
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design and in vitro testing of new urease inhibitors to prevent urinary catheter blockage†
Rachel A.
Heylen
 a,
Nicola
Cusick
a,
Tom
White
a,
Emily J.
Owen
a,
Bethany L.
Patenall
a,
Martin
Alm
c,
Peter
Thomsen
c,
Maisem
Laabei
b and
A. Toby A.
Jenkins
*a
a,
Nicola
Cusick
a,
Tom
White
a,
Emily J.
Owen
a,
Bethany L.
Patenall
a,
Martin
Alm
c,
Peter
Thomsen
c,
Maisem
Laabei
b and
A. Toby A.
Jenkins
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: a.t.a.jenkins@bath.ac.uk
bSchool of Cellular and Molecular Medicine, University of Bristol, BS8 1TD, Bristol, UK
cBiomodics ApS, Fjeldhammervej 15, 2610 Rødovre, Denmark
First published on 12th September 2024
Abstract
Catheter associated urinary tract infections (CAUTI) caused by urease-positive organisms can lead to catheter blockage: urease metabolizes urea in urine to ammonia causing an increase in pH and hence precipitation of struvite and apatite salts into the catheter lumen and bladder leading to blockage. Acetohydroxamic acid (AHA) is the only urease inhibitor currently approved for patient use, however, it is rarely used owing to its side effects. Here, we report the identification and development of new urease inhibitors discovered using a rational in silico drug design approach. A series of compounds were designed, the compounds were screened and filtered to identify three compounds which were tested in in vitro urease activity assays. N,N′-Bis(3-pyridinylmethyl)thiourea (Bis-TU) outperformed AHA in activity assays and was tested in an in vitro bladder model, where it significantly extended the lifetime of the catheter compared to AHA. Bis-TU was delivered via a diffusible balloon catheter directly to the site of activity, thus demonstrating localized drug delivery. This cost-effective drug design approach allowed the identification of a potent urease inhibitor, which could be improved through iterative repeats of the method, and the process of design could be utilized to target other diseases.
Introduction
Catheter-associated urinary tract infections (CAUTI) are widely recognized as a consequence of using a long-term indwelling urinary catheter (Foley).1 After 4 weeks, over 90% of catheter users are likely to have bacteriuria (bacteria present in the urine).1 As a result, CAUTI increases morbidity and economic burden; in the UK, CAUTI accounted for 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 717 excess NHS bed days between 2016–2017 and costs the NHS £1.50–2.25 billion per year.2–4 Globally, UTIs affect approximately 150–250 million patients per year.5 The catheter increases the risk of infection because it bypasses the bladder's natural ability to fill and void urine, therefore causing a residual pool of urine in the bladder around the tip of the catheter and the balloon. This pool of urine provides nutrients for bacterial colonization, thus increasing the likelihood of CAUTI.6 The Foley catheter is the most commonly prescribed prosthetic medical device.6 In patients, asymptomatic bacterial colonization is common; approximately 12.5% of patients develop symptomatic CAUTI, and given the high number of catheter users, this results in significant morbidity globally.7,8 Around 1 in 3 long-term urinary catheter users experience at least one, and often many, urinary catheter blockage every year; the majority of these blockages are due to urease-positive infections.7,9 Urease, the virulence factor associated with catheter blockage, metabolizes urea to ammonia and carbonic acid.10 This liberation of ammonia increases the pH within the bladder, causing precipitation of urinary salts, apatite and struvite, thus leading to the formation of crystals.11 Encrustations form on and around the catheter, resulting in occlusion of the catheter.11Proteus mirabilis is the most common urease-positive microorganism associated with catheter blockage, and is found in 80% of blocked catheters.7 A blocked urinary catheter causes painful distention of the bladder, and forces the urine up the ureters towards the kidneys, thus increasing the risk of pyelonephritis.12 Consequently, a blocked catheter increases the risk of a bloodstream infection, urosepsis, and ultimately is associated with increased mortality.12,13
717 excess NHS bed days between 2016–2017 and costs the NHS £1.50–2.25 billion per year.2–4 Globally, UTIs affect approximately 150–250 million patients per year.5 The catheter increases the risk of infection because it bypasses the bladder's natural ability to fill and void urine, therefore causing a residual pool of urine in the bladder around the tip of the catheter and the balloon. This pool of urine provides nutrients for bacterial colonization, thus increasing the likelihood of CAUTI.6 The Foley catheter is the most commonly prescribed prosthetic medical device.6 In patients, asymptomatic bacterial colonization is common; approximately 12.5% of patients develop symptomatic CAUTI, and given the high number of catheter users, this results in significant morbidity globally.7,8 Around 1 in 3 long-term urinary catheter users experience at least one, and often many, urinary catheter blockage every year; the majority of these blockages are due to urease-positive infections.7,9 Urease, the virulence factor associated with catheter blockage, metabolizes urea to ammonia and carbonic acid.10 This liberation of ammonia increases the pH within the bladder, causing precipitation of urinary salts, apatite and struvite, thus leading to the formation of crystals.11 Encrustations form on and around the catheter, resulting in occlusion of the catheter.11Proteus mirabilis is the most common urease-positive microorganism associated with catheter blockage, and is found in 80% of blocked catheters.7 A blocked urinary catheter causes painful distention of the bladder, and forces the urine up the ureters towards the kidneys, thus increasing the risk of pyelonephritis.12 Consequently, a blocked catheter increases the risk of a bloodstream infection, urosepsis, and ultimately is associated with increased mortality.12,13
Ideally, a patient using a long-term Foley catheter will be well assessed and the ‘pattern of catheter life’ recorded by community nurses or at the clinic. This assessment, in theory, is used to track the catheter's performance and therefore ensure replacement of the catheter prior to complications.14 In reality, this does not always occur and, rather than a planned catheter removal, catheters often have to be removed when blocked, this is often referred to as ‘crisis care’.15 A blocked catheter is generally treated by one of two methods, either complete removal of the catheter and re-insertion of a new catheter, or a bladder washout is performed. Removal of the catheter is often painful for patients; encrustations anchor the catheter within the bladder and removal can damage the urethra. In serious cases catheters have to be surgically removed.11,13 Bladder washout is a general term used for three types of treatment: bladder irrigation, bladder washouts, and bladder instillations. Overall, the aim of this treatment is to remove the encrustations from the bladder and therefore extend the lifetime of the catheter.14 The solutions used for washouts can vary from sterile saline to citric acid solutions which aim to decrease the pH of the bladder to dissolve the encrustations and prevent further blockage. Evidence for the efficacy of these treatments is lacking, and some reports claim that the solutions damage the mucosa lining of the bladder.13,14 Alternative care pathways, such as prescription of antibiotics (prophylactic or treatment), are generally discouraged, owing to risks of developing resistant bacteria.13,16 A long-term catheter is often colonized by more than one species of bacteria, therefore antibiotic prescription should be carefully controlled due to the increased risk of developing antimicrobial resistance in multiple bacterial species.17 However, in many clinics, antibiotics are still prescribed for long-term catheter users.13 As described in a recent Lancet review, antibiotic resistant infection was determined as a leading cause of death worldwide; this is an additional challenge for treating CAUTI.18 If possible, antibiotic treatment should be avoided to prevent resistance occuring.1 Alternative treatments for catheter blockage are: emergency catheter change or catheter maintenance by nurses.13 If the catheter blockage is not treated quickly enough then the patient's morbidity increases, and the likelihood of hospitalization increases.19
Alternative treatments to catheter changes, washouts, and antibiotics are thus of great interest. One such therapy uses urease inhibitors. Urease inhibitors act as an anti-virulence treatment, disarming the virulence factor of blockage, urease. The concept is such that urease inhibitors do not kill the bacteria, unlike antibiotics, but disarm the ability of the bacteria to metabolize urease, therefore there is no elevation in pH, no formation of crystalline biofilm and no catheter blockage.20 Acetohydroxamic acid (AHA) is the only registered urease inhibitor; it has been used to treat recurrent catheter blockages and the formation of bladder stones, as well as to treat hyperammonemia caused by Helicobacter pylori infections in the stomach.21–23 AHA has been registered in the USA, under the name Lithostat; and in Kuwait and Spain under the name Uronefrex.21,24,25 However, AHA exhibits toxic side effects including hemolytic anemia and teratogenesis (birth defects formed in an embryo/fetus).26 Therefore, the use of AHA in the clinic has decreased and it has been withdrawn from the general market.22,24 Other urease inhibitors have been explored, not just as a treatment for urease-positive bacterial infections, but also for use in agriculture to prevent nitrogen loss by ammonia volatilization.27 These identified urease inhibitors have been well reviewed by Rego et al., Kafarski and Talma, and Kappaun et al.24,28,29
In this study, we introduce an alternative methodology to identify new urease inhibitors. We propose the use of in silico docking experiments to examine the interaction of known inhibitors with urease, followed by virtual screens to identify new potential inhibitors. These inhibitors can then be sourced/synthesized for in vitro testing against urease. This method has been described as rational drug design because the screen is designed based on known inhibitors and decision points throughout the process. This is different to a traditional drug discovery approach where generally millions of random compounds are screened against a target. The hypothesis is this molecular modelling approach will yield a series of new potent inhibitors without the laborious physical synthesis and screening of many compounds.
Another important element of blockage treatment is the delivery of the drug to the target site. If administered systemically, the drug must be excreted in the urine into the bladder at a therapeutic level, but achieving this level can lead to additional side effects and the drugs can be metabolized in the liver.30 Intravesical drug delivery systems deliver the active pharmaceutical ingredient (API) directly into the bladder, where it is often used in the treatment of bladder cancer and is the principle behind bladder washouts.30 Biomodics ApS, Denmark, have developed a novel drug delivery catheter, where the drug is delivered through the catheter balloon, which is composed of an interpenetrating polymer network (IPN). A hydrophilic poly(2-hydroxyethyl methacrylate-co-poly(ethylene glycol) methyl ether acrylate) (poly(HEMA-co-PEGMEA)) network was integrated into the silicone elastomer of the catheter balloon. This formation of an IPN makes the balloon permeable, so it can be filled with a treatment solution, allowing sustained delivery into the bladder. This has been shown to be effective against Escherichia coli CAUTI and treatment of bladder cancer using a porcine model.31,32 Here we show the identification of a new urease inhibitor, identified by rational in silico drug design and subsequent validation through in vitro experiments. This inhibitor is more potent than the industrial standard, AHA, and can be delivered directly into the bladder using Biomodics' IPN catheter.
Results and discussion
In silico docking results
The compound screen was designed based on known urease inhibitors, allowing for an informed drug design process. In silico docking experiments were performed using a high resolution crystal structure of urease from Sporosarcina pasteurii (Protein Data Bank (PDB) = 4UBP).33 Ligands were docked using Cresset Flare™ software. The docking score used was a Lead Finder (LF) dG score, which has been optimized for protein–ligand binding energy, ΔG, on the assumption that the pose of the compound is correct. A more negative score predicts a better binding. Initially, to check the docking efficacy, urea and AHA were docked. This confirmed whether the docking was performing as expected and allowed comparison to the literature (Fig. S1†). Compounds were designed based on known inhibitors, then the docking score and contacts with the protein were assessed and used to design further compound series (Fig. 1 and 8, Table S1†). Care was taken with the interpretation of the docking score. | ||
| Fig. 1 Chemical structures of the series of compounds docked. Series (A) based around thiourea, (B) and (C) 2-MA, (D) quercetin, and (E) 2-MA and quercetin. Full structures can be found in Fig. S3,† compounds are drawn using ChemDraw 19.1.1.21 (PerkinElmer Informatics, Waltham, Massachusetts, US). | ||
Compounds with much higher molecular weights appear to have bias and therefore predict higher docking scores.34 Initial docking of the known inhibitor, thiourea, identified the formation of hydrogen bonds between the amine hydrogen on the ligand and Asp-383 and Gly-280, Fig. 2A (Fig. S2†). Therefore, in the design of series A, an amine hydrogen was kept, maintaining its H-bonding capacity. When compounds A1–A17 were docked, a carboxylic derivative appeared to hydrogen bond with His-222 and aligned the compound between the two Ni2+ ions. From the literature, a carboxyl group had been previously identified as coordinating with the Ni center, leading to increased urease inhibition.28 The pyridine ring also appeared to form hydrogen bond interactions between the His-222 and cation–pi interactions with N of Arg-339 (Fig. 2B). Series B was based on 2-mercaptoacetamide (2-MA), which has been shown to increase catheter lifetime in in vitro bladder models of a catheterized urinary tract.20 Furthermore, the pyridine ring from series A was also incorporated into the compounds, as it was found to increase the docking score. Surprisingly, B17 had the most negative score, with two pyridine rings (Fig. 2C). Series C was an optimization of series B. It was hypothesized that an increase in the length of the compound would improve hydrophobic interactions within the active site. Additionally, it was noted that the sulphide did not appear to be involved in contacts, so was replaced with a carbonyl. It was predicted that an oxygen would be less toxic than a sulphur and lead to fewer unwanted interactions35 (C21–24, Table S1†). The substitution of the sulphide with a carbonyl did not significantly affect the docking score, (C10 LF dG = −10.195 kcal mol−1vs. C24 (S) LF dG = −8.526 kcal mol−1), despite more contacts being identified visually, hence emphasizing the importance of visual assessments as well as analysis of the docking score (Fig. 2D) (Table S1†).
 | ||
| Fig. 2 A. Thiourea docked into the active site of urease, distance measured between the amine hydrogen and D398 and G280, indicates hydrogen bond formation. B. A11 docked into the active site, showing the hydrogen bond formation between the pyridine ring and the H222 as well as the interactions with the Ni2+ ions. C. Results from the 2-mercaptoacetamide (2-MA) series (B); docking of B17 (R) (C2 (R)) show that the pyridine ring improved docking scores as seen in series A. D. Docking of C24 (S) indicating the interactions between the surrounding amino acids: D224, R339, and H323. Ni2+ ions shown as pink spheres. Urease, PDB 4UBP, shown as green ribbon, selected amino acids as thin sticks and compound docked as thick sticks. Molecules docked with Cresset Flare v. 4.0.2. Images generated using Flare™ from Cresset®. | ||
Flavonoids have been identified as potential urease inhibitors.36 To allow comparison to in vitro literature data, five flavonoids were docked and their docking score compared to reported in vitro IC50 data (concentration of the compound which reduces the activity of urease by half) and docking experiments using Canavalia ensiformis urease.37 A more negative docking score correlated with a lower IC50 and agreed with the trend of the docking score from C. ensiformis experiments reported by Kataria and Khatkar (Fig. 3A).37 This supported the docking methodology. Chlorogenic acid appears to have greater potency, compared to the other flavonoids; the extra length was thought to improve potency, owing to the greater likelihood of more contacts in the active site being made. Therefore, for series D an extra ring was added to quercetin to increase interactions. Compound Diii2 had the most negative docking score of −11.171 kcal mol−1; it had interactions with residues within the active site and outwards towards the edge of the protein (Fig. 3B). This supported the hypothesis that flavonoids bind more favorably to the active site flap, rather than directly into the active site center, as described for quercetin by Xiao et al. (Fig. S2†).33,36
 | ||
| Fig. 3 A. LF dG (blue) is the calculated docking score based on the docking results for the flavonoids, acetohydroxamic acid (AHA), and thiourea; docking score taken from Katrina et al.37 (red) calculated against urease from Canavalia ensiformis (PDB: 3LA4); in vitro IC50 taken from Xiao et al.,36 (black). Urease (PDB: 4UBP) was used for docking experiments. Molecules docked with Cresset Flare™ v. 4.0.2. B. Compound Diii2 docked into the active site of urease, with interactions with the active site flap, C322 and H323; as well as into the active site: H222, D363, and M367. C. Compound E5 (R) docked to urease, contacts made with the active site: D363, and R339; and the active site flap: C322 and H323. Urease (PDB: 4UBP) shown as green ribbon, selected amino acids as thin sticks and compound docked as thick sticks. Molecules docked and images created using Cresset Flare v. 4.0.2. D. Lead thiourea based compounds A5 (N-phenylthiourea), A6 (benzylthiourea) and A11N,N′-bis(3-pyridinylmethyl)thiourea (Bis-TU). | ||
Analysis of docking scores from series C and D led to the conclusion that structures based on 2-MA could be used as a warhead, directly interacting within the active site and binding to residues involved in the catalytic mechanism, whilst flavonoid-based compounds interact with the active site flap. Series E incorporated the warhead, 2-MA, with the flavonoid scaffold. Compound E5 (S) had the most negative docking score (−12.902 kcal mol−1), and made interactions with Cys-322, known to be present in the mobile active site flap (Fig. 3C).38 The docking of these larger ligands was slightly limited by the size of the grid box; if these compounds are further investigated as inhibitors a larger grid box would be required for in silico experiments.
Filtering the in silico screen: identifying structure–activity relationships (SAR) and determining commercial availability. As described in Fig. 8, the compounds were then screened using Lipinski's rule of 5.39 The empirical ‘rule of 5’ states that drug-like compounds should have <5 hydrogen bond donors (HBD), <10 hydrogen bond acceptors (HBA), molecular weight <500 Da and logP <5. These are good parameters for oral drug delivery, but also apply for local delivery through the Biomodics IPN catheters; as delivery through a membrane depends on permeability, which is a product of diffusivity and solubility.40 Lipinski's rule of 5 provided a filter for the results of the in silico screen. SAR were identified; all top 20 compounds contained a carbonyl group, top compounds from series C, D, and E all contained a catechol moiety, and a pyridine ring was found in 18 of the top 20 compounds.
Hydrophobic domains were common in compounds with high docking scores, agreeing with previous studies showing hydrophobic behavior to be successful in urease inhibition.28 The top compounds identified were predominantly found in the flavonoid series (D), however assessment of the accessibility of these compounds, either by purchasing or synthesizing, highlighted that these compounds are difficult to produce. As this is the first run of what could be an iterative screening process, it was decided to focus on the thiourea series as these compounds could be purchased (Fluorochem, UK). The following compounds were purchased for in vitro testing: A5 (N-phenylthiourea), A6 (benzylthiourea), and A11 (N,N′-bis(3-pyridinylmethyl)thiourea (Bis-TU)).
In vitro testing
The three compounds: A5 (N-phenylthiourea), A6 (benzylthiourea) and A11N,N′-bis(3-pyridinylmethyl)thiourea (Bis-TU) were initially tested against urease-expressing whole cell P. mirabilis cultures. Urease in P. mirabilis is intracellular so this assay also tests the ability of the compound to cross the outer bacterial membrane and access the urease within the periplasm, moreover P. mirabilis is the principal pathogen implicated in urinary catheter blockage.41 IC50 was determined for each compound; known urease inhibitors were also run to compare against the newly identified compounds: AHA,21 2-MA,20 and quercetin (Fig. 4 and Table 1), fitting results shown in Table S3.†36 Results from the whole cell P. mirabilis urease showed that both quercetin and A11 (Bis-TU) had a ca. 25-fold lower IC50 than AHA and again A5 and A6 outperformed the control compound AHA (Table 1), although A6 was not statistically significantly lower. As A11 (Bis-TU) had the lowest IC50 of the thiourea compounds, it was selected for further study. 2-MA did not fit to a sigmoidal dose–response curve, hence IC50 could not be estimated. | ||
| Fig. 4 A. Proteus mirabilis urease activity measured with compounds: acetohydroxamic acid (AHA) (red), 2-mercaptoacetamide (2-MA) (green), quercetin (purple), A6 (benzylthiourea, brown), A5 (N-phenylthiourea, dark green), and A11 (N,N′-bis(3-pyridinylmethyl)thiourea (Bis-TU), blue). IC50 calculated by fitting response to a sigmoidal dose–response curve (see Table S3†). Experiments were completed with three biological repeats, two technical repeats, graphs show the mean of the repeats with error bars representing standard deviation. B. Plot of IC50 from fitting in A for tested compounds. Note, 2-MA did not fit to the sigmoidal dose–response curve. Graphs and fitting using Origin Pro. Error bars represent the standard deviation of four independent replicates, analyzed on GraphPad using an unpaired t test: p < 0.01 (**); p < 0.001 (***). | ||
| Proteus mirabilis | p vs. AHA | |
|---|---|---|
| IC50/mol dm−3 | ||
| AHA | 3.66 × 10−5 ± 1.58 × 10−5 | |
| A5 | 9.66 × 10−6 ± 2.54 × 10−6 | 0.0021 |
| Quercetin | 8.66 × 10−7 ± 6.47 × 10−7 | 0.0002 |
| A6 | 2.27 × 10−5 ± 9.13 × 10−6 | NS (0.0916) |
| A11 (Bis-TU) | 1.40 × 10−6 ± 8.86 × 10−7 | 0.0003 |
| 2-MA | No fit |
The urease inhibitors were designed to be anti-virulence compounds and are used to knock out the urease function and not antimicrobials. The ability of the compounds to inhibit bacteria was assessed using P. mirabilis and E. coli, common causes of CAUTI (Fig. S4, Table S2†).42 AHA and Bis-TU appeared to have only a modest inhibitory effect on E. coli and P. mirabilis growth. As almost all long-term catheter users have CAUTI (asymptomatic or symptomatic), the aim was to prevent catheter blockage rather than inhibit bacterial growth: thus reducing selection pressure for evolution of resistant bacteria.
Drug delivery
Biomodics ApS have developed a drug-delivery urinary catheter, whereby the balloon of the catheter is made of an IPN. The IPN consists of silicone, the usual material for long-term catheters, modified with the hydrogel poly(HEMA-co-PEGMEA).43 The hydrogel imparts a degree of permeability into the catheter, providing a sustained drug delivery when incorporated into the IPN.31 Therefore, the delivery system prevents immediate exposure to potentially toxic concentrations of the drug. Critically, release of the API is sustained locally to the site of infection. In comparison to oral delivery, this enables a low dose to achieve the same therapeutic effect in addition to ensuring greater bioavailability for lower solubility formulations.Additionally, local delivery can reduce unwanted side effects; AHA is toxic when delivered systemically. To demonstrate that AHA and A11 (Bis-TU) had the capability to diffuse across the balloon and be delivered into the bladder, UV-vis spectroscopy experiments were used to detect the delivery of the drugs across the balloon in artificial urine (Fig. 5). The release rate from the Biomodics IPN catheter was compared to a standard silicone catheter used by long-term catheter users. For both compounds there was no release from the standard silicone catheters above that of the limit of detection (LoD), whilst the Biomodics catheters demonstrate zero order kinetics release over the 12 h experimental period. Therefore, Bis-TU and AHA can be delivered through the Biomodics IPN catheter.
 | ||
| Fig. 5 A. Release of acetohydroxamic acid (AHA). B. Release of N,N′-bis(3-pyridinylmethyl)thiourea (Bis-TU (A11)). Measured across the balloon of the Biomodics catheter and silicone catheter over 12 h. Experiments were completed with three repeats, graphs show the mean of the repeats with error bars representing standard deviation, simple linear regression analysis has been completed to get a line of best fit and limit of detection (LOD) is shown. Graphs produced using GraphPad Prism v. 9.4.1. | ||
In vitro bladder models
The in vitro bladder model of a catheterized urinary tract enables the simulation of the crystalline biofilm and measurement of the blockage of the catheter caused by a P. mirabilis infection.44,45 The artificial bladders were infected with a high inoculum of bacteria to mimic a late-stage CAUTI, average inoculation was 3.36 × 107 CFU mL−1. Each model was catheterized with a Biomodics IPN catheter, and the balloons were inflated with 20 mM AHA in a 9:1 mixture of saline and DMSO; 20 mM Bis-TU in a 9:1 mixture of saline and DMSO; and the control bladder a 9:1 mixture of saline and DMSO. The artificial bladders were allowed to equilibrate over 18 h before inoculation. There is approximately a four-fold dilution during the diffusion of the compound in the artificial urine (∼5 mM at the start), however, this concentration changes once the pumps start because of the continual dilution from the filling of the bladder from the ‘kidneys’ and the drainage via the catheter. Monitoring of the pH and CFU mL−1 in the bladder allowed tracking of the experiment (Fig. 6A and B). Bis-TU was able to keep the pH lower than that of the AHA and control bladders (Fig. 6A), whilst the quantity of bacteria were comparable across all bladders (Fig. 6B). The blockage of the catheters was the end of the experiment and allowed determination of whether the compounds could increase the lifetime of the catheter (Fig. 6C). Bis-TU significantly outperformed the clinical standard AHA, and the control, indicating that Bis-TU has anti-ureolytic activity (unpaired t-test, GraphPad Prism 9.4.1, p = 0.0366 and p = 0.426 respectively).
 | ||
| Fig. 6 A. Monitoring of the in vitro bladder models' pH over time, comparing bladders treated with: acetohydroxamic acid (AHA), N,N′-bis(3-pyridinylmethyl)thiourea (Bis-TU) and no treatment (control). B. Monitoring the CFU mL−1 of the models over time. C. Comparing the blockage time between each of the bladder models. Experiments were completed with three repeats, graphs show the mean of the repeats with error bars representing standard deviation, * indicates a p = 0.0426 calculated by unpaired t-test. Graphs and calculations produced using GraphPad Prism v. 9.4.1. | ||
Cytotoxicity analysis
The ability of AHA and Bis-TU to cause haemolysis was assessed using an ex vivo haemolytic assay. Results indicate that Bis-TU and AHA do not cause haemolytic activity (Fig. 7A). The methyl thiazolyl tetrazolium (MTT) assay was used to test the effect of the compounds on HepG2 cells; these liver cells are often used to evaluate toxicity during pharmacological research.46 At high concentrations, 10 mM, both Bis-TU and AHA affect the survival of HepG2 cells within the 24 h of incubation. As the concentration decreases AHA appears less cytotoxic, however at concentrations below 1.25 mM there is no difference between the effect of AHA and Bis-TU (Fig. 7B). Although the concentrations used in the in vitro bladder model experiment where higher than 1.25 mM (∼5 mM) the Biomodics IPN catheter delivery mechanism means that the effective concentration in the bladder is below the toxic concentration and hence the effect of the toxicity is reduced. | ||
| Fig. 7 A. Ex vivo haemolytic assay to test the toxicity of acetohydroxamic acid (AHA) and N,N′-bis(3-pyridinylmethyl)thiourea (Bis-TU). B. HepG2 mammalian cell cytotoxicity experiment with AHA and Bis-TU assessed over 24 h. Experiments were completed with three biological repeats, graphs show the mean of the repeats with error bars representing standard deviation. Graphs and calculations produced using GraphPad Prism v. 9.4.1. | ||
Conclusions
Bis-TU is a newly identified urease inhibitor, which significantly extends the lifetime of a urinary catheter compared to AHA. It was discovered by a rationally designed in silico screen coupled with rigorous in vitro methods, thus enabling a cost-effective drug discovery process. This methodology is underpinned by the following: (1) strong previous literature to design the screen,28,29 (2) a high-resolution crystal structure, and (3) a physiologically representative in vitro model. This approach could be used to target other virulence factors to treat further diseases. Overall, we believe the targeting of bacterial virulence factors, rather than the bacteria themselves may be a pragmatic approach to development of new drugs for infection control, since it could reduce selection pressure on bacteria to evolve resistance, in contrast to antibiotic drugs.Additionally, the screening method can be repeated in an iterative manner to improve the outcome. For example, the learning from the first screen could be fed back into another round, therefore improving the potency of Bis-TU, and potentially incorporating the learning from the flavonoid screen to produce compounds which can easily be synthesized. The limitation of Bis-TU is its relatively low solubility: future work could involve the addition of an excipient which could improve solubility and could be incorporated into the balloon inflation solution to allow more effective delivery at higher concentrations. Here, we have also shown a drug delivery technique which allows directed local delivery of Bis-TU into the bladder. It should be noted that the data shows that quercetin, found in plants including watercress, has an IC50 comparable to Bis-TU, which suggests that it may have utility as a nature derived urease inhibitor suggesting the utility of a parallel drug development approach.
Experimental
Computational docking methodology
A summary of the inhibitor discovery process is shown in Fig. 8. The urease crystal structure from Sporosarcina pasteurii was used to carry out the in silico docking experiments; this had a high resolution of 1.55 Å (PDB code: 4UBP).33 Although this is not the crystal structure from the P. mirabilis urease, it has a conserved active site and has been used previously for modelling compound docking.20 Cresset Flare™ 4.0.2 (revision: 40719, Cresset, Litlington, Cambridgeshire, UK) software was used to prepare the crystal structure and complete the docking experiments.47–49 The ‘accurate but slow’ docking setting was used by the Flare software. The compound structures were prepared in ChemDraw 19.1.1.21 (PerkinElmer Informatics, Waltham, Massachusetts, US).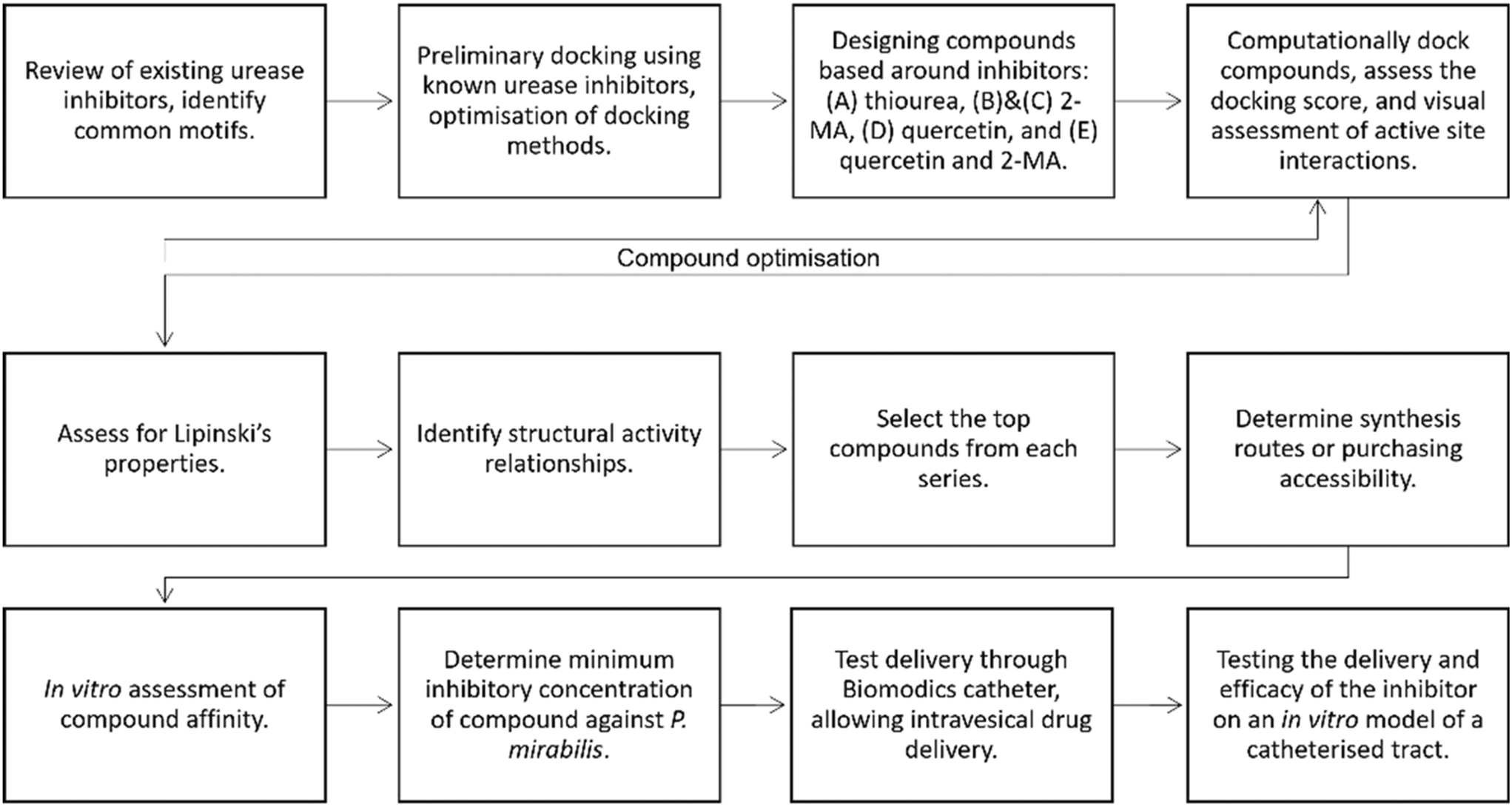 | ||
| Fig. 8 Flow diagram depicting the strategy for identifying a new urease inhibitor. | ||
A 10 Å grid box was prepared around Ni 6057 (C Ni 798) found in the active site of the protein. The crystal structure contains crystallized AHA which allowed for control docking experiments. AHA was computationally docked onto the protein, the docked coordinates of the compound were compared to the crystallized coordinates of AHA, and the root mean squared deviation (RMSD) was used to compare the coordinates. The RMSD was 0.997 Å, which indicates the AHA is well docked, as any compound with a RMSD <2 Å is considered to be performing well and would accurately predict the docking of further compounds.50 Known urease inhibitor 2-MA and the substrate, urea, were docked into the active site, allowing optimization of the method.
Designing the compound series
A series of compounds were designed with inspiration from known urease inhibitors; (A) thiourea,24,29,51,52 (B) & (C) 2-MA,20 (D) quercetin,36,53 and (E) quercetin and 2-MA.20 Compounds were assessed for their HBD and HBA groups, hydrophobic chains, aromatic groups, and sulphur-containing groups. Any compound containing chiral centers was docked in all configurations. Structures of all compounds can be found in ESI Fig. S3;† this has been summarized in Fig. 1. Contacts to the active site were assessed, and number of contacts <3.5 Å were counted. Compounds were optimized with additional changes to the R-groups depending on docking score and docking pose in the active site (Fig. 1). The design of the compounds was not limited by synthetic or purchasing accessibility, such that the design of the compounds was not constrained.In vitro assays
Experiments were run in triplicate of two technical replicates (n = 6), with 10-fold dilutions of the compounds including a positive and negative control. To each well, 20 μL of 60 mM sodium hydroxide is added. Solution A (50 μL, 106 mM phenol, 191 μM sodium nitroprusside) and solution B (50 μL, 125 mM sodium hydroxide, 125 mM sodium hypochlorite) were added to each well and the plate was incubated for 30 min at 37 °C. The absorbance of the wells was read at 636 nm (SPECTROstar Omega BMG LabTech, Germany). Urease activity was calculated using eqn (1). IC50 was determined using non-linear regression (GraphPad Prism v9).
 | (1) |
 | (2) |
:1 mixture of saline (150 mM sodium chloride) and DMSO; Bis-TU bladder: 20 mM Bis-TU in a 9:1 mixture of saline and DMSO; and AHA bladder: 20 mM AHA in a 9:1 mixture of saline and DMSO. The models were set up and filled with artificial urine until they started to drain, the pump was then paused, and the compound solution allowed to equilibrate with the artificial urine in the bladder for 18 h. P. mirabilis B4 (from the Jenkins Group bacteria collection) was grown at 37 °C overnight with 200 rpm shaking in 10 mL of LB (Merck, Germany). After 18 h of growth the culture was centrifuged at 3100g, 10 min, 4 °C (5810 R Eppendorf); the supernatant was discarded, and the pellet resuspended in PBS (Fisher Scientific, UK). Resuspended P. mirabilis was diluted ×100 with PBS, volume of 5 mL, and a sample was taken. An average of 3.7 × 107 CFU mL−1 of P. mirabilis was added to the artificial bladder, to mimic a late-stage CAUTI. Inoculation and CFU was determined by the Miles and Misra quantification technique.56 At regular time intervals during the experiment, a sample was taken aseptically from each artificial bladder for bacterial quantification and to measure the pH. The end of the experiment was determined when the bladders blocked. Additionally, the artificial bladders were monitored by time-lapse photography overnight using a Nikon D3100 camera with photos taken every 2 min.
Cytotoxicity testing
 | (3) |
Compounds, AHA and Bis-TU, were prepared at 10 mM with 1% DMSO in DMEM media. Varying concentrations of compounds were prepared by serial dilution across at 96-well plate (100 μL). MTT (Invitrogen) assay was used to measure the metabolic activity of the HepG2 cells. MTT (1 mg mL−1) in media was filter sterilized and cell culture plates, 96-well (Nunc), were seeded with 1 × 104 cells per well and grown for 24 h. Media/compound was removed and MTT (100 μL) was added for 60 min at 37 °C and then removed. Isopropanol (150 μL) was added and the plate was incubated in foil for 15 min on an orbital shaker. Absorbance was measured at 590 nm (reference filter at 620 nm) (Sunrise, Tecan). The % survival was calculated as per eqn (4).
 | (4) |
Ethics statement
All experiments involving human tissue were performed in accordance with the Declaration of Helsinki and Guidelines of the University of Bath Research Ethics Approval Committee for Health reference number: EP 18/19 108, and experiments were approved by this ethics committee at the University of Bath. Informed consents were obtained from human participants of this study, who were over 18 years of old and able to consent.Data availability
Data provided in the ESI† and on request to the corresponding author.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
The authors wish to thank their funders; Heylen, the Annette Trust and EPSRC; Patenall, the James Tudor Foundation; and Owen, Convatec Ltd. Cresset Ltd. kindly provided the software for the in silico screen.References
- S. Choong, S. Wood, C. Fry and H. Whitfield, Int. J. Antimicrob. Agents, 2001, 17, 305–310 CrossRef CAS PubMed.
- K. Getliffe and T. Newton, Age Ageing, 2006, 35, 477–481 CrossRef PubMed.
- D. R. M. Smith, K. B. Pouwels, S. Hopkins, N. R. Naylor, T. Smieszek and J. V. Robotham, J. Hosp. Infect., 2019, 103, 44–54 CrossRef CAS PubMed.
- R. C. L. Feneley, I. B. Hopley and P. N. T. Wells, J. Med. Eng. Technol., 2015, 39, 459–470 CrossRef PubMed.
- H. M. Zowawi, P. N. A. Harris, M. J. Roberts, P. A. Tambyah, M. A. Schembri, M. D. Pezzani, D. A. Williamson and D. L. Paterson, Nat. Rev. Urol., 2015, 12, 570–584 CrossRef CAS PubMed.
- D. Stickler and R. Feneley, The Indwelling Bladder Catheter: Attempts to Prevent Infection and the Development of Bacterial Biofilms, Springer Science+Business Media New York, New York, 2013 Search PubMed.
- L. E. Nicolle, Antimicrob. Resist. Infect. Control, 2014, 3, 1–8 CrossRef.
- R. Pickard, T. Lam, G. MacLennan, K. Starr, M. Kilonzo, G. McPherson, K. Gillies, A. McDonald, K. Walton, B. Buckley, C. Glazener, C. Boachie, J. Burr, J. Norrie, L. Vale, A. Grant and J. N'Dow, Lancet, 2012, 380, 1927–1935 CrossRef CAS PubMed.
- M. H. Wilde, J. M. McMahon, H. F. Crean and J. Brasch, J. Clin. Nurs., 2017, 26, 2558–2571 CrossRef PubMed.
- A. N. Norsworthy and M. M. Pearson, Trends Microbiol., 2017, 25, 304–315 CrossRef CAS PubMed.
- D. J. Stickler and R. C. L. Feneley, Spinal Cord, 2010, 48, 784–790 CrossRef CAS PubMed.
- C. E. Armbruster, H. L. T. Mobley and M. M. Pearson, EcoSal Plus, 2018, 8, 1–73 CrossRef CAS PubMed.
- T. M. Hooton, S. F. Bradley, D. D. Cardenas, R. Colgan, S. E. Geerlings, J. C. Rice, S. Saint, A. J. Schaeffer, P. A. Tambayh, P. Tenke and L. E. Nicolle, Clin. Infect. Dis., 2010, 10, 321–324 Search PubMed.
- A. Evans and H. Godfrey, Br. J. Nurs., 2000, 9, 111–114 Search PubMed.
- K. A. Getliffe, J. Adv. Nurs., 1994, 20, 140–149 CrossRef CAS PubMed.
- B. S. Niël-Weise and P. J. Van Den Broek, Cochrane Database Syst. Rev., 2012, 1–36 Search PubMed.
- M. Ramstedt, I. A. C. Ribeiro, H. Bujdakova, F. J. M. Mergulhão, L. Jordao, P. Thomsen, M. Alm, M. Burmølle, T. Vladkova, F. Can, M. Reches, M. Riool, A. Barros, R. L. Reis, E. Meaurio, J. Kikhney, A. Moter, S. A. J. Zaat and J. Sjollema, Macromol. Biosci., 2019, 19, 1–26 CrossRef PubMed.
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- J. Kohler-Ockmore and R. C. L. Feneley, Br. J. Urol., 1996, 77, 347–351 CAS.
- S. Milo, R. A. Heylen, J. Glancy, G. T. Williams, B. L. Patenall, H. J. Hathaway, N. T. Thet, S. L. Allinson, M. Laabei and A. T. A. Jenkins, Sci. Rep., 2021, 11, 3726 CrossRef CAS PubMed.
- J. R. Burns and J. F. Gauthier, J. Urol., 1984, 132, 455–456 CrossRef CAS PubMed.
- P. Kosikowska and Ł. Berlicki, Expert Opin. Ther. Pat., 2011, 21, 945–957 CrossRef CAS PubMed.
- A. Zullo, V. Rinaldi, C. Hassan, S. Folino, S. Winn, A. Pinto and A. F. Attili, Ital. J. Gastroenterol. Hepatol., 1998, 30, 496384 Search PubMed.
- K. Kappaun, A. Regina, C. Regina and R. Ligabue-braun, J. Adv. Res., 2018, 13, 3–17 CrossRef CAS PubMed.
- Food and Drug Administration FDA, Approved Drug Products with Therapeutic Equivalence Evaluations, Food and Drug Administration FDA, 40th edn, 2020 Search PubMed.
- D. P. Griffith, M. J. Gleeson, H. Lee, R. Longuet, E. Deman and N. Earle, Eur. Urol., 1991, 20, 243–247 CrossRef CAS PubMed.
- K. C. Cameron, H. J. Di and J. L. Moir, Ann. Appl. Biol., 2013, 162, 145–173 CrossRef CAS.
- Y. F. Rego, M. P. Queiroz, T. O. Brito, P. G. Carvalho, V. T. De Queiroz, Â. De Fátima and F. Macedo, J. Adv. Res., 2018, 13, 69–100 CrossRef CAS PubMed.
- P. Kafarski and M. Talma, J. Adv. Res., 2018, 13, 101–112 CrossRef CAS PubMed.
- H. Y. Yoon, H. M. Yang, C. H. Kim, Y. T. Goo, M. J. Kang, S. Lee and Y. W. Choi, Expert Opin. Drug Delivery, 2020, 17, 1555–1572 CrossRef CAS PubMed.
- K. Stærk, R. B. Grønnemose, Y. Palarasah, H. J. Kolmos, L. Lund, M. Alm, P. Thomsen and T. E. Andersen, Front. Microbiol., 2021, 12, 1–14 Search PubMed.
- K. Stærk, J. S. Hjelmager, M. Alm, P. Thomsen and T. E. Andersen, Urol. Oncol.: Semin. Orig. Invest., 2022, 40, 409.e19–409.e26 CrossRef PubMed.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, J. Biol. Inorg. Chem., 2000, 5, 110–118 CrossRef CAS PubMed.
- G. Carta, A. J. S. Knox and D. G. Lloyd, J. Chem. Inf. Model., 2007, 47, 1564–1571 CrossRef CAS PubMed.
- G. M. F. Batista, P. P. De Castro, J. A. Dos Santos, T. Skrydstrup and G. W. Amarante, Org. Chem. Front., 2021, 8, 326–368 RSC.
- Z. P. Xiao, X. D. Wang, Z. Y. Peng, S. Huang, P. Yang, Q. S. Li, L. H. Zhou, X. J. Hu, L. J. Wu, Y. Zhou and H. L. Zhu, J. Agric. Food Chem., 2012, 60, 10572–10577 CrossRef CAS.
- R. Kataria and A. Khatkar, BMC Chem., 2019, 13, 1–17 CrossRef PubMed.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, Structure, 1999, 7, 205–216 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- A. Dahan and J. M. Miller, AAPS J., 2012, 14, 244–251 CrossRef CAS.
- R. J. C. Mclean, K. J. Cheng, W. D. Gould, J. C. Nickel and W. J. Costerton, Can. J. Microbiol., 1986, 32, 772–778 CrossRef CAS PubMed.
- S. M. Jacobsen, D. J. Stickler, H. L. T. Mobley and M. E. Shirtliff, Clin. Microbiol. Rev., 2008, 21, 26–59 CrossRef CAS PubMed.
- S. L. Steffensen, M. H. Vestergaard, E. H. Møller, M. Groenning, M. Alm, H. Franzyk and H. M. Nielsen, J. Biomed. Mater. Res., Part B, 2016, 104, 402–410 CrossRef CAS PubMed.
- D. J. Stickler, N. S. Morris and C. Winters, Methods Enzymol., 1999, 310, 494–501 CAS.
- J. Nzakizwanayo, H. Pelling, S. Milo and B. V. Jones, Methods Mol. Biol., 2019, 2021, 139–158 CrossRef CAS PubMed.
- M. T. Donato, L. Tolosa and M. J. Gomez-Lechon, Methods Mol. Biol., 2015, 1250, 77–93 CrossRef CAS PubMed.
- T. Cheeseright, M. Mackey, S. Rose and A. Vinter, J. Chem. Inf. Model., 2006, 46, 665–676 CrossRef CAS PubMed.
- M. R. Bauer and M. D. Mackey, J. Med. Chem., 2019, 62, 3036–3050 CrossRef CAS PubMed.
- M. Kuhn, S. Firth-Clark, P. Tosco, A. S. J. S. Mey, M. MacKey and J. Michel, J. Chem. Inf. Model., 2020, 60, 3120–3130 CrossRef CAS PubMed.
- H. Gohlke, M. Hendlich and G. Klebe, Perspect. Drug Discovery Des., 2000, 20, 115–144 CrossRef CAS.
- L. Mazzei, F. Musiani and S. Ciurli, The Biological Chemistry of Nickel, Royal Society of Chemistry, 2017, ch. 5, Urease Search PubMed.
- K. M. Khan, F. Naz, M. Taha, A. Khan, S. Perveen, M. I. Choudhary and W. Voelter, Eur. J. Med. Chem., 2014, 74, 314–323 CrossRef CAS PubMed.
- R. Kataria and A. Khatkar, Curr. Pharm. Biotechnol., 2019, 20, 410–421 CAS.
- M. W. Weatherburn, Anal. Chem., 1967, 39, 971–974 CrossRef CAS.
- P. L. Searle, Analyst, 1984, 109, 549–568 RSC.
- A. A. Miles, S. S. Misra and J. O. Irwin, J. Hyg., 1938, 38, 732–749 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00378k |
| This journal is © The Royal Society of Chemistry 2024 |