
Realizing long-term cycling stability of O3-type layered oxide cathodes for sodium-ion batteries†
Guohua
Zhang
a,
Yuheng
Gao
b,
Ping
Zhang
a,
Yuheng
Gao
a,
Jingrong
Hou
a,
Xuemin
Shi
a,
Jiwei
Ma
 a,
Renyuan
Zhang
*a and
Yunhui
Huang
*b
a,
Renyuan
Zhang
*a and
Yunhui
Huang
*b
aInstitute of New Energy for Vehicles, Shanghai Key Laboratory for R&D and Application of Metallic Functional Materials, School of Materials Science and Engineering, Tongji University, Shanghai 201804, China. E-mail: ryzhang@tongji.edu.cn
bState Key Laboratory of Material Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China. E-mail: huangyh@hust.edu.cn
First published on 10th June 2024
Abstract
O3-type layered oxide cathodes are promising for practical sodium-ion batteries (SIBs) owing to their high theoretical capacity, facile synthesis, and sufficient Na+ storage. However, they face challenges such as rapid capacity loss and poor cycling stability, mainly attributed to irreversible phase transitions. To address these challenges, a novel cathode material, Li/Sn co-substituted O3-Na0.95Li0.07Sn0.01Ni0.22Fe0.2Mn0.5O2 (LSNFM), has been designed by regulating the electronic structure, in which Li+ activates more redox reactions of Ni2+/3+ and Fe3+/4+ above 2.5 V and suppresses the redox reactivity of Mn3+/4+ below 2.5 V, while Sn4+ can prevent the charge delocalization in the transition metal layer, contributing to structural stability. Due to this synergistic effect, the as-prepared LSNFM electrode with high structural reversibility displays a 27.2% capacity increase contributed by the high-voltage transition metal ion redox activity and exhibits excellent long-term cycling stability, an 84.0% capacity retention after 500 cycles at 1 C and an 84.7% capacity retention after 2000 cycles at 5 C. The fundamental mechanism is fully investigated using systematic in situ/ex situ characterization techniques and density functional theory computations. This work provides a paradigm for designing long-term cycle life cathode materials by synergistically regulating the electronic structure in practical SIBs.
New conceptsO3-type Mn-based layered oxide cathodes have shown great application potential in practical sodium ion batteries (SIBs). However, they usually suffer from rapid capacity decay and poor cycle life, primarily due to irreversible phase transitions caused by the Jahn–Teller effect of Mn3+. To address this challenge, we designed a novel Na0.95Li0.07Sn0.01Ni0.22Fe0.2Mn0.5O2 (LSNFM) cathode to effectively inhibit the irreversible phase transition by regulating the electronic structure. This approach activated more transition metal ion (Ni2+/3+ and Fe3+/4+) redox reactions in the high-voltage area and suppressed the redox reactivity of Mn3+/4+ below 2.5 V, eventually resulting in a 27.2% capacity increase. Besides, Sn4+ can prevent the charge delocalization in the transition metal layer and contribute to improved structural stability. Due to this synergistic effect, the LSNFM electrode exhibited high structural reversibility and excellent long-term cycle life. This work reveals the structure–performance relationship between the electronic structure, structural reversibility, and electrochemical properties, which provides a paradigm for designing long-term cycle life cathode materials by regulating the electronic structure in practical SIBs. |
Introduction
Sodium-ion batteries (SIBs) have shown great application potential in large-scale energy storage systems because of their low cost, high safety, and rich sodium resource, which have attracted unprecedented attention.1–3 Among the crucial components of SIBs, cathode materials, including layered oxides, polyanionic frameworks, Prussian blue analogues, and organic compounds, have been extensively studied.4–8 In particular, layered oxide cathode materials stand out as promising candidates due to their high theoretical capacity, flexible composition, and straightforward synthesis process.9 However, they face challenges such as progressive capacity degradation and poor cycling stability, especially evident in long-term cycling performance, which significantly impedes their commercialization applications.2,10According to the coordination environment of sodium ions and the oxygen stacking sequence, layered oxides (NaxTMO2, 0 < x ≤ 1, TM = transition metal) can be mainly classified into P2 type and O3 type.11 By comparison, O3-type layered oxides are more suitable for practical full-cell systems due to the sufficient Na+ storage in the initial framework.12 However, they generally suffer from irreversible phase transitions, poor structural stability and sluggish kinetics during sodiation/desodiation processes, leading to rapid degradation of the electrochemical performance, especially over long-term cycling.2,4,13,14 Various strategies have been attempted to address these challenges, including surface coating,15,16 doping,14,17 high entropy design,18–21 and construction of biphase structures.22–24 Nonetheless, the O3–P3 phase transition occurs in nearly all O3-type layered oxides during Na+ deinsertion/insertion processes, as it is easily triggered by a simple gliding of TMO2 layers without breaking TM–O bonds.25 It is almost impossible to avoid this phase transition during Na+ (de)intercalation processes, making the improvement of structural reversibility crucial. In Mn-based oxide cathodes, structural irreversibility is always associated with the Jahn–Teller effect of Mn3+.13,26 The substitution of monovalent Li+ has been proven to effectively reduce the electrochemical activity of Mn3+ below 2.5 V and activate more redox reactions of TM ions in the high-voltage region by regulating the electronic structure. For example, Ding et al. reported a high-entropy Na0.95Li0.07Cu0.11Ni0.11Fe0.3Mn0.41O1.97F0.03 cathode, in which the Li+ substitution increased the ratio to facilitate the high voltage TM redox activity, resulting in an approximately 29% increase in capacity.27 Furthermore, the Sn elemental substitution strategy has been demonstrated to achieve high structural reversibility by suppressing the gliding of TM layers and regulate redox potential through effectively manipulating hybridization between the orbitals of oxygen and TM atoms. Sn4+ with filled d10 electrons ([Kr] 4d10), a non-transition metal ion, is formed when the four outermost electrons are lost. According to Hund's rule, prior to d orbital splitting, the five d orbitals of Sn4+ are degenerate, and the electrons occupy different orbitals with parallel spins, resulting in only one type of arrangement. The various ligand environments that influence the characteristics of TM complexes can be observed in d orbital splitting diagrams. When d orbital splitting occurs, there are no single electrons on the Sn eg and Sn t2g orbitals. Therefore, Sn4+ lacks single d electrons and is thus unable to participate in hybridization with O 2p orbitals, hence reducing orbital overlap and inhibiting the charge delocalization in the metallic layer. This effect benefits the increase of bond iconicity and the redox potential, as well as the improvement of structural stability.25,28,29 Therefore, doping Sn4+ into NaxTMO2 has been recognized as an effective approach to raise the cell voltage and suppress phase transitions.30–33 Hu et al. investigated the effect of Sn substitution on the O3-NaNi0.5Mn0.5−xSnxO2 cathode, which not only effectively restrained irreversible multiphase transformation but also manipulated redox potential and stabilized anion redox reactions, resulting in competitive electrochemical performance.34 However, the Li/Sn synergistic effect based on the electronic structure regulation of O3-type Mn-based oxides remains unexplored.
Herein, we designed a novel Na0.95Li0.07Sn0.01Ni0.22Fe0.2Mn0.5O2 (LSNFM) cathode by regulating the electronic structure through Li/Sn co-substitution. The low-valence Li+ was strategically introduced to activate more redox reactions of Ni2+/3+ and Fe3+/4+ above 2.5 V and suppress the Jahn–Teller effect of Mn3+ below 2.5 V, while Sn4+ prevents charge delocalization in the TM layer and contributes to improved structural stability. The synergistic effect of Li/Sn co-substitution was comprehensively investigated via systematic in situ/ex situ characterization and density functional theory computations. Consequently, the LSNFM cathode exhibited high structural reversibility, exceptional long-term cycling stability, and significantly enhanced rate performance. This work presents a practical strategy to synergistically regulate the electronic structure of O3-type layered oxides and provides a new perspective for designing cathode materials with long-term cycle life.
Results and discussion
Physical characterization
Na0.95Ni0.22Fe0.28Mn0.5O2 (NFM), Na0.95Sn0.01Ni0.22Fe0.27Mn0.5O2 (SNFM), Na0.95Li0.07Ni0.22Fe0.21Mn0.5O2 (LNFM), and Na0.95Li0.07Sn0.01Ni0.22Fe0.2Mn0.5O2 (LSNFM) cathode materials were synthesized via a simple solid-state method. Fig. 1a–d display the corresponding XRD Rietveld refinement patterns, and all the diffraction peaks could be well indexed to the rhombohedral symmetry O3-type structure (α-NaFeO2, JCPDS no. 01-082-1495) with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The specific refined results are listed in Table S1 (ESI†). All the R-variance factors are within reasonable ranges, indicating the reliability of the calculated results. The comparisons of the lattice parameters among the four samples are shown in Table S2 (ESI†). The a values of the NFM, SNFM, LNFM, and LSNFM samples are 2.9486, 2.9522, 2.9455, and 2.9506 Å, respectively, while the c values of the four samples are 16.3253, 16.3579, 16.2028, and 16.2188 Å, respectively. The results demonstrate that incorporating Sn4+ in Fe sites causes a left shift of the (003) diffraction peaks, resulting in an expansion of the a- and c-lattice parameters, which can be attributed to the higher bonding energy of Sn–O (528 kJ mol−1) than that of Fe–O (407 kJ mol−1) and the larger radius of Sn4+ (0.69 Å) than that of Fe3+ (0.645 Å).30,34,35 As shown in Fig. 1e, the (003) diffraction peak shifts to a higher degree after Li substitution, leading to the contraction of the c-lattice parameter, because of the increased electrostatic attraction to the neighboring oxygen layers and reduced electrostatic repulsion among oxygen anions in the NaO2 layer,17,27 consistent with the calculated results shown in Fig. 1f. The lengths of Mn–O bonds and distances of Na+ layers from Rietveld refinement results are displayed in Table S3 (ESI†). The illustration of lattice evolution in Fig. 1g demonstrates that the interlayer spacing of TM slabs (dTM), dO–Na–O, and the ratio of dTM/dO–Na–O all decrease after Li/Sn co-substitution, suggesting the enhanced structure stability.17 Additionally, the decrease of dMn–O from 2.0335 to 2.0305 Å signifies that Li/Sn co-substitution suppresses the Jahn–Teller distortion of Mn3+.
m. The specific refined results are listed in Table S1 (ESI†). All the R-variance factors are within reasonable ranges, indicating the reliability of the calculated results. The comparisons of the lattice parameters among the four samples are shown in Table S2 (ESI†). The a values of the NFM, SNFM, LNFM, and LSNFM samples are 2.9486, 2.9522, 2.9455, and 2.9506 Å, respectively, while the c values of the four samples are 16.3253, 16.3579, 16.2028, and 16.2188 Å, respectively. The results demonstrate that incorporating Sn4+ in Fe sites causes a left shift of the (003) diffraction peaks, resulting in an expansion of the a- and c-lattice parameters, which can be attributed to the higher bonding energy of Sn–O (528 kJ mol−1) than that of Fe–O (407 kJ mol−1) and the larger radius of Sn4+ (0.69 Å) than that of Fe3+ (0.645 Å).30,34,35 As shown in Fig. 1e, the (003) diffraction peak shifts to a higher degree after Li substitution, leading to the contraction of the c-lattice parameter, because of the increased electrostatic attraction to the neighboring oxygen layers and reduced electrostatic repulsion among oxygen anions in the NaO2 layer,17,27 consistent with the calculated results shown in Fig. 1f. The lengths of Mn–O bonds and distances of Na+ layers from Rietveld refinement results are displayed in Table S3 (ESI†). The illustration of lattice evolution in Fig. 1g demonstrates that the interlayer spacing of TM slabs (dTM), dO–Na–O, and the ratio of dTM/dO–Na–O all decrease after Li/Sn co-substitution, suggesting the enhanced structure stability.17 Additionally, the decrease of dMn–O from 2.0335 to 2.0305 Å signifies that Li/Sn co-substitution suppresses the Jahn–Teller distortion of Mn3+.
 | ||
| Fig. 1 Rietveld refinements of the XRD patterns of the as-synthesized cathode materials: (a) NFM, (b) SNFM, (c) LNFM and (d) LSNFM. (e) Enlarged view of the (003) diffraction peaks in the 2θ range from 15.9° to 16.9°. (f) Changes of the lattice parameters. (g) Illustration of the crystallographic evolution after Li/Sn co-substitution. | ||
High-resolution transmission electron microscopy (HR-TEM) was utilized to further confirm the change of the lattice structure at the atomic level after substitution. The HR-TEM images in Fig. 2a and b reveal that the interlayer spacing along the (003) plane of the NFM sample is 5.461 Å, whereas that of the LSNFM sample slightly reduces to 5.409 Å. This contraction aligns with the findings from X-ray diffraction (XRD) refinement results. Additionally, the scanning electron microscopy–energy dispersive spectroscopy (SEM–EDS) analysis was conducted to examine the local morphology and elemental distribution of NFM and LSNFM, as depicted in Fig. 2c and Fig. S1 (ESI†). The SEM–EDS images show that both NFM and LSNFM consist of flaky particles with sizes ranging from 3 to 5 μm. The elements Na, Sn, Ni, Fe, and Mn are evenly dispersed across the entire secondary particles with no discernible segregation observed.
 | ||
| Fig. 2 High-resolution TEM images of (a) NFM and (b) LSNFM samples. (c) SEM–EDS mapping images of the LSNFM sample. | ||
Electrochemical performances
The electrochemical performances of the as-prepared samples were evaluated in half cells versus sodium metal over the potential range of 2.0 to 4.0 V. As depicted in Fig. 3a, the LSNFM electrode exhibits a 27.2% capacity increase after Li/Sn co-substitution with the lowest polarization (85.6 mA h g−1 for NFM, 89.3 mA h g−1 for SNFM, 100.1 mA h g−1 for LNFM, and 109 mA h g−1 for LSNFM). Compared to the smooth galvanostatic charge/discharge (GCD) curves of NFM and SNFM electrodes, those of LNFM and LSNFM cathodes display a noticeable voltage plateau at 2.85 V, indicating that more TM ions participate in redox reactions in the high-voltage region, as evidenced by the following dQ/dV and cyclic voltammetry results. As shown in the inset of Fig. 3a, the region below 2.5 V can be considered as the redox reaction of Mn3+/4+.36,37 The LSNFM electrode exhibits a significantly lower specific capacity below 2.5 V compared to the NFM electrode, indicating that fewer Mn3+/4+ redox pairs participate in the electrochemical redox reaction. The combination of these two phenomena not only improves the reversible specific capacity of the LSNFM electrode, but also inhibits the Jahn–Teller distortion of Mn3+ to enhance the structural stability. | ||
| Fig. 3 (a) Galvanostatic charge/discharge curves in the first cycle at 0.1 C for the four as-prepared electrodes. Inset: The capacity contributions at different voltage stages. Galvanostatic charge/discharge profiles and the corresponding dQ/dV curves of (b) and (d) NFM and (c) and (e) LSNFM electrodes at 0.1 C in the first 20 cycles. (f) Rate performance at various rates. CV curves of (g) NFM and (h) LSNFM electrodes at a scan rate of 0.1 mV s−1 during the first ten cycles. (i) and (j) Cycling performances at 1 C and 5 C, respectively. | ||
The charge/discharge curves of NFM and LSNFM electrodes for the first 20 cycles at 0.1 C are shown in Fig. 3b and c. It is clearly demonstrated that the capacity attenuation of the LSNFM electrode is much slower than that of the NFM electrode, revealing enhanced reversibility and cycling stability after Li/Sn co-substitution. The corresponding differential capacity (dQ/dV) curves are shown in Fig. 3d and e. Clearly, the strength of the redox peaks at 2.87/3.00 V for the LSNFM electrode is much higher than that for the NFM electrode, indicating that more Ni2+ participates in redox reactions in the high-voltage region, coinciding with the GCD curves. In addition, the voltage gap between the oxidation and reduction peaks of Ni2+/3+ is only 0.10 V for the LSNFM electrode, much smaller than that for the NFM electrode (0.18 V), suggesting less polarization and better reversibility after Li/Sn co-substitution. On the other hand, the Mn3+/4+ redox peaks of the NFM electrode below 2.5 V are much stronger than those of the LSNFM electrode, indicating that the Li/Sn co-substitution inhibits the reaction activity of Mn3+/4+, then suppresses the Jahn–Teller distortion of Mn3+ and improves the structural stability.
Cyclic voltammetry (CV) was further used to confirm the abovementioned conclusions. Fig. 3g and h depict the CV profiles of NFM and LSNFM electrodes at a scan rate of 0.1 mV s−1 within the voltage range of 2.0–4.0 V during the initial ten cycles. Notably, the intensity of cathodic and anodic peaks in the LSNFM electrode is obviously elevated at above 2.5 V after Li/Sn co-substitution, suggesting an enhancement of redox activity. The redox peaks observed at 3.18 V/2.67 V can be attributed to the Ni2+/3+ couple,36 while those at 3.65 V/3.52 V can be identified as the Fe3+/4+ couple.37,38 The redox peaks below 2.5 V attributed to the Mn3+/4+ couple are barely visible for the LSNFM electrode, whereas the NFM electrode exhibits relatively stronger peaks.36,37 In addition, the voltage gap between the oxidation and reduction peaks of Ni2+/3+ is only 0.28 V after 10 cycles for the LSNFM electrode, much smaller than that for the NFM electrode (0.40 V), suggesting less polarization. Evidently, Li/Sn co-substitution effectively stimulates more TM ion redox reactions (Ni2+/3+ and Fe3+/4+) above 2.5 V, decreases the polarization and suppresses the activity of Mn3+ below 2.5 V, which is consistent with the abovementioned electrochemical results shown in Fig. 3a–e.
The impact of Li/Sn co-substitution on Na+ diffusion kinetics was further investigated using the galvanostatic intermittent titration technique (GITT). The charge/discharge GITT curves in the first cycle are displayed in Fig. S3 (ESI†), and the corresponding DNa+ values calculated as a function of testing time during the charge/discharge process are plotted in Fig. S4 (ESI†). Notably, apart from the late stage of discharge, the LSNFM electrode exhibits higher DNa+ values compared to the NFM electrode throughout almost all the tested periods, indicating that Li/Sn co-substitution enhances Na+ diffusion kinetics in the high-voltage region. Conversely, at the late stage of discharge, the NFM electrode exhibits higher DNa+ values, which could be ascribed to the stronger Jahn–Teller effect to accelerate Na+ diffusion.39 Besides, inflection points at approximately 3.00 V during charging and 2.86 V during discharging corresponded to the phase transitions evidenced by in situ XRD analysis below. The decrease of DNa+ within these two periods can be attributed to the sluggish Na+ transfer within the O3 phase relative to the P3 phase. Therefore, as shown in Fig. 3f, the specific capacities of the LSNFM electrode are 108.5, 106.4, 101.6, 96.7, 89.9, 75.3, and 57.3 mA h g−1 at 0.1, 0.2, 0.5, 1, 2, 5, and 10 C, respectively. By contrast, the LSNFM electrode exhibits the best rate performance with an 82.9% capacity retention (2 C/0.1 C), whereas the NFM, SNFM, and LNFM electrodes show only 61.8%, 63.2% and 81.1%, respectively.
Furthermore, the LSNFM electrode presents a capacity retention of 84.0% after 500 cycles at 1 C and exhibits the best cycling performance compared with the other three electrodes (Fig. 3i). In addition, the excellent long-term cycling performance of the LSNFM electrode at a high rate is demonstrated in Fig. 3j. Even after 2000 cycles at 5 C, the LSNFM electrode exhibits an 84.7% capacity retention, whereas the NFM, SNFM and LNFM electrodes show only 33.6%, 58.3%, and 71.4% retention, respectively. Besides, the coulombic efficiency of the NFM electrode fluctuates continuously with cycling, whereas the LSNFM electrode keeps a steady trend. The cycling performances of the recent representative and important layered oxide cathode materials are listed in Table S4 (ESI†). By comparison, the LSNFM material in our work exhibits the best long-term cycling performance among all the reported literature studies to date.
In a word, the above results indicate that the Li/Sn co-substitution suppresses the Jahn–Teller distortion of Mn3+ and activates more high-voltage TM ion redox reactions, resulting in the enhancement of structural stability and electrochemical performances. Meanwhile, it is evidenced that Li/Sn co-substitution improves the dynamic performance. It is important to emphasize that the LSNFM material has not undergone any form of optimization or modification, such as surface coatings, implying that there is considerable potential for further enhancement in its electrochemical performance to meet the requirement of practical SIBs.
Charge transfer and structural evolution
Ex situ XPS measurements were conducted to investigate the redox mechanism and charge compensation during the charge/discharge process by observing the valence state changes of surface elements. Fig. 4a and b display the Mn 2p spectra of NFM and LSNFM samples, respectively. The deconvolutions of these spectra reveal four characteristic peaks within the Mn 2p1/2 and Mn 2p3/2 regions. The 642.0 and 653.6 eV peaks are attributed to Mn3+, while the 643.3 and 654.8 eV peaks correspond to Mn4+.40–42 Both NFM and LSNFM samples are found to contain Mn3+ and Mn4+ in their initial state with the proportion of Mn3+ being greatly lower in the LSNFM sample, suggesting that Li/Sn co-substitution could suppress Jahn–Teller distortion by elevating the valence state of manganese ions. Upon charging to 4.0 V, the peak area corresponding to Mn4+ increases in both NFM and LSNFM samples, demonstrating that Mn3+ participates in the charge transfer process and contributes to the capacity. When fully discharged to 2.0 V, Mn4+ is reduced. By comparison, the Mn3+/4+ redox pairs participating in the reaction of the LSNFM sample are much fewer than those of the NFM sample, suggesting that Li/Sn co-substitution suppresses the reactivity of Mn3+/4+, consistent with the previous electrochemical results. Fig. 4c and d present the Ni 2p spectra, where the main peaks appear at 854.7 and 872.3 eV, accompanied by satellite peaks, indicating that Ni solely exists in a divalent form initially.12,41 Upon charging to 4.0 V, an additional Ni3+ 2p3/2 peak emerges at 857.6 eV, demonstrating the oxidation of a portion of Ni2+ to Ni3+. With the following discharge back to 2.0 V, the newly formed Ni3+ is fully reverted to Ni2+, showing that Ni2+ participates in electrochemical redox reactions with excellent reversibility during sodium (de)intercalation. Significantly, more Ni3+ ions are activated in the LSNFM sample during charging and discharging processes. In the Fe 2p region shown in Fig. 4e and f, the peaks at 710.3 and 723.8 eV correspond to Fe3+ 2p3/2 and Fe3+ 2p1/2, each accompanied by a satellite peak.43 Similar to the Ni 2p spectra, upon charging to 4.0 V, a peak indicative of Fe4+ 2p3/2 emerged at 713.8 eV, evidencing that Fe also engages in the charge compensation during sodium (de)insertion. Meanwhile, the Fe3+/4+ redox activity is enhanced after Li/Sn co-substitution. The above XPS results confirm that Li/Sn co-substitution suppresses the Mn3+/4+ redox activity while activating more Ni2+/3+ and Fe3+/4+ redox pairs, which agrees with the preceding electrochemical test results.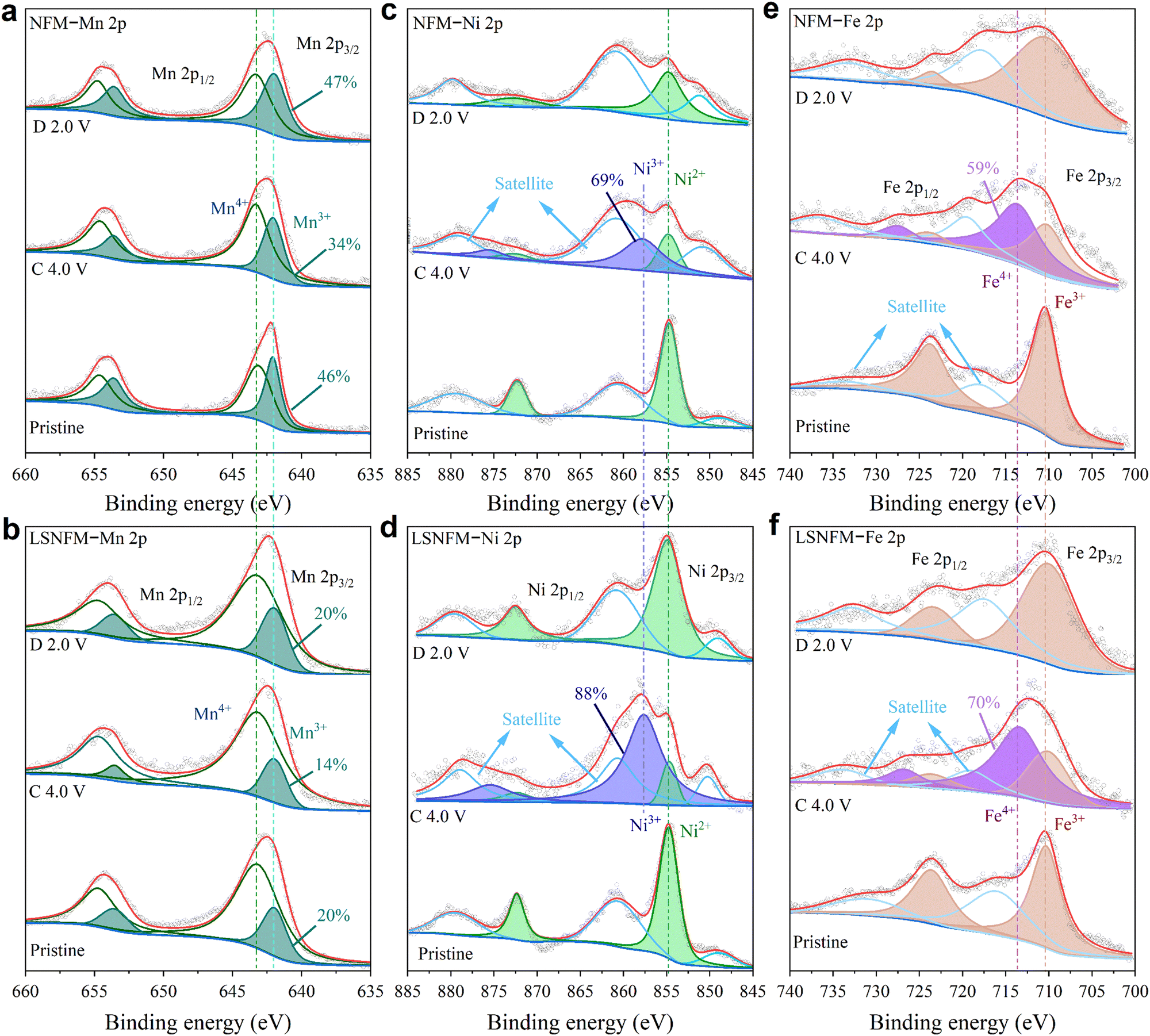 | ||
| Fig. 4 Comparison of the XPS spectra of NFM and LSNFM samples: (a) and (b) Mn 2p region, (c) and (d) Ni 2p region and (e) and (f) Fe 2p region. | ||
In situ XRD measurements were carried out to further monitor the structural evolution during the first cycle within the voltage range of 2.0–4.0 V. In Fig. 5a and b, the peaks corresponding to the Al current collector and beryllium window are denoted by asterisks and diamonds, respectively. The intensity contour maps for all diffraction peaks are displayed in Fig. S2 and S3 (ESI†). During the initial charging process, when Na+ is extracted, the (003), (006), and (108) peaks progressively shift to lower angles, while the (101) and (102) peaks migrate to higher angles, implying the contraction of the ab-plane and the elongation along the c-axis due to the enhanced repulsion between adjacent TM layers and the weakening of TM–TM bond energy. This demonstrates that the O3 phase begins to transform to the P3 phase. Notably, upon reaching 3.12 V, the (104) peak abruptly disappears and all the diffraction peaks can be indexed to a hexagonal P3 phase, indicating the completion of the O3–P3 transition. Upon fully charging to 4.0 V, the P3 structure remains unchanged, concurrent with a smooth curve in the electrochemical measurements, suggesting that this stage mainly involves a solution reaction. During the discharge process, the peaks associated with the LSNFM material undergo a nearly complete reversible reaction, whereas irreversible transitions occurred in the NFM electrode. Remarkably, a portion of the P3 phase persists till the discharge reaches 2.0 V in the NFM electrode, indicative of the structural irreversibility. In contrast, the LSNFM electrode completely reverts to the O3 phase at the end of discharging, thereby demonstrating that Li/Sn co-substitution effectively boosts structural robustness and inhibits the detrimental irreversible phase transitions, contributing to improving the electrochemical performance, especially in terms of long-term cycling stability.
 | ||
| Fig. 5 In situ XRD patterns of (a) NFM and (b) LSNFM electrodes and the corresponding contour plots during the first charge/discharge process at 0.1 C within 2.0–4.0 V. | ||
Theoretical computations
To gain a deeper understanding of the fundamental mechanism behind the exceptional electrochemical performance of LSNFM, migration energy and projected density of states (PDOS) calculations were conducted using first-principles based on density functional theory (DFT). Fig. 6a displays the optimized atomic structure of LSNFM, modeled in line with experimental atomic ratios and site occupancies. As shown in Fig. 6b, the calculated Na+ migration energy barrier in LSNFM is 0.46 eV, significantly lower than that in NFM (0.83 eV). This supports the notion that Li/Sn co-substitution could facilitate easier Na+ diffusion, thereby enhancing rate performance. Additionally, Fig. 6c compares the PDOS for NFM and LSNFM materials. It is evident that the Mn PDOS in LSNFM exhibits a gradual positive shift, suggesting a reduction in the d-band filling of manganese ions (oxidation of manganese ions) due to the charge balancing effect of the low-valence Li.27 Simultaneously, the 3d band centers of Ni and Fe shift from −2.17 eV and 1.53 eV to −2.01 eV and 1.73 eV, respectively. These findings are intuitively visualized in the schematic representation of the electronic structure shown in Fig. 6d. The positive shift of Mn, Ni and Fe PDOSs significantly optimizes the localized electronic structure, leading to a decrease in the activity of Mn3+/4+ and an increase in the activity of Ni2+/3+ and Fe3+/4+ within the voltage range of 2.0–4.0 V. The theoretical computation results align well with the previous electrochemical data, demonstrating that the synergistic effect of Li/Sn co-substitution effectively suppresses the Jahn–Teller effect of Mn3+ below 2.5 V and activates more TM ion redox reactions above 2.5 V. Consequently, the LSNFM material exhibits high structural stability and excellent electrochemical performance. | ||
| Fig. 6 DFT calculations. (a) Crystal structure of LSNFM. (b) Calculated Na+ migration energy barriers in NFM and LSNFM. (c) Calculated projected density of states of TM ions for NFM and LSNFM, EF: Fermi level. (d) Schematic illustration of the electronic adjustment of NFM and LSNFM. | ||
Conclusions
We have designed a novel Li/Sn co-substituted O3-Na0.95Li0.07Sn0.01Ni0.22Fe0.2Mn0.5O2 (LSNFM) cathode using a simple solid-state method to realize the long-term cycling stability by regulating the electronic structure. The synergistic effect of Li/Sn co-substitution not only activates more redox reactions of Ni2+ and Fe3+ in the high-voltage region and suppresses the reaction activity of Mn3+/4+ below 2.5 V, but also inhibits charge delocalization in the TM layer and effectively suppresses phase transitions. Significantly, this synergistic mechanism is fully understood using systematic in situ/ex situ characterization and density functional theory computations. It is demonstrated that this synergistic effect effectively inhibits the irreversible phase transition and significantly improves structural stability. Consequently, the LSNFM electrode exhibits a 27.2% capacity increase contributed by high-voltage TM ion redox activity and excellent long-term cycle life (84.0% capacity retention after 500 cycles at 1 C and 84.7% capacity retention after 2000 cycles at 5 C). This work provides a paradigm for designing long-term cycle life cathode materials by regulating the electronic structure in practical SIBs.Experimental section
Materials preparation
Na0.95Ni0.22Fe0.28Mn0.5O2 (NFM), Na0.95Sn0.01Ni0.22Fe0.27Mn0.5O2 (SNFM), Na0.95Li0.07Ni0.22Fe0.21Mn0.5O2 (LNFM), and Na0.95Li0.07Sn0.01Ni0.22Fe0.2Mn0.5O2 (LSNFM) materials were synthesized by a solid-state method. The metal oxides, including Mn2O3 (Aladdin, 99%), Fe2O3 (Aladdin, 99.9%), NiO (Aladdin, 99%), SnO2 (Aladdin, 99.99%), carbonate Na2CO3 (Aladdin, 99.8%) and Li2CO3 (Aladdin, 99%), were mixed via planetary ball-milling at 500 rpm for 8 h with an appropriate stoichiometric ratio. The resulting mixture was then dried, pressed into tablets, and calcined at 500 °C for 5 h, followed by sintering at 920 °C for 12 h in an oxygen atmosphere. Subsequently, the samples were cooled to room temperature in a furnace and then transferred to a glove box filled with argon gas.Materials characterization
High precision X-ray powder diffraction patterns were obtained at room temperature using a Bruker D8 X-ray diffractometer operating with Cu Kα radiation (λ = 1.5406 Å) and then refined using the FullProf program. Field-emission scanning electron microscopy (SEM, JSM-6390) equipped with energy dispersive spectroscopy (EDS) and high-resolution transmission electron microscopy (HR-TEM, JEM 2100F, 200 kV) were employed to examine the morphologies and crystal structures of the materials. XPS measurements were conducted using an Escalab 250Xi (Thermo Fisher Scientific) with Al Kα radiation (1486.6 eV).Electrochemical characterization
Cathode electrodes were prepared by coating the slurries, consisting of 80 wt% active substance, 10 wt% conductive agents (Super P), and 10 wt% polyvinylidene fluoride (PVDF) binder, with an appropriate amount of N-methyl-2-pyrrolidone (NMP) solution onto an aluminum foil. The prepared electrodes were then punched into disks with a diameter of 12 mm and a mass loading of approximately 3.5 mg cm−2. Sodium metal was used as an anode electrode, and glass fiber (GF/B, Whatman) was used as a separator. Subsequently, Na half cells (CR2032) were assembled in a glove box filled with argon gas. The galvanostatic charge/discharge tests were performed using both LAND (CT2001A, Wuhan Jinnuo Electronics Co., Ltd) and Neware battery testing systems within the voltage range of 2.0–4.0 V vs. Na+/Na0 at 25 °C. Cyclic voltammetry measurements were carried out using an electrochemical workstation with a scan rate of 0.1 mV s−1. Galvanostatic intermittent titration technique (GITT) experiments were conducted using a Neware battery testing system, with the cells tested at a rate of 0.1 C for 30 minutes, followed by a 10-hour rest period. The Na+ diffusion coefficients (DNa+) were calculated using the following formula:where τ is defined as the relaxation time of a single current pulse, mB, Vm and MB represent the mass (g), molar volume (cm3 mol−1), and molar mass (g mol−1) of the active substance in the electrode, respectively, S represents the effective contact area between the electrode and the electrolyte, ΔEs signifies the value change of the adjacent steady-state voltage, and ΔEτ represents the voltage change induced by a current pulse. Fig. S2 (ESI†) illustrates the voltage versus time profile for a single titration event.

Theoretical calculations
Density functional theory (DFT) calculations were conducted using the VASP code.44 The Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA)45 was employed for exchange–correlation processing, while the projector-augmented-wave (PAW) pseudopotential46 was utilized with a kinetic energy cut-off of 500 eV to describe the expansion of the electronic eigenfunctions. A vacuum thickness of 25 Å was set to minimize interlayer interactions. The Brillouin-zone integration was sampled using a Γ-centered 5 × 5 × 1 Monkhorst–Pack k-point. All atomic positions were fully relaxed until the energy and force reached a tolerance of 1 × 10−5 eV and 0.03 eV Å−1, respectively. The dispersion-corrected DFT-D method was employed to account for long-range interactions.47 The climbing image nudged elastic band (CI-NEB) method was used to compute the minimum energy pathway of the cyclization reaction and its corresponding activation barrier.Author contributions
Guohua Zhang: conceptualization, methodology, investigation, data curation, formal analysis, visualization, and writing – original draft. Yuheng Gao, Ping Zhang, Yuheng Gao, Jingrong Hou and Xuemin Shi: investigation and validation. Jiwei Ma: resources and validation. Renyuan Zhang and Yunhui Huang: supervision, validation, funding acquisition, and writing – review and editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant No. 22279092 and 5202780089).Notes and references
- A. Rudola, R. Sayers, C. J. Wright and J. Barker, Nat. Energy, 2023, 8, 215–218 CrossRef.
- H. Zhang, Y. Gao, X. H. Liu, L. F. Zhou, J. Y. Li, Y. Xiao, J. Peng, J. Z. Wang and S. L. Chou, Adv. Energy Mater., 2023, 13, 2300149 CrossRef CAS.
- R. Usiskin, Y. X. Lu, J. Popovic, M. Law, P. Balaya, Y. S. Hu and J. Maier, Nat. Rev. Mater., 2021, 6, 1020–1035 CrossRef CAS.
- P. F. Wang, Y. You, Y. X. Yin and Y. G. Guo, Adv. Energy Mater., 2018, 8, 1701912 CrossRef.
- Y. Liu, C. Sun, Y. Li, H. B. Jin and Y. J. Zhao, Energy Storage Mater., 2023, 57, 69–80 CrossRef.
- J. Peng, W. Zhang, Q. N. Liu, J. Z. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Mater., 2022, 34, 2108384 CrossRef CAS PubMed.
- J. H. Hu, Y. Hong, M. C. Guo, Y. Hu, W. Tang, S. Xu, S. Jia, B. S. Wei, S. H. Liu, C. Fan and Q. C. Zhang, Energy Storage Mater., 2023, 56, 267–299 CrossRef.
- J. A. K. Satrughna, A. Kanwade, A. Srivastava, M. K. Tiwari, S. C. Yadav, S. Teja Akula and P. M. Shirage, Mater. Today, 2023 DOI:10.1016/j.mattod.2023.06.013.
- X. Liang, J. Y. Hwang and Y. K. Sun, Adv. Energy Mater., 2023, 13, 2301975 CrossRef CAS.
- N. Li, W. Yin, B. T. Wang, F. W. Wang, X. L. Xiao, J. K. Zhao and E. Y. Zhao, Energy Environ. Mater., 2023, e12671 CrossRef.
- C. Delmas, C. Fouassier and P. Hagenmuller, Physica B+C, 1980, 99, 81–85 CrossRef CAS.
- X. Liang and Y. K. Sun, Adv. Funct. Mater., 2022, 32, 2206154 CrossRef CAS.
- K. Wang, H. X. Zhuo, J. T. Wang, F. Poon, X. L. Sun and B. W. Xiao, Adv. Funct. Mater., 2023, 33, 2212607 CrossRef CAS.
- B. Peng, G. L. Wan, N. Ahmad, L. Yu, X. Y. Ma and G. Q. Zhang, Adv. Energy Mater., 2023, 13, 2300334 CrossRef CAS.
- J. M. Kim, X. H. Zhang, J. G. Zhang, A. Manthiram, Y. S. Meng and W. Xu, Mater. Today, 2021, 46, 155–182 CrossRef CAS.
- W. Xu, R. Dang, L. Zhou, Y. Yang, T. Lin, Q. Guo, F. Xie, Z. Hu, F. Ding, Y. Liu, Y. Liu, H. Mao, J. Hong, Z. Zuo, X. Wang, R. Yang, X. Jin, X. Hou, Y. Lu, X. Rong, N. Xu and Y. S. Hu, Adv. Mater., 2023, 35, 2301314 CrossRef CAS PubMed.
- Z. H. Wu, Y. X. Ni, S. Tan, E. Y. Hu, L. H. He, J. D. Liu, M. C. Hou, P. X. Jiao, K. Zhang, F. Y. Cheng and J. Chen, J. Am. Chem. Soc., 2023, 145, 9596–9606 CrossRef CAS PubMed.
- X. Y. Du, Y. Meng, H. Y. Yuan and D. Xiao, Energy Storage Mater., 2023, 56, 132–140 CrossRef.
- X. D. Gao, X. Y. Zhang, X. Y. Liu, Y. F. Tian, Q. Y. Cai, M. Jia and X. H. Yan, Small Methods, 2023, 7, e2300152 CrossRef PubMed.
- F. X. Ding, C. L. Zhao, D. D. Xiao, X. H. Rong, H. B. Wang, Y. Q. Li, Y. Yang, Y. X. Lu and Y. S. Hu, J. Am. Chem. Soc., 2022, 144, 8286–8295 CrossRef CAS PubMed.
- F. Fu, X. Liu, X. G. Fu, H. W. Chen, L. Huang, J. J. Fan, J. B. Le, Q. X. Wang, W. H. Yang, Y. Ren, K. Amine, S. G. Sun and G. L. Xu, Nat. Commun., 2022, 13, 2826 CrossRef CAS PubMed.
- Z. Cheng, X. Y. Fan, L. Yu, W. Hua, Y.-J. Guo, Y.-H. Feng, F. D. Ji, M. Liu, Y.-X. Yin, X. Han, Y.-G. Guo and P.-F. Wang, Angew. Chem., Int. Ed., 2022, 134, e202117728 CrossRef.
- A. K. Paidi, W. B. Park, P. Ramakrishnan, S. H. Lee, J. W. Lee, K. S. Lee, H. Ahn, T. C. Liu, J. Gim, M. Avdeev, M. Pyo, J. I. Sohn, K. Amine, K. S. Sohn, T. J. Shin, D. Ahn and J. Lu, Adv. Mater., 2022, 34, e2202137 CrossRef PubMed.
- X. Gao, H. Q. Liu, H. Y. Chen, Y. Mei, B. W. Wang, L. Fang, M. Z. Chen, J. Chen, J. Q. Gao, L. S. Ni, L. Yang, Y. Tian, W. T. Deng, R. Momen, W. F. Wei, L. B. Chen, G. Q. Zou, H. S. Hou, Y. M. Kang and X. B. Ji, Sci. Bull., 2022, 67, 1589–1602 CrossRef CAS PubMed.
- P. F. Wang, H. Xin, T. T. Zuo, Q. Li, X. Yang, Y. X. Yin, X. Gao, X. Yu and Y. G. Guo, Angew. Chem., Int. Ed., 2018, 57, 8178–8183 CrossRef CAS PubMed.
- G. H. Zhang, J. Y. Li, Y. X. Fan, Y. K. Liu, P. Zhang, X. Y. Shi, J. W. Ma, R. Y. Zhang and Y. H. Huang, Energy Storage Mater., 2022, 51, 559–567 CrossRef.
- F. X. Ding, H. B. Wang, Q. H. Zhang, L. R. Zheng, H. Guo, P. F. Yu, N. Zhang, Q. B. Guo, F. Xie, R. B. Dang, X. H. Rong, Y. X. Lu, R. J. Xiao, L. Q. Chen and Y. S. Hu, J. Am. Chem. Soc., 2023, 145, 13592–13602 CrossRef CAS PubMed.
- Y. Xiao, H. R. Wang, H. Y. Hu, Y. F. Zhu, S. Li, J. Y. Li, X. W. Wu and S. L. Chou, Adv. Mater., 2022, 34, e2202695 CrossRef PubMed.
- T. F. Song and E. Kendrick, J. Phys. Mater., 2021, 4, 032004 CrossRef CAS.
- M. Sathiya, Q. Jacquet, M. L. Doublet, O. M. Karakulina, J. Hadermann and J. M. Tarascon, Adv. Energy Mater., 2018, 8, 1702599 CrossRef.
- X. H. Rong, F. Gao, F. X. Ding, Y. X. Lu, K. Yang, H. Li, X. J. Huang, L. Q. Chen and Y. S. Hu, J. Mater. Sci. Technol., 2019, 35, 1250–1254 CrossRef CAS.
- X. Rong, X. Qi, Y. Lu, Y. Wang, Y. Li, L. Jiang, K. Yang, F. Gao, X. Huang, L. Chen and Y.-S. Hu, J. Energy Chem., 2019, 31, 132–137 CrossRef.
- C. Zhao, F. Ding, Y. Lu, L. Chen and Y. S. Hu, Angew. Chem., Int. Ed., 2019, 59, 264–269 CrossRef PubMed.
- H. Y. Hu, H. R. Wang, Y. F. Zhu, J. Y. Li, Y. F. Liu, J. Q. Wang, H. X. Liu, X. B. Jia, H. W. Li, Y. Su, Y. Gao, S. Q. Chen, X. W. Wu, S. X. Dou, S. L. Chou and Y. Xiao, ACS Nano, 2023, 17, 15871–15882 CrossRef CAS PubMed.
- S. Q. Yuan, L. Yu, G. N. Qian, Y. Y. Xie, P. H. Guo, G. J. Cui, J. Ma, X. Y. Ren, Z. X. Xu, S. J. Lee, J. S. Lee, Y. J. Liu, Y. Ren, L. S. Li, G. Q. Tan and X. Z. Liao, Nano Lett., 2023, 23, 1743–1751 CrossRef CAS PubMed.
- Y. Liu, Q. Shen, X. Zhao, J. Zhang, X. Liu, T. Wang, N. Zhang, L. Jiao, J. Chen and L. Z. Fan, Adv. Funct. Mater., 2019, 30, 1907837 CrossRef.
- Q. Y. Shen, X. D. Zhao, Y. C. Liu, Y. P. Li, J. Zhang, N. Zhang, C. H. Yang and J. Chen, Adv. Sci., 2020, 7, 2002199 CrossRef CAS PubMed.
- Q. Yang, P. F. Wang, J. Z. Guo, Z. M. Chen, W. L. Pang, K. C. Huang, Y. G. Guo, X. L. Wu and J. P. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 34272–34282 CrossRef CAS PubMed.
- X. Li, Y. Wang, D. Wu, L. Liu, S. H. Bo and G. Ceder, Chem. Mater., 2016, 28, 6575–6583 CrossRef CAS.
- Q. Q. Zhao, F. K. Butt, Z. F. Guo, L. Q. Wang, Y. Q. Zhu, X. Y. Xu, X. L. Ma and C. B. Cao, Chem. Eng. J., 2021, 403, 126308 CrossRef CAS.
- J. R. Fu, H. J. Huang, K. Shi, F. Chen, Z. H. Yang and W. X. Zhang, Electrochim. Acta, 2020, 349, 136347 CrossRef CAS.
- H. W. Nesbitt and D. Banerjee, Am. Mineral., 1998, 83, 305–315 CrossRef CAS.
- T. T. Wei, X. Liu, S. J. Yang, P. F. Wang and T. F. Yi, J. Energy Chem., 2023, 80, 603–613 CrossRef CAS.
- G. Kresse and J. Furthmiiller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00333k |
| This journal is © The Royal Society of Chemistry 2024 |