DOI:
10.1039/D4MD00543K
(Research Article)
RSC Med. Chem., 2024,
15, 4180-4192
Strong in vitro anticancer activity of copper(II) and zinc(II) complexes containing naturally occurring lapachol: cellular effects in ovarian A2780 cells†‡
Received
15th July 2024
, Accepted 29th August 2024
First published on 9th September 2024
Abstract
Copper(II) and zinc(II) complexes with lapachol (HLap) of the composition [M(Lap)2(N–N)] and [Cu(Lap)(H2O)(terpy)]NO3 (4), where M = Cu (1–3) or Zn (for 5–7), and N–N stands for bathophenanthroline (1 and 5), 5-methyl-1,10-phenanthroline (2 and 6), 2,2′-bipyridine (3), 2,2′;6′,2′′-terpyridine (terpy, 4) and 1,10-phenanthroline (7), were synthesised and characterised. Complexes 1–5 revealed strong in vitro antiproliferative effects against A2780, A2780R, MCF-7, PC-3, A549 and HOS human cancer lines and MRC-5 normal cells, with IC50 values above 0.5 μM, and reasonable selectivity index (SI), with SI > 3.8 for IC50(MRC-5)/IC50(A2780). Considerable time-dependent cytotoxicity in A2780 cells was observed for complexes 6 and 7, with IC50 > 50 μM (24 h) to ca. 4 μM (48 h). Cellular effects of complexes 1, 5 and 7 in A2780 cells were investigated by flow cytometry revealing that the most cytotoxic complexes (1 and 5) significantly perturbed the mitochondrial membrane potential and the interaction with mitochondrial metabolism followed by the triggering of the intracellular pathway of apoptosis.
Introduction
Lapachol (HLap), 2-hydroxy-3-(3-methylbut-2-en-1-yl)naphthalene-1,4-dione, has long been known as a naturally occurring organic compound that can be isolated from the bark of the lapacho tree. Chemically, it contains a 1,4-naphthoquinone moiety, similar to the fat-soluble vitamin K (see Scheme 1).
 |
| | Scheme 1 The structural formulas of lapachol (HLap) used as a proligand for the preparation of heteroleptic Cu(II) and Zn(II) complexes 1–7 in this study, together with vitamin K, showing the structural similarity of both systems. | |
Due to the mentioned moiety's presence in its structure, lapachol reveals a broad spectrum of interesting biological activities associated with its ability to interfere with enzymes critical for DNA replication in cells and/or to induce the production of the so-called reactive oxygen species (ROS). Thus, lapachol itself is related to a wide spectrum of therapeutic effects, such as antiseptic, antileishmanial, anticancer, antiedemic, anti-inflammatory, antiviral, bactericidal, fungicidal, etc. These features have been reviewed by Hussain et al. in ref. 1. As for the anticancer activity of lapachol, several authors have used different human cancer cell lines showing diverse IC50 values, depending on the incubation times. For instance, S. Fiorito et al. published data,2 where IC50 ≈ 8 μM on U373, A549, Hs683, SKMEL-28, PC3 and LoVo cancer cells after 72 h of incubation, while Atolani et al. found IC50 = 19.04, 20.51 and 6.15 μg mL−1 on HeLa, PC3, and 3T3, respectively, after 72 h of incubation.3 On the other hand, lapachol displayed an IC50 value of 25 μM on HL60 cells following 48 h of incubation.4 Overall, positive findings regarding the in vitro anticancer features of lapachol have been reviewed by E. R. de Almeida and by F. Epifano et al.5 Moreover, it has been proposed that 1,4-naphthoquinones interfere with biological electron-transfer processes, and the redox potential is the most important physicochemical property tuning the antitumour activity of a series of quinones, including lapachol. Lapachol is also a strong inhibitor of glyoxalase I, an enzyme which has been suggested as a target for anticancer drug action. These properties and an apparent steric requirement of planarity are important for effective inactivators of the small protein. Moreover, lapachol is a feasible ligand for the preparation of coordination or organometallic compounds.6 Interesting cytotoxic properties have been reported for lapachol complexes, including Ru(II), Co(III), and Cu(II) complexes. In fact, there are some findings showing that lapachol–metal complexes are biologically more active than the free molecule.7 Particularly, ruthenium complexes are quite promising types of metal compounds for cancer treatment due to their ability in ligand exchange (i.e. kinetic lability), octahedral geometry and variability of oxidation states.8 Different structures of Ru-based complexes were also investigated in this context by Barbosa et al.,9 such as [RuII(Lap)(PPh3)2(bpy)]PF6, [RuII(Lap)(PPh3)2(Me-bpy)]PF6·MeOH, [RuII(Lap)(PPh3)2(MeO-bpy)]PF6, [RuII(Lap)(PPh3)2(phen)]PF6 and [RuIIICl2(Lap)(dppb)], where dppb stands for 1,4-bis(diphenylphosphine)butane. The latter exhibited activity against promastigotes, becoming the most potent and selective antiparasitic compound among the five ruthenium compounds mentioned above. Other ruthenium(II) naphthoquinone derivatives with p-cymene as the ligand were synthesised and tested,10 highlighting a higher selectivity against some of the cell lines. Notably, the complex [Ru(η6-p-cymene)(Lap)(PTA)](PF6) showed the best cytotoxic effect with IC50 values even better than those determined for cisplatin against HepG-2, MCF-7, LoVo, A2780 and HCT-8 cell lines. The recently reported Ru(II)–diphosphine complex (i.e. [Ru(Lap)(dppm)2]PF6), where dppm stands for 1,1-bis(diphenylphosphino)methane), showed an anti-melanoma effect.11 Overall, the mechanisms underlying the antitumor activity of all the complexes probably include interaction with DNA and the induction of apoptosis.
Moving to selected 3d metals, iron, copper, and zinc are essential elements for most aerobic organisms, which employ them as principal constituents of thousands of enzymes, playing both a structural and a catalytic role. Iron can participate in several biological processes, such as respiration, photosynthesis,12 N2 fixation13 and H2 metabolism.14 In particular, the main role of Fe is for dioxygen transport during respiration operated by myoglobin and haemoglobin, where iron is surrounded by the heme group. In biological systems, zinc can be tetra-, penta- or hexacoordinated to N, O or S donor atoms comprising histidine, glutamate/aspartate, and cysteine residues, or to water molecules.15 Due to these elements' role in different biological pathways, it is not surprising that they are crucial for increasing cell survival. Notably, their complexes have shown a broad spectrum of anticancer activities demonstrating good selectivity and low toxicity, and exhibit mechanisms of action different from those of platinum metallodrugs. A new coordination complex containing lapachol and an Fe(II) atom was synthesised and characterised ([Fe(Lap)2(H2O)2]), showing the coordination of lapachol to the Fe(II) central atom in a keto–enolic way.16 The complex has been shown to be effective against the tumor line of glioma (U251). Copper and zinc have a long history of medical application, but their potential antitumor properties have boosted dramatically in the past few decades as can be demonstrated by a search within the Web of Science database (http://www.isiknowledge.com) using a combination of suitable keywords localised in titles of published papers, i.e. “copper” and “cancer” and “review”, and references given therein, where the significance of copper and zinc complexes in this field can be seen. These metals have been shown to possess a broad spectrum of activities and relatively low toxicity and are suggested to be able to overcome the resistance to cisplatin. Cu(II) and Zn(II) complexes of lapachol derivatives are known and well-characterized in the literature.17,18 In 2017, Cu(II), Co(II) and Ni(II) complexes of lapachol, containing the 1,10-phenanthroline ligand and having the composition of [M(Lap)2(phen)], were reported by L. Tabrizi et al.19 and their cytotoxicity was also investigated. The results suggested that the complexes could interact with DNA via the intercalation mode. All three complexes possess considerable cytotoxicity (IC50 ≈ 0.15–2.41 μM) against HeLa, HepG-2 and HT-29 cancer cell lines in comparison with cisplatin. In this regard, the cleavage activity of the Cu(II) complex was higher than the other two metal complexes, resulting in a higher cytotoxicity.
The results obtained and described for the metal complexes described above are relevant from the bioinorganic point of view since they demonstrate the potential of coordination chemistry in the development of new and effective compounds possibly utilisable in cancer treatment. This general aspect motivated us to conduct the current study. Based on the biological features of lapachol, this compound may be considered an excellent candidate for the design of new biologically active metal complexes. That is why we decided to prepare a series of complexes based on relatively nontoxic metals and to evaluate their antiproliferative activity and cellular effects in selected human cancer cells. Our initial idea was to generate square-planar mixed-ligand ionic systems of the type [M(Lap)(N–N)]NO3, where M = Cu(II) and Zn(II), and N–N stands for a bidentate N-donor aromatic imine, with the aim to prepare systems with good solubility in water and possibility to influence the cell cycle of cancer cells. However, we were not successful in this intent, because all our attempts led only to the formation of compounds of the general formula [M(Lap)2(N–N)], although we screened different reaction conditions. Thus, a series of seven complexes of the composition [M(Lap)2(N–N)] and [Cu(Lap)(H2O)(terpy)]NO3 has been prepared, characterised and evaluated for their anticancer features at the in vitro level.
Results and discussion
General characterisation of complexes 1–7
Copper(II) (1–4) and zinc(II) (5–7) mixed-ligand complexes with lapachol and the corresponding N-donor aromatic-imine-based ligand were synthesised through a general reaction pathway as shown in Scheme 2. All the complexes were characterised by elemental analysis, mass spectrometry and infrared spectroscopy. Moreover, the structures of 4, 5 and 6 were confirmed by single-crystal X-ray analysis. Each of the complexes revealed one of the dominant peaks in the positive ionisation mass spectra at m/z = 636.33 (1); 498.25 (2); 460.23 (3); 537.35 (4); 637.36 (5); 499.29 (6); and 485.27 (7) corresponding to the [MII(Lap)(N–N)]+ ion, followed by the peaks belonging to the [MI(N-N)2]+ ions and usually less intensive [MII(Lap)(N–N)2]+ ions. The ESI mass spectra of complexes 1–7 and the agreement of the assigned compositions of the pseudomolecular ions with the theoretical monoisotopic m/z values are shown in Fig. S1–S7 in the ESI,‡ together with isotopic-resolved parts of the spectra in the regions corresponding to the identified species compared with the theoretical isotopic patterns obtained using the on-line tool at https://www.sisweb.com/mstools/isotope.htm (see Fig. S1a, S1b, S2a, S3a, S4a, S5a, S5b, S6a, S6b, S7a, and S7b in the ESI‡). The presence of the corresponding organic ligands in complexes 1–7 can be seen from the characteristic absorption bands observed in the infrared spectra around 3060, 2870, 2850, 1650, 1590, and 1430 cm−1, which can be attributed to the stretching vibrations of ν(CH)ar, ν(CH2)aliph, ν(CH3)aliph, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ν(C–N)ar, and ν(C–C)ring, respectively.20 For comparative purposes, the spectra of free lapachol (HLap) and complexes 1–7 are depicted in Fig. S8 and S9–S15, respectively, in the ESI.‡ In trying to uncover the stability of the complexes in MeOH and water-containing systems, electronic spectra of the representative complex 1 were measured in the solid state and MeOH, MeOH/water (1
O), ν(C–N)ar, and ν(C–C)ring, respectively.20 For comparative purposes, the spectra of free lapachol (HLap) and complexes 1–7 are depicted in Fig. S8 and S9–S15, respectively, in the ESI.‡ In trying to uncover the stability of the complexes in MeOH and water-containing systems, electronic spectra of the representative complex 1 were measured in the solid state and MeOH, MeOH/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and water, however, the concentrations of complex 1 in MeOH/water and water were too low to obtain absorption maxima in the visible region and thus, only the solid state and MeOH spectra are shown in Fig. S16 in the ESI.‡ The maxima observed in the spectra (at ca. 485 and 645 nm) are nearly identical pointing out that the stereochemistry remains without significant changes in MeOH as compared to the solid phase. In addition to that, the spectra of Cu-complexes 2, 3 and 4 measured in MeOH are shown in Fig. S16 in the inset,‡ revealing the d–d absorption maxima at ca. 435–480 nm (for 2, 3 and 4), 630 nm (for 2) and 719 nm (for 4), pointing out a similar geometry in the vicinity of the copper(II) atoms. Additional experiments were performed using ESI mass spectra in MeOH/water medium. The stability of complexes 1 and 5 in water-containing medium (i.e. the 1:1 mixture of MeOH:H2O) was confirmed by comparing the ESI mass spectra of freshly prepared solutions and the same solutions after 24 h. As can be seen from the spectra of copper(II) complex 1 (Fig. S17 and S18 in ESI‡) and zinc(II) complex 5 (Fig. S19 and S20 in ESI‡), the specific peaks could be attributed to the ions representing the main structural motifs of the complexes, i.e. in the case of complex 1, the main peak at 636.20 m/z corresponds to [Cu(Lap)(bphen)]+ and in the case of complex 5, the main peaks at 637.20 m/z and 969.34 m/z correspond to [Zn(Lap)(bphen)]+ and [Zn(Lap)(bphen)2]+, respectively. These results clearly showed that the solutions containing water involve the coordination species with structural motifs specific for the studied copper(II) and zinc(II) complexes even after standing at laboratory temperature for 24 h.
:1 v/v) and water, however, the concentrations of complex 1 in MeOH/water and water were too low to obtain absorption maxima in the visible region and thus, only the solid state and MeOH spectra are shown in Fig. S16 in the ESI.‡ The maxima observed in the spectra (at ca. 485 and 645 nm) are nearly identical pointing out that the stereochemistry remains without significant changes in MeOH as compared to the solid phase. In addition to that, the spectra of Cu-complexes 2, 3 and 4 measured in MeOH are shown in Fig. S16 in the inset,‡ revealing the d–d absorption maxima at ca. 435–480 nm (for 2, 3 and 4), 630 nm (for 2) and 719 nm (for 4), pointing out a similar geometry in the vicinity of the copper(II) atoms. Additional experiments were performed using ESI mass spectra in MeOH/water medium. The stability of complexes 1 and 5 in water-containing medium (i.e. the 1:1 mixture of MeOH:H2O) was confirmed by comparing the ESI mass spectra of freshly prepared solutions and the same solutions after 24 h. As can be seen from the spectra of copper(II) complex 1 (Fig. S17 and S18 in ESI‡) and zinc(II) complex 5 (Fig. S19 and S20 in ESI‡), the specific peaks could be attributed to the ions representing the main structural motifs of the complexes, i.e. in the case of complex 1, the main peak at 636.20 m/z corresponds to [Cu(Lap)(bphen)]+ and in the case of complex 5, the main peaks at 637.20 m/z and 969.34 m/z correspond to [Zn(Lap)(bphen)]+ and [Zn(Lap)(bphen)2]+, respectively. These results clearly showed that the solutions containing water involve the coordination species with structural motifs specific for the studied copper(II) and zinc(II) complexes even after standing at laboratory temperature for 24 h.
 |
| | Scheme 2 The reaction pathway leading to the preparation of complexes 1–7, together with their structural formulas. | |
Single crystal X-ray analysis
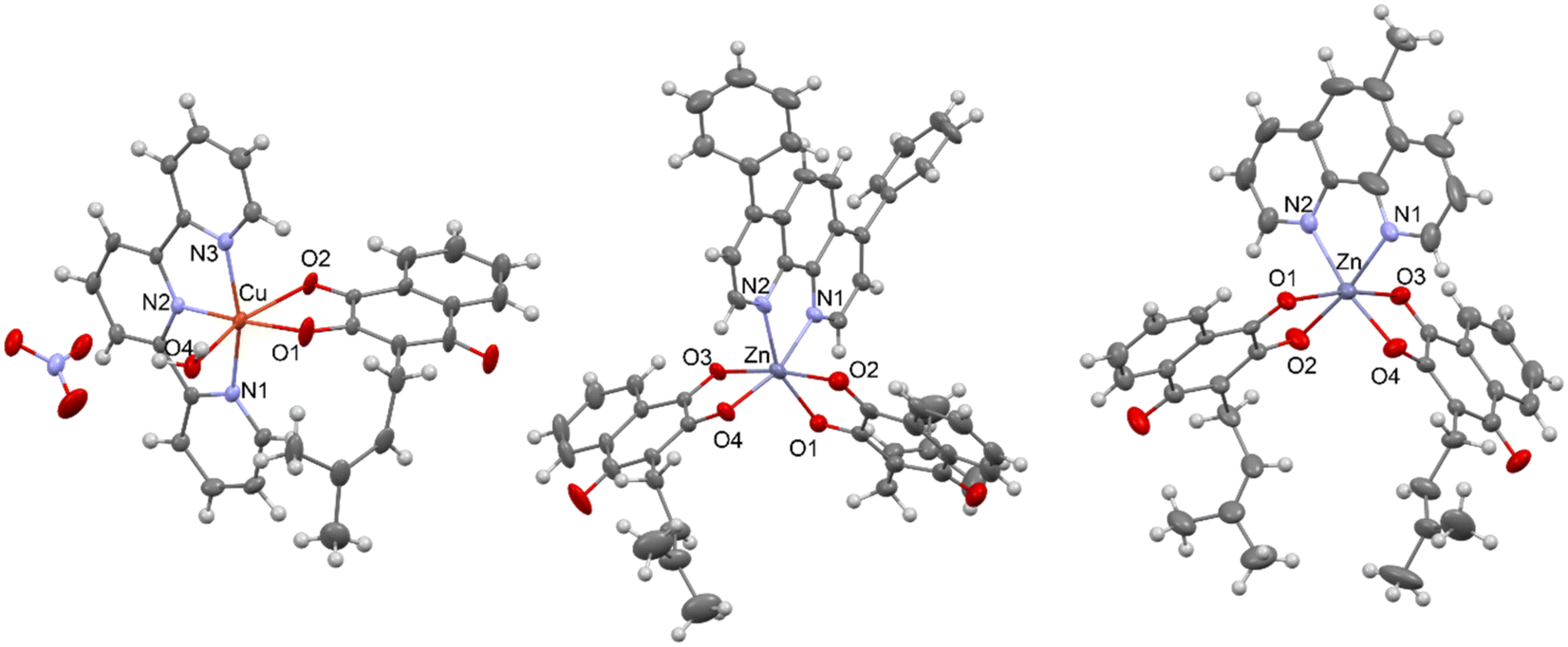
The molecular structures of [Cu(Lap)(H2O)(terpy)]NO3 (4), [Zn(Lap)2(bphen)] (5) and [Zn(Lap)2(mphen)] (6) were determined using single crystal X-ray diffraction analysis, and they are shown in Fig. 1. The crystal data and structure refinements are summarised in Table S1 in the ESI.‡ In 4, the copper(II) atom is octahedrally coordinated in a distorted fashion, having an N3O3 donor arrangement, while the zinc(II) atoms in 5 and 6 are octahedrally bonded, showing the N2O4 donor sets. The selected interatomic parameters in the vicinity of the central metal atoms in complexes 4, 5 and 6 are summarised in Table 1. To date, only one X-ray structure of a copper(II) complex containing the lapachol moiety in combination with a bidentate N-donor ligand, namely 2,2′-bipyridine, of the composition [Cu(Lap)2(bpy)], has been deposited with the Cambridge Structural Database (CSD version 5.44, April 2023, Refcode NEZPIR).21 As can be seen from Table 1, the bond lengths and angles differ significantly between those belonging to copper(II) complex 4 and zinc(II) complexes 5 and 6. This conclusion can be anticipated because it is associated mainly with the structural features of the tridentate terpy ligand in comparison with the bidentate bphen and mphem ligands, and in addition to that, also with different types and extent of hydrogen bonding in the crystal packing of the complexes. On the other hand, the interatomic parameters in the vicinity of the zinc(II) atoms in 5 and 6 are very close and comparable. Each crystal structure of 4, 5 and 6 is stabilised by various hydrogen bonds and/or other types of non-covalent bonding. In 4, the two [Cu(Lap)(H2O)(terpy)]+ moieties are centro-symmetrically and non-covalently connected through two nitrate anions, as depicted in Fig. S21 in the ESI.‡ Moreover, the C–HO contacts also stabilise the crystal structure of 4. For the hydrogen bond parameters in 4, see Table S2 in the ESI.‡ The crystal structures of 5 and 6 are stabilised by the C–H⋯C and C–H⋯O, and C–H⋯N, C–H⋯C and C⋯C non-covalent contacts, respectively. More detailed information can be found in Fig. S22 and S23, as well as Tables S3 and S4 in the ESI.‡
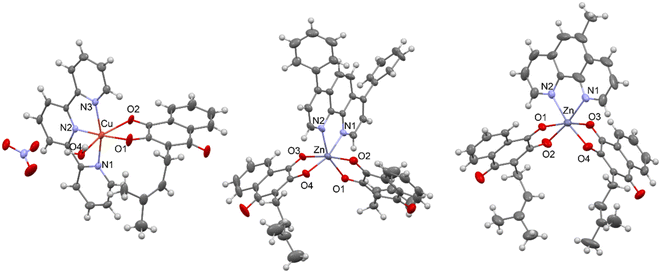 |
| | Fig. 1 X-ray structures of [Cu(Lap)(H2O)(terpy)]NO3 (4) – left, [Zn(Lap)2(bphen)] (5) – middle, and [Zn(Lap)2(mphen)] (6) – right. The labelling of the carbon (dark grey) and hydrogen (light grey) atoms, as well as some nitrogen (blue) and oxygen (red) atoms, is omitted for the sake of clarity. | |
Table 1 Selected bond lengths [Å] and angles [°] for complexes 4, 5 and 6
| Bond length, Å/angle, [°] |
4 (Cu) |
5 (Zn) |
6 (Zn) |
| Cu/Zn–N |
1.849(4) |
2.160(3) |
2.145(6) |
| 2.200(4) |
2.143(3) |
2.138(6) |
| 2.236(5) |
— |
— |
| Cu/Zn–O (Lap) |
1.837(4) |
2.000(3) |
2.013(5) |
| 2.425(4) |
2.024(3) |
2.022(5) |
| — |
2.190(3) |
2.197(5) |
| — |
2.223(3) |
2.202(5) |
| Cu–O (H2O) |
2.298(4) |
— |
— |
| O–Cu/Zn–N |
172.37(14) |
157.67(11) |
169.3(2) |
| — |
162.66(11) |
170.8(2) |
| N–Cu/Zn–N |
162.41(14) |
76.55(12) |
77.8(2) |
| O–Cu/Zn–O |
165.43(12) |
171.99(11) |
168.37(17) |
In vitro cytotoxicity studies
The in vitro cytotoxicity of complexes 1–7, along with lapachol and cisplatin for comparative purposes, was determined on A2780, A2780R, MCF-7, PC-3, A549 and HOS human cancer cell lines (see Table 2). The obtained results revealed significant in vitro antiproliferative effects for the complexes with the bphen ligand (for 1 and 5) against A2780 and A2780R, and with the mphen ligand (for 2) in A2780, with sub-micromolar IC50 values. Moreover, complexes 1, 2 and 5 also showed significant effects in the MCF-7, PC-3, A549 and HOS cells, with IC50 ≈ 1–6 μM. All these complexes are also considerably more active than the free HLap ligand and the reference drug cisplatin. Regarding bidentate N-donor aromatic imine ligands, it is well-known from the literature that they show no significant cytotoxicity on cancer cell lines at 24 h incubation time, as can be observed for phen, mphen and bphen against A2780, A2780R, G361, A549, HeLa, LNCaP and THP-1 cell lines, with IC50 values > 50 μM.22 Overall, the antiproliferative effects of the copper(II) complexes 1–4 are higher in comparison to those of the zinc(II) ones (6 and 7), except for 5, which shows comparable efficacy to copper(II) complex 1. The optimal anticancer effects of complexes 1 and 5 can be associated with the presence of the bulkiest bphen ligand in their structures. This conclusion is in good accordance with previous findings regarding other Cu(II)-complexes, having the CuN2O2 chromophore, and containing a combination of similar N-donor ligands with bidentate O-donor ones.23,24 Moreover, complexes 1–7 revealed a relatively good selectivity index (SI), defined as IC50(MRC-5)/IC50(A2780), with SI > 3.8, which is slightly better than that for cisplatin (SI ≈ 3).
Table 2
In vitro cytotoxicity against selected human cancer cell lines (A2780, A2780R, MCF-7, PC-3, A549 and HOS) and normal (MRC-5) cells as determined after 24 h of incubation. IC50 values are given in μM together with the standard deviations based on triplicate experiments
| Compound |
IC50 ± SD [μM] |
| A2780 |
A2780R |
MCF-7 |
PC-3 |
A549 |
HOS |
MRC-5 |
SIa |
|
SI = the selectivity index expressed as IC50(MRC-5)/IC50(A2780).
|
| [Cu(Lap)2(bphen)] 1 |
0.53 ± 0.13 |
0.6 ± 0.1 |
0.9 ± 0.3 |
1.2 ± 0.1 |
1.3 ± 0.3 |
1.4 ± 0.4 |
3.2 ± 0.1 |
6.0 |
| [Cu(Lap)2(mphen)]·H2O 2 |
0.55 ± 0.18 |
0.9 ± 0.1 |
2.6 ± 0.1 |
6.2 ± 1.6 |
2.7 ± 0.1 |
3.2 ± 0.2 |
3.0 ± 0.1 |
5.8 |
| [Cu(Lap)2(bpy)]·H2O 3 |
9.4 ± 3.0 |
22.0 ± 2.8 |
31.9 ± 1.5 |
34.1 ± 0.6 |
33.9 ± 1.6 |
31.9 ± 3.1 |
36.0 ± 2.8 |
3.8 |
| [Cu(Lap)(H2O)(terpy)]NO34 |
7.1 ± 0.6 |
19.5 ± 2.6 |
39.2 ± 4.3 |
>50 |
>50 |
42.8 ± 3.3 |
44.1 ± 2.8 |
6.2 |
| [Zn(Lap)2(bphen)] 5 |
0.65 ± 0.12 |
0.75 ± 0.03 |
1.2 ± 0.2 |
1.4 ± 0.3 |
2.5 ± 0.2 |
1.6 ± 0.2 |
3.5 ± 0.1 |
5.4 |
| [Zn(Lap)2(mphen)]·H2O 6 |
>50 |
>50 |
37.8 ± 3.1 |
>50 |
>50 |
>50 |
>50 |
— |
| [Zn(Lap)2(phen)]·2H2O 7 |
>50 |
>25 |
>25 |
>25 |
>25 |
>25 |
>25 |
— |
| Lapachol (HLap) |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
— |
| Cisplatin |
15.2 ± 1.1 |
42.7 ± 3.8 |
37.0 ± 2.2 |
>50 |
>50 |
>50 |
46.1 ± 2.4 |
3.0 |
Time-dependent in vitro cytotoxicity and cellular uptake of the title complexes in A2780 cells
To understand more deeply the dynamics of the antiproliferative effect of the title copper(II) and zinc(II) complexes in A2780 cells, time-dependent experiments were performed during three incubation times (24, 48 and 72 h (see Table 3)). In the case of the most effective complexes 1, 2 and 5, with IC50 ≈ 0.6 μM, the cytotoxicity decreases slightly between 24 h and 48 h and stays nearly constant between 48 h and 72 h. On the other hand, the increase in cytotoxicity of the copper(II) complexes 3 and 4 after 24 h is significant and remains nearly unchanged between 48 and 72 h. Surprisingly, the zinc(II) complexes 6 and 7, with IC50 > 50 μM at 24 h incubation, showed strongly increased cytotoxicity, reaching IC50 values of ca. 3.6 μM after 48 h and ca. 2.1 μM after 72 h incubation. A very similar course can be seen for lapachol, changing IC50 from >50 μM (24 h, 48 h) to ca. 6 μM (72 h). Cisplatin showed a typical increase in cytotoxicity with increasing time, showing IC50 from 15.4 μM (24 h), through 6.5 μM (48 h) to 3.7 μM (72 h).
Table 3 The IC50 values of time-dependent in vitro cytotoxicity of the studied compounds on the A2780 cell line
| Compound |
Incubation time (A2780 cell line, IC50 ± SD [μM]) |
| 24 h |
48 h |
72 h |
| [Cu(Lap)2(bphen)] 1 |
0.53 ± 0.13 |
0.32 ± 0.05 |
0.33 ± 0.01 |
| [Cu(Lap)2(mphen)]·H2O 2 |
0.55 ± 0.18 |
0.3 ± 0.1 |
0.31 ± 0.03 |
| [Cu(Lap)2(bpy)]·H2O 3 |
9.4 ± 3.0 |
1.0 ± 0.3 |
1.3 ± 0.6 |
| [Cu(Lap)(H2O)(terpy)]NO34 |
7.1 ± 0.6 |
0.5 ± 0.1 |
0.38 ± 0.01 |
| [Zn(Lap)2(bphen)] 5 |
0.65 ± 0.12 |
0.4 ± 0.1 |
0.35 ± 0.01 |
| [Zn(Lap)2(mphen)]·H2O 6 |
>50 |
3.7 ± 0.6 |
2.0 ± 0.1 |
| [Zn(Lap)2(phen)]·2H2O 7 |
>50 |
3.5 ± 0.6 |
2.3 ± 0.2 |
| Lapachol (HLap) |
>50 |
>50 |
6.4 ± 0.8 |
| Cisplatin |
15.4 ± 0.3 |
6.5 ± 3.2 |
3.7 ± 0.2 |
Cellular uptake in A2780 cells
The ability of the complexes to internalise into the A2780 cells after 2 h, 6 h, 12 h, 24 h, 48 h, and 72 h of incubation was determined by the ICP-MS method after mineralisation with 65% HNO3. Due to the heterogeneous nature of the tested group of compounds involving both copper(II) and zinc(II) complexes, the determination was divided into two independent treatments of A2780 cells, first with copper(II) complexes 1 and 2, and consecutively with zinc(II) complexes 5, 6, and 7. After treatment with complexes 1 and 2, the intracellular copper levels rose slowly, with a maximum after 24 h reaching 76 and 72.5 ng 10−6 cells (i.e. ca. 4-times higher concentrations than basal levels in untreated cells, see Fig. 2).
 |
| | Fig. 2 Cellular uptake of copper(II) complexes 1 and 2 as compared with zinc(II) complexes 5, 6, and 7 in A2780 cells after 2–72 h of incubation as determined by ICP-MS. | |
Unpredictable results (see Fig. 2) were achieved after the treatment of A2780 cells with zinc(II) complexes 5, 6, and 7, as there was a significant difference between the effectivity of complexes 5 (containing bathophenanthroline) and 6 (containing 5-methyl-1,10-phenanthroline) as compared to complex 7 with one unsubstituted 1,10-phenanthroline ligand. This feature probably allowed complex 7 to rapidly reach the intracellular concentration of zinc in A2780 cells between 2 h and 24 h of incubation, reaching up to a 10-fold higher concentration than that determined in untreated cells.
Cellular effects of the selected complexes 1, 5, and 7 in A2780 cells
Modification of the A2780 cell cycle by the selected complexes.
To gain a deeper understanding of the cellular and molecular mechanisms involved in the biological actions of the studied complexes, we examined the modification of the cell cycle in A2780 cells by representative complexes 1, 5, and 7, along with cisplatin as the reference drug, using flow cytometry. The compounds were applied at the half-cytotoxic concentration and incubated for 24 hours. Fig. 3A illustrates how each compound affects the cell cycle in the A2780 cells. The most cytotoxic complexes, 1 and 5, modified the cell cycle of the A2780 cells by diminishing cells in the S phase and simultaneously increasing the number of cells in the G2/M phase, which is the behaviour previously described for a number of copper(II) complexes, e.g. the Schiff base complex derived from S-benzyldithiocarbazate and 3-acetylcoumarin [Cu(SBCM)2],25 the copper(II) complex of tolfenamic acid,26 copper(II) terpyridine complexes,27 which were able to interact with DNA and cause its breakdown, and copper complexes acting as topoisomerase I and II inhibitors.28
 |
| | Fig. 3 (A) Modifications of the cell cycle of A2780 cells caused by the 24 h incubation of complexes 1, 5 and 7, and the reference drug cisplatin, applied at the half-cytotoxic concentration levels. (B) The ability of 1, 5, and 7 and the reference drug cisplatin to damage the cellular structures of living A2780 cells (annexin V−/PI−) towards early apoptosis (annexin V+/PI−) and late-stage apoptosis (annexin V+/PI+) after 24 h of incubation with half-cytotoxic concentrations of the compounds. (C) The ability of complexes 1, 5 and 7 and the reference drug cisplatin to activate the executioner caspases 3/7 in A2780 cells after 24 h of incubation with the half-cytotoxic concentrations of the compounds. The positive control represents the untreated cells heated to 60 °C for 10 minutes. (D) The effectiveness of complexes 1, 5 and 7 and the reference drug cisplatin to induce autophagy of A2780 cells after 24 h of incubation with half-cytotoxic concentrations of the compounds. The positive control represents the cells treated with the mixture of chloroquine and rapamycin (CQ + Rap), a known strong inducer of autophagy. The statistical significance was considered at the following levels: * p < 0.05, ** p < 0.01, *** p < 0.001 with respect to the untreated control group. | |
On the other hand, complex 7 caused a significant arrest of the cells in the G0/G1 phase of the cell cycle, probably by suppressing the cellular metabolism. As shown below, complex 7 significantly induced autophagy (see Fig. 3D) and apoptosis (see Fig. 3B and C). These cellular effects resemble the effect of zinc on esophageal squamous cancer cell line EC109, leading to the induction of autophagy and apoptosis by the upregulation of wild-type p53-induced gene 1 (WIG-1), a kind of zinc finger protein, which can further be altering the expression of Bax, p21 (WAF), and cyclin D1 genes.29
General comments to the induction of cell death in A2780 cells.
Several flow cytometry experiments were conducted to elucidate the mechanism of cytotoxicity induced by the studied complexes in A2780 cells. These experiments entailed annexin V/PI fluorescent staining to identify cellular structural damage characteristic of apoptosis (see Fig. 3B). We also assessed the activation of executioner caspases 3/7 (see Fig. 3C), indicative of late-stage apoptosis, and the induction of autophagy (see Fig. 3D), which is considered a normal cellular response to cellular metabolic stressors. These experiments were carried out on A2780 cells following a 24 hour exposure to the half-cytotoxic concentrations of the compounds under investigation. The results were compared with the effects of the platinum-based cisplatin used as a positive control. The most effective inducer of cellular death in A2780 cells was complex 1, a copper(II) complex involving the bulky bathophenanthroline ligand, which was able to induce both early and late-stage apoptosis in the target cells. The less effective pro-apoptotic agent was complex 7, a zinc(II) complex involving two phenanthroline ligands, which utilizes an alternate mix of cellular effects leading to cellular death; concretely, it acts both through the induction of apoptosis and the autophagy caused probably by the dysregulation of zinc homeostasis by extremely elicited intracellular levels of zinc in the A2780 cells.30,31
The effect of the complexes on intracellular ROS levels and damage to the mitochondrial membranes.
Copper(II) and zinc(II) complexes usually act by multifaceted mechanisms of action, one of which is connected with the intracellular redox-based generation of ROS, damaging the cellular structures and organelles. Another mechanism relates to the interaction of the complexes with intracellular thiols, disrupting the intracellular thiol-recovery systems, depletion of reduced glutathione and oxidative stress. The subsequent outcome of this process involves impaired mitochondrial metabolism, degradation of mitochondrial membranes, and initiation of the intrinsic apoptotic pathway. In all cases, the complexes lowered the ROS levels (Fig. 4), even after the co-incubation of pyocyanin. Zinc(II) complexes lack the ability to directly scavenge the radical species and reduce them to less deleterious forms, yet zinc can elicit the biosynthesis of several antioxidant molecules, such as metallothioneins, and activate antioxidant genes, such as metal regulatory transcription factor 1 (MTF-1), as well as redox ones (via nuclear factor erythroid 2-related factor 2 (NRF2)).32 In addition to the cell-based models, we strived to understand the redox-processes that are involved in the interaction of the complexes, and therefore we performed measurements of cyclic voltammograms (see Fig. S24 and Table S5 in the ESI‡) of the representative complexes. However, the obtained data showed an intriguing tapestry of redox processes occurring during the oxidation and reduction of the copper(II) complexes 2 and 3, zinc(II) complex 5, and free lapachol. The identified maxima/shoulders of the anodic and cathodic waves are summarised in Table S5 in the ESI.‡
 |
| | Fig. 4 (A) The effectiveness of complexes 1, 5 and 7 and the reference drug cisplatin to induce the production of ROS in A2780 cells after 24 h of co-incubation with complexes 1, 5, and 7 in the absence (pyocyanin−) or the presence of the bacterial ROS inducer pyocyanin (pyocyanin+). (B) Damage to mitochondrial membranes in A2780 cells after 24 h of incubation with half-cytotoxic concentrations caused by complexes 1, 5 and 7 and the reference drug cisplatin. The positive control represents the cells treated with carbonyl cyanide chlorophenylhydrazone (CCCP), a well-documented agent causing mitochondrial function disruption. The statistical significance was considered at the following levels: ** p < 0.01, *** p < 0.001 with respect to the untreated control group. | |
Experimental section
Materials and methods used for characterisation
Chemicals and solvents were purchased from commercial sources (Sigma-Aldrich Co., Acros Organics Co., Fluka Co. and Lachema Co.) and used without further purification. Complexes 1–7 were characterised using CHN elemental analyses (CHN analyser Flash Smart CHN, Thermo Scientific), infrared spectroscopy (IR) performed using an ATR technique and in the range of 400–4000 cm−1 (Nicolet iS5 FT-IR, Thermo Nicolet), and mass spectrometry (MS) performed using the ESI+ technique (Bruker amaZon SL, Bruker). Electronic spectra of complex 1 were measured using a Jasco V-750 UV-vis spectrophotometer (Jasco, Easton, MD, USA). ICP-MS analyses were performed using an Agilent 7700x inductively-coupled plasma mass spectrometer (ICP)-MS (Agilent, Santa Clara, CA, USA). Single crystal X-ray diffraction experiments were realised by means of a D8 Quest diffractometer (Bruker) at 293 K. Cyclic voltammetry measurements of complexes 2, 3, 5, and ligand lapachol were performed in 0.1 mM CH2Cl2 solutions, containing 0.1 M Bu4NPF6 as a supporting electrolyte using a Gamry Series G300 potentiostat (Gamry Instruments, Warminster, PA, USA) equipped with a platinum foil working and counter electrode and Ag/AgCl in saturated KOH reference electrode. The potentials (V) of the maxima/shoulders of the anodic and cathodic waves (Epa – anodic, Epc – cathodic) vs. reference electrode Ag/AgCl in saturated KOH were obtained by the analysis of the raw data using the Gamry Echem Analyst software (ver. 6.33).
Preparation of complexes 1–7
Complexes 1–3 and 5–7 were prepared by a slightly modified procedure reported in the literature.19 At first, 2 molar equivalents of free lapachol (HLap) were dissolved in 5 mL of MeOH, and then, a methanolic solution of NaOH (in a 1:1 molar ratio) was added to it under stirring. The reaction system was stirred at room temperature for the next 1 hour, forming a dark-red solution. In the meantime, the commercial precursor Cu(NO3)2·3H2O (or Zn(NO3)2·6H2O) and the corresponding N–N ligand were dissolved in a MeOH/H2O mixture volume (1:1 v/v) in a 1:1 molar ratio and stirred at room temperature for 20 minutes, giving a light blue solution. Then, the methanolic solution of the deprotonated Lap was added dropwise, and the mixture was allowed to react for 3 hours. The precipitate obtained was collected by filtration, washed with deionised water (3 × 10 mL), methanol (3 × 10 mL), and Et2O (3 × 10 mL), and dried under vacuum.
Complex 4 was prepared as follows: 1 molar equivalent of HLap was dissolved in 5 mL of MeOH, and then, a methanolic solution of NaOH (in a 1:1 molar ratio) was added to it with stirring at room temperature (1 hour). In the meantime, Cu(NO3)2·3H2O (20 mg, 0.089 mmol) and the terpyridine (20.8 mg, 0.089 mmol) were dissolved in a MeOH/H2O mixture (1:1 v/v) in a 1:1 molar ratio and stirred at room temperature for 1 hour, giving a light blue solution. Consequently, the methanolic solution of the deprotonated Lap was added, and the reaction was stirred at 70 °C for 24 hours, resulting in a red solution. Volatile solvents were evaporated under reduced pressure, and the residue was washed with Et2O (3 × 10 mL) and toluene (2 × 10 mL), and suspended in pentane for 1 night. Reprecipitation of the solid in a CH2Cl2/petroleum ether (ca. 1:15 v/v) mixture allowed an orange powder to be obtained, which was collected by filtration and dried under vacuum.
For [Cu(Lap)2(bphen)] (1).
From Cu(NO3)2·3H2O (20 mg, 0.089 mmol), bphen (29.7 mg, 0.089 mmol), HLap (43.1 mg, 0.178 mmol) and NaOH (7.11 mg, 0.178 mmol). Dark-red solid: 46 mg (yield 59%). Anal. calcd. (%) for C54H42CuN2O6 (Mr = 878.5): C 73.8, H 4.8, N 3.2. Found (%): C 73.5, H 4.6, N 3.0. ESI mass spectra (m/z, corresponding species (theor. m/z)): 636.33 m/z [Cu(Lap)(bphen)]+ (636.15); 727.41 m/z [Cu(bphen)2]+ (727.19). IR (ATR; cm−1): 3076, 3041, 2958, 2908, 2853, 1650, 1621, 1589, 1545, 1418, 1349, 1272, 1237, 952, 850, 734, 699, 667, 634, 549.
For [Cu(Lap)2(mphen)]·H2O (2).
From Cu(NO3)2·3H2O (20 mg, 0.089 mmol), mphen (17.3 mg, 0.089 mmol), HLap (43.1 mg, 0.178 mmol) and NaOH (7.11 mg, 0.178 mmol). Dark-red solid: 30.2 mg (yield 46%). Anal. calcd. (%) for C43H36CuN2O6·(H2O) (Mr = 758.3): C 68.1, H 5.1, N 3.7. Found (%): C 67.6, H 4.7, N 3.7. ESI mass spectra (m/z, corresponding species (theor. m/z)): 498.25 m/z [Cu(Lap)(mphen)]+ (498.10). IR (ATR; cm−1): 3068, 3021, 2958, 2909, 2852, 1647, 1588, 1541, 1424, 1369, 1350, 1269, 1238, 1221, 951, 880, 807, 731, 698, 664, 552, 436.
For [Cu(Lap)2(bpy)]·H2O (3).
From Cu(NO3)2·3H2O (20 mg, 0.089 mmol), bpy (14 mg, 0.089 mmol), HLap (43.1 mg, 0.178 mmol) and NaOH (7.11 mg, 0.178 mmol). Dark-red solid: 32.5 mg (yield 52%). Anal. calcd. (%) for C40H34CuN2O6·(H2O) (Mr = 720.3): C 66.7, H 5.0, N 3.9. Found (%): C 67.0, H 4.9, N 3.9. ESI mass spectra (m/z, corresponding species (theor. m/z)): 460.23 m/z [Cu(Lap)(bpy)]+ (460.08). IR (ATR; cm−1): 3107, 3077, 3033, 2959, 2903, 1639, 1615, 1587, 1540, 1489, 1442, 1349, 1269, 1238, 1223, 1101, 1053, 1030, 954, 768, 733, 693, 664, 553, 496.
For [Cu(Lap)2(H2O)(terpy)]NO3 (4).
Yield: 18.5 mg (35%). X-ray quality crystals were obtained by recrystallizing the product from MeOH. Anal. calcd. (%) for C30H26CuN4O7 (Mr = 618.1): C 58.3, H 4.2, N 9.1. Found (%): C 57.6, H 4.1, N 8.7. ESI mass spectra (m/z, corresponding species (theor. m/z)): 537.35 m/z [Cu(Lap)(terpy)]+ (537.11). IR (ATR; cm−1): 3429, 3362, 3286, 3075, 3062, 3039, 2989, 2963, 2908, 2876, 2848, 1635, 1587, 1557, 1478, 1451, 1400, 1325, 1272, 1245, 1220, 1166, 1097, 1062, 1038, 1022, 953, 879, 783, 728, 672, 650, 547, 504, 442.
For [Zn(Lap)2(bphen)] (5).
From Zn(NO3)2·6H2O (20 mg, 0.067 mmol), bphen (22.2 mg, 0.067 mmol), HLap (32.4 mg, 0.134 mmol) and NaOH (5.4 mg, 0.134 mmol). The product was washed with cold H2O (3 × 10 mL) and MeOH (2 × 5 mL). Then, the residue was suspended in pentane for 2 hours and reprecipitated with a CH2Cl2/petroleum ether (ca. 1:15 v/v) mixture. The resulting red powder was collected by filtration and dried under vacuum. Red solid: 34 mg (58%). X-ray quality crystals were obtained from a THF solution layered with pentane and settled at room temperature. Anal. calcd. (%) for C54H42ZnN2O6 (Mr = 880.3): C 73.7, H 4.8, N 3.2. Found (%): C 73.9, H 4.9, N 3.2. ESI mass spectra (m/z, corresponding species (theor. m/z)): 637.36 m/z [Zn(Lap)(bphen)]+ (637.15); 969.58 m/z [Zn(Lap)(bphen)2]+ (969.28). IR (ATR; cm−1): 3059, 2962, 2909, 2878, 2852, 1622, 1585, 1547, 1432, 1352, 1277, 1242, 955, 842, 765, 737, 701, 666, 631, 546, 466, 501, 546, 436.
For [Zn(Lap)2(mphen)]·H2O (6).
From Zn(NO3)2·6H2O (20 mg, 0.067 mmol), mphen (13 mg, 0.067 mmol), HLap (32.4 mg, 0.134 mmol) and NaOH (5.4 mg, 0.134 mmol). The product was filtered with cold H2O (3 × 10 mL) and Et2O (3 × 10 mL). The residue was suspended in pentane for 2 hours and reprecipitated with a CH2Cl2/petroleum ether (ca. 1:15 v/v) mixture. The resulting purple powder was collected and dried under vacuum. Dark-red solid: 32.3 mg (65%). X-ray quality crystals were obtained from a THF solution layered with pentane and settled aside at 30 °C. Anal. calcd. (%) for C43H36ZnN2O6·H2O (Mr = 760.2): C 67.9, H 5.0, N 3.7. Found (%): C 67.9, H 5.0, N 3.8. ESI mass spectra (m/z, corresponding species (theor. m/z)): 499.29 m/z [Zn(Lap)(mphen)]+ (499.10); 693.39 m/z [Zn(Lap)(mphen)2]+ (693.18). IR (ATR; cm−1): 3066, 2956, 2908, 2852, 1625, 1583, 1544, 1422, 1352, 1289, 1276, 1239, 1152, 1056, 955, 881, 848, 808, 697, 660, 642, 547, 507, 429.
For [Zn(Lap)2(phen)]·2H2O (7).
From Zn(NO3)2·6H2O (20 mg, 0.067 mmol), phen (12.10 mg, 0.067 mmol), HLap (32.4 mg, 0.134 mmol) and NaOH (5.4 mg, 0.134 mmol). The product was filtered with cold H2O (3 × 10 mL), cold MeOH (2 × 10 mL) and Et2O (3 × 10 mL). Purple solid: 36 mg (74%). Anal. calcd. (%) for C42H34ZnN2O6·2(H2O) (Mr = 764.1): C 66.0, H 5.0, N 3.7. Found (%): C 66.0, H 4.8, N 3.7. ESI mass spectra (m/z, corresponding species (theor. m/z)): 485.27 m/z [Zn(Lap)(phen)]+ (485.08); 665.35 m/z [Zn(Lap)(phen)2]+ (665.15). IR (ATR; cm−1): 3566, 3068, 2959, 2920, 2875, 2847, 1624, 1584, 1546, 1429, 1349, 1293, 1275, 1244, 1223, 1142, 952, 847, 728, 696, 542, 498, 439.
Single-crystal X-ray diffraction analysis
X-ray data of complexes 4, 5 and 6, suitable for single-crystal X-ray analysis, were collected on a Bruker D8 QUEST diffractometer equipped with a PHOTON 100 CMOS detector using Mo-Kα radiation (λ = 0.71073 Å) at 293 K. The APEX3 software package33 was used for data collection and reduction. The molecular structure was solved by direct methods (SHELXS) and refined by a full-matrix least-squares procedure (SHELXL).34 Hydrogen atoms were found in the difference Fourier maps and refined using a rigid model, with C–H = 0.93 Å (CH)ar, C–H = 0.99 Å (CH2), C–H = 0.96 Å (CH3) and O–H = 0.83 Å (OH), and with Uiso(H) = 1.2Ueq(CH, CH2, OH) and Uiso(H) = 1.5Ueq(CH3). The figures were drawn, and additional structural calculations were performed, using MERCURY35 software. The crystal data and structure refinement details for complexes 4, 5 and 6 are summarised in Table S1 in the ESI.‡
In vitro cytotoxicity against human cancerous and normal cell lines
The in vitro cytotoxicity of complexes 1–7, and free lapachol and cisplatin for comparative purposes, was determined by the MTT assay at various incubation times (24, 48 and 72 h) in the case of the human ovarian carcinoma (A2780) cells. Moreover, the following human carcinoma cells were also used for studies with 24 h incubation time: cisplatin-resistant ovarian carcinoma (A2780R), breast adenocarcinoma (MCF-7), prostate adenocarcinoma (PC-3), lung adenocarcinoma (A549) and bone osteosarcoma (HOS), which were obtained from ATCC collection of cell lines and cultivated according to the producer's instructions. The reference normal cell line of human foetal fibroblasts (MRC-5) was obtained from the same commercial source and maintained according to the producer's instructions. The half-maximal inhibitory concentrations (IC50) were calculated from dose–response curves by means of the GraphPad Prism 6 software (GraphPad Software, San Diego, USA).
Cell cycle analysis
The A2780 cells (Sigma, 93112519-1VL) were grown according to the manufacturer's instructions and seeded at 104 cells per well in 96-well plates. After 24 hours, the cells were treated with the solutions of complexes 1, 5, and 7 (at half-cytotoxic concentrations), and incubated for an additional 24 hours. Subsequently, the cells were washed once with PBS (0.1 M, pH 7.4), and cell cycle analysis was conducted using the BD Cycletest™ Plus DNA kit (Becton Dickinson, USA) following the manufacturer's protocol. The data were collected using a BD FACSVerse flow cytometer (Becton Dickinson, USA) in three separate experiments, each performed in duplicate, with at least 5 × 103 events recorded for each sample.
Induction of cell death and related processes
The cell death induced by complexes 1, 5, and 7 (at half-cytotoxic concentrations) was examined using two methods in the A2780 cell line. Firstly, we tested the induction of apoptosis using the annexin V–FITC/PI commercial kit (V13242, Thermo Fisher Scientific, USA). Secondly, we monitored the activation of caspase 3/7 using the CellEvent™ Caspase-3/7 Green Flow Cytometry Assay Kit (C10427, Thermo Fisher Scientific, USA). Both assays were carried out in accordance with the manufacturers' protocols, with a modification in the caspase induction assay: we used the CellEvent™ Caspase-3/7 green detection reagent only for detecting caspase-3/7 activation. The general procedure involved seeding 5 × 104 cells per well in a 24-well cell culture plate, treating the cells with the complex solution, and cultivating for the next 24 hours. Subsequently, we washed the cells once with PBS (0.1 M, pH 7.4) and trypsinized them using 0.25% trypsin–EDTA (Gibco™). We then blocked trypsin by adding cell culture medium to a final volume of 500 μL. The resulting cell suspensions were divided into two separate 250 μL aliquots. The first set of aliquots was utilized for the annexin V–FITC/PI assay, while the second set was used for the caspase 3/7 activation assay. Both assays were performed using the BD FACSVerse flow cytometer (Becton Dickinson, USA) in three independent experiments, each conducted in duplicate.
Induction of autophagy
Cells were seeded in a 24-well cell culture plate at 5 × 104 cells per well and allowed to grow for 24 hours. The next day, the cells were treated with complexes 1, 5, and 7 at the concentrations of 0.5, 0.7, and 50 μM, respectively. After a 24 hour incubation period, the cells were washed once with PBS (0.1 M, pH 7.4), trypsinized using 0.25% trypsin–EDTA, and resuspended in the cell culture medium. Subsequently, the cells were stained with CYTO-ID® Green stain solution and incubated for 30 minutes in the dark. After a final wash with PBS, the cell suspensions were analysed using the BD FACSVerse flow cytometer in three separate experiments, with at least 10000 events recorded for each sample prepared in duplicate. As a positive control, cells incubated with a combination of chloroquine (10 μM) and rapamycin (0.5 μM) for 18 hours were used. This analysis was conducted using the CYTO-ID® Autophagy detection kit 2.0 (ENZO, USA) according to the manufacturer's protocol to assess the induction of autophagy by complexes 1, 5, and 7.
Induction of intracellular ROS/superoxide production
ROS induction in A2780 cells was investigated after incubation with complexes 1, 5, and 7 (at half-cytotoxic concentrations). The ROS-ID® Total ROS/Superoxide detection kit (Enzo Life Sciences, US) was used according to the manufacturer's protocol. The cells were initially seeded in 96-well cell culture plates at 104 cells per well and incubated for 24 hours, followed by a 24 hour experimental treatment. Subsequently, the cells were stained with a ROS/superoxide probe mixture and then incubated at 37 °C in the dark for 60 minutes. Finally, the fluorescence was measured using a microplate reader, specifically the Infinite M200Pro (Tecan, Switzerland). The control samples contained 500 μM pyocyanin, and the experiment was conducted three times, each with triplicate.
Mitochondrial membrane potential analysis
The A2780 cells were seeded in 24-well cell culture plates at a density of 5 × 104 cells per well and allowed to cultivate for 24 hours. The following day, complexes 1, 5, and 7 were introduced to the cells at half-cytotoxic concentrations. The cells were then further incubated for an additional 24 hours. Subsequent to the experimental treatment, the cells underwent a PBS wash, detachment using 0.25% trypsin–EDTA (Gibco™), and resuspension in the cell culture medium. The cells were subsequently isolated by centrifugation and suspended in a staining solution prepared in accordance with the manufacturer's protocol using the MITO-ID® Membrane potential detection kit (Enzo Life Sciences, USA). Finally, the cells were analysed using the BD FACSVerse flow cytometer (Becton Dickinson, USA). The control group was treated with 2 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP). The entire process was conducted across three independent experiments, each with duplicate samples.
Cellular uptake analysis
The A2780 cells were seeded at the density 106 cells per well and co-incubated under standard growing conditions with the half-effective concentrations of the representative complexes 1, 2, 5, 6 and 7 dissolved in 0.1% DMF for 2, 6, 12, 24, 48, or 72 h. The respective controls for the normal content of Cu and Zn in the A2780 cells were treated by the vehicle only. After each time period, the cells were detached, harvested, washed twice with PBS (0.1 M, pH 7.4) and isolated by centrifugation. The cell pellets were digested with 500 μL of concentrated nitric acid for ICP-MS (65%, at laboratory temperature for 24 h). Prior to the ICP-MS analysis, the solutions were adjusted to suitable concentration with 4.5 mL of ultrapure water. The intracellular Cu and Zn contents were determined using ICP-MS (ICP-MS spectrometer 7700x, Agilent) using external calibration (transition metal mix 1 for ICP, Merck). The results were corrected for adsorption effects.
Conclusions
Copper(II) and zinc(II) complexes, 1–7, containing a naturally occurring ligand (lapachol), in combination with various N-donor bidentate/tridentate ligands, were prepared, characterized, and evaluated for their in vitro anticancer effects against a panel of six human cancer cell lines (A2780, A2780R, MCF-7, PC-3, A549, and HOS). The obtained IC50 values (≈1–5 μM) are better than those found for conventional platinum-based drug cisplatin. The selected complexes 1 and 5, which represent the most cytotoxic ones, and complex 7, which was one of the least cytotoxic ones, were studied by a series of flow-cytometry experiments, including the evaluation of the effects of the complexes on the cell cycle, induction of cell death (by apoptosis/necrosis, induction of Cas 3/7, and autophagy), ROS levels and mitochondrial membrane potential disruption, in A2780 cells after 24 h of incubation with half-cytotoxic concentrations of the complexes. Complexes 1 and 5 increased the number of cells arrested in the G2/M phase, while 7 arrested the majority of the A2780 cells in the G0/G1 phase. All the complexes induced apoptosis in the early and late stages. Complex 7 induced significant autophagy in A2780 cells; therefore, the most probable mechanism of its action relates to the induction of metabolic stress in A2780 cells. On the other hand, 1 and 5 significantly perturbed the mitochondrial membrane potential, and the destruction of the mitochondrial membranes is probably their main mechanism of action, followed by the activation of the intrinsic pathway of apoptosis. Moreover, the time-dependent cytotoxicity studies revealed that a significant increase in cytotoxicity between 24 and 48 h proceeds for zinc(II) complexes 6 and 7, from IC50 > 50 M (24 h) to ca. 4 μM (48 h).
Data availability
Supplementary Information (SI) available: ESI-MS spectra in Fig. S1–S7,‡ IR spectra in Fig. S8–S15,‡ electronic spectra in Fig. S16,‡ ESI-MS of complex 5 in a mixture of MeOH/water in Fig. S17–S20,‡ crystal data and structure refinements in Table S1,‡ hydrogen bonding in Tables S2–S4 and Fig. S21–S23,‡ see DOI: https://doi.org/10.1039/d4md00543k. Cambridge Crystallographic Database contains the supplementary crystallographic data for complexes 4, 5 and 6, the CCDC deposition numbers: 2369949–2369951, respectively.
Author contributions
The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript. Conceptualisation, Z. T.; methodology, Z. T., J. V., J. B., Z. D.; validation, Z. T., J. V., J. B., Z. D. and S. S.; investigation Z. T., J. V., J. B., S. S. and Z. D.; resources, Z. T. and Z. D.; writing – original draft preparation, Z. T., J. V., J. B.; writing – review and editing, Z. T., J. V., S. S., Z. D.; visualisation, Z. T., J. V., J. B.; supervision, Z. T.; project administration, Z. T.; funding acquisition, Z. T.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the Czech Sciences Foundation (GAČR), a grant number 21-19060S. The authors thank Ms Marta Rešová for in vitro cytotoxicity testing, Ms Renata Jakubcová for infrared spectra measurements, Ms Barbora Komendová for CHN elemental analysis and electronic spectra measurements, and Dr. Hejazi Seyyedmohammadhossein for the cyclic-voltammetry measurements. The authors would also like to thank Prof. Fabio Marchetti from the University of Pisa, Italy, for useful discussions and suggestions during the preparation of the manuscript.
References
- H. Hussain, K. Krohn, V. Uddin Ahmad, G. A. Miana and I. R. Green, ARKIVOC, 2007, ii, 145–171, DOI:10.3998/ark.5550190.0008.204.
- S. Fiorito, F. Epifano, C. Bruyere, V. Mathieu, R. Kiss and S. Genovese, Bioorg. Med. Chem. Lett., 2014, 24, 454–457, DOI:10.1016/j.bmcl.2013.12.049.
- O. Atolani, G. A. Olatunji and O. S. Adeyemi, Arab. J. Sci. Eng., 2021, 46, 5307–5312, DOI:10.1007/s13369-020-05113-1.
- L. B. Marques, F. M. Ottoni, M. C. X. Pinto, J. M. Ribeiro, F. S. de Sousa, R. Weinlich, N. Cruz de Victo, J. Kisitu, A.-K. Holzer, M. Leist, R. J. Alves and E. M. Souza-Fagundes, Toxicol. In Vitro, 2020, 65, 104772, DOI:10.1016/j.tiv.2020.104772.
-
(a) E. R. de Almeida, Open Nat. Prod. J., 2009, 2, 42–47 CrossRef;
(b) F. Epifano, S. Genovese, S. Fiorito, V. Mathieu and R. Kiss, Phytochem. Rev., 2014, 13, 37–49, DOI:10.1007/s11101-013-9289-1.
- L. Todorov and I. Kostova, Molecules, 2023, 28, 1959, DOI:10.3390/molecules28041959.
- W. Kandioller, E. Balsano, S. M. Meier, U. Jungwirth, S. Göschl, A. Roller, M. A. Jakupec, W. Berger, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2013, 49, 3348–3350, 10.1039/c3cc40432c.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 9055–9068, 10.1039/c0dt01816c.
- M. I. F. Barbosa, R. S. Correa, K. M. de Olveira, C. Rodriguez, J. Ellena, O. R. Nascimento, V. P. C. Rocha, F. R. Nonato, T. S. Macedo, J. M. Barbosa-Filho, M. B. P. Soares and A. A. Batista, J. Inorg. Biochem., 2014, 136, 33–39, DOI:10.1016/j.jinorgbio.2014.03.009.
- L. Tabrizi and H. Chiniforoshan, J. Organomet. Chem., 2016, 822, 211–220, DOI:10.1016/j.jorganchem.2016.09.003.
- F. Rinaldi-Neto, A. B. Ribeiro, N. H. Ferreira, I. S. Squarisi, K. M. Oloveira, R. P. Orenha, R. L. T. Parreira, A. A. Batista and D. C. Tavares, J. Inorg. Biochem., 2021, 222, 111497, DOI:10.1016/j.jinorgbio.2021.111497.
- S. Zaidi, Md. I. Hassan, A. Islam and F. Ahmad, Cell. Mol. Life Sci., 2014, 71, 229–255, DOI:10.1007/s00018-013-1341-1.
- G. Hanke and P. Mulo, Plant, Cell Environ., 2013, 36, 1071–1084, DOI:10.1111/pce.12046.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749, DOI:10.1021/acs.chemrev.6b00180.
-
W. Kaim, B. Schwederski and A. Klein, Bioinorganic chemistry: inorganic elements in the chemistry of life: an introduction and guide, Wiley, 2013 Search PubMed.
- T. D. Oliveira, N. A. Cabeza, G. T. S. T. da Silva, A. L. T. G. Ruiz, A. R. L. Caires, R. G. da Silveira, D. C. M. Rodrigues, A. R. Fiorucci and A. dos Anjos, Transition Met. Chem., 2021, 46, 111–120, DOI:10.1007/s11243-020-00427-3.
- R. H. Molina, I. Kalinina, P. Esparza, M. Sokolov, J. G. Platas, A. E. Braun and E. P. Sacau, Polyhedron, 2007, 26, 4860–4864, DOI:10.1016/j.poly.2007.06.022.
- M. A. Martínez, M. C. L. de Jimenez, E. E. Castellano, O. E. Piro and P. J. Aymonino, J. Coord. Chem., 2003, 56, 803–816, DOI:10.1080/0095897031000113959.
- L. Tabrizi, F. Talaie and H. Chiniforoshan, J. Biomol. Struct. Dyn., 2017, 35, 3330–3341, DOI:10.1080/07391102.2016.1254118.
-
C. J. Pouchert, The Aldrich Library of Infrared Spectra, Aldrich Chemical Company Press, Milwaukee, USA, 3rd edn, 1981, pp. 1–1850 Search PubMed.
-
The Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, United Kingdom Search PubMed.
- Z. Trávníček, J. Vančo, J. Hošek, R. Buchtík and Z. Dvořák, Chem. Cent. J., 2012, 6, 160, DOI:10.1186/1752-153X-6-160.
- J. Vančo, Z. Trávníček, J. Hošek, T. Malina and Z. Dvořák, Int. J. Mol. Sci., 2021, 22, 7626, DOI:10.3390/ijms22147626.
- J. Vančo, Z. Trávníček, J. Hošek, T. Malina and Z. Dvořák, J. Inorg. Biochem., 2022, 226, 111639, DOI:10.1016/j.jinorgbio.2021.111639.
- J. B. Foo, L. S. Ng, J. H. Lim, P. X. Tan, Y. Z. Lor, J. S. Ee Loo, M. L. Low, L. C. Chan, C. Y. Beh, S. W. Leong, L. S. Yazan, Y. S. Tor and C. W. How, RSC Adv., 2019, 9, 18359, 10.1039/c9ra03130h.
- M. Hurtado, U. T. Sankpal, A. Kaba, S. Mahammad, J. Chhabra, D. T. Brown, R. K. Gurung, A. A. Holder, J. K. Vishwanatha and R. Basha, Cell. Physiol. Biochem., 2018, 51, 1894–1907, DOI:10.1159/000495715.
- K. Choroba, B. Machura, K. Erfurt, A. R. Casimiro, S. Cordeiro, P. V. Baptista and A. R. Fernandes, J. Med. Chem., 2024, 67, 5813–5836, DOI:10.1021/acs.jmedchem.4c00119.
- C. Molinaro, A. Martoriati, L. Pelinski and K. Cailliau, Cancers, 2020, 12, 2863, DOI:10.3390/cancers12102863.
- W. Guo, Y.-B. Zou, Y.-G. Jiang, R.-W. Wang, Y.-P. Zhao and Z. Ma, Tumor Biol., 2011, 32, 801–808, DOI:10.1007/s13277-011-0182-5.
- J. Wang, H. Zhao, Z. Xu and X. Cheng, Cancer Biol. Med., 2020, 17, 612–625, DOI:10.20892/j.issn.2095-3941.2020.0106.
- H. Kocdor, H. Ates, S. Aydin, R. Cehreli, F. Soyarat, P. Kemanli, D. Harmanci, H. Cengiz and M. A. Kocdor, Drug Des., Dev. Ther., 2015, 9, 3899–3909, DOI:10.2147/DDDT.S87662.
- Ch. Hubner and H. Haase, Redox Biol., 2021, 41, 101916, DOI:10.1016/j.redox.2021.101916.
-
APEX3 Software Suite, © 2016 Bruker AXS Inc., 5465 East Cheryl Parkway, Madison, WI 53711 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470, DOI:10.1107/S0021889807067908.
Footnotes |
| † The authors would like to dedicate this work to Jan Belza, our beloved colleague and friend, who passed away unexpectedly on February 15, 2024, at the age of 31 years. |
| ‡ Electronic supplementary information (ESI) available: ESI-MS spectra in Fig. S1–S7, IR spectra in Fig. S8–S15, electronic spectra in Fig. S16, ESI-MS of complex 5 in a mixture of MeOH/water in Fig. S17–S20, crystal data and structure refinements in Table S1, hydrogen bonding in Tables S2–S4 and Fig. S21–S23. Cambridge Crystallographic Database contains the supplementary crystallographic data for complexes 4, 5 and 6, the CCDC deposition numbers: 2369949–2369951, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4md00543k |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Ján
Vančo
ab,
Ján
Vančo