
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extraction and untargeted analysis of metabolome from undemineralised cortical bone matrix†
Andrea
Bonicelli
a,
George
Taylor
b and
Noemi
Procopio
 *a
*a
aSchool of Law and Policing, Research Centre for Field Archaeology and Forensic Taphonomy, University of Central Lancashire, Preston, UK. E-mail: nprocopio@uclan.ac.uk
bBiological Mass Spectrometry (BioMS) Core Facility, Faculty of Biology, Medicine and Health, The University of Manchester, Manchester, M13 9PT, UK
First published on 22nd July 2024
Abstract
Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) untargeted metabolomics has become the gold standard for the profiling of low-molecular-weight compounds. Recently, this discipline has raised great interest in forensic sciences, especially in the field of toxicology and for post-mortem interval estimation. The current study aims at evaluating three extraction protocols and two LC-MS/MS assays run in both positive and negative modes, to identify the most suitable method to conduct post-mortem metabolomic profiling of bone tissue. A fragment of the anterior tibia of a 82 years-old male sampled from a human taphonomy facility was powdered via freeze-milling. The powdered sub-samples were extracted in five replicates per protocol. Methods tested were (I) a biphasic chloroform–methanol–water protocol, (II) a single phase methanol–water protocol, and (III) a single phase methanol–acetonitrile–water protocol. LC-MS/MS analyses were carried out via high performance liquid chromatography, either on hydrophilic interaction (HILIC) or on reversed-phase (C18) columns in both positive and negative ionisation modes, coupled with a Q-TOF mass spectrometer. Results suggest that the highest consistency between replicates and quality control samples was obtained with the single phase extractions (i.e., methanol–acetonitrile–water), whilst the ideal combination of instrumental set up HILIC chromatography in positive ionisation mode and of C18 chromatography in negative ionisation mode. For the purpose of forensic investigations, a combination of a single phase extraction and the two aforementioned chromatographic and mass spectrometry modes could represent an ideal set up for obtaining bone metabolomic profiles from taphonomically altered bones.
1. Introduction
The aim of metabolomics is to profile the entirety of low-molecular-weight compounds in a biological system. Untargeted approaches for metabolomics focus on the identification of the highest number of compounds with a mass below 1500 Da from a single tissue or fluid. However, despite the significant technological advances in the field of metabolomics, there are currently no analytical platforms that can comprehensively achieve this goal.2 Therefore, untargeted metabolomics commonly focuses on identifying smaller groups of compounds linked to specific biological processes, which are then further validated and investigated using targeted experiments.2,3Sample-preparation strategy plays a crucial role in planning an efficient and successful experiment.4–6 An efficient sample preparation protocol is characterised by four main attributes: (I) lack of selectivity, (II) simplicity, (III) reproducibility, and (IV) consideration of any chemical or enzymatic reactions that could affect the compound's stability after their extraction.3 The metabolomic workflow includes sample collection and extraction, experimental analysis, data pre-treatment, and statistical analysis.7 Proper storage of samples is essential to reduce post-collection instability of the metabolomic profiles. Quenching, which limits or removes chemical or enzymatic interactions after sampling, is a critical step, and the choice of quenching strategy depends on the matrix being analysed.7 The extraction process involves homogenization to increase the surface area exposed to the solvent and the selection of an appropriate solvent based on factors such as toxicity, solubilisation, selectivity, dissolution rate, chemical reactivity, and pH.3,7
High-throughput metabolomics approaches have gained popularity in bone research for understanding bone physiology and the connection between metabolite expression and tissue biomechanical properties.8 Elucidating bone metabolic pathways is valuable for investigating disease development and creating diagnostic and prognostic tools.9 For example, Zhao et al.10 studied the bone lipidome and metabolome in relation to bone mass changes in ovariectomised mice, and identified metabolic pathways associated with bone loss.
Recently, metabolic profiling of bone material has gained interest in the fields of archaeology and forensic science.11–14 Archaeological dental calculus analysis revealed changes in compounds and degradation products over time.11 Metabolomics applied to skeletal remains also aids to capture the significant correlations between the abundance and presence of specific compounds and the time elapsed since death (post-mortem interval, PMI) of the person.12 It is important to consider that the process of post-mortem decomposition, in fact, alters the metabolomic profile of the tissues ante mortem, due to the leaching of biomolecules in the environment surrounding the body, the biomolecular degradation of larger molecules into smaller metabolites due to taphonomic processes, and the introduction of new metabolites resulting from the decomposition process led by microbial decomposers. This results in profiles significantly different from the ante mortem ones, or from those extracted from preserved specimens from fresh cadavers, therefore requiring ad hoc protocols for their analysis and interpretation.
Another limitation encountered by forensic and archaeological experts dealing with bones samples from curated skeletal collections (e.g., from forensic osteological collections at human taphonomy facilities, HTFs, or from museums) is the processing that bones undergo prior to their long term storage. 'Bone maceration', the way in which such bone processing is defined, consists in the submersion of skeletal elements in high temperature water baths with the addition of chemical agents to degrease the bone surface, and was shown to significantly impact the bone metabolomic profiles by reducing the compounds coverage and by introducing contaminating features.13,14
This study aims to investigate three different extraction protocols, selected amongst existing ones for murine bone samples,10 plasma15 and bacteria,16 on a taphonomically altered and macerated bone sample collected from an HTF: an adapted biphasic extraction method (Chlor_Meth),15 and two single-phase extraction methods using methanol and water (Meth_Water)10 and methanol, acetonitrile, and water (Meth_ACN)16 extraction solvents. Two LC-MS/MS assays (HILIC and reversed-phase (C18) chromatography run in both positive and negative ionization modes) are investigated to determine the most appropriate protocol for undemineralised bone metabolomics of challenging forensic samples.
2. Materials and methods
A bone fragment (∼1 cm3) of the anterior midshaft of the tibia of an 82 years-old male donor was collected at the Forensic Anthropology Center of Texas State (FACTS) University by means of a 12 V Dremel cordless lithium-ion drill with a diamond impregnated wheel drill used at maximum 5000 revolutions. The bone was previously macerated following the standard procedure used at FACTS (submersion at 87 °C in a covered waterbath with laundry detergent for two days).13 After the collection of the bone fragment, the sample was further powdered using a Spex SamplePrep 6775-115 freezer/mill small cryogenic grinder operated in liquid nitrogen at speed 10 with 3 min pre-cooling, 2 min run and 2 min cooling protocol between the two grinding cycles. The powder was stored in a cryovial at −80 °C until further processing. Two sets of samples were created to be able to perform the two different types of chromatographies. In total five replicates for each extraction protocol were collected and processed using the three protocols described in Sections 2.1.1, 2.1.2.2.1. Metabolite extraction material
Chloroform (Chlor) AnalaR NORMAPUR ACS was purchased from VWR Chemicals (Lutterworth, UK). Water optima LC/MS grade, methanol (MeOH) optima LC/MS grade, pierce acetonitrile (ACN), optima LC/MS grade were purchased from Thermo Scientific (Hemel Hempstead, United Kingdom). All chemical were kept ice cold prior to extraction. Eppendorf protein LoBind tubes (Eppendorf UK Limited, Stevenage, UK) were used during the extraction.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (% v/v) Chlor:MeOH were added. Samples were vortexed for 30 s and and homogenised (4 × 20 s bursts at 5854 g, pause 2 min between bursts) in a Precellys Evolution Touch Homogenizer. To induce phase separation, 400 μL of LC-MS grade water were added and homogenised (4 × 20 s bursts at 5854 g, pause 2 min between bursts). The samples were then centrifuged at 4 °C for 10 min at 452 g and left in ice for 5 min. 600 μL of the lower fraction (organic) was collected and transferred to a fresh microtube tube, then samples were re-extracted for a second time using 500 μL of 2:1 (% v/v) Chlor:MeOH and homogenised (4 × 20 s bursts at 5854 g, pause 2 min between bursts). 600 μL of the lower fraction (organic) were collected and transferred into the previous microtube tube. This was centrifuged at 18213 g at 4 °C for 10 min and 1 mL of the supernatant was collected and dried under nitrogen flow. 350 μL of the aqueous phase were transferred to a fresh microtube tube and centrifuged at 18213 g at 4 °C for 10 min and 300 μL were transferred to a fresh tube and dried under nitrogen flow. Dry extracts were stored at −80 °C until testing. Only the aqueous phase was submitted for LC-MS/MS analysis.
:2 (% v/v) MeOH:Water were added. Samples were vortexed for 30s and and homogenised (4 × 20 s bursts at 5854 g, pause 2 min between bursts) in a Precellys Evolution Touch Homogenizer. The homogenisation tube was centrifuged at 18213 gs at 4 °C for 10 min, then 700 μL were moved to a fresh tube. 750 μL of 8:2 (% v/v, Meth_Water) MeOH:Water were added and the homogenisation step was repeated. The homogenisation tube was centrifuged at 18213 gs at 4 °C for 10 min, then 700 μL were moved to the same collection tube. The tube with the two extracts was centrifuged at 18213 gs at 4 °C for 10 min and 1.2 mL of supernatant were transferred in a fresh tube and dried under nitrogen flow. Dry extracts were stored at −80 °C until testing. The same protocol was performed using methanol–acetonitrile–water 2:2:1 (% v/v/v, Meth_ACN) as solvent.
:1 (% v/v) Chlor:MeOH were added. Samples were vortexed for 30 s and and homogenised (4 × 20 s bursts at 5854 g, pause 2 min between bursts) in a Precellys Evolution Touch Homogenizer. To induce phase separation, 400 μL of LC-MS grade water were added and homogenised (4 × 20 s bursts at 5854 g, pause 2 min between bursts). The samples were then centrifuged at 4 °C for 10 min at 452 g and left in ice for 5 min. 600 μL of the lower fraction (organic) was collected and transferred to a fresh microtube tube, then samples were re-extracted for a second time using 500 μL of 2:1 (% v/v) Chlor:MeOH and homogenised (4 × 20 s bursts at 5854 g, pause 2 min between bursts). 600 μL of the lower fraction (organic) were collected and transferred into the previous microtube tube. This was centrifuged at 18213 g at 4 °C for 10 min and 1 mL of the supernatant was collected and dried under nitrogen flow. 350 μL of the aqueous phase were transferred to a fresh microtube tube and centrifuged at 18213 g at 4 °C for 10 min and 300 μL were transferred to a fresh tube and dried under nitrogen flow. Dry extracts were stored at −80 °C until testing. Only the aqueous phase was submitted for LC-MS/MS analysis.
:2 (% v/v) MeOH:Water were added. Samples were vortexed for 30s and and homogenised (4 × 20 s bursts at 5854 g, pause 2 min between bursts) in a Precellys Evolution Touch Homogenizer. The homogenisation tube was centrifuged at 18213 gs at 4 °C for 10 min, then 700 μL were moved to a fresh tube. 750 μL of 8:2 (% v/v, Meth_Water) MeOH:Water were added and the homogenisation step was repeated. The homogenisation tube was centrifuged at 18213 gs at 4 °C for 10 min, then 700 μL were moved to the same collection tube. The tube with the two extracts was centrifuged at 18213 gs at 4 °C for 10 min and 1.2 mL of supernatant were transferred in a fresh tube and dried under nitrogen flow. Dry extracts were stored at −80 °C until testing. The same protocol was performed using methanol–acetonitrile–water 2:2:1 (% v/v/v, Meth_ACN) as solvent.
2.2. Liquid chromatography mass spectrometry
LC-MS analyses were performed using a Thermo-Fisher Ultimate 3000 HPLC system (HPG-3400RS high pressure gradient pump, TCC 3000SD column compartment and WPS 3000 Autosampler) coupled with a SCIEX 6600 TripleTOF Q-TOF mass spectrometer with TurboV ion source. The system was controlled by SCIEX Analyst 1.7.1, DCMS Link and Chromeleon Xpress software. Samples for HILIC were reconstituted in 4:1 (% v/v) acetonitrile/water, and samples for RP were reconstituted in 95:5 (% v/v) water/acetonitrile. For all run types, a sample volume of 5 μL was injected by pulled loop onto a 5 μL sample loop with 150 μL post-injection needle wash. Injection cycle time was 1 min per sample. The mass spectrometer was ran under the following source conditions: curtain gas pressure, 50 psi; temperature, 400 °C; ESI nebuliser gas pressure, 50 psi; heater gas pressure, 70 psi; declustering potential, 80 V. All method-dependent variable instrument and data processing parameters are given in the Table S1 (ESI†).
Mass spectrometry data was acquired in a data-dependent manner. Features were selected for fragmentation automatically on a basis of the top 10 most intense ions with a charge state of 1–2 and minimum threshold of 10 cps. Isotopes within 4 Da were excluded from the scan. The accumulation time for each scan was 100 ms and the accumulation time for the TOF survey scan was 250 ms. Total cycle time was 1.3 s. Collision energy was determined using the formula CE (V) = 0.084 × m/z +12 up to a maximum of 55 V. Isotopes within 4 Da were excluded from the scan. Acquired data were checked in PeakView 2.2 and imported into Progenesis QI 2.4 for metabolomics, where they were aligned, peaks were picked, normalised to all compounds and deconvoluted according to standard Progenesis QI workflows. Peak picking parameters were set to automatic with default sensitivity level and a minimum peak width of 0.1 min. Ions were ignored before 1.3 min and after 24 min for HILIC runs and before 0.9 min and after 10 min for C18 runs. Adducts and specific of all assays are given in ESI† Table S1. MSI level 2 annotations were made by searching the accurate mass, MS/MS spectrum and isotope distribution ratios of acquired data against the NIST MS/MS metabolite library, and additionally by searching retention times and accurate masses against an in-house made library of chemical standards using Progenesis QI (using a 0.5 min retention time tolerance, Table S2, ESI†). Metabolites with a score higher than 40 were accepted. MSI level 1 identifications were made when both libraries were in agreement and MS/MS spectra matching the NIST library entries were present. Table S3 (ESI†) includes the list of putatively annotated compounds and their annotation scores.
2.3. Extract blank and quality control (QC)
Extraction blanks were prepared by repeating each extraction protocol without bone material. Pooled QC samples were prepared for each instrumental assay by aliquoting a fraction of 10 μL from each sample excluding the blank. The QC pool was vortexed, centrifuged at 21000 g for 10 min at 4 °C and 100 μL of the supernatant was aliquoted in low recovery vials and analysed at the beginning and end of the run.
2.4. Data processing and statistical analysis
Data processing and statistical analysis was carried out in R version 4.3.1 (2023-06-16) using the ‘StructToolbox’ package17 only on putatively annotated features. Features were retained if they were present in 60% of the samples. Features with fold change less than 15 compared to the blanks signal were removed. Corresponding metabolomics pooled quality control sample (QC) were used to assess for instrumental drifts. Any feature with relative standard deviation above 30% compared to the QCs was removed. This was repeated for each extraction protocol separately to evaluate overall number of compounds profiled and common compounds between assays and globally for the entire study using Venn diagram to compare compounds was created using ‘ggvenn (https://CRAN.R-project.org/package=ggvenn)’. Visualisation of intersecting sets across different assays was performed using the ‘UpSetR’18 package. Relative standard deviation (RSD) percentage values were calculated across extraction replicates (QCs) for repeatability and between type of extraction protocol for extraction efficiency prior to data normalisation. Further, the three extraction protocols were processed together, according to the same parameters previously described, and missing data were imputed using k-nearest neighbor algorithm based on three neighbours. Data normalisation was performed via generalised logarithmic transformation, probabilistic quotient normalisation. Exploratory data analysis and outlier detection were performed by principal component analysis (PCA) after autoscaling the processed data.3. Results & discussion
Standardisation of extraction and analysis in metabolomic workflows is of paramount importance in order to optimise the comparability between studies and to obtain an ideal metabolic coverage. In forensic science, bone is all that remains of the body after prolonged PMIs or in advanced decomposition stages,19 therefore improved analytical approaches are necessary to gather the largest amount of information possible to aid investigations. In contrast with metabolomic approaches applied on fresh skeletal tissue in biological and medical contexts, in forensic contexts further issues such as taphonomic alterations and maceration should be considered when developing tailored metabolomic protocols.13,14 Additionally, when applying LC-MS/MS metabolomics to bone analysis in forensic contexts, it is vital to design an experiment that is clear and easy to be replicated, as well as that provides appropriate measurements of the errors for the estimations. In the present study, we tested three extraction protocols, two based on the use of a single phase protocol (i.e., Meth_ACN and Meth_Water) and one based on an adapted version of the biphasic Chlor_Meth protocol, and evaluated their advantages and disadvantages. The results from the single phase protocols demonstrated a consistent pattern between the two. Conversely, the Chlor_Meth protocol yielded markedly distinct outcomes, showcasing notable variations both in terms of replicability and in the overall compound coverage (Fig. S3, ESI†). Furthermore, different LC-MS/MS assays showed considerably different results; after data processing, the suggested combination for optimal compound coverage was HILIC ESI+ and RP ESI− when applied in conjunction with the Meth_ACN protocol. To summarise, we presented the outcome of three distinct metabolite extraction and four analytical protocols and their relevance to bone research in forensic contexts. Our findings can be summarised in the following key points: (a) improved replicability for single-phase extraction protocols, (b) higher number of compounds obtained using single-phase extractions compared to the biphasic extraction method, and (c) most robust compound profiling and more reliable data analysis utilising a combination of HILIC ESI+ and C18 ESI− techniques.It is appropriate to introduce certain limitations of the current study. First, QC samples were injected only before and after the 15 samples injected, in every assay. Ideally, QC samples should be analysed at regular intervals in the run to better control for instrumental drifts. Another limitation is that samples from each class were analysed consecutively and not in a randomised order, which could have introduced a bias. Despite the above mentioned limitations, we believe that the differences observed among extraction protocols are genuine and related with the procedure chosen and the solvents employed.
Fig. 1A–D reports the RSD for both extraction replicates and QCs across the four experiments (RSD for single compounds are available in ESI† Table S4). In HILIC ESI+ the RSD values for the total peak area amongst the three extractions showed acceptable median values for Meth_ACN (5.93%) and QCs (9%). Higher values were found for Meth_Water and Chlor_Meth, respectively 14.6% and 18.9%. For HILIC ESI− the highest RSD remained for Chlor_Meth (57.6%), followed by Meth_Water (30.0%) and Meth_ACN (21.7%). The lowest RSD was calculated for QCs (18.9%). All these values for HILIC ESI− are considered to be unacceptable in terms of replicates' agreement. C18 ESI+ gave the highest RSD with Chlor_Meth (18.4%), followed by QCs (11.9%), Meth_Water (8.36%), and Meth_ACN (5.28%). C18 ESI− showed similar results to HILIC ESI+ with the highest RSD being the one for Chlor_Meth (10.1%), followed by Meth_Water (8.36%). Lower RSD were those of QCs (6.66%) and Meth_ACN (6.04%). Fig. S1 (ESI†) shows the distribution of single metabolite's RSD for each extraction protocol and for the QCs, confirming the better suitability of Meth_ACN in combination with HILIC chromatography and the overall appropriate replicability of both single phase extractions with C18. QCs RSD distributions confirmed the repeatability of the all the assays. The plots in Fig. 1E–H provide an overview of the instrumental stability of the each assay across the entire run. It is clear that the two HILIC experiments presented the same trend with a drift towards the end of the experiment. This drift was effectively corrected via data normalisation (see also Fig. S2, ESI†) and transformation as showed in Fig. 1I–L. In contrast, higher stability in the system was shown for C18 ESI+ and ESI− as noticeable both in extraction replicates and in QCs. These results suggest that overall Meth_ACN is the most repeatable extraction protocol, and that HILIC ESI− and C18 ESI+ has the lowest instrumental stability across the QCs. All the remaining assays were characterised by an acceptable RSD range.
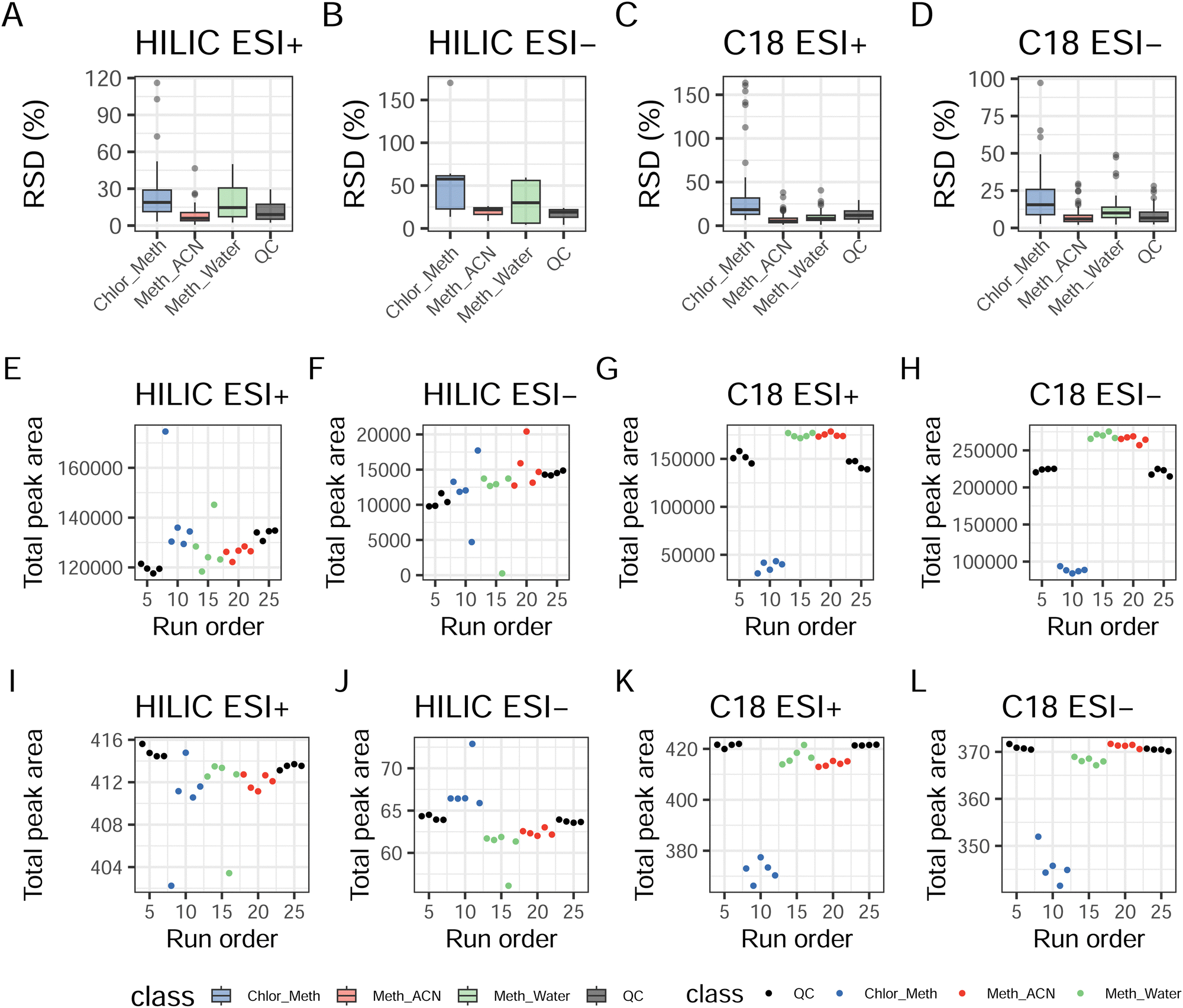 | ||
| Fig. 1 Evaluation of the experimental runs. (A)–(D) Median and interquartile values for the the RSD across extraction replicates for extraction suitability and across QCs for the analysis of replicability. (E)–(H) Total ion count (TIC) for the four assays to evaluate instrumental drift. (I)–(L) TIC after transformation and normalisation, showing efficient processing and reduction of instrumental drift. | ||
Considering the number of annotated compounds profiled with the three different protocols post data processing performed separately, for HILIC ESI+ 74 compounds were retained when extracted with Meth_ACN, 77 with Chlor_Met and 71 with Meth_Water, with 63 of those being shared between the three extraction protocols as shown in ESI† Fig. S3. A much lower number of compounds was obtained using HILIC ESI−, where a maximum of 11 compounds were found with Chlor_Meth and Meth_ACN extractions, and 8 with Meth_Water. Coverage for C18 ESI+ was deeper, with a maximum of 97 compounds obtained using Meth_ACN, 95 using Chlor_Meth, and 87 using Meth_Water. This assay allowed the profiling of the highest number of compounds. C18 ESI− allowed the profiling of 71, 70, and 64 compounds respectively for Meth_ACN, Chlor_Met, and Meth_ACN. After data processing, none of the assays were able to profile more than 97 putatively annotated compounds. The limited number of compounds identified in this study stems from the unique composition of the bone tissue and on the nature of the bone under analysis (decomposed and macerated). Bones are constituted approximately by 60–70% of mineral matrix, and the remaining organic fraction is made up by approximately 90% type I collagen, 5% non-collagenous proteins (NCPs), 2% lipids and metabolites by weight.20 Additionally, bones subjected to taphonomic processes (such as harsh climate conditions) and maceration are further depleted of small molecules and organic components, therefore explaining the reduced number of compounds identified in this analysis.13
The three extraction methods tested were then processed together in order to evaluate the overall number of compounds identified. Prior to data processing, 110 compounds were putatively annotated in HILIC ESI+, 18 in HILIC ESI−, 228 in C18 ESI+, and 106 in C18 ESI−. After data processing, results showed the highest number for C18 ESI− (n = 87), followed by HILIC ESI+ (n = 66), C18 ESI− (n = 64), and HILIC ESI− (n = 9). Fig. 2 shows the compounds shared by the four experimental modes. HILIC ESI+ allowed the profiling of 54 unique compounds, 11 shared with C18 ESI+ and one only shared with C18 ESI−. HILIC ESI− had only 6 unique compounds, one shared with C18 ESI+ and two shared with C18 ESI−. C18 ESI+ had 73 unique compounds with two compounds being exclusively shared with C18 ESI−. Finally, C18 ESI− had 58 compounds profiled only with this assay. No putatively annotated compounds were shared across all four assays. This suggests that no clear advantage in the identification and relative quantification of polar compounds can be obtained by the removal of the lipids from the polar phase.21
 | ||
| Fig. 2 Venn diagram showing the intersection of common metabolites between assays. | ||
PCA for HILIC ESI+ (Fig. 3) showed extremely good instrumental stability, with QCs forming a close cluster and principal component (PC) one explaining 66.9% of the sample variance responsible for the separation between single and biphasic extractions. Furthermore, it can be seen in Fig. 3 that, with the exception of one outlier for the Meth_Water extraction (B4), the two clusters for the single phase extractions almost overlap, suggesting great similarity in the two profiles. HILIC ESI− results are notably different, with PC1, accounting for 68.2% of the total variance, failing to explain the difference between the different types of extractions. This components seems to be heavily influenced by the presence of two outliers for Chlor_Meth and Meth_Water. In contrast, 18.0% of the variance explained by PC2 seems to capture the variation between single phase and biphasic extraction protocols. C18 experiments in both ESI+ and ESI− showed very similar trends. In ESI+, 88.8% of the variance described the separation between Chlor_Meth and the two single phase extractions, while in ESI− the variance accounted for 92.7%. There was one outliers for Chlor_Meth. Due to the acceptable RSD values for the QC samples, outliers for all assays may be attributed to issues related to the extraction phase rather than with instrumental instability. Overall, the profiles obtained by the two single phase extractions across all experiments seem to have minimal differences. The higher volume for the extraction solvent for the single phase protocols (1:15 compared to 1:8 for the biphasic protocol) could have aided the operator into more accurate pipetting.22 Furthermore, the biphasic protocol is considerably slower than then single phase one, although it might be ideal in cases of low amounts of starting material. Furthermore, the interaction between the two solvents allows the selective removal of less polar lipid compounds and optimises deproteinisation.22 However, the present study shows that when applied to bone powder material and it might increase technical variance.22 Furthermore, due to the the complexity of the protocol and the use of bead homogenisation, the biphasic extraction is not advisable for large scale studies as suggested also by the low degree of agreement obtained between the biological replicates. Finally, several classes are consistently found across multiple assays (such as amino acids and fatty acyls), some others are found in two out of three protocols (alkaloids and bases for HILIC ESI+ and C18 ESI+, carboxylic acids and vitamins for C18 ESI+ and C18 ESI−), and some classes are unique for specific protocols (glycerophospholipids and phenylpropanoids for HILIC ESI+, sphingolipids for C18 ESI+ and monosaccharides and sterol lipids for C18 ESI−). For the HILIC ESI+ runs, we detected significant differences between the biphasic protocol and the two monophasic ones for glycerophospholipids, where the monophasic extractions gave higher abundances than the biphasic one, and for phenylpropanoids, where all three protocols were significantly different with the higher intensity being the one obtained using the biphasic method. In C18 ESI+ runs, alkaloids abundance was higher in the biphasic protocol and significantly lower in the Meth_ACN one, carboxylic acids intensities were lower in the two monophasic protocols, while for sphingolipids and vitamins the abundance was significantly higher with the two monophasic protocols than in the biphasic one. In C18 ESI− runs, higher intensities for carboxylic acids, monosaccharides, sterol lipids and vitamins were found using the biphasic protocol in comparison with the monophasic ones, whereas fatty acyls were more abundant in the monophasic protocols.
 | ||
| Fig. 3 PCA score plot showing the clustering of the three extractions based on all putatively metabolites after data processing when using (A) HILIC ESI+, (B) HILIC ESI−, (C) C18 ESI+ and (D) C18 ESI− runs. Samples labelled and outside the dashed line are to be considered as outliers. | ||
4. Conclusions
In the present study we considered three different extraction protocols (Chlor_Meth, Meth_ACN, and Meth_Water) and four LC-MS/MS settings (HILIC ESI+, HILIC ESI−, C18 ESI+, and C18 ESI−) to investigate the effect that the combination of extraction and analytical method would have had in the metabolic profiling of undemineralised, decomposed and macerated bone matrix. According to the results presented, the single-phase protocols seem to be preferable in comparison with the biphasic one in terms of number of compounds and repeatability. Furthermore, when selecting the appropriate chromatographic strategy, we advise the combined use of HILIC ESI+ and C18 ESI− for maximising the number of compounds identifiable. We also considered here the identification of common compounds across assays, the RSD and instrumental stability, as well as the number of features lost during data processing. The two assays overall offer the best compromise and resulted in being the most complementary between the four considered here. We also suggest to adjust data processing according to the specific needs for the experiment. Finally, we would like to emphasise that these results only represent an indication for further investigation, and that future evaluations should take into account inter-sex variability and inter- and intra-skeletal variability as well as other factors such as age and pathological conditions of the tissue donor.Author contributions
Andrea Bonicelli: conceptualisation, methodology, formal analysis, software, investigation, data curation, writing – original draft, review and editing, project administration. George Taylor: methodology, investigation, writing – review and editing. Noemi Procopio: conceptualization, writing – review and editing, supervision, project administration.Institutional review
This study did not involve human living subjects but only human tissues from deceased individuals who gave their consent prior to death. The institutional review board is not required in these circumstances. The ethics code of FACTS was adhered and an approval for the study was obtained from the FACTS Ethics review board as well as from the Ethics committee at UCLan (ref. SCIENCE 0223).Data availability
Data for this paper, including metabolomics raw data files, are available at Metabolomics Workbench at https://doi.org/10.21228/M8DX4F.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was funded by the UKRI Future Leaders Fellowship (N.P.) under grant number MR/S032878/1. The authors also gratefully acknowledge the donors and their next of kin for allowing the use of donated bodies to perform this research.Notes and references
- M. Sud, E. Fahy, D. Cotter, K. Azam, I. Vadivelu, C. Burant, A. Edison, O. Fiehn, R. Higashi, K. S. Nair, S. Sumner and S. Subramaniam, Nucleic Acids Res., 2015, 44, D463–D470 CrossRef PubMed.
- Q. Yang, A. Hua Zhang, J. Hua Miao, H. Sun, Y. Han, G. Li Yan, F. Fang Wu and X. Jun Wang, RSC Adv., 2019, 9, 37245–37257 RSC.
- D. Vuckovic, Anal. Bioanal. Chem., 2012, 403, 1523–1548 CrossRef CAS PubMed.
- S. Moco, J. Vervoort, S. Moco, R. J. Bino, R. C. D. Vos and R. Bino, TrAC, Trends Anal. Chem., 2007, 26, 855–866 CrossRef CAS.
- B. Álvarez-Sánchez, F. Priego-Capote and M. L. de Castro, TrAC, Trends Anal. Chem., 2010, 29, 111–119 CrossRef.
- B. Álvarez-Sánchez, F. Priego-Capote and M. L. de Castro, TrAC, Trends Anal. Chem., 2010, 29, 120–127 CrossRef.
- M. Y. Mushtaq, Y. H. Choi, R. Verpoorte and E. G. Wilson, Phytochem. Anal., 2014, 25, 291–306 CrossRef CAS PubMed.
- J. Fan, V. Jahed and K. Klavins, Metabolites, 2021, 11, 434 CrossRef CAS PubMed.
- C. Guijas, J. R. Montenegro-Burke, B. Warth, M. E. Spilker and G. Siuzdak, Nat. Biotechnol., 2018, 36, 316–320 CrossRef CAS PubMed.
- H. Zhao, X. Li, D. Zhang, H. Chen, Y. Chao, K. Wu, X. Dong and J. Su, Sci. Rep., 2018, 8, 16456 CrossRef PubMed.
- I. M. Velsko, K. A. Overmyer, C. Speller, L. Klaus, M. J. Collins, L. Loe, L. A. F. Frantz, K. Sankaranarayanan, C. M. Lewis, J. B. R. Martinez, E. Chaves, J. J. Coon, G. Larson and C. Warinner, Metabolomics, 2017, 13, 134 CrossRef PubMed.
- A. Bonicelli, H. L. Mickleburgh, A. Chighine, E. Locci, D. J. Wescott and N. Procopio, eLife, 2022, 11, e83658 CrossRef CAS PubMed.
- A. Bonicelli, W. Cheung, S. Hughes, D. J. Wescott and N. Procopio, Metabolites, 2022, 12, 1020 CrossRef CAS PubMed.
- D. Badillo-Sanchez, M. S. Ruber, A. M. Davies-Barrett, J. K. Sandhu, D. J. L. Jones, M. Hansen and S. A. Inskip, Sci. Rep., 2023, 13, 696 CrossRef CAS PubMed.
- M. W. K. Wong, N. Braidy, R. Pickford, P. S. Sachdev and A. Poljak, Front. Neurol., 2019, 10, 879 CrossRef PubMed.
- S. K. P. Lau, C.-W. Lam, S. O. T. Curreem, K.-C. Lee, W.-N. Chow, C. C. Y. Lau, S. Sridhar, S. C. Y. Wong, P. Martelli, S.-W. Hui, K.-Y. Yuen and P. C. Y. Woo, Cell Biosci., 2015, 5, 26 CrossRef PubMed.
- G. R. Lloyd, A. Jankevics and R. J. M. Weber, Bioinformatics, 2020, 36, 5551–5552 CrossRef CAS PubMed.
- A. Lex, N. Gehlenborg, H. Strobelt, R. Vuillemot and H. Pfister, IEEE Trans. Vis. Comput. Graph., 2014, 20, 1983–1992 Search PubMed.
- B. Madea, C. Henßge, S. Reibe and M. Tsokos, Handbook of Forensic Medicine, 2022, pp. 91–149 Search PubMed.
- A. L. Boskey, BoneKEy Rep., 2013, 2, 447 CrossRef PubMed.
- S. Tulipani, R. Llorach, M. Urpi-Sarda and C. Andres-Lacueva, Anal. Chem., 2012, 85, 341–348 CrossRef PubMed.
- A. D. Southam, L. D. Haglington, L. Najdekr, A. Jankevics, R. J. M. Weber and W. B. Dunn, Analyst, 2020, 145, 6511–6523 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Supplementary tables and figures (PDF); (2) Table S2 (Microsoft Excel file): retention time and neutral mass values for in-house standard library; (3) Table S3 (Microsoft Excel file): additional information on the annotation quality for each experiment (assigned annotation, compound, neutral mass (Da), m/z, retention time (min), chromatographic peak width (min), identifications, identifier, MSI classification, adducts, formula, score, fragmentation score, absolute mass error (ppm), isotope similarity); (4) Table S4 (Microsoft Excel file): RSD values for all compounds across extraction protocols and for quality control samples for each experiment. All raw data is available on-line at Metabolomics Workbench1 using identifier PR001650. See DOI: https://doi.org/10.1039/d4mo00015c |
| This journal is © The Royal Society of Chemistry 2024 |