
Hydrophobic CPP/HDO conjugates: a new frontier in oligonucleotide-warheaded PROTAC delivery†
Miyako
Naganuma
 ab,
Nobumichi
Ohoka
*c,
Motoharu
Hirano
ab,
Daishi
Watanabe
ab,
Genichiro
Tsuji
a,
Takao
Inoue
c and
Yosuke
Demizu
*abd
ab,
Nobumichi
Ohoka
*c,
Motoharu
Hirano
ab,
Daishi
Watanabe
ab,
Genichiro
Tsuji
a,
Takao
Inoue
c and
Yosuke
Demizu
*abd
aDivision of Organic Chemistry, National Institute of Health Sciences, Kanagawa, Japan. E-mail: demizu@nihs.go.jp; Fax: +81 44 270 6578; Tel: +81 44 270 6578
bGraduate School of Medical Life Science, Yokohama City University, Kanagawa, Japan
cDivision of Molecular Target and Gene Therapy Products, National Institute of Health Sciences, Kanagawa, Japan. E-mail: n-ohoka@nihs.go.jp; Tel: +81 44 270 6537
dGraduate School of Medicine, Dentistry and Pharmaceutical Sciences, Division of Pharmaceutical Science of Okayama University, Japan
First published on 26th September 2024
Abstract
Proteolysis-targeting chimeras (PROTACs) have emerged as a potent strategy for inducing targeted degradation of proteins, offering promising therapeutic potential to treat diseases such as cancer. However, oligonucleotide-based PROTACs face significant delivery challenges because of their anionic nature and chemical instability. To address these issues, we developed a novel hydrophobic cell-penetrating peptide (CPP) and heteroduplex oligonucleotide (HDO)-conjugated PROTAC, CPP/HDO-PROTAC, to enhance intracellular delivery and degradation efficiency. CPP/HDO-PROTAC was designed to enter the cell through the activity of the conjugated hydrophobic CPP and release decoy oligonucleotide-based PROTACs by RNase H-mediated RNA strand breaks. Our findings demonstrated that CPP/HDO-PROTAC binds to the estrogen receptor α (ERα) with higher affinity than previous constructs, significantly degrades ERα in MCF-7 human breast cancer cells and inhibits cell proliferation at 10 μM. This research highlights the potential of CPP/HDO-PROTAC as a viable method for delivering and activating decoy oligonucleotide-based PROTACs within cells, overcoming the limitations of traditional transfection methods and paving the way for their clinical application.
1. Introduction
Over the past two decades, proteolysis-targeting chimeras (PROTACs) have become a potent strategy for addressing severe illnesses such as cancer.1 These molecules induce the targeted destruction of proteins through the ubiquitin-proteasome system, showing versatility by targeting non-enzymatic proteins, including transcription factors (TFs) and scaffold proteins.2 Building on the success of small molecule and peptide PROTACs designed for specific proteins, recent advances have generated decoy oligonucleotide-based PROTACs capable of targeting a wide range of TFs.3 In 2021, Wei et al. reported a decoy oligonucleotide-based PROTAC targeting the TFs NF-κB and E2F, TF-PROTAC.4 In the same year, an oligonucleotide PROTAC (O′-PROTAC), developed by Huang et al., was shown to degrade LEF1 and ERG and reported to be effective in vivo.5 In addition to decoy oligonucleotide-based PROTAC, several PROTACs using aptamers as warheads have been reported.3,6–9 We previously reported decoy oligonucleotide-based PROTACs targeting estrogen receptor α (ERα), introducing PROTACs that combine three E3 ligase ligands — LCL161 for inhibitor of apoptosis protein (IAP), VH032 for von Hippel–Lindau and pomalidomide for cereblon.10,11 Each variant (LCL-ER(dec), VH-ER(dec) and POM-ER(dec)) efficiently degraded ERα. In addition, we have modified decoy oligonucleotides to enhance the chemical stability of PROTAC. Our findings revealed that this modified PROTAC, incorporating a standard nucleic acid modification, is more resistant to nucleases and maintains ERα-degrading activity longer than its unmodified counterpart.11 Thus, in recent years, the development of several decoy oligonucleotide-based PROTACs has underscored their potential utility.2,3 However, the delivery of oligonucleotide-based PROTACs to the plasma membrane poses challenges because of their low chemical stability with native sequences and their anionic properties, which complicate transport. In addition, the delivery of all reported oligonucleotide-based PROTACs into cells necessitates the use of transfection reagents, presenting a significant obstacle in their clinical deployment.Nucleic acid therapeutics are gaining attention for their high therapeutic efficacy and specificity; however, efficient delivery poses a significant challenge. Developments in drug delivery systems (DDS) for these drugs, including antisense oligonucleotides (ASOs) and small interfering RNA (siRNA), aim to overcome this challenge and facilitate their practical application.12,13 In particular, it has been reported that double-stranded RNA has a lower tissue distribution and cellular uptake efficiency than single-stranded ASO. Therefore, strategies for siRNAs include formulation with polymers or lipid nanoparticles14 and binding to targeting ligands, including ligands for lipids and cell surface receptors. Although single-stranded ASOs can distribute to tissues and enter cells without special formulations or targeting ligands, reaching target mRNAs efficiently remains challenging. Ligand-conjugated ASOs have been explored to address this issue. Yokota et al. introduced heteroduplex oligonucleotides (HDOs) in 2015, comprising a DNA/LNA gapmer paired with its complementary RNA, enhanced by α-tocopherol as a DDS unit.12,15 This configuration increased the activity of the gapmer-type ASO over single-stranded ASOs significantly by facilitating lipid release from the oligonucleotides for targeted mRNA delivery.
In this study, we introduced a cell-penetrating peptide (CPP) and heteroduplex oligonucleotide (HDO)-conjugated PROTAC (CPP/HDO-PROTAC) strategy, applying an CPP/HDO to enhance the delivery of a decoy oligoncleotide-based PROTAC into cells. This innovation enabled successful intracellular delivery and degradation of the target protein ERα, eliminating the need for transfection reagents. The CPP/HDO-PROTAC, a novel molecule, combines a 30-mer DNA linked to an LCL161 derivative (LCL-HDO-F) and a 30-mer DNA with a 9-mer RNA (P4-HDO-R) attached to a hydrophobic CPP (Fig. 1A and B). This design allows the CPP/HDO-PROTAC to enter cells via the hydrophobic CPP, P4 peptide (LGAQSNF), and the potential release of the decoy oligonucleotide-based PROTAC through RNase H-mediated RNA strand cleavage and CPP detachment. The P4 peptide was discovered through screening a 7-mer phage display peptide library for Duchenne muscular dystrophy treatment, aiming to deliver ASOs to targeted tissues.16–19 Given the aggregation problems posed by the anionic nature of oligonucleotides when combined with cationic CPPs, the strategy shifted towards employing CPPs made of hydrophobic residues to facilitate efficient conjugation and delivery. The study assessed the effectiveness of the CPP/HDO-PROTAC in RNase H-mediated cleavage, cellular uptake efficiency, ERα degradation activity and inhibition of cell proliferation.
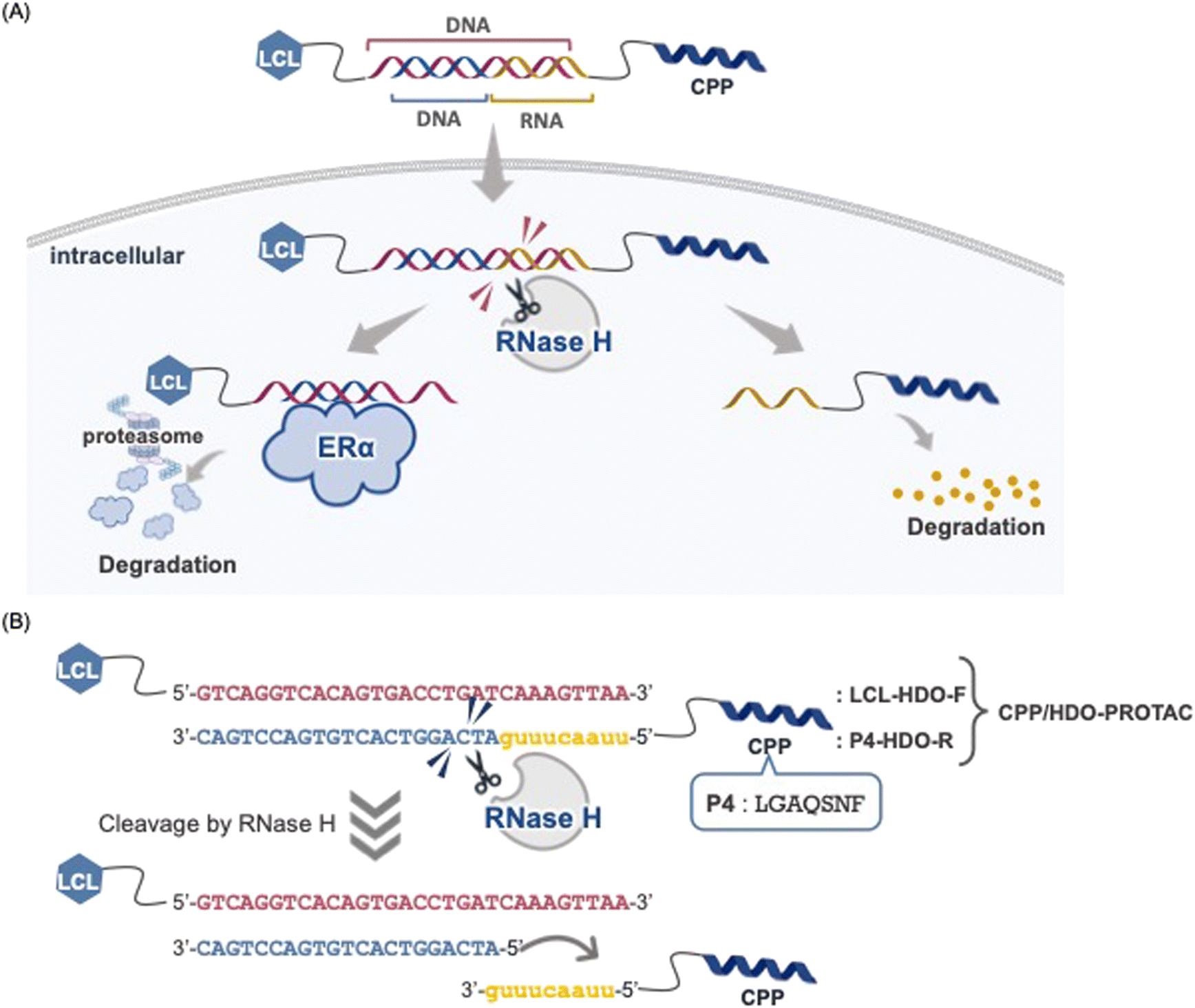 | ||
| Fig. 1 (A) Schematic of the CPP/HDO-PROTAC responsible for degrading ERα. (B) Detailed sequence of the CPP/HDO-PROTAC. | ||
2. Results and discussion
Based on the estrogen response element, a sequence for ER(dec) was extended by nine bases, and a sense strand with the sequence 5′-GTCAGGTCACAGTGACCTGATCAAAGTTAA-3′ was designed.10,20 Additionally, an HDO-F modified with a hexynyl group at the 5′ end was created for this sequence.LCL-HDO-F was synthesized by conjugating HDO-F and LCL161 (ref. 21) with an azidized PEG3 linker via a copper-catalyzed click reaction. In contrast, for the antisense strand, HDO-R was designed by modifying the 5′ end with a hexynyl group for the sequence containing nine bases of RNA (5′-uuaacuuugATCAGGTCACTGTGTGACCTGAC-3′). HDO-R was conjugated with the P4 peptide via a copper-catalyzed click reaction to afford P4-HDO-R (Fig. 2 and Scheme 1). The synthesized LCL-HDO-F and P4-HDO-R were mixed and annealed by heating from room temperature to 95 °C and returning to room temperature to prepare CPP/HDO-PROTAC. In addition, an antisense strand HDOdna-R (5′-TTAACTTTGATCAGGTCACTGTGACCTGAC-3′) was designed as a negative control, in which all complementary strands of HDO-F were replaced with DNA sequences. LCL-HDO-F and HDOdna-R were annealed in a similar manner to generate LCL-HDOdna, and LCL-HDOcl was annealed with HDO-F to generate ER(dec)-R when the RNA strand was cleaved.
 | ||
| Fig. 2 Decoy-warheaded PROTACs and related molecules used in this study. | ||
 | ||
| Scheme 1 Synthesis of (A) LCL-HDO-F and (B) P4-HDO-R. | ||
The binding affinities of LCL-ER(dec), LCL-HDOcl, LCL-HDOdna and CPP/HDO-PROTAC against ERα were evaluated by a competitive fluorescence polarization (FP) assay. For evaluation, compounds were added to a buffer system containing recombinant ERα and the FP-probe, a FAM-labeled ER(dec) at the 5′-end and 3′-end, and FP was measured (Fig. S5†).10 The half inhibitory concentration (IC50) values of the compounds were determined by calculating, which were 148 nM for LCL-ER(dec), 24.6 nM for LCL-HDOcl and 1.2 nM for LCL-HDOdna (Table 1 and Fig. S5†). The increased binding affinity of LCL-HDOcl and LCL-HDOdna toward ERα may be attributed to additional interaction points provided by their longer nucleotide lengths. The CPP/HDO-PROTAC also exhibited an IC50 value of 3.0 nM, indicating that the binding activity is preserved even after conjugation with the P4 peptide sequence.
| Entry | Compound | IC50 (nM) |
|---|---|---|
| 1 | LCL-ER(dec) | 148.0 ± 10.9 |
| 2 | LCL-HDOcl | 24.6 ± 3.93 |
| 3 | LCL-HDOdna | 1.2 ± 0.98 |
| 4 | CPP/HDO-PROTAC | 3.0 ±0.45 |
The susceptibility of the HDO to RNase H-mediated cleavage was assessed by conducting experiments using a recombinant RNase H enzyme. After 2 h RNase H treatment, the decoys were analyzed using 20% denaturing polyacrylamide gel electrophoresis. The findings indicated that RNase H specifically degraded RNA in CPP/HDO-PROTAC and LCL-HDO with DNA–RNA heteroduplexes, leaving LCL-HDO-F, HDO-R, LCL-HDOdna and ER(dec)-R intact (Fig. 3). Therefore, it was hypothesized that CPP/HDO-PROTAC and LCL-HDO, upon cellular introduction, should be effectively targeted by RNase H, resulting in the degradation of the RNA component.
 | ||
| Fig. 3 Evaluation of HDO cleavage by RNase H. CPP/HDO-PROTAC, LCL-HDO, LCL-HDO-F, P4-HDO-R, LCL-HDOdna and ER(dec)-R were incubated with RNase H for 2 h. Full-length and digested oligonucleotides were resolved on 20% denaturing polyacrylamide gels. | ||
The ERα degradation activity of CPP/HDO-PROTAC was evaluated by western blotting in MCF-7 cells. CPP/HDO-PROTAC and a comparator, LCL-HDOdna, were added to cells, and the expression levels of ERα were evaluated after 24 h treatment. The results showed that CPP/HDO-PROTAC exhibited ERα degradation activity at 10 μM, suggesting that CPP/HDO-PROTAC penetrates cells and functions as a PROTAC without requiring transfection reagents (Fig. 4a). The ERα-degrading activity was also observed in the LCL-ER(dec) + P4 co-treatment group at a concentration of 10 μM, even in the absence of transfection (Fig. 4b). At high concentrations, some of LCL-ER(dec) might have penetrated the cell membrane along with the hydrophobic CPP P4 and been taken up into the cell. Conversely, ERα was not degraded when adding only P4, indicating P4 does not enable ERα degradation (Fig. 4b). By contrast, LCL-ER(dec)-P4, which was directly conjugated with P4 to LCL-ER(dec), showed little ERα degrading activity (Fig. S6†).
 | ||
| Fig. 4 Degradation of ERα by the synthesized PROTACs. Whole-cell lysates were analyzed by western blotting with the indicated antibodies (representative data are shown). The numbers below the ERα panels represent the ERα/β-actin ratios, normalized by designating the expression using the vehicle control (condition without a PROTAC) as 100%. (a) Degradation of ERα by CPP/HDO-PROTAC. MCF-7 cells were treated for 24 h with the indicated concentrations of CPP/HDO-PROTAC and LCL-HDOdna. (b) Degradation of ERα by LCL-ER(dec). MCF-7 cells were transiently transfected for 24 h with the indicated concentrations of LCL-ER(dec). An LCL-ER(dec) + P4 mixture or P4 alone was added to MCF-7 cells for 24 h. | ||
High ERα expression, associated with poor prognosis and increased cell proliferation in breast cancer. Under transfection conditions, all PROTACs showed concentration-dependent inhibitory activity (Fig. S7†). In contrast, under non-transfection conditions, inhibitory activity was observed only for CPP/HDO-PROTAC (Fig. 5 and S8†). LCL-ER(dec) showed slight inhibitory activity at 10 μM, possibly because its sequence is shorter and its molecular weight is smaller than that of LCL-HDOdna, LCL-HDO, and LCL-HDOcl, therefore allowing partial cell penetration and activity at high concentrations. These results suggest that CPP/HDO-PROTAC was capable of efficiently entering cells and exerting its inhibitory effect even under non-transfection conditions, due to the presence of the P4 sequence in CPP.
 | ||
| Fig. 5 Effects of CPP/HDO-PROTAC on the proliferation of ERα-positive breast cancer cells. Growth inhibition of ERα-positive breast cancer cells by CPP/HDO-PROTAC. MCF-7 cells were treated with 1–10 μM of CPP/HDO-PROTAC or LCL-HDOdna for 72 h, and cell proliferation was then evaluated using a cell viability assay. Data represent the mean ± standard deviation (n = 5). | ||
Intracellular transduction was observed using fluorescence microscopy with HDO-fluorescently labeled decoys; cells treated with FAM-HDO-P4 exhibited brighter fluorescence than those treated with FAM-HDOdna (Fig. 6). After 2 h treatment with FAM-HDO-P4, fluorescence exhibited a point-like distribution and co-localized with the endosomal/lysosomal marker LysoTracker Red, indicating that most internalized decoys remained within endosomes or lysosomes.
 | ||
| Fig. 6 Fluorescence microscopy images of MCF-7 cells treated with 10 μM decoy for 2 h. Nuclei and endosomes/lysosomes were stained by Hoechst 33342 and LysoTracker Red, respectively. The fluorescent microscopy observations were performed with a 100× objective lens. Scale bar: 20 μm. | ||
3. Conclusions
Decoy oligonucleotide-based PROTACs offer a novel strategy for targeting proteins where suitable small-molecule ligands are unavailable. Specifically, these PROTACs are highly promising for targeting TFs as their design leverages the motif sequences of TF DNA-binding domains. This method facilitates the development of PROTACs capable of targeting an extensive array of TFs. However, delivering all reported decoy oligonucleotide-based PROTACs into cells currently relies on transfection agents, highlighting the need for DDS technology integration to advance their pharmaceutical application. Furthermore, the use of native oligonucleotide-based decoy PROTACs face challenges related to chemical instability, necessitating substantial enhancement for their use in pharmaceutical applications. To address this issue, we developed HDO-PROTAC, adapting the HDO used for ASO delivery to decoy oligonucleotide-based PROTACs. We designed CPP/HDO-PROTAC based on the previously developed ERα-targeted lead sequence LCL-ER(dec). CPP/HDO-PROTAC consists of LCL-HDO-F with LCL at the 5′ end of a 30-mer DNA and its complementary strand P4-HDO-R. P4-HDO-R was designed by extending the ER(dec)-R sequence with a 9-mer RNA and attaching the hydrophobic CPP P4 to the 5′-end. Although general cationic CPPs have difficulty conjugating with anionic nucleic acids, the P4 used in this study is a hydrophobic peptide and its conjugates could be synthesized through a click reaction.CPP/HDO-PROTAC has been proposed to be susceptible to RNase H-mediated degradation in vitro, but RNase H only cleaves double-stranded DNA and RNA and not other single or double strands. The binding activity of ERα by the assumed post-RNase H degradation product, LCL-HDOcl, and the control, LCL-HDOdna, demonstrated enhanced binding compared with LCL-ER(dec). Evaluation of the ERα degradation activity of CPP/HDO-PROTAC revealed that CPP/HDO-PROTAC was effective at a concentration of 10 μM. Furthermore, the ability of CPP/HDO-PROTAC to inhibit MCF-7 cell proliferation indicates it may suppress estrogen signaling. This study confirms that intracellular delivery of decoy oligonucleotide-based PROTACs is possible by combining the hydrophobic CPP P4 with an HDO, allowing for the first time the transfection-independent introduction of PROTACs into cells. Improving transport efficiency and tissue specificity is vital for unlocking the therapeutic potential of these PROTACs. The introduction of artificial nucleic acids that increase the chemical stability of the RNA strand is expected to improve its stability after intracellular transport. Continued efforts in DDS optimization are essential for their successful clinical application.
4. Experimental section
General chemistry methods
All chemicals were purchased from Sigma-Aldrich, FUJIFILM Wako Pure Chemicals, and Tokyo Chemical Industry, and were used without further purification. High-resolution mass spectra were measured using a Shimadzu IT-TOF MS equipped with an electrospray ionization source.Oligonucleotide synthesis
Decoys were synthesized on a 1.0 μmol scale using an automated DNA synthesizer (T-Series, Nihon Techno Service) by means of phosphoramidite chemistry with 5′-dimethoxytrityl-2′-deoxynucleoside phosphoramidites (Glen Research). To synthesize a sequence containing a hexynyl group at the 5′ end, synthesis continued according to the standard protocol and 5′-hexynyl phosphoramidite (Glen Research) was used in the last coupling step to introduce a hexynyl group at the 5′-terminus. To synthesize fluorescein-labeled oligonucleotides, synthesis continued according to the standard protocol and 6-Fluorescein phosphoramidite (Sigma-Aldrich) was used in the last coupling step to introduce FAM dye at the 5′-terminus. DNA on solid support (CPG) were treated with 28% ammonium hydroxide for 15 h at 55 °C and concentrated in vacuo. RNA Deprotected decoys were purified using reverse phase HPLC. HPLC conditions were as follows; column: CAPCELL PAK MG-II (C18, 10 × 250 mm, 5 μm; OSAKA soda), mobile phase: A = 0.1 M triethylammonium acetate (TEAA) buffer (pH 7.0), B = CH3CN. Gradient: B% = 10–40 over 20 min, 40–100 over 5 min. Flow rate: 1 mL min−1, detection: 260 nm, column temperature: 35 °C. Concentration of the solution of decoys was determined using NanoDrop OneC (Thermo Fisher Scientific) by absorbance at 260 nm and molar absorbance coefficient (ε260) calculated from OligoAnalyzer (IDT).Peptide synthesis
The peptides were synthesized by Fmoc-based solid-phase methods. A representative coupling and deprotection cycle are described as follows. Rink Amide ChemMatrix resin was soaked for 30 min in CH2Cl2. After the resin had been washed with DMF, the Fmoc protecting group was removed by treatment with 25% piperidine in DMF for 15 min at room temperature. Amino acids were coupled for 1 hour using 4 equiv. of Fmoc-protected amino acid, 4 equiv. of (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholinocarbenium hexafluorophosphate (COMU) as the activating agent, and 8 equiv. of diisopropylethylamine (DIPEA) in NMP.The peptide was suspended in cleavage cocktail (95% TFA, 2.5% water, 2.5% triisopropylsilane) at 42 °C for 30 min on Razor (CEM corp.) to cleave from the resin. TFA was evaporated to a small volume under a stream of N2 and dripped into cold ether to precipitate the peptide. The peptides were dissolved in dimethyl sulfoxide and purified using reverse-phase HPLC (Waters) using a Discovery® BIO Wide Pore C18 column (Supelco, Bellefonte, PA, USA) (25 cm × 21.2 mm solvent A: 0.1% TFA/water, solvent B: 0.1% TFA/CH3CN, flow rate: 10.0 mL min−1, gradient: 10–90% gradient of solvent B over 40 min). After purification, the peptide solutions were lyophilized, and peptide purity was assessed using UPLC (Waters) and a ACQUITY UPLC® BEH C18 1.7 μm column (2.1 × 50 mm; solvent A: 0.1% TFA/water, solvent B: 0.1% TFA/CH3CN, flow rate: 0.5 mL min−1, gradient: 10–90% gradient of solvent B over 4 min).
Synthesis of chimeric molecules by click reaction
To a solution of IAP ligand (LCL-PEG3-N3) in MeOH (10 mM, 168 μL, 1.68 μmol) was added the ERα-binding decoy 5′-hexynyl-HDO-F in H2O (1.6 mM, 500 μL, 0.8 μmol), TEA in DMSO (32 mM, 250 μL, 8 μmol), and CuIP(OEt)3 in DMSO (35.8 mM, 250 μL, 11.2 μmol). The mixture was incubated at 40 °C for 3 h. The reaction mixture was purified using HPLC and lyophilized. The collected oligo was precipitated in a solution of 0.3 M sodium acetate (pH 5.2)/70% ethanol to give the single strand of LCL-HDO-F, which was resuspended in nuclease-free water to prepare a stock solution (1 mM, 232 μL, 0.232 μmol, 29%).To a solution of decoys in water and DMSO (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) were added an azide-peptide (3 equiv.), CuSO4 (10 equiv.), sodium ascorbate (5 equiv.), and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (10 equiv.). The mixture was incubated at 45 °C for 2 h. The reaction mixture was purified using HPLC and lyophilized. The collected oligo was precipitated in a solution of 0.3 M sodium acetate (pH 5.2)/70% ethanol to give the single strand of P4-HDO-R, which was resuspended in nuclease-free water to prepare a stock solution.
:3) were added an azide-peptide (3 equiv.), CuSO4 (10 equiv.), sodium ascorbate (5 equiv.), and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (10 equiv.). The mixture was incubated at 45 °C for 2 h. The reaction mixture was purified using HPLC and lyophilized. The collected oligo was precipitated in a solution of 0.3 M sodium acetate (pH 5.2)/70% ethanol to give the single strand of P4-HDO-R, which was resuspended in nuclease-free water to prepare a stock solution.
Finally, LCL-HDO-F and the corresponding antisense strand (P4-HDO-R) was mixed in an aqueous solution and hybridized by heating at 95 °C for 10 min and gradually returning to room temperature to give the double-stranded title compound CPP/HDO-PROTAC.
Abbreviations
| PROTAC | Proteolysis targeting chimera |
| IAP | Inhibitor of apoptosis protein |
| TF | Transcription factor |
| ERα | Estrogen receptor α |
| FAM | Fluorescein |
| DDS | Drug delivery systems |
| CPP | Cell-penetrating peptide |
| TEA | Triethylamine |
| DMF | N,N-Dimethylformamide |
Data availability
Raw data were generated at National Institute of Health Sciences. Derived data supporting the findings of this study are available from the corresponding author Y. D. on request.Author contributions
M. N., N. O., M. H., D. W., and G. T. performed the experiments and analyzed results. M. N., N. O., T. I., and Y. D. designed the research and wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This study was supported in part by grants from AMED under grant numbers 24mk0121286, 24ama221127, and 24ak0101185 (all to Y. D.); 24ak0101186 (to T. I. and N. O.); 24fk0310504 (to N. O.). The study also received support from the Japan Society for the Promotion of Science (KAKENHI, grants 21K05320 and 23H04926, both to Y. D.; 18K06567 and 21K06490, both to N. O.) and JSPS Fellows 21J23036 to Miyako Naganuma. We thank Edanz (https://www.jp.edanz.com/ac) for editing a draft of this manuscript.References
- K. Li and C. M. Crews, PROTACs: past, present and future, Chem. Soc. Rev., 2022, 51(12), 5214–5236, 10.1039/D2CS00193D.
- W. Wang, S. He, G. Dong and C. Sheng, Nucleic-Acid-Based Targeted Degradation in Drug Discovery, J. Med. Chem., 2022, 65(15), 10217–10232, DOI:10.1021/acs.jmedchem.2c00875.
- Y. Liu, X. Qian, C. Ran, L. Li, T. Fu, D. Su, S. Xie and W. Tan, Aptamer-Based Targeted Protein Degradation, ACS Nano, 2023, 17(7), 6150–6164, DOI:10.1021/acsnano.2c10379.
- J. Liu, H. Chen, H. U. Kaniskan, L. Xie, X. Chen, J. Jin and W. Y. Wei, TF-PROTACs Enable Targeted Degradation of Transcription Factors, J. Am. Chem. Soc., 2021, 143(23), 8902–8910, DOI:10.1021/jacs.1c03852.
- Y. Yan, J. Shao, D. Ding, Y. Pan, P. Tran, W. Yan, Z. Wang, H. Y. Li and H. Huang, 3-Aminophthalic acid, a new cereblon ligand for targeted protein degradation by O'PROTAC, Chem. Commun., 2022, 58(14), 2383–2386, 10.1039/d1cc06525d.
- L. Zhang, L. Li, X. Wang, H. Liu, Y. Zhang, T. Xie, H. Zhang, X. Li, T. Peng and X. Sun, et al., Development of a novel PROTAC using the nucleic acid aptamer as a targeting ligand for tumor selective degradation of nucleolin, Mol. Ther. – Nucleic Acids, 2022, 30, 66–79, DOI:10.1016/j.omtn.2022.09.008.
- H. Tsujimura, M. Naganuma, N. Ohoka, T. Inoue, M. Naito, G. Tsuji and Y. Demizu, Development of DNA Aptamer-Based PROTACs That Degrade the Estrogen Receptor, ACS Med. Chem. Lett., 2023, 14(6), 827–832, DOI:10.1021/acsmedchemlett.3c00126.
- Y. Wang, G. Yang, X. Zhang, R. Bai, D. Yuan, D. Gao, Q. He, Y. Yuan, X. Zhang and J. Kou, et al., Antitumor Effect of Anti-c-Myc Aptamer-Based PROTAC for Degradation of the c-Myc Protein, Adv. Sci., 2024, 11(26), e2309639, DOI:10.1002/advs.202309639.
- L. Kong, F. Meng, P. Zhou, R. Ge, X. Geng, Z. Yang, G. Li, L. Zhang, J. Wang and J. Ma, et al., An engineered DNA aptamer-based PROTAC for precise therapy of p53-R175H hotspot mutant-driven cancer, Sci. Bull., 2024, 69(13), 2122–2135, DOI:10.1016/j.scib.2024.05.017.
- M. Naganuma, N. Ohoka, G. Tsuji, H. Tsujimura, K. Matsuno, T. Inoue, M. Naito and Y. Demizu, Development of Chimeric Molecules That Degrade the Estrogen Receptor Using Decoy Oligonucleotide Ligands, ACS Med. Chem. Lett., 2022, 13(1), 134–139, DOI:10.1021/acsmedchemlett.1c00629.
- M. Naganuma, N. Ohoka, G. Tsuji, T. Inoue, M. Naito and Y. Demizu, Structural Optimization of Decoy Oligonucleotide-Based PROTAC That Degrades the Estrogen Receptor, Bioconjugate Chem., 2023, 34(10), 1780–1788, DOI:10.1021/acs.bioconjchem.3c00332.
- K. Nishina, W. Piao, K. Yoshida-Tanaka, Y. Sujino, T. Nishina, T. Yamamoto, K. Nitta, K. Yoshioka, H. Kuwahara and H. Yasuhara, et al., DNA/RNA heteroduplex oligonucleotide for highly efficient gene silencing, Nat. Commun., 2015, 6, 7969, DOI:10.1038/ncomms8969.
- S. M. Hammond, A. Aartsma-Rus, S. Alves, S. E. Borgos, R. A. M. Buijsen, R. W. J. Collin, G. Covello, M. A. Denti, L. R. Desviat and L. Echevarria, et al., Delivery of oligonucleotide-based therapeutics: challenges and opportunities, EMBO Mol. Med., 2021, 13(4), e13243, DOI:10.15252/emmm.202013243.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda and J. K. Narayanannair, et al., Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo, Angew. Chem., Int. Ed., 2012, 51(34), 8529–8533, DOI:10.1002/anie.201203263.
- H. Kaburagi, T. Nagata, M. Enomoto, T. Hirai, M. Ohyagi, K. Ihara, K. Yoshida-Tanaka, S. Ebihara, K. Asada and H. Yokoyama, et al., Systemic DNA/RNA heteroduplex oligonucleotide administration for regulating the gene expression of dorsal root ganglion and sciatic nerve, Mol. Ther. – Nucleic Acids, 2022, 28, 910–919, DOI:10.1016/j.omtn.2022.05.006.
- S. M. Jirka, H. Heemskerk, C. L. Tanganyika-de Winter, D. Muilwijk, K. H. Pang, P. C. de Visser, A. Janson, T. G. Karnaoukh, R. Vermue and P. A. t Hoen, et al., Peptide conjugation of 2′-O-methyl phosphorothioate antisense oligonucleotides enhances cardiac uptake and exon skipping in mdx mice, Nucleic Acid Ther., 2014, 24(1), 25–36, DOI:10.1089/nat.2013.0448.
- A. Gonzalez-Barriga, B. Nillessen, J. Kranzen, I. D. G. van Kessel, H. J. E. Croes, B. Aguilera, P. C. de Visser, N. A. Datson, S. A. M. Mulders and J. C. T. van Deutekom, et al., Intracellular Distribution and Nuclear Activity of Antisense Oligonucleotides After Unassisted Uptake in Myoblasts and Differentiated Myotubes In Vitro, Nucleic Acid Ther., 2017, 27(3), 144–158, DOI:10.1089/nat.2016.0641.
- D. Honcharenko, K. Druceikaite, M. Honcharenko, M. Bollmark, U. Tedebark and R. Stromberg, New Alkyne and Amine Linkers for Versatile Multiple Conjugation of Oligonucleotides, ACS Omega, 2021, 6(1), 579–593, DOI:10.1021/acsomega.0c05075.
- S. M. G. Jirka, P. A. C. t Hoen, V. Diaz Parillas, C. L. Tanganyika-de Winter, R. C. Verheul, B. Aguilera, P. C. de Visser and A. M. Aartsma-Rus, Cyclic Peptides to Improve Delivery and Exon Skipping of Antisense Oligonucleotides in a Mouse Model for Duchenne Muscular Dystrophy, Mol. Ther., 2018, 26(1), 132–147, DOI:10.1016/j.ymthe.2017.10.004.
- V. Kumar and P. Chambon, The Estrogen-Receptor Binds Tightly To Its Responsive Element As A Ligand-Induced Homodimer, Cell, 1988, 55(1), 145–156, DOI:10.1016/0092-8674(88)90017-7.
- N. Ohoka, K. Okuhira, M. Ito, K. Nagai, N. Shibata, T. Hattori, O. Ujikawa, K. Shimokawa, O. Sano and R. Koyama, et al., In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs), J. Biol. Chem., 2017, 292(11), 4556–4570, DOI:10.1074/jbc.M116.768853.
Footnote |
| † Electronic supplementary information (ESI) available: The synthetic procedures for all compounds listed in this manuscript and the protocols for the in vitro assays (binding and protein degradation assays) (PDF). See DOI: https://doi.org/10.1039/d4md00546e |
| This journal is © The Royal Society of Chemistry 2024 |