other solvents, including toluene, dimethyl sulfoxide (DMSO), tetrahydrofuran (THF) and acetonitrile (CH3CN), were also screened. However, all of them led to lower product yields. Thus, it is obvious that the present mechanochemical solvent-free protocol shows advantages and uniqueness compared to the corresponding liquid-phase reaction in terms of product yield, reaction time and selectivity, likely due to more intimate and efficient intermolecular interactions of the “highly concentrated” reagents and/or absence of the solvation effect under the current ball-milling conditions. On the basis of the above results, the optimized conditions were determined as follows: 1a (0.20 mmol), KOtBu (0.40 mmol), H2O (0.60 mmol), a milling frequency of 1800 rpm and a milling time of 1.5 h at room temperature.
:1 led to a similar result to that of using NaOH only, and no reaction occurred when K2CO3 was employed alone (Table 1, entries 13 and 14). Other inorganic and organic bases, such as Cs2CO3, KOAc and Et3N, were also examined. Nevertheless, all these bases were ineffective in this reaction (Table 1, entries 15–17). The above experimental results showed that the type and amount of the employed base exhibited strong influences on the reaction efficiency, stereoselectivity and product distribution. The exact reason remains to be clarified. To compare the present solvent-free reaction with its solution-based counterpart, the reaction of 1a (0.20 mmol) with KOtBu (0.40 mmol) and H2O (0.60 mmol) performed in 1.5 mL of diethyl ether (Et2O) at 25 °C for 48 h provided 2a (22%), 1,3,5-tribenzoyl-2,4,6-triphenylcyclohexane7 (3a, 27%) and other unidentified byproducts, showing much lower selectivity (Table 1, entry 18). Other solvents, including toluene, dimethyl sulfoxide (DMSO), tetrahydrofuran (THF) and acetonitrile (CH3CN), were also screened. However, all of them led to lower product yields. Thus, it is obvious that the present mechanochemical solvent-free protocol shows advantages and uniqueness compared to the corresponding liquid-phase reaction in terms of product yield, reaction time and selectivity, likely due to more intimate and efficient intermolecular interactions of the “highly concentrated” reagents and/or absence of the solvation effect under the current ball-milling conditions. On the basis of the above results, the optimized conditions were determined as follows: 1a (0.20 mmol), KOtBu (0.40 mmol), H2O (0.60 mmol), a milling frequency of 1800 rpm and a milling time of 1.5 h at room temperature.
|
|
||||
|---|---|---|---|---|
| Entry | Base | Molar ratiob | Yield of 2a (%) | Yield of 2a′ (%) |
| a Unless otherwise noted, all reactions were performed with 0.20 mmol of chalcone, KOtBu and H2O together with 4 stainless steel balls (5 mm in diameter) in a stainless steel jar (5 mL) and milled vigorously (1800 rpm) in a Retsch MM 400 mixer mill at room temperature for 1.5 h. b Molar ratio referred to chalcone/KOtBu/H2O. c The reaction time was 1 h. d The reaction time was 2 h. e 0.20 mmol of 18-crown-6 was added as the additive. f 0.20 mmol of 15-crown-5 was added as the additive. g Molar ratio referred to chalcone/NaOH/K2CO3/H2O. h The reaction was performed in Et2O at 25 °C for 48 h. | ||||
| 1 | KOtBu | 1:2:0 |
Trace | 34 |
| 2 | KOtBu | 1:2:2 |
86 | Trace |
| 3 | KO t Bu |
1:2:3 |
95 | Trace |
| 4 | KOtBu | 1:2:4 |
89 | Trace |
| 5 | KOtBu | 1:3:3 |
82 | Trace |
| 6 | KOtBu | 1:1:3 |
21 | 58 |
| 7c | KOtBu | 1:2:3 |
87 | Trace |
| 8d | KOtBu | 1:2:3 |
96 | Trace |
| 9e | KOH | 1:2:3 |
90 | Trace |
| 10f | NaOH | 1:2:3 |
87 | Trace |
| 11 | KOH | 1:2:3 |
13 | 41 |
| 12 | NaOH | 1:2:3 |
16 | 37 |
| 13 | NaOH/K2CO3 | 1:2:1:3g |
11 | 34 |
| 14 | K2CO3 | 1:2:3 |
n.r. | n.r. |
| 15 | Cs2CO3 | 1:2:3 |
n.r. | n.r. |
| 16 | KOAc | 1:2:3 |
n.r. | n.r. |
| 17 | Et3N | 1:2:3 |
n.r. | n.r. |
| 18h | KOtBu | 1:2:3 |
22 | 0 |
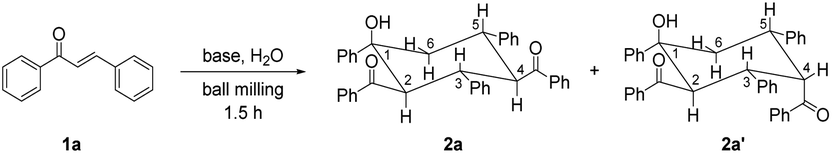
 | ||
| Fig. 1 ORTEP diagram for one enantiomer of racemic 2a with 15% thermal ellipsoids. The solvent molecule CHCl3 is omitted for clarity. | ||
With the optimized reaction conditions in hand, we proceeded to study the substrate scope of the trimerization of enones to provide 1,3,5-triaryl-2,4-acyl-cyclohexanols (Scheme 2). Enones with different functional groups on the aromatic substituent (Ar1) next to the carbonyl group and the β-aryl substituent (Ar2) as the same phenyl group (1b–1g) were investigated. Chalcones with Ar1 bearing electron-donating (p-OMe and m-OMe) groups afforded trimers 2b and 2c in yields of 86% and 47%, respectively. Enones with Ar1 containing electron-withdrawing (p-Cl, m-Cl and p-Br) groups proceeded well and provided products 2d–2f in 71–90% yields. Furthermore, the enone with Ar1 as a heteroaryl (2-thienyl) group was also suitable and gave product 2g in 80% yield. We then studied chalcones with Ar1 as the same phenyl group and different Ar2 substituents (1h–1n) and found that the reactions proceeded smoothly to afford trimers 2h–2n in moderate to good yields. Chalcones with Ar2 bearing electron-donating groups (p-OMe and p-Me) afforded products 2h and 2i in 87% and 63% yields, respectively. Substrates with Ar2 containing electron-withdrawing groups (p-Cl, p-Br and m-Br) underwent trimerization and generated cyclohexanols 2j–2l in 79–88% yields. In addition, chalcones with Ar2 as the naphthyl or heteroaryl (2-furyl) group were compatible and provided products 2m and 2n in 45% and 63% yields, respectively. Alternatively, enones with both Ar1 and Ar2 carrying an electron-donating p-Me or p-OMe group gave products 2o and 2p in 76% and 75% yields, respectively. Substrates with both Ar1 and Ar2 containing an electron-withdrawing p-Cl or p-Br group could be used and converted into trimers 2q and 2r in 63% and 72% yields, respectively. Finally, enones with Ar1 and Ar2 as a combination of an electron-donating group and an electron-withdrawing group were examined, and products 2s and 2t could be obtained in 89% and 73% yields, respectively.
 | ||
| Scheme 2 Results for the mechanochemical trimerization of enones 1a–1t mediated by KOtBu. All reactions were performed with 0.20 mmol of 1, KOtBu (2 equiv.) and H2O (3 equiv.) together with 4 stainless steel balls (5 mm in diameter) in a stainless steel jar (5 mL) and milled (1800 rpm) at room temperature for 1.5 h. | ||
To further evaluate the practicability of the present mechanochemical protocol, a scale-up trimerization of chalcone 1a (1 mmol) was carried out under our optimal reaction conditions for 1.5 h. To our delight, trimer 2a was obtained in a satisfactory yield of 74%, indicating that the present method could be performed for a larger-scale preparation.
To gain insights into the reaction pathway, we performed several control experiments (Table 2). When 3 equiv. of the radical scavenger 2,2,6,6-tetramethylpiperidinooxy (TEMPO) was added to the reaction mixture of 1a, KOtBu and H2O under the optimized conditions, the yield of 2a was 47% (Table 2, entry 1). Increasing the amount of TEMPO to 10 equiv. led to a similar yield of 41% (Table 2, entry 2). These experiments with TEMPO suggested that our reaction was significantly different from the previous solution reaction, which occurred through SET radical pathways.7 To explore whether molecular oxygen under an air atmosphere played a role during the milling process, the jar containing 1a, KOtBu and H2O was filled with nitrogen (N2) and tightened in a glovebox. Then, the reaction mixture was milled under the optimal conditions, and 2a was isolated in 83% yield (Table 2, entry 3). There was no obvious effect on the reaction, indicating that oxygen was not involved in our reaction.
|
|
||
|---|---|---|
| Entry | Condition | Yield of 2a (%) |
| a All reactions were performed with 0.20 mmol of 1a, KOtBu (2 equiv.) and H2O (3 equiv.) together with 4 stainless steel balls (5 mm in diameter) in a stainless steel jar (5 mL) and milled (1800 rpm) at room temperature for 1.5 h. | ||
| 1 | 3 equiv. of TEMPO | 47 |
| 2 | 10 equiv. of TEMPO | 41 |
| 3 | Nitrogen atmosphere | 83 |
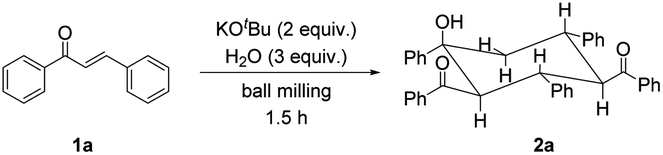
The rarely investigated retro-aldol reaction of chalcones could take place in the presence of water, a secondary amine as an organocatalyst and trifluoroacetic acid as a cocatalyst in MeCN10a or catalyzed by tetrabutylammonium hydroxide at 150 °C under microwave irradiation in aqueous solution.10b Benzaldehyde was detected from the solvent-free reaction of 1a with KOtBu and H2O in a molar ratio of 1:2:3 after milling for 0.5 h (Fig. S1†), hinting that the challenging retro-aldol reaction could be facilitated solely by the inorganic base and water under mechanochemical conditions.
Based on the above experimental results and previous literature,11 a plausible reaction mechanism is proposed and shown in Scheme 3. Initially, the retro-aldol reaction of enone 1 with the aid of KOtBu and H2O generates an aldehyde and the carboanionic species A. The latter undergoes Michael addition to the second enone 1. The resulting intermediate B undergoes a second Michael addition to the third enone 1 to afford intermediate C. Subsequently, intermediate C cyclizes via an intramolecular aldol reaction to give anion D, followed by protonation with H2O to afford trimer 2.
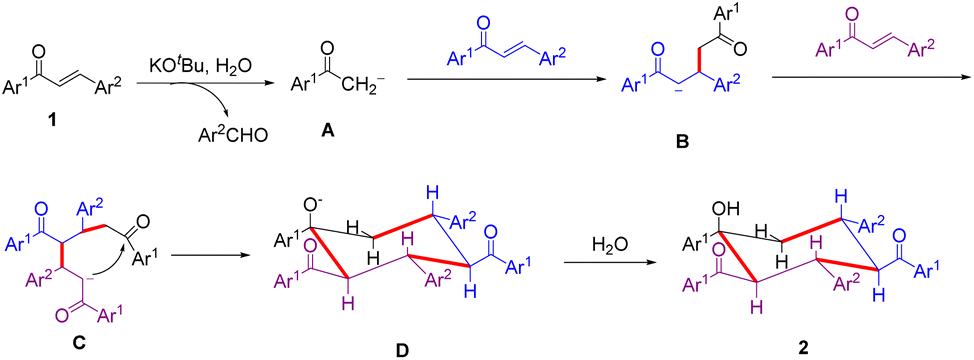 | ||
| Scheme 3 Proposed reaction mechanism. | ||
To verify whether the carboanionic species A can indeed be generated from the retro-aldol reaction of enones, 4′-methoxyacetophenone was added as the competing precursor to the reaction mixture of 1a, KOtBu and H2O. Interestingly, 2a (37%) and inseparable isomeric products 2u and 2v in a 1:1 ratio (a combined yield of 41%) were obtained (Scheme 4). In this case, 2a could still be formed in a comparable yield to the combined yield of 2u and 2v. 4′-Methoxyacetophenone could be deprotonated by KOtBu to give a similar carboanionic A, which underwent Michael addition to 1a, resulting in anion B and equally stable anion B′ by 1,3-proton transfer. The following steps were the same as those shown in Scheme 3 and provided isomeric products 2u and 2v (for details, see Scheme S1†).
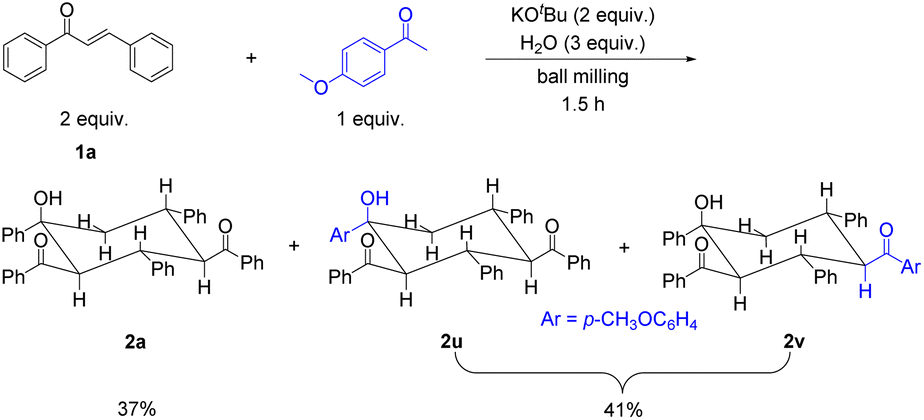 | ||
| Scheme 4 Mechanochemical reaction of 1a and 4′-methoxyacetophenone with KOtBu and H2O. The reaction was performed with 1a (0.20 mmol), 4′-methoxyacetophenone (0.10 mmol), KOtBu (0.40 mmol) and H2O (0.60 mmol) together with 4 stainless steel balls (5 mm in diameter) in a stainless steel jar (5 mL) at 1800 rpm and room temperature for 1.5 h. | ||
In the cyclohexene rings of trimers 2, there exists an axial hydroxyl group, which can be eliminated by further dehydration. Trimers 2a and 2q were employed as representative examples (Scheme 5). As desired, cyclohexene derivative 4a was obtained in 93% yield when 2a was treated with 3 equiv. of trifluoromethanesulfonic acid (TfOH) for 1.5 h under ball-milling conditions.6d Similarly, 4q could be successfully isolated in 89% yield. The dehydration of 2a and 2q caused an unusual stereoselective elimination, resulting in the formation of β,γ-unsaturated cyclohexenes instead of the more stable α,β-unsaturated cyclohexenes. This regioselectivity may be a result of the axial positioning of the hydroxyl group in the cyclohexanols, which preferentially underwent elimination with an equatorial hydrogen to form a six-membered transition state. In addition, the dehydration reaction under basic conditions, including KOtBu, KOAc, K2CO3 or 4-dimethylaminopyridine (DMAP), was attempted to afford the more stable α,β-unsaturated cyclohexenes. However, the desired dehydration products could not be produced.
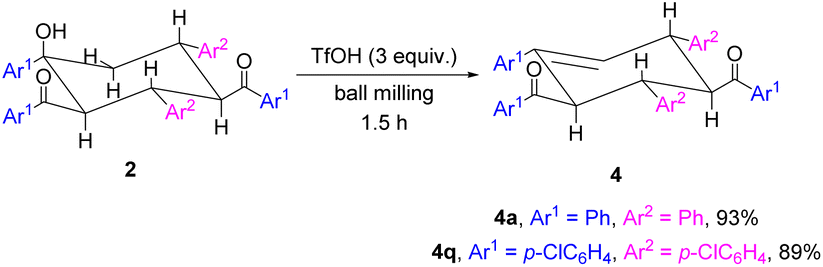 | ||
| Scheme 5 Dehydration reactions. Both reactions were performed with 0.10 mmol of 2 and TfOH (3 equiv.) together with 4 stainless steel balls (5 mm in diameter) in a stainless steel jar (5 mL) and milled (1800 rpm) at room temperature for 1.5 h. | ||
Conclusions
In summary, we have developed a solvent-free stereoselective trimerization of enones with KOtBu as the base and water as the proton source under ball-milling conditions, affording 1,3,5-triaryl-2,4-acyl-cyclohexanols with all bulky substituents located at equatorial positions. These unusual trimerization products can be straightforwardly obtained from enones by a cascade process of a retro-aldol reaction, double Michael additions and intramolecular aldol reaction. Other stereoisomers with most bulky substituents situated at equatorial sites except for a 4-acyl group at the axial position can also be obtained in moderate yields by reducing the amount of KOtBu or changing KOtBu to a relatively weaker base KOH or NaOH. It should be noted that the challenging retro-aldol reaction can be mechanochemically achieved solely with an inorganic base and water and is the critical initiation step. In addition, the formed cyclohexanol derivatives can be efficiently transformed into the corresponding cyclohexene derivatives via a dehydration reaction with TfOH. Our protocol features solvent-free and ambient conditions, a short reaction time, and easily available and inexpensive reagents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21372211).Notes and references
- For reviews, see: (a) G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC; (b) M. Leonardi, M. Villacampa and J. C. Menéndez, Chem. Sci., 2018, 9, 2042–2064 RSC; (c) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC; (d) J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC; (e) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed; (f) G.-W. Wang, Chin. J. Chem., 2021, 39, 1797–1803 CrossRef CAS; (g) S. Hwang, S. Grätza and L. Borchardt, Chem. Commun., 2022, 58, 1661–1671 RSC; (h) A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- For a review, see: (a) J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed , For recent examples, see:; (b) M. Turberg, K. J. Ardila-Fierro, C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2018, 57, 10718–10722 CrossRef CAS PubMed; (c) K. Chen, C. Niu and G.-W. Wang, Molecules, 2020, 25, 3719 CrossRef CAS PubMed; (d) Y. Liu, F.-Z. Liu and K. Yan, Angew. Chem., Int. Ed., 2022, 61, e202116980 CrossRef CAS PubMed.
- (a) Z. Zhang, Y.-W. Dong, G.-W. Wang and K. Komatsu, Chem. Lett., 2004, 33, 168–169 CrossRef CAS; (b) Z. Zhang, Y.-W. Dong, G.-W. Wang and K. Komatsu, Synlett, 2004, 61–64 Search PubMed; (c) Y. Li, Y. Cao, F. Xu, W. Fang, W. Yu, J. Jia and J. Gao, Sci. China: Chem., 2012, 55, 1252–1256 CrossRef CAS.
- (a) G.-W. Wang and X.-L. Wu, Adv. Synth. Catal., 2007, 349, 1977–1982 CrossRef CAS; (b) X.-L. Wu, J.-J. Xia and G.-W. Wang, Org. Biomol. Chem., 2008, 6, 548–553 RSC.
- (a) G.-W. Wang, Y.-W. Dong, P. Wu, T.-T. Yuan and Y.-B. Shen, J. Org. Chem., 2008, 73, 7088–7095 CrossRef CAS PubMed; (b) Z. Liu, G.-P. Fan and G.-W. Wang, Chem. Commun., 2012, 48, 11665–11667 RSC.
- (a) G. Kaupp, J. Schmeyers, A. Kusec and A. Atfeh, Angew. Chem., Int. Ed., 1999, 38, 2896–2899 CrossRef CAS; (b) X. Zhu, Z. Li, C. Jin, L. Xu, Q. Wu and W. Su, Green Chem., 2009, 11, 163–165 RSC; (c) H. Xu, H.-W. Liu, K. Chen and G.-W. Wang, J. Org. Chem., 2018, 83, 6035–6049 CrossRef CAS PubMed; (d) H.-W. Liu, H. Xu, G. Shao and G.-W. Wang, Org. Lett., 2019, 21, 2625–2628 CrossRef CAS PubMed.
- Y. Zhang, X. Wu, L. Hao, Z.-R. Wong, S. J. L. Lauw, S. Yang, R. D. Webster and Y. R. Chi, Org. Chem. Front., 2017, 4, 467–471 RSC.
- For recent examples, see: (a) H. Xu and G.-W. Wang, J. Org. Chem., 2022, 87, 8480–8491 CrossRef CAS PubMed; (b) R.-K. Fang, J.-S. Chen and G.-W. Wang, Green Chem. Lett. Rev., 2022, 15, 519–528 CrossRef CAS; (c) L. Li, C. Niu and G.-W. Wang, Chin. J. Chem., 2022, 40, 2539–2545 CrossRef CAS; (d) G. Shao, C. Niu, H.-W. Liu, H. Yang, J.-S. Chen, Y.-R. Yao, S. Yang and G.-W. Wang, Org. Lett., 2023, 25, 1229–1234 CrossRef CAS PubMed.
- (a) X. Luo and Z. Shan, Tetrahedron Lett., 2006, 47, 5623–5627 CrossRef CAS; (b) Z.-X. Shan, X.-X. Luo, L. Hu and X.-Y. Hu, Sci. China: Chem., 2010, 53, 1095–1101 CrossRef CAS.
- (a) D. Enders and T. V. Nguyen, Tetrahedron Lett., 2012, 53, 2091–2095 CrossRef CAS; (b) H. Lee and C.-H. Jun, RSC Adv., 2014, 4, 48331–48335 RSC.
- H. Gezegen and M. Ceylan, Synth. Commun., 2015, 45, 2344–2349 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data, NMR spectra of products 2a–2v, 2a′, 3a, 4a and 4q, and X-ray data of product 2a. CCDC 2277780. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3mr00010a |
| This journal is © The Royal Society of Chemistry 2024 |