
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-covalent planarizing interactions yield highly ordered and thermotropic liquid crystalline conjugated polymers†
Sina
Sabury‡
 a,
Zhuang
Xu‡
c,
Shamil
Saiev
b,
Daniel
Davies
c,
Anna M.
Österholm
a,
Joshua M.
Rinehart
a,
Motahhare
Mirhosseini
d,
Benedict
Tong
a,
Sanggyun
Kim
e,
Juan-Pablo
Correa-Baena
e,
Veaceslav
Coropceanu
b,
Oana D.
Jurchescu
d,
Jean-Luc
Brédas
b,
Ying
Diao
c and
John R.
Reynolds
*a
a,
Zhuang
Xu‡
c,
Shamil
Saiev
b,
Daniel
Davies
c,
Anna M.
Österholm
a,
Joshua M.
Rinehart
a,
Motahhare
Mirhosseini
d,
Benedict
Tong
a,
Sanggyun
Kim
e,
Juan-Pablo
Correa-Baena
e,
Veaceslav
Coropceanu
b,
Oana D.
Jurchescu
d,
Jean-Luc
Brédas
b,
Ying
Diao
c and
John R.
Reynolds
*a
aSchool of Chemistry and Biochemistry, School of Materials Science and Engineering, Center for Organic Photonics and Electronics, Georgia Tech Polymer Network, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: reynolds@chemistry.gatech.edu
bDepartment of Chemistry and Biochemistry, The University of Arizona, Tucson, AZ 85721-0041, USA
cDepartment of Chemical and Biomolecular Engineering, Department of Chemistry, Beckman Institute for Advanced Science and Technology, University of Illinois Urbana–Champaign, 600 S. Mathews Avenue, Urbana, Illinois 61801, USA
dDepartment of Physics and Center for Functional Materials, Wake Forest University, Winston-Salem, NC 27109, USA
eSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
First published on 30th April 2024
Abstract
Controlling the multi-level assembly and morphological properties of conjugated polymers through structural manipulation has contributed significantly to the advancement of organic electronics. In this work, a redox active conjugated polymer, TPT–TT, composed of alternating 1,4-(2-thienyl)-2,5-dialkoxyphenylene (TPT) and thienothiophene (TT) units is reported with non-covalent intramolecular S⋯O and S⋯H–C interactions that induce controlled main-chain planarity and solid-state order. As confirmed by density functional theory (DFT) calculations, these intramolecular interactions influence the main chain conformation, promoting backbone planarization, while still allowing dihedral rotations at higher kinetic energies (higher temperature), and give rise to temperature-dependent aggregation properties. Thermotropic liquid crystalline (LC) behavior is confirmed by cross-polarized optical microscopy (CPOM) and closely correlated with multiple thermal transitions observed by differential scanning calorimetry (DSC). This LC behavior allows us to develop and utilize a thermal annealing treatment that results in thin films with notable long-range order, as shown by grazing-incidence X-ray diffraction (GIXD). Specifically, we identified a first LC phase, ranging from 218 °C to 107 °C, as a nematic phase featuring preferential face-on π–π stacking and edge-on lamellar stacking exhibiting a large extent of disorder and broad orientation distribution. A second LC phase is observed from 107 °C to 48 °C, as a smectic A phase featuring sharp, highly ordered out-of-plane lamellar stacking features and sharp tilted backbone stacking peaks, while the structure of a third LC phase with a transition at 48 °C remains unclear, but resembles that of the solid state at ambient temperature. Furthermore, the significance of thermal annealing is evident in the ∼3-fold enhancement of the electrical conductivity of ferric tosylate-doped annealed films reaching 55 S cm−1. More importantly, thermally annealed TPT–TT films exhibit both a narrow distribution of charge-carrier mobilities (1.4 ± 0.1) × 10−2 cm2 V−1 s−1 along with a remarkable device yield of 100% in an organic field-effect transistor (OFET) configuration. This molecular design approach to obtain highly ordered conjugated polymers in the solid state affords a deeper understanding of how intramolecular interactions and repeat-unit symmetry impact liquid crystallinity, solution aggregation, solution to solid-state transformation, solid-state morphology, and ultimately device applications.
New conceptsThe macromolecular assembly of conjugated polymers is determined by π–π stacking, main-chain planarity, side-chain structure, and placement, regioregularity, and symmetry. In this study, we have designed a conjugated polymer with regiosymmetric repeat units that we intentionally engineer to enable intramolecular through-space interactions along the backbone. The novelty of our design concept resides in its seeking to serve simultaneously the following purposes: (1) regulating the polymer backbone's conformation, (2) managing solubility and aggregation behavior, and (3) creating a highly organized and ordered solid-state structure. While there exists a considerable body of literature detailing the integration of non-covalent interactions into the structural framework of conjugated materials, both small molecules and polymers, the primary focus has typically been on inducing planarity without the solubility challenges associated with other planarization strategies, such as ladder-type designs. To date, the investigation of the reversibility of these non-covalent intramolecular interactions in the solid state has received significantly less attention. For instance, to the best of our knowledge, there are no reports concerning the utilization of intramolecular reversible conformational locks to induce liquid crystallinity. Thus, the present work contributes significantly to the fundamental design principles aimed at achieving heightened control over the self-assembly of conjugated polymers. |
Introduction
Polymers that assemble into ordered macromolecular structures have greatly contributed to the advancement of organic electronics, including the development of highly performing photovoltaic devices, light emitting diodes, and field-effect transistors, as well as the creation of highly conductive and electroactive materials.1–4 The long-range order in conjugated polymers, described as the extended spatial coherence of the polymer chains over considerable distances, contributes significantly to the electronic and optical properties of these materials. Within the ordered regions, the extended π-electron conjugation and minimal energetic disorder substantially reduces energy barriers for charge transport, resulting in improved energy and charge transfer properties.5–8 Hence, achieving long-range order in conjugated polymers through precise molecular design is an important goal as it optimizes their performance in (opto)electronic devices.Backbone π–π stacking and chain planarity, as well as side-chain length and bulkiness, regioregularity, and symmetry are recognized as primary factors controlling chain organization in conjugated polymers.1,3 Regioregularity, as exemplified by poly(3-hexylthiophene) (P3HT), plays a crucial role in promoting the formation of well-ordered and semicrystalline polymer structures as the material transitions from a solution phase to the solid state.9,10 Moreover, using symmetrically substituted monomers to form polymers with regiosymmetric repeat units minimizes chain defects and promotes longer-range ordering.11–14 For instance, poly(1,4-bis(2-thienyl)phenylene) (TPT) with symmetrical bis-dodecyloxy side-chains (–OC12H25, –OC12H25) is highly crystalline, whereas the asymmetric homolog with unsymmetrical substituent placement (–OCH3, –OC12H25) has an amorphous solid-state morphology.12 In addition to structures similar to poly(1,4-bis(2-thienyl)phenylene),15,16 regiosymmetry in dioxythiophene-based and other high-mobility conjugated polymers is also shown to promote ordered chain packing.14,17,18 Conversely, repeat unit asymmetry reduces the molecular packing tendency and aggregation behaviors in both solid and solution states.19,20 Various approaches such as the incorporation of fused rings,21 ladder-type structures,22 as well as non-covalent intramolecular interactions23 have been taken to promote backbone planarity to improve the solid-state order in conjugated polymers along with making torsion-free highly performing amorphous conjugated polymers.24–26 While fused ring and ladder-type structures often impose solubility limitations, backbone planarization through non-covalent interactions (such as S⋯O and S⋯F) leads to highly planar yet solution processable polymers.23,27–30 Non-covalent interactions impacting oxidation potential and conductivity in doped complexes are exemplified in the poly(3,4-ethylenedioxythiophene)'s (PEDOT, Scheme 1a) rigid and co-planar backbone and attributed to the through-space short contacts between neighboring sulfur and oxygen atoms.31,32 This conformational control through non-covalent heteroatom interactions notably influences, e.g., the transport gap (corresponding to the difference between the polymer ionization potential and electron affinity or, roughly speaking, to the energy gap between the highest-occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO)). Variations in the transport gap as large as ∼1 eV have been correlated to this effect.32 Due to the reversible formation-dissociation nature of these non-bonding interactions,33–36 the resulting polymers retain solution processability.23 Once processed, these non-covalent interactions gain greater significance in the solid state as they promote the formation of more ordered structures through conformational locking.37
 | ||
| Scheme 1 Structure of PEDOT (a), PBTTT (b), and TPT–TT (c) polymers. Schematic representation of non-covalent interactions contributing to chain conformations in solution and solid states (d). | ||
In addition to regiosymmetry, regioregularity, and backbone planarity, side-chain intercalation and crystallization also contribute to the solid-state morphology and order.38–40 For example, regiosymmetric poly[2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophene] (PBTTT, Scheme 1b) exhibits high hole mobility (on the order of 0.1–1 cm2 V−1 s−1), high conductivity (exceeding 400 S cm−1) upon chemical doping, as well as liquid crystalline behavior owing to its distinctive side-chain interdigitation.41–43 While the as-cast PBTTT-C16 (PBTTT with hexadecyl side-chain, n = 16 in Scheme 1b) has moderate mobilities ∼0.069 cm2 V−1 s−1, further post-processing treatments such as annealing and high-temperature rubbing leads to mobilities as high as ∼1 cm2 V−1 s−1 owing to highly ordered morphologies in optimized transistor devices.44,45 Likewise, significant enhancement in the electrical conductivity of the sequentially doped PBTTT films has been reported following post-processing treatments.46,47 These performance improvements stemming from post-processing treatments are attributed to a high degree of crystallinity and/or alignment brought by thermotropic liquid crystallinity that improves interchain transport.
Here, our aim was to create a thermotropic, liquid crystalline, redox active material with the potential to form highly ordered structures. To achieve this, we combined the regiosymmetric 1,4-(2-thienyl)-2,5-dialkoxyphenylene (TPT) with two non-covalent interactions per unit (S⋯O and S⋯H–C) with thienothiophene (TT) as the π-bridge to obtain TPT–TT (Scheme 1c). Scheme 1d illustrates how the non-bonding interactions allow this polymer to switch between a torsionally flexible form and a conformationally locked state that dictates the chain arrangement. Photophysical measurements show temperature-dependent formation of polymer aggregates in solution, whereas multiple thermal transitions (observed by differential scanning calorimetry, DSC) and the birefringent optical response confirm liquid crystalline behavior. This liquid crystalline behavior is then exploited to further enhance the solid-state order by thermally annealing the thin films, resulting in diffraction patterns indicating long-range order through in situ grazing incidence X-ray diffraction (GIXD) measurements. Ordered phase unit-cell determination demonstrates that side-chain interdigitation during the annealing process boosts the coherent solid-state ordering. The impact of enhanced solid-state order upon annealing is exemplified by the improvement in the electrical conductivity (from 18 to 55 S cm−1) in ferric tosylate doped TPT–TT films. Field-effect transistors with TPT–TT as the transport layer material exhibited hole mobilities of (1.4 ± 0.1) × 10−2 cm2 V−1 s−1. More importantly, the narrow distribution of the measured field-effect mobility values and the high device yield (100%) are particularly promising in the context of future device applications.
Results and discussion
To investigate the impact of intra-chain interactions on backbone conformation, we carried out density functional theory (DFT) calculations to examine the dihedral potentials along the TPT–TT repeat unit in isolated chains, i.e., in the “gas phase” (see Fig. 1a). The reference energy, set at 0 kcal mol−1, corresponds to the anti-conformation of the T–TT group (as illustrated by the green bonds in the inset of Fig. 1a). We note that the alkyl side-chains were modeled by ethyl groups, an approach that effectively circumvents the computational complexities associated with longer chains while enabling the assessment of the influence of side-chain bulkiness on torsional barriers. As evidenced by the low-energy conformers at 150°–200° in the phenylene–thienylene (P–T) dihedral (purple bonds in the inset of Fig. 1a, 0° representing anti-conformation for oxygen and sulfur in P–T bond), the through-space non-covalent S⋯O interactions play a determining factor in inducing backbone planarity. Stabilization of this dihedral is also seen at approximately 30° and 330°, which is attributed to S⋯H–C interactions. These two sets of minima display comparable energy levels and are separated by an energy barrier ΔE1 = 3.17 kcal mol−1, which points to the strong contributions of the intra-chain interactions in the stabilization of the optimal conformations. The rotation of thienylene–thienothiophene (T–TT) is less restricted, with a significantly smaller anti to syn rotational barrier of ΔE2 = 1.63 kcal mol−1, making this transition more energetically accessible. It is worth noting that the stabilized T–TT conformations yield a slight reduction in the ring coplanarity (∼40°) compared to P–T conformer. With lower energy barriers and a capacity for wider twist angles, this (T–TT) dihedral angle is expected to facilitate chain coiling. | ||
| Fig. 1 (a) Torsional potentials around the phenylene–thienylene (P–T) and thienylene–thienothiophene (T–TT) groups; the most stable conformers for both T–TT and P–T dihedrals are shown in the repeat unit structure. (b) Time-dependent evolution of the radius of gyration of the TPT–TT chain, as obtained from molecular dynamics simulations of the transformation of a linear rod TPT–TT polymer chain to stacked-rod and toroid structures. | ||
In addition to the segmental planarity induced by intramolecular contacts, π–π stacking interactions of the constituent rings contribute to the backbone configuration. Specifically, thienothiophene units with their fused-ring structure (and no side-chains) are reported to promote interchain interactions.40,48,49 Our molecular dynamics simulations conducted at 300 K in the gas phase, employing the NVT (constant number of particles, volume, and temperature) ensemble, point to the folding of a single chain. We observe the transitions from linear rod conformations to coiled stacked-rod and toroid structures (Fig. 1b) an evolution driven by the preference to maximize π–π stacking in combination with the nature of the dihedral potential energy surfaces.
Expanding on the single-chain simulations, the solvation behavior of the TPT–TT polymer was investigated in toluene and chlorobenzene using a pair of trimeric chains (dihedral scans for optimized potentials for liquid simulations−all atom, OPLS-AA, force field is presented in Fig. S1, ESI†) at a concentration of 15 mg mL−1 to closely mimic the experimental solubility values. Starting from chains being in close vicinity of each other (about 5 Å apart), as shown in Fig. S2 (ESI†), simulations ran for 150 ns or until the centers of mass were separated by 20 Å, a criterion established to indicate irreversible separation. The results for simulations conducted between 280 and 330 K reveal pronounced separation of the chains in chlorobenzene, even at room temperature. In marked contrast, in toluene, the chains maintain their association throughout the simulation, although there are some indications of chain de-aggregation above 320 K. As we will show, these results are consistent with the temperature- and solvent-dependent aggregation tendencies observed for TPT–TT. Overall, the MD simulations indicate the potential for the TPT–TT polymer to exhibit an ordered assembly due to the interplay between chain planarity and π–π stacking, promoted by non-covalent interactions and the thienothiophene unit, respectively. The reversible nature of the non-covalent intra-chain interactions points to the microstructural tunability that TPT–TT could display.
TPT–TT (Scheme 1c), with octyloxy and decyl sidechains attached to the phenyl and thiophene rings, respectively, was synthesized via Stille cross-coupling polymerization (synthetic and general characterization details found in the ESI,† Fig. S3–S11). The polymer has a number-average molecular weight of 24.6 kg mol−1 and a dispersity (Đ) of 1.93 with a monomodal distribution. Based on matrix-assisted laser desorption ionization (MALDI) time-of-flight (TOF) mass spectrometry, the incorporation ratio of TPT vs. TT units was equivalent, as expected for an alternating co-polymer. There is no evidence of homocoupling, as shown in Fig. S12 (ESI†), and the end group analysis in Fig. S13 (ESI†) shows the expected H–H, Br–Br, and H–Br chain ends. The photographs of polymer solutions and normalized UV-vis absorption spectra in Fig. 2a and b, demonstrate that TPT–TT is well solvated in dilute (∼0.1 mg mL−1) room-temperature chlorobenzene, based on the featureless band with a λmax of 470 nm (2.64 eV). However, when dissolved in toluene, two low-energy shoulders appear around 520 nm and 558 nm, which are expected to originate from the presence of aggregates or strong intermolecular interactions between polymer segments.50–55 Time-dependent density functional theory (TD-DFT) calculations, based on a TPT–TT trimer and considering implicit solvents, indicate that the main absorption band of a polymer chain in both chlorobenzene and toluene is predominantly due to the S0 → S1 electronic transition, corresponding to λ = 482 nm (2.56 eV), which closely aligns with the experimentally determined value (see Fig. S14, ESI†).
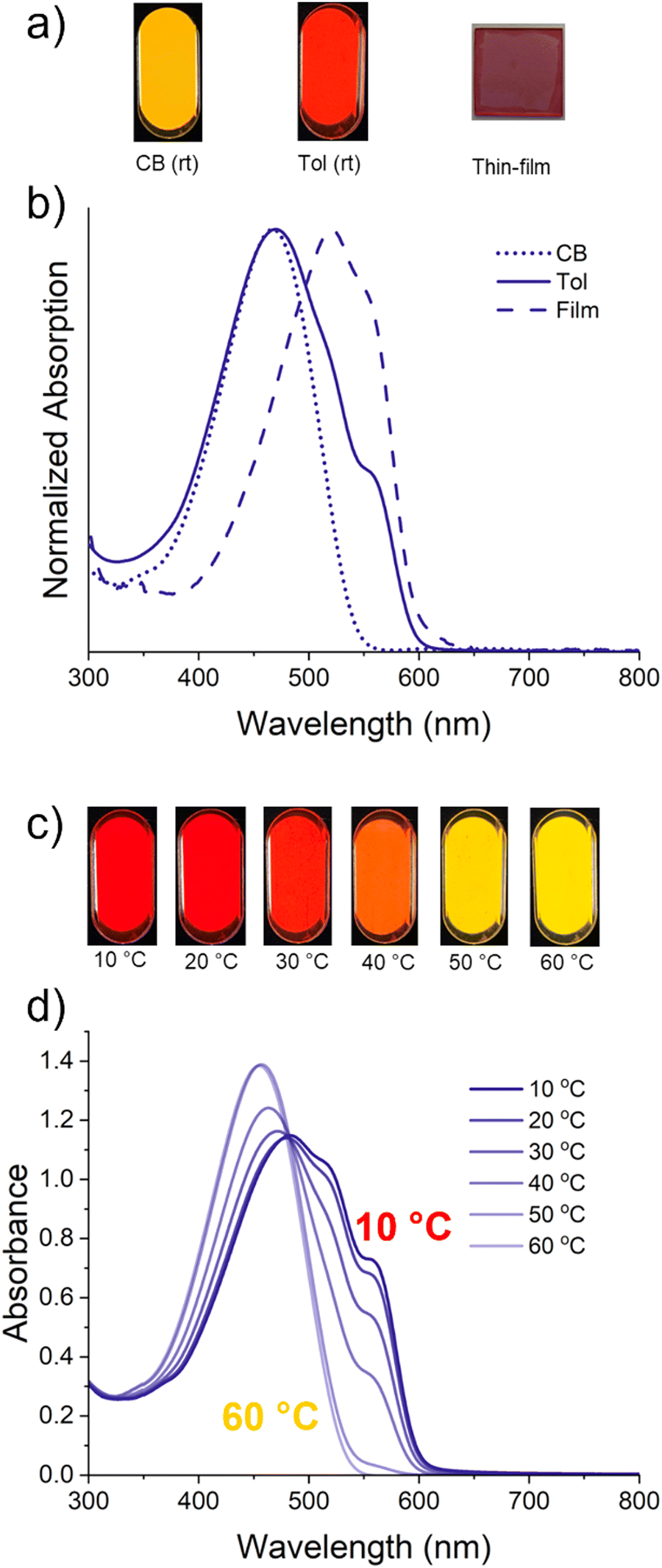 | ||
| Fig. 2 Photographs (a) and spectra (b) of room-temperature 0.1 mg mL−1 TPT–TT solutions in chlorobenzene (CB, dotted line) and toluene (Tol, solid line), as well as a thin film UV-vis spectrum (dashed line). (c) and (d) Thermochromism experiment from 10–60 °C demonstrating the temperature-dependent solution aggregation behavior of TPT–TT in toluene at a concentration of 0.1 mg mL−1. | ||
The thin-film UV-vis absorption spectrum (films being spin coated from ∼5 mg mL−1 chlorobenzene solution) is red-shifted (onset of absorption of 605 nm) compared to solution and shows two transitions around 522 nm and 558 nm, which resemble a profile similar to that of the aggregated species in toluene. The strong thermochromism (Fig. 2c and d) and the solution aggregation behavior highlight how modulation of the planarizing intramolecular non-covalent contact using solvent quality (toluene vs. chlorobenzene) or temperature dictates the chain organization, changing from a highly aggregated solution at 10 °C to a well-dissolved solution at 50 °C in toluene.
Subsequently, we evaluated if these temperature- and solvent-dependent structural locking interactions can lead to multi-phase ordered assemblies. We note (Fig. S15, ESI†) that TPT–TT is thermally stable, and the onset of the decomposition process (405 °C at 5% weight loss) is well above the thermal transitions observed by DSC. The DSC scan of TPT–TT (Fig. 3) shows three well-defined endotherms in the second heating cycle, occurring at 48 °C, 107 °C, and 218 °C, indicative of liquid crystalline (LC) behavior with multiple phases. The thermal transitions at 107 °C and 218 °C are assigned to side-chain and backbone order–disorder transitions, respectively, based on our GIXD analysis (see below) and previous literature reports.44,48,56,57 The thermotropic liquid crystalline behavior of TPT–TT was directly observed using cross-polarized optical microscopy (CPOM) for thin films (Fig. 3 insets) and for polymer powders (Fig. S16, ESI†). When heating to temperatures above the 218 °C transition, which results in a molten isotropic state, the sample appears dark under CPOM. Subsequent LC–LC transitions between 150 °C and 25 °C did not show appreciative changes under CPOM possibly due to the subtlety of the structural changes that did not lead to significant refractive index change. These thermal transitions are reversible, as evidenced by the cooling cycle in DSC.
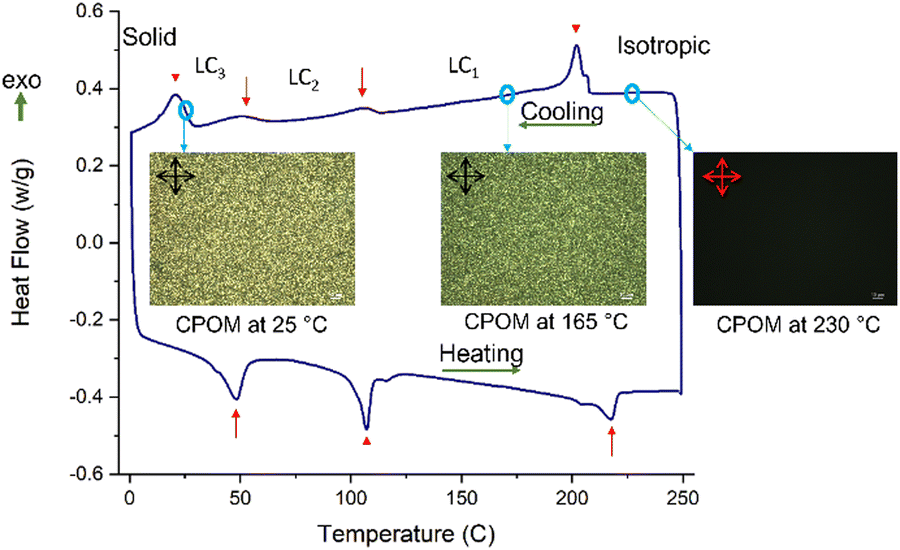 | ||
| Fig. 3 DSC profile with multiple thermal transitions and phases for the TPT–TT powder along with CPOM images (white scale bars = 20 μm.) of thin films at different temperatures during the cooling cycle which was performed after the heating the sample to 250 °C. Dark CPOM image at 230 °C shows isotropy at temperatures above highest thermal transition while brightness and patterns of CPOM images during the cooling process indicates the solidification through ordered intermediate phases. | ||
To elucidate the structural basis to the phase transitions observed in DSC, we performed synchrotron-based GIXD to investigate the evolution of molecular-scale packing structures in spin-coated films during thermal annealing. Initial observations during the first heating cycle revealed that the crystals formed during spin-coating underwent melting, a process captured in both GIXD and CPOM images (Fig. S17, ESI†). This crystal melting is supported by the observed expansion of the lamellar and π-stacking distances, from 16.36 to 19.21 Å and 4.14 to 4.60 Å respectively (Fig. S18, ESI†). Additionally, the intensity of π-stacking peaks, observed both face-on (χ = 0°) and at a χ = 65° azimuthal tilt, diminishes progressively with increasing temperature. Intriguingly, the intensity of the lamellar stacking peak exhibits a notable increase from 35 °C to 110 °C before dropping to zero as temperatures reach from 110 °C to 215 °C, accompanied by substantial peak broadening over the entire temperature range. This behavior is indicative of a phase transition near 110 °C, consistent with DSC. We surmise that the structural transition near 110 °C arise from side-chain melting. This inference aligns with findings from a thiophene-based polymer PBTTT-C14, which possesses long alkyl side-chains of 14 carbons and exhibits a side-chain melting temperature of 150 °C.57,58 We note that the absence of structural transitions other than melting during the first heating run may be due to the fact that the spin-coated films are originally entrapped in non-equilibrium phases. Therefore, transitions from the stable structure at room temperature were not observable upon initial heating. This point is well supported by our observation that the film structures are drastically different before and after thermal annealing, as shown later (Fig. 6).
During cooling from 215 °C to 125 °C, we identified the liquid crystalline (LC) phase I as a nematic phase via in situ GIXD and CPOM. Between 215–185 °C, we observed a broad edge-on (001) lamellar stacking peak (qxy ∼ 0.33 Å−1) corresponding to a stacking distance of 19.0 Å (Fig. 4a and b). We also observed diffuse isotropic scattering centered at qr = 1.34 Å−1, which likely arises from disordered backbone stacking (a.k.a. amorphous ring) at an average spacing of 4.69 Å. Around 185 °C, a broad face-on (010) π–π stacking arc appears on top of the amorphous ring at a π–π stacking distance of 4.30 Å. Further lowering temperature from 185 °C to 125 °C, the π–π stacking distance gradually decreased from 4.30 Å to 4.15 Å, while the lamellar stacking distance slightly contracts from 19.0 Å to 18.8 Å. The broad face-on π–π stacking along with the broad edge-on lamellar stacking are characteristic of a nematic liquid crystal with an in-plane director along the polymer backbone direction. Indeed, the observed scattering pattern closely resembles those of previously reported nematic liquid crystal phases.59,60 We further confirmed the nematic nature using in situ CPOM analysis on sandwiched polymer powder samples. Initially, spindle-shaped nematic tactoids, approximately 2–5 μm in length and 1 μm in width, nucleated from the isotropic phase upon cooling to 200 °C (Fig. 4c and Fig. S16, ESI†). Further cooling led to the merging of these tactoids into a continuous birefringent domain featuring Schlieren-like textures. This indicates a classic isotropic-to-biphasic (isotropic + nematic)-to-nematic phase transition observed in many material systems,61–64 thereby confirming the nematic nature of LC phase I.
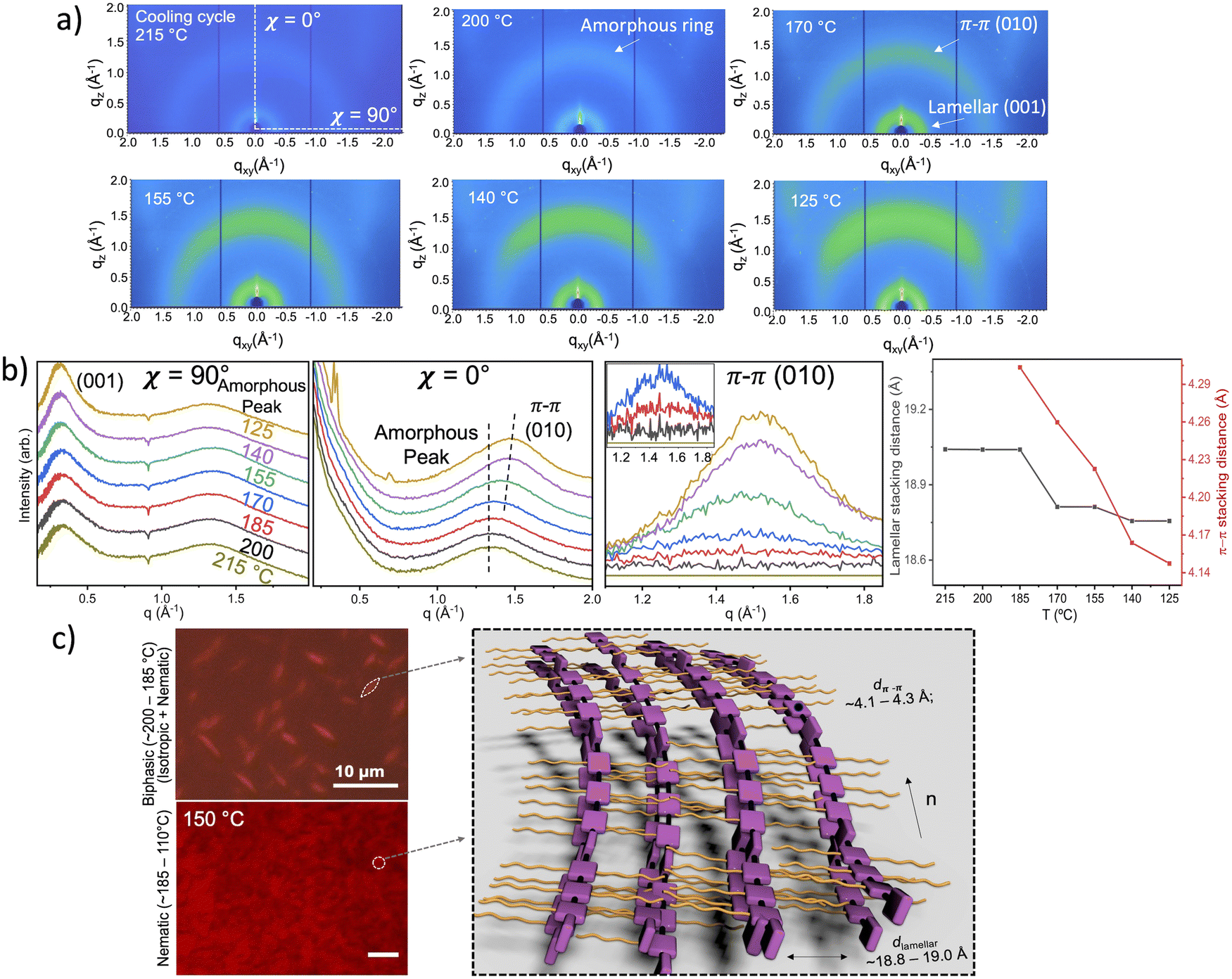 | ||
| Fig. 4 Identification of the nematic phase in the first cooling cycle (215 °C to 125 °C). (a) In situ GIXD images at various temperatures. (b) In-plane and out-of-plane linecuts (left two); π-stacking peaks extracted by subtracting the 215 °C amorphous peak at each temperature (middle); calculated lamellar and π-stacking distances (right). (c) CPOM images of TPT–TT liquid crystal samples with schematic illustration of the nematic phase structure. | ||
Upon further cooling from 125 °C to 50 °C, we identified LC phase II as a Smectic A phase. At 125 °C, a new (001) lamellar stacking peak appears in the out-of-plane direction at ∼0.35 Å−1. Upon further cooling to 110 °C near the DSC transition temperature of 107 °C, there emerged second and third lamellar stacking peaks at 0.70 Å−1 and 1.05 Å−1, with a fourth-order peak becoming apparent at qz = 1.40 Å−1 when cooled to 95 °C (Fig. 5a). These peaks are assigned as (002), (003), and (004) respectively. These sharp out-of-plane lamellar stacking peaks are distinct from the broad lamellar stacking peak from the nematic LC phase I centered around 0.33 Å−1 and oriented in-plane. The new lamellar stacking peaks are characterized by much narrower full-width-half-maximum (FWHM) values of 0.02 Å−1, in contrast to 0.15 Å−1 for the nematic lamellar stacking peak. These observations suggest that the new lamellar stacking feature has alkyl chains standing on the substrate rather than lying flat, possesses closer lamellar stacking distance, and is far more ordered than that of the nematic phase. These features suggest the formation of a highly ordered smectic-like structure with a layer spacing of ∼18.0 Å in the out-of-plane direction.65 Besides the new out-of-plane peaks, at 110 °C two sharp peaks appear at qxy = ±1.35 Å−1 forming a Bragg rod, which we infer to be originated from π–π stacking of tilted polymer backbones and assigned as (020) and (030) respectively. The lowest order (010) peak is likely buried by background scattering due to the overlapping amorphous ring. This inference is based on how the two peaks further evolve during cooling all the way until crystallization near room temperature. The inference is consistent with the peak indexing result for annealed crystalline thin films discussed below (Fig. 6, Fig. S20, ESI†). Based on the peak assignment, we estimate a π-stacking distance of 4.24 Å calculated based on the qr position of the (010) peak. We note that the (020) and (030) peaks appear along the same qz = 0.70 Å−1 and 1.05 Å−1 as the (002) and (003) lamellar stacking peaks, together suggesting an orthorhombic unit cell with alkyl chains standing normal to the substrate, hence the inference of smectic A phase. Further cooling results in spreading of (020) and (030) peaks along azimuthal angles, giving rise to two sharp and intense arcs at qr = 1.48 Å−1 and qr = 1.63 Å−1, atop the broad nematic π-stacking peak (Fig. 5d). The two arcs appear at similar qr positions as the (020) and (030) peaks respectively suggesting the same structural origin (i.e., π–π stacking). We speculate that such peak spreading from a sharp dot to an arc is due to interfacial nucleation of the smectic A phase followed by growth of this phase into the bulk. A linecut along χ angle (−90° to 90°) at fixed qr = 1.48 Å−1 reveals that the backbone stacking peak is tilted at an χ angle of ±45° (Fig. 5e). We thus propose a smectic A packing structure in LC II, as illustrated in Fig. 5f: the side-chains are layered up in the out-of-plane direction, forming ordered smectic layers with a thickness of nearly 18.0 Å; the backbones are tilted 45° relative to the layer normal. With further cooling to below the DSC transition temperature at 48 °C, we observed that GIXD patterns at 35 °C and 25 °C exhibit numerous sharp diffraction peaks (Fig. S19, ESI†). These peaks indicate a highly ordered structure obtained in LC phase III and the crystalline thin film. Based on this diffraction pattern, we successfully solved the unit cell structure for annealed thin film, as discussed below.
 | ||
| Fig. 5 Identification of Smectic A phase in the first cooling cycle (125 °C to 50 °C). (a) In situ GIXD images across temperatures. (b) Linecut along qz at qxy = 1.35 Å−1 revealing (020), (030) π-stacking peaks at 0.70 Å−1 and 1.05 Å−1, respectively. Out-of-plane (χ = 0°) (c) and χ = 40° (d) linecuts revealing smectic layer and side-chain tilt peaks. (e) Linecuts along azimuthal angle at qr = 1.48 Å−1 for tilted peaks 1. (f) Proposed schematic of smectic A phase structure. | ||
Since thermotropic LC polymers are responsive to thermal annealing,4,66 due to their ability to align while having fluidic behavior, we evaluated the effect of thermal annealing on the solid-state structure of TPT–TT films. Fig. 6a and b present the CPOM images of a spin-coated film before and after thermal annealing across the various LC phases described earlier. The as-spun film displays negligible birefringence under CPOM, suggesting a low degree of crystallinity. In contrast, the annealed film demonstrates a high birefringence, indicating a notable increase in the number and/or size of crystallites.67–69 Furthermore, the CPOM images of the annealed film (Fig. 6b and Fig. S16, ESI†) reveal Schlieren textures,70,71 further corroborating the liquid-crystalline phase assignments discussed above. The enhanced crystallinity is also confirmed by GIXD measurements. The 2D GIXD pattern of the as-spun film shown in Fig. 6c displays three diffraction peaks along qz at 0.39 Å−1, 0.78 Å−1, and 1.17 Å−1, corresponding to out-of-plane lamellar stacking at spacings closer than those in the smectic A phase. We also observed a broad peak at qz = 1.51 Å−1, which we ascribe to a π-stacking distance of 4.16 Å, comparable to that of the nematic phase (4.15 to 4.30 Å). This stacking appears primarily in the face-on orientation, with the conjugated core parallel to the substrate surface. These features suggest that this broad face-on π-stacking is inherited from the nematic phase. There is a weaker peak at qxy = −1.20 Å−1, qz = 1.00 Å−1 appearing at comparable qr position (1.56 Å−1) as the face-on π-stacking peak. We believe this peak arises from π-stacking of tilted backbones also likely inherited from the smectic A phase due to the resemblance to the (020) π-stacking arc. Such tiled π-stacks in the as-spun film may have served as the precursor to nucleating highly ordered crystallites during thermal annealing when the same peak significantly intensifies and shifts to a slightly higher q position. In contrast to a few broad diffraction features in as-spun films, numerous sharp diffraction peaks appear (Fig. 6d) after the film undergoes thermal annealing through the liquid crystalline phase (i.e., heating to 250 °C followed by slow cooling to room temperature at which point the GIXD was taken), signifying the formation of highly ordered crystalline structures with long-range order. We observed new lamellar stacking peaks appearing at higher qz positions after annealing. This gives a closer lamellar stacking distance of 14.96 Å after annealing compared to 16.11 Å before annealing, which possibly arises from side chain interdigitation during liquid crystal mediated crystallization during the annealing process.
 | ||
| Fig. 6 CPOM (a) and (b) and GIXD (c) and (d) images for as-cast (top) and annealed (bottom) thin-films and the schematic representation of the observed chain ordering and side-chain interdigitation upon annealing (e). The thin films are annealed by heating to 250 °C followed by a slow cooling (∼10 °C min−1) to room temperature, similar to the DSC and CPOM experiments. The GIXD of the annealed films are obtained at room temperature. | ||
Unit-cell parameters were obtained from GIXD by minimizing the least square error between the diffraction peak positions simulated from the fitted unit cell and the measured positions (a = 14.2 Å, b = 13.9 Å, c = 16.2 Å, α = 71.1°, β = 86.8°, γ = 64.8°). A detailed description of the fitting process can be found in the ESI.† We note that certain peaks of the initial structure still persist after annealing, this is noticeable in the out-of-plane lamellar stacking peaks that were initially excluded from the fitting. Based on this fitted unit cell, we identified each of the Bragg rods - vertical diffraction lines with equivalent h and k indices – which characterize the in-plane crystal structure along the a- and b-axes (Fig. S20, ESI†). We found good agreement between the measured and simulated diffraction patterns. By fitting the unit cell, we can determine the indices of the π-stacking peak as (3![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 2), corresponding to a qr of 1.60 Å−1 and a reduced π-stacking distance of 3.97 Å in the annealed film. Compared to the nematic face-on and smectic 45° tilted π-stacking peak, which has spacings of 4.15–4.30 Å and 4.21–4.25 Å, respectively, the (32) peak exhibits a more closely packed and tilted nature, with a tilting angle of 35° with respect to the substrate normal (Fig. S20, ESI†). Based on this analysis, we developed a schematic showing how the packing and chain alignment changes upon annealing (Fig. 6e). It appears as if the polymer chains in as-spun films are initially deposited in layers of disordered π-stacks with bimodal distribution inherited from both the nematic and smectic A LC phases. Then, upon cooling from the melt state, the polymer backbone initially forms predominantly face-on stacking in the nematic phase. This is followed by the formation of tilted stacking in the smectic A phase, which then transitions to closely spaced π-stacks less tilted with respect to the substrate normal in the solid state. Meanwhile, alkyl chains undergo order–disorder transitions with a higher extent of interdigitation to result in a highly crystalline phase that displays exceptional long-range order unusual for conjugated polymers.
2), corresponding to a qr of 1.60 Å−1 and a reduced π-stacking distance of 3.97 Å in the annealed film. Compared to the nematic face-on and smectic 45° tilted π-stacking peak, which has spacings of 4.15–4.30 Å and 4.21–4.25 Å, respectively, the (32) peak exhibits a more closely packed and tilted nature, with a tilting angle of 35° with respect to the substrate normal (Fig. S20, ESI†). Based on this analysis, we developed a schematic showing how the packing and chain alignment changes upon annealing (Fig. 6e). It appears as if the polymer chains in as-spun films are initially deposited in layers of disordered π-stacks with bimodal distribution inherited from both the nematic and smectic A LC phases. Then, upon cooling from the melt state, the polymer backbone initially forms predominantly face-on stacking in the nematic phase. This is followed by the formation of tilted stacking in the smectic A phase, which then transitions to closely spaced π-stacks less tilted with respect to the substrate normal in the solid state. Meanwhile, alkyl chains undergo order–disorder transitions with a higher extent of interdigitation to result in a highly crystalline phase that displays exceptional long-range order unusual for conjugated polymers.
The ionization energy (IE) and doping susceptibility of TPT–TT was estimated by differential pulse voltammetry (DPV) as shown in Fig. S21a (ESI†). The oxidation onset for TPT–TT was determined to be 0.1 V vs. Fc/Fc+, equivalent to an IE of 5.22 eV (assuming Fc/Fc+ is −5.12 eV vs. vacuum). The IE and electron affinity estimates are described in the ESI.† TPT–TT can be reversibly electrochemically doped in tetra-n-butylammonium hexafluorophosphate (TBAPF6)/propylene carbonate as evidenced by the cyclic voltammetry and potential dependent UV-vis-NIR results in Fig. S21a and b (ESI†). The doped form of TPT–TT can also be accessed chemically in solution, evidenced by the bleaching of the π–π* transition and the growth of the carrier absorption just past 700 nm (Fig. S22, ESI†), or via sequential doping of the thin films with iron(III) p-toluenesulfonate (ferric tosylate). Doping the as-cast samples with ferric tosylate results in an electrical conductivity of 18.5 S cm−1 (± 3.9 S cm−1) and a Seebeck coefficient of 43.3 μV K−1 (± 4.8 μV K−1). Thermal annealing at 250 °C followed by slow cooling of the samples prior to the doping, similar to the GIXD and DSC experiments, raises the conductivity ∼threefold to 54.9 S cm−1 (±4.6 S cm−1) and reduces the Seebeck coefficient to 26.9 μV K−1 (±2.5 μV K−1) as demonstrated in Fig. S23 (ESI†). The Seebeck coefficient serves as an indicator of the asymmetry of the electronic density of states around the Fermi level. A reduction in the Seebeck coefficient points to a decrease in energetic disorder in the material, and by extension, a potential reduction in the number of carriers trapped by energetic disorder. Increasing the doping level, on the other hand, would raise the Fermi level but also influence the symmetry of the density of states.72,73 Combined, our results suggest that thermal annealing creates a a more ordered microstructure that reduces the energetic disorder in the film and/or enhances the efficiency of the chemical doping by generating more charge carriers in annealed films.
To further evaluate the charge transport properties of TPT–TT, we incorporated it as the active transport material in top-gate, bottom contacts organic field-effect transistors (OFETs).74–76 The device fabrication details can be found in the ESI† and the device structure is depicted in the inset in Fig. 7a. Along with the “as cast” reference films, a set of TPT–TT films has been annealed at 250 °C for 10 min in a glovebox, and then cooled slowly at 10 °C min−1 to resemble the thermal history of the DSC, CPOM, and GIXD experiments. The devices were characterized in dark and under mild vacuum.77Fig. 7a and b show typical transfer and output current–voltage characteristics for a device that has been annealed prior to characterization. The corresponding plots for an unannealed device are shown in Fig. S24 (ESI†). All OFETs exhibited p-type (hole) transport (i.e., enhancement in the drain current ID for negative gate–source VGS and drain–source VDS voltages).78 The device yield was 100% and the spread in electrical properties was very narrow. The OFETs exhibited almost ideal I–V curves, as can be observed in Fig. 7a and b, which is critical for the accuracy in the extraction of the figures of merit.79,80 The average charge carrier mobility for the OFETs made with as-cast TPT–TT films was μ = (8.5 ± 2.6) × 10−3 cm2 V−1 s−1, while the average mobility extracted on devices fabricated using annealed films was μ = (1.4 ± 0.1) × 10−2 cm2 V−1 s−1. We complemented this in-plane mobility by space-charge-limited current (SCLC) out-of-plane measurements, the details of which are included in the ESI† (Fig. S25). Similarly, SCLC mobilities (μSCLC = (2.43 ± 0.01) × 10−4 cm2 V−1 s−1 for the as-spun samples and μSCLC = (1.82 ± 0.02) × 10−4 cm2 V−1 s−1 for the annealed samples) are in the same range for the as-prepared and annealed films. The large observed variation in the measured mobility depending on the direction of charge transport, where we observe lower mobilities in out-of-plane SCLC regime compared to the OFET measured in-plane transport, is likely due to interfacial effects such as edge-on to face-on ratio throughout the film thickness,81 and the different morphological environments carriers experience along their transport pathways.82 While the increase in the average in-plane mobility for the OFETs after annealing is not large, note the narrower distribution in the annealed devices and the fact that the lowest mobility in the annealed devices is higher than the average value obtained in the samples that have not been annealed in devices with as-cast TPT–TT films (Fig. 7c). It is important to highlight the impact of this result in a more general context: the value of mobility, along with contact resistance and geometrical factors, impacts the operating frequency of the device, which, in turn, dictates the type of application that a certain FET technology can enable.83–85 For a large area transistor array, the mobilities of the lowest performing device determine the application that the ensemble can address. Thus, having a low variability in device properties and high yields is critical.
 | ||
| Fig. 7 Electrical properties of TPT–TT transistors. (a) Transfer curve in the saturation regime (VDS = −50 V) for a device that was annealed prior to testing, having L = 30 μm and W = 400 μm. The drain current is plotted on a log scale in black (left axis), while in blue we show the square root of the current (right axis). The inset shows the device structure. (b) Output curves for the same device. (c) Mobility values for the OFETs fabricated with as-cast (blue) and annealed (orange) films. | ||
In summary, by incorporating temperature and solvent-dependent non-covalent intramolecular interactions into the structure of a symmetric π-conjugated unit (TPT), we have successfully induced liquid crystalline behavior in the TPT–TT polymer. The combination of π–π stacking and backbone planarity has resulted in a controllable solution aggregation and chain configuration properties. Through extensive GIXD studies, we identified: (i) a first LC phase, ranging from 218 °C to 107 °C, as a nematic phase featuring preferential face-on π–π stacking and edge-on lamellar stacking exhibiting a large extent of disorder and broad orientation distribution; (ii) a second LC phase, from 107 °C to 48 °C, as a smectic A phase featuring sharp, highly ordered out-of-plane lamellar stacking features and sharp tilted backbone stacking peaks; and (iii) a third LC phase with a transition at 48 °C whose structure remains unclear but resembles that of the solid state at ambient temperature. This liquid crystalline behavior leads to a significantly higher degree of order in the solid-state, achieved through thermal annealing. Due to the tendency of the backbone to planarize while having higher torsional freedom at elevated temperatures, the thermal annealing process promotes chain rearrangement into highly birefringent and ordered microstructures, which is reflected in the improved electrical conductivity of ferric tosylate-doped films and the observed low dispersity and high (100%) device yield in OFET devices. Moreover, we propose that the utilization of intra-chain interactions in combination with repeat unit symmetry is a powerful approach for promoting liquid crystallinity and designing highly ordered organic semiconductors.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge and appreciate funding from US Office of Naval Research (J. R. R. Award No. N00014-22-1-2185, J.L.B. Award No. N00014-22-1-2178, Y. D. Award No. N00014-22-1-2202) and US Department of Energy (DOE) (Award No. EE-0009524). The work at Wake Forest University was supported by the National Science Foundation under Award ECCS-1810273.References
- A. Khasbaatar, Z. Xu, J.-H. Lee, G. Campillo-Alvarado, C. Hwang, B. N. Onusaitis and Y. Diao, Chem. Rev., 2023, 123, 8395–8487 CrossRef CAS PubMed.
- L. Ding, Z.-D. Yu, X.-Y. Wang, Z.-F. Yao, Y. Lu, C.-Y. Yang, J.-Y. Wang and J. Pei, Chem. Rev., 2023, 123, 7421–7497 CrossRef CAS PubMed.
- Z.-F. Yao, J.-Y. Wang and J. Pei, Prog. Polym. Sci., 2023, 136, 101626 CrossRef CAS.
- L. Zhang, K. Zhao, H. Li, T. Zhang, D. Liu and Y. Han, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1572–1591 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- Z. Peng, L. Ye and H. Ade, Mater. Horiz., 2022, 9, 577–606 RSC.
- N. A. Kukhta and C. K. Luscombe, Chem. Commun., 2022, 58, 6982–6997 RSC.
- K. Gu and Y.-L. Loo, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1559–1571 CrossRef CAS.
- K. A. Mazzio, A. H. Rice, M. M. Durban and C. K. Luscombe, J. Phys. Chem. C, 2015, 119, 14911–14918 CrossRef CAS.
- N. Kleinhenz, N. Persson, Z. Xue, P. H. Chu, G. Wang, Z. Yuan, M. A. McBride, D. Choi, M. A. Grover and E. Reichmanis, Chem. Mater., 2016, 28, 3905–3913 CrossRef CAS.
- H. Lee, S. B. Lee, Y.-S. Kim, H. Kim, M.-J. Kim, T. W. Yoon, D. Lee, J. H. Cho, Y.-H. Kim and B. Kang, Chem. Eng. J., 2023, 468, 143654 CrossRef CAS.
- J. P. Ruiz, J. R. Dharia, J. R. Reynolds and L. J. Buckley, Macromolecules, 1992, 25, 849–860 CrossRef CAS.
- M. He, J. Li, A. Tandia, M. Sorensen, F. Zhang, H. H. Fong, V. A. Pozdin, D.-M. Smilgies and G. G. Malliaras, Chem. Mater., 2010, 22, 2770–2779 CrossRef CAS PubMed.
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS.
- C. Li, X. Gu, Z. Chen, X. Han, N. Yu, Y. Wei, J. Gao, H. Chen, M. Zhang, A. Wang, J. Zhang, Z. Wei, Q. Peng, Z. Tang, X. Hao, X. Zhang and H. Huang, J. Am. Chem. Soc., 2022, 144, 14731–14739 CrossRef CAS PubMed.
- K. Chung, D. S. Yang, W.-H. Sul, B.-G. Kim, J. Kim, G. Jang, M. S. Kwon, M. Barłóg, T. S. Lee, S.-Y. Park, M. Al-Hashimi and J. Kim, Macromolecules, 2019, 52, 6485–6494 CrossRef CAS.
- C. R. G. Grenier, W. Pisula, T. J. Joncheray, K. Müllen and J. R. Reynolds, Angew. Chem., Int. Ed., 2007, 46, 714–717 CrossRef CAS PubMed.
- J. Zhu, Y. Liu, S. Huang, S. Wen, X. Bao, M. Cai and J. Li, Phys. Chem. Chem. Phys., 2022, 24, 17462–17470 RSC.
- S. Ming, S. Zhen, H. Zhang, X. Han, Y. Zhang, J. Xu and J. Zhao, Eur. Polym. J., 2022, 163, 110938 CrossRef CAS.
- D. Liu, Y. Lei, X. Ji, Y. Wu, Y. Lin, Y. Wang, S. Zhang, Y. Zheng, Y. Chen, J.-C. Lai, D. Zhong, H.-W. Cheng, J. A. Chiong, X. Gu, S. Gam, Y. Yun, J. B. H. Tok and Z. Bao, Adv. Funct. Mater., 2022, 32, 2203527 CrossRef CAS.
- Y. Liu, Y. Liu and X. Zhan, Macromol. Chem. Phys., 2011, 212, 428–443 CrossRef CAS.
- C. Zhu, A. J. Kalin and L. Fang, Acc. Chem. Res., 2019, 52, 1089–1100 CrossRef CAS PubMed.
- A. Haque, K. M. Alenezi, M. S. Khan, W.-Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2023, 52, 454–472 RSC.
- K. S. Park, J. J. Kwok, R. Dilmurat, G. Qu, P. Kafle, X. Luo, S.-H. Jung, Y. Olivier, J.-K. Lee, J. Mei, D. Beljonne and Y. Diao, Sci. Adv., 2019, 5, eaaw7757 CrossRef CAS PubMed.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Nat. Mater., 2020, 19, 491–502 CrossRef CAS PubMed.
- D. Venkateshvaran, M. Nikolka, A. Sadhanala, V. Lemaur, M. Zelazny, M. Kepa, M. Hurhangee, A. J. Kronemeijer, V. Pecunia, I. Nasrallah, I. Romanov, K. Broch, I. McCulloch, D. Emin, Y. Olivier, J. Cornil, D. Beljonne and H. Sirringhaus, Nature, 2014, 515, 384–388 CrossRef CAS PubMed.
- N. E. Jackson, B. M. Savoie, K. L. Kohlstedt, M. Olvera de la Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, J. Am. Chem. Soc., 2013, 135, 10475–10483 CrossRef CAS PubMed.
- Y. Cheng, Y. Qi, Y. Tang, C. Zheng, Y. Wan, W. Huang and R. Chen, J. Phys. Chem. Lett., 2016, 7, 3609–3615 CrossRef CAS PubMed.
- Z.-D. Yu, Y. Lu, J.-Y. Wang and J. Pei, Chem. – Eur. J., 2020, 26, 16194–16205 CrossRef CAS PubMed.
- B. Liu, D. Rocca, H. Yan and D. Pan, JACS Au, 2021, 1, 2182–2187 CrossRef CAS PubMed.
- T. Kharandiuk, E. J. Hussien, J. Cameron, R. Petrina, N. J. Findlay, R. Naumov, W. T. Klooster, S. J. Coles, Q. Ai, S. Goodlett, C. Risko and P. J. Skabara, Chem. Mater., 2019, 31, 7070–7079 CrossRef CAS.
- G. Conboy, H. J. Spencer, E. Angioni, A. L. Kanibolotsky, N. J. Findlay, S. J. Coles, C. Wilson, M. B. Pitak, C. Risko, V. Coropceanu, J.-L. Brédas and P. J. Skabara, Mater. Horiz., 2016, 3, 333–339 RSC.
- Y. Zhang, C. Yu, T. Shan, Y. Chen, Y. Wang, M. Xie, T. Li, Z. Yang and H. Zhong, Cell Rep. Phys. Sci., 2022, 3, 100765 CrossRef CAS.
- S. Sabury, G. S. Collier, M. N. Ericson and S. M. Kilbey, Polym. Chem., 2020, 11, 820–829 RSC.
- S. Sabury, B. LaRiviere, M. N. Ericson and S. M. Kilbey, II, ACS Appl. Polym. Mater., 2021, 3, 1012–1021 CrossRef CAS.
- M. Liu, X. Han, H. Chen, Q. Peng and H. Huang, Nat. Commun., 2023, 14, 2500 CrossRef CAS PubMed.
- W. J. Mullin, S. A. Sharber and S. W. Thomas III, J. Polym. Sci., 2021, 59, 1643–1663 CrossRef CAS.
- P. J. W. Sommerville, A. H. Balzer, G. Lecroy, L. Guio, Y. Wang, J. W. Onorato, N. A. Kukhta, X. Gu, A. Salleo, N. Stingelin and C. K. Luscombe, ACS Polym. Au, 2023, 3, 59–69 CrossRef CAS PubMed.
- K. S. Ahn, H. Jo, J. B. Kim, I. Seo, H. H. Lee and D. R. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 1142–1150 CrossRef CAS PubMed.
- Y. Yang, Z. Liu, G. Zhang, X. Zhang and D. Zhang, Adv. Mater., 2019, 31, 1903104 CrossRef CAS PubMed.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed.
- C. Poelking, E. Cho, A. Malafeev, V. Ivanov, K. Kremer, C. Risko, J.-L. Brédas and D. Andrienko, J. Phys. Chem. C, 2013, 117, 1633–1640 CrossRef CAS.
- V. Vijayakumar, E. Zaborova, L. Biniek, H. Zeng, L. Herrmann, A. Carvalho, O. Boyron, N. Leclerc and M. Brinkmann, ACS Appl. Mater. Interfaces, 2019, 11, 4942–4953 CrossRef CAS PubMed.
- P. Boufflet, Y. Han, Z. Fei, N. D. Treat, R. Li, D.-M. Smilgies, N. Stingelin, T. D. Anthopoulos and M. Heeney, Adv. Funct. Mater., 2015, 25, 7038–7048 CrossRef CAS.
- B. H. Hamadani, D. J. Gundlach, I. McCulloch and M. Heeney, Appl. Phys. Lett., 2007, 91, 243512 CrossRef.
- K. Kang, S. Schott, D. Venkateshvaran, K. Broch, G. Schweicher, D. Harkin, C. Jellett, C. B. Nielsen, I. McCulloch and H. Sirringhaus, Mater. Today Phys., 2019, 8, 112–122 CrossRef.
- W. A. Memon, Y. Zhang, J. Zhang, Y. Yan, Y. Wang and Z. Wei, Macromol. Rapid Commun., 2022, 43, 2100931 CrossRef CAS PubMed.
- T. Qu, Y. Li, L. Li, C. Zhang, X. Wang, W. Chen, G. Xue, E. Zhuravlev, S. Luo and D. Zhou, Macromolecules, 2023, 56, 6407–6418 CrossRef CAS.
- Y. Pan, J. Huang, D. Gao, Z. Chen, W. Zhang and G. Yu, Polym. Chem., 2021, 12, 2471–2480 RSC.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- N. Schopp, S. Sabury, T. Chaney, J. Zhang, H. Wakidi, B. M. Kim, R. Sankar, H. M. Luong, P. Therdkatanyuphong, V. V. Brus, S. Marder, M. F. Toney, J. R. Reynolds and T.-Q. Nguyen, ACS Energy Lett., 2023, 8, 3307–3313 CrossRef CAS.
- B. Xu, I. Pelse, S. Agarkar, S. Ito, J. Zhang, X. Yi, Y. Chujo, S. Marder, F. So and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2018, 10, 44583–44588 CrossRef CAS PubMed.
- X. Yi, Z. Peng, B. Xu, D. Seyitliyev, C. H. Y. Ho, E. O. Danilov, T. Kim, J. R. Reynolds, A. Amassian, K. Gundogdu, H. Ade and F. So, Adv. Energy Mater., 2020, 10, 1902430 CrossRef CAS.
- S. Sabury, T. J. Adams, M. Kocherga, S. M. Kilbey and M. G. Walter, Polym. Chem., 2020, 11, 5735–5749 RSC.
- Z. Du, H. Luong, S. Sabury, P. Therdkatanyuphong, S. Chae, C. Welton, A. Jones, J. Zhang, Z. Peng, Z. Zhu, S. Nanayakkara, V. Coropceanu, D. G. Choi, S. Xiao, A. Yi, H. J. Kim, J.-L. Bredas, H. Ade, G. N. Manjunatha Reddy, S. R. Marder, J. R. Reynolds and T.-Q. Nguyen, Mater. Horiz., 2023, 10, 5564–5576 RSC.
- Z. Fei, P. Pattanasattayavong, Y. Han, B. C. Schroeder, F. Yan, R. J. Kline, T. D. Anthopoulos and M. Heeney, J. Am. Chem. Soc., 2014, 136, 15154–15157 CrossRef CAS PubMed.
- D. M. DeLongchamp, R. J. Kline, Y. Jung, D. S. Germack, E. K. Lin, A. J. Moad, L. J. Richter, M. F. Toney, M. Heeney and I. McCulloch, ACS Nano, 2009, 3, 780–787 CrossRef CAS PubMed.
- D. M. DeLongchamp, R. J. Kline, Y. Jung, E. K. Lin, D. A. Fischer, D. J. Gundlach, S. K. Cotts, A. J. Moad, L. J. Richter, M. F. Toney, M. Heeney and I. McCulloch, Macromolecules, 2008, 41, 5709–5715 CrossRef CAS.
- R. J. Mandle, S. J. Cowling and J. W. Goodby, Sci. Rep., 2017, 7, 13323 CrossRef PubMed.
- R. J. Mandle, C. T. Archbold, J. P. Sarju, J. L. Andrews and J. W. Goodby, Sci. Rep., 2016, 6, 36682 CrossRef PubMed.
- M. Bagnani, G. Nyström, C. De Michele and R. Mezzenga, ACS Nano, 2019, 13, 591–600 CrossRef CAS PubMed.
- Z. S. Davidson, Y. Huang, A. Gross, A. Martinez, T. Still, C. Zhou, P. J. Collings, R. D. Kamien and A. G. Yodh, Nat. Commun., 2017, 8, 15642 CrossRef CAS PubMed.
- G. Nyström, M. Arcari and R. Mezzenga, Nat. Nanotechnol., 2018, 13, 330–336 CrossRef PubMed.
- P.-X. Wang and M. J. MacLachlan, Philos. Trans. R. Soc., A, 2017, 376, 20170042 CrossRef PubMed.
- Y. Shi, F. G. Tournilhac and S. Kumar, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 55, 4382–4385 CrossRef CAS.
- S.-H. Yang and C.-S. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2713–2733 CrossRef CAS.
- M. Chang, J. Lee, N. Kleinhenz, B. Fu and E. Reichmanis, Adv. Funct. Mater., 2014, 24, 4457–4465 CrossRef CAS.
- Y. Minowa, Y. Yabuuchi, S. Nagano, S. Nagamatsu, A. Fujii and M. Ozaki, ACS Appl. Mater. Interfaces, 2022, 14, 50112–50119 CrossRef CAS PubMed.
- J. Kuebler, T. Loosbrock, J. Strzalka and L. Fernandez-Ballester, Macromolecules, 2023, 56, 3083–3094 CrossRef CAS.
- J. Nehring and A. Saupe, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 1–15 RSC.
- D. Trefz, Y. M. Gross, C. Dingler, R. Tkachov, A. Hamidi-Sakr, A. Kiriy, C. R. McNeill, M. Brinkmann and S. Ludwigs, Macromolecules, 2019, 52, 43–54 CrossRef CAS.
- K. Broch, D. Venkateshvaran, V. Lemaur, Y. Olivier, D. Beljonne, M. Zelazny, I. Nasrallah, D. J. Harkin, M. Statz, R. D. Pietro, A. J. Kronemeijer and H. Sirringhaus, Adv. Electron. Mater., 2017, 3, 1700225 CrossRef.
- M. Lögdlund, R. Lazzaroni, S. Stafström, W. R. Salaneck and J. L. Brédas, Phys. Rev. Lett., 1989, 63, 1841–1844 CrossRef PubMed.
- Z. A. Lamport, H. F. Haneef, S. Anand, M. Waldrip and O. D. Jurchescu, J. Appl. Phys., 2018, 124, 071101 CrossRef.
- Z. A. Lamport, K. J. Barth, H. Lee, E. Gann, S. Engmann, H. Chen, M. Guthold, I. McCulloch, J. E. Anthony, L. J. Richter, D. M. DeLongchamp and O. D. Jurchescu, Nat. Commun., 2018, 9, 5130 CrossRef PubMed.
- H. F. Iqbal, Q. Ai, K. J. Thorley, H. Chen, I. McCulloch, C. Risko, J. E. Anthony and O. D. Jurchescu, Nat. Commun., 2021, 12, 2352 CrossRef CAS PubMed.
- H. F. Iqbal, M. Waldrip, H. Chen, I. McCulloch and O. D. Jurchescu, Adv. Electron. Mater., 2021, 7, 2100393 CrossRef CAS.
- Y. Mei, D. Fogel, J. Chen, J. W. Ward, M. M. Payne, J. E. Anthony and O. D. Jurchescu, Org. Electron., 2017, 50, 100–105 CrossRef CAS.
- H. H. Choi, K. Cho, C. D. Frisbie, H. Sirringhaus and V. Podzorov, Nat. Mater., 2018, 17, 2–7 CrossRef CAS PubMed.
- E. G. Bittle, J. I. Basham, T. N. Jackson, O. D. Jurchescu and D. J. Gundlach, Nat. Commun., 2016, 7, 10908 CrossRef CAS PubMed.
- M. Saito, T. Koganezawa and I. Osaka, ACS Appl. Polym. Mater., 2019, 1, 1257–1262 CrossRef CAS.
- J. K. Wenderott, B. X. Dong and P. F. Green, Phys. Chem. Chem. Phys., 2021, 23, 27076–27102 RSC.
- A. Perinot, M. Giorgio, V. Mattoli, D. Natali and M. Caironi, Adv. Sci., 2021, 8, 2001098 CrossRef CAS PubMed.
- H. Klauk, Adv. Electron. Mater., 2018, 4, 1700474 CrossRef.
- M. Waldrip, O. D. Jurchescu, D. J. Gundlach and E. G. Bittle, Adv. Funct. Mater., 2020, 30, 1904576 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01974h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |