
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stretchable and biodegradable plant-based redox-diffusion batteries†
Aiman
Rahmanudin
 *ab,
Mohsen
Mohammadi
ab,
Patrik
Isacsson
abc,
Yuyang
Li
a,
Laura
Seufert
a,
Nara
Kim
ae,
Saeed
Mardi
ad,
Isak
Engquist
ab,
Reverant
Crispin
abe and
Klas
Tybrandt
*abe
*ab,
Mohsen
Mohammadi
ab,
Patrik
Isacsson
abc,
Yuyang
Li
a,
Laura
Seufert
a,
Nara
Kim
ae,
Saeed
Mardi
ad,
Isak
Engquist
ab,
Reverant
Crispin
abe and
Klas
Tybrandt
*abe
aLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, 602 21 Norrköping, Sweden. E-mail: aiman.rahmanudin@liu.se; klas.tybrandt@liu.se
bWallenberg Wood Science Center, ITN, Linköping University, Norrköping, Sweden
cAhlstrom Group Innovation, 38140 Apprieu, France
dÅngström Laboratory, Department of Chemistry, Uppsala University, 751 21 Uppsala, Sweden
eWallenberg Initiative Materials Science for Sustainability, Department of Science and Technology, Linköping University, 601 74 Norrköping, Sweden
First published on 19th June 2024
Abstract
The redox-diffusion (RD) battery concept introduces an environmentally friendly solution for stretchable batteries in autonomous wearable electronics. By utilising plant-based redox-active biomolecules and cellulose fibers for the electrode scaffold, separator membrane, and current collector, along with a biodegradable elastomer encapsulation, the battery design overcomes the reliance on unsustainable transition metal-based active materials and non-biodegradable elastomers used in existing stretchable batteries. Importantly, it addresses the drawback of limited attainable battery capacity, where increasing the active material loading often leads to thicker and stiffer electrodes with poor mechanical properties. The concept decouples the active material loading from the mechanical structure of the electrode, enabling high mass loadings, while retaining a skin-like young's modulus and stretchability. A stretchable ion-selective membrane facilitates the RD process, allowing two separate redox couples, while preventing crossovers. This results in a high-capacity battery cell that is both electrochemically and mechanically stable, engineered from sustainable plant-based materials. Notably, the battery components are biodegradable at the end of their life, addressing concerns of e-waste and resource depletion.
New conceptsHere, we demonstrate a conceptual advancement in battery technology intended to power next generation wearables that require mechanical functions beyond flexibility such as softness and stretchability. Notably, the concept emphasises on environmental sustainability by incorporating the use of biodegradable plant-based materials throughout the fabrication of the battery components to address the issue of resource depletion and electronic waste. Our concept overcomes both the battery performance and sustainability drawbacks in existing stretchable electrode designs. Typically, it relies on the mechanical coupling of the redox-active species with the conductive filler for efficient energy storage processes, but this limits the attainable battery capacity as increasing the redox-active content leads to thicker and stiffer electrodes with undesired mechanical properties. The stretchable redox-diffusion battery concept decouples the electrochemical function from the electrical components and mechanical structure of the electrode. The process of storing energy is mediated by the diffusion of dissolved redox-active biomolecules in an electrolyte within a cellulose-based electrically conductive scaffold, and a stretchable ion-selective membrane facilitates the two separate redox couples. This enabled a stretchable and biodegradable battery that is electrochemicaly and mechanically stable where a skin-like Young's modulus is retained despite increasing the active mass loadings in the electrodes to achieve higher battery capacity. |
1. Introduction
Next generation electronics interfaces intimately with the human body as skin patches, implants, or smart clothing.1 They will advance neuroscience, medical diagnostics and therapy, and enable new consumer wearable technologies such as electronic (e) skin, e-textiles and soft robotics.2 A prerequisite for conformal and imperceptible integration with the body is the need for soft and stretchable electronics. These devices contain multiple electrical components to perform intricate functions, and advances have been made to enable their stretchability during operation, but they are typically designed to accommodate rigid and bulky battery components.3–5 Integrating a stretchable and compact battery would alleviate this problem. However, increasing the redox-active material content in existing stretchable battery designs often leads to stiffer electrodes with lower stretchability.6–8 Furthermore, they are predominantly based on unsustainable transition metal-oxide active materials, non-biodegradable petroleum-based elastomers (silicone, styrene block copolymers etc.), and expensive conductive metal nano-fillers (Au, Ag, etc.) in the current collector.9 Commercialization of such batteries will contribute to the depletion of finite resources and the growing problem of e-waste.10 Therefore, new design concepts are required to achieve sustainable batteries with softer and stretchable electrodes while retaining charge storage capacity.To design a stretchable battery cell, one must consider each component (Fig. 1a) as they play a separate yet interconnected function that requires good electrochemical performance and the ability to accommodate mechanical stress during operation. There are two common ways to induce softness and stretchability in the various components of a battery cell. In the first approach, structural design is used to isolate rigid components from major strain. This includes forming layered twisted fibers of each battery component,11 integrating stiff battery cells onto deformable substrates with stretchable interconnects,12 creating wavy/buckled13,14 kirigami,15 or ultrathin16 structures. The second approach involves the design of each material with stretchability that is intrinsic to the bulk of the system such as the mixing of rigid filler materials with elastomers to form composites,17–22 gels,23,24 liquid metals,25–28 or porous structures.29,30 Combinatory approaches also exist, including wavy31 or kirigami-patterned current collectors combined with intrinsically stretchable separators and electrodes.32 Despite the advancements in most stretchable battery designs, to promote efficient energy storage processes, (e.g., ion intercalation, redox reaction and electron transfer) the redox-active species that is embedded within a solid porous electrode network must have high solid interfacial contact with the conductive filler and exposure to the electrolyte (Fig. 1b). This dependency on a solid-to-solid contact limits active material loading within the electrode since increasing the active material content tends to lead to thicker and stiffer electrodes, i.e., the coupling of the electrochemical and mechanical properties.
 | ||
| Fig. 1 Stretchable battery architecture and its components. Schematic representation: (a) the full cell, (b) conventional (coupled) and (c) the redox-diffusion electrode design (decoupled). (d) Biomaterials derived from various parts of a plant and the molecular structure of the cellulose nanofibrils (CNFs), alizarin and lignin. (*) indicates that the CNFs used were carboxymethylated cellulose and sulfonated ARS and LS were prepared post extraction of the materials. (e) The infiltration process. The 3D porous conductive scaffold is made from the conducting polymer PEDOT:PSS and CNF, with the dissolved redox-active biomolecules, LS (catholyte) and ARS (anolyte) in PVA/1 M H2SO4 within the pores. (f) The redox processes of LS and ARS. (g) Schematic of the selective ion conducting membrane with a hydrophobic elastomer and hydrophilic CNF:PSS network. (h) Schematic of the kirigami-patterned current collector made from nanographite and cellulosic fibers, and PGS (R = H or crosslinked chains) as the biodegradable elastomer encapsulation. | ||
Similarly, addressing the sustainability aspects of stretchable batteries has been challenging. Biodegradable batteries have been reported but generally have limited mechanical flexibility and stretchability.10 The active materials are typically based on degradable metallic electrodes such as Zn, Mg, Fe and Mo, and their oxide counterparts, and a degradable encapsulation polymer. Only two examples have incorporated stretchability using structural engineering via kirigami patterning of the rigid metal electrodes.15,32 These degradable metals are highly abundant but they are still a finite resource, and the ethical, social and environmental concerns of mining and their energy-intensive processing are still problematic.33,34 Furthermore, the reported cells are non-rechargeable primary batteries. Recently, organic redox-active materials have been poised as a cost-effective and sustainable alternative for battery technology, especially if the precursors come from renewable biomass feedstocks,35–37 similarly for the passive battery components.38,39 The handful of reports showing the use of organic active materials for stretchable batteries are still petrochemical-based including their passive components.14,16,40 So far, no effective holistic strategy has been put forward for a sustainable stretchable battery.7 It is imperative that future designs of all components within a battery cell not only focus on high performance (mechanical and electrochemical) but also encompass the primary use of sustainable materials and processing methods that are human- and environment-friendly.
Here, we developed a stretchable redox-diffusion (RD) battery concept that uses both structural- and material-level approaches to enable stretchability, while maximizing sustainable plant-based biomaterials into all components of the cell to ensure degradability at the end of their use (Fig. 1d). The RD concept is a hybrid between a redox flow battery (which requires pumps41 or capillary forces42,43 to flow the electrolyte) and a conventional battery. Instead of an active “flow” of the redox-active species, the process of storing (and delivering) energy is mediated by diffusion of the dissolved redox-active biomolecules within a cellulose-based electrically conductive 3D porous scaffold (Fig. 1c and e). The RD design decouples the redox-active species from the mechanical structure of the electrically conducting electrode. It resulted in high mass loadings (≈40 mg cm−2) of the dissolved redox-active biomaterials, while reducing the amount of conductive filler. The “sponge-like” structure and high water content of the porous hydrogel scaffold is able to dissipate strain to achieve stretchability (∼250%) and softness (∼110 kPa) within the range of the Young's modulus of human skin despite being thicker.44 Additionally, to facilitate the RD design, a stretchable ion-selective membrane separator was developed to allow the utilisation of two separate redox couples, while preventing crossovers (Fig. 1e and f). The remaining battery components were engineered to achieve sustainability and biodegradability (Fig. 1g), and their electrochemical and mechanical properties were characterised individually and together in the full cell.
2. Results & discussion
2.1. Redox-diffusion electrode concept
In an ideal conventional electrode, high pore volume and surface area reduces tortuosity and facilitates ionic transport to access the active material embedded within the solid electrode matrix to promote redox reactions (Fig. 1b).45,46 As discussed earlier, the dependency on high interfacial contact between the conductive filler and active material will result in stiffening of the electrode with increasing mass loading. This issue becomes pertinent for organic redox-active materials as they are generally insulating in nature (as compared to their inorganic counterparts) and therefore require high loadings of conductive fillers (CF) with respect to redox-active (RA) material content to facilitate electronic transport in the electrode. For example, battery electrodes using redox-active biomass-derived biopolymer lignin requires a high weight ratio (CF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RA) of 1:1 with carbon,47,48 and 1:2 and 3:2 with a conducting polymer as the CF.49,50 Similarly, the electrodes based on plant-derived RA biomolecules only achieved a maximum capacity at a weight ratio of 1:4, which decreased with higher RA loading, presumably due to less interfacial contact with the CF.40,51 Moreover, the high solubility of redox-active biomolecules results in dissolution into the aqueous electrolytes that degrades the capacity over time. In contrast, the RD electrode design exploits the high solubility of the RA biomolecules to achieve a higher CF:RA ratio of 1:285 with a significantly higher mass loading of ≈ 40 mg cm−2. Further details are discussed in Section 2.6. There have been reports on electrochemical energy storage (EES) systems using dissolved RA species in polymer hydrogels, but most are rigid or flexible, and the low surface area electrodes and a lack of an ion-selective separator membrane leads to relatively low capacities.52–54 This validates the ability of the RD concept to maximize the mass loading of the redox-active component in the active electrode to achieve substantially higher capacities, all while remaining soft and stretchable. Table S1 in the ESI† summarises the comparison with the relevant EES systems discussed above.
:RA) of 1:1 with carbon,47,48 and 1:2 and 3:2 with a conducting polymer as the CF.49,50 Similarly, the electrodes based on plant-derived RA biomolecules only achieved a maximum capacity at a weight ratio of 1:4, which decreased with higher RA loading, presumably due to less interfacial contact with the CF.40,51 Moreover, the high solubility of redox-active biomolecules results in dissolution into the aqueous electrolytes that degrades the capacity over time. In contrast, the RD electrode design exploits the high solubility of the RA biomolecules to achieve a higher CF:RA ratio of 1:285 with a significantly higher mass loading of ≈ 40 mg cm−2. Further details are discussed in Section 2.6. There have been reports on electrochemical energy storage (EES) systems using dissolved RA species in polymer hydrogels, but most are rigid or flexible, and the low surface area electrodes and a lack of an ion-selective separator membrane leads to relatively low capacities.52–54 This validates the ability of the RD concept to maximize the mass loading of the redox-active component in the active electrode to achieve substantially higher capacities, all while remaining soft and stretchable. Table S1 in the ESI† summarises the comparison with the relevant EES systems discussed above.
2.2. Sustainable material selection
The structural element of the porous conductive scaffold is cellulose, which is the most abundant biopolymer on earth (Fig. 1c).55 Cellulose fibers are extracted from wood in the macro to nanoscale dimensions with high surface area and mechanical strength. Our 3D scaffold employed carboxymethylated (–COOH) cellulose nanofibrils (CNFs) as the structural component with poly(3,4-ethylenedioxythiophene):poly(styrene-sulfonate) (PEDOT:PSS) as the conductive filler to construct a porous conductive aerogel scaffold.56,57 Two plant-based redox couples were selected for charge storage, lignosulfonate (LS) as the catholyte and alizarin red sulfonate (ARS) as the anolyte. These biomolecules contain quinone groups that can undergo an electrochemically induced reversible two-electron/two-proton redox reaction to permit energy storage and have been used for non-stretchable battery systems (Fig. 1e).37,47–49,58–65 Lignin is the second most abundant biopolymer on earth with LS being a common water soluble by-product from paper manufacturing.66,67 ARS can be extracted from the roots of the plant Rubia tinctorum in its non-sulfonated form, and is traditionally used as a red dye in textiles.68 The redox-active biomolecules were dissolved in polyvinyl alcohol (PVA) and soaked into the porous conductive scaffolds, resulting in an interpenetrative hydrogen bonded network between the PEDOT:PSS:CNF scaffold and the redox-active hydrogel electrolyte (Fig. 1d).52 The intermolecular interactions between the hydroxyl (–OH), carboxylate (–CO2−), and sulfonate (–SO3−) groups present in the molecular structures of the various material components physically crosslinks the entire active electrode to dissipate strain, which strengthens the hydrogel and induces stretchability. The electrodes are separated by a CNF-based stretchable ion-selective separator membrane, which prevents crossover between the catholyte and anolyte (Fig. 1f). To avoid conventional metallic current collectors, we engineered a stretchable kirigami-patterned current collector based on a composite of environmentally benign nanographite (NG) and cellulosic fibers (CFs) to electrically connect the 3D scaffold to the external circuit. Lastly, a biomass-derived biodegradable elastomer packaging based on poly(glycerol sebacate) (PGS) was used to hold the battery components together (Fig. 1g.)2.3. Stretchable nanographite paper kirigami current collector
The paper current collector was fabricated using a standardized lab scale sheet forming technique according to a previous report with a NG filler loading of 50 wt%.69 A calendaring step was added to compact the composite structure and improve conductivity. PGS was used as the elastomer packaging. It is commonly used in tissue engineering for biomedical applications due to its intrinsic stretchability, biocompatibility, and biodegradability.70,71 PGS was polymerized using a two-step polycondensation reaction of equimolar amounts of non-toxic glycerol and sebacic acid monomer (Fig. 2a). This forms a covalently crosslinked elastomer with a dense three-dimensional hydrogen-bonded network due to the hydroxyl groups along the polymer chain. The hydroxyl groups induce tackiness that facilitated the adhesion of the current collector. To engineer stretchability, laser patterning of a kirigami structure was performed on the NG paper composite embedded on the PGS elastomer. Tuning of the kirigami pattern affects the stretchability and the sheet resistance (Rs) of the current collector. The horizontal (x) and vertical (y) distances between the slits were fixed to x = 0.75 mm and y = 0.5 mm, and the slit length (L) was varied between 0 and 2.0 mm (Fig. 2b.). Rs increases with a longer slit length from an initial Rs = 1.4 ± 0.3 Ω □−1 to 19.8 ± 0.2 Ω □−1 at L = 2.0 mm. This is expected as the subtraction of conductive materials lengthens the conductive path.72 Non-patterned current collectors mechanically break at a low elongation of 2% strain (Fig. 2c.). Increasing the slit length improves stretchability but increases initial resistance. As a compromise, a slit length of 1.5 mm was chosen as it combined a reasonable initial Rs of 7.45 ± 0.8 Ω □−1 with less than 2-fold increase in normalised resistance at 150% strain. This current collector was tested under 1000 strain cycles and there were no significant changes in normalised resistance after 1000 cycles at 20% strain. Even at 50% and 100% strain the resistance was stable and remained below twice its initial value (Fig. 2d). For 150% strain, the resistance increased to above twice its initial resistance, but it remained stable over 1000 stretching cycles (Fig. S1, ESI†). Compared to other carbon-based stretchable current collectors,18 the cellulose-based kirigami patterned current collector exhibits lower sheet resistance and higher electromechanical durability under stretching. | ||
| Fig. 2 Current collector fabrication and characterization. (a) Schematic of the fabrication process of the stretchable current collector: Step 1 - Synthesis of PGS at 120 °C under nitrogen for 12 h, R = H or crosslinked chains. Step 2 - Casting the pre-polymer into a mould and crosslinking at 120 °C for a further 48 h under a vacuum. Step 3 – Embedding of the cellulose graphite paper. Step 4 – Laser kirigami patterning. (b) Sheet resistance as a function of slit length (L) in the kirigami-patterned NG current collectors. The inset shows the kirigami-pattern used for electrode processing where x = 0.75 mm, y = 0.5 mm, and L = 0, 1.0 1.25 1.5, 1.75, or 2.0 mm. (c) Normalized resistance as a function of strain for different slit lengths. (d) Cyclic stretching to 20, 50, and 100% strain for L = 1.5 mm. Inset images are of the current collector at the respective strains. | ||
2.4. Stretchable ion-selective membrane
Commercial ion-selective membranes are typically based on fluoropolymers (e.g. Nafion), which have an environmentally problematic production, are expensive (∼$ 500 per m2), and not stretchable.73 As an alternative, cellulose-based membranes have been reported74–76 for non-stretchable large-scale battery applications. To induce stretchability in our membranes, a hybrid hydrophobic (PU) and hydrophilic (CNF:PSS) nanocomposite was developed. The selected material has good interfacial compatibility due to the hydrogen bonded intermolecular interactions between the hydrophilic anionic groups on PU, the carboxylate and hydroxyl groups on CNFs and the sulfonated groups on PSS.77 Stable emulsions were formed as shown by the dynamic light scattering data where the main peaks of the particle size remained stable at 170 nm even after 6 months of storage with no observable agglomeration (Fig. 3a). Upon casting, the membrane had a transmittance of 70% at 550 nm as compared to pristine films of PU and CNF, which obtained a transmittance of 88% and 83% (Fig. 3b). The increase in light scattering indicates a structural change occurring within the PU:CNF:PSS matrix.78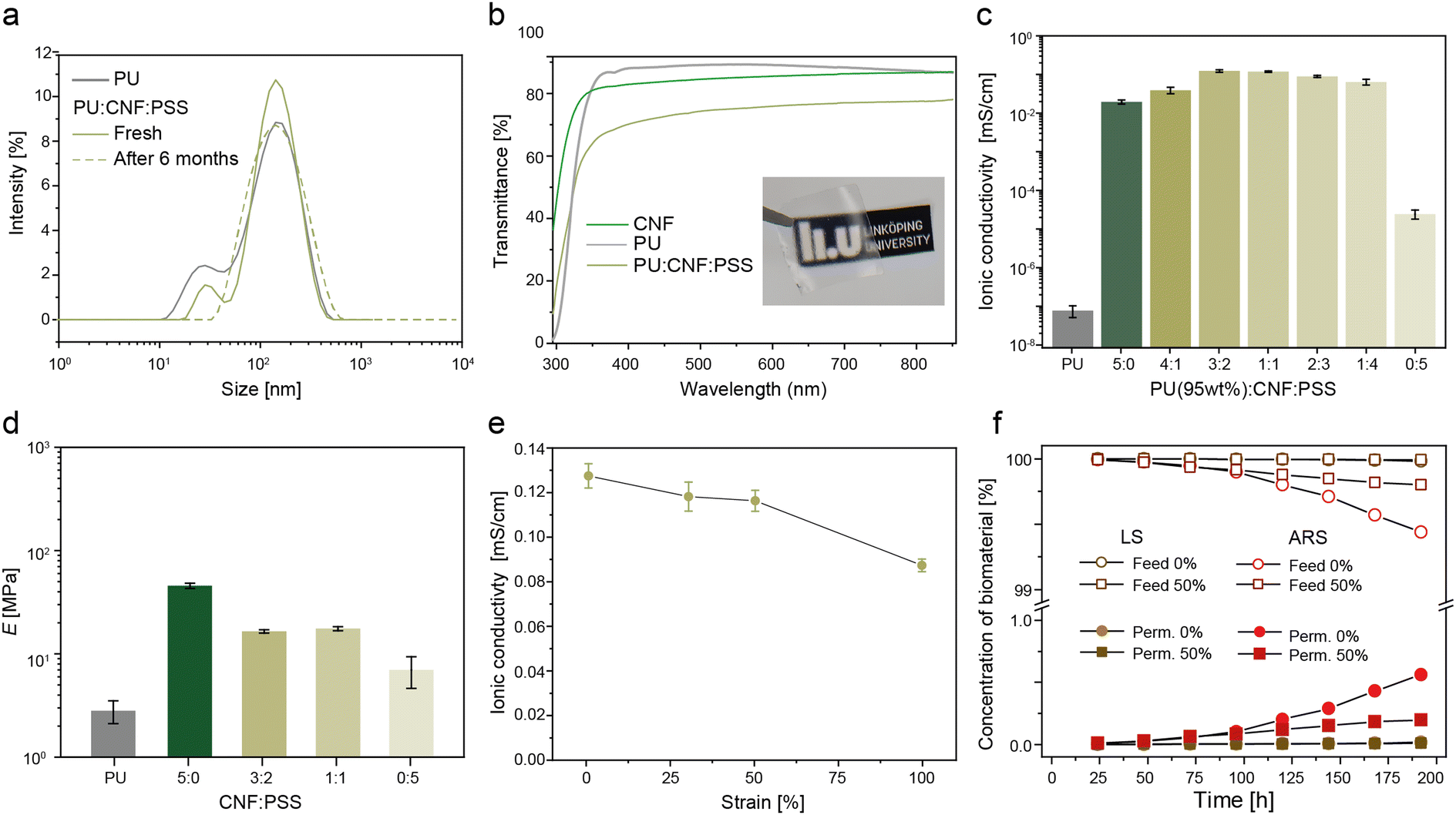 | ||
| Fig. 3 Stretchable separator membrane. (a) The particle size distribution of the PU:CNF:PSS emulsions before casting. (b) Transmittance of the films of PU, CNF and the optimized PU(95 wt%):CNF:PSS(3:2) membrane including a photograph of the membrane. (c) Plots of the ionic conductivity as a function of the CNF:PSS ratio with a fixed PU loading at 95 wt%. (d) Young's moduli of the membranes with different CNF:PSS weight ratios after soaking in the electrolyte solution (1 M H2SO4) for 12 h. (e) The ionic conductivity as a function of strain. (f) Percentage of cross-over of LS and ARS as a function of time over a period of 192 h. | ||
To optimise the membrane properties, the weight ratios of the hydrophilic CNF:PSS phase were modulated with a fixed amount of hydrophobic PU phase at 95 wt%. The ionic (proton) conductivity was measured using a four-point electrochemical impedance spectroscopy (EIS) technique.18 As expected, pristine PU was highly resistive with a proton conductivity of 7.7 × 10−8 mS cm−1 due its dense hydrophobic film structure (Fig. 3c and ESI† Fig. S2 for the respective EIS Bode plots). The addition of CNF into the PU matrix significantly improved the conductivity by six orders of magnitude to 0.019 ± 0.002 mS cm−1. To improve the proton conduction further, sulfonate groups were added via incorporation of PSS.79 CNF:PSS ratios of 3:2 and 1:1 resulted in maximum conductivities of 0.127 ± 0.007 and 0.118 ± 0.004 mS cm−1, respectively. As the ratio decreased to 0:1, where only PSS and PU were present in the membrane, the conductivity decreased by four orders of magnitude to 2.4 × 10−5 ± 7 × 10−6 mS cm−1. This behaviour suggests that the rigid fibril shape of the CNFs is essential in the formation of a highly interconnected ionic channel through the hydrophobic PU matrix. The connectivity of the CNFs within the PU matrix can be elucidated by the Young's modulus of the membranes (Fig. 3d and ESI† Fig. S3 for the stress–strain curves), where membranes with CNFs have approximately twice the modulus (16–17 MPa) compared to composites with only PSS (7 MPa). Furthermore, the trend observed for the conductivity of membrane as a function of CNF:PSS corresponded with the electrolyte uptake measurements (Fig. S4, ESI†).
Based on these results, the ionic conductivity as a function of tensile strain was studied for membranes with a CNF:PSS ratio of 3:2 (Fig. 3e). As the membranes were stretched to 30% and 50% strain, minimal changes (≈ 7%) in the ionic conductivity were observed from 0.127 to 0.117 and 0.115 mS cm−1, respectively. At a 100% strain, the conductivity decreased by 37% to 0.08 mS cm−1, which was attributed to the alignment of the CNFs along the stretching direction, which may disrupt the ionic percolation pathways through the membrane.77 To investigate the cross-over of the redox-biomaterials through the membrane, a diffusion permeability test over a 192 h period was conducted (Fig. 3f and ESI† Fig. S5 for concentration calibration curves and the permeate concentrations). The permeate concentration of the biomaterials remained below 1% of the feed concentrations after 192 h (5.4 μM vs. 30 mM for LS and 560 μM vs. 100 mM for ARS). The permeate concentrations were similar for membranes at 50% strain (2.6 μM vs. 30 mM for LS and 196 μM vs. 100 mM). This suggests that no large defects occurred in the membranes under deformation and the results corresponded with the decrease in ionic conductivity of the membranes under strain caused by the alignment of the CNFs (Fig. 3e). Thus, the membranes showed promising performance in terms of conductivity, mechanical robustness, and prevention of cross-over of the redox-biomaterials.
2.5. Conductive 3D porous scaffolds
The 3D scaffolds were prepared by shear mixing a dispersion of PEDOT:PSS, CNF and a silane cross-linker at different water:ethanol (H2O:EtOH) contents, followed by lyophilisation and thermal crosslinking (see ESI† methods section for details).57 Upon mixing, PEDOT:PSS nanoparticles are known to undergo non-solvent induced phase separation (NIPS) in the presence of short chain alcohols within the emulsion, leading to densely packed PEDOT, chains which improves conductivity.80,81 The electrical conductivity of the aerogels was extracted from the slope of I–V curves (Fig. 4a) and showed a gradual increase from 0.82 ± 0.2 mS cm−1 to 6.88 ± 0.4 mS cm−1 for H2O:EtOH ratios from 1:0 to 7:3 (Fig. 4b). The electrochemical properties of the aerogels were characterised using a three-electrode set-up with 1 M H2SO4 as the electrolyte. Cyclic voltammetry (CV) of the aerogels generally displayed a capacitive behaviour as indicated from the quasi-rectangle-shaped voltammograms in the 0 to 0.8 V (vs. Ag/AgCl, 3 M KCl) potential range (Fig. 4c). The aerogel prepared with only water (H2O:EtOH, 1:0) had a blunt and slanted voltammogram, which indicated a less conductive scaffold network.82 The bare current collector had lower CV currents, indicating that the capacitive contribution mainly arose from the porous scaffold. Galvanostatic charge–discharge (GCD) experiments (1 to 20 mA cm−2) showed that the capacitance was better retained at higher current densities for aerogels with high ethanol content (Fig. 4d, ESI† Fig. S6 for full GCD curves). X-ray diffraction patterns (Fig. 4e) of aerogels with increasing ethanol content showed two peaks at 2θi = 24.89° and 2θii = 25.8° corresponding to both the CNF and the π–π stacking of the PEDOT chains within the aerogel scaffold.83 Similarly, scanning electron microscopy (SEM) images of the aerogels revealed the formation of a denser fibril microstructure network as the volume ratio of the ethanol increased (Fig. 4f). The NIPS effect caused by ethanol during the mixing of the components clearly improves the electrical percolation pathways of the conducting polymers, which enhances the charge transport within the scaffold and in turn improves charge storage at higher current rates. Aside from this, it is likely that the freezing point of the H2O:EtOH mixture decreased with increasing ethanol content from 0 °C for pure water and to −5, −11 and −20 °C.84 The change in the rate of freezing and the size of the water:ethanol crystals may have resulted in denser fibril formation that affected packing of the PEDOT chains and the resulting microstructure of the aerogel.85 Based on these results, aerogels prepared using a H2O:EtOH of 7:3 were used for the subsequent experiments.
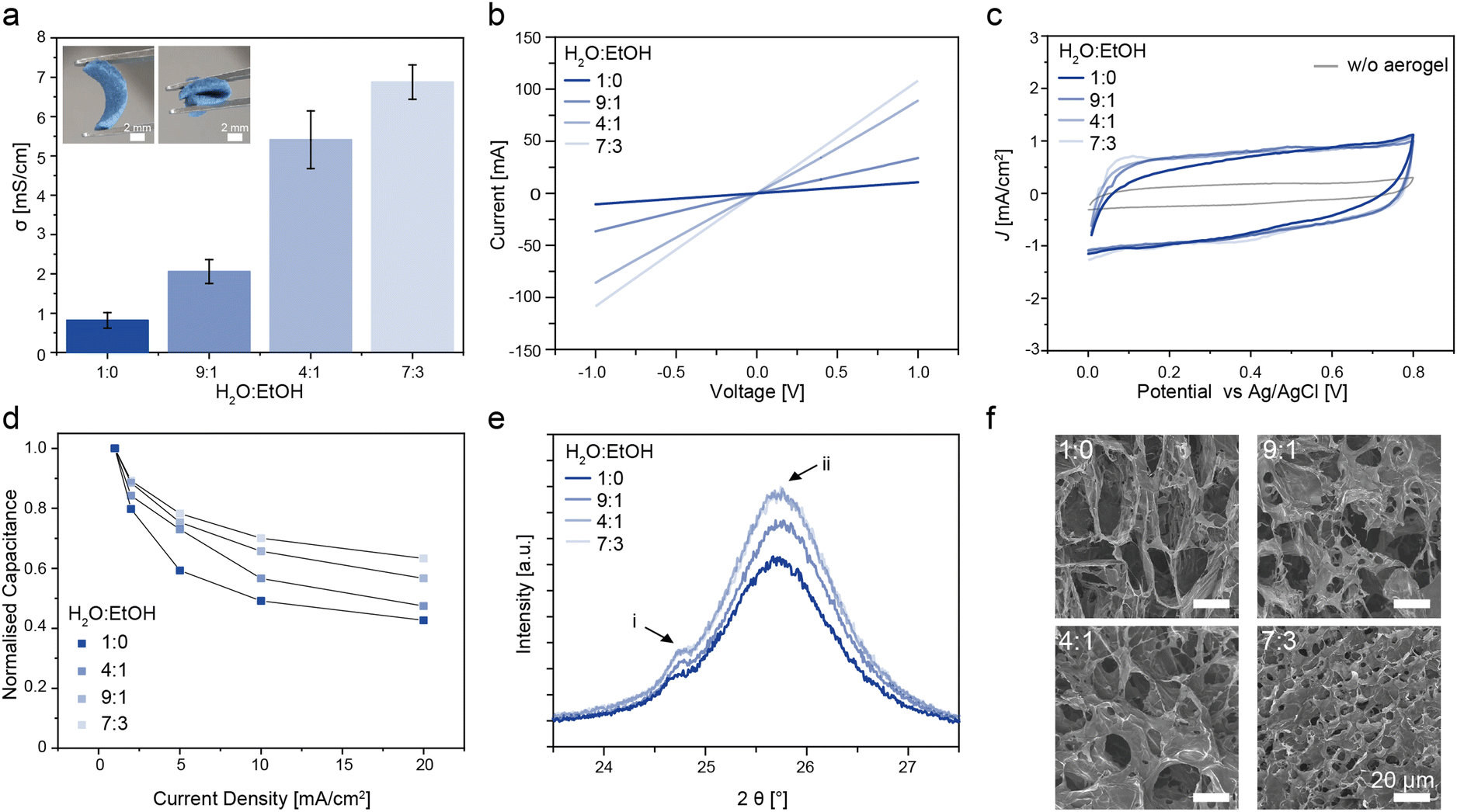 | ||
| Fig. 4 3D electrode characterisation. (a) Electrical conductivity of the PEDOT:PSS-CNF aerogel. Insets are photographs of the prepared aerogel in its initial and compressed/bent states. (b) I–V curves and (c) cyclic voltammetry (20 mV s−1) of the 3D porous scaffolds prepared with different H2O:EtOH volume ratios. (d) Normalised capacitance vs. applied current density. (e) Grazing incidence X-ray diffraction patterns with the peaks corresponding to both PEDOT:PSS and CNF. (f) Scanning electron microscopy images of PEDOT:PSS-CNF aerogels. Scale bar (white) = 20 μm. | ||
2.6. Stretchable active electrode design and characterisation
Stretchable cathodes and anodes were developed by incorporating PVA hydrogels with dissolved redox-active biomolecules into the 3D porous scaffold. The electrochemical properties were determined in a half-cell configuration. The solubility of the anolyte redox-active biomolecule (ARS) in the electrolyte (PVA in 1M H2SO4) was improved from 0.1 M to 2 M by the exchange of the sodium ions to protons.86 GCD characterisation of the anode was performed at various anolyte concentrations using the optimized membrane and current collector in a three electrode set-up (Fig. 5a). A potential window of −0.4 to 0.4 V vs. Ag/AgCl was selected as it had been shown that ARS was prone to chemical degradation via a Michael addition reaction and gem diol formation in its fully oxidized state at higher positive potentials.87 The solubility of ARS-Na is limited to 0.1 M and at 1 mA cm−2, a discharge capacity of 1.09 mA h cm−3 is obtained. Protonated ARS-H of the same concentration achieved more than twice the capacity of 2.24 mA h cm−3 (Fig. 5a.). Due to the higher solubility of ARS-H, 2.0 M loading achieved a maximum capacity of 28.6 ± 6.4 mA h cm−3. As discussed earlier, a previous report using a similar material composite of CNF:(PEDOT:PSS):ARS reported a ARS mass loading of ≈ 7 mg cm−2 with a maximum PEDOT:ARS weight ratio of 1:10 (Fig. 5b.).51 Comparatively, our RD electrode design can reach a PEDOT:ARS ratio of 1:285 and a significantly higher mass loading of ≈ 40 mg cm−2, all while remaining ultra-soft and stretchable.
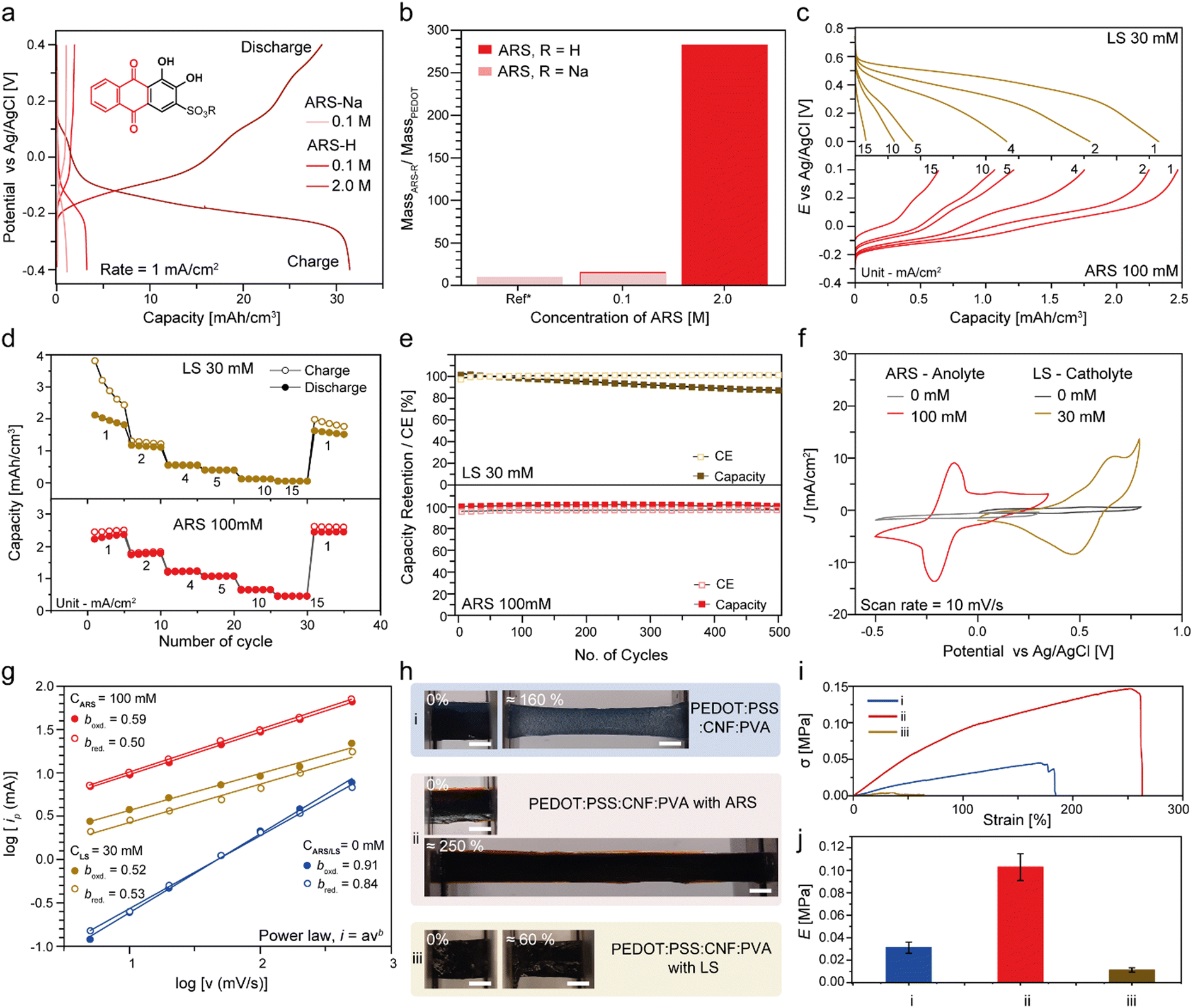 | ||
| Fig. 5 Stretchable active electrode characterisation. (a) GCD curves of electrodes containing various concentrations of ARS-R (R = H or Na). (b) Mass ratio of ARS-R/PEDOT as a function of the molar concentration loading of ARS in the electrode. (c) Representative discharge curves from the first cycle of the cathode (LS) and anode (ARS) at various current densities, unit = mA cm−2. (d) Rate capability of the respective electrodes. (e) GCD cyclic performance at 5 mA cm−2 over 500 cycles (capacity (solid square), coulombic efficiency (CE, void square)). (f) CV curves of electrodes at a scan rate of 10 mV s−1. (g) Plots of peak current vs. scan rate on a logarithmic scale. Mechanical properties of the hydrogel electrodes. (h) (i) Without the redox biomaterial, (ii) with 100 mM of ARS and (iii) with 30 mM of LS. (i) Photographs in the initial unstretched state and under elongation. Scale bar (white) = 500 μm. Stress–strain curves and (j) Young's moduli. | ||
Next, a stretchable cathode based on dissolved LS in the PVA gel electrolyte was developed and characterised in the half-cell configuration. To balance the electrode capacity in the full cell, the concentrations of ARS and LS were optimized to 0.1 M and 30 mM, respectively. The GCD curves for the cathode (LS) and anode (ARS) half-cells delivered a maximum discharge capacity of 2.12 mA h cm−3 and 2.24 mA h cm−3, respectively at a current density of 1 mA cm−2 (Fig. 5c). Both cathode and anode demonstrated reasonable rate capabilities with the discharge capacity recovering close to its initial value after cycling at higher current densities (Fig. 5d). The cycling performance of the electrodes over 500 cycles at 5 mA cm−2 showed stable capacity retention of approximately 80% and 98% for the cathode and anode, respectively (Fig. 5e). The difference in capacity retention can be attributed to instabilities of the fully oxidized quinone units in LS, which are prone to Michael addition reactions with water.87
CV curves of the cathode and anode showed two characteristic pairs of the reversible redox peaks related to ARS and LS in a potential range between −0.25 V and 0.6 V vs. Ag/AgCl (Fig. 5f), whereas capacitive quasi-rectangle-shaped voltammograms were observed for the half-cells without any redox biomaterials (Fig. 5f). Based on the power-law relationship between the peak currents (ip) and scan rates (υ), the b-values obtained from the slope of log(ip) vs. log(υ) provided insights into the charge storage kinetics of the electrodes (Fig. 5e and Fig. S7 for the CV curves at various scan rates, ESI†).88 The half-cells containing ARS and LS obtained boxd/bred values of 0.59/0.50 and 0.52/0.53, respectively, indicating a battery-like faradaic diffusion-controlled process. As expected, the b-values of half-cells without any redox biomaterials were 0.91/0.84, implying a mainly capacitive charge storage process. The conductivities of the cathode and anode were then measured using electrochemical impedance spectroscopy (EIS) and the impedance was extracted at the high frequency region that correlates to the resistive regime (See ESI† Fig. S8). The conductivities of the cathode and anode were 20.5 ± 4.3 and 20.0 ± 3.1 mS cm−1, respectively, and were relatively similar to those of the hydrogels without the PEDOT:PSS-CNF scaffold (ESI† Fig. S8a). Since the inclusion of the PEDOT:PSS-CNF aerogel scaffold into the hydrogels did not significantly affect the overall conductivity, it suggested that the ionic conduction was dominant in the electrodes.
The mechanical properties of the hydrogel electrodes with optimised concentrations of ARS and LS were characterised (Fig. 5h–j). The addition of 100 mM of ARS into the electrolyte improved the fracture strain of the electrodes from 180% to 250% strain, with an increase in the Young's modulus from 30 kPa to 110 kPa (Fig. 5i.). The improvement in the mechanical properties of the hydrogel electrodes can be attributed to the extended hydrogen-bonded network due to the increase in hydrophilic groups (–OH, SO4−) present in the ARS molecular structure. On the contrary, the electrodes containing 30 mM of LS showed a fracture strain of 60% with a significantly lower E of 10 kPa. This was due to a lower concentration of PVA in the electrolyte, as the original PVA concentration resulted in viscous gels that were difficult to homogenously infiltrate into the PEDOT:CNF porous scaffold (see ESI† Fig. S9).
2.7. Stretchable and biodegradable batteries
The optimised components were assembled into a full cell configuration and its electrochemical, mechanical and degradation properties were characterised. CVs of the cell at 2 and 5 mV s−1 exhibited reversible redox peaks between 0.43 V and 0.55 V (Fig. 6a), which correspond to the reversible two-electron/two-proton redox reactions of LS and ARS (Fig. 1d). Interestingly, multiple peaks were observed in the CV of the full cell. Similarly, both ARS and LS showed multiple redox peaks in their CV measured in the half-cell configuration at slower scan rates (see ESI† Fig. S10). The exact mechanism is complex and warrants further investigation that is beyond the scope of this study, but it has been suggested that smaller peaks are attributed to the two-electron transfer occurring in sequential steps.89 Galvanostatic discharge capacities of the battery at different current densities exhibited a voltage plateau between 0.55 V and 0.32 V (Fig. 6b). A volumetric capacity of 2.68 mA h cm−3 at a current density of 0.2 mA cm−2 was achieved when considering a cell volume of 0.056 cm3 comprising the current collectors, the separator membrane, and the respective active electrodes. Minimal changes to the discharge capacities were observed at smaller current densities (0.2 to 0.5 mA cm−2), while a larger voltage drop and a significant reduction in the capacity to 1.67 and 0.85 mA h cm−3 was observed as the current was increased to 1.0 and 2.0 mA cm−2 respectively. Since electron transport is typically faster than ion transport, the diffusion kinetics of the large redox-species (LS and ARS) within the porous scaffold likely gives rise to ohmic losses at higher current densities.90 The GCD cyclic performance of the cell showed a reasonable capacity retention of 58% over 500 cycles at a current density of 0.5 mA cm−2 (Fig. 6c). The decrease in capacity is likely caused by the electrochemical instability of the LS catholyte as discussed earlier. | ||
| Fig. 6 Characterisation of the full battery cell. (a) CV curves at various scan rates. (b) Discharge capacities at various current densities. (c) Capacity retention and coulombic efficiency vs. number of GCD cycles. (d) Discharge capacity as a function of strain. The inset images are photographs of the cell at various strains. (e) Nyquist plots of the cells at various strains. (f) Discharge capacity of the cells mechanically cycled at 30% strain. (g) A pie chart representing the weight percentages of the materials used in the cell. (h) PGS mass loss [%] as a function of time degrading in PBS at various temperatures over a period of 8 weeks. (i) Photographs of the cell degradation process in PBS at 85 °C. | ||
At 30% strain, the discharge capacity showed minimal changes, while at 50% and 100% strain, the capacity decreased to 35% and 20% of the original value, respectively (Fig. 6d). The corresponding CV of the full cells under the respective strains showed a similar trend (ESI† Fig. S11a). Minimal changes in the CV curves with a slight broadening of the peaks occurred at 30% strain. However, at 50% and 100% strain, a significant decrease in the redox peaks was observed. Based on the characterisation of the individual battery components, it is likely that the lower stretchability of the cathode limits overall performance at higher strains. EIS measurements of the cells at the respective stretched states showed that the internal resistances at higher frequencies increased accordingly from 48 Ω and 51 Ω at 0% and 30% strain to 114 Ω and 141 Ω at 50% and 100% strain, respectively. Nonetheless, the cell showed stable mechanical cycling at 30% strain over 200 cycles where the discharge capacity was measured upon recovery from its stretched state (Fig. 6f) and showed a capacity retention of 55% over 500 GCD cycles at a current density of 0.5 mA cm−2 under 30% strain (see ESI† Fig. S11b). As the biomechanical environment for majority of on-skin or implanted devices experience mechanical strains of < 30%, the developed cell constitutes a suitable power supply unit for such applications.44
There are two broad categories for biodegradable materials; type I materials that partially break down and are typically classified as bio-disintegrable, and type II materials that completely degrade into its monomeric or oligomeric building blocks.91 In both cases, the degradation products should be environmentally benign. For our battery cell, the PGS encapsulation constitutes 56% in weight of the overall cell, and it must be degraded first before the breakdown of the internal materials can be initiated (Fig. 6g). Recently, it has been established that the ultimate aerobic biodegradability of PGS is approximately 70 days based on a standardized test (ISO 14851 2004-10), which is comparable to the 60 days for cellulose.92 To demonstrate degradation of the battery, we used a phosphate buffer solution (PBS) to initiate the de-esterification of the PGS polymer chains.71 We first studied the degradation of PGS by immersing it in PBS at various temperatures (Fig. 6h). Minimal mass loss (< 30%) was observed at room temperature and at 37 °C after 8 weeks, while at 85 °C the polymer completely degraded into its starting monomers as seen from the formation of white flakes (ESI† Fig. S12).
The accelerated condition was used to study the degradation of the full cell. After the second day, we observed the separation of the individual half-cells, which led to the leakage of the internal components (Fig. 6i). Here, the continued degradation of the individual components would vary at different time scales and pathways (See ESI† Table S2). Approximately 15.8% of the cell is composed of wood-based materials (CNFs, CFs and LS) that are known to degrade naturally and under aerobic conditions.93 The graphite present in the current collectors is naturally stable and non-toxic to the environment, while graphene up to a few layers thick has been reported to degrade in the presence of a human enzyme.94 PVA (0.06%) is biocompatible and can be degraded enzymatically through the oxidation of the alcohol linkages.95 ARS (0.36%) is a known wastewater dye molecule and various methods have been established to degrade the biomolecule under enzymatic96 and photocatalytic97 conditions. PEDOT:PSS (0.18%) is biocompatible but its biodegradability at molecular level is not well established. However, a recent report suggests that PEDOT:PSS can be consumed and digested by a larva of the darkling beetle, Zophobas Morio into smaller molecular building blocks.98 The commercial biomass-derived PU (4.05%) degrades into smaller oligomeric units under acidic conditions of the electrolyte at elevated temperatures but is stable at room temperature over an extended period (see ESI† Fig. S3) Based on the degradation behaviour of the individual materials, the full cell would degrade via type I and type II pathways. Further evaluation of the biodegradable properties of the individual materials would be required, preferably using standard methods such as the DIN EN ISO 14851 (2004-10) and will be in the scope of the follow-up work.
3. Conclusions
In this study, a holistic design approach for soft, stretchable, and biodegradable batteries has been developed – the redox-diffusion battery. It maximizes the utilisation of sustainable plant-based materials in all components of the cell, from cellulose fibers, plant-based redox-molecules (alizarin) and polymers (lignin), and biomass-derived elastomers (PGS and PU). The unique cellulose-based 3D electrode method enabled high mass loadings of the soluble redox-active component (ARS ≈ 40 mg cm−2), which led to a high capacity of 28.6 mA h cm−3, while retaining a low Young's modulus of 110 kPa. The negligible stiffening effect of the dissolved redox components allows for combining high mass loadings with outstanding softness, something which typically is not possible using conventional approaches. A stretchable ion-selective membrane was developed where CNFs play an important role in promoting the formation of the hydrophilic ionic transport channels within the membrane. For the current collectors we circumvented the need of metals by laser structuring cellulose graphite composite into kirigami patterns to induce stretchability. The full cell showed an operating voltage of 0.6 V with a maximum volumetric capacity of 2.68 mA h cm−3 at a current density of 0.2 mA cm−2. It showed stable capacity retention after 200 cycles at 30% strain, which demonstrates the mechanical robustness of all the battery components under stretching. Furthermore, at the end-of-use, we show that the encapsulation was chemically triggered to disintegrate under mild conditions, and briefly discussed the potential biodegradability of the individual materials used in the respective components of the cell. The 3D porous electrode design should be applicable for a wide range of dissolvable redox-active organic compounds. The full cell performance is mainly limited by the electrochemical and mechanical properties of the LS-based catholyte with respect to its solubility limitations and redox stability. Further improvement can be made by investigating alternative redox-active biomaterials with high solubility, stable redox activity, higher potential window, and preferably operation under neutral conditions.86,99 Ultimately, we believe that our work will encourage future battery designs to consider the mechanical softness and stretchability, the sustainability of the material used, its processing conditions, and its biodegradability at the end-of-life.Author contributions
A. R. and K. T. conceived the project and designed the experiments. P. I. prepared and processed the paper current collector. A. R., M. M. and P. I. designed and developed the kirigami patterned current collectors. A. R. and M. M designed and characterized the membranes. A. R. and M. M. synthesized and investigated the properties of the aerogel scaffolds. A. R., S. M., and N. K investigated and characterized the mechanical and electrochemical properties of the active electrodes and full cells. A. R. performed the biodegradation experiments. Y. L. and L. S performed the SEM experiments. Y. L. and N. K. performed and analysed the DLS and XRD experiments. A. R., I. E., R. C and K. T. supervised the work. A. R. wrote the first draft of the manuscript, and all authors contributed to the finalisation of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A.R acknowledges the funding support from the Marie Skłodowska-Curie Actions Seal of Excellence Fellowship program from the Swedish Governmental Agency for Innovation Systems, VINNOVA (grant 2021-01668). A. R., M. M., P. I., I. E., R. C., and K. T. were financially supported by the Knut and Alice Wallenberg Foundation, Linköping University, and industry through the Wallenberg Wood Science Centre. N. K. acknowledges the Swedish Research Council (Starting Grant No. 2020-05218). S. M. and R. C. are supported by the Swedish Energy Agency (P52023-1). N. R., R. C., and K. T. acknowledge the Swedish Government Strategic Research Area in Materials Science on Advanced Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No. 2009-00971). This work was partially supported by the Wallenberg Initiative Materials Science for Sustainability (WISE) funded by the Knut and Alice Wallenberg Foundation.Notes and references
- W. J. Song, S. Yoo, G. Song, S. Lee, M. Kong, J. Rim, U. Jeong and S. Park, Batteries Supercaps, 2019, 2, 181–199 CrossRef.
- J. C. Yang, J. Mun, S. Y. Kwon, S. Park, Z. Bao and S. Park, Adv. Mater., 2019, 31, 1904765 CrossRef CAS PubMed.
- H. C. Ates, P. Q. Nguyen, L. Gonzalez-Macia, E. Morales-Narváez, F. Güder, J. J. Collins and C. Dincer, Nat. Rev. Mater., 2022, 7, 887–907 CrossRef PubMed.
- Y. Luo, M. R. Abidian, J.-H. Ahn, D. Akinwande, A. M. Andrews, M. Antonietti, Z. Bao, M. Berggren, C. A. Berkey, C. J. Bettinger, J. Chen, P. Chen, W. Cheng, X. Cheng, S.-J. Choi, A. Chortos, C. Dagdeviren, R. H. Dauskardt, C.-A. Di, M. D. Dickey, X. Duan, A. Facchetti, Z. Fan, Y. Fang, J. Feng, X. Feng, H. Gao, W. Gao, X. Gong, C. F. Guo, X. Guo, M. C. Hartel, Z. He, J. S. Ho, Y. Hu, Q. Huang, Y. Huang, F. Huo, M. M. Hussain, A. Javey, U. Jeong, C. Jiang, X. Jiang, J. Kang, D. Karnaushenko, A. Khademhosseini, D.-H. Kim, I.-D. Kim, D. Kireev, L. Kong, C. Lee, N.-E. Lee, P. S. Lee, T.-W. Lee, F. Li, J. Li, C. Liang, C. T. Lim, Y. Lin, D. J. Lipomi, J. Liu, K. Liu, N. Liu, R. Liu, Y. Liu, Y. Liu, Z. Liu, Z. Liu, X. J. Loh, N. Lu, Z. Lv, S. Magdassi, G. G. Malliaras, N. Matsuhisa, A. Nathan, S. Niu, J. Pan, C. Pang, Q. Pei, H. Peng, D. Qi, H. Ren, J. A. Rogers, A. Rowe, O. G. Schmidt, T. Sekitani, D.-G. Seo, G. Shen, X. Sheng, Q. Shi, T. Someya, Y. Song, E. Stavrinidou, M. Su, X. Sun, K. Takei, X.-M. Tao, B. C. K. Tee, A. V.-Y. Thean, T. Q. Trung, C. Wan, H. Wang, J. Wang, M. Wang, S. Wang, T. Wang, Z. L. Wang, P. S. Weiss, H. Wen, S. Xu, T. Xu, H. Yan, X. Yan, H. Yang, L. Yang, S. Yang, L. Yin, C. Yu, G. Yu, J. Yu, S.-H. Yu, X. Yu, E. Zamburg, H. Zhang, X. Zhang, X. Zhang, X. Zhang, Y. Zhang, Y. Zhang, S. Zhao, X. Zhao, Y. Zheng, Y.-Q. Zheng, Z. Zheng, T. Zhou, B. Zhu, M. Zhu, R. Zhu, Y. Zhu, Y. Zhu, G. Zou and X. Chen, ACS Nano., 2023, 17(6), 5211–5295 CrossRef CAS PubMed.
- L. Portilla, K. Loganathan, H. Faber, A. Eid, J. G. D. Hester, M. M. Tentzeris, M. Fattori, E. Cantatore, C. Jiang, A. Nathan, G. Fiori, T. Ibn-Mohammed, T. D. Anthopoulos and V. Pecunia, Nat. Electron., 2023, 6, 10–17 Search PubMed.
- D. G. Mackanic, M. Kao and Z. Bao, Adv. Energy Mater., 2020, 2001424, DOI:10.1002/aenm.202001424.
- A. Rahmanudin, Z. Khan, K. Tybrandt and N. Kim, J. Mater. Chem. A, 2023, 11, 22718–22736 RSC.
- S. H. Lee, J. Hwang, C. Song, C. Park, H. S. Kim and H. Ahn, Adv. Funct. Mater., 2024, 34, 2310571 CrossRef CAS.
- D. G. Mackanic, T.-H. Chang, Z. Huang, Y. Cui and Z. Bao, Chem. Soc. Rev., 2020, 49, 4466–4495 RSC.
- N. Mittal, A. Ojanguren, M. Niederberger and E. Lizundia, Adv. Sci., 2021, 8, 2004814 CrossRef CAS.
- Y. Zhang, W. Bai, J. Ren, W. Weng, H. Lin, Z. Zhang and H. Peng, J. Mater. Chem. A, 2014, 2, 11054–11059 RSC.
- S. Xu, Y. Zhang, J. Cho, J. Lee, X. Huang, L. Jia, J. A. Fan, Y. Su, J. Su, H. Zhang, H. Cheng, B. Lu, C. Yu, C. Chuang, T.-I. Kim, T. Song, K. Shigeta, S. Kang, C. Dagdeviren, I. Petrov, P. V. Braun, Y. Huang, U. Paik and J. A. Rogers, Nat. Commun., 2013, 4, 1543 CrossRef PubMed.
- W. Liu, J. Chen, Z. Chen, K. Liu, G. Zhou, Y. Sun, M.-S. Song, Z. Bao and Y. Cui, Adv. Energy Mater., 2017, 7, 1701076 CrossRef.
- C. Wang, W. Zheng, Z. Yue, C. O. Too and G. G. Wallace, Adv. Mater., 2011, 23, 3580–3584 CrossRef CAS PubMed.
- Z. Wang, X. Li, Z. Yang, H. Guo, Y. J. Tan, G. J. Susanto, W. Cheng, W. Yang and B. C. K. Tee, EcoMat, 2021, 3, e12073 CrossRef CAS.
- K. Hatakeyama-Sato, H. Wakamatsu, K. Yamagishi, T. Fujie, S. Takeoka, K. Oyaizu and H. Nishide, Small, 2019, 15, 1805296 CrossRef.
- X. Chen, H. Huang, L. Pan, T. Liu and M. Niederberger, Adv. Mater., 2019, 31, 1904648 CrossRef CAS.
- T. N. Nguyen, B. Iranpour, E. Cheng and J. D. W. Madden, Adv. Energy Mater., 2022, 12, 2103148 CrossRef CAS.
- D. G. Mackanic, X. Yan, Q. Zhang, N. Matsuhisa, Z. Yu, Y. Jiang, T. Manika, J. Lopez, H. Yan, K. Liu, X. Chen, Y. Cui and Z. Bao, Nat. Commun., 2019, 10, 5384 CrossRef PubMed.
- W. J. Song, M. Kong, S. Cho, S. Lee, J. Kwon, H. B. Son, J. H. Song, D. G. Lee, G. Song, S. Y. Lee, S. Jung, S. Park and U. Jeong, Adv. Funct. Mater., 2020, 30, 2003608 CrossRef CAS.
- L. Yin, M. Cao, K. N. Kim, M. Lin, J.-M. Moon, J. R. Sempionatto, J. Yu, R. Liu, C. Wicker, A. Trifonov, F. Zhang, H. Hu, J. R. Moreto, J. Go, S. Xu and J. Wang, Nat. Electron., 2022, 5, 694–705 CrossRef.
- R. Kumar, J. Shin, L. Yin, J.-M. You, Y. S. Meng and J. Wang, Adv. Energy Mater., 2017, 7, 1602096 CrossRef.
- T. Ye, J. Wang, Y. Jiao, L. Li, E. He, L. Wang, Y. Li, Y. Yun, D. Li, J. Lu, H. Chen, Q. Li, F. Li, R. Gao, H. Peng and Y. Zhang, Adv. Mater., 2022, 34, 2105120 CrossRef CAS.
- M. Kaltenbrunner, G. Kettlgruber, C. Siket, R. Schwödiauer and S. Bauer, Adv. Mater., 2010, 22, 2065–2067 CrossRef CAS PubMed.
- S. Park, G. Thangavel, K. Parida, S. Li and P. S. Lee, Adv. Mater., 2019, 31, 1805536 CrossRef PubMed.
- G. Liu, J. Y. Kim, M. Wang, J.-Y. Woo, L. Wang, D. Zou and J. K. Lee, Adv. Energy Mater., 2018, 8, 1703652 CrossRef.
- G. Costa, P. A. Lopes, A. L. Sanati, A. F. Silva, M. C. Freitas, A. T. De Almeida and M. Tavakoli, Adv. Funct. Mater., 2022, 32, 2113232 CrossRef CAS.
- D. Liu, L. Su, J. Liao, B. Reeja-Jayan and C. Majidi, Adv. Energy Mater., 2019, 9, 1902798 CrossRef CAS.
- W. Liu, Z. Chen, G. Zhou, Y. Sun, H. R. Lee, C. Liu, H. Yao, Z. Bao and Y. Cui, Adv. Mater., 2016, 28, 3578–3583 CrossRef CAS PubMed.
- H. Li, Y. Ding, H. Ha, Y. Shi, L. Peng, X. Zhang, C. J. Ellison and G. Yu, Adv. Mater., 2017, 29, 1700898 CrossRef PubMed.
- D. Danninger, F. Hartmann, W. Paschinger, R. Pruckner, R. Schwödiauer, S. Demchyshyn, A. Bismarck, S. Bauer and M. Kaltenbrunner, Adv. Energy Mater., 2020, 10, 2000467 CrossRef CAS.
- M. Karami-Mosammam, D. Danninger, D. Schiller and M. Kaltenbrunner, Adv. Mater., 2022, 2204457, DOI:10.1002/adma.202204457.
- É. Lèbre, M. Stringer, K. Svobodova, J. R. Owen, D. Kemp, C. Côte, A. Arratia-Solar and R. K. Valenta, Nat. Commun., 2020, 11, 4823 CrossRef PubMed.
- F. P. Carvalho, Food Energy Secur., 2017, 6, 61–77 CrossRef.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2023, 8, 54–70 CrossRef.
- C. Liedel, ChemSusChem, 2020, 13, 2110–2141 CrossRef CAS PubMed.
- H. Wang, Y. Yang and L. Guo, Adv. Energy Mater., 2017, 7, 1700663 CrossRef.
- Z. Wang, Y. H. Lee, S. W. Kim, J. Y. Seo, S. Y. Lee and L. Nyholm, Adv. Mater., 2021, 33, 2000892 CrossRef CAS PubMed.
- L. Ma, Z. Bi, Y. Xue, W. Zhang, Q. Huang, L. Zhang and Y. Huang, J. Mater. Chem. A, 2020, 8, 5812–5842 RSC.
- N. Kim, S. Lienemann, Z. Khan, G. Greczynski, A. Rahmanudin, M. Vagin, F. Ahmed, I. Petsagkourakis, J. Edberg, X. Crispin and K. Tybrandt, J. Mater. Chem. A, 2023, 11, 25703–25714 RSC.
- X. Wei, W. Pan, W. Duan, A. Hollas, Z. Yang, B. Li, Z. Nie, J. Liu, D. Reed, W. Wang and V. Sprenkle, ACS Energy Lett., 2017, 2, 2187–2204 CrossRef CAS.
- M. Navarro-Segarra, C. Tortosa, C. Ruiz-Díez, D. Desmaële, T. Gea, R. Barrena, N. Sabaté and J. P. Esquivel, Energy Environ. Sci., 2022, 15, 2900–2915 RSC.
- M. Navarro-Segarra, P. P. Alday, D. Garcia, O. A. Ibrahim, E. Kjeang, N. Sabaté and J. P. Esquivel, ChemSusChem, 2020, 13, 2394–2401 CrossRef CAS PubMed.
- G. Balakrishnan, J. Song, C. Mou and C. J. Bettinger, Adv. Mater., 2022, 34, 2106787 CrossRef CAS PubMed.
- H. Sun, J. Zhu, D. Baumann, L. Peng, Y. Xu, I. Shakir, Y. Huang and X. Duan, Nat. Rev. Mater., 2019, 4, 45–60 CrossRef.
- Y. Kuang, C. Chen, D. Kirsch and L. Hu, Adv. Energy Mater., 2019, 9, 1901457 CrossRef.
- U. Ail, J. Nilsson, M. Jansson, I. A. Buyanova, Z. Wu, E. Björk, M. Berggren and X. Crispin, Adv. Sustainable Syst., 2023, 7, 2200396 CrossRef CAS.
- U. Ail, J. Phopase, J. Nilsson, Z. U. Khan, O. Inganäs, M. Berggren and X. Crispin, ACS Sustainable Chem. Eng., 2020, 8, 17933–17944 CrossRef CAS.
- J. Edberg, O. Inganäs, I. Engquist and M. Berggren, J. Mater. Chem. A, 2018, 6, 145–152 RSC.
- F. N. Ajjan, N. Casado, T. Rębiś, A. Elfwing, N. Solin, D. Mecerreyes and O. Inganäs, J. Mater. Chem. A, 2016, 4, 1838–1847 RSC.
- J. Edberg, R. Brooke, H. Granberg, I. Engquist and M. Berggren, Adv. Sustainable Syst., 2019, 3, 1900050 CrossRef.
- K. Sun, F. Ran, G. Zhao, Y. Zhu, Y. Zheng, M. Ma, X. Zheng, G. Ma and Z. Lei, RSC Adv., 2016, 6, 55225–55232 RSC.
- J. Zhong, L.-Q. Fan, X. Wu, J.-H. Wu, G.-J. Liu, J.-M. Lin, M.-L. Huang and Y.-L. Wei, Electrochim. Acta, 2015, 166, 150–156 CrossRef CAS.
- F. N. Crespilho, G. C. Sedenho, D. De Porcellinis, E. Kerr, S. Granados-Focil, R. G. Gordon and M. J. Aziz, J. Mater. Chem. A, 2019, 7, 24784–24787 RSC.
- T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 590, 47–56 CrossRef CAS PubMed.
- S. Han, F. Jiao, Z. U. Khan, J. Edberg, S. Fabiano and X. Crispin, Adv. Funct. Mater., 2017, 27, 1703549 CrossRef.
- Z. U. Khan, J. Edberg, M. M. Hamedi, R. Gabrielsson, H. Granberg, L. Wågberg, I. Engquist, M. Berggren and X. Crispin, Adv. Mater., 2016, 28, 4556–4562 CrossRef PubMed.
- M. Vagin, C. Che, V. Gueskine, M. Berggren and X. Crispin, Adv. Funct. Mater., 2020, 30, 2007009 CrossRef CAS.
- J. Yang, H. Wang, Y. Yang, J. Wu, P. Hu and L. Guo, Nanoscale, 2017, 9, 9879–9885 RSC.
- A. Mukhopadhyay, J. Hamel, R. Katahira and H. Zhu, ACS Sustainable Chem. Eng., 2018, 6, 5394–5400 CrossRef CAS.
- Z. Khan, U. Ail, F. Nadia Ajjan, J. Phopase, Z. Ullah Khan, N. Kim, J. Nilsson, O. Inganäs, M. Berggren and X. Crispin, Adv. Energy Sustainability Res., 2022, 3, 2100165 CrossRef CAS.
- D. Kumar, U. Ail, Z. Wu, E. M. Björk, M. Berggren, V. Gueskine, X. Crispin and Z. Khan, Adv. Sustainable Syst., 2022, 2200433, DOI:10.1002/adsu.202200433.
- C. Che, M. Vagin, U. Ail, V. Gueskine, J. Phopase, R. Brooke, R. Gabrielsson, M. P. Jonsson, W. C. Mak, M. Berggren and X. Crispin, Adv. Sustainable Syst., 2019, 3, 1900039 CrossRef CAS.
- L. Tong, Y. Jing, R. G. Gordon and M. J. Aziz, ACS Appl. Energy Mater., 2019, 2, 4016–4021 CrossRef CAS.
- Y. Liu, S. Lu, S. Chen, H. Wang, J. Zhang and Y. Xiang, ACS Appl. Energy Mater., 2019, 2(4), 2469–2474 CrossRef CAS.
- E. Melro, A. Filipe, D. Sousa, B. Medronho and A. Romano, New J. Chem., 2021, 45, 6986–7013 RSC.
- I. Gomez and E. Lizundia, Adv. Sustainable Syst., 2021, 2100236, DOI:10.1002/adsu.202100236.
- T. N. Williams, Nat. Chem., 2022, 14, 966 CrossRef CAS PubMed.
- P. Isacsson, X. Wang, A. Fall, D. Mengistie, E. Calvie, H. Granberg, G. Gustafsson, M. Berggren and I. Engquist, ACS Appl. Mater. Interfaces, 2020, 12, 48828–48835 CrossRef CAS PubMed.
- L. Vogt, F. Ruther, S. Salehi and A. R. Boccaccini, Adv. Healthcare Mater., 2021, 10, 2002026 CrossRef CAS PubMed.
- Y. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, Nat. Biotechnol., 2002, 20, 602–606 CrossRef CAS PubMed.
- T. C. Shyu, P. F. Damasceno, P. M. Dodd, A. Lamoureux, L. Xu, M. Shlian, M. Shtein, S. C. Glotzer and N. A. Kotov, Nat. Mater., 2015, 14, 785–789 CrossRef CAS PubMed.
- R. Lohmann, I. T. Cousins, J. C. Dewitt, J. Glüge, G. Goldenman, D. Herzke, A. B. Lindstrom, M. F. Miller, C. A. Ng, S. Patton, M. Scheringer, X. Trier and Z. Wang, Environ. Sci. Technol., 2020, 54, 12820–12828 CrossRef CAS PubMed.
- S. Lander, M. Vagin, V. Gueskine, J. Erlandsson, Y. Boissard, L. Korhonen, M. Berggren, L. Wågberg and X. Crispin, Adv. Energy Sustainability Res., 2022, 2200016, DOI:10.1002/aesr.202200016.
- A. Mukhopadhyay, Z. Cheng, A. Natan, Y. Ma, Y. Yang, D. Cao, W. Wang and H. Zhu, Nano Lett., 2019, 19, 8979–8989 CrossRef CAS PubMed.
- L. Hu, L. Gao, M. Di, X. Jiang, X. Wu, X. Yan, X. Li and G. He, Energy Storage Mater., 2021, 34, 648–668 CrossRef.
- P. Xi, F. Quan, Y. Sun and Y. Jiang, Composites, Part B, 2022, 242, 110078 CrossRef CAS.
- H. Yang, J. Edberg, V. Gueskine, M. Vagin, M. G. Say, J. Erlandsson, L. Wågberg, I. Engquist and M. Berggren, Carbohydr. Polym., 2021, 118938, DOI:10.1016/j.carbpol.2021.118938.
- F. Jiao, J. Edberg, D. Zhao, S. Puzinas, Z. U. Khan, P. Mäkie, A. Naderi, T. Lindström, M. Odén, I. Engquist, M. Berggren and X. Crispin, Adv. Sustainable Syst., 2018, 2, 1700121 CrossRef.
- S. B. Rauer, D. J. Bell, P. Jain, K. Rahimi, D. Felder, J. Linkhorst and M. Wessling, Adv. Mater. Technol., 2022, 7, 2100836 CrossRef CAS.
- X. Dong, X. Zhou, Y. Liu, S. Xiong, J. Cheng, Y. Jiang and Y. Zhou, Energy Environ. Sci., 2023, 16, 1511–1519 RSC.
- D. Boonpakdee, C. F. Guajardo Yévenes, W. Surareungchai and C. La-O-Vorakiat, J. Mater. Chem. A, 2018, 6, 7162–7167 RSC.
- A. Malti, J. Edberg, H. Granberg, Z. U. Khan, J. W. Andreasen, X. Liu, D. Zhao, H. Zhang, Y. Yao, J. W. Brill, I. Engquist, M. Fahlman, L. Wågberg, X. Crispin and M. Berggren, Adv. Sci., 2016, 3, 1500305 CrossRef PubMed.
- M. Lubert-Martin, M. Darbouret and J.-M. Herri, Récents Progrès en Génie des Procédés, Saint-Nazaire, France, 2003 Search PubMed.
- J. Nemoto, T. Saito and A. Isogai, ACS Appl. Mater. Interfaces, 2015, 7, 19809–19815 CrossRef CAS PubMed.
- J. Carretero-González, E. Castillo-Martínez and M. Armand, Energy Environ. Sci., 2016, 9, 3521–3530 RSC.
- D. P. Tabor, R. Gómez-Bombarelli, L. Tong, R. G. Gordon, M. J. Aziz and A. Aspuru-Guzik, J. Mater. Chem. A, 2019, 7, 12833–12841 RSC.
- T. S. Mathis, N. Kurra, X. Wang, D. Pinto, P. Simon and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1902007 CrossRef CAS.
- W. Wang, W. Xu, L. Cosimbescu, D. Choi, L. Li and Z. Yang, Chem. Commun., 2012, 48, 6669 RSC.
- M. O. Bamgbopa, J. Edberg, I. Engquist, M. Berggren and K. Tybrandt, J. Mater. Chem. A, 2019, 7, 23973–23980 RSC.
- V. R. Feig, H. Tran and Z. Bao, ACS Cent. Sci., 2018, 4, 337–348 CrossRef CAS PubMed.
- M. Held, A. Pichler, J. Chabeda, N. Lam, P. Hindenberg, C. Romero-Nieto and G. Hernandez-Sosa, Adv. Sustainable Syst., 2022, 6, 2100035 CrossRef CAS.
- N. B. Erdal and M. Hakkarainen, Biomacromolecules, 2022, 23, 2713–2729 CrossRef CAS PubMed.
- R. Kurapati, S. P. Mukherjee, C. Martín, G. Bepete, E. Vázquez, A. Pénicaud, B. Fadeel and A. Bianco, Angew. Chem., Int. Ed., 2018, 57, 11722–11727 CrossRef CAS PubMed.
- N. Ben Halima, RSC Adv., 2016, 6, 39823–39832 RSC.
- M. Rani, U. Shanker and A. K. Chaurasia, J. Environ. Chem. Eng., 2017, 5, 2730–2739 CrossRef CAS.
- S. K. Kansal, R. Lamba, S. K. Mehta and A. Umar, Mater. Lett., 2013, 106, 385–389 CrossRef CAS.
- S. Lee, Y. Hong and B. S. Shim, Adv. Sustainable Syst., 2022, 6, 2100056 CrossRef CAS.
- I. K. Ilic, V. Galli, L. Lamanna, P. Cataldi, L. Pasquale, V. F. Annese, A. Athanassiou and M. Caironi, Adv. Mater., 2023, 2211400, DOI:10.1002/adma.202211400.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00170b |
| This journal is © The Royal Society of Chemistry 2024 |