
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aluminothermic reduction of CeO2: mechanism of an economical route to aluminum–cerium alloys†
Alfred
Amon
 *a,
Emily E.
Moore
*a,
Hunter B.
Henderson
a,
Jibril
Shittu
a,
Martin
Kunz
b,
Shane
Kastamo
c,
Nikolai
Huotari
c,
Adam
Loukus
c,
Ryan
Ott
d,
David
Weiss
c and
Scott K.
McCall
a
*a,
Emily E.
Moore
*a,
Hunter B.
Henderson
a,
Jibril
Shittu
a,
Martin
Kunz
b,
Shane
Kastamo
c,
Nikolai
Huotari
c,
Adam
Loukus
c,
Ryan
Ott
d,
David
Weiss
c and
Scott K.
McCall
a
aLawrence Livermore National Laboratory, 7000 East Ave, Livermore, CA 94550, USA. E-mail: amon1@llnl.gov; moore255@llnl.gov
bLawrence Berkeley National Laboratory, 1 Cyclotron Rd, Berkeley, CA 94720, USA
cLoukus Technologies Inc., 58390 Centennial Number 6 Road, Calumet, MI 49913, USA
dAmes National Laboratory, 2415 Pammel Dr, Ames, IA 50011, USA
First published on 19th March 2024
Abstract
Cerium oxide is a low-value byproduct of rare-earth mining yet constitutes the largest fraction of the rare earth elements. The reduction of cerium oxide by liquid aluminum is proposed as an energy- and cost-efficient route to produce high-strength Al–Ce alloys. This work investigated the mechanism of a multi-step reduction reaction to facilitate the industrial adaptation of the process. Differential scanning calorimetry in combination with time-resolved synchrotron diffraction data uncovered the rate-limiting reaction step as the origin of the reported temperature dependence of reduction efficiency. This is the first in situ study of a metallothermic reaction mechanism and will serve as guidance for cost- and energy efficient industrial process control.
New conceptsAluminothermic reduction of cerium oxide can significantly reduce the cost and energy-consumption associated with Al–Ce alloy production compared to conventional processes. The direct use of cerium compounds accrued during rare earth element (REE) mining makes lightweight REE a value-added co-product instead of a waste product. In this study we demonstrate a novel approach to produce Al–Ce alloys by reducing CeO2 in liquid aluminum at 95% reduction efficiency. This work provides the first ever mechanistic insight into the reaction kinetics and intermediate products of an aluminothermic reaction, which reveal a multi-step reaction mechanism with complex temperature dependency, details that are necessary for scaling on an industrial level. This work provides the basis of an economic route to produce high strength Al–Ce alloys with enhanced dispersion strengthening from embedded Al2O3 particles. |
Nearly 200 years ago, H. C. Oersted revolutionized metallurgy, as he prepared metallic aluminum through the reduction of AlCl3 by potassium amalgam, inventing the metallothermic reaction.1 Since then, many elements have been isolated for the first time by reaction of their compounds with a more reactive metal to form a more stable compound and the sought-after elemental metal.2 Metallothermic reactions remain a crucial technology applied in the iron thermite reaction for welding of train tracks and the industrial production of metals such as Be, Ti, Ta and the rare earth elements through the reduction of their halides by Mg, Na or Ca.3,4
Rare earth elements tend to be co-located in ore deposits and are treated together in extraction. Cerium is the majority element in most deposits, and the growing need for Nd, Pr and the heavy lanthanides in permanent magnets and other energy transition technologies results in costly stockpiling of cerium oxide that has low demand.5–7 Aluminum alloys with up to 10 wt% cerium have been developed in the last decade as a new class of light-weight, high-temperature materials, exploiting formation of the high-melting intermetallic compound Ce3Al11 in the microstructure.6,8,9 As such, Al–Ce alloys are a competitive high strength alloy class that creates a high-value demand for excess cerium to help stabilize the rare earth market.10,11
Al–Ce alloys are produced by alloying Al melts with metallic Ce, which itself is produced by calciothermic reduction of CeCl3 in a prior energy-intensive step. This step can potentially be avoided by leveraging the following reaction (1), with a standard reaction enthalpy of −30.1 kJ mol−1.
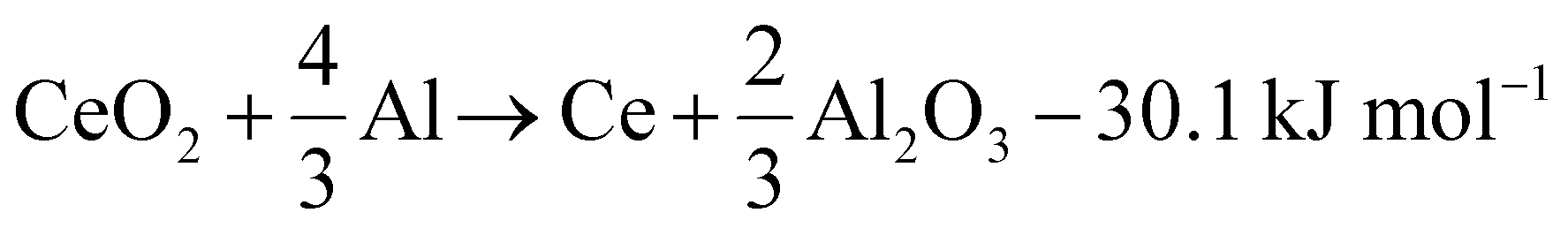 | (1) |
Mechanistic understanding of metallothermic reactions supports the effective parameter optimization in industry but is still in its infancy due to the harsh reaction conditions of these highly exothermic reactions. Aluminothermic reactions with ZrO2,12 MoO3,13,14 NaVO3,15 MgO,16 and Cr2O3,17 have been studied for industrial alloy production, while reduction of ZnO,18,19 was investigated by preparing Al/Al2O3 matrix composites.
Early investigations of the aluminothermic reduction of CeO2,20 and cerium carbonate,21 reported a strong temperature dependence of the conversion efficiency. No study has reported data on intermediate reaction products or mechanistic details.
To further fundamental understanding and accelerate industrial adaptation, we have investigated the reaction kinetics and mechanism of the aluminothermic reduction of CeO2 for the preparation of Al–Ce alloys, by means of differential scanning calorimetry (DSC), metallography, time-resolved synchrotron X-ray diffraction, and thermodynamic calculations.
The reaction between liquid aluminum and CeO2 particles was investigated via isothermal holds (800 °C ≤ Thold ≤ 950 °C) in DSC on pressed pellets of aluminum–CeO2 powder mixtures (Fig. 1A, experimental details in ESI†). The onset time of the exotherms (shaded areas under curves in Fig. 1A) was reduced with increasing reaction temperature from 15 min to less than 1 minute for the reactions at 825 °C and 950 °C, respectively.
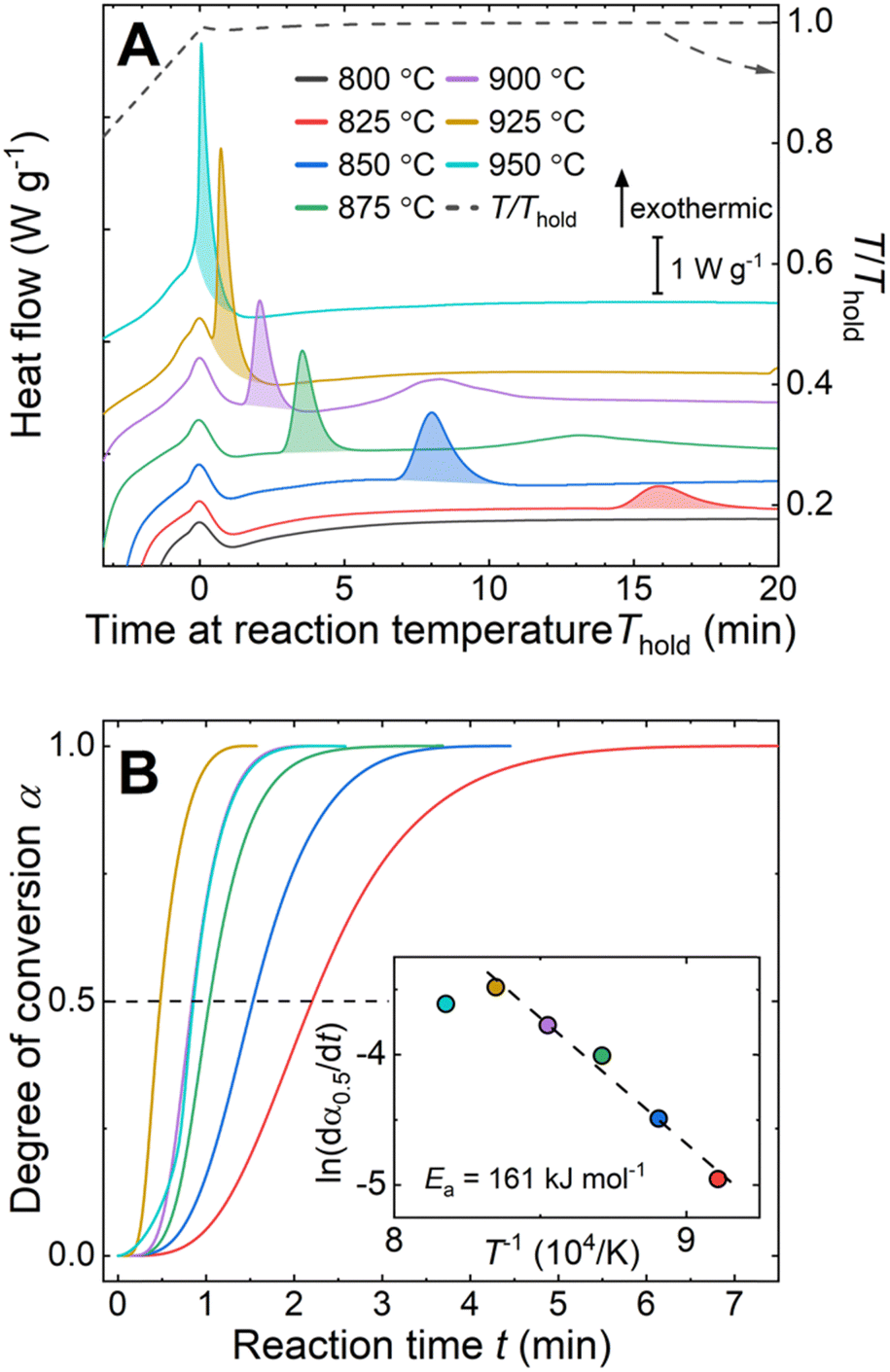 | ||
| Fig. 1 Calorimetry of Al–CeO2 mixtures with color legend in the top panel. (A) Detailed view of isothermal holds where the shaded areas reflect the heat of reaction. Curves are offset for clarity. (B) Corresponding kinetic curves of degree of conversion vs. reaction time for aluminothermic reduction reactions, derived from peak integration in (A). Inset to (B): Arrhenius-plot with fit (dashed line) to the data from 825 °C to 925 °C provides Ea. | ||
The kinetics of the reaction were further examined using the isothermal kinetic curves, i.e. normalized integral curves of the integrated peak areas, as the integral curves were found less sensitive to uncertainties in reaction onset time and baseline.22,23 The kinetic curves (Fig. 1B) revealed an accelerated reaction rate with increasing temperature up to 925 °C, while the rate at 950 °C did not increase further. The reaction rate 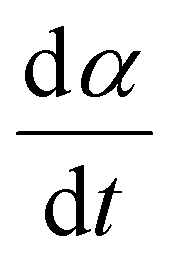 determined at half conversion (α = 0.5) confirmed the increase of the reaction rate with temperature and, using Friedman's isoconversional formulation of the Arrhenius equation (inset to Fig. 1B),22,24,25 allowed estimation of an effective activation energy Ea = 161 kJ mol−1. A second broad exothermic effect was observed with onset about 20, 13, 4 and 2 minutes after the completion of the initial reaction at 825, 850 °C, 875 °C and 900 °C, respectively (see Fig. 1A and Fig. S1, ESI†). This process of unclear origin appeared accelerated at higher temperatures.
determined at half conversion (α = 0.5) confirmed the increase of the reaction rate with temperature and, using Friedman's isoconversional formulation of the Arrhenius equation (inset to Fig. 1B),22,24,25 allowed estimation of an effective activation energy Ea = 161 kJ mol−1. A second broad exothermic effect was observed with onset about 20, 13, 4 and 2 minutes after the completion of the initial reaction at 825, 850 °C, 875 °C and 900 °C, respectively (see Fig. 1A and Fig. S1, ESI†). This process of unclear origin appeared accelerated at higher temperatures.
The reaction products after the DSC measurement at 850 °C were identified by powder X-ray diffraction (PXRD, see Fig. S2, ESI†) as Al (Cu structure, Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, a(300 K) = 4.045(2) Å), CeO2−x‡(CaF2 structure, Fmm, a(300 K) = 5.4054(2) Å), Al2O3 (Corundum structure, Rc, a(300 K) = 4.753(3) Å, c(300 K) = 12.984(4) Å) and Ce3Al11 (La3Al11 structure, Immm, a(300 K) = 4.3879(2) Å, b(300 K) = 10.0513(6) Å, c(300 K) = 13.0079(7) Å) confirming successful reduction of CeIVO2−x to Ce03Al11 (arguendo, we assume oxidation state Ce0 in the intermetallic compounds). After reaction at 950 °C, Al, Al2O3 and Ce3Al11 but no residual CeO2−x were observed, and the relative amounts of Ce3Al11 and Al2O3 doubled and tripled, respectively, compared to the reaction at 850 °C (see Fig. S3 and Table S1 for details, ESI†).
m, a(300 K) = 4.045(2) Å), CeO2−x‡(CaF2 structure, Fmm, a(300 K) = 5.4054(2) Å), Al2O3 (Corundum structure, Rc, a(300 K) = 4.753(3) Å, c(300 K) = 12.984(4) Å) and Ce3Al11 (La3Al11 structure, Immm, a(300 K) = 4.3879(2) Å, b(300 K) = 10.0513(6) Å, c(300 K) = 13.0079(7) Å) confirming successful reduction of CeIVO2−x to Ce03Al11 (arguendo, we assume oxidation state Ce0 in the intermetallic compounds). After reaction at 950 °C, Al, Al2O3 and Ce3Al11 but no residual CeO2−x were observed, and the relative amounts of Ce3Al11 and Al2O3 doubled and tripled, respectively, compared to the reaction at 850 °C (see Fig. S3 and Table S1 for details, ESI†).
Scanning electron micrographs of the pellet after reaction at 950 °C (Fig. 2) showed large agglomerates of Ce3Al11 grains with 30 µm to 50 µm diameter in an aluminum matrix, as identified by energy-dispersive X-ray spectroscopy (EDS) (Table S2 and Fig. S4, ESI†). Higher magnification revealed small crystallites of Al2O3 with a narrow size distribution around 5 µm embedded in the Ce3Al11 and Al phases (red arrows in Fig. 2B–E). These are significantly larger than the Al2O3 particles with 200 nm diameter observed in composite materials produced by reduction of Ce2(CO3)3.21
 | ||
| Fig. 2 Scanning electron microscopy on Al–CeO2 sample after holding at 950 °C for 20 minutes. (A) Secondary electron image showing large grains of Ce3Al11 (bright phase) in an Al matrix (dark). (B) High-magnification backscatter-electron image reveals small Al2O3 crystals (red arrows) embedded between Ce3Al11 and Al phases, as well as unreacted oxides. (C)–(E) EDS elemental maps of Al, O, and Ce distribution in the microstructure. | ||
After reaction at 850 °C, the micrographs (Fig. S5, ESI†) showed significant amounts of unreacted CeO2−x particles as well as Al and Ce3Al11 grains. Detailed investigation of the micrographs (Fig. S5B–E, ESI†) revealed that the Ce3Al11 grains appear to crystallize only from within the aluminum matrix.
To better understand the kinetics and mechanism of the aluminothermic reduction of CeO2 as well as the origin of the observed second exotherm in DSC, time-resolved synchrotron diffraction data were recorded on pressed pellets of aluminum–CeO2 powder mixtures contained in glass capillaries (12.2.2, Advanced Light Source/LBNL, USA). Diffraction data were recorded while ramping the sample temperature to the isothermal hold temperature (Thold = 850 °C, 900 °C and 950 °C) to monitor the formation and decomposition of crystalline phases by means of the peak areas of selected peaks (details in ESI†). At selected times, the relative phase fractions were determined by full pattern Rietveld refinement of the diffraction data (insets to Fig. 3).
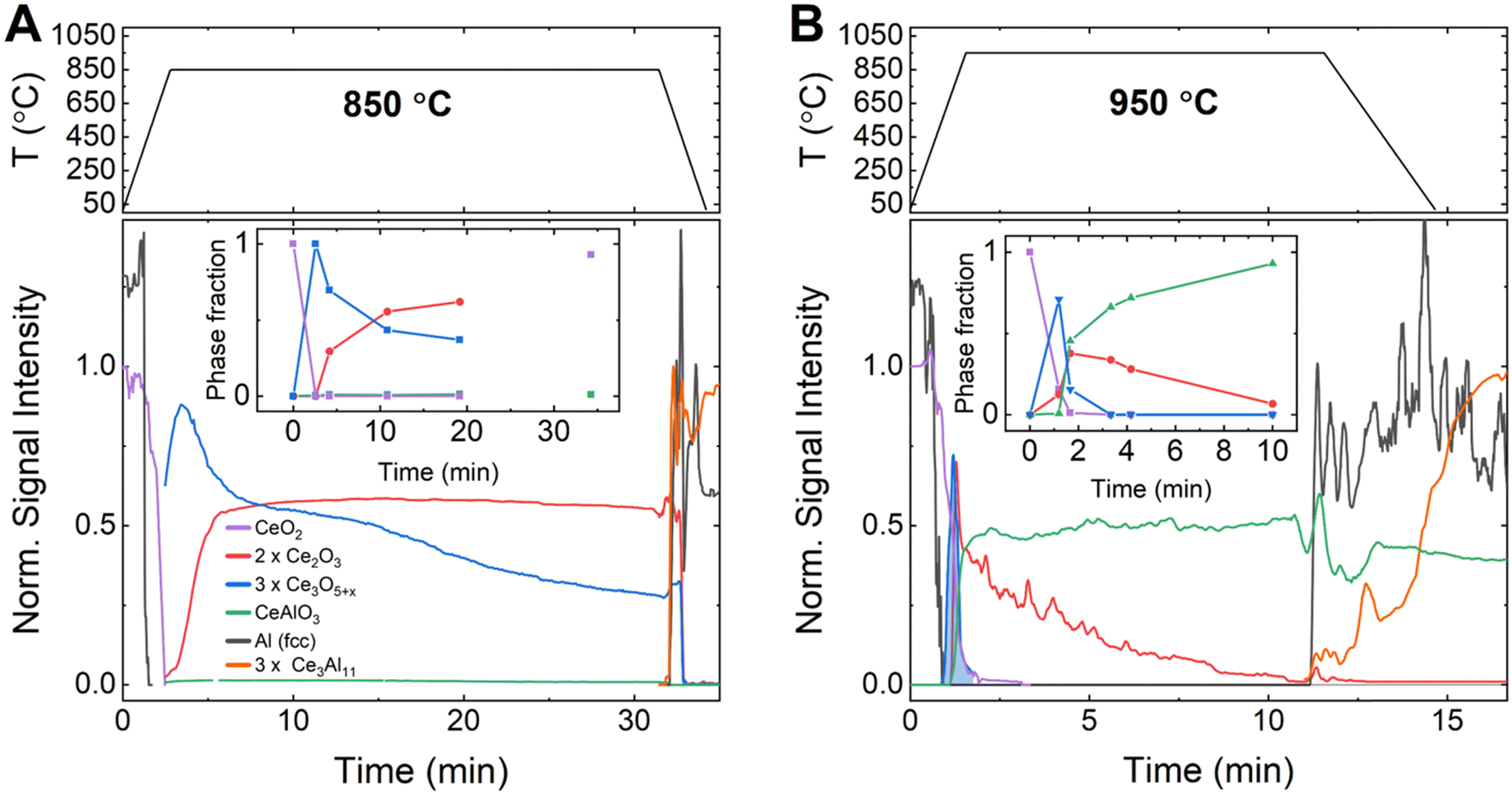 | ||
| Fig. 3 Phase formation (lower panels) observed by time-resolved synchrotron diffraction for the reaction of Al with CeO2 at (A) 850 °C and (B) 950 °C, together with temperature programs (upper panels). Curves for respective phases are normalized using results from Rietveld refinements to approximately reflect the evolution of relative phase amounts over time. Insets: Relative amounts of Ce-oxide phases determined at selected times, normalized by Ce content. | ||
As soon as melting of aluminum was complete, a rapid conversion of CeO2−x (CaF2 structure, Fmm, lattice parameter a(300 K) = 5.4280(2) Å) to the defect oxide Ce3O5+x (Bixbyite structure, space group Ia, a(1123 K) ≈ 11.37 Å) was observed at all temperatures.27 At 850 °C (Fig. 3A), Ce3O5+x reacted then slowly to form the sesquioxide Ce2O3 (HT-La2O3 structure, Pm, a(1123 K) ≈ 3.94 Å, c(1123 K) ≈ 6.17 Å). After 20 min of reaction time, a molar ratio of Ce3O5+x to Ce2O3, normalized by cerium content (i.e. CeO1.67+0.5x to CeO1.5), of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7 was determined by Rietveld refinement (inset to Fig. 3A, Table S4, ESI†). Upon cooling of the reaction mixture, the signal for both phases, Ce3O5+x and Ce2O3, disappeared and together with the solidification of the excess aluminum, the crystallization of Ce3Al11 and formation of CeO2−x was observed, in line with observations after DSC.
:1.7 was determined by Rietveld refinement (inset to Fig. 3A, Table S4, ESI†). Upon cooling of the reaction mixture, the signal for both phases, Ce3O5+x and Ce2O3, disappeared and together with the solidification of the excess aluminum, the crystallization of Ce3Al11 and formation of CeO2−x was observed, in line with observations after DSC.
The reactions were strongly accelerated at 950 °C (Fig. 3B) as the melting of aluminum initiated the rapid conversion of CeO2−x to Ce3O5+x which then decomposed rapidly to form Ce2O3.
Almost instantly, the ternary oxide CeAlO3 (CaTiO3 structure, Pmm, a(1223 K) ≈ 3.82 Å) formed and the evolution of relative phase amounts suggests that CeAlO3 was formed by consumption of Ce2O3. After 10 min, a ratio of Ce2O3 to CeAlO3, normalized by cerium content (i.e. CeO1.5 to CeAlO3), of about 1:93 was observed (Inset to Fig. 3B, Table S4, ESI†). Upon cooling of the reaction mixture, the residual Ce2O3 signal dropped to zero, while CeAlO3 remained constant, and Al and Ce3Al11 crystallized from the melt. The experiment at 900 °C (Fig. S7, ESI†), showed essentially a similar order of reactions as at 950 °C. After melting of Al and conversion of CeO2−x to Ce3O5+x, the latter phase reacted quickly to form Ce2O3 and CeAlO3. The relative amount of these phases, however, remained nearly constant over time at a 1:10 molar ratio (CeO1.5 to CeAlO3). The crystallization of small amounts of Ce3Al11 was observed prior to cooling down from 900 °C.
Thermal effects in DSC appeared delayed compared to the reactions observed in synchrotron data, most likely due to a difference in heating rate and sample mass. The first thermal effect coincides in time with the observed rapid conversion of CeO2−x to Ce3O5+x and Ce2O3 for reactions at 850 °C and 900 °C. Synchrotron data at 900 °C revealed a broad peak in the signal for Ce2O3 (Fig. S1, ESI†), suggesting accelerated formation of Ce2O3, coinciding with the delayed second signal in the DSC curve. Above 900 °C, the first and second thermal effects seem to overlap in one peak. As the time-resolved synchrotron data indicate the continuous decrease of Ce2O3 amount over an extended duration, this suggests that the reduction of Ce2O3 to CeAlO3 and subsequently metallic cerium is too slow to show a discernible DSC signal.
The reaction cascade taking place can then be summarized through eqn (2)–(5), omitting non-stoichiometry in the oxides or the dissolution of Ce−Al intermetallic phases in excess Al(l), where the enthalpies were calculated using the thermodynamic database developed for this work. Due to enhanced diffusion in the liquid phase and the strongly negative mixing enthalpy28 of Ce in Al(l), the excess liquid aluminum present will enrich in Ce content by rapidly dissolving any formed Ce-containing intermetallic compounds. This will remove any formed metallic cerium quickly from the equilibrium reaction, up to the solubility limit (about 7 at% at 900 °C), and precludes the observation of CeAl2 and CeAl3 intermetallic compounds by synchrotron diffraction.
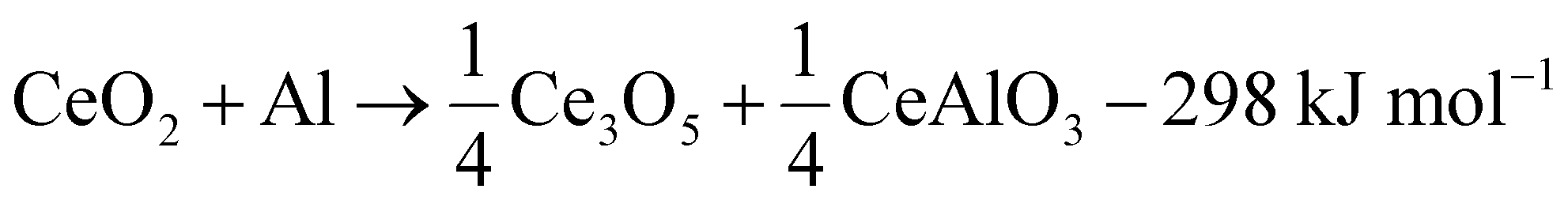 | (2) |
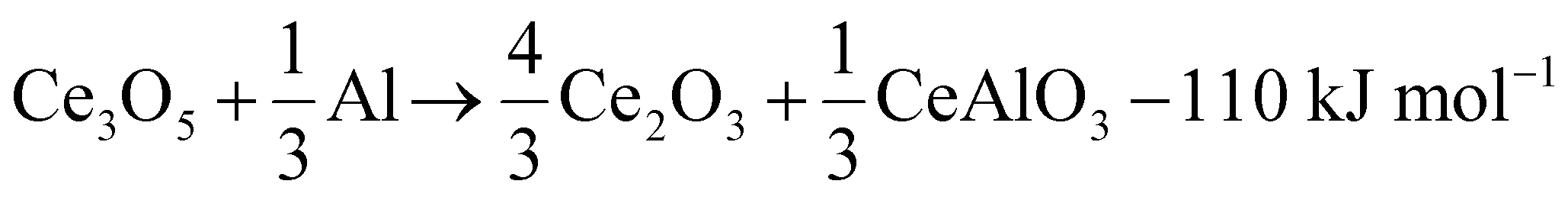 | (3) |
| Ce2O3 + 3Al → CeAl2 + CeAlO3 − 123 kJ mol−1 | (4) |
| CeAlO3 + 4Al → CeAl3 + Al2O3 − 23.8 kJ mol−1 | (5) |
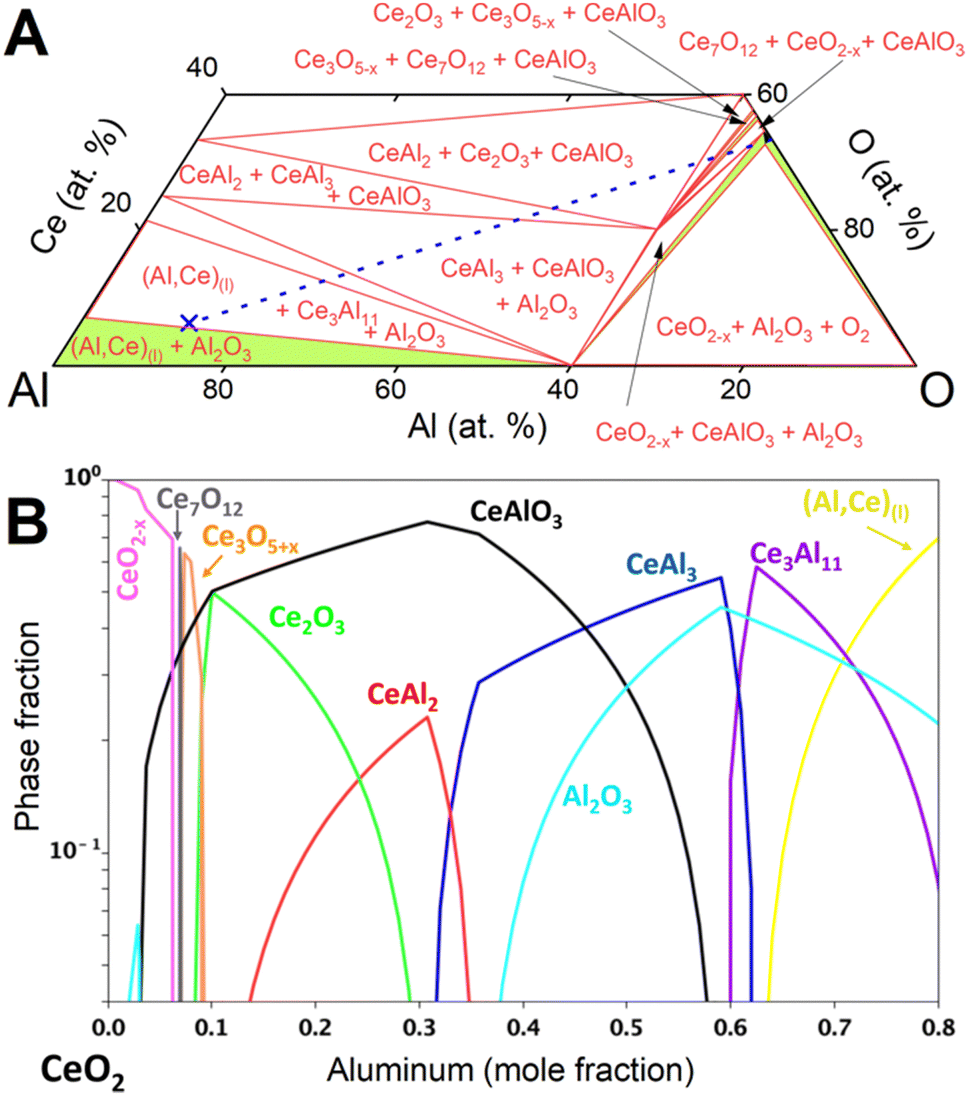 | ||
| Fig. 4 (A) Calculated isothermal section of the Al–Ce–O system (xCe ≤ 0.4) at 950 °C. Two- and three-phase equilibria are given as green and white areas (equilibrium phases indicated), respectively. Narrow two-phase equilibria have been omitted. Composition of synchrotron samples (blue cross) and connecting line (blue, dashed) from CeO2−x to Al are indicated. (B) Phases in equilibrium at 950 °C for compositions along the join from CeO2−x to Al (dashed line in panel A). | ||
As a new phase is formed and expands into the particle from the particle–Al(l) interface, the newly formed phase and excess Al(l) at the particle surface will tend to form the next stable phases in the next local equilibrium. The relative phase amounts observed in a particle over time depend on the competing rates of phase formation (advance of reaction front into particle) and consumption of the phase by the subsequent reaction.
The observed evolution of phase fractions (Fig. 3) deviates significantly from the calculated phase amounts along the Al–CeO2 vertical section (dashed line in Fig. 4A), which assume immediate and bulk equilibrium conditions (Fig. 4B).
During the first minutes, the rapid conversion of CeO2−x to Ce3O5+x (eqn (2)) and then Ce2O3 (eqn (3)) was observed at all temperatures, in line with the thermodynamic predictions (Fig. 4B). Only the Ce7O12 phase was not observed experimentally, possibly due to the narrow stability window. The fast reactions between CeO2−x, Ce3O5+x and Ce2O3 at all temperatures are in line with the high oxygen diffusion rate and the structural similarity of the cerium oxides.29–31
The consequent reaction of Ce2O3 with Al to form CeAlO3 (eqn (4)) shows markedly different dynamics depending on temperature, as outlined below. The observed strong temperature dependence of observed phase amounts of Ce2O3 and CeAlO3 can be explained by the competition of the diffusion-limited reaction of Ce2O3 to CeAlO3 and the consumption of CeAlO3 to form Ce0 as liquid solution (Al,Ce)(l), and solid Al2O3(s).
At 850 °C, Ce3O5+x reacts slowly to Ce2O3 (eqn (3)) but the consequent conversion to CeAlO3 is extremely slow compared to its reduction to Ce0 (eqn (5)) and only small quantities of CeAlO3 are therefore observed at any time. The Ce2O3 amount remained nearly constant during the observation period.
Rietveld phase fraction analysis of samples after DSC showed that about 63 at% of cerium was reduced to Ce03Al11, about 7 at% was found as CeIIIAlO3 and 30 at% as unreacted CeIVO2.
At 900 °C, a steady state between conversion of Ce2O3 to CeAlO3 and reduction to Ce0 is rapidly achieved, although at a much higher ratio of CeAlO3 to Ce2O3 than at 850 °C, as diffusion through the product layer is accelerated. About 95 at% of cerium was reduced to Ce03Al11, 2 at% was found as CeIIIAlO3 and 3% as unreacted CeO2.
At 950 °C, formation of CeAlO3 from Ce2O3 and reduction to Ce0 progress at similar rates. Ce2O3 was mostly consumed over the observation period, but the amount of CeAlO3 had not started to decrease by the time the reaction mixture was cooled down.
In the sample reacted during DSC, about 92 at% of cerium was reduced to Ce03Al11 and 8 at% was found as CeIIIAlO3.
Equilibrium calculations (Fig. 4) predict reduction of CeIVO2−x to Ce0 in the form of compounds CeAl2, CeAl3, Ce3Al11 and finally the liquid solution (Al,Ce)(l). Synchrotron diffraction data showed no evidence of crystalline CeAl2 or CeAl3 and only Ce3Al11 was crystallizing from the liquid solution (Al,Ce)(l). The large excess of Al(l), rapid diffusion in the liquid, and highly negative mixing enthalpy28 of Ce in Al(l) facilitated rapid removal of cerium from the particle surface into the bulk of the melt.
Conclusions
The results demonstrate the aluminothermic reduction of cerium as a viable, potentially economical, and direct route for the production of Al–Ce alloys, avoiding the costly and energy intensive isolation of cerium metal and providing a high-value use for excess cerium oxide from mine tailings. Material generated in the direct reduction process has a large amount of Al2O3 particles embedded in the aluminum matrix expected to contribute to dispersion strengthening in the final alloy. Al–Ce master alloys produced by this process can be used in the Al casting industry after addition of pure Al and other alloying elements (e.g. Mg, Si) to tune the desired composition (below 3 at% Ce).8 The rapid cooling rates observed in typical casting operations can then produce the high-strength eutectic microstructures of Ce3Al11 particles in the Al matrix.Time-resolved monitoring of the intermediate products revealed a complex multi-step reaction mechanism and clarified the importance of temperature control to maximize reaction yield while minimizing heating costs, which will be crucial for industrial implementation of this process.§
Author contributions
Conceptualization by A. A., D. W., R. T. O, and S. K. M. Methodology, validation by A. A. E. E. M and H. B. H. Investigation and experimentation by A. A., E. E. M, J. S., M. K., S. K., N. H. and A. L. Writing – original draft by A. A. and E. E. M. Writing – review and editing – A. A., E. E. M, H. B. H., J. S., M. K., S. K., N. H., A. L., R. T. O., D. W., and S. K. M. Visualization by A. A.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was sponsored by the Critical Materials Institute, an Energy Innovation Hub funded by the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy and Advanced Manufacturing Office. Work performed at Ames National Laboratory under contract DE-AC02-07CH11358, LLNL under contract DE-AC52-07NA27344. Beamline 12.2.2 at the Advanced Light Source is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.Notes and references
- H. C. Örsted, K. Dan. Vidensk. Selsk., 1825 Search PubMed.
- F. Wöhler, Ann. Phys., 1828, 89, 577–582 CrossRef.
- A. Holleman, Band 2 Nebengruppenelemente, Lanthanoide, Actinoide, Transactinoide, De Gruyter, 2016 Search PubMed.
- F. W. Hall, Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2000 Search PubMed.
- K. Binnemans, P. T. Jones, K. Van Acker, B. Blanpain, B. Mishra and D. Apelian, JOM, 2013, 65, 846–848 CrossRef.
- Z. C. Sims, M. S. Kesler, H. B. Henderson, E. Castillo, T. Fishman, D. Weiss, P. Singleton, R. Eggert, S. K. McCall and O. Rios, J. Sustainable Metall., 2022, 8, 1225–1234 CrossRef PubMed.
- D. J. Cordier, U.S. Geological Survey (2023) Minerals yearbook 2023, can be found under https://www.usgs.gov/centers/national-minerals-information-center/rare-earths-statistics-and-information, 2023.
- Z. C. Sims, O. R. Rios, D. Weiss, P. E. A. Turchi, A. Perron, J. R. I. Lee, T. T. Li, J. A. Hammons, M. Bagge-Hansen, T. M. Willey, K. An, Y. Chen, A. H. King and S. K. McCall, Mater. Horiz., 2017, 4, 1070–1078 RSC.
- H. B. Henderson, D. Weiss, Z. C. Sims, M. J. Thompson, E. E. Moore, A. Perron, F. Meng, R. T. Ott and O. Rios, in Light Metals, ed. A. Tomsett, Springer International Publishing, Cham, 2020, pp. 227–232 Search PubMed.
- R. T. Nguyen, D. D. Imholte, O. R. Rios, D. Weiss, Z. Sims, E. Stromme and S. K. McCall, Resour., Conserv. Recycl., 2019, 144, 340–349 CrossRef.
- T. Wu, A. Plotkowski, A. Shyam and D. C. Dunand, Mater. Sci. Eng., 2022, 833, 142551 CrossRef CAS.
- L. Chen, J. Yang, Y. Yang, Y. Zhang and Z. Wang, Mater. Today Commun., 2022, 31, 103714 CrossRef CAS.
- K. Sheybani, M. H. Paydar and M. H. Shariat, Int. J. Refract. Met. Hard Mater., 2019, 82, 245–254 CrossRef CAS.
- K. Sheybani, M. H. Paydar and M. H. Shariat, Trans. Indian Inst. Met., 2020, 73, 2875–2888 CrossRef CAS.
- Y. Zhang, X. Hu, F. Liu, J. Yang, L. Chen, W. Tao, A. Liu, Z. Shi and Z. Wang, J. Alloys Compd., 2023, 945, 169252 CrossRef CAS.
- J. Yang, M. Kuwabara, Z. Liu, T. Asano and M. Sano, Tetsu-to-Hagane, 2006, 92, 239–245 CrossRef CAS PubMed.
- K. Yoshitaka, J. Yang, Z. Liu and M. Kuwabara, J. High Temp. Soc., 2009, 34, 20–25 CrossRef.
- A. Maleki, M. Panjepour, B. Niroumand and M. Meratian, J. Mater Sci., 2010, 45, 5574–5580 CrossRef CAS.
- A. Maleki, N. Hosseini and B. Niroumand, Ceram. Int., 2018, 44, 10–23 CrossRef CAS.
- J. S. Luna A, A. Flores V, R. Muñiz V, A. F. Fuentes, J. Torres, N. Rodríguez R, J. C. Ortiz and P. Orozco, J. Rare Earths, 2011, 29, 74–76 CrossRef CAS.
- H. Wang, G. Li, Y. Zhao and G. Chen, Mater. Sci. Eng., 2010, 527, 2881–2885 CrossRef.
- S. Vyazovkin, Isoconversional Kinetics of Thermally Stimulated Processes, Springer International Publishing, Cham, 2015 Search PubMed.
- H. S. Ray and S. Ray, Kinetics of Metallurgical Processes, Springer, Singapore, 2018 Search PubMed.
- H. L. Friedman, J. Polym. Sci., Part C: Polym. Symp., 1964, 6, 183–195 CrossRef.
- S. Vyazovkin and C. A. Wight, Int. Rev. Phys. Chem., 1998, 17, 407–433 Search PubMed.
- D. J. M. Bevan, J. Inorg. Nucl. Chem., 1955, 1, 49–59 CrossRef CAS.
- M. Zinkevich, D. Djurovic and F. Aldinger, Solid State Ionics, 2006, 177, 989–1001 CrossRef CAS.
- M. I. Ivanov, V. V. Berezutskii, M. A. Shevchenko, V. G. Kudin and V. S. Sudavtsova, Powder Metall. Met. Ceram., 2015, 54, 80–92 CrossRef CAS.
- S. Ackermann, J. R. Scheffe and A. Steinfeld, J. Phys. Chem. C, 2014, 118, 5216–5225 CrossRef CAS.
- N. V. Skorodumova, S. I. Simak, B. I. Lundqvist, I. A. Abrikosov and B. Johansson, Phys. Rev. Lett., 2002, 89, 166601 CrossRef CAS PubMed.
- H. Charlton, G. Baldinozzi and M. Patel, Front. Nucl. Eng., 2023, 1, 1096142 CrossRef.
- M. Kunz, A. A. MacDowell, W. A. Caldwell, D. Cambie, R. S. Celestre, E. E. Domning, R. M. Duarte, A. E. Gleason, J. M. Glossinger, N. Kelez, D. W. Plate, T. Yu, J. M. Zaug, H. A. Padmore, R. Jeanloz, A. P. Alivisatos and S. M. Clark, J. Synchrotron Radiat., 2005, 12, 650–658 CrossRef PubMed.
- A. Doran, L. Schlicker, C. M. Beavers, S. Bhat, M. F. Bekheet and A. Gurlo, Rev. Sci. Instrum., 2017, 88, 013903 CrossRef CAS PubMed.
- L. Schlicker, A. Doran, P. Schneppmüller, A. Gili, M. Czasny, S. Penner and A. Gurlo, Rev. Sci. Instrum., 2018, 89, 033904 CrossRef PubMed.
- M. Basham, J. Filik, M. T. Wharmby, P. C. Y. Chang, B. El Kassaby, M. Gerring, J. Aishima, K. Levik, B. C. A. Pulford, I. Sikharulidze, D. Sneddon, M. Webber, S. S. Dhesi, F. Maccherozzi, O. Svensson, S. Brockhauser, G. Náray and A. W. Ashton, J. Synchrotron Radiat., 2015, 22, 853–858 CrossRef PubMed.
- J. Filik, A. W. Ashton, P. C. Y. Chang, P. A. Chater, S. J. Day, M. Drakopoulos, M. W. Gerring, M. L. Hart, O. V. Magdysyuk, S. Michalik, A. Smith, C. C. Tang, N. J. Terrill, M. T. Wharmby and H. Wilhelm, J. Appl. Cryst., 2017, 50, 959–966 CrossRef CAS PubMed.
- B. H. Toby and R. B. Von Dreele, J. Appl. Cryst., 2013, 46, 544–549 CrossRef CAS.
- L. Kaufman and H. Bernstein, Computer Calculation of Phase Diagrams With Special Reference to Refractory Metals, Academic Press Inc., United States, 1970 Search PubMed.
- N. Saunders and A. P. Miodownik, CALPHAD (Calculation of Phase Diagrams): A Comprehensive Guide, Elsevier, 1998 Search PubMed.
- H. Lukas, S. G. Fries and B. Sundman, Computational Thermodynamics: The Calphad Method, Cambridge University Press, Cambridge, 2007 Search PubMed.
- M. C. Gao, N. Ünlü, G. J. Shiflet, M. Mihalkovic and M. Widom, Metall. Mater. Trans. A, 2005, 36, 3269–3279 CrossRef.
- H. Mao, M. Selleby and O. Fabrichnaya, Calphad, 2008, 32, 399–412 CrossRef CAS.
- C. Guéneau, N. Dupin, L. Kjellqvist, E. Geiger, M. Kurata, S. Gossé, E. Corcoran, A. Quaini, R. Hania, A. L. Smith, M. H. A. Piro, T. Besmann, P. E. A. Turchi, J. C. Dumas, M. J. Welland, T. Ogata, B. O. Lee, J. R. Kennedy, C. Adkins, M. Bankhead and D. Costa, Calphad, 2021, 72, 102212 CrossRef.
- W. T. Fu and D. J. W. Ijdo, J. Solid State Chem., 2006, 179, 2732–2738 CrossRef CAS.
- G. Grimvall, Thermophysical Properties of Materials, North-Holland, 1986 Search PubMed.
- O. Kubaschewski, Materials Thermochemistry, Pergamon Press, New York, 1993 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. Experimental methods as well as details on diffraction experiments, metallography, and DSC. The authors cite additional ref. 32–46. See DOI: https://doi.org/10.1039/d4mh00087k |
| ‡ Significant homogeneity ranges were reported for the oxides CeO2−x and Ce3O5+x.26,27 |
| § Please refer to the ESI† for the Methods section, which includes details of sample preparation, DSC, PXRD, and electron micrograph and spectroscopy data collection, Rietveld refinement, and equilibrium calculations.32–46 |
| This journal is © The Royal Society of Chemistry 2024 |