
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PerSEveML: a web-based tool to identify persistent biomarker structure for rare events using an integrative machine learning approach†
Sreejata
Dutta
 a,
Dinesh Pal
Mudaranthakam
ab,
Yanming
Li
ab and
Mihaela E.
Sardiu
*abc
a,
Dinesh Pal
Mudaranthakam
ab,
Yanming
Li
ab and
Mihaela E.
Sardiu
*abc
aDepartment of Biostatistics & Data Science, University of Kansas Medical Center, Kansas City, Kansas, USA. E-mail: msardiu@kumc.edu
bUniversity of Kansas Cancer Center, Kansas City, USA
cKansas Institute for Precision Medicine, University of Kansas Medical Center, Kansas City, Kansas, USA
First published on 16th April 2024
Abstract
Omics data sets often pose a computational challenge due to their high dimensionality, large size, and non-linear structures. Analyzing these data sets becomes especially daunting in the presence of rare events. Machine learning (ML) methods have gained traction for analyzing rare events, yet there has been limited exploration of bioinformatics tools that integrate ML techniques to comprehend the underlying biology. Expanding upon our previously developed computational framework of an integrative machine learning approach, we introduce PerSEveML, an interactive web-based tool that uses crowd-sourced intelligence to predict rare events and determine feature selection structures. PerSEveML provides a comprehensive overview of the integrative approach through evaluation metrics that help users understand the contribution of individual ML methods to the prediction process. Additionally, PerSEveML calculates entropy and rank scores, which visually organize input features into a persistent structure of selected, unselected, and fluctuating categories that help researchers uncover meaningful hypotheses regarding the underlying biology. We have evaluated PerSEveML on three diverse biologically complex data sets with extremely rare events from small to large scale and have demonstrated its ability to generate valid hypotheses. PerSEveML is available at https://biostats-shinyr.kumc.edu/PerSEveML/ and https://github.com/sreejatadutta/PerSEveML.
1. Introduction
With the continuous expansion of omics and related fields, machine learning (ML) techniques are gaining importance in extracting meaningful insights and advancing our understanding of complex biological systems.1–4 Omics data sets encompass large-scale biological data from various disciplines, including genomics, transcriptomics, proteomics, and metabolomics. High-throughput technologies enable researchers to gather a wealth of data from biological samples relatively quickly. Traditional methodologies frequently falter when confronted with the daunting challenges posed by the immense dimensionality, expansive scale, and intricate non-linear structures inherent in omics data.5 In this context, ML plays a crucial role in unraveling the intricacies of these vast and complex data sets by deciphering patterns, extracting meaningful insights, and providing actionable intelligence from these multifaceted data repositories.ML methods excel at discovering concealed patterns within complex data sets without extensive human involvement, making them invaluable in fields like omics data analysis. Their scalability and ability to process vast amounts of information, especially in high-throughput technologies, enhance their appeal.6 Unlike traditional methods relying on predefined assumptions, ML models learn directly from data, capturing intricate relationships and patterns that might be overlooked. However, analyzing rare events in omics data through ML poses challenges.2 Most ML algorithms struggle with imbalanced data, focusing on the majority class and overlooking patterns in rare events (minority class). ML models require sufficient data to learn meaningful patterns, yet in omics data sets, rare events like specific mutations or low-abundance molecules are often outnumbered by common events.7
Rare events in cancer-genomic studies refer to occurrences of infrequent disease outcomes compared to the controls—for instance, onsite rare cancers like gallbladder cancer and hairy cell leukemia. In quantitative trait studies, rare events could refer to the expression status of rarely expressed genes or low-abundance proteins.8,9 Rarely expressed genes and rare alternative splicing transcripts can provide insights into unique biological processes,10,11 low-abundance proteins may indicate specialized biological functions,12,13 and rare post-translational modifications (PTMs) can play critical roles in cellular signaling pathways.14,15 Analyzing rare events in omics data using ML methods can present several complications due to the inherent challenges posed by the scarcity of these events.
Past research suggested various approaches to deal with the problem of class imbalance, which include data-level, algorithm-based, and hybrid methods.16,17 The data-level approaches involve under-sampling the majority class or over-sampling the minority class before training the model, which is an additional step in data preprocessing. Some of the well-known sampling techniques include ADASYN (adaptive synthetic sampling)18 and SMOTE (synthetic minority over-sampling technique).19 Both SMOTE and ADASYN generate synthetic samples for the minority class. SMOTE creates synthetic samples along line segments between existing minority class instances, while ADASYN adjusts the synthetic sample generation based on the density of the minority class.
Algorithm-based techniques leverage robust algorithms to address class imbalance, employing methods such as cost-sensitive frameworks, where a higher penalty for misclassification is applied to the minority class for enhanced classification performance. These techniques also encompass optimizing hyperparameters through cross-validation, a procedure involving training models on subsets of the training set and evaluating them on unseen data subsets. Another approach to handling class imbalance is the hybrid method: A combination of data-level and algorithm-based approaches. Notable hybrid methods include SMOTEBoost20 and RUSBoost.21 Despite the comparable effectiveness of all these approaches, algorithm-based techniques are favored by ML practitioners due to their simplicity and systematic enhancement of ML performance. However, they come at the cost of significantly increased training time for ML models.
Several analytical tools have been developed in recent years for high-dimensional omics data, providing simplicity of implementation and results in a comprehensible format.22–26 For instance, HTPmod,22 introduced in 2018, is a web-based shiny application offering various ML methods and visualization choices for high-dimensional data sets. In 2021, multiSLIDE23 was introduced, enabling the visualization of interconnected features in omics data sets and aiding biologists in understanding underlying biological relationships. Enrichr-KG,25 developed in 2023, enhances enrichment analysis and visualization using knowledge graphs, serving as a valuable resource for gene enrichment analysis. Despite the availability of these advanced analysis tools, there remains a need for ML tools that specifically address the computational challenges associated with rare events and corresponding visualization techniques that can aid researchers in formulating meaningful hypotheses.
Analyzing rare events demands meticulous data preprocessing, thoughtful algorithm selection, and rigorous validation methods to ensure reliable results. However, analyzing rare events poses computational challenges due to limited data availability for the minority class or ML methods being overwhelmed by the majority class. To address this problem of rare data analysis, we have created an interactive tool called PerSEveML. PerSEveML allows users to predict rare events and visualize the contribution of input features to these predictions. Fig. 1 represents the tool's functioning and computational framework.
 | ||
| Fig. 1 Graphical representation of the computational framework for PerSEveML. | ||
PerSEveML addresses common challenges in analyzing omics data sets, offering twelve ML methods suitable for small and large data sets. PerSEveML uses normalization techniques to handle non-linear data structures before training ML models. Six different normalization techniques have been integrated into the interface to ensure a wide application of this tool. These normalization techniques include log transformation for skewed data, standardization for feature scaling, and TopS, a normalization based on topological scoring, which effectively accentuates extreme data points for omics data with rare events.27,28 To comprehend the effect of normalization, ML practitioners often rely on data visualization tools such as boxplots. Therefore, we have incorporated box plots into the PerSEveML interface to visualize post-normalization data distribution.
PerSEveML is a versatile tool for various classification problems, uniquely capable of handling rare events through an integrated ML approach. While other ML toolkits like SuperLearner24 and HTPmod22 have attempted to tackle classification problems using multiple ML algorithms, these methods depend only on one best-performing model for feature selection. Our goal in adopting an integrative approach was to capitalize on the learning abilities of all top-performing models. Each ML algorithm is influenced by various factors, including decision boundaries, cost functions, sampling models, and hyperparameters; thus, suggesting that different models may identify distinct features that contribute to predicting rare events. Decision trees, for example, exhibit high variability with low biases, while models like logistic regression or linear discriminant analysis (LDA) have higher biases but lower variances. Proper training of each model is crucial to prevent underfitting or overfitting, considering the impact of biases and variances on ML performance.
PerSEveML is specifically tailored for complex biological data, such as large protein complex networks with multiple modules and shared subunits, where every feature holds biological significance. The tool allows users to assess, compare, and download the performance of the integrative ML approach with individual models using evaluation metrics. PerSEveML employs cross-validation for each selected ML model, enabling users to specify the number of folds, k, for cross-validation. Cross-validation evaluates a model's generalization by dividing data into training and validation subsets, ensuring reliable assessment across various partitions. Cross-validation serves two main objectives: optimizing model hyperparameters to prevent overfitting, and improving model performance for reliable predictions on unseen data.
In scenarios involving rare events, past research has employed ML methods with cross-validation.29,30 However, in order for cross-validation to work on rare event prediction, adequate information on the minority class need to be present. Thus, for scenarios where analysts require a sophisticated solution to deal with class imbalance, they can choose SMOTE or ADASYN. Our PerSEveML interface integrates SMOTE and ADASYN techniques, enabling users to incorporate these resampling methods for data analysis.
The PerSEveML interface allows users to visualize the correlation between input features and the persistent feature structure created using the integrative ML approach. Dutta et al.31 introduced the utilization of cut-point analysis to combine feature importance derived from diverse ML methods by utilizing entropy and rank scores to formulate a persistent feature structure. The determination of the persistent feature structure relies significantly on cut-point analysis, with the cut-off point defined as the percentage of features hypothesized by users to encapsulate the utmost information about the rare event of interest. PerSEveML offers users the chance to change the cut-off, thus allowing them to select an optimal cut-off point that works for their data set. The proposed feature structure is segregated into persistently selected, fluctuating, and unselected categories. These three categories can be used to select important features from the selected categories or generate a hypothesis using the fluctuating category to understand the association of a weak signaling feature with the rare event being studied. Moreover, the feature structure can serve as a metric for feature reduction. This involves excluding features from the unselected category, facilitating further experiments during the exploratory stage. The users are provided with the option to download the entropy and rank scores, alongside the persistent feature structure for further analysis.
We highlight the capabilities of PerSEveML by presenting three examples that utilize multi-omics data sets. Each of these data sets has varying sizes and rarity. The first data set is from a study of polychromatic flow cytometry on the rare population of human hematopoietic stem cells (HSCs).32 The cells are derived from human bone marrow cells from a single healthy donor. This data set has 44,140 data points and utilizes expression levels from thirteen (13) surface protein biomarkers to determine the presence or absence of HSC. The second data set is from a high-dimensional flow cytometry and mass cytometry (CyTOF) study on a rare population of activated (cytokine-producing) memory CD4 T cells.33 The cells in this data set are derived from human peripheral blood cells exposed to influenza antigens. To determine the presence and absence of T cells, this data set utilizes expression levels from fourteen (14) biomarkers and has 396, 460 data points. The third set of data evaluated on PerSEveML consists of proteomics data from Adams et al.34 focusing on SIN3/HDAC complexes. In this data set, the bait proteins are the features, and the prey proteins are listed in rows. The significance of bait proteins in the complex prediction of SIN3/HDAC complexes has been assessed through protein expression analysis and profiling of the interaction networks of SIN3/HDAC subunits. In summary, we demonstrated the capabilities of PerSEveML as a web tool that simplifies omics data analysis for data sets of different sizes with rare events, and enhances the understanding of biological systems.
2. Experimental
The integrative approach employed in this study unfolds in four distinct phases which are dictated by the user defined choices in the app interface. The first phase is normalization, followed by the selection of ML methods, proceeding to calculate entropy and rank scores, and culminating in the final stage—utilizing the cut-point analysis method to derive the persistent feature structure.1.1 Data preprocessing
PerSEveML, developed on the R Shiny framework, offers a user-friendly interface accommodating various file formats, including the preferred rds (R data serialization) format for handling large data sets. The users need to ensure that the binary outcome variable in their data set is in the format of zero (0) and one (1), with one (1) representing the rare event. Users can select percentages for training and test sets as part of the data preprocessing, followed by selecting the normalization techniques that are better suited for their data. Normalization plays a critical role in data preprocessing as it can reduce overfitting in ML models. PerSEveML accommodates six data normalization methods, including techniques specifically developed for particular omics data. Each of the six methods is briefly described as follows.1. Hyperbolic arcsine transformation: Hyperbolic arcsine transformation (with cofactor) is specifically designed for cytometry data to allow linearity around zero.35 Users can adjust the cofactor, although the default value is set at 150 based on previous studies.32,36 PerSEveML also allows users to perform regular arcsine transformation.
2. TopS normalization: PerSEveML features topological scoring, a method of normalization that is suitable for multi-omics data.27,28 TopS is a topological scoring method that accentuates extreme data points, thereby aiding the segregation of rare cell populations from abundant cells. TopS can effectively reduce the number of clusters for rare events; thus making it effective for analyzing biologically complex omics data sets. Let, Tij be the normalized value of ith biomarker of jth observation and Qij be the expression level of ith biomarker of jth observation. Then, TopS can be mathematically described by eqn (1).
 | (1) |
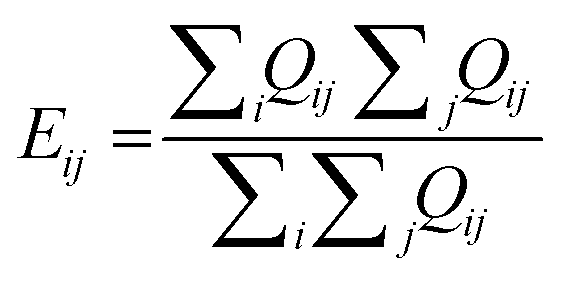 . The definitions of Tij and Qij remain consistent for the remainder of the manuscript. It is important to highlight that TopS normalization, specifically designed for biological data, uniquely addresses both between-sample and within-sample normalization needs. TopS achieves this through comprehensive row, column, and total summations. Unlike other methods that often estimate row, column, or total sums from the training set and apply them to the test set, TopS normalization operates directly on the entire data set. Thus, TopS normalization should be applied prior to train-test splitting.
. The definitions of Tij and Qij remain consistent for the remainder of the manuscript. It is important to highlight that TopS normalization, specifically designed for biological data, uniquely addresses both between-sample and within-sample normalization needs. TopS achieves this through comprehensive row, column, and total summations. Unlike other methods that often estimate row, column, or total sums from the training set and apply them to the test set, TopS normalization operates directly on the entire data set. Thus, TopS normalization should be applied prior to train-test splitting.
3. Percentage row normalization: This normalization is expected to work similarly to TopS but is designed specifically for proteomics data sets. The percentage row normalization can be defined by eqn (2).
 | (2) |
4. Log transformation: The log transformation is particularly beneficial when dealing with skewed data, as it has the capability to render transformed features resembling a Gaussian distribution.37 In the context of PerSEveML, users are granted the flexibility to introduce a constant (≥ 0.001) to their data points, ensuring the log transformation does not yield null values; thereby preserving the integrity of the analysis. This adjustment is also crucial for proper functioning within PerSEveML. Furthermore, log transformation finds application in cytometry data sets, especially when confronted with higher positive and negative intensities commonly observed in high-density multicolor flow cytometry (MFC).35
5. Min–max scaling: Min–max normalization usually scales the features between zero (0) and one (1).37,38 Min–max normalization is a common preprocessing technique among ML practitioners. The mathematical formulation of max–min normalization can be described by eqn (3).
 | (3) |
6. Standard scaling or standardization: Standardization transforms individual features into a standard normal distribution with a mean of zero (0) and a standard deviation of one (1).39 However, this type of normalization fails when the features are skewed.38 PerSEveML also included standardization or the z-score normalization.37,38Eqn (4) defines the z-score normalization mathematically.
 | (4) |
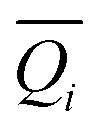 is the mean of the ith biomarker and σ(Qi) is the standard deviation of ith biomarker.
is the mean of the ith biomarker and σ(Qi) is the standard deviation of ith biomarker.
Users can choose not to normalize their data if it is already normalized or considered inappropriate for the data set. The selected normalization method is integrated into the data preprocessing stage only after the data has been divided into training and test sets, with the exception of TopS and percentage row normalization. PerSEveML extracts the mean and standard deviation for standardization, while the minimum and maximum values for min–max normalization from the training set to normalize the test set. This ensures that ML models are less prone to overfitting. Additionally, PerSEveML provides correlation plots and the option to download the correlation matrix for further analysis. These plots are valuable for comprehending the distribution of individual features within a data set and understanding the impact of the chosen normalization method. We furthermore advise users to perform external imputation methods such as KNNImput before uploading the data set into PerSEveML.
2.2. Data analysis
To strategize the performance of the integrative approach for binary classification of rare events and feature selection, PerSEveML can train twelve (12) different ML methods while utilizing the k-fold cross-validation as per user choice. The twelve (12) ML methods incorporated into PerSEveML can be categorized into three classes: Tree-based, non-tree-based, and linear classifiers. The tree-based classifiers include decision tree, random forest, XgBoost, and AdaBoost. The non-tree-based methods include naïve Bayes, linear, non-linear, and polynomial support vector machines (SVM). The four linear classifiers in the model include LDA, logistic regression, and penalized regression methods such as lasso and ridge.37–39 Based on the user selection, individual models are tuned to find the optimal hyperparameters using k-fold cross-validation. The user can select the number of folds and accept values between two (2) and ten (10). During cross-validation, it is typical to opt for either five (5) or (10) folds, as empirical evidence suggests that these values strike a balance, offering test error rate estimates that are not excessively biased or prone to high variance.Upon training the user-selected models, the model performances can be evaluated based on evaluation metrics such as sensitivity, specificity, accuracy, kappa, and ROC–AUC. In addition, based on the predictive performance of all the selected models, PerSEveML internally performs a voting classification based on the highest number of predicted classes for individual observations on the test set; thus, constructing an integrative prediction incorporating predictions from all selected models. This prediction is also compared to the observed classes on the test set. If all the selected models perform well, the integrative ML shows performance metrics closer to one, while the performance of the integrative ML model lowers when one or more models do not show good performance. PerSEveML consolidates the performance metrics of the chosen ML methods into a unified table, presenting a comprehensive overview that includes both individual ML methods and the integrated ML model. Additionally, users can download this consolidated data for in-depth analysis.
2.3. Calculation of entropy and rank scores
After training the user-selected ML models, PerSEveML computes the importance of features (variable importance) through the inherent R caret package.2,38 PerSEveML determines entropies and ranks of individual features based on their feature importance. To calculate the entropy score across the different ML methods, we adopted the mathematical formula expressed in eqn (5). | (5) |
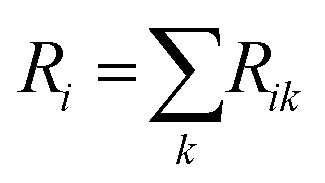 | (6) |
2.4. Persistent feature structure
Cut-point analysis is crucial in determining the persistent feature structure in PerSEveML. PerSEveML empowers users to specify the cut-off as a percentage and can be defined as the users’ interpretation of the proportion of features considered important in their study. The feature structure constructed by PerSEveML consists of three categories: Persistently selected, fluctuating, and unselected. Based on the cut-off c, PerSEveML selects the top c% of features across entropy and rank scores separately. Suppose a feature is in the top c% across both the entropy and rank scores. Then, it is designated as the persistently selected feature since it is a top predictor across many ML methods and two different methods of scoring feature importance. If a feature is not selected in the top c%, it is considered persistently unselected. Whereas features that show up in the top c% in either one of the scoring methods but fail to show up in the other are categorized as persistently fluctuating.The persistent feature structure serves as a feature selection method where the user can use the persistently selected categories to represent the features that emerged as important features in most of the ML algorithms, suggesting that the feature provides constructive information on the rare event prediction. The persistently unselected categories represent features that provided minimal information regarding the rare event, as the different ML methods indicated. Therefore, assisting researchers in formulating a plausible explanation for feature reduction. However, the most interesting category belongs to the group of fluctuating features. These dynamic features are pivotal in predicting rare events for certain methods but do not emerge as significant predictors for others. This variation suggests that the complexity of biological processes within these data sets leads different ML models to capture distinct patterns based on their computational algorithm and decision boundaries. Hence, these features provide significant hypotheses for future testing.
The application defaults to 40% as the optimal value for the cutoff for cut-point analysis. After testing various data sets within PerSEveML, we found that the optimal range for cut-point analysis is between 40–60%. Fig. 2 serves as an illustrative guide, shedding light on the working mechanism seamlessly integrated into the app.
 | ||
| Fig. 2 Workflow of PerSEveML. | ||
3. Results
3.1. Case study 1: Nilsson rare
Nilsson rare represents one large data set with 44, 140 observations and thirteen (13) biomarker expression levels. This data set was introduced in Nilsson et al.32 as manually gated data to identify the rare cell population of HSC from human bone marrow. The thirteen surface protein biomarkers included in the study are CD10, CD110, CD11b, CD123, CD19, CD3, CD34, CD38, CD4, CD45, CD45RA, CD49fpur, and CD90bio. 0.8% of the observations, or 358 instances, indicate the presence of rare HSC cells within the 44,140 total observations. Out of these thirteen (13) biomarkers, past studies have confirmed that CD90bio, CD38, and CD45RA play a significant role in identifying HSCs.40–42 While surface proteins such as CD11b are expressed on the surface of many leukocytes,43 CD45 is mainly expressed in immune cells.44Using PerSEveML, boxplots revealed that TopS, arcsine transformation with a cofactor of 150, percentage row normalization, and standard scaling yielded good results when applied to an 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20% train-test split. We utilized TopS normalization, and hyperbolic arcsine transformation with a cofactor of 150 to facilitate performance comparison within PerSEveML. Tree-based algorithms, specifically XgBoost, demonstrated commendable performance on this data set, regardless of the normalization method employed. Non-tree-based models followed in performance, with linear models showing the least favorable results. To reach these conclusions, we assessed evaluation metrics such as AUC, sensitivity, specificity, and kappa.
:20% train-test split. We utilized TopS normalization, and hyperbolic arcsine transformation with a cofactor of 150 to facilitate performance comparison within PerSEveML. Tree-based algorithms, specifically XgBoost, demonstrated commendable performance on this data set, regardless of the normalization method employed. Non-tree-based models followed in performance, with linear models showing the least favorable results. To reach these conclusions, we assessed evaluation metrics such as AUC, sensitivity, specificity, and kappa.
The performance of the integrative ML approach was highly dependent on the individual models' performance. For instance, combining a linear model like logistic regression with XgBoost negatively impacted the integrative ML's performance. However, combining XgBoost with another tree-based model, such as AdaBoost, yielded significantly better results. The feature selection process displayed variations when altering parameters like the train-test split percentage, “k” value for k-fold cross-validation, and the cut-off for cut-point analysis. However, CD90bio and CD45RA consistently appeared in the selected feature category, while CD11b and CD123 consistently fell into the unselected category. These findings align with our existing knowledge of HSCs and suggest that the combination of CD90bio, CD34, and CD45RA can reliably identify the presence of HSCs in human bone marrow.45,46 Furthermore, we noted that the majority of the biomarkers for Nilsson rare remained within similar persistent categories, as illustrated by Dutta et al.31 The persistent biomarker structure utilizing 80:20 split, TopS normalization, 5-fold cross-validation, and 40% cut-off for cut-point analysis using three ML models (XgBoost, naïve Bayes, and LDA) on ADASYN incorporated Nilsson rare data set is presented in Fig. 3a.
 | ||
| Fig. 3 (a) Persistent feature (or biomarker) structure calculated on ADASYN incorporated TopS normalized Nilsson rare data with 80:20 split, 5-fold cross-validation, and 40% cut-off for cut-point analysis using three ML models: XgBoost, naïve Bayes, and LDA. (b) Persistent feature (or biomarker) structure found with Mosmann rare data with percentage row normalization, 80:20 split, 5-fold cross-validation, and 40% cut-off for cut-point analysis using XgBoost and decision tree as ML models. (c) Persistent feature structure illustrated using TopS normalization on SIN3 protein network data, while implementing XgBoost and naïve Bayes as the preferred ML method and setting a 4-fold cross-validation with a cut-point of 30%. | ||
3.2. Case study 2: Mosmann rare
This extensive data set represents a vast flow cytometry data set comprising 396, 460 observations derived from a manually gated data set focusing on a rare population of activated (cytokine-producing) memory CD4 T cells. The data set encompasses fourteen (14) distinct biomarker expression levels. Among these biomarkers, seven (7) pertain to surface proteins, including CCL4, CD14, CD3, CD4, CD45RA, CD69, and CD8a, while the remaining seven (7) are signaling biomarkers, namely CXCR5, GZB.SA, IFNg, IL17A, IL2, IL5, and TNFa.Hundred and nine (109) cells from the Mosmann rare data set detected the presence of memory-activated CD4 T cells, highlighting an extreme class imbalance with a rarity of 0.03%. Prior research has underscored the crucial role of signaling biomarkers in identifying rare events,47–49 with biomarkers such as CD69 proving invaluable in identifying T lymphocytes and natural killer (NK) cells.50
The application of PerSEveML to the Mosmann rare data set revealed that five (5) of the six (6) normalization techniques yielded satisfactory results, with the sole exception being the log transformation. Similar to the Nilsson rare data set, our focus centered on TopS, percentage row, and hyperbolic arcsine transformations utilizing a cofactor 150 as normalization techniques. Notably, XgBoost consistently demonstrated superior performance compared to all other models.
In general, tree-based models outperformed non-tree-based or linear models. Throughout our iterations, it became evident that signaling biomarkers exhibited superior predictive capabilities compared to surface protein biomarkers. Signaling biomarkers such as IFNg, CXCR5, and TNFa consistently stood out as members of the selected category, while CD4 and GZB.SA often found themselves in the unselected category. This underscores the robust performance of PerSEveML in elucidating the underlying biology, as previously suggested by past researchers. The persistent biomarker structure using 80:20 split, percentage row normalization, 5-fold cross-validation, and 40% cut-off for cut-point analysis on the Mosmann data set using two ML methods (XgBoost and decision tree) is presented in Fig. 3b. To investigate the impact of SMOTE application on the persistent feature structure, we employed SMOTE with percentage row normalization, while utilizing 80:20 split, 5-fold cross-validation, and a 40% cut-off for cut-point analysis on the Mosmann data set. We selected XgBoost and naïve Bayes as our preferred ML methods based on their predictive performance. The ESI† Fig. S1 illustrates the feature structure. While the majority of the persistent feature structure remained similar across the two iterations, we observed differences for biomarkers CD3, CD69, IL2, and IL5, as they transitioned between persistently selected and unselected categories.
3.3. Case study 3: SIN3/HDAC proteomics network
Our latest analysis examined SIN3/HDAC data using bait proteins as features and listing prey proteins in rows. The data set contains eighteen (18) bait, and four hundred and seventy-six (476) prey proteins, representing their interactions. In this case, rare events refer to prey proteins that are subunits of the SIN3/HDAC complex, which comprise 5.8% of the data. We evaluated the importance of bait proteins in predicting SIN3/HDAC complexes by analyzing protein abundance in interaction networks of SIN3/HDAC complex using data from Adams et al.34The task of predicting protein complexes with ML is a complex and challenging one in the fields of bioinformatics and computational biology. Protein complexes are important for various cellular processes, and knowing their composition can provide valuable insights into the functioning of biological systems. However, despite numerous attempts, identifying which human proteins exist in protein complexes and how they are organized on a proteome-wide scale remains challenging.51 Recently, ML approaches such as deep learning have been recognized for their potential to predict protein complexes from protein abundances.51 SIN3/HDAC contains seven homologous pairs: SAP30/SAP30-LIKE, ING1/ING2 (1-like), BRMS1/BRMS1-LIKE, RBBP4/RBBP7, HDAC1/HDAC2, SIN3A/SIN3B, and ARID4A/ARID4B.34
The authors of Adams et al.34 showed that proteins in homologous pairs exist in mutually exclusive pairs. Additionally, there are two distinct forms of SIN3 complexes in S. cerevisiae: RPD3L (SIN3 large) and RPD3S (SIN3 small). Higher eukaryote genes encode proteins similar to components of the SIN3 complex in S. cerevisiae. In humans, there are proteins like HDAC1/HDAC2, SIN3A/SIN3B, and RBBP4/RBBP7 that have similarities to the core SIN3 complex components RPD3, SIN3, and UME1 in S. cerevisiae. Additionally, humans have proteins similar to components specific to Rpd3L and Rpd3S. For example, SUDS3/BRMS1/BRMS1L, SAP30/SAP30L, and ING1/ING2 have similarities to RPD3L-specific components SDS3, SAP30, and Pho23, respectively. Within RPD3S, components like Rco1 and Eaf3 have similarities to human PHF12 and MORF4L1, respectively. This organization of the SIN3/HDAC complex highlights its complexity, making it an excellent system for ML analysis.
It can be difficult to differentiate between persistently selected and unselected baits when predicting SIN3/HDAC subunits. We experimented with various ML techniques and normalization methods to overcome this challenge. Our findings reveal that we can effectively distinguish between baits by using TopS alongside XgBoost and naïve Bayes while setting an 80:20 train-test split, cut-point of 30% and 4-fold cross-validation (Fig. 3c). As illustrated in Fig. 3c, we successfully separated mutually exclusive pairs within our data. For instance, ARID4B was persistently unselected while ARID4A was selected. Similarly, BRMS1L and SAP130 were persistently unselected while BRMS1 was selected. For the pair SIN3A/SIN3B, SIN3A was in the fluctuating group along with SAP30 in the SAP30/SAP30L pair. Although the ING1/ING2 pair is traditionally considered mutually exclusive, our data set includes both proteins in the purifications, explaining their presence in persistently selected features. Based on the criteria mentioned earlier, it was discovered that one of the subunits of the large complex, identified as SAP130, was not selected in the persistent group. This particular subunit could not pull down some subunits that compose the SIN3/HDAC complex, distinguishing it from other baits and placing it in the persistently unselected group. In the case of the small complex, the MORF4L1 bait was separated from the PHF12 bait in the persistently unselected group, as it pulled lower abundance proteins overall compared to PHF12 bait.
Thus, these results show that our ML approach applied to protein abundances can reveal hidden features for protein complex prediction that are not easy to detect without prior knowledge.
3.4. Stability and robustness of PerSEveML
Our study builds upon the work of Dutta et al.31 by focusing on a single normalization technique. It aligns with the original findings concerning normalization techniques, the ML method selection, and the persistence of biomarker structure. Hyperbolic arcsine transformation and TopS normalization techniques proved effective for Mosmann and Nilsson rare data sets, particularly with tree-based ML algorithms, outperforming non-tree-based approaches and linear models.4. Discussion
Our research aimed to bridge the gap between the vast resources of ML methods and their applicability to rare events in complex biological processes. The distribution of these rare events presents a computational challenge as they are generally spread across multiple clusters of non-rare events. Topological methods, such as TopS normalization, can condense information related to rare events into a more manageable format for ML models. Additionally, PerSEveML integrates multiple ML methods for prediction and feature selection and offers flexibility regarding the train-test split, cross-validation folds, and cut-point analysis. PerSEveML provides complete access to figures and tables for further analysis or publication.To demonstrate the robustness of PerSEveML in modeling and visualizing results from a crowd-sourced intelligence ML approach, we used three data sets varying in size, number of biomarkers, and rarity percentage. Each data set had unique complexities due to differences in the distribution of rare events across biomarkers. However, by normalizing the data and utilizing high-performing ML methods, we drew informative conclusions on the predictive properties of biomarkers using the persistent feature structure. PerSEveML's use of entropy and rank scores allows for less rigid results than feature importance generated by individual models. PerSEveML stands out to SuperLearner24 and HTPmod,22 which are similar tools, in the context of a more robust feature selection method since it not only utilizes multiple ML methods to predict but combines the strength of pattern recognition from many ML methods to perform feature selection using the persistent feature structure.
One of the focal points of PerSEveML is the persistent feature structure. Therefore, it is crucial for users to understand the implications of this structure and its pivotal role in understanding the underlying biology. It is worth noting that certain ML methods, such as LDA and logistic regression, capture linear associations among features, while tree-based algorithms, naïve Bayes, and SVM (employing non-linear basis functions like polynomial kernel or radial basis function) consider non-linear decision boundaries. As a result, the overall persistent feature structure may comprise a combination of these linear and non-linear decision boundaries, essentially encapsulating different facets of the data. Furthermore, in situations where the feature structure is utilized to generate hypotheses for future experiments or for feature selection during the exploratory stage, users must recognize that features in the persistently fluctuating category possess some signals necessary for rare event prediction; however, due to the structure of the decision boundary of various algorithms, these features may not exhibit signals as robust as those in the persistently selected category. Thus, it is important to consider the features from the fluctuating category alongside the features from the selected categories to avoid misleading conclusions.
PerSEveML automates data analysis with a point-and-click interface. The application requires users to input display-ready data sets organized with observations in rows and features in columns, and does not offer preprocessing functionality, such as handling missing data. Based on preferences, we suggest the users perform imputation methods such as KNNImput, prior to uploading the data set into PerSEveML. Users must also select ML methods that work best for their data sets and assess multicollinearity prior to training final models to avoid drawing invalid conclusions. Understanding the model performances and deciding whether to include individual ML models in the final analysis is at the discretion of the user.
Even though PerSEveML was built on our previous work of Dutta et al.31 PerSEveML's computational framework focuses on faster computation and easy ML implementation in analyzing rare events. For instance, we decided to work extensively with a single normalization method. Even though implementing TopS and percentage row normalization is time-consuming for very large data sets, we decided to include them in the application since these normalization techniques are extremely important when working with omics data sets. In addition, unlike Dutta et al.31 we have not included KNN as a part of PerSEveML due to two reasons: during the app development stage, we found that KNN takes a significantly longer time to tune parameters; secondly, for none of our test cases the algorithm showed optimal performance. Another deviation from the original work is related to the two methods of calculating feature importance—one via the inbuilt feature importance method from the caret package, and the other using the stepwise ROC method. To enhance the user experience of our application, we have removed the stepwise ROC analysis feature from PerSEveML due to its considerable computational demands.
As demonstrated through the examples of Nilsson and Mosmann rare data sets, we found that PerSEveML could capture all the major findings from past articles.31 Users can leverage PerSEveML in combination with different ML methods and various normalization techniques to uncover hidden patterns. For users keen on exploring the original computational framework with two normalizations, PerSEveML offers easy access to entropy and rank scores. By readily downloading these scores, users can seamlessly implement the original author's approach31 and discover a more robust version of the persistent biomarker structure.
Our study affirms the robustness of PerSEveML in identifying relevant biomarker structures and detecting subtle shifts in their categorization. Additionally, it showcases PerSEveML's ability to analyze intricate data structures such as stem cells that belong to multiple clustering groups and protein complexes consisting of modules with shared subunits and mutually exclusive pairs. By refining and expanding our understanding of these persistent features, PerSEveML stands as a valuable tool for unraveling the complexities of biomarker-driven phenomena in various domains of network-based research.
We envision that additional applications will be added to the PerSEveML app in the near future. These include perturbation data prediction, disease survival outcome prediction based on omics data sets, and neuroimaging data with genomics profiles.
Author contributions
SD developed the PerSEveML R Shiny application. MS, YL, and DM performed application testing. SD, MS, and YL wrote the manuscript. DM deployed the application on the server. All authors reviewed the manuscript.Data availability
The mentioned Nilsson and Mosmann data sets are available publicly in FlowRepository (https://flowrepository.org/), repository FR-FCM-ZZPH in FSC format and can be accessed through Spidlen et al.52 All FSC files were converted into CSV format prior to usage in PerSEveML. While SIN3 data set can be accessed directly from the supplementary materials associated with Adams et al.34The PerSEveML tool is accessible for free at https://biostats-shinyr.kumc.edu/PerSEveML/. For handling larger data sets, we recommend downloading the application from GitHub (https://github.com/sreejatadutta/PerSEveML) and running it locally on your system. Note that performing resampling methods on large data sets require more computation time.
Conflicts of interest
The author(s) declare no competing interests.Acknowledgements
The research reported in this publication was supported by the University of Kansas Cancer Center (KUCC) pilot project. The high-performance computing resources used in this study was supported by the K-INBRE Bioinformatics Core, which is supported in part by the National Institute of General Medical Science award (P20 GM103418), the Biostatistics and Informatics Shared Resource, supported by the National Cancer Institute Cancer Center Support Grant (P30 CA168524), and the Kansas Institute for Precision Medicine COBRE, supported by the National Institute of General Medical Science award (P20 GM130423).References
- N. Erfanian, A. A. Heydari, A. M. Feriz, P. Iañez, A. Derakhshani, M. Ghasemigol, M. Farahpour, S. M. Razavi, S. Nasseri, H. Safarpour and A. Sahebkar, Biomed. Pharmacother., 2023, 165, 115077 CrossRef CAS PubMed.
- Z. Hu, S. Bhattacharya and A. J. Butte, Front. Immunol., 2022, 12 DOI:10.3389/fimmu.2021.787574.
- E. Sussano, Electronic Theses and Dissertations Search PubMed.
- W. Choe, O. K. Ersoy and M. Bina, Bioinformatics, 2000, 16, 1062–1072 CrossRef CAS PubMed.
- H. S. R. Rajula, G. Verlato, M. Manchia, N. Antonucci and V. Fanos, Medicina, 2020, 56, 455 CrossRef PubMed.
- L. Zhou, S. Pan, J. Wang and A. V. Vasilakos, Neurocomputing, 2017, 237, 350–361 CrossRef.
- C. M. Micheel, S. J. Nass and G. S. Omenn, Evolution of translational omics lessons learned and the path forward, National Academies Press, 2021 Search PubMed.
- S. Girirajan, J. A. Rosenfeld, B. P. Coe, S. Parikh, N. Friedman, A. Goldstein, R. A. Filipink, J. S. McConnell, B. Angle, W. S. Meschino, M. M. Nezarati, A. Asamoah, K. E. Jackson, G. C. Gowans, J. A. Martin, E. P. Carmany, D. W. Stockton, R. E. Schnur, L. S. Penney, D. M. Martin, S. Raskin, K. Leppig, H. Thiese, R. Smith, E. Aberg, D. M. Niyazov, L. F. Escobar, D. El-Khechen, K. D. Johnson, R. R. Lebel, K. Siefkas, S. Ball, N. Shur, M. McGuire, C. K. Brasington, J. E. Spence, L. S. Martin, C. Clericuzio, B. C. Ballif, L. G. Shaffer and E. E. Eichler, N. Engl. J. Med., 2012, 367, 1321–1331 CrossRef CAS PubMed.
- R. Redon, S. Ishikawa, K. R. Fitch, L. Feuk, G. H. Perry, T. D. Andrews, H. Fiegler, M. H. Shapero, A. R. Carson, W. Chen, E. K. Cho, S. Dallaire, J. L. Freeman, J. R. González, M. Gratacòs, J. Huang, D. Kalaitzopoulos, D. Komura, J. R. MacDonald, C. R. Marshall, R. Mei, L. Montgomery, K. Nishimura, K. Okamura, F. Shen, M. J. Somerville, J. Tchinda, A. Valsesia, C. Woodwark, F. Yang, J. Zhang, T. Zerjal, J. Zhang, L. Armengol, D. F. Conrad, X. Estivill, C. Tyler-Smith, N. P. Carter, H. Aburatani, C. Lee, K. W. Jones, S. W. Scherer and M. E. Hurles, Nature, 2006, 444, 444–454 CrossRef CAS PubMed.
- E. T. Wang, R. Sandberg, S. Luo, I. Khrebtukova, L. Zhang, C. Mayr, S. F. Kingsmore, G. P. Schroth and C. B. Burge, Nature, 2008, 456, 470–476 CrossRef CAS PubMed.
- N. L. Barbosa-Morais, M. Irimia, Q. Pan, H. Y. Xiong, S. Gueroussov, L. J. Lee, V. Slobodeniuc, C. Kutter, S. Watt, R. Çolak, T. Kim, C. M. Misquitta-Ali, M. D. Wilson, P. M. Kim, D. T. Odom, B. J. Frey and B. J. Blencowe, Science, 1979, 2012(338), 1587–1593 Search PubMed.
- T. Maier, M. Güell and L. Serrano, FEBS Lett., 2009, 583, 3966–3973 CrossRef CAS PubMed.
- S. Ghaemmaghami, W.-K. Huh, K. Bower, R. W. Howson, A. Belle, N. Dephoure, E. K. O’Shea and J. S. Weissman, Nature, 2003, 425, 737–741 CrossRef CAS PubMed.
- M. Mann and O. N. Jensen, Nat. Biotechnol., 2003, 21, 255–261 CrossRef CAS PubMed.
- C. Choudhary, C. Kumar, F. Gnad, M. L. Nielsen, M. Rehman, T. C. Walther, J. V. Olsen and M. Mann, Science, 1979, 2009(325), 834–840 Search PubMed.
- H. He and E. A. Garcia, IEEE Trans. Knowl. Data Eng., 2009, 21, 1263–1284 Search PubMed.
- S. Yadav and G. P. Bhole, in 2020 IEEE Pune Section International Conference (PuneCon), 2020, pp. 38–43.
- H. He, Y. Bai, E. A. Garcia and S. Li, in 2008 IEEE International Joint Conference on Neural Networks (IEEE World Congress on Computational Intelligence), 2008, pp. 1322–1328.
- N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, J. Art. Intell. Res., 2002, 16, 321–357 Search PubMed.
- N. V. Chawla, A. Lazarevic, L. O. Hall and K. W. Bowyer, in Knowledge Discovery in Databases: PKDD 2003:7th European Conference on Principles and Practice of Knowledge Discovery in Databases, Cavtat-Dubrovnik, Croatia, September 22-26, 2003. Proceedings 7, 2003, pp. 107–119.
- C. Seiffert, T. M. Khoshgoftaar, J. Van Hulse and A. Napolitano, IEEE Trans. Syst. Man Cybern, 2010, 40, 185–197 Search PubMed.
- D. Chen, L.-Y. Fu, D. Hu, C. Klukas, M. Chen and K. Kaufmann, Commun. Biol., 2018, 1, 89 CrossRef PubMed.
- S. Ghosh, A. Datta and H. Choi, Nat. Commun., 2021, 12, 2279 CrossRef CAS PubMed.
- M. J. van der Laan, E. C. Polley and A. E. Hubbard, Stat. Appl. Genet. Mol. Biol., 2007, 6 DOI:10.2202/1544-6115.1309.
- J. E. Evangelista, Z. Xie, G. B. Marino, N. Nguyen, D. J. B. Clarke and A. Ma’ayan, Nucleic Acids Res., 2023, 51, W168–W179 CrossRef CAS PubMed.
- I. Walsh, D. Fishman, D. Garcia-Gasulla, T. Titma, G. Pollastri, E. Capriotti, R. Casadio, S. Capella-Gutierrez, D. Cirillo, A. Del Conte, A. C. Dimopoulos, V. D. Del Angel, J. Dopazo, P. Fariselli, J. M. Fernández, F. Huber, A. Kreshuk, T. Lenaerts, P. L. Martelli, A. Navarro, P. Ó. Broin, J. Piñero, D. Piovesan, M. Reczko, F. Ronzano, V. Satagopam, C. Savojardo, V. Spiwok, M. A. Tangaro, G. Tartari, D. Salgado, A. Valencia, F. Zambelli, J. Harrow, F. E. Psomopoulos and S. C. E. Tosatto, Nat. Methods, 2021, 18, 1122–1127 CrossRef CAS PubMed.
- M. E. Sardiu, J. M. Gilmore, B. D. Groppe, A. Dutta, L. Florens and M. P. Washburn, Nat. Commun., 2019, 10, 1118 CrossRef PubMed.
- M. E. Sardiu, A. C. Box, J. S. Haug and M. P. Washburn, Mol. Omics, 2021, 17, 59–65 RSC.
- J. Hancock and T. M. Khoshgoftaar, in 2021 IEEE 22nd International Conference on Information Reuse and Integration for Data Science (IRI), IEEE, 2021, pp. 348–354.
- B. H. Shekar and G. Dagnew, in 2019 Second International Conference on Advanced Computational and Communication Paradigms (ICACCP), IEEE, 2019, pp. 1–8.
- S. Dutta, A. C. Box, Y. Li and M. E. Sardiu, Proteomics, 2023, 23 DOI:10.1002/pmic.202200290.
- A. Rundberg Nilsson, D. Bryder and C. J. H. Pronk, Cytometry, Part A, 2013, 83A, 721–727 CrossRef PubMed.
- L. M. Weber and M. D. Robinson, Cytometry, Part A, 2016, 89, 1084–1096 CrossRef CAS PubMed.
- M. K. Adams, C. A. S. Banks, J. L. Thornton, C. G. Kempf, Y. Zhang, S. Miah, Y. Hao, M. E. Sardiu, M. Killer, G. L. Hattem, A. Murray, M. L. Katt, L. Florens and M. P. Washburn, Mol. Cell. Proteomics, 2020, 19, 1468–1484 CrossRef CAS PubMed.
- R. Folcarelli, S. van Staveren, G. Tinnevelt, E. Cadot, N. Vrisekoop, L. Buydens, L. Koenderman, J. Jansen and O. F. van den Brink, Cytometry, Part A, 2022, 101, 72–85 CrossRef CAS PubMed.
- L. M. Weber and M. D. Robinson, Cytometry, Part A, 2016, 89, 1084–1096 CrossRef CAS PubMed.
- J. Gareth, D. Witten, T. Hastie and R. Tibshirani, An Introduction to Statistical Learning: with Applications in R, Springer Texts in Statistics, 2nd edn, 2021 Search PubMed.
- A. Géron, Hands-On Machine Learning with Scikit-Learn, Keras, and TensorFlow: Concepts, Tools, and Techniques to Build Intelligent Systems, O’Reilly, 2nd edn, 2019 Search PubMed.
- G. Casella and R. L. Berger, Statistical Inference, 2nd edn, 2002 Search PubMed.
- C. Sauzay, K. Voutetakis, A. Chatziioannou, E. Chevet and T. Avril, Front. Cell Dev. Biol., 2023, 11 DOI:10.3389/fcell.2019.00066.
- Q. Cui, C. Qian, N. Xu, L. Kang, H. Dai, W. Cui, B. Song, J. Yin, Z. Li, X. Zhu, C. Qu, T. Liu, W. Shen, M. Zhu, L. Yu, D. Wu and X. Tang, J. Hematol. Oncol., 2021, 14, 82 CrossRef CAS PubMed.
- B. Kersten, M. Valkering, R. Wouters, R. van Amerongen, D. Hanekamp, Z. Kwidama, P. Valk, G. Ossenkoppele, W. Zeijlemaker, G. Kaspers, J. Cloos and G. J. Schuurhuis, Br. J. Haematol., 2016, 173, 219–235 CrossRef CAS PubMed.
- Y.-L. Zhang, J. Bai, W.-J. Yu, Q.-Y. Lin and H.-H. Li, J. Adv. Res., 2024, 55 DOI:10.1016/j.jare.2023.02.010.
- D. Leitenberg, R. Falahati, D. D. Lu and A. Takeda, Immunology, 2007, 121, 545–554 CrossRef CAS PubMed.
- S. Radtke, L. Colonna, A. M. Perez, M. Hoffman, L. S. Kean and H.-P. Kiem, Transplant. Direct, 2020, 6, e579 CrossRef CAS PubMed.
- D. Wisniewski, M. Affer, J. Willshire and B. Clarkson, Blood Cancer J., 2011, 1, e36–e36 CrossRef CAS PubMed.
- S. A. Ghanekar, L. E. Nomura, M. A. Suni, L. J. Picker, H. T. Maecker and V. C. Maino, Clin. Diagn. Lab. Immunol., 2001, 8, 628–631 CrossRef CAS PubMed.
- F. Castro, A. P. Cardoso, R. M. Gonçalves, K. Serre and M. J. Oliveira, Front. Immunol., 2018, 9 DOI:10.3389/fimmu.2018.00847.
- W. Cao, Y. Chen, S. Alkan, A. Subramaniam, F. Long, H. Liu, R. Diao, T. Delohery, J. McCormick, R. Chen, D. Ni, P. S. Wright, X. Zhang, S. Busch and A. Zilberstein, Eur. J. Immunol., 2005, 35, 2709–2717 CrossRef CAS PubMed.
- S. F. Ziegler, F. Ramsdell and M. R. Alderson, Stem Cells, 1994, 12, 456–465 CrossRef CAS PubMed.
- B. Li, M. Altelaar and B. van Breukelen, Int. J. Mol. Sci., 2023, 24, 7884 CrossRef CAS PubMed.
- J. Spidlen, K. Breuer, C. Rosenberg, N. Kotecha and R. R. Brinkman, Cytometry, Part A, 2012, 81A, 727–731 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mo00008k |
| This journal is © The Royal Society of Chemistry 2024 |