
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering the interaction of short antimicrobial peptides with bacterial barriers
Costanza
Montis
 ab,
Elisa
Marelli
c,
Francesco
Valle
bd,
Francesca
Baldelli Bombelli
c and
Claudia
Pigliacelli
*c
ab,
Elisa
Marelli
c,
Francesco
Valle
bd,
Francesca
Baldelli Bombelli
c and
Claudia
Pigliacelli
*c
aDepartment of Chemistry and CSGI, University of Florence, Florence, Italy
bConsorzio Interuniversitario per lo Sviluppo dei Sistemi a Grande Interfase, 50019 Firenze, Italy
cLaboratory of Supramolecular and Bio-Nanomaterials (SupraBioNano Lab), Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Via Luigi Mancinelli 7, 20131 Milan, Italy. E-mail: claudia.pigliacelli@polimi.it
dConsiglio Nazionale delle Ricerche, Istituto per lo Studio dei Materiali Nanostrutturati, 40129 Bologna, Italy
First published on 19th March 2024
Abstract
While the rise of superbugs and new resistance mechanisms continues decreasing the effectiveness of classical antibiotics, antimicrobial peptides (AMPs) are emerging as a new class of antimicrobials. Still, several drawbacks limit their transition to the clinic, including high production cost, haemolytic activity and possible inactivation by proteases. Here, we give an overview of the most recent work on short AMPs, which are currently a minority in the AMP databases, and of the main AMP design rules, describing their application for short sequences. We also summarize the techniques that can serve to investigate the key steps of the antimicrobial action and that can aid in the engineering of a tuned AMP interaction with bacterial barriers. Particular emphasis is given to the relationship between peptide sequence features and interfacial behaviour, highlighting the role of AMPs self-assembly in the interaction with membranes and their antimicrobial activity.
 Costanza Montis | Costanza Montis is an Associate Professor of Physical Chemistry at the Department of Chemistry, University of Florence, and a member of the Italian Consortium for Colloid and Surface Science (CSGI). Her main research activity focuses on the understanding of complex phenomena occurring at interfaces, from a physicochemical perspective. Her scientific interests are in Physical Chemistry of Soft Matter and include the biophysical understanding of nano–bio interfaces, the design of lipid–nanoparticle hybrid materials for biomedical applications, and the engineering/characterisation of biogenic extracellular vesicles. Her current research is focused on the development of advanced lipid formulations for delivery of actives, RNA technologies and cosmetic applications, and on the design and development of complex biomimetic systems via microfluidics. |
 Elisa Marelli | Elisa Marelli is a PhD student in Chemical Engineering and Industrial Chemistry at Politecnico di Milano. She graduated in Materials Engineering and Nanotechnology at the same university with a thesis on biocomposites based on fibrillated nanocellulose and amyloidogenic peptides. Her current research activity is focused on investigating the self-assembly mechanisms of short amino acid sequences and exploring their potential in designing novel hybrid biomaterials for diverse applications, from biomedicine to nanotechnology. |
 Francesco Valle | Francesco Valle is a Senior Researcher at the Institute for the Study of Nanostructured Materials of Consiglio Nazionale delle Ricerche. He received his Master's degree in Physics from the University of Roma “La Sapienza” in 1999, and his PhD in Science from the University of Lausanne, Switzerland. He was a Post-doctoral Fellow at the Ecole Polytechnique Federale de Lausanne (EPFL) and at the University of Bologna. He joined CNR-ISMN Bologna in 2009 as a Research Scientist. His research interests include nanomechanics, polymer physics, single-molecule techniques, protein folding and extracellular vesicles. |
 Francesca Baldelli Bombelli | Francesca Baldelli Bombelli is an Associate Professor of Chemistry at Politecnico di Milano. She was a Group Leader at the European Centre of Nanomedicine (CEN – https://nanomedicen.eu/) in 2013–2015 and a Lecturer in Nanotechnology and Colloid Science at the School of Pharmacy, UEA, Norwich, UK in 2011–2014. Her research interests are focused on the development of engineered nanomaterials (ENMs) for the diagnosis and treatment of untreatable diseases such as cancer and neurodegenerative pathologies. She has recently developed fluorinated nanomaterials (FNMs) with exceptional multiscale F-MRI and Raman microscopy imaging properties. She is also interested in studying the effect of fluorination on the promotion of cytosolic delivery of biomolecules and nanoparticles. Moreover, her research also aims at investigating the interactions between ENMs and cellular machinery to improve their in vivo efficiency and evaluate possible toxicity effects. |
 Claudia Pigliacelli | Claudia Pigliacelli is a Tenure-Track Assistant Professor of Chemistry at the Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta” of Politecnico di Milano. She received her PhD in Pharmacy from the University of East Anglia (UK) in 2015. She was a research assistant and postdoc researcher at Politecnico di Milano (2013–2016), and worked as a postdoc at Aalto University (Finland, 2016–2019) and at the Centro de Investigacións Científicas Avanzadas (CICA, Universidade da Coruña, Spain, 2020). Her research interests focus on the design of nanoscale systems based on biomolecules (peptides and proteins) and inorganic nanoparticles, whose applications are not limited to but mainly fall within the biomedical field. |
Design, System, ApplicationAntimicrobial peptides (AMPs) are expected to be key players in the fight against bacterial infections. Their amino acid and chemical composition clearly determine the physicochemical properties of AMPs in terms of charge, amphipathicity, hydrophobicity, flexibility and non-covalent interaction capacity, features that define their mode of action and selectivity toward microbial cells. The growing knowledge on bacterial membrane structures and susceptibility to AMPs has led to establishment of some recognized design rules, which aid in the development of novel AMPs. Here, we give an overview of well-established design rules and highlight the structure–activity relationship for short AMP design. Particular insight is given into AMP interfacial behaviour and its role in the engineering of a tuned interaction with bacterial barriers and antimicrobial activity. |
Introduction
As antimicrobial resistance (AMR) rages, flanked by the exhausted pipeline of new antibiotics, the search for alternative therapeutic options for the treatment of bacterial infections is ever more topical.1–4 The era of antibiotics is, indeed, coming to its end, fuelled by the rise of “superbugs” and the continuous global spread of new resistance mechanisms that are making available antibiotics increasingly ineffective.5,6 In this scenario, antimicrobial peptides (AMPs) are emerging as a new class of antimicrobials, as shown by the thousands of entries in the AMP databases.7,8Peptides are used as natural defensive molecules in all domains of life, and according to their primary sequence and secondary structure, they exert antimicrobial activity through multiple mechanisms, as summarized in Fig. 1.9 Among them, disruption of the bacterial membrane is the most reported one, followed by the triggering of bacterial DNA damage upon membrane translocation. Achieving fine control over AMP interfacial behaviour is key.10 Indeed, for both membrane disrupting and translocating peptides, their interaction with the bacterial membrane and wall represents a first and crucial step.11,12 Generally, two main physical features need to be tuned to properly engineer this interaction: cationic charge content, that promotes selectivity for negatively charged microbial cytoplasmic membranes over zwitterionic eukaryotic ones, and a significant proportion of hydrophobic residues that facilitate interactions with the fatty acyl chains in the lipid bilayer.13–15
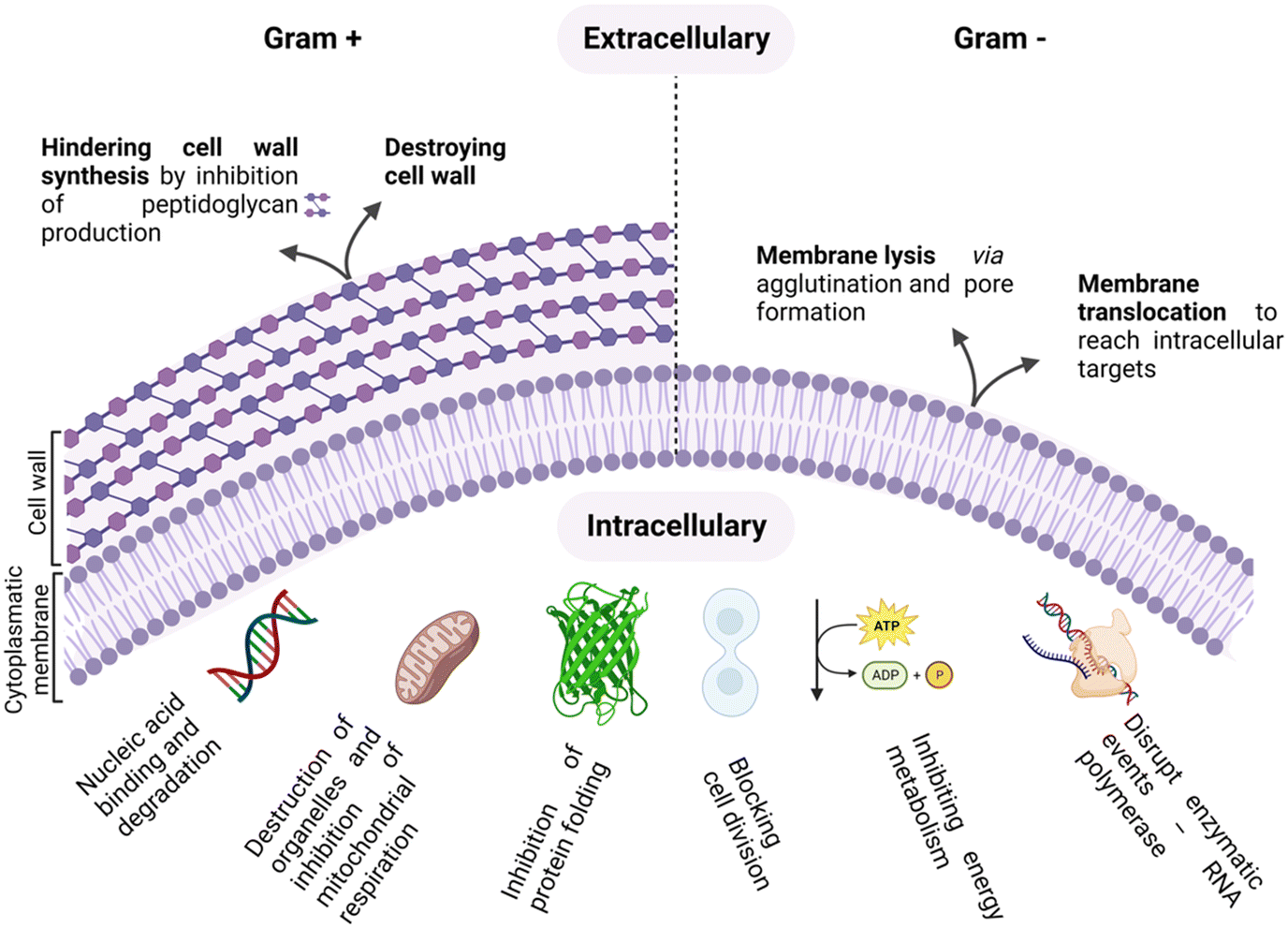 | ||
| Fig. 1 Summary of AMP intracellular and extracellular modes of action. | ||
Despite the definition of design rules and the high number of AMPs discovered to date, their translation into the clinic is still slow, and several limitations hamper their confirmation as new therapeutics.8,16–18 Among them, high cost of production, when compared to classical antibiotics, and susceptibility to proteases represent critical issues. The cost-effectiveness of AMPs could potentially be improved by decreasing the length of the polypeptide sequences. Also, the presence of a good proline (Pro, P) or tryptophan (Trp, W) content, the chemical modification of peptides aimed at increasing hydrophobicity,19 and the use of D-amino acids have shown to strongly improve AMP resistance to proteolysis. But translating the key features that have been identified as essential to control AMP behaviour in a shorter sequence can be challenging, and such a difficulty is reflected in the low number of AMPs shorter than 10 amino acids (AAs) that can be found in the database.
In this minireview, we aim to give an overview of the most recent work describing short AMPs. We will first introduce the bacterial wall and membrane structures and the AMP–membrane interaction mechanisms reported to date, and the techniques that can serve to investigate this key step of the antimicrobial action. Particular emphasis is given to the relationship between sequence features and interfacial behaviour, highlighting the role of AMPs self-assembly in the interaction with bacterial barriers and their antimicrobial activity.
Bacterial membranes and walls: disruption and translocation
The integrity and selective permeability of the lipid bilayer membrane are essential features for cell viability and functionality of every living organism. Membrane permeabilizing and/or disruptive biomolecules play a critical role in both defensive and offensive strategies of living systems and many of them, such as defensins, amphipathic peptides used as defensive molecules by eukaryotes and plants, and melittin, the main component of the bee venom, have been extensively studied as possible antimicrobial drugs.20–22 Defensins and melittin are examples of membrane-disruptive and permeating peptides, respectively, and the study of these and other naturally-occurring peptides has paved the way towards the understanding of AMP interaction with bacterial membranes and the possible mechanism of action both in Gram+ (GP) and Gram− (GN) species.The cell structure and membrane composition vary significantly between GP and GN bacteria.23 GP species are surrounded by a cell wall composed of a peptidoglycan (PG) layer with a thickness of tens of nanometres. PG is a single macromolecule made of glycan chains crosslinked by peptide side branches, and by enclosing the bacterial cell, it acts as a constraint that provides mechanical strength to the bacteria.24 Given its porous structure, PG can easily be crossed by many drugs and chemicals. Thus, the main barrier to external agents or molecules is represented by the lipid bilayer membrane (Fig. 2).
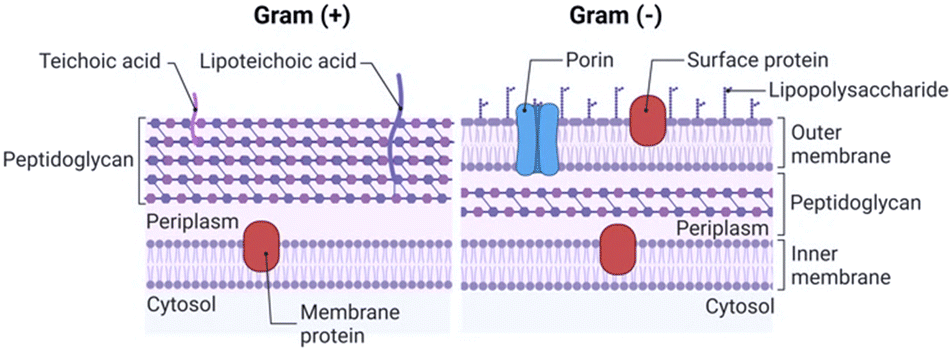 | ||
| Fig. 2 Schematic representation of Gram+ and Gram− bacterial walls and membranes. | ||
Differently, the GN bacteria cell wall comprises an outer membrane (OM), a thin PG layer and an inner membrane (IN). Notably, like other biological membranes, the OM is a lipid bilayer, but it is composed of glycolipids, mostly lipopolysaccharides (LPSs), and phospholipids (PLs) are confined in the inner leaflet of the membrane (Fig. 2).25 Together with contributing to the stiffness and strength of the bacterial cell,26 the OM acts as an efficient barrier, with highly selective porins controlling the uptake of external molecules.27 Importantly, prokaryotic membranes bear negatively charged lipids namely phosphatidylglycerol, cardiolipin and phosphatidylserine and have, therefore, a net negative charge.28
AMP interaction with the bacterial membrane has widely been studied, with the OM being frequently overlooked as a first obstacle for peptides to exert their antimicrobial activity.29 Many AMPs kill bacteria by inducing membrane disruption and leakage of bacterial content, and several models describing the possible mechanism behind this action have been proposed, to date, and already extensively reviewed.30,31 Still, the understanding of the interaction mechanisms remains poor for many AMPs, for which an unambiguous mechanistic insight is lacking. Fig. 3 summarizes the models proposed to date, with the barrel-stave (a), carpet (b), and toroidal pore (c) models being the most frequently used to describe the origin of the membrane-lytic action.
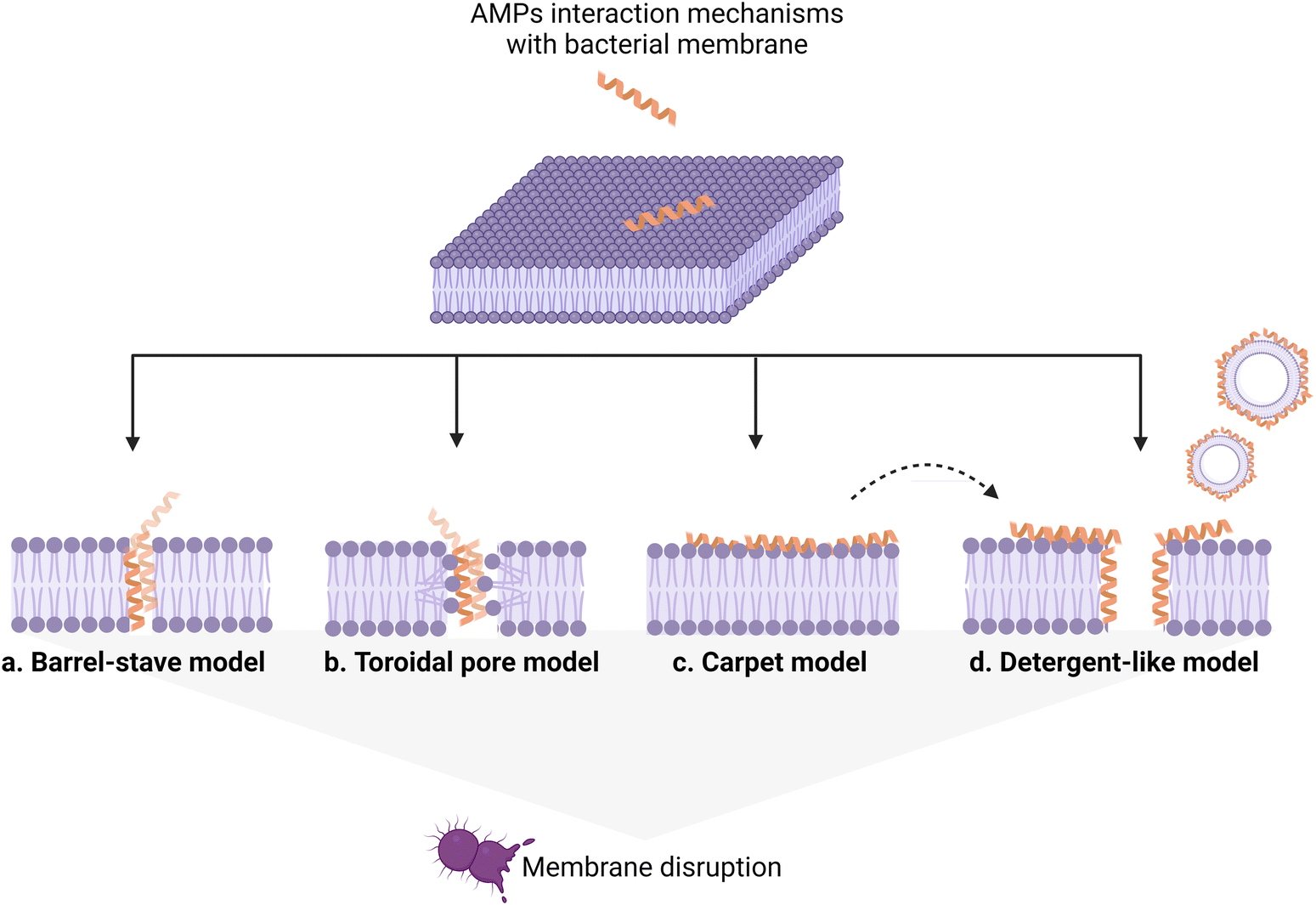 | ||
| Fig. 3 Proposed models for the AMP–bacterial membrane disruption mechanism. a) Barrel-stave model: after AMP insertion into the membrane, a transmembrane pore is formed with the peptides lining the pore lumen in a parallel direction relative to the phospholipid chains, which remain perpendicular to the bilayer plane. b) Toroidal pore model: peptide insertion induces a local membrane curvature in such a way that the pore lumen is lined partly by peptides and partly by phospholipid head groups. c) Carpet model: AMPs interact with the membrane surface and disrupt its integrity by covering it like a carpet. This model does not involve the formation of transmembrane pores; instead, above a minimum AMP concentration, it leads to a detergent-like effect (d) causing membrane breakdown and fragmentation. | ||
Not all antimicrobial peptides exert their major action on membranes. Indeed, an increasing number of peptides have been proved to act on intracellular targets in bacteria. Successful membrane translocation of an AMP strictly depends on its ability to interact with the bacterial membrane and create a passage across the lipid bilayer hydrocarbon core, without causing irreversible membrane damage. This process can happen through different pathways. In detail, the peptide can enter either via autonomous membrane translocation and/or through a specific transporter mechanism. In the first case, transient membrane permeabilization occurs. The mechanism behind this process is still under debate, but the leading hypothesis is that, upon their addition, AMPs accumulate on the outer monolayer of the targeted membrane, causing an imbalance of mass, charge, surface tension and lateral pressure. This is followed by a stochastic local dissipation event that relieves the asymmetry caused by peptides, rendering the membrane transiently permeable with a rapid burst of cell content leakage accompanying the peptide translocation. The leakage event is fast and membrane re-sealing takes place in a time frame that allows the bacterial cell survival.21,32
Among specific membrane carriers, ABC transporters SbmA and BacA, belonging to the peptide uptake permease family, are the most known ones.33 Although essential for bacterial survival and host colonization, these transporters serve well as an aid for AMP membrane translocation,34 particularly for Pro-rich ones.35
Given the multiple possible modes of action of AMPs, tailored design of their primary sequence and secondary structure is essential for engineering a specific antimicrobial mechanism. In the next paragraph, the main design rules established, to date, will be summarized.
Interaction with bacterial membranes – design rules
The amino acid composition clearly determines the physicochemical properties of peptides in terms of charge, amphipathicity, hydrophobicity, flexibility and non-covalent interaction capacity, features that define their mode of action and selectivity toward microbial cells. The growing knowledge on bacterial membrane structures and susceptibility to AMPs has led to establishment of some well-recognized design rules, which aid in the development of novel AMPs. Here, we will give an overview of well-established design rules and highlight the structure–activity relationship for AMP design, including the key points that are still not fully elucidated.Charge
Given the net negative charge of the bacterial membrane, AMPs are generally cationic, a feature that facilitates their adsorption on bacterial cells. Negatively charged AMPs have also been discovered, but they represent a minority.36,37 The peptides' positive charge is directly related to their arginine (Arg; R) and lysine (Lys; K) content, as these cationic AAs mediate the formation of strong electrostatic interactions with anionic lipids of GN and GP bacteria. Notably, both Arg and Lys bear one single positive charge, but with respect to Arg, Lys lacks a guanidinium group, resulting in lower toxicity to eukaryotic cells.38 Several studies have been performed to better understand the impact of charge content on peptides' antimicrobial activity and obtain quantitative information about the minimum charge content needed for an effective bactericidal action.39 In general, an increase in positive charge content leads to a boost in the antimicrobial activity, but increases, at the same time, the AMP haemolytic action, which represents one of the main limitations in AMP confirmation.40In a recent study by López Cascales et al., the antimicrobial activity of two short AMPs, RQWRRWWQR-NH2 (P4) and RKFRRKFKK-NH2 (P7) with charges +4 and +7, respectively, was compared. The Edmundson wheels obtained for these two peptides (Fig. 4a) clearly showed the presence of two perfectly differentiated portions of similar size, a cationic face (marked with blue cut lines) and a more hydrophobic face (represented by solid yellow lines), for P4, while P7 was characterized by a large cationic portion covering most of the peptide surface and a small hydrophobic moiety. Notably, only P4 showed antimicrobial activity. Thermodynamic studies about peptide insertion in simulated bacterial membranes were performed and revealed that P7 is unable to reach the threshold concentration necessary to induce membrane disruption. This effect was associated with the poor insertion of the peptide into the lipid bilayer, which is essential to screen out the electrostatic repulsion between neighbouring peptides and favour their subsequent adsorption on the membrane. Differently, P4, owing to its wider hydrophobic portion, could protrude into the lipid bilayer, limiting the electrostatic repulsion between neighbouring peptides. This interfacial behaviour allowed P4 to reach the threshold concentration needed to induce membrane disruption.41 More recently, a study by Wu et al. investigated the impact of the Lys content on the activity of short Fmoc-cationic AMPs. Among three designed peptides, Fmoc-KF, Fmoc-KKF and Fmoc-KKKF, Fmoc-KKF showed the strongest antimicrobial activity against both GP bacteria and GN bacteria, as well as low haemolytic activity. Thus, this work proved that systematically increasing the number of Lys residues in short Fmoc-F AMPs could not improve the antibacterial activity, highlighting the need to properly engineer a combined effect of cationicity, hydrophobicity, and secondary conformation.42
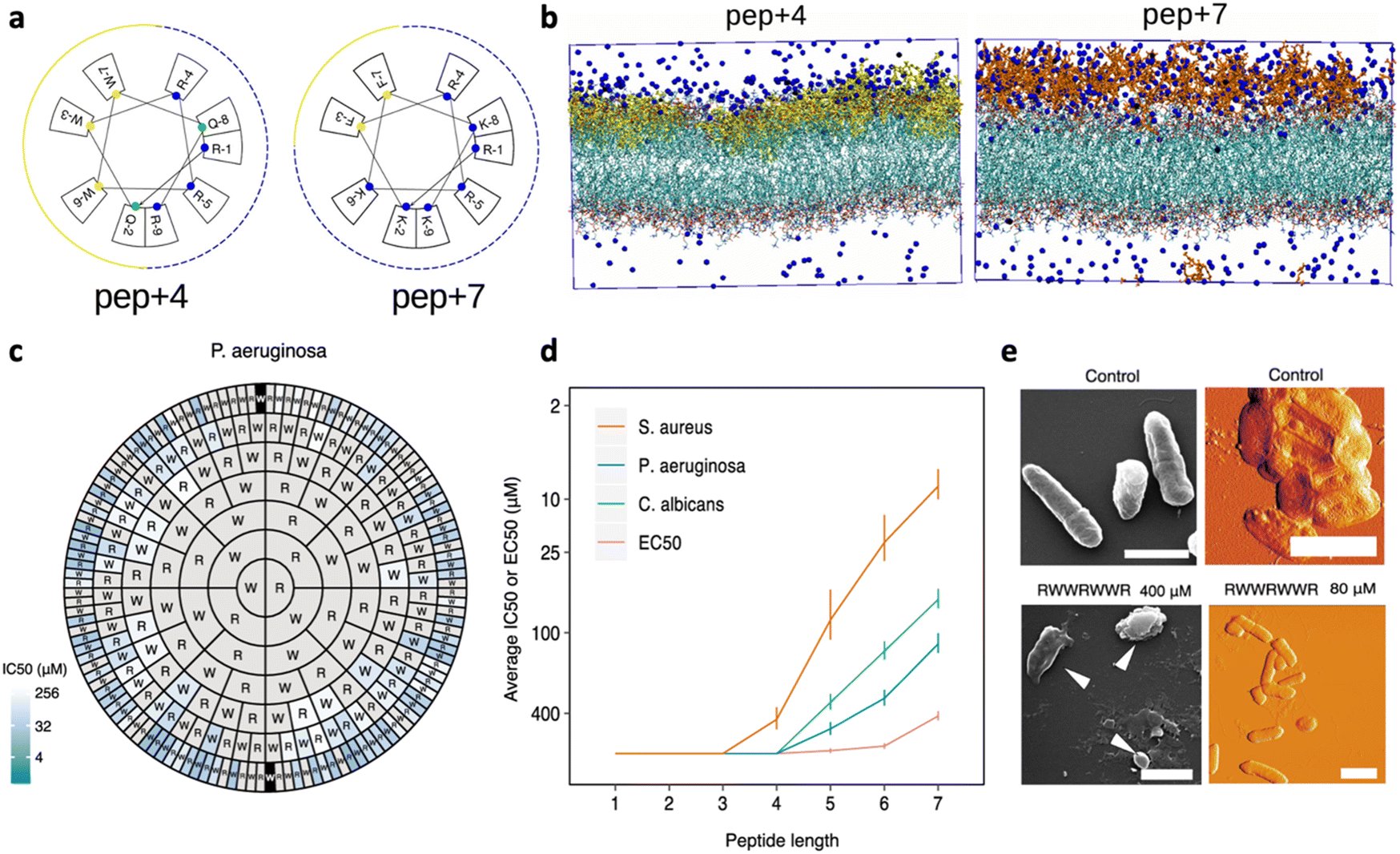 | ||
| Fig. 4 a) Edmundson representation obtained for P4 (RQWRRWWQR-NH2) and P7 (RKFRRKFKK-NH2). b) Molecular dynamics (MD) simulation of the starting configuration of phospholipid bilayers of dipalmitoylphosphatidylcholine (DPPC) in the presence of P4 (yellow) and P7 (red). Blue beads correspond to chloride ions used to balance the total charge existing in the system. Water has been removed for clarity. Panels a and b are reproduced with permission from ref. 41. Copyright © 2018 American Chemical Society. c) Harris–Clark diagram showing the inhibitory activities of peptide sequences against P. aeruginosa. Each peptide can be identified by reading the sequence from the N-terminal residue in each ring towards the C-terminal residue at the center of the chart; inhibitory activity (IC50) is indicated for each peptide by the colour of shading in the outermost compartment (i.e. the compartment identifying the N-terminal of each peptide). Grey sections represent peptides which did not exhibit an IC50 within the range of concentrations assayed (0.8–400 μM). d) Effect of peptide length on harmonic means and standard deviations for IC50 and EC50. e) Appearance of P. aeruginosa bacteria treated with the peptide RWWRWWR at concentrations of 80 and 400 μM, visualized by SEM (left: scale bars shown are 1 μm in length) and AFM (right: scale bars shown are 2 μM in length). A control image of bacteria not treated with peptide is also shown in both cases. Panels c–e are adapted and reproduced with permission under a Creative Commons License (CC-BY 4.0) from ref. 43. Copyright © 2021 Springer Nature. | ||
Importantly, also the position of positively charged residues plays a role in AMP activity, as it affects contact frequency between the peptides and cell membrane. Indeed, positive residues at a peptide's extremity (i.e., N- and C-terminal) are more exposed to the solvent and, thus, encounter the membrane with a higher frequency. In contrast, positive residues located in the middle of the peptide sequence tend to be less exposed to the solvent and bind to the lipid bilayer with a lower frequency.44
Hydrophobicity
Insertion of the AMP hydrophobic portion into the bacterial membrane core is an essential step for achieving either membrane rupture or permeabilization. Therefore, hydrophobic content is another crucial parameter to be tuned when engineering AMPs.45 Notably, while peptides with carbon chain AAs (leucine (Leu), valine (Val)) are energetically favoured for membrane insertion, membrane interfaces tend to prefer AAs with aromatic rings (tryptophan (Trp), phenylalanine (Phe)). Thus, the position of hydrophobic residues is an additional determinant for AMP interaction with membranes, with sequences bearing aromatic residues at the extremity of the peptide sequence having a higher tendency to be adsorbed.46As for the charge content, the AMP hydrophobicity needs to be properly balanced.47 Indeed, while its increase usually tends to boost antimicrobial activity, favouring membrane insertion, it can, at the same time, result in a decreased selectivity for bacterial cells, often leading to haemolysis.48 Yang et al. reported on the antimicrobial activity of a set of nonapeptides designed based on the sequence RXRXRXRXL-NH by introducing different ratios (W![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :I = 1:3, 2:2, and 3:1) of aromatic residues (Trp, W) and branched-chain residues (Ile, I) in the X position.49 They found that the presence of Trp at the 4th and 6th loci of the nonapeptide sequence facilitated the formation of a β-pleated sheet with a certain turn conformation, indicating that these two positions determine structural flexibility (Fig. 4a and b). The nonapeptides (3IW, W2IW, and IW2I) with the β-pleated sheet conformation showed optimal antimicrobial activity, suggesting a correlation between antimicrobial activity and structural conformation.
:I = 1:3, 2:2, and 3:1) of aromatic residues (Trp, W) and branched-chain residues (Ile, I) in the X position.49 They found that the presence of Trp at the 4th and 6th loci of the nonapeptide sequence facilitated the formation of a β-pleated sheet with a certain turn conformation, indicating that these two positions determine structural flexibility (Fig. 4a and b). The nonapeptides (3IW, W2IW, and IW2I) with the β-pleated sheet conformation showed optimal antimicrobial activity, suggesting a correlation between antimicrobial activity and structural conformation.
In general, amphiphilicity has been found to be far more important for interfacial binding than simple hydrophobicity.50 This is confirmed by the work by Clark et al., who recently studied a complete set of all possible peptides, up to 7 residues long, composed of positively charged Arg (R) and/or hydrophobic Trp.43 Peptide sequences comprising only Trp or only Arg were inactive against selected bacteria, highlighting the importance of a mixture of the two residues underpinning activity. Antimicrobial efficacy was typically higher for heptamer peptides containing 3R and 4W, but, importantly, small differences in the sequence could be associated with large differences in activity, with the heptapeptide WWWRRRR showing an IC50 value 65-fold higher than WWWRRRW. Overall, for penta-, hexa- and heptapeptides, those with ∼60% Trp content exhibited the lowest IC50 concentration against all the tested organisms, but consistently with what was reported by other research groups, a further increase in Trp content, and thus an overall 70–80% hydrophobicity, led to an increased haemolytic activity.48,51
Secondary structure
The formation of an ordered secondary structure upon AMP interaction with phospholipid biomolecules is another key step defining peptide antimicrobial activity. Most AMPs assume an alpha-helix conformation when in contact with membrane components, but lately β-sheet structures have also been reported for some AMPs. The majority of natural host defence peptides spontaneously assume an alpha conformation when exerting their antimicrobial activity. Thus, the design of α-helix AMPs has mostly relied, to date, on the identification of common sequence patterns in numerous naturally occurring peptides.52 Recently, Lohan et al. designed a set of de novo short helical peptides with broad-range bactericidal activity and selectivity toward bacterial cells.53 All peptides showed an irregular spatial structure in aqueous solution. The most effective peptide of the series, NH2-KWLKKWLKWWKK-CONH2, formed an amphipathic helix upon interaction with the lipid bilayer, confirming the correlation of the α-helix content with its membranolytic action.Teixobactin, an undecapeptide containing five non-canonical AAs discovered for the first time in 2015, represents an innovative AMP effective against several superbugs.54 It exerts its antimicrobial action by targeting lipid II, a precursor of peptidoglycan. Teixobactin specifically binds to the pyrophosphate-sugar moiety of lipid II, whereas the N terminus coordinates to the pyrophosphate of another lipid II molecule. Shukla et al. recently proved that this configuration favours the formation of a β-sheet for teixobactins bound to the target, creating a supramolecular fibrillar structure, key to antimicrobial action. In detail, small β-sheets are formed upon binding of lipid II (Fig. 5). Then, these assemblies elongate into fibrils that eventually associate into lateral fibrillar sheets, obstructing the biosynthesis of peptidoglycan and causing membrane defects.55
 | ||
| Fig. 5 a) Chemical structure of teixobactin. b) Mode of action model for teixobactin. Teixobactin first forms small β-sheets upon binding of lipid II, and then elongates into fibrils that eventually associate into lateral fibrillar sheets, obstructing biosynthesis of peptidoglycan and causing membrane defects. c) Snapshots of a timelapse HS-AFM video following the assembly of teixobactin–lipid II fibrils. Images were obtained on a supported lipid bilayer containing 1% (mol) lipid II in the presence of 800 nM teixobactin, added after 24 s. Image acquisition rate of 0.5 frames per second. d) Zoomed in view of an HS-AFM image of a fibrillar sheet on the membrane surface, as marked by a white rectangle in (c) at 624 s. The inset in the lower right corner shows the height profile at the dashed line. e) HS-AFM image of a lipid bilayer deformed by teixobactin–lipid II fibrils below the membrane surface, 50 min after the addition of 800 nM teixobactin. The inset shows the height profile at the dashed line. Panels a–e are reproduced and adapted with permission under a Creative Commons License (CC-BY 4.0) from ref. 55. Copyright © 2022 Springer Nature. | ||
Importantly, secondary structure formation is crucial also for cell-penetrating AMPs having an intracellular target.54 Most cell-penetrating peptides (CPPs) have a disordered/random coil conformation in solution, but the presence of α-helices and β-sheets when in contact with the bacterial membrane and cells has been reported. In general, it can be argued that peptides undergoing a greater extent of helicity are more likely to cause large phase perturbations in a membrane and are, therefore, more likely to kill the bacteria through membrane damage. This is consistent with the widely reported tendency of Pro, known as a helix-breaker, in promoting peptide translocation ability. Due to its cyclic structure, the Pro amide group lacks the proton needed to form hydrogen bond (HB) interactions, which are essential for stabilising α-helix and β-sheet structures.56 Therefore, Pro usually induces regions of helix distortions with higher flexibility in transmembrane helices.57 Typically, the cell penetration efficiency of peptides tends to increase with their amphipathicity content, but when a high value of hydrophobicity is reached, peptides predominantly remain on plasma cell membranes.58 Overall it is not possible, from the reported literature, to correlate a specific secondary structure with an efficient passage through the membrane, as the uptake process was obtained with peptides characterized by an α-helical, a β-sheet, or a random coil structure (Table 1).
| Feature | General rule | Short AMPs | Exceptions |
|---|---|---|---|
| Charge | Cationic content favors the interaction with bacterial cells and antimicrobial activity (up to +9). Positive charges mainly from Arg and Lys content | High charge content hinders membrane insertion. Optimal activity between +2 and +4 | Neutral AMPs, as Fmoc-F,59 or specific anionic peptides37 |
| Hydrophobicity | Hydrophobic residues favor AMPs insertion into the lipid bilayer. Aromatic AAs (Trp, Phe) have higher tendency for adsorption | Peptide hydrophobic content should be balanced to prevent haemolysis. Optimal activity with hydrophobic content ≤ 60% | Highly hydrophobic59 or hydrophilic AMPs60 |
| Secondary structure | Ordered secondary structure (α-helix, β-sheet) enhances interaction with lipid bilayer. Optimal secondary structure for efficient peptide translocation not well defined | As for longer peptides. Short AMPs may lack clearly defined secondary structures | Some CPPs with random coil structures58 |
| Non-covalent interaction capacity | Electrostatic interactions, hydrogen bonding, π–π interactions crucial for secondary structure stabilization and membrane perturbation | Optimal activity when Arg and Trp residues are combined (Arg ≥ 40% and multiple adjacent Trp). Extended non-covalent interactions can lead to AMPs self-assembly | – |
Non-covalent interaction capacity
AMP interaction with bacterial component membranes is based on an interplay of weak interactions, namely electrostatic interactions, hydrophobic effect and HBs. Besides being crucial for the stabilization of the peptide secondary structure, HBs are a determinant for direct AMP interaction with the bacterial membrane. Indeed, under physiological conditions, basic residues such as Lys and Arg are HB donors, enabling HB formation with the sulfate and carboxylate moieties of the bilayer phospholipid headgroups. Arg, in particular, can bind more favorably to the membrane interface, owing to the multidentate HB mediated by its guanidine side group, leading to multiple interactions that contribute to membrane perturbation and destabilization.61 The extensive HB capacity of Arg strongly influences its overall contribution to the creation of a negative membrane curvature, which is topologically necessary for pore formation and disruption of the membrane. A recent study by Arora et al. evidenced the direct correlation between membrane deformation and relative amounts of Arg, Lys and Leu in the twelve amino acid-long peptide KLLLRLRKLLRR.62 Considering the modification in the membrane spontaneous curvature that can be induced by cationic and hydrophobic AAs (negative and positive, respectively), a higher Arg content was combined with fewer Leu residues, given the additional positive contribution induced by the formation of Arg-mediated multidentate HBs with the membrane. When combined with other hydrophobic residues such as Trp, however, Arg-rich peptides have often been associated with a stronger overall activity, as evidenced by the recurring presence of Arg and Trp even in short AMPs.43 In addition to a balanced cationic/hydrophobic content, the high activity of these peptides has been attributed to the complex interaction pattern that is created between Trp, Arg and the cell membrane.63 Trp itself is able to form HBs with the bilayer components due to its aromatic side chain. Moreover, the negatively charged clouds of its π–electron system can mediate the interaction with the Arg guanidium group, enabling the formation of ion-pair–π interactions.64 This allows for a deeper insertion of Arg inside the lipid bilayer, being shielded by the Trp residue. Notably, in contrast to Lys, the unique HB capability of Arg is maintained while engaging in cation–π interactions, facilitating its interaction with the negatively charged lipid bilayer and promoting disruption of the pathogen membrane.65Non-canonical amino acids and chemical modifications
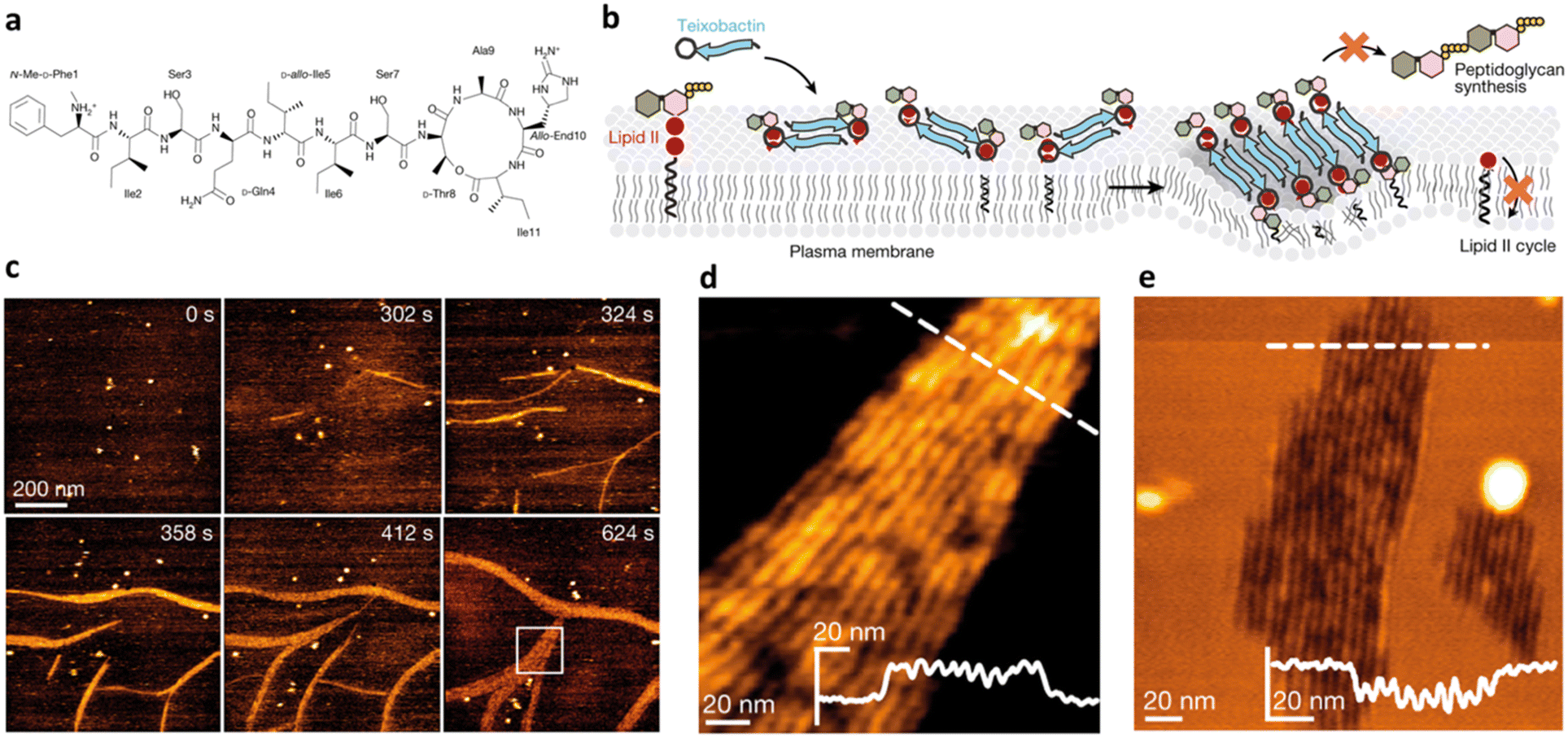
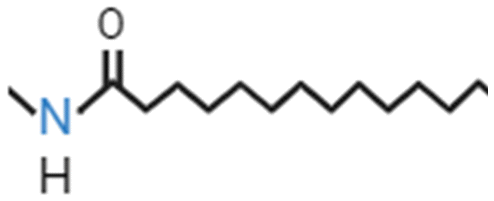
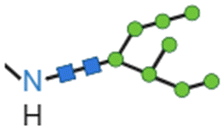
In an attempt to face drawbacks of AMPs limiting their confirmation as new antimicrobials, several chemical strategies have been employed, to date, to tune their chemical–physical properties and optimize their antimicrobial activity (Table 2). These include N- and/or C-terminal modification, incorporation of unnatural AAs, cyclization and use of non-peptide backbones (peptidomimetics).66 Among different N-terminal post-modifications (e.g., glycosylation, PEGylation…), lipidation is one of the most explored approaches, as evidenced by the fact that the most successful AMPs under clinical studies are cyclic lipopeptides, such as daptomycin and polymyxins E and B.67 Lipidation, i.e. the attachment of a fatty acid moiety to N-terminal residues or Lys side chains, increases peptide hydrophobicity, favouring interfacial binding and membrane permeability. This results in an overall improvement in the antimicrobial activity of peptides without altering their essential properties. Starting from the landmark work of Makovitzki et al. on ultrashort cationic lipopeptides, several studies focusing on the optimal combination in terms of both peptide length and fatty acid chain length have been reported.68 For instance, Narayana et al. identified different peptides ranging from four to twelve AAs derived from the KR12 fragment of the human cathelicidin LL-37 and modified the terminal amine with fatty acid chains with varying length (C6–C14).69 Results showed that C10-KR8 (KRIWQRIK) is the most cell selective lipopeptide, with strong activity against S. aureus, E. coli, and P. aeruginosa (MIC 1.6–12.5 μM) and poor haemolytic activity (HC50 > 200 μM), while longer fatty acid chains (>C10) lead to higher haemolysis. Increased haemolysis induced by lipopeptides in a fatty acid length-dependent manner is generally known, but the specific peptide sequence plays an essential role in determining the optimal acyl length for the overall AMP activity.68,70,71 Narayana et al. also observed that by substituting the L-amino acids with their D-enantiomeric form, C10-KR8 displayed significantly higher stability under physiological conditions and improved resistance to five different proteases.69 AA L-to-D conversion is indeed another common strategy used to improve AMP antimicrobial activity, even though a complete substitution may lead to highly stable peptides, with a consequent undesirable increase of cytotoxicity. Thus, only partial D-amino acid substitution of key residues was proven to be sufficient to enhance the half-life of peptides without compromising the AMP pharmacological profile.72| Chemical modification | Residues modified/involved | Impact on antimicrobial activity | Serum stability and resistance to proteolytic degradation | Cell selectivity and haemolysis activity | Ref. | |
|---|---|---|---|---|---|---|
| Lipidation |
|
N-terminal or Lys | Enhanced due to increased membrane affinity | Generally improved | Haemolysis increase in a fatty acid length-dependent manner | 68–70 |
| N-Glycosylation |
|
N-terminal, Lys, Asn | Variable effect | Generally improved | Negligible haemolytic activity | 67 |
| D-Amino acids |
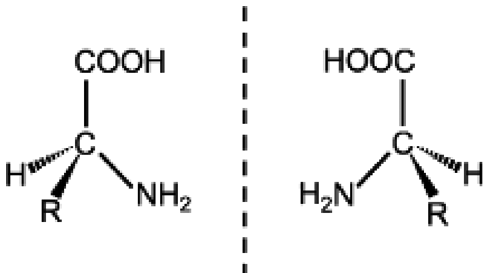
|
– | Variable effect dependent on peptide sequence | High stability at physiological conditions. Significantly improved resistance to enzymatic degradation | Variable effect | 72, 73 |
| Halogenation |
|
Phe, Tyr, Trp | Enhanced membrane affinity and disruption, but overall effect dependent on the halogen type and peptide sequence | Generally improved | Variable effect | 74–76, 77 |
| Dimerization and cyclization via disulfide bonds |
|
Cys | Generally improved | Stable peptide structure with improved resistance to proteolytic degradation | Possible haemolysis increase | 51, 78 |
Halogenation is another valuable and novel approach for optimizing physicochemical and functional features of a wide range of bioactive compounds, including AMPs. Several small halogenated biomolecules with antibacterial properties have been found in nature, where halogen atoms are usually incorporated at phenolic and indolic moieties catalyzed by substrate-specific halogenases.79 Similar building blocks are generally used in synthetic halogenated AMPs, leading to an increase of the hydrophobic volume of Phe and Trp residues.73 Molchanova et al. demonstrated that inactive peptoids can be converted into effective antibacterials by bromination of Phe residues without leading to increased haemolytic activity or in vitro cytotoxicity.74 Similarly, a study by Jia et al. evidenced the improved proteolytic stability of several halogenated derivatives of the honeybee peptide Jelleine-1 (PFKLSLHL-NH2), with the iodinated analogue displaying an eightfold increase in the antimicrobial activity with respect to the parent peptide.75 Notably, AMPs modified with fluorinated sulfono-γ-AA were recently tested in antimicrobial in vivo studies, showing excellent efficacy and safety.76 While the impact of halogenation on the physicochemical and structural properties of AMPs is evident (but still, strongly dependent on the halogen type and peptide sequence),80–82 the exact mechanisms behind the enhanced antimicrobial activity are still under investigation.77
Finally, in terms of increasing AMP stability, intramolecular disulfide bonds are known to improve the conformational rigidity of peptides.51 Thus, introducing a cysteine (Cys) residue in the sequence can lead to the formation of peptide dimers or cyclic structures via disulfide bridges, which direct and stabilize the supramolecular properties of AMPs, which, in turn, contributes to their antimicrobial activity.78
The role of peptide self-assembly
Peptides hold unique self-assembly properties, being able to form highly ordered and hierarchical nanoscale structures. With 20 AAs available and the possibility to further functionalize the peptides' primary sequence, a huge toolbox of self-assembling building blocks is accessible and such a versatility has led to a plethora of peptide nanostructures with different sizes, shapes and functionalities.83 Self-assembly is a key step also for the antimicrobial action of many AMPs.83–85 Indeed, as previously discussed in this review, upon contact with microbial membranes, AMPs often undergo structural changes, oligomerizing into aggregates that also account considerably for the diversity of the antimicrobial mode of action.43 Moreover, nanoscale systems of several peptides have been recently proposed as effective antimicrobials.85–87 Importantly, supramolecular organization of peptides not only enhances their antimicrobial activity, but can also result in the formation of highly dynamic suprastructures, which are often responsive to external stimuli (such as pH, ionic strength, etc.) and more stable (resistant to proteolysis) in the biological environment.88In this regard, the scientific community has made many efforts in designing, with the help of in silico simulations, biomimetic peptide sequences forming self-assembled nanostructures with enhanced resistance to enzymatic degradation and able to selectively destabilize bacteria cell membranes.83 The self-assembly behaviour of AMPs is driven by the onset of different types of non-covalent interactions, previously described in this review. To control and predict AMP self-assembly, it is pivotal to carefully design and select distinct peptide building blocks. Several self-assembling peptides have been proposed and studied to date: cyclic peptides, peptides functionalized with hydrophobic chains (alkyl and lipid chains) and surfactant-like peptides.3
About three decades ago, Ghadiri et al. first reported that cyclic D,L-a-peptides based on alternating L-Trp and D-Leu self-associated in nanotubular β-sheet-like structures, with exposure of the AAs side groups to the environment.89,90 It has been shown that these nanotubes, upon interaction with the bacterial membrane, lay parallel to the lipid plane. This supramolecular arrangement afforded superior proteolytic stability, thanks to the ring rigidity, and enhanced anti-bacterial activity by increasing membrane permeability and destabilizing transmembrane ion potentials.54 The majority of self-assembling cyclic peptides are rich in Trp and Arg residues that provide hydrophobic and positively charged moieties, respectively, and that are key factors for antibacterial activity.91,92 Substitution with glutamic acid, instead, seems to reduce this activity due to the electrostatic repulsion with the bacterial membrane.93 More recently, Parang et al. have deeply investigated, for example, the assembly and antibacterial activities of cyclic R4W4 and W4KR5 showing their selectivity and bactericidal ability against both GP and GN species.94–96 Further, in a recent study, Granja's group designed a modular approach for the preparation of cyclic peptides, having a small hydrophobic core and large hydrophilic surface. Such features led to the parallel stacking of the peptides and to the formation of effective antimicrobial nanotubes.60 All these studies highlighted the importance of achieving optimal balance between hydrophobicity and cationic charge for enabling the formation of the nanotubes and obtaining a broad-spectrum antimicrobial action.
Another common strategy used for promoting self-assembly of AMPs is their functionalization with hydrophobic moieties, such as alkyl and lipid chains. Such functionalization leads to higher hydrophobicity and stronger tendency to self-assemble minimizing the number of AAs (PA).97 Of note, nature-derived lipopeptides, isolated by fungi and bacteria, have shown to have antimicrobial properties against diverse pathogens.98 Thus, peptide lipidation, or more generally functionalization with hydrophobic moieties, has been largely used to promote the antibacterial activity of short peptides.99 These peptides, composed of a hydrophilic peptide portion linked to a hydrophobic chain, have an amphiphilic nature and usually self-assemble in nanostructures of different shapes (micelles, vesicles or nanofibers) depending on the packing parameter of the obtained molecular adduct, maintaining or assuming a β-sheet conformation upon membrane interaction.97 This strategy has been pioneered by Stupp and coworkers, who have designed many amphiphilic peptides forming supramolecular aggregates for different biomedical applications.100,101 Independent of the shape of the final aggregate, alkylation or lipidation of peptide sequences usually ends up in an increased resistance to proteolysis and enhanced activity against the bacterial membranes, thanks to the multivalence provided by the formation of a nanostructure at the bionanointerface. However, as previously mentioned, the length of the lipid chains was found to be correlated to cytotoxicity as increasing hydrophobicity decreases the specificity to bacterial membranes, with the optimal value in the range of C8 and C16.102,103 Lipidated tripeptides, containing a minimal cationic sequence (two Lys residues as either L or D) and a C16 alkyl chain, were reported to form positively charged spherical micelles showing good viability and promising antibacterial activity against both GN and GP bacteria in vitro.104 Interestingly, double-sited lipidated peptides were synthesized using shorter chain lengths (C2–C10) showing high antibacterial activity and lower cytotoxicity with respect to analogues with longer single lipid chains.105 Further, bolaamphiphiles with a generic structure Arg–(Ala)x–Arg exhibited different self-association properties as a function of the number of Ala residues with correlated antimicrobial activities against both GP and GN species, thus representing a promising class of potential AMPs (Fig. 6a).106
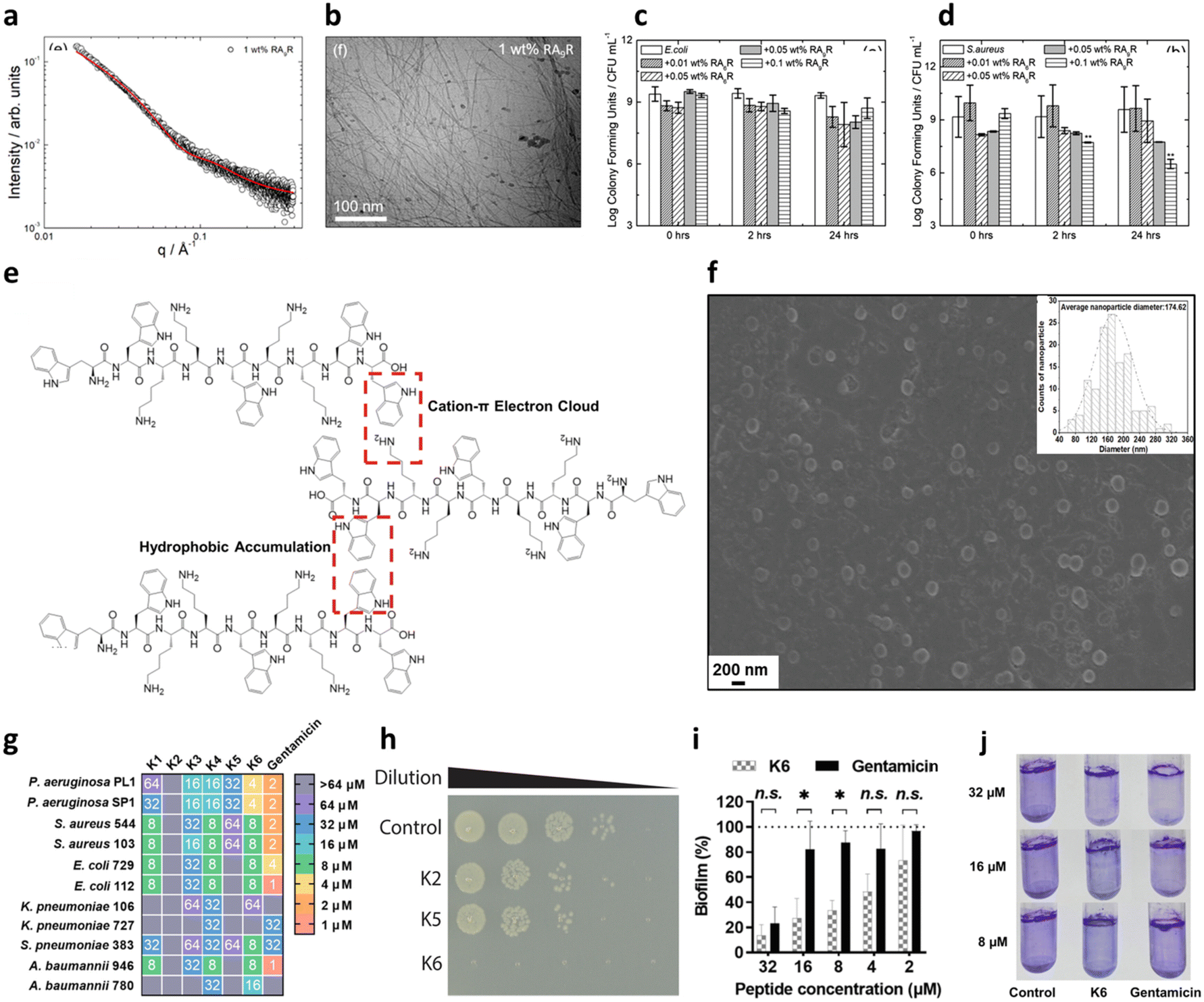 | ||
| Fig. 6 a) SAXS data and b) cryo-TEM image of 1 wt% amphiphile RA9R in water. The red line in panel a corresponds to the fitting using the form factor of a long cylindrical shell. c and d) Antimicrobial activity against c) E. coli and d) S. aureus. Time 0 is ∼5 min from when the peptide is added to the solution. Panels a–d are reproduced with permission under a Creative Commons License (CC-BY) from ref. 106. Copyright © 2019 American Chemical Society. e) Peptide K6 chemical structure and conceptual graph of its main self-assembly driving force. f) SEM image of peptide K6 taken at its minimal inhibitory concentration (MIC) for P. aeruginosa (4 μM) (insert: average size of peptide K6 nanoparticles). g) Minimal inhibitory concentration (MIC) values of peptides K1–K6 and gentamicin against bacteria. The color code indicates the dilution of a standard stock of the peptide from 1 to 64 μM, which showed complete growth inhibition. h) 10-fold dilutions of E. coli OMNIMAX were plated after 30 min of treatment with the respective peptides or in PBS at 37 °C, showing the rapid effect of peptide K6. i and j) Antibiofilm activity of peptide K6 with respect to gentamicin, measured through crystal violet staining. Panels e–j are reproduced with permission under a Creative Commons License (CC-BY-NC-ND 4.0) from ref. 107. Copyright © 2023 PNAS. | ||
Another interesting approach for promoting AMP self-assembly is fluorination. In fact, it has been shown that fluorination of gene and protein vectors enhances cellular internalization, endosome escape and plasma stability with respect to alkylated ones (i.e. conventional lipids or polymers).108,109 In the same way, the functionalization of peptide sequences of different lengths (7–15 AAs) with linear fluoroalkyl chains resulted in the formation of peptide nanoparticles of about 200 nm with increased proteolytic stability and ability to successfully deliver the peptide intracellularly.110 Thus, with a correct design of the peptide sequence, fluoroalkylation could be exploited for promoting the self-assembly of AMPs in more resistant nanostructures, likely facilitating their interaction with the bacteria lipid membrane and thus enhancing their therapeutic effect.
It is also possible to design a pure peptide sequence (i.e. only composed of AAs) bearing intrinsic amphiphilic properties. This strategy has led to the development of short peptides with surface activity able to form nanostructures, such as nanotubes, vesicles and micelles, in aqueous solutions.111 In this regard, Chou et al. have recently reported promising antimicrobial and antibiofilm activities for a panel of peptides containing WWW or WW motifs.107 One of the sequences, termed K6, containing two central KK motifs and two positively charged WW moieties at the edges, could engage in a network of hydrophobic and π-electron cloud interactions that led to the formation of spherical aggregates of 150–200 nm in size (Fig. 6e and f). Such a self-assembly pattern was correlated with the highest antimicrobial ability of the series (Fig. 6g and h). Even more interestingly, K6 also demonstrated a strong biofilm-disrupting ability against mixed P. aeruginosa and S. aureus biofilms in vitro at low concentrations (Fig. 6i and j). Bacterial biofilms are clusters of bacteria that are attached to a surface and/or to each other and embedded in a self-produced matrix consisting of proteins (e.g., fibrin) and polysaccharides (e.g., alginate), as well as eDNA, and the use of peptides to dismantle such a structure is gaining increasing attention.112 K6 was also successfully tested in a mouse infection model, showing no acute toxicity.
Other examples of amphiphilic peptides were reported by Hamley and co-workers, who have extensively studied the self-assembly behaviour of Ala/Arg peptides, along with their interaction with lipid membranes and their potential use as antimicrobial agents for both GP and GN pathogens.101,111,113–115
Another common category of peptides with surfactant-like behaviour, forming hydrogels with antimicrobial properties is represented by (Fmoc)-protected ultrashort peptides.116 Notably, even Fmoc-Phe, despite not bearing positive charges, exhibits antibacterial activity against GP bacteria.59 Moreover, the incorporation of D-AAs in these compounds was beneficial for the resistance to proteolytic degradation.117
Probing AMP interaction with bacterial membranes
A physicochemical approach aimed at establishing clear relationships between the structural and physical features of therapeutics and their functional properties can be instrumental to define the guidelines for a tailored design. Such an approach, which can be of general applicability, is particularly advantageous in the design of AMPs with their action being strongly dependent on their interfacial behaviour.21,118 In this framework, an emerging research topic related to AMPs is the investigation of their interaction with synthetic proxies of bacterial membranes. Compared to the direct in vitro investigation of bacteria challenged by AMPs, the employment of bacterial membrane biomimetic systems, made of either synthetic lipids or bacterial membrane extracts, has multiple advantages.119 First, the membrane composition of the artificial proxies can be controlled and finely tuned to investigate and determine the role of selected components in the interaction with AMPs. Furthermore, the geometry and structural arrangement of the biomimetic membrane can be varied at will, to match the requirement of different experimental techniques. Fig. 7 reports some examples of biomimetic membranes.120 From a compositional standpoint, the two general reference bacterial membrane structures are those of GP and GN bacteria, reported in Fig. 2 and here discussed in a previous section. Accordingly, the composition of displayed biomimetic membranes can be finely modulated in a controlled manner, by adding specific components, such as LPSs, lipid A, and zwitterionic or anionic lipids, in tuned proportions. Further, from a structural standpoint, depending on the specific phenomenon under investigation and on the available experimental technique, diverse biomimetic systems can be employed, from nanometric or micrometric vesicles (large and giant unilamellar vesicles (LUVs and GUVs)) enclosing an aqueous pool, to planar supported (SLBs) or freestanding lipid bilayers or monolayers.121 As a general consideration, while the effects of AMPs on bacteria can be strongly dependent on the bacterial type/strain and environmental conditions, and can be investigated through limited experimental tools, probing the interaction of AMPs with biomimetic systems of bacterial membranes gives access to a host of fundamental information gained via diverse complementary experimental techniques. Membrane adhesion to a bacterial membrane can be quantified via quartz crystal microbalance with dissipation monitoring (QCM-D), nuclear magnetic resonance (NMR) spectroscopy and null ellipsometry,122 and structurally resolved at the nanoscale through neutron and X-ray reflectivity (NR, XRR) on SLBs or suspended bilayers,123 and through small-angle X-ray and neutron scattering (SAXS, SANS) on LUVs.124 For instance, QCM-D provides information on the amount of adhered AMPs on the membrane, while NR and XRR allow reconstructing the profile of the bilayer along the z-axis (i.e., perpendicularly to the membrane), allowing thus the adhesion “mode” of AMPs to the membrane to be determined, with simple surface adhesion or a significant penetration degree inside the outer or the inner leaflet of the bilayer. Membrane disruption effects can be quantified at the molecular-to-nano length scale through spectroscopic or scattering methods on nanometric vesicles, or through NR, XRR, and atomic force microscopy (AFM) on SLBs.125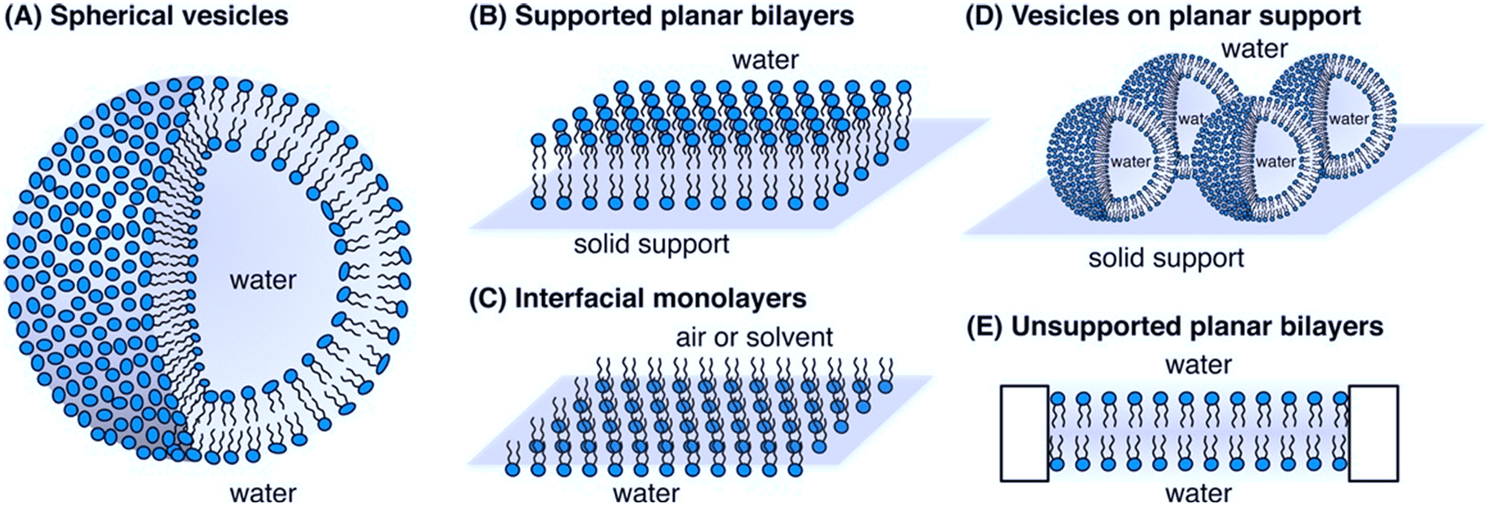 | ||
| Fig. 7 Schematic representation of bacterial biomimetic membrane models employed to study the mechanism of action of AMPs: A) spherical vesicles, B) supported planar bilayers, C) interfacial monolayers, D) vesicles on planar support and E) unsupported planar bilayers. Reprinted (adapted) with permission from ref. 120. Copyright © 2014 American Chemical Society. | ||
Specifically, both NR and XRR allow estimating the order/thickness decrease of the lipid bilayer upon incubation with AMPs, as well as its overall hydration increase, suggesting the occurrence of membrane perturbation/disruption effects. As a complementary approach, AFM allows visualizing in 2D the membrane disruption effects at the nanoscale. From a design perspective, these techniques allow determining how the balance between the hydrophobicity and cationic charge of AMPs drives their binding to model bacterial membranes and influences localized disruption, and how topological characteristics of self-assembled AMPs impact bacterial membrane integrity and, ultimately, lead to bacterial death.126,127 More extensive structural modifications of the target membrane, such as membrane disruption or morphological alterations occurring at a micrometric length scale, can be detected, for instance, through light microscopy-based techniques, such as fluorescence microscopy, laser scanning confocal microscopy (LSCM),128 and total internal reflection fluorescence microscopy (TIRF) on GUVs and fluorescently-labeled SLBs.129,130 For instance, by employing fluorescently labelled lipid probes marking the biomimetic membrane, it is possible to determine how incubation of the biomimetic membranes with AMPs affects fluorescence distribution homogeneity, which is a clear hallmark of extensive membrane disruption. The interaction mechanism, either of membrane disrupting or of membrane translocating AMPs, can be resolved on LUVs at a molecular lengthscale through time-resolved SAXS and SANS. For instance, in a recent publication, Lund et al. have investigated at a molecular level the membrane impact of natural AMPs with nanometric lipid vesicles.131 By combining SAXS and time-resolved SANS experiments, they explored the hypothesis of membrane pore formation as a consequence of AMP incubation with lipid vesicles, studying both the overall structure of AMP insertion in the lipid membrane (Fig. 8a) and the impact of AMPs on the kinetic mechanisms of lipid transport within the lipid membrane. Interestingly, a similar effect of most AMPs was found, suggesting the possibility of finding a unique description for AMP-induced destabilization of bacterial membranes. Besides the well-established experimental techniques and methods, the versatility of artificial bacterial membrane proxies allows designing and setting-up specific experimental tools to investigate AMP–bacterial membrane proxy interactions. For instance, Dittrich and coworkers developed a double emulsion-based system designed with the aid of microfluidics as a high-throughput tool for fast screening of AMP interaction with LUVs of mammalian-like and bacterial-like cell composition (Fig. 8b).132
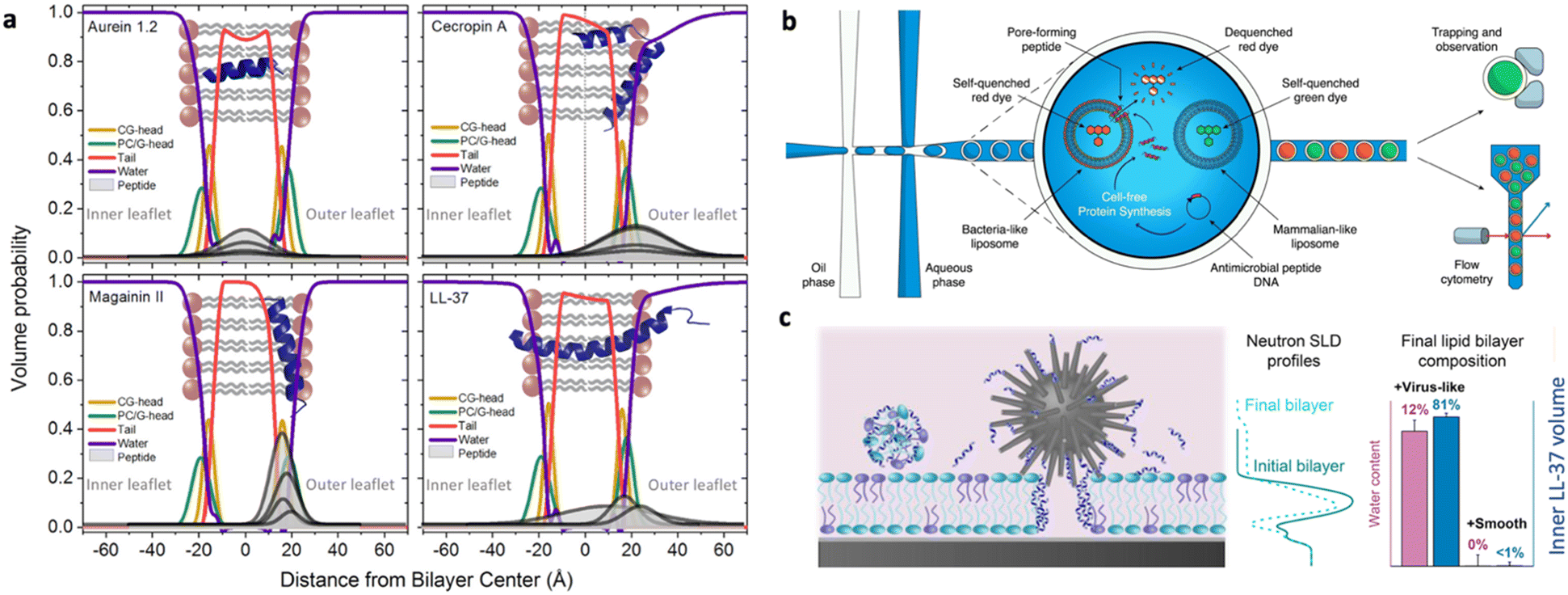 | ||
| Fig. 8 a) Volume probability distributions for the lipid membrane of vesicles with various amounts of natural peptides, showing the insertion of peptides in the membrane, calculated from small angle X-ray scattering curve fitting of biomimetic lipid vesicle/peptide systems in solution. Reprinted with permission from ref. 131. Copyright 2021 Elsevier. b) High throughput microfluidic system to monitor the membrane interaction of AMPs with large unilamellar vesicles (LUVs): double emulsions are formed on a microfluidic device, each containing a particular AMP. The peptide may or may not interact with the co-encapsulated LUVs, disrupting their membranes. As the LUVs are loaded with a self-quenching concentration of fluorescent dye, their disruption causes release and dilution of the dye, generating a fluorescence signal, which can be detected. Reprinted with permission from ref. 132. Copyright 2022 Wiley. c) Interaction of vectorized AMPs with planar biomimetic membranes (supported lipid bilayers, SLBs): silica NPs of different topologies (smooth, virus-like, mesoporous) are co-incubated with a SLB and their combined interaction with SLBs is monitored, highlighting a prominent impact of the vector topology in driving the interaction at the nano–bio interfaces. Reprinted with permission from ref. 133. Copyright 2021 American Chemical Society. | ||
The difficulties in achieving a thorough understanding and description of AMP interaction with bacterial membranes are further enhanced in the case of vectorized AMPs. Associating AMPs with nanoparticles or nanostructured delivery systems can be a valuable strategy to enhance AMP ability to interact with cells, or to improve AMP pharmacokinetic properties. For these complex nanosystems, fundamental studies with biomimetic membranes are key to decouple the interfacial effects due to AMPs and to the vector itself and to derive some fundamental information to improve the formulation of AMP–nanovector systems. In a recent study, the topology of different silica-based nanovectors (smooth, mesoporous or virus-like) was explored in terms of capability to enhance LL37 AMP interaction with a target membrane (Fig. 8c).133 Similarly, the ability of a soft lipid nanocarrier having a cubic structure to efficiently deliver AMPs without hampering their membrane adhesion/disruption function was tested on different mammalian and bacterial mimicking membranes, to determine the relative affinity of the system for different cells.129
Overall, these examples show how recent efforts of researchers in the field of AMP development are aimed at exploiting the potentialities of bacterial membrane biomimetic systems both to derive specific information on the behaviour of selected AMPs and, at the same time, to build up a core of new general concepts on the interaction of AMPs with bacterial membranes, which could boost the design of novel systems of superior efficacy, and, ultimately, the full translation of synthetic AMPs into medical practice.
The aid of microscopy
Direct visualization by nanoscale microscopy techniques has represented, in the last few years, the natural complement to the reciprocal space analysis tools described in the previous section.In the characterization of peptide–membrane interaction, atomic force microscopy (AFM) and electron microscopy (EM) have both played a crucial role in different frames. While EM, taking advantage of its high resolution over several magnification ranges, has been precious to elucidate the membrane permeabilization and bacterial damage at the cellular level, AFM has rather served to gain insight into the molecular basis of peptide–membrane interaction.134
In the last two decades, AFM has been the most suitable technique for imaging biological molecules at the nanoscale, under conditions close to the native ones. Indeed, while when employing EM techniques biomolecules have to be imaged in a vacuum or under controlled humidity conditions, single proteins have been imaged and manipulated in a fully liquid environment by AFM, thus allowing their conformation to be monitored during their activity.135 Moreover, proteins and polypeptides displaying a symmetric three-dimensional quaternary structure, such as membrane proteins, have allowed gaining extremely high resolution.136–138 Further, AFM represents an effective tool to visualize many of the membrane-mimicking structures described in the previous section. In particular, this microscopy technique has been widely used to study tethered, polymer-cushioned, standing over pores and supported lipid bilayers (SLBs), allowing the structural and physical–chemical properties of their interface to be probed at the nanoscale such as their stability, phase separation, and their interaction with biomolecules.139
In the specific frame of AMPs, direct visualization of the fine structure assumed upon their interaction with membranes has contributed to unveiling the different mechanisms of actions. Several membrane disruption models have been identified by AFM, such as barrel-stave and toroidal pore models, by imaging the pores formed at different length scales, while membrane roughening has been used to support the carpet models.140 When membrane thinning has been thought to be the mechanism of action, a direct measurement of the bilayer thickness has been performed with extremely high accuracy to decipher the role of peptide-induced changes in membrane interfacial tension.141 In addition, when preparing bilayers with different lipid phases, AFM can reveal differences in peptide preference for binding onto them or, when accessible, on the phase boundaries.142 Importantly, together with probing the membrane bilayer structure, AFM also allows tracking of the AMP self-assembly. Such an approach has been recently employed by Shen and coworkers to investigate the activity of a new short AMP, KRRFFRRK (termed FF8), designed to target the negatively charged lipid membrane and self-assemble into nanofibers on it.143
Besides its main role of imaging, AFM is also widely used to mechanically manipulate biomolecules. In the case of peptide–membrane interaction, this has a twofold implication: (1) measuring the interaction force between the peptide and the phospholipids, and thus correlating such force with the AMP impact on the membrane, and (2) characterizing the bilayer fluidity and mechanical properties. In the first case, the interaction force can be directly measured by bringing into contact a peptide tethered to the AFM probe with a bilayer formed on the substrate surface and then measuring the load necessary to break the formed bond.144 In the second one, by indenting the membrane with the AFM tip, one can measure the force necessary to punch through the lipid layers, which is related to their cohesive energy, their packing and the overall fluidity.145 Such a nanomechanical approach has been applied by Manioglu and coworkers to discover new molecular insights into the mechanism of action of polymyxins, a group of cyclic heptapeptides bearing an N-terminal fatty acid, currently used in the clinic against the superbugs Acinetobacter baumannii and Pseudomonas aureginosa.146 In particular, they utilized outer membrane vesicles (OMVs) of Gram-negative E. coli to form native membrane patches containing LPSs in the outer leaflet. Their study revealed that, upon exposure to the membrane patches, polymyxins arrange the LPSs into hexagonal assemblies to form crystalline structures. This led to a decreased membrane thickness and an increased membrane area and stiffness, proving that the crystalline hexagonal assembly formation constitutes the mechanism of action of polymyxins (Fig. 9a–c).
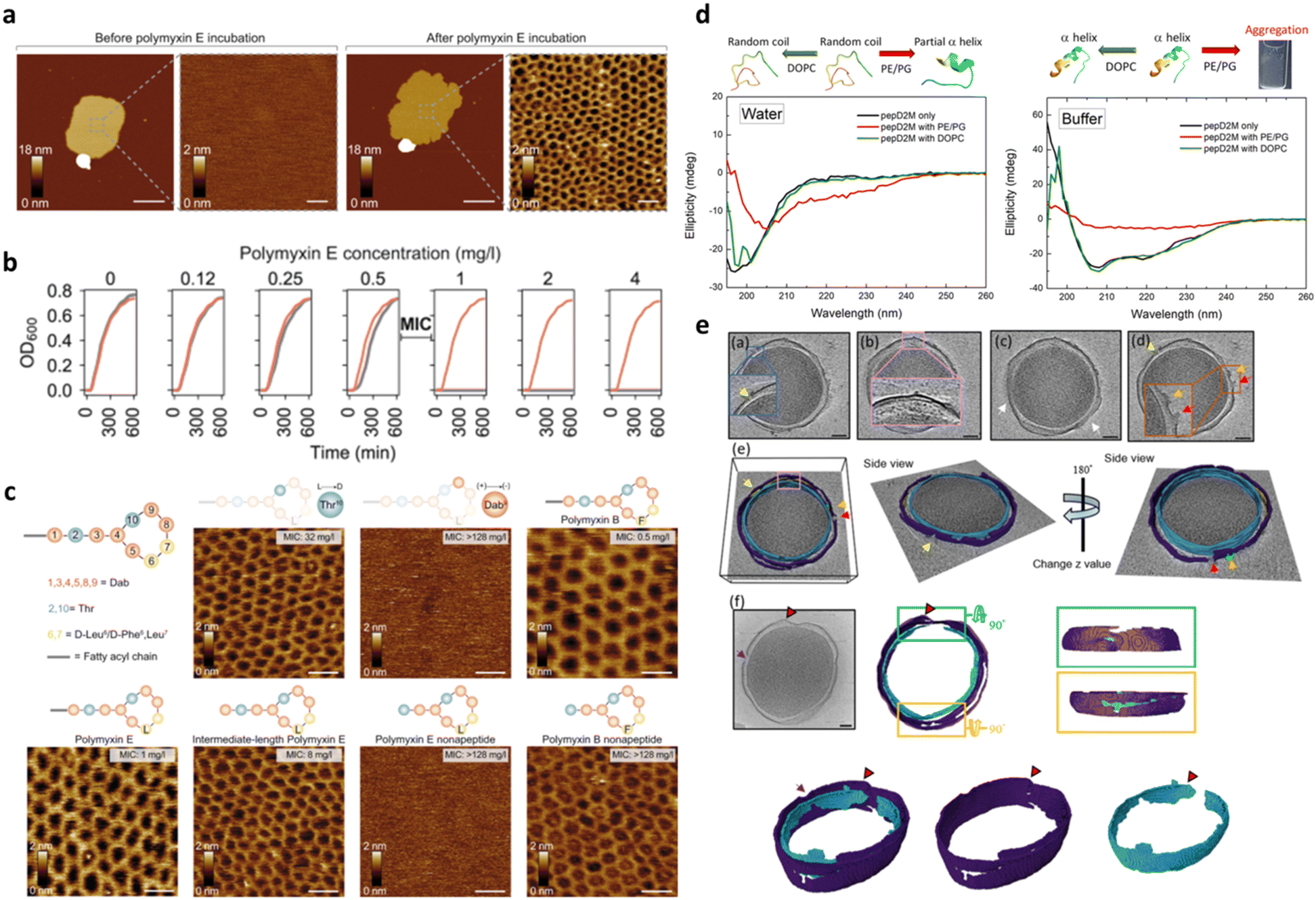 | ||
| Fig. 9 a) AFM topographs of an OM patch before and after incubation with polymyxin E. Scale bars, 200 nm (overview) and 20 nm (zoom-in). b) Growth curves of the E. coli MG1655 WT (gray) and MG1655MCR-1 (red) strains at different polymyxin concentrations. Above the minimal inhibitory concentration (MIC) of 0.5–1 mg L−1, the growth of the E. coli MG1655 WT strain is inhibited. c) Schematic representation of the polymyxin structure, where polymyxin E has D-Leu (L) and polymyxin B has D-Phe (F) in the 6th position and AFM topographs of OM patches from the E. coli MG1655 WT strain upon incubation with different polymyxin E and polymyxin B variants (50 mg L−1). Crystalline structures are formed with the enantiomer variant Thr10 (L → D) and with full and intermediate-length polymyxin E variants. Structures are not formed in the polymyxin variant Dab9 (+) → (−) and polymyxin E nonapeptide variant. With polymyxin B variants, crystalline structures are formed in all cases. Panels a–c are reproduced and adapted with permission under a Creative Commons License (CC-BY 4.0) from ref. 146. Copyright © 2022 Springer Nature. d) CD spectra of pepD2M in water and in buffer. Black line: without liposomes; red line: mixed with PE/PG liposomes; green line: mixed with DOPC liposomes. Illustrations of the structural change that occurs upon conditional changes are shown above the spectra. e) Cryo-ET slices of two minicells treated with pepD2M and their inner and outer membranes. IM: cyan; OM: violet; PG: yellow; released material: green. Scale bar = 100 nm. Pores are pointed by arrows with different colors. Two volcanic-crater-like pores are indicated by the red and orange arrows. Panels d and e are reproduced and adapted with permission under a Creative Commons License (CC-BY 4.0) from ref. 147. Copyright © 2023 Springer Nature. | ||
It is important to mention that high resolution AFM imaging is not limited to ideal samples, such as synthetic lipid bilayers on flat surfaces, but is still informative when working with the entire living bacteria showing nanoscale changes in their membrane.148,149 AMPs alter cell physical properties, specifically morphology, volume, surface roughness, and stiffness. These effects can be fully investigated by AFM working with living cells in their physiological environment and further corroborated by EM techniques, which can be complementary to AFM to fully elucidate the AMP mechanism of action and impact on the bacterial morphology.150 The role of EM techniques in AMP design and characterization becomes even more relevant when employed under cryogenic conditions that allow samples to be preserved in their frozen-hydrated (native-like) state. More recently, cryo-electron tomography (cryo-ET) has been emerging as an excellent method for studying the three-dimensional (3D) structure of living cells. In this framework, Chen and coworkers reported on the use of cryo-ET and AFM to study how the de novo-designed peptide composed of fourteen AAs (Myr-WKKLKKLLKKLKKL-NH2; Myr: myristoylation), termed pepD2M, interacted with the natural membrane of Gram-negative bacteria.147 This study pioneered the use of cryo-ET in AMP design. As shown in Fig. 9, tomography reconstruction of cryo-EM images allowed visualizing the severe disruption exerted by pepD2M on the E. coli membrane, and imaging and measuring the size of the formed pores. AFM was instead used to prove that pepD2M effectively removes the lipids from the bacterial OM. The combination of the experimental data obtained with the two microscopy approaches allowed the authors to identify the exact mechanism of action of pepD2M, which differed from the one predicted for an amphipathic and α-helix forming peptide which usually forms transmembrane pores. Indeed, the removal of lipids from the OM by pepD2M leads to the formation of irregular pores destabilizing the bacterial wall. This allows pepD2M to reach the IM where the membranolytic action is fully exerted, leading to loss of the cytoplasmic material and shrinkage of the bacterial cell.
Future outlook and conclusions
AMPs are expected to become future key players in the fight against antimicrobial resistance and superbug emergence. This is proved by the intense research effort made by scientists from several scientific communities to better understand the mechanism of action of these peptides and optimize their design towards new effective therapeutic options.To date, AMPs have mostly been studied as agents able to kill bacteria targeting intracellular components or bacterial walls and membranes. Still, as described in the work by Chou and coworkers previously discussed in this review,107 additional antimicrobial effects have recently been reported, with AMPs shown to be able to suppress biofilm formation and even induce the dissolution of existing biofilms.151,152 A recent example of the role of AMPs as antibiofilm agents was reported by Harding et al., who designed cyclic-ERWGHDFIK, a potent inhibitor of the P. aeruginosa aminopeptidase (PaAP), highly abundant in the biofilm matrix where it contributes to its formation and nutrition.153 Notably, the antibiofilm action shown by many AMPs also opens up to their use in the development of biomaterials endowed with intrinsic antimicrobial activity, which could limit the insurgence of infections.154,155
In this review, we have highlighted the main steps of the AMP engineering process and we chose to focus on short AMPs which, we believe, have a more realistic chance to replace classical antibiotics, given their lower cost and faster production, when compared to longer sequences. We summarized the main design rules that can aid in developing effective AMPs by modulating physicochemical determinants and obtain the desired biological parameters.156 As recently stated by Gagat et al., the engineering of novel AMPs could significantly be eased by a database of non-AMPs, currently not available, reporting on peptide sequences that were found to be inactive for specific bacterial species.157 Access to such a database would undoubtedly reduce the number of studies and peptide sequences tested and would provide a precious tool in the identification of effective AMPs and the optimization of their features. Further, given the charged nature of almost all AMPs, we believe that the study of their interaction, not only with bacterial membranes and cells, but also with the host biological fluids, would be functional for the identification of effective peptide sequences. This is currently an underexplored side of AMP research, but probing the bio–nano interaction taking place in the physiological environment would be key, in particular, to nanoscale AMP systems, which could be deeply changed in terms of surface charge, composition and features upon contact with physiological proteins, salts and biosurfactants.158
Overall, we have provided an overview of the most recent work on short AMPs and on the techniques that are key to studying their chemical and physical features, as well as for the assessment of their interaction with bacterial walls and membranes. Bearing in mind the multiple opportunities offered by AMP design, in terms of peptide sequence and post-synthetic chemical modification, we expect that many novel peptides of relevance in the fight against microbial infection and resistance will be identified and that the number of AMPs tested in the clinic will soon increase.
Author contributions
Costanza Montis: writing – original draft, review & editing and conceptualization; Elisa Marelli: writing – original draft, review & editing; Francesco Valle: writing – original draft, review & editing; Francesca Baldelli Bombelli: funding acquisition and writing – original draft, review & editing; Claudia Pigliacelli: writing – original draft, review & editing and conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
FBB acknowledges the project Lancelot (PRIN 2022 PNRR n P2022RBF5P) funded by MUR (European Union – Next Generation EU).References
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS.
- M. McKenna, Nature, 2020, 584, 338–341 CrossRef CAS PubMed.
- J. A. Doolan, G. T. Williams, K. L. F. Hilton, R. Chaudhari, J. S. Fossey, B. T. Goult and J. R. Hiscock, Chem. Soc. Rev., 2022, 51, 8696–8755 RSC.
- U. Theuretzbacher, K. Outterson, A. Engel and A. Karlén, Nat. Rev. Microbiol., 2020, 18, 275–285 CrossRef PubMed.
- M. A. Cook and G. D. Wright, Sci. Transl. Med., 2022, 14, eabo7793 CrossRef CAS PubMed.
- G. Sun, Q. Zhang, Z. Dong, D. Dong, H. Fang, C. Wang, Y. Dong, J. Wu, X. Tan, P. Zhu and Y. Wan, Front. Public Health, 2022, 10, 1002015 CrossRef PubMed.
- L. Lombardi, A. Falanga, V. Del Genio and S. Galdiero, Pharmaceutics, 2019, 11, 166 CrossRef CAS PubMed.
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- A. H. Benfield and S. T. Henriques, Front. Med. Technol., 2020, 2, 25–28 Search PubMed.
- N. G. J. Oliveira, M. H. Cardoso, N. Velikova, M. Giesbers, J. M. Wells, T. M. B. Rezende, R. de Vries and O. L. Franco, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- F. Savini, M. R. Loffredo, C. Troiano, S. Bobone, N. Malanovic, T. O. Eichmann, L. Caprio, V. C. Canale, Y. Park, M. L. Mangoni and L. Stella, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183291 CrossRef CAS PubMed.
- M. H. Cardoso, B. T. Meneguetti, B. O. Costa, D. F. Buccini, K. G. N. Oshiro, S. L. E. Preza, C. M. E. Carvalho, L. Migliolo and O. L. Franco, Int. J. Mol. Sci., 2019, 20, 4877 CrossRef CAS PubMed.
- D. J. Trojanowska, G. Suarato, C. Braccia, A. Armirotti, F. Fiorentini, A. Athanassiou and G. Perotto, ACS Appl. Nano Mater., 2022, 5, 15272–15287 CrossRef CAS PubMed.
- H. Gong, J. Zhang, X. Hu, Z. Li, K. Fa, H. Liu, T. A. Waigh, A. McBain and J. R. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 34609–34620 CrossRef CAS PubMed.
- A. Hollmann, M. Martínez, M. E. Noguera, M. T. Augusto, A. Disalvo, N. C. Santos, L. Semorile and P. C. Maffía, Colloids Surf., B, 2016, 141, 528–536 CrossRef CAS.
- S. Kim, J. Lee, S. Lee, H. Kim, J.-Y. Sim, B. Pak, K. Kim and J. Il Kim, Commun. Biol., 2022, 5, 1199 CrossRef CAS PubMed.
- F. H. Waghu and S. Idicula-Thomas, Protein Sci., 2020, 29, 36–42 CrossRef CAS PubMed.
- C. Wang, T. Hong, P. Cui, J. Wang and J. Xia, Adv. Drug Delivery Rev., 2021, 175, 113818 CrossRef CAS PubMed.
- S. He, Z. Yang, X. Li, H. Wu, L. Zhang, A. Shan and J. Wang, Acta Biomater., 2023, 164, 175–194 CrossRef CAS.
- T. Ganz, Nat. Rev. Immunol., 2003, 3, 710–720 CrossRef CAS.
- S. Guha, J. Ghimire, E. Wu and W. C. Wimley, Chem. Rev., 2019, 119, 6040–6085 CrossRef CAS PubMed.
- P. Askari, M. H. Namaei, K. Ghazvini and M. Hosseini, BMC Pharmacol. Toxicol., 2021, 22, 1–12 CrossRef PubMed.
- J. S. Depelteau, S. Brenzinger and A. Briegel, Bacterial and Archeal Cell Structure, in Encyclopedia of Microbiology, ed. T. M. B. T.-E. of M. and E. Schmidt, Academic Press, Oxford, 4th edn, 2019, pp. 348–360 Search PubMed.
- L. Pasquina-Lemonche, J. Burns, R. D. Turner, S. Kumar, R. Tank, N. Mullin, J. S. Wilson, B. Chakrabarti, P. A. Bullough, S. J. Foster and J. K. Hobbs, Nature, 2020, 582, 294–297 CrossRef CAS PubMed.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, 1–16 Search PubMed.
- E. R. Rojas, G. Billings, P. D. Odermatt, G. K. Auer, L. Zhu, A. Miguel, F. Chang, D. B. Weibel, J. A. Theriot and K. C. Huang, Nature, 2018, 559, 617–621 CrossRef CAS PubMed.
- K. L. May and M. Grabowicz, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8852–8854 CrossRef CAS PubMed.
- D. Poger, S. Pöyry and A. E. Mark, ACS Omega, 2018, 3, 16453–16464 CrossRef CAS PubMed.
- A. Ebbensgaard, H. Mordhorst, F. M. Aarestrup and E. B. Hansen, Front. microbiol., 2018, 9, 1–13 CrossRef PubMed.
- J. Li, J. J. Koh, S. Liu, R. Lakshminarayanan, C. S. Verma and R. W. Beuerman, Front. Neurosci., 2017, 11, 1–18 CAS.
- D. I. Chan, E. J. Prenner and H. J. Vogel, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1184–1202 CrossRef CAS PubMed.
- Z. Yang, H. Choi and J. C. Weisshaar, Biophys. J., 2018, 114, 368–379 CrossRef CAS PubMed.
- D. J. Slotboom, T. W. Ettema, M. Nijland and C. Thangaratnarajah, FEBS Lett., 2020, 594, 3898–3907 CrossRef CAS PubMed.
- D. Ghilarov, S. Inaba-Inoue, P. Stepien, F. Qu, E. Michalczyk, Z. Pakosz, N. Nomura, S. Ogasawara, G. C. Walker, S. Rebuffat, S. Iwata, J. G. Heddle and K. Beis, Sci. Adv., 2021, 7, eabj5363 CrossRef CAS PubMed.
- J. Frimodt-Møller, C. Campion, P. E. Nielsen and A. Løbner-Olesen, Curr. Genet., 2022, 68, 83–90 CrossRef PubMed.
- J. S. M. Svendsen, T. M. Grant, D. Rennison, M. A. Brimble and J. Svenson, Acc. Chem. Res., 2019, 52, 749–759 CrossRef CAS PubMed.
- G. Wang, Curr. Biotechnol., 2012, 1, 72–79 CrossRef CAS PubMed.
- J. Andrä, D. Monreal, G. M. De Tejada, C. Olak, G. Brezesinski, S. S. Gomez, T. Goldmann, R. Bartels, K. Brandenburg and I. Moriyon, J. Biol. Chem., 2007, 282, 14719–14728 CrossRef PubMed.
- Z. Jiang, A. I. Vasil, J. D. Hale, R. E. W. Hancock, M. L. Vasil and R. S. Hodges, Biopolym. - Pept. Sci. Sect., 2008, 90, 369–383 CrossRef CAS PubMed.
- I. Greco, N. Molchanova, E. Holmedal, H. Jenssen, B. D. Hummel, J. L. Watts, J. Håkansson, P. R. Hansen and J. Svenson, Sci. Rep., 2020, 10, 1–13 CrossRef PubMed.
- J. J. López Cascales, S. Zenak, J. García De La Torre, O. G. Lezama, A. Garro and R. D. Enriz, ACS Omega, 2018, 3, 5390–5398 CrossRef PubMed.
- Y. Wu, Q. He, X. Che, F. Liu, J. Lu and X. Kong, Biochem. Biophys. Res. Commun., 2023, 648, 66–71 CrossRef CAS PubMed.
- S. Clark, T. A. Jowitt, L. K. Harris, C. G. Knight and C. B. Dobson, Commun. Biol., 2021, 4, 605 CrossRef CAS PubMed.
- Y. Yang and C. L. Dias, J. Phys. Chem. B, 2023, 127, 912–920 CrossRef CAS PubMed.
- B. Mattei, A. Miranda, K. R. Perez and K. A. Riske, Langmuir, 2014, 30, 3513–3521 CrossRef CAS PubMed.
- K. Wang, J. D. Keasling and S. J. Muller, Int. J. Biol. Macromol., 2005, 36, 232–240 CrossRef CAS PubMed.
- I. A. Edwards, A. G. Elliott, A. M. Kavanagh, J. Zuegg, M. A. T. Blaskovich and M. A. Cooper, ACS Infect. Dis., 2016, 2, 442–450 CrossRef CAS PubMed.
- B. H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820–7880 RSC.
- Z. Yang, Y. Wei, W. Wu, L. Zhang, J. Wang and A. Shan, Food Funct., 2023, 14, 3139–3154 RSC.
- M. Fernández-Vidal, S. Jayasinghe, A. S. Ladokhin and S. H. White, J. Mol. Biol., 2007, 370, 459–470 CrossRef PubMed.
- T. Wang, C. Zou, N. Wen, X. Liu, Z. Meng, S. Feng, Z. Zheng, Q. Meng and C. Wang, J. Pept. Sci., 2021, 27, e3306 CrossRef CAS PubMed.
- I. Zelezetsky and A. Tossi, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1436–1449 CrossRef CAS PubMed.
- S. Lohan, A. G. Konshina, R. G. Efremov, I. Maslennikov and K. Parang, J. Med. Chem., 2023, 66, 855–874 CrossRef CAS PubMed.
- S. Fernandez-Lopez, H. Kim, E. C. Choi, M. Delgado, J. R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. A. Weinberger, K. M. Wilcoxen and M. R. Ghadiri, Nature, 2001, 412, 452–455 CrossRef CAS PubMed.
- R. Shukla, F. Lavore, S. Maity, M. G. N. Derks, C. R. Jones, B. J. A. Vermeulen, A. Melcrová, M. A. Morris, L. M. Becker, X. Wang, R. Kumar, J. Medeiros-Silva, R. A. M. van Beekveld, A. M. J. J. Bonvin, J. H. Lorent, M. Lelli, J. S. Nowick, H. D. MacGillavry, A. J. Peoples, A. L. Spoering, L. L. Ling, D. E. Hughes, W. H. Roos, E. Breukink, K. Lewis and M. Weingarth, Nature, 2022, 608, 390–396 CrossRef CAS PubMed.
- D. Glatzová, H. Mavila, M. C. Saija, T. Chum, L. Cwiklik, T. Brdička and M. Cebecauer, FEBS J., 2021, 288, 4039–4052 CrossRef PubMed.
- R. P. Bywater, D. Thomas and G. Vriend, J. Comput.-Aided Mol. Des., 2001, 15, 533–552 CrossRef CAS PubMed.
- A. Gori, G. Lodigiani, S. G. Colombarolli, G. Bergamaschi and A. Vitali, ChemMedChem, 2023, 18, e202300236 CrossRef CAS PubMed.
- A. Y. Gahane, P. Ranjan, V. Singh, R. K. Sharma, N. Sinha, M. Sharma, R. Chaudhry and A. K. Thakur, Soft Matter, 2018, 14, 2234–2244 RSC.
- E. González-Freire, F. Novelli, A. Pérez-Estévez, R. Seoane, M. Amorín and J. R. Granja, Chem. – Eur. J., 2021, 27, 3029–3038 CrossRef PubMed.
- A. Rice and J. Wereszczynski, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1941–1950 CrossRef CAS PubMed.
- A. Arora, S. Majhi and A. Mishra, Mater. Today: Proc., 2022, 49, 2392–2396 CAS.
- A. K. Mishra, J. Choi, E. Moon and K. H. Baek, Molecules, 2018, 23, 1–23 CrossRef PubMed.
- A. Walrant, A. Bauzá, C. Girardet, I. D. Alves, S. Lecomte, F. Illien, S. Cardon, N. Chaianantakul, M. Pallerla, F. Burlina, A. Frontera and S. Sagan, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183098 CrossRef CAS PubMed.
- S. K. Straus, Biochim. Biophys. Acta, Biomembr., 2024, 1866, 184260 CrossRef CAS PubMed.
- W. Li, F. Separovic, N. M. O'Brien-Simpson and J. D. Wade, Chem. Soc. Rev., 2021, 50 Search PubMed.
- E. Grimsey, D. W. P. Collis, R. Mikut and K. Hilpert, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183195 CrossRef CAS PubMed.
- A. Makovitzki, D. Avrahami and Y. Shai, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15997–16002 CrossRef CAS PubMed.
- J. L. Narayana, R. Golla, B. Mishra, X. Wang, T. Lushnikova, Y. Zhang, A. Verma, V. Kumar, J. Xie and G. Wang, ACS Infect. Dis., 2021, 7, 1795–1808 CrossRef PubMed.
- Y. Wang, M. Xue, R. Gao, S. Chakraborty, S. Wang, X. Zhao, M. Gu, C. Cao, X. Sun and J. Cai, Int. J. Mol. Sci., 2023, 24, 6407 CrossRef CAS PubMed.
- C. Zhong, N. Zhu, Y. Zhu, T. Liu, S. Gou, J. Xie, J. Yao and J. Ni, Eur. J. Pharm. Sci., 2020, 141, 105123 CrossRef CAS PubMed.
- Y. Zai, Y. Ying, Z. Ye, M. Zhou, C. Ma, Z. Shi, X. Chen, X. Xi, T. Chen and L. Wang, Antibiotics, 2020, 9, 1–19 CrossRef PubMed.
- A. J. Craig, Y. Ermolovich, A. Cameron, A. Rodler, H. Wang, J. A. Hawkes, M. Hubert, F. Björkling, N. Molchanova, M. A. Brimble, L. W. K. Moodie and J. Svenson, ACS Med. Chem. Lett., 2023, 14, 802–809 CrossRef CAS PubMed.
- N. Molchanova, J. E. Nielsen, K. B. Sørensen, B. K. Prabhala, P. R. Hansen, R. Lund, A. E. Barron and H. Jenssen, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- F. Jia, Y. Zhang, J. Wang, J. Peng, P. Zhao, L. Zhang, H. Yao, J. Ni and K. Wang, Peptides, 2019, 112, 56–66 CrossRef CAS PubMed.
- X. Guo, X. Miao, Y. An, T. Yan, Y. Jia, B. Deng, J. Cai, W. Yang, W. Sun, R. Wang and J. Xie, Eur. J. Med. Chem., 2024, 264, 116001 CrossRef CAS PubMed.
- M. Mardirossian, M. Rubini, M. F. A. Adamo, M. Scocchi, M. Saviano, A. Tossi, R. Gennaro and A. Caporale, Molecules, 2021, 26, 7401 CrossRef CAS PubMed.
- D. Neubauer, M. Jaśkiewicz, E. Sikorska, S. Bartoszewska, M. Bauer, M. Kapusta, M. Narajczyk and W. Kamysz, Int. J. Mol. Sci., 2020, 21, 1–30 Search PubMed.
- A. Pizzi, C. Pigliacelli, G. Bergamaschi, A. Gori and P. Metrangolo, Coord. Chem. Rev., 2020, 411, 213242 CrossRef CAS.
- A. Marchetti, A. Pizzi, G. Bergamaschi, N. Demitri, U. Stollberg, U. Diederichsen, C. Pigliacelli and P. Metrangolo, Chem. – Eur. J., 2022, 28, e202104089 CrossRef CAS PubMed.
- D. Maiolo, A. Pizzi, A. Gori, G. Bergamaschi, C. Pigliacelli, L. Gazzera, A. Consonni, F. Baggi, F. Moda, F. Baldelli Bombelli, P. Metrangolo and G. Resnati, Supramol. Chem., 2020, 32, 247–255 CrossRef CAS.
- A. Pizzi, L. Sori, C. Pigliacelli, A. Gautieri, C. Andolina, G. Bergamaschi, A. Gori, P. Panine, A. M. Grande, M. B. Linder, F. Baldelli Bombelli, M. Soncini and P. Metrangolo, Small, 2022, 18, 2200807 CrossRef CAS PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- L. Lombardi, Y. Shi, A. Falanga, E. Galdiero, E. de Alteriis, G. Franci, I. Chourpa, H. S. Azevedo and S. Galdiero, Biomacromolecules, 2019, 20, 1362–1374 CrossRef CAS PubMed.
- S. Malekkhaiat Häffner and M. Malmsten, Curr. Opin. Colloid Interface Sci., 2018, 38, 56–79 CrossRef.
- Z. Ye and C. Aparicio, Nanoscale Adv., 2019, 1, 4679–4682 RSC.
- F. Cao, G. Ma, M. Song, G. Zhu, L. Mei and Q. Qin, Soft Matter, 2021, 17, 4445–4451 RSC.
- A. P. McCloskey, B. F. Gilmore and G. Laverty, Pathogens, 2014, 3, 792–821 CrossRef PubMed.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS PubMed.
- L. Motiei, S. Rahimipour, D. A. Thayer, C. H. Wong and M. R. Ghadiri, Chem. Commun., 2009, 3693–3695 RSC.
- D. J. Schibli, R. F. Epand, H. J. Vogel and R. M. Epand, Biochem. Cell Biol., 2002, 80, 667–677 CrossRef CAS PubMed.
- S. E. Blondelle and R. A. Houghten, Trends Biotechnol., 1996, 14, 60–65 CrossRef CAS PubMed.
- Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS PubMed.
- E. H. M. Mohammed, S. Lohan, T. Ghaffari, S. Gupta, R. K. Tiwari and K. Parang, J. Med. Chem., 2022, 65, 15819–15839 CrossRef CAS PubMed.
- E. H. M. Mohammed, S. Lohan, R. K. Tiwari and K. Parang, Eur. J. Med. Chem., 2022, 235, 114278 CrossRef CAS PubMed.
- D. Oh, J. Sun, A. Nasrolahi Shirazi, K. L. LaPlante, D. C. Rowley and K. Parang, Mol. Pharmaceutics, 2014, 11, 3528–3536 CrossRef CAS PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- K. Fa, H. Liu, H. Gong, L. Zhang, M. Liao, X. Hu, D. Ciumac, P. Li, J. Webster, J. Petkov, R. K. Thomas and J. R. Lu, Langmuir, 2022, 38, 6623–6637 CrossRef CAS PubMed.
- R. Kowalczyk, P. W. R. Harris, G. M. Williams, S.-H. Yang and M. A. Brimble, Peptide Lipidation - a Synthetic Strategy to Afford Peptide Based Therapeutics, in Peptides and Peptide-Based Biomaterials and Their Biomedical Applications, ed. A. Sunna and P. L. Bergquist, Springer International Publishing, Cham, 2017, pp. 185–227 Search PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- A. Dehsorkhi, V. Castelletto and I. W. Hamley, J. Pept. Sci., 2014, 20, 453–467 CrossRef CAS PubMed.
- E. Kamysz, E. Sikorska, M. Jaśkiewicz, M. Bauer, D. Neubauer, S. Bartoszewska, W. Barańska-Rybak and W. Kamysz, Int. J. Mol. Sci., 2020, 21, 887 CrossRef CAS PubMed.
- D. A. Heesterbeek, B. W. Bardoel, E. S. Parsons, I. Bennett, M. Ruyken, D. J. Doorduijn, R. D. Gorham, E. T. Berends, A. L. Pyne, B. W. Hoogenboom and S. H. Rooijakkers, EMBO J., 2019, 38, e99852 CrossRef PubMed.
- A. Adak, V. Castelletto, A. de Sousa, K.-A. Karatzas, C. Wilkinson, N. Khunti, J. Seitsonen and I. W. Hamley, Biomacromolecules, 2024, 25, 1205–1213 CrossRef CAS PubMed.
- Z. Lai, H. Chen, X. Yuan, J. Tian, N. Dong, X. Feng and A. Shan, Front. microbiol., 2022, 13, 1–14 Search PubMed.
- C. J. C. Edwards-Gayle, V. Castelletto, I. W. Hamley, G. Barrett, F. Greco, D. Hermida-Merino, R. P. Rambo, J. Seitsonen and J. Ruokolainen, ACS Appl. Bio Mater., 2019, 2, 2208–2218 CrossRef CAS PubMed.
- S. Chou, H. Guo, F. G. Zingl, S. Zhang, J. Toska, B. Xu, Y. Chen, P. Chen, M. K. Waldor, W. Zhao, J. J. Mekalanos and X. Mou, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2219679120 CrossRef CAS PubMed.
- Y. Wan, Y. Yang, M. Wu and S. Feng, Expert Opin. Drug Delivery, 2022, 19, 1435–1448 CrossRef CAS PubMed.
- Z. Zhang, W. Shen, J. Ling, Y. Yan, J. Hu and Y. Cheng, Nat. Commun., 2018, 9, 1377 CrossRef PubMed.
- G. Rong, C. Wang, L. Chen, Y. Yan and Y. Cheng, Sci. Adv., 2020, 6, eaaz1774 CrossRef CAS PubMed.
- S. Vauthey, S. Santoso, H. Gong, N. Watson and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355–5360 CrossRef CAS PubMed.
- L. K. Vestby, T. Grønseth, R. Simm and L. L. Nesse, Antibiotics, 2020, 9, 59 CrossRef CAS PubMed.
- V. Castelletto, R. H. Barnes, K.-A. Karatzas, C. J. C. Edwards-Gayle, F. Greco, I. W. Hamley, R. Rambo, J. Seitsonen and J. Ruokolainen, Biomacromolecules, 2018, 19, 2782–2794 CrossRef CAS PubMed.
- A. Dehsorkhi, V. Castelletto, I. W. Hamley, J. Seitsonen and J. Ruokolainen, Langmuir, 2013, 29, 14246–14253 CrossRef CAS PubMed.
- I. W. Hamley, A. Dehsorkhi and V. Castelletto, Chem. Commun., 2013, 49, 1850 RSC.
- S. Debnath, A. Shome, D. Das and P. K. Das, J. Phys. Chem. B, 2010, 114, 4407–4415 CrossRef CAS PubMed.
- N. Chauhan and Y. Singh, ACS Biomater. Sci. Eng., 2020, 6, 5507–5518 CrossRef CAS PubMed.
- M. A. Sani and F. Separovic, Acc. Chem. Res., 2016, 49, 1130–1138 CrossRef CAS PubMed.
- D. Ciumac, H. Gong, X. Hu and J. R. Lu, J. Colloid Interface Sci., 2019, 537, 163–185 CrossRef CAS PubMed.
- K. L. Chen and G. D. Bothun, Environ. Sci. Technol., 2014, 48, 873–880 CrossRef CAS PubMed.
- C. Montis, P. Joseph, C. Magnani, A. Marín-Menéndez, F. Barbero, A. R. Estrada, R. Nepravishta, J. Angulo, A. Checcucci, A. Mengoni, C. J. Morris and D. Berti, Colloids Surf., B, 2020, 195, 111266 CrossRef CAS PubMed.
- H. Ilyas, M. J. A. Van Der Plas, M. Agnoletti, S. Kumar, A. K. Mandal, H. S. Atreya, A. Bhunia and M. Malmsten, Bioconjugate Chem., 2021, 32, 1729–1741 CrossRef CAS PubMed.
- S. E. Ayscough, L. A. Clifton, M. W. A. Skoda and S. Titmuss, J. Colloid Interface Sci., 2023, 633, 1002–1011 CrossRef CAS PubMed.
- J. E. Nielsen, V. R. Koynarev and R. Lund, Curr. Opin. Colloid Interface Sci., 2023, 66, 101709 CrossRef CAS.
- H. Gong, X. Hu, M. Liao, K. Fa, D. Ciumac, L. A. Clifton, M. A. Sani, S. M. King, A. Maestro, F. Separovic, T. A. Waigh, H. Xu, A. J. Mcbain and J. R. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 16062–16074 CrossRef CAS PubMed.
- H. Gong, X. Hu, L. Zhang, K. Fa, M. Liao, H. Liu, G. Fragneto, M. Campana and J. R. Lu, J. Colloid Interface Sci., 2023, 637, 182–192 CrossRef CAS PubMed.
- H. Gong, M. A. Sani, X. Hu, K. Fa, J. W. Hart, M. Liao, P. Hollowell, J. Carter, L. A. Clifton, M. Campana, P. Li, S. M. King, J. R. P. Webster, A. Maestro, S. Zhu, F. Separovic, T. A. Waigh, H. Xu, A. J. McBain and J. R. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 55675–55687 CrossRef CAS PubMed.
- S. Anselmo, G. Sancataldo, H. Mørck Nielsen, V. Foderà and V. Vetri, Langmuir, 2021, 37, 13148–13159 CrossRef CAS PubMed.
- B. P. Dyett, H. Yu, B. Lakic, N. De Silva, A. Dahdah, L. Bao, E. W. Blanch, C. J. Drummond and C. E. Conn, J. Colloid Interface Sci., 2021, 600, 14–22 CrossRef CAS PubMed.
- M. Hasan, F. Hossain, H. Dohra and M. Yamazaki, Biochem. Biophys. Res. Commun., 2022, 630, 50–56 CrossRef CAS PubMed.
- J. E. Nielsen, V. A. Bjørnestad, V. Pipich, H. Jenssen and R. Lund, J. Colloid Interface Sci., 2021, 582, 793–802 CrossRef CAS PubMed.
- N. Nuti, P. Rottmann, A. Stucki, P. Koch, S. Panke and P. S. Dittrich, Angew. Chem., Int. Ed., 2022, 61, e202114632 CrossRef CAS PubMed.
- S. M. Häffner, E. Parra-Ortiz, K. L. Browning, E. Jørgensen, M. W. A. Skoda, C. Montis, X. Li, D. Berti, D. Zhao and M. Malmsten, ACS Nano, 2021, 15, 6787–6800 CrossRef PubMed.
- R. Brasseur, M. Deleu, M. P. Mingeot-Leclercq, G. Francius and Y. F. Dufrêne, Surf. Interface Anal., 2008, 40, 151–156 CrossRef CAS.
- M. Radmacher, Methods Cell Biol., 2007, 83, 347–372 CAS.
- Y. F. Dufrêne, T. Ando, R. Garcia, D. Alsteens, D. Martinez-Martin, A. Engel, C. Gerber and D. J. Müller, Nat. Nanotechnol., 2017, 12, 295–307 CrossRef PubMed.
- F. Valle, J. A. DeRose, G. Dietler, M. Kawe, A. Plückthun and G. Semenza, Ultramicroscopy, 2002, 93, 83–89 CrossRef CAS PubMed.
- A. Ridolfi, M. Brucale, C. Montis, L. Caselli, L. Paolini, A. Borup, A. T. Boysen, F. Loria, M. J. C. Van Herwijnen, M. Kleinjan, P. Nejsum, N. Zarovni, M. H. M. Wauben, D. Berti, P. Bergese and F. Valle, Anal. Chem., 2020, 92, 10274–10282 CrossRef CAS PubMed.
- F. Perissinotto, V. Rondelli, B. Senigagliesi, P. Brocca, L. Almásy, L. Bottyán, D. G. Merkel, H. Amenitsch, B. Sartori, K. Pachler, M. Mayr, M. Gimona, E. Rohde, L. Casalis and P. Parisse, Nanoscale, 2021, 13, 5224–5233 RSC.
- F. Abbasi, J. Alvarez-Malmagro, Z. Su, J. J. Leitch and J. Lipkowski, Langmuir, 2018, 34, 13754–13765 CrossRef CAS PubMed.
- J. M. Henderson, A. J. Waring, F. Separovic and K. Y. C. Lee, Biophys. J., 2016, 111, 2176–2189 CrossRef CAS PubMed.
- V. K. Sharma and S. Qian, Langmuir, 2019, 35, 4152–4160 CrossRef CAS PubMed.
- Z. Shen, Z. Guo, L. Zhou, Y. Wang, J. Zhang, J. Hu and Y. Zhang, Biomater. Sci., 2020, 8, 2031–2039 RSC.
- V. Montana, W. Liu, U. Mohideen and V. Parpura, Croat. Chem. Acta, 2008, 81, 31–40 CAS.
- J. D. Unsay, K. Cosentino and A. J. García-Sáez, J. Visualized Exp., 2015, e52867 Search PubMed.
- S. Manioglu, S. M. Modaresi, N. Ritzmann, J. Thoma, S. A. Overall, A. Harms, G. Upert, A. Luther, A. B. Barnes, D. Obrecht, D. J. Müller and S. Hiller, Nat. Commun., 2022, 13, 6195 CrossRef CAS PubMed.
- E. H. L. Chen, C. H. Wang, Y. T. Liao, F. Y. Chan, Y. Kanaoka, T. Uchihashi, K. Kato, L. Lai, Y. W. Chang, M. C. Ho and R. P. Y. Chen, Nat. Commun., 2023, 14, 1–12 Search PubMed.
- T. O. Paiva, A. Viljoen and Y. F. Dufrêne, Nat. Commun., 2022, 13, 10–12 CrossRef PubMed.
- G. Benn, A. L. B. Pyne, M. G. Ryadnov and B. W. Hoogenboom, Analyst, 2019, 144, 6944–6952 RSC.
- H. Rudilla, A. Merlos, E. Sans-Serramitjana, E. Fuste, J. M. Sierra, A. Zalacain, T. Vinuesa and M. Vinas, AIMS Microbiol., 2018, 4, 522–540 CAS.
- H. Shahrour, R. Ferrer-Espada, I. Dandache, S. Bárcena-Varela, S. Sánchez-Gómez, A. Chokr and G. Martinez-de-Tejada, AMPs as Anti-biofilm Agents for Human Therapy and Prophylaxis, in Antimicrobial Peptides, ed. K. Matsuzaki, 2019, vol. 1117, pp. 257–279 Search PubMed.
- A. P. McCloskey, E. R. Draper, B. F. Gilmore and G. Laverty, J. Pept. Sci., 2017, 23, 131–140 CrossRef CAS PubMed.
- C. J. Harding, M. Bischoff, M. Bergkessel and C. M. Czekster, Nat. Chem. Biol., 2023, 19, 1158–1166 CrossRef CAS PubMed.
- S. Parhi, S. Pal, S. K. Das and P. Ghosh, Biotechnol. Bioeng., 2021, 118, 4590–4622 CrossRef CAS PubMed.
- A. K. Tripathi, J. Singh, R. Trivedi and P. Ranade, J. Funct. Biomater., 2023, 14, 539 CrossRef CAS PubMed.
- C. D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CrossRef CAS PubMed.
- P. Gagat, A. Duda-Madej, M. Ostrówka, F. Pietluch, A. Seniuk, P. Mackiewicz and M. Burdukiewicz, Int. J. Mol. Sci., 2023, 24, 804 CrossRef CAS PubMed.
- S. J. Lam, E. H. H. Wong, N. M. O'Brien-Simpson, N. Pantarat, A. Blencowe, E. C. Reynolds and G. G. Qiao, ACS Appl. Mater. Interfaces, 2016, 8, 33446–33456 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |