
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of selective LATS inhibitors via scaffold hopping: enhancing drug-likeness and kinase selectivity for potential applications in regenerative medicine†
Guldana
Issabayeva
 ab,
On-Yu
Kang
a,
Seong Yun
Choi
ab,
Ji Young
Hyun
ab,
Seong Jun
Park
*ab,
Hei-Cheul
Jeung
*c and
Hwan Jung
Lim
*ab
ab,
On-Yu
Kang
a,
Seong Yun
Choi
ab,
Ji Young
Hyun
ab,
Seong Jun
Park
*ab,
Hei-Cheul
Jeung
*c and
Hwan Jung
Lim
*ab
aData Convergence Drug Research Center, Korea Research Institute of Chemical Technology, 141 Gajeong-ro, Daejeon 34114, Republic of Korea. E-mail: sjunpark@krict.re.kr; indium@krict.re.kr
bDepartment of Medicinal Chemistry and Pharmacology, University of Science & Technology, 217 Gajeong-ro, Daejeon 34113, Republic of Korea
cDepartment of Medical Oncology, Yonsei University College of Medicine, 211 Eonju-ro, Gangnam-gu, Seoul 06273, Republic of Korea. E-mail: jeunghc1123@yuhs.ac
First published on 14th September 2024
Abstract
Due to its essential roles in cell proliferation and apoptosis, the precise regulation of the Hippo pathway through LATS presents a viable biological target for developing new drugs for cancer and regenerative diseases. However, currently available probes for selective and highly drug-like inhibition of LATS require further improvement in terms of both activity, selectivity and drug-like properties. Through scaffold hopping aided by docking studies and AI-assisted prediction of metabolic stabilities, we successfully identified an advanced LATS inhibitor demonstrating potent kinase activity, exceptional selectivity against other kinases, and superior oral pharmacokinetic profiles.
1. Introduction
The Hippo pathway,1,2 initially identified in Drosophila, plays a critical role in governing organ size and tissue homeostasis by modulating cell proliferation and apoptosis.3–11 Core components of the Hippo pathway include the large tumor suppressor kinases (LATS1 and LATS2), which phosphorylate and inhibit the transcriptional co-activators YAP (Yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif).12 Dysregulation of the Hippo pathway has been implicated in various human diseases, including cancer and regenerative disorders.13Recent studies have shed light on the potential therapeutic utilities of targeting the Hippo pathway for promoting tissue regeneration.14–20 Among the key effectors of this pathway, LATS kinases have received significant attention as attractive pharmacological targets.21 Inhibition of LATS activity has been shown to unleash the regenerative potential of stem and progenitor cells, facilitating tissue repair and regeneration in preclinical models of diverse pathological conditions.22–26
Harnessing LATS inhibition as a therapeutic strategy for regenerative diseases presents both challenges and opportunities. Key challenges include the need for improved kinase selectivity and the development of drugs with good pharmacokinetic profiles. Finally, we believe that the development of exceptionally selective LATS inhibitors with good pharmacokinetic properties can pave the way to apply them from bench to bedside.
Until now, several LATS inhibitors have been discovered.23–27 Mostly, the reported compounds have pyrrolo[2,3-b]pyridine (7-azaindole) structure23,24,26 (Fig. 1). Existing LATS inhibitors often suffer from limitations such as a lack of kinase selectivity, poor metabolic stability, and low water solubility, hampering the further development of these compounds for target diseases. Especially, low pharmacokinetic profiles due to poor metabolic stability24 are one of the major issues to overcome in developing these compounds for clinical purposes. To address these challenges, the development of novel LATS kinase inhibitors with enhanced drug-like properties and improved selectivity has become imperative.
 | ||
| Fig. 1 Structures of the reported LATS inhibitors. | ||
In this study, we report the design, synthesis, and characterization of a novel class of LATS kinase inhibitors. Employing rational drug design strategies centered on scaffold hopping,28,29 our aim was to improve drug-likeness while maintaining high potency and selectivity of these inhibitors, thereby addressing the limitations of previous compounds.23,24 Our findings open avenues for exploring LATS pharmacological modulators as targeted therapeutic approaches for treating cancer and other diseases characterized by dysregulated cell proliferation and survival.
2. Results & discussion
To identify an optimal core structure with potent inhibitory activity, excellent kinase selectivity, and drug-likeness, we systematically designed potential structures distinct from the 1H-pyrrolo[2,3-b]pyridine utilized in prior studies.23,24 Initially, we dissected the reported inhibitor 1 into its bicyclic core and the carboxamide group at the 3-position (Fig. 2). Subsequently, 1H-pyrrolo[2,3-b]pyridine was substituted by various 5,6-bicyclic heterocycles30,31 to elucidate crucial structural features for effective LATS inhibition. Modifications such as ring deletions, amine relocations or substitutions, and heteroatom additions were implemented to craft alternative scaffolds. | ||
| Fig. 2 Types of the designed structures. | ||
We conducted subsequent molecular docking studies to compare the binding pose and predict affinities of the designed compounds.32,33
Additionally, we used PredMS, an AI-based tool developed by KRICT (Korea Research Institute of Chemical Technology), to assess the predicted metabolic stability of the compounds. PredMS is a web-based model designed to support early drug discovery for small organic molecules (https://predms.netlify.app/).34 According to the output, a compound is considered ‘stable’ if more than 50% of it remains after 30 minutes in human liver microsomes. A value closer to one indicates higher stability.
Due to the unavailability of a reported crystal structure of a LATS-inhibitor complex, the most similar structure, the ROCK1-7-azaindole crystal structure,35 was utilized for docking simulations to predict the binding pose of the designed compounds to further compare them with Truli's reported binding interactions (Fig. 3). 1, Truli, as reported,23 exhibited a favorable binding score (−8.2 kcal mol−1) and moderate human metabolic stability (0.457). The presence and positioning of both amines in the pyrrolopyridine ring appeared crucial for achieving a good binding pose. In this molecular docking simulation model, the pyrrolopyridine ring forms a bidentate hydrogen bond with the backbone hinge residues Met156 and Glu154. These hydrogen bond interactions are similarly preserved in the pyrrolopyrimidine of compound 5k and the pyrazolopyridine of compound 5l, indicating that the addition of an amine to the pyrrolopyridine ring retains a favorable binding pose. The thiazolamine moiety is predicted to interact with Asp216 in 1 which is also the case for 5k and 5l. Moreover, the benzyl group interacts with Ile82 in both 1 and 5l, although these van der Waals interactions were not observed in 5k.
 | ||
| Fig. 3 Docking simulation and predicted human metabolic stabilities of 1, 5k and 5l. | ||
The designed compounds 5a–m and further derivatized compounds 6a–e were synthesized from commercially available carboxylic acids with 2-aminothiazole along with N-substituted thiazol-2(3H)-imine 4a and oxazol-2(3H)-imine 4b using the reported procedures36,37 (Scheme 1). The desired compounds were thoroughly analyzed by 1H NMR and LC–MS for structural confirmation. The purity of the synthesized compounds was assessed using HPLC.
 | ||
| Scheme 1 Reagents and conditions: (i) HATU, DIPEA, DMF, RT; (i′) HBTU, pyridine, 80–100 °C; (ii) NaH, benzyl bromide, DMF, RT; (iii) benzyl bromide, DMF, 60 °C. | ||
The in vitro kinase inhibitory activities of the new compounds were carefully assessed (Table 1). Monocyclic structures, such as phenyl- and 3-pyridyl-substituted compounds 5a–b, completely lost their inhibitory activities upon deletion (entries 2–3). Similarly, bicyclic structures lacking an amine in the 6-membered ring, such as indole structures 5c–d (entries 4–5), showed negligible inhibition, regardless of the presence of an N-substituent in the 5-membered ring.
|
|
||||
|---|---|---|---|---|
| Entry | Compound | Scaffold | % enzymatic activity (at 1 μM of a compound) | |
| LATS1(h) | LATS2(h) | |||
| 1 | 1 (Truli) |

|
3 ± 1 | 1 ± 1 |
| 2 | 5a |

|
106 ± 0 | 101 ± 4 |
| 3 | 5b |

|
134 ± 2 | 109 ± 1 |
| 4 | 5c |

|
145 ± 15 | 92 ± 7 |
| 5 | 5d |

|
103 ± 2 | 90 ± 0 |
| 6 | 5e |
|
114 ± 10 | 109 ± 7 |
| 7 | 5f |

|
111 ± 1 | 102 ± 7 |
| 8 | 5g |

|
106 ± 2 | 86 ± 4 |
| 9 | 5h |

|
104 ± 0 | 104 ± 0 |
| 10 | 5i |
|
94 ± 4 | 55 ± 1 |
| 11 | 5j |

|
57 ± 10 | 23 ± 3 |
| 12 | 5k |

|
21 ± 2 | 21 ± 2 |
| 13 | 5l |

|
8 ± 0 | 1 ± 1 |
| 14 | 5m |

|
99 ± 4 | 111 ± 2 |

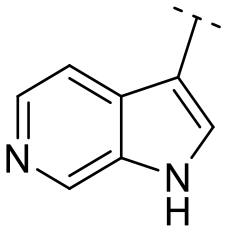
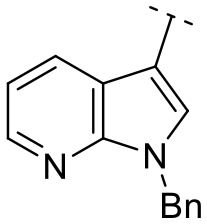
Subsequently, the importance of the position and substitution of amines in the 6-membered ring was investigated. Unlike the original structure of 1H-pyrrolo[2,3-b]pyridine (1), which bears an N at the 7-position in the 6-membered ring, both relocation structures with N at the 6-position (1H-pyrrolo[2,3-c]pyridine, 5e) and at the 5-position (1H-pyrrolo[3,2-c]pyridine, 5f) significantly diminished their inhibitory activities, irrespective of the presence of an N-substituent (entries 6–9). However, substitutions without relocating the original N-position showed decreased activities, but not universally (entries 10–11).
In summary, the positions of the two amines in 5,6-bicyclic heterocycles were crucial for demonstrating potent inhibitory activities, as confirmed by docking studies using similar X-ray structures. The data is available in ESI.†
We further explored the addition or attachment of an additional heteroatom. Synthesized compounds with an additional amine incorporated into either the 5- or 6-membered ring (5k–l) exhibited good to excellent activities (entries 12–13). However, the introduction of both amine and oxygen on 6-membered ring (5m) did not result in effective inhibition (entry 14). These results suggest that adding an amine to a ring structure could be valuable for discovering improved scaffolds.30,31 Therefore, we selected two scaffolds, 7H-pyrrolo[2,3-d]pyrimidine and 1H-pyrazolo[3,4-b]pyridine, for further validation of structure–activity relationships and drug-likeness.
Following scaffold optimization, the carboxamide part was also studied (Table 2). Compounds missing a benzyl group on the thiazole (6a and 6b) did not exhibit good activity (entries 1–2). Compounds with oxygen (6c and 6d) instead of sulfur (5k and 5l) were slightly less active (entries 3–4 in Table 2versus entries 12–13 in Table 1). Attachment of the carboxamide group at the 2-position (6e) resulted in a significant loss of activity (entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Compound | Scaffold | R | X | % enzymatic activity (at 1 μM of a compound) | |
| LATS1(h) | LATS2(h) | |||||
| 1 | 6a |

|
H | S | 82 ± 4 | 78 ± 4 |
| 2 | 6b |

|
H | S | 100 ± 4 | 103 ± 4 |
| 3 | 6c | Bn | O | 52 ± 2 | 61 ± 2 | |
| 4 | 6d |

|
Bn | O | 30 ± 3 | 28 ± 3 |
| 5 | 6e |

|
Bn | S | 96 ± 0 | 107 ± 2 |

We selected the two most active compounds to determine their IC50 values against LATS1 and LATS2. Pyrrolopyrimidine 5k exhibited nearly ten-fold lower activity, while pyrazolopyridine structure 5l demonstrated comparable results to the reference compound 1 (Table 3). Isoform selectivity was not significant in any of the cases.
| Compound | Kinase | IC50 (nM) |
|---|---|---|
| 1 (Truli) | LATS1(h) | 22 |
| LATS2(h) | 6 | |
| 5k | LATS1(h) | 265 |
| LATS2(h) | 395 | |
| 5l | LATS1(h) | 43 |
| LATS2(h) | 24 |
The results of kinase profiling for the most potent compound, 5l, demonstrated exceptional selectivity towards other protein kinases in comparison to the reported LATS inhibitors. Apart from the intended targets, only three kinases (DMPK, YSK4 and FLT3(D835V)) showed activity out of 468 kinases tested at 100 nM of 5l (Fig. 4) resulting in an excellent kinase selectivity, S(35) score of 0.007. The selectivity value of 5l is comparable to the selectivity score of the recently reported LATS inhibitor, 2, NIBR-LTSi.26 The selectivity score for Truli has not been reported, but it has been noted that Truli exhibited better binding affinity for more than 30 protein kinases out of the 314 tested, compared to its affinity for LATS1 and LATS2.23 For another reported LATS inhibitor, 3, GA-017, it was noted that it exhibited high potency against 16 out of 321 tested protein kinases.25
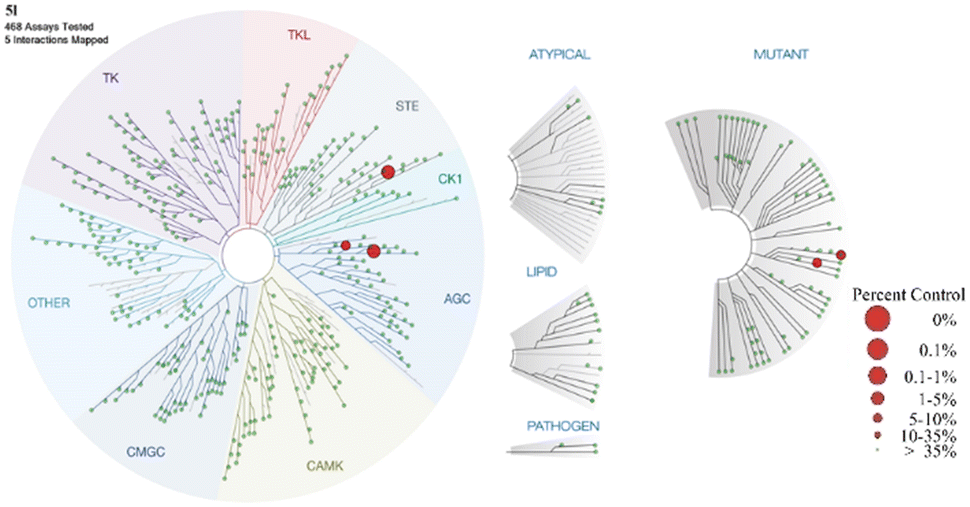 | ||
| Fig. 4 Kinome tree with 5l kinase selectivity data (data are generated using Discoverex Treespot). A large red circle indicates higher-affinity binding of numerous kinases. | ||
The drug-likeness of the two selected compounds was assessed. Compared to reference compound 1, both compounds 5k and 5l showed improved thermodynamic water solubility. According to the AI-predicted metabolic stability values obtained with using PredMS (please refer to Fig. 3), compounds 5k (HLM: stable (0.592)) and 5l (HLM: stable (0.561)) exhibited higher stability in human liver microsomes than 1, Truli (HLM: unstable (0.457)) did. In case of experimentally obtained metabolic stability values (Table 4), both compounds (5k and 5l) exhibited more than ten times higher stability in human liver microsomes than 1, Truli, consistent with the AI-predicted metabolic stabilities. It was evident that adding an amine to the ring positively influenced the compounds' drug-likeness,30,31,38 potentially making them more suitable for in vivo studies.
| Compound | 1 (Truli) | 5k | 5l |
|---|---|---|---|
| MLM phase I stability (% remaining after 30 min) | 0.18 ± 0.03 | 14 ± 0.6 | 0.69 ± 0.17 |
| HLM phase I stability (% remaining after 30 min) | 2.7 ± 0.5 | 43 ± 1.1 | 35 ± 1.3 |
| Solubility (mg mL−1) | 0.14 ± 0.026 | 0.68 ± 0.089 | 0.29 ± 0.015 |
In vivo pharmacokinetic profiles of 5k and 5l were tested using male mice. Both compounds demonstrated high bioavailability upon oral administration and excellent pharmacokinetic (PK) profile. Pharmacokinetic data on compound 5k can be found in ESI† (Fig. S42, Table S5). The reported PK data for both compounds was comparable to the recently reported LATS inhibitor, 2, NIBR-LTSi.26 Other reported LATS inhibitors, including 1, Truli, and 3, GA-017, did not provide information on their PK profiles. The selected PK parameters for 5l including volume of distribution (Vss), clearance rate (CL), half-life (T½) and bioavailability (F) are depicted in Fig. 5.
 | ||
| Fig. 5 Pharmacokinetic profile of 5l obtained using male mice. | ||
3. Conclusion
We have developed novel selective LATS inhibitors with high oral bioavailability. Using docking studies and AI-assisted prediction of metabolic stability, we identified alternative scaffolds that substantially improved drug-likeness. Interestingly, these modifications also increased selectivity towards other kinases. Biological studies on these compounds for potential applications in regenerative medicine are currently underway, specifically focusing on their ability to modulate the Hippo pathway for treating regenerative diseases. Detailed results from these studies will be reported in due course.4. Experimental part
4.1. Chemistry
For more detailed information see ESI.†The compounds 4a–b, 5a–m, 6a–e were synthesized according to the reported procedure.1,2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane (DCM) = 10:90), providing 4a as yellow oil.
:dichloromethane (DCM) = 10:90), providing 4a as yellow oil.
Yield: 67%; 1H NMR (300 MHz, CDCl3) δ 7.40–7.27 (m, 5H), 6.35 (d, J = 5.0 Hz, 1H), 5.79 (d, J = 5.0 Hz, 1H), 4.91 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 164.89, 136.71, 128.97, 127.93, 127.85, 127.08, 97.96, 49.11; LCMS (ESI) m/z: 191.00 [M + H]+.
:DCM = 10:90) or by preparatory TLC (methanol:DCM = 5:95), providing compounds 5a–f, 5k–m and 6c–e.
4.1.4.1. (Z)-N-(3-Benzylthiazol-2(3H)-ylidene)benzamide (5a). Yield: 56%; m.p.: 68–69 °C; HPLC: 95.9% (tR = 10.0 min); 1H NMR (300 MHz, CDCl3) δ 8.40–8.33 (m, 2H), 7.51–7.41 (m, 3H), 7.39–7.31 (m, 5H), 6.96 (d, J = 4.8 Hz, 1H), 6.65 (d, J = 4.7 Hz, 1H), 5.50 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 174.29, 168.28, 137.04, 135.73, 133.60, 131.61, 130.26, 129.44, 129.20, 128.58, 128.31, 128.18, 125.72, 109.54, 52.12; LC–MS (ESI) m/z: 295.1 [M + H]+.
4.1.4.2. (Z)-N-(3-Benzylthiazol-2(3H)-ylidene)nicotinamide (5b). Yield: 56%; m.p.: 69–70 °C; HPLC: 99.0% (tR = 2.7 min); 1H NMR (300 MHz, CDCl3) δ 9.54 (dd, J = 2.1, 0.9 Hz, 1H), 8.70 (dd, J = 4.8, 1.8 Hz, 1H), 8.54 (dt, J = 7.9, 2.1 Hz, 1H), 7.41–7.31 (m, 6H), 7.02 (d, J = 4.7 Hz, 1H), 6.72 (d, J = 4.7 Hz, 1H), 5.51 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 172.58, 168.27, 151.88, 151.08, 136.85, 135.38, 132.50, 129.25, 128.69, 128.23, 126.06, 123.28, 109.95, 52.30; LC–MS (ESI) m/z: 296.10 [M + H]+.
4.1.4.3. (Z)-N-(3-Benzylthiazol-2(3H)-ylidene)-1H-indole-3-carboxamide (5c). Yield: 27%; m.p.: 209–210 °C; HPLC: 99.6% (tR = 14.9 min); 1H NMR (400 MHz, CDCl3) δ 9.08 (s, 1H), 8.58–8.49 (m, 1H), 8.08 (d, J = 2.9 Hz, 1H), 7.42–7.30 (m, 6H), 7.24–7.16 (m, 2H), 6.87 (d, J = 4.8 Hz, 1H), 6.55 (d, J = 4.8 Hz, 1H), 5.48 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 173.07, 166.91, 136.88, 135.88, 131.37, 129.11, 128.38, 128.16, 126.38, 125.44, 122.54, 122.22, 121.39, 116.06, 111.77, 108.85, 51.91; LC–MS (ESI) m/z: 334.00 [M + H]+.
4.1.4.4. (Z)-N-(3-Benzylthiazol-2(3H)-ylidene)-1-methyl-1H-indole-3-carboxamide (5d). Yield: 9%; m.p.: 184–185 °C; HPLC: 99.3% (tR = 17.3 min); 1H NMR (400 MHz, CDCl3) δ 8.52 (d, J = 7.1 Hz, 1H), 7.96 (s, 1H), 7.40–7.31 (m, 5H), 7.30–7.20 (m, 3H), 6.88 (d, J = 4.8 Hz, 1H), 6.57 (d, J = 4.8 Hz, 1H), 5.51 (s, 2H), 3.84 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 172.67, 166.88, 137.73, 136.08, 135.16, 129.19, 128.46, 128.22, 127.25, 125.25, 122.68, 122.36, 121.40, 114.90, 109.58, 108.82, 51.92, 33.47; LC–MS (ESI) m/z: 348.1 [M + H]+.
4.1.4.5. (Z)-N-(3-Benzylthiazol-2(3H)-ylidene)-1H-pyrrolo[2,3-c]pyridine-3-carboxamide (5e). Yield: 29%; m.p.: 149–150 °C; HPLC: 98.2% (tR = 4.5 min); 1H NMR (500 MHz, MeOD-d4) δ 8.68 (s, 1H), 8.25 (s, 1H), 8.23 (d, J = 5.6 Hz, 1H), 8.09 (d, J = 5.6 Hz, 1H), 7.39–7.26 (m, 5H), 7.24 (d, J = 4.6 Hz, 1H), 6.83 (d, J = 4.6 Hz, 1H), 5.50 (s, 2H); 13C NMR (101 MHz, MeOD-d4) δ 173.27, 168.70, 138.20, 137.69, 137.63, 135.28, 134.12, 133.80, 129.95, 129.17, 128.78, 128.22, 118.13, 116.82, 110.51, 52.87; LC–MS (ESI) m/z: 335.00 [M + H]+.
4.1.4.6. (Z)-N-(3-Benzylthiazol-2(3H)-ylidene)-1H-pyrrolo[3,2-c]pyridine-3-carboxamide (5f). Yield: 41%; HPLC: 98.8% (tR = 4.7 min); 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H), 8.28 (d, J = 5.8 Hz, 1H), 8.25 (s, 1H), 7.62 (d, J = 4.7 Hz, 1H), 7.53 (d, J = 5.8 Hz, 1H), 7.43 (d, J = 7.1 Hz, 2H), 7.36 (t, J = 7.4 Hz, 2H), 7.29 (t, J = 7.3 Hz, 1H), 7.01 (d, J = 4.7 Hz, 1H), 5.55 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 170.43, 166.05, 143.04, 140.66, 139.45, 136.73, 132.95, 128.74, 127.84, 127.79, 127.31, 122.66, 115.31, 108.79, 107.78, 50.94; LC–MS (ESI) m/z: 335.00 [M + H]+.
4.1.4.7. (Z)-N-(3-Benzylthiazol-2(3H)-ylidene)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide (5k). Yield: 76%; m.p.: 224–225 °C; HPLC: 99.2% (tR = 3.1 min); 1H NMR (300 MHz, CDCl3) δ 10.22 (s, 1H), 9.73 (s, 1H), 8.94 (s, 1H), 8.18 (s, 1H), 7.32–7.40 (m, 5H), 6.97 (d, J = 4.7 Hz, 1H), 6.68 (d, J = 4.7 Hz, 1H), 5.53 (s, 2H); 13C NMR (101 MHz, CDCl3 + MeOD-d4) δ 171.19, 167.55, 151.85, 151.00, 150.91, 135.22, 132.07, 129.03, 128.37, 127.65, 126.09, 117.29, 114.90, 109.66, 51.99; HRMS (EI) calcd. for C17H13N5OS m/z: 335.0841, found m/z: 335.0837 [M]+.
4.1.4.8. (Z)-N-(3-Benzylthiazol-2(3H)-ylidene)-1H-pyrazolo[3,4-b]pyridine-3-carboxamide (5l). Yield: 28%; m.p.: 244–245 °C; HPLC: 98.8% (tR = 9.5 min); 1H NMR (300 MHz, CDCl3) δ 12.13 (s, 1H), 8.79 (dd, J = 8.1, 1.6 Hz, 1H), 8.62 (dd, J = 4.6, 1.6 Hz, 1H), 7.33–7.37 (m, 5H), 7.25 (tr, J = 6 Hz, 1H), 6.98 (d, J = 4.7 Hz, 1H), 6.74 (d, J = 4.7 Hz, 1H), 5.60 (s, 2H); 13C NMR (101 MHz, CDCl3 + MeOD-d4) δ 169.58, 168.24, 152.28, 148.84, 135.24, 132.92, 129.12, 128.54, 128.07, 126.22, 118.32, 115.09, 110.28, 52.24; HRMS (EI) calcd. for C17H13N5OS m/z: 335.0841, found m/z: 335.0837 [M]+.
4.1.4.9. (Z)-2-Amino-N-(3-benzylthiazol-2(3H)-ylidene)-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidine-5-carboxamide (5m). Yield: 7%; HPLC: 95.0% (tR = 1.5 min); 1H NMR (300 MHz, MeOD-d4) δ 7.66 (s, 1H), 7.34 (s, 6H), 6.61 (s, 1H), 5.42 (s, 2H); 13C NMR (101 MHz, MeOD-d4) δ 191.11, 165.76, 147.19, 135.96, 129.16, 128.63, 128.53, 127.75, 126.85, 126.19, 119.49, 106.63, 39.38; LC–MS (ESI) m/z: 367.37 [M + H]+.
4.1.4.10. (Z)-N-(3-Benzyloxazol-2(3H)-ylidene)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide (6c). Yield: 38%; m.p.: 210–211 °C; HPLC: 95.4% (tR = 1.3 min); 1H NMR (400 MHz, DMSO-d6) δ 12.52 (s, 1H), 9.39 (s, 1H), 8.79 (s, 1H), 8.13 (d, J = 2.5 Hz, 1H), 7.72 (d, J = 1.7 Hz, 1H), 7.54 (d, J = 1.7 Hz, 1H), 7.45–7.29 (m, 5H), 5.13 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 168.39, 156.38, 151.95, 151.42, 150.21, 136.00, 132.19, 132.07, 128.80, 128.00, 127.74, 117.69, 116.76, 114.81, 47.98; LC–MS (ESI) m/z: 320.10 [M + H]+.
4.1.4.11. (Z)-N-(3-Benzyloxazol-2(3H)-ylidene)-1H-pyrazolo[3,4-b]pyridine-3-carboxamide (6d). Yield: 16%; m.p.: 170–171 °C; HPLC: 99.7% (tR = 4.5 min); 1H NMR (400 MHz, DMSO-d6) δ 8.52 (d, J = 4.4 Hz, 1H), 8.48 (dd, J = 8.1, 1.6 Hz, 1H), 7.80 (d, J = 1.7 Hz, 1H), 7.61 (d, J = 1.7 Hz, 1H), 7.45–7.30 (m, 5H), 7.21 (dd, J = 8.1, 4.5 Hz, 1H), 5.16 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 166.67, 157.09, 152.34, 148.67, 141.86, 135.80, 132.55, 131.83, 128.83, 128.03, 127.61, 118.07, 118.00, 114.04, 48.17; LC–MS (ESI) m/z: 320.00 [M + H]+.
4.1.4.12. (Z)-N-(3-Benzylthiazol-2(3H)-ylidene)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide (6e). Yield: 46%; m.p.: 249–250 °C; HPLC: 95.0% (tR = 4.3 min); 1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1H), 9.12 (s, 1H), 8.84 (s, 1H), 7.70 (d, J = 4.6 Hz, 1H), 7.47 (d, J = 7.0 Hz, 2H), 7.42–7.24 (m, 5H), 7.14 (d, J = 4.6 Hz, 1H), 5.66 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 166.81, 166.51, 152.71, 151.38, 151.26, 137.08, 136.68, 128.71, 128.29, 128.23, 127.94, 127.83, 118.50, 110.04, 103.15, 50.93; LC–MS (ESI) m/z: 336.37 [M + H]+.
4.1.5.1. (Z)-N-(Thiazol-2(3H)-ylidene)-1H-pyrrolo[2,3-b]pyridine-3-carboxamide (6a). Yield: 56%; m.p.: 232–233 °C; HPLC: 97.1% (tR = 2.4 min); 1H NMR (300 MHz, DMSO-d6) δ 12.45 (s, 1H), 12.27 (s, 1H), 8.68 (d, J = 2.7 Hz, 1H), 8.54 (dd, J = 7.9, 1.7 Hz, 1H), 8.34 (dd, J = 4.7, 1.7 Hz, 1H), 7.52 (d, J = 3.6 Hz, 1H), 7.25 (dd, J = 7.9, 4.7 Hz, 1H), 7.21 (d, J = 3.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.80, 158.67, 148.62, 144.12, 137.48, 130.59, 129.36, 118.76, 117.64, 113.03, 107.08; LC–MS (ESI) m/z: 245.00 [M + H]+.
4.1.5.2. (Z)-N-(Thiazol-2(3H)-ylidene)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide (6b). Yield: 69%; m.p.: 302–303 °C; HPLC: 99.3% (tR = 0.98 min); 1H NMR (400 MHz, DMSO-d6) δ 12.86 (s, 1H), 12.46 (s, 1H), 9.47 (s, 1H), 8.89 (s, 1H), 8.71 (d, J = 2.6 Hz, 1H), 7.54 (d, J = 3.5 Hz, 1H), 7.25 (d, J = 3.5 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.03, 158.45, 152.25, 151.88, 150.18, 137.57, 131.41, 117.04, 113.36, 107.63; LC–MS (ESI) m/z: 246.00 [M + H]+.
:DCM = 10:90), providing 5i as a white solid.
Yield: 61%; m.p.: 95–96 °C; HPLC: 98.1% (tR = 17.7 min); 1H NMR (400 MHz, CDCl3) δ 8.68 (dd, J = 7.9, 1.6 Hz, 1H), 8.34 (dd, J = 4.7, 1.6 Hz, 1H), 8.03 (s, 1H), 7.35–7.20 (m, 10H), 7.15 (dd, J = 7.9, 4.7 Hz, 2H), 6.87 (d, J = 4.8 Hz, 1H), 6.56 (d, J = 4.8 Hz, 1H), 5.50 (s, 2H), 5.44 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 172.09, 167.13, 148.43, 143.70, 137.08, 135.80, 133.60, 130.84, 129.19, 128.93, 128.51, 128.12, 127.97, 127.85, 125.46, 119.67, 117.75, 114.00, 109.08, 52.02, 48.34; LC–MS (ESI) m/z: 425.3 [M + H]+.
:DCM = 10:90), providing corresponding compounds 5g–h and 5j.
4.1.7.1. (Z)-6-Benzyl-N-(3-benzylthiazol-2(3H)-ylidene)-6H-pyrrolo[2,3-c]pyridine-3-carboxamide (5g). Yield: 9%; HPLC: 97.2% (tR = 10.4 min); 1H NMR (400 MHz, CDCl3) δ 9.33 (s, 1H), 8.73 (d, J = 6.7 Hz, 1H), 8.66 (s, 1H), 8.10 (s, 1H), 7.95 (d, J = 6.7 Hz, 1H), 7.50–7.27 (m, 10H), 7.02 (d, J = 4.7 Hz, 1H), 6.72 (d, J = 4.7 Hz, 1H), 5.61 (s, 2H), 5.47 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 170.85, 169.85, 142.60, 135.82, 135.18, 132.58, 131.42, 130.44, 130.09, 129.64, 129.37, 128.80, 128.74, 127.98, 126.22, 119.85, 118.27, 109.76, 65.00, 52.36; LC–MS (ESI) m/z: 425.20 [M + H]+.
4.1.7.2. (Z)-5-Benzyl-N-(3-benzylthiazol-2(3H)-ylidene)-5H-pyrrolo[3,2-c]pyridine-3-carboxamide (5h). Yield: 64%; m.p.: 189–190 °C; HPLC: 99.0% (tR = 10.1 min); 1H NMR (300 MHz, CDCl3) δ 9.23 (s, 1H), 8.86 (s, 1H), 7.69 (d, J = 6.8 Hz, 1H), 7.52 (dd, J = 6.8, 1.9 Hz, 1H), 7.43–7.27 (m, 8H), 7.16 (dd, J = 6.8, 2.9 Hz, 2H), 6.90 (d, J = 4.8 Hz, 1H), 6.54 (d, J = 4.8 Hz, 1H), 5.44 (s, 2H), 5.37 (s, 2H); 13C NMR (101 MHz, CDCl3 + MeOD-d4) δ 172.20, 166.70, 135.75, 135.61, 134.54, 129.33, 129.21, 129.1, 129.03, 128.33, 127.86, 127.59, 125.70, 113.94, 108.75, 62.96, 51.79; LC–MS (ESI) m/z: 425.10 [M + H]+.
4.1.7.3. (Z)-7-Benzyl-N-(3-benzylthiazol-2(3H)-ylidene)-7H-pyrrolo[2,3-b]pyridine-3-carboxamide (5j). Yield: 63%; m.p.: 107–108 °C; HPLC: 96.5% (tR = 10.4 min); 1H NMR (400 MHz, CDCl3) δ 8.87 (dd, J = 7.5, 1.2 Hz, 1H), 8.75 (s, 1H), 7.59 (dd, J = 6.3, 1.2 Hz, 1H), 7.38–7.27 (m, 10H), 6.95 (dd, J = 7.5, 6.3 Hz, 1H), 6.87 (d, J = 4.8 Hz, 1H), 6.50 (d, J = 4.8 Hz, 1H), 5.88 (s, 2H), 5.44 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 172.42, 166.39, 151.60, 150.81, 136.03, 134.82, 133.59, 129.56, 129.30, 129.15, 129.03, 128.80, 128.51, 128.28, 128.16, 125.33, 115.80, 112.22, 108.37, 55.72, 51.86; LC–MS (ESI) m/z: 425.00 [M + H]+.
4.2. Molecular docking simulation and AI-prediction of metabolic stability
The molecular docking was performed on the HyperLab (https://www.hyperlab.hits.ai/en) online platform with the homology model created from the crystal structure of kinase ROCK1 bound to azaindole thiazole inhibitor34 as there is no known crystal structure of the LATS kinases. The X-ray crystal structure of ROCK1 kinase complex (PDB ID: 5KKS) was downloaded from the protein data bank (https://www.rcsb.org). The 2D structure of the analyzed compounds 1, 5a–m and 6a–e were drawn using ChemDraw software. Binding energies were automatically calculated after registering the molecular structures of the analyzed compounds.32,33KRICT-AI (pre-trained machine learning model, PredMS) platform was accessed online viahttps://predms.netlify.app/ and used to get predicted values of metabolic stabilities34 of the analyzed compounds 1, 5a–m and 6a–e. PredMS predicts metabolic stability for a given compound as stable (≥50% remaining at 30 min) or unstable (<50% remaining at 30 min) in human liver microsomes. The chemical structures of the compounds were presented in the simplified molecular-input line-entry system (SMILES) format and were further submitted for evaluation.
4.3. In vitro kinase activity assay
The Eurofins Kinase Profiler service was requested and used for obtaining in vitro kinase activity data. Kinase activity was determined at 1 μM compound concentration and at 90 μM ATP concentration for LATS1(h) and at 155 μM for LATS2(h) kinases for compounds 1, 5a–m and 6a–e. For LATS1(h) and LATS2(h), the selected kinase was incubated with 8 mM MOPS (pH 7.0), 0.2 mM EDTA, 250 μM RLGRDKYKTLRQIRQ, 10 mM magnesium acetate, and [γ-33P-ATP]. The reaction was initiated by adding the Mg/ATP mixture. After 40 minutes of incubation at room temperature, the reaction was halted by adding phosphoric acid to a final concentration of 0.5%. A 10 μL of the reaction mixture were then spotted onto a P30 filtermat, washed four times for 4 minutes each in 0.425% phosphoric acid, and one time in methanol before drying and scintillation counting. The values represent the average of two independent experiments. The IC50 data for compounds 1, 5k, and 5l was also determined using the Eurofins IC50 Profiler service at 90 μM ATP concentration for LATS1(h) and at 155 μM for LATS2(h) kinases. The IC50 curves (refer to Fig. S39–S41†) were generated using 9 test compound concentrations, diluted in half-log increments from 10 μM to 1 nM, alongside vehicle control wells. The values represent the mean of two independent experiments.4.4. Kinase profiling
A set of 468 kinase inhibitory tests was performed on the compound 5l at a concentration of 100 nM by using the scanMAX kinase assay panel of KINOMEscan. In this study, the kinase binding maps of 5l demonstrated the strength and relative specificity of kinase-binding interactions. TREEspot interaction map for compound 5l was generated online using the TREEspot™ software tool.4.5. Experimental determination of metabolic stability and thermodynamic solubility
Microsomes diluted with potassium phosphate buffer were incubated at 37 °C for 5 minutes, then the tested compound and NADPH were added and reacted at 37 °C for 30 minutes (test compound final conc.: 1 μM, microsome final conc.: 0.5 mg mL−1). To terminate the reaction, cold acetonitrile containing an internal standard was added and then treated with deproteinization. After centrifugation (4000 rpm, 4 °C, 15 min), the supernatant was analyzed by LC–MS/MS (mass spectrometry (Agilent 6460) with HPLC (Agilent 1260)).39,40 To measure solubility, 3 mg of each compound was weighed into 1.5 mL Eppi tubes. 700 μL of D2O (deuterium oxide) was then added and the sample was sonicated for 5 min and was subjected to shaking on a high-speed vibrating mixer for 24 h at rt and was centrifuged at 10000 rpm for 5 min. For analysis, the supernatant was filtered using 0.45 μm PVDF syringe filter. For the quantitation, DMSO (dimethyl sulfoxide) was used as an internal standard. The concentration of each compound in D2O was calculated based on the integration ratio of the compound signal to the internal standard DMSO signal41 (2.71 ppm, 6H) (n = 3).
4.6. In vivo pharmacokinetics
Pharmacokinetic profiles of 5k and 5l were obtained using male mice. Blood was centrifuged to separate plasma, and 9× the volume of cold acetonitrile containing an internal standard is added, followed by deproteinization. After centrifugation (13000 rpm, 4 °C, 10 min), the supernatant was analyzed by LC–MS/MS (mass spectrometry (Agilent 6460) with HPLC (Agilent 1260)).42 For the calibration curve of the compound, a 10× higher concentration solution (0.5–8000 ng mL−1) was prepared by adding it to blank plasma, and it was further prepared in the same manner as the tested compounds. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Korea Research Institute of Chemical Technology and approved by the Animal Ethics Committee of Korea Research Institute of Chemical Technology.
Data availability
All data generated or analysed during this study are included in this published article [and its ESI† files].Author contributions
Guldana Issabayeva: formal analysis, investigation, writing (original draft). On-Yu Kang: formal analysis, investigation, methodology. Seong Yun Choi: formal analysis, investigation, methodology. Ji Young Hyun: investigation, writing (review and editing). Seong Jun Park: investigation, writing (review and editing). Hei-Chul Jeung: funding acquisition, investigation, writing (review and editing). Hwan Jung Lim: funding acquisition, investigation, supervision, writing (original draft, review and editing). G. I. and O.-Y. K. contributed equally.Conflicts of interest
The authors declare that they have no known competing financial interests. The content and writing are solely responsible to the authors.Acknowledgements
This work was supported by KRICT (KK2431-20) and the National Research Foundation of Korea (NRF-2019M3E5D5066177).References
- R. W. Justice, O. Zilian, D. F. Woods, M. Noll and P. J. Bryant, The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation, Genes Dev., 1995, 9, 534–546 Search PubMed.
- T. Xu, W. Wang, S. Zhang, R. A. Stewart and W. Yu, Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase, Development, 1995, 121, 1053–1063 Search PubMed.
- M. Kango-Singh, R. Nolo, C. Tao, P. Verstreken, P. R. Hiesinger and H. J. Bellen, et al., Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila, Development, 2002, 129, 5719–5730 Search PubMed.
- N. Tapon, K. F. Harvey, D. W. Bell, D. C. Wahrer, T. A. Schiripo and D. Haber, et al., salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines, Cell, 2002, 110, 467–478 Search PubMed.
- K. F. Harvey, C. M. Pfleger and I. K. Hariharan, The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis, Cell, 2003, 114, 457–467 Search PubMed.
- J. Jia, W. Zhang, B. Wang, R. Trinko and J. Jiang, The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis, Genes Dev., 2003, 17, 2514–2519 Search PubMed.
- S. Pantalacci, N. Tapon and P. Leopold, The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila, Nat. Cell Biol., 2003, 5, 921–927 Search PubMed.
- R. S. Udan, M. Kango-Singh, R. Nolo, C. Tao and G. Halder, Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway, Nat. Cell Biol., 2003, 5, 914–920 Search PubMed.
- S. Wu, J. Huang, J. Dong and D. Pan, Hippo encodes a Ste20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts, Cell, 2003, 114, 445–456 Search PubMed.
- Z. C. Lai, X. Wei, T. Shimizu, E. Ramos, M. Rohrbaugh and N. Nikolaidis, et al., Control of cell proliferation and apoptosis by mob as tumor suppressor, mats, Cell, 2005, 120, 675–685 Search PubMed.
- J. Huang, S. Wu, J. Barrera, K. Matthews and D. Pan, The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP, Cell, 2005, 122, 421–434 Search PubMed.
- Z. Meng, T. Moroishi and K. L. Guan, Mechanisms of Hippo pathway regulation, Genes Dev., 2016, 30(1), 1–17, DOI:10.1101/gad.274027.115.
- P. C. Calses, J. J. Crawford, J. R. Lill and A. Dey, Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities, Trends Cancer, 2019, 5(5), 297–307, DOI:10.1016/j.trecan.2019.04.001.
- R. Johnson and G. Halder, The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment, Nat. Rev. Drug Discovery, 2013, 13, 63–79 Search PubMed.
- F. Fan, Z. He, L. L. Kong, Q. Chen, Q. Yuan and S. Zhang, et al., Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration, Sci. Transl. Med., 2016, 8(352), 352ra108, DOI:10.1126/scitranslmed.aaf2304.
- Y. Morikawa, T. Heallen, J. Leach, Y. Xiao and J. F. Martin, Dystrophin–glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation, Nature, 2017, 547, 227–231 Search PubMed.
- E. Bassat, Y. E. Mutlak, A. Genzelinakh, I. Y. Shadrin, K. Baruch Umansky and O. Yifa, et al., The extracellular matrix protein agrin promotes heart regeneration in mice, Nature, 2017, 547(7662), 179–184, DOI:10.1038/nature22978.
- H. Hashimoto, E. N. Olson and R. Bassel-Duby, Therapeutic approaches for cardiac regeneration and repair, Nat. Rev. Cardiol., 2018, 15, 585–600 Search PubMed.
- T. O. Monroe, M. C. Hill, Y. Morikawa, J. P. Leach, T. Heallen and S. Cao, et al., YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo, Dev. Cell, 2019, 48(6), 765–779, DOI:10.1016/j.devcel.2019.01.017.
- I. M. Moya and G. Halder, Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine, Nat. Rev. Mol. Cell Biol., 2019, 20, 211–226 Search PubMed.
- T. Moroishi, T. Hayashi, W. W. Pan, Y. Fujita, M. V. Holt and J. Qin, et al., The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity, Cell, 2016, 167(6), 1525–1539, DOI:10.1016/j.cell.2016.11.005.
- M. A. Rudolf, A. Andreeva, M. M. Kozlowski, C. E. Kim, B. A. Moskowitz and A. Anaya-Rocha, et al., YAP Mediates Hair Cell Regeneration in Balance Organs of Chickens, But LATS Kinases Suppress Its Activity in Mice, J. Neurosci., 2020, 40(20), 3915–3932, DOI:10.1523/JNEUROSCI.0306-20.2020.
- N. Kastan, K. Gnedeva, T. Alisch, A. A. Petelski, D. J. Huggins and J. Chiaravalli, et al., Small-molecule inhibition of Lats kinases may promote Yap-dependent proliferation in postmitotic mammalian tissues, Nat. Commun., 2021, 12(1), 3100, DOI:10.1038/s41467-021-23395-3.
- N. R. Kastan, S. Oak, R. Liang, L. Baxt, R. W. Myers and J. Ginn, et al., Development of an improved inhibitor of Lats kinases to promote regeneration of mammalian organs, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(28), e2206113119, DOI:10.1073/pnas.2206113119.
- A. Aihara, T. Iwawaki, N. Abe-Fukasawa, K. Otsuka, K. Saruhashi and T. Mikashima, et al., Small molecule LATS kinase inhibitors block the Hippo signaling pathway and promote cell growth under 3D culture conditions, J. Biol. Chem., 2022, 298(4), 101779, DOI:10.1016/j.jbc.2022.101779.
- K. Namoto, C. Baader, V. Orsini, A. Landshammer, E. Breuer and K. T. Dinh, et al., NIBR-LTSi is a selective LATS kinase inhibitor activating YAP signaling and expanding tissue stem cells in vitro and in vivo, Cell Stem Cell, 2024, 31(4), 554–569, DOI:10.1016/j.stem.2024.03.003.
- S. Ma, T. Tang, G. Probst, A. Konradi, C. Jin and F. Li, et al., Transcriptional repression of estrogen receptor alpha by YAP reveals the Hippo pathway as therapeutic target for ER+ breast cancer, Nat. Commun., 2022, 13, 1061, DOI:10.1038/s41467-022-28691-0.
- Y. Hu, D. Stumpfe and J. Bajorath, Recent Advances in Scaffold Hopping, J. Med. Chem., 2017, 60(4), 1238–1246, DOI:10.1021/acs.jmedchem.6b01437.
- A. Acharya, M. Yadav, M. Nagpure, S. Kumaresan and S. K. Guchhait, Molecular medicinal insights into scaffold hopping-based drug discovery success, Drug Discovery Today, 2024, 29(1), 103845, DOI:10.1016/j.drudis.2023.103845.
- D. J. Baillache and A. Unciti-Broceta, Recent developments in anticancer kinase inhibitors based on the pyrazolo [3,4-d] pyrimidine scaffold, RSC Med. Chem., 2020, 11(10), 1112–1135 Search PubMed.
- P. Halder, A. Rai, V. Talukdar, P. Das and N. R. Lakkaniga, Pyrazolopyridine-based kinase inhibitors for anti-cancer targeted therapy, RSC Med. Chem., 2024, 15(5), 1452–1470 Search PubMed.
- J. Lim, S. Ryu, K. Park, Y. J. Choe, J. Ham and W. Y. Kim, Predicting Drug-Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation, J. Chem. Inf. Model., 2019, 59(9), 3981–3988, DOI:10.1021/acs.jcim.9b00387.
- S. Moon, W. Zhung, S. Yang, J. Lim and W. Y. Kim, PIGNet: a physics-informed deep learning model toward generalized drug-target interaction predictions, Chem. Sci., 2022, 13(13), 3661–3673, 10.1039/d1sc06946b.
- J. Y. Ryu, J. H. Lee, B. H. Lee, J. S. Song, S. Ahn and K. S. Oh, PredMS: a random forest model for predicting metabolic stability of drug candidates in human liver microsomes, Bioinformatics, 2022, 38(2), 364–368, DOI:10.1093/bioinformatics/btab547.
- U. K. Bandarage, J. Cao, J. H. Come, J. J. Court, H. Gao and M. D. Jacobs, et al., ROCK inhibitors 3: Design, synthesis and structure-activity relationships of 7-azaindole-based Rho kinase (ROCK) inhibitors, Bioorg. Med. Chem. Lett., 2018, 28(15), 2622–2626, DOI:10.1016/j.bmcl.2018.06.040.
- K. Gnedeva, A. J. Hudspeth, N. Kastan, R. Liang, P. T. Meinke and D. J. Huggins, et al., Pyrrolo [2,3-b]pyridine-3-carboxamide compositions and methods for ameliorating hearing loss, WO Pat., WO2021158936A1, 2021 Search PubMed.
- M. E. Dalziel, J. J. Patel, M. K. Kaye, J. L. Cosman, M. O. Kitching and V. Snieckus, Regioselective Functionalization of 7-Azaindole by Controlled Annular Isomerism: The Directed Metalation-Group Dance, Angew. Chem., Int. Ed., 2019, 58(22), 7313–7317, DOI:10.1002/anie.201901724.
- S. Zhao, Y. Wang, X. Zhang, L. Qiao, S. Wang and Y. Jin, et al., Discovery of carboxyl-containing heteroaryldihydropyrimidine derivatives as novel HBV capsid assembly modulators with significantly improved metabolic stability, RSC Med. Chem., 2023, 14(11), 2380–2400 Search PubMed.
- R. S. Obach, Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes, Drug Metab. Dispos., 1999, 27(11), 1350–1359 Search PubMed.
- L. Di, E. H. Kerns, Y. Hong, T. A. Kleintop, O. J. McConnell and D. M. Huryn, Optimization of a higher throughput microsomal stability screening assay for profiling drug discovery candidates, J. Biomol. Screening, 2003, 8(4), 453–462, DOI:10.1177/1087057103255988.
- M. Lin, M. Tesconi and M. Tischler, Use of (1)H NMR to facilitate solubility measurement for drug discovery compounds, Int. J. Pharm., 2009, 369(1–2), 47–52, DOI:10.1016/j.ijpharm.2008.10.038.
- L. Di and E. H. Kerns, Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization, Academic press, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00492b |
| This journal is © The Royal Society of Chemistry 2024 |