
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Sulfonylphenoxazines as neuronal calcium ion channel blockers†
Matthieu
Schmit
 ab,
Md. Mahadhi
Hasan‡
c,
Yashad
Dongol
c,
Fernanda C.
Cardoso
c,
Michael J.
Kuiper
d,
Richard J.
Lewis
c,
Peter J.
Duggan
*be and
Kellie L.
Tuck
*a
ab,
Md. Mahadhi
Hasan‡
c,
Yashad
Dongol
c,
Fernanda C.
Cardoso
c,
Michael J.
Kuiper
d,
Richard J.
Lewis
c,
Peter J.
Duggan
*be and
Kellie L.
Tuck
*a
aSchool of Chemistry, Monash University, Victoria 3800, Australia. E-mail: Kellie.Tuck@monash.edu
bCSIRO Manufacturing, Research Way, Clayton, Victoria 3168, Australia. E-mail: Peter.Duggan@csiro.au
cInstitute for Molecular Bioscience, The University of Queensland, St. Lucia, QLD 4072, Australia
dCSIRO Data 61, Clunies Ross Street, Acton ACT 2601, Australia
eCollege of Science and Engineering, Flinders University, Adelaide, South Australia 5042, Australia
First published on 12th June 2024
Abstract
Neuropathic pain is a type of chronic pain, usually caused by nerve damage, that responds poorly to traditional pain therapies. The N-type calcium channel (CaV2.2) is a well-validated pharmacological target to treat this condition. In order to further improve the inhibition of the N-type calcium channel relative to previously described inhibitors, and also address their problematic instability in blood plasma, the development of N-sulfonylphenoxazines as new calcium channel inhibitors was pursued. A series of N-sulfonylphenoxazines bearing ammonium side chains were synthesised and tested for their ability to inhibit both CaV2.2 and CaV3.2 (T-type) neuronal ion channels. Compounds with low micromolar activity in CaV2.2 were identified, equivalent to the most effective reported for this class of bioactive, and calculations based on their physical and chemical characteristics suggest that the best performing compounds have a high likelihood of being able to penetrate the blood–brain barrier. Representative N-sulfonylphenoxazines were tested for their stability in rat plasma and were found to be much more resilient than the previously reported N-acyl analogues. These compounds were also found to be relatively stable in an in vitro liver microsome metabolism model, the first time that this has been investigated for this class of compound. Finally, molecular modelling of the CaV2.2 channel was used to gain an understanding of the mode of action of these inhibitors at a molecular level. They appear to bind in a part of the channel, in and above its selectivity filter, in a way that hinders its ability to undergo the conformational changes required to open and allow calcium ions to pass through.
Introduction
Neuropathic pain is a condition stemming from nerve damage caused by surgery, trauma, infection or disease, which results in pain, most often chronic, even in the absence of stimuli. The prevalence of severe neuropathic pain in the general population has been estimated to be as high as 5%.1 The most commonly studied types of neuropathic pain in clinical trials include peripheral diabetic neuropathy, which is caused by chronically high blood glucose levels damaging nerves and affecting up to 70% of type 2 diabetics; postherpetic neuralgia, a complication encountered in up to 20% of varicella zoster virus infections; and neuropathic cancer pain, which is attributable to nerve damage caused by the cancer itself, therapy or surgery. Other occurrences of neuropathic pain include phantom limb pain, post-stroke pain, and as a result of multiple sclerosis or spinal cord injury.2–4Neuropathic pain does not respond well to traditional pain management therapies that employ non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin or ibuprofen; in about half of the cases, only 30 to 50% pain relief is achieved. Safe and effective therapies are lacking. First line treatments typically rely on antidepressants or anticonvulsant drugs used outside of their initial indication,5 while in more severe cases opioids are employed.4,6,7 In recent decades, voltage-gated calcium channels (VGCC), in particular the N-type (CaV2.2) and T-type (CaV3.1, 3.2 and 3.3) subtypes, have emerged as promising and valid targets and have become a major focus of research into the treatment of neuropathic pain.8,9 These channels are expressed by cells involved in the transmission of action potentials characteristic of neuropathic pain; thus it is hypothesised that these inhibitors owe their pain blocking effect to shutting down such signals.10 While there are many new inhibitors undergoing clinical trials, there are currently only three drugs targeting VGCCs approved by the United States Food and Drug Administration for the treatment of neuropathic pain – gabapentin, pregabalin and ziconotide. Gabapentin and pregabalin are small amino acids, initially designed as gamma-amino butyrate (GABA) analogues for the treatment of epilepsy, and have limited effectiveness. Ziconotide, on the other hand, is a synthetic version of the peptide ω-conotoxin MVIIA found in the venom of the marine snail Conus magus and is a selective inhibitor of CaV2.2 channels. Treatment with ziconotide requires that it is injected intrathecally into the spinal fluid of the patient. While it was shown to deliver superior pain relief to that provided by morphine, with no addictive symptoms, the severe side effects and the invasive mode of administration makes ziconotide a less than ideal drug.8
Two molecules currently undergoing clinical trials are PP353 (Phase 1b, Persica Pharmaceuticals), an antibiotic formulation for treating chronic lower back pain,11 and STA363 (Phase 2b, Stayble Therapeutics), aimed at treating pain from herniated discs.12 The structures of both molecules are currently not in the public domain.
The present study stemmed from a series of iterations beginning with mimics of ω-conotoxin GVIA and proceeding towards smaller, open chain aromatic compounds,13 and then more constrained analogues, from which the acylphenoxazines 1a–1d (Fig. 1) were obtained.14 These compounds showed promising channel-blocking activity relative to positive controls, and in the case of 1b–1d, favourable central nervous system multiparameter optimisation (CNS MPO)15,16 scores. However, 1d in particular was found to be quite unstable in rat plasma, readily undergoing diacylation, and hence making these acyl derivatives unsuitable for further development. In a recently published study, a similar instability in open chain phenoxyanilides was overcome through the substitution of the amide with a more robust sulfonamide link. Interestingly, this was also associated with a marked improvement in the functional inhibition of the target CaV2.2 ion channel.17 In the current work, the acyl link in the acylphenoxazines 1b–1d was substituted for a sulfonyl moiety in a similar way. In addition, the importance of the nature of the side chain on channel blocking activity was further explored with an expanded set of terminal amines, and for the first time, an in silico model based on a recently reported CaV2.2 Cryo-EM structure18 was used to rationalise the observed results obtained with this class of compound. Finally, the stability of significant compounds in in vitro plasma and liver microsome assays was also assessed.
 | ||
| Fig. 1 Chemical structures and ion channel inhibition activity, determined by calcium influx fluorescence imaging assays, of previously reported14 acylphenoxazines 1a–1d. | ||
Results and discussion
Chemistry
The first set of sulfonylphenoxazines to be prepared had the side chain linked via an aryl ether, analogous to 1a–1d, and were synthesised in three steps from 4-(3-chloropropoxy)benzenesulfonyl chloride (2, see ESI†), as described in Scheme 1. Sulfonylation of phenoxazine with 2, was readily achieved in pyridine to yield the chloride 3 and a microwave-assisted Finkelstein reaction was subsequently used to prepare an iodide precursor. Treatment of the iodide with six different amines gave compounds 4a–4f as mono-TFA salts following RP-HPLC purification. | ||
| Scheme 1 Synthesis of sulfonylphenoxazine analogues 4a–4f. | ||
A series of sulfonylphenoxazines was also prepared where the oxygen link to the sidechain was replaced with a nitrogen, as described in Scheme 2. Sulfonylation of phenoxazine was again readily achieved in pyridine, this time with p-nitrosulfonyl chloride (5), to yield the nitro compound 6. Hydrogenation gave the aniline 7, which was acylated with 3-chloropropionyl chloride to give the amide 8. Borane reduction yielded the substituted aniline 9, which was converted to a series of diamines (10a–10f) in a one-pot Finkelstein reaction followed by alkylation of the appropriate amine. The N-methylated aniline 12 was prepared via a reductive amination to give 11, which was used to alkylate dimethylamine. Reductive amination of the aniline 7 with Boc-protected piperidone gave the Boc-protected piperidine 13, which was deprotected the give the free amine 14. All final compounds (10a–10f, 12 and 14) were purified by RP-HPLC and obtained as di-TFA salts.
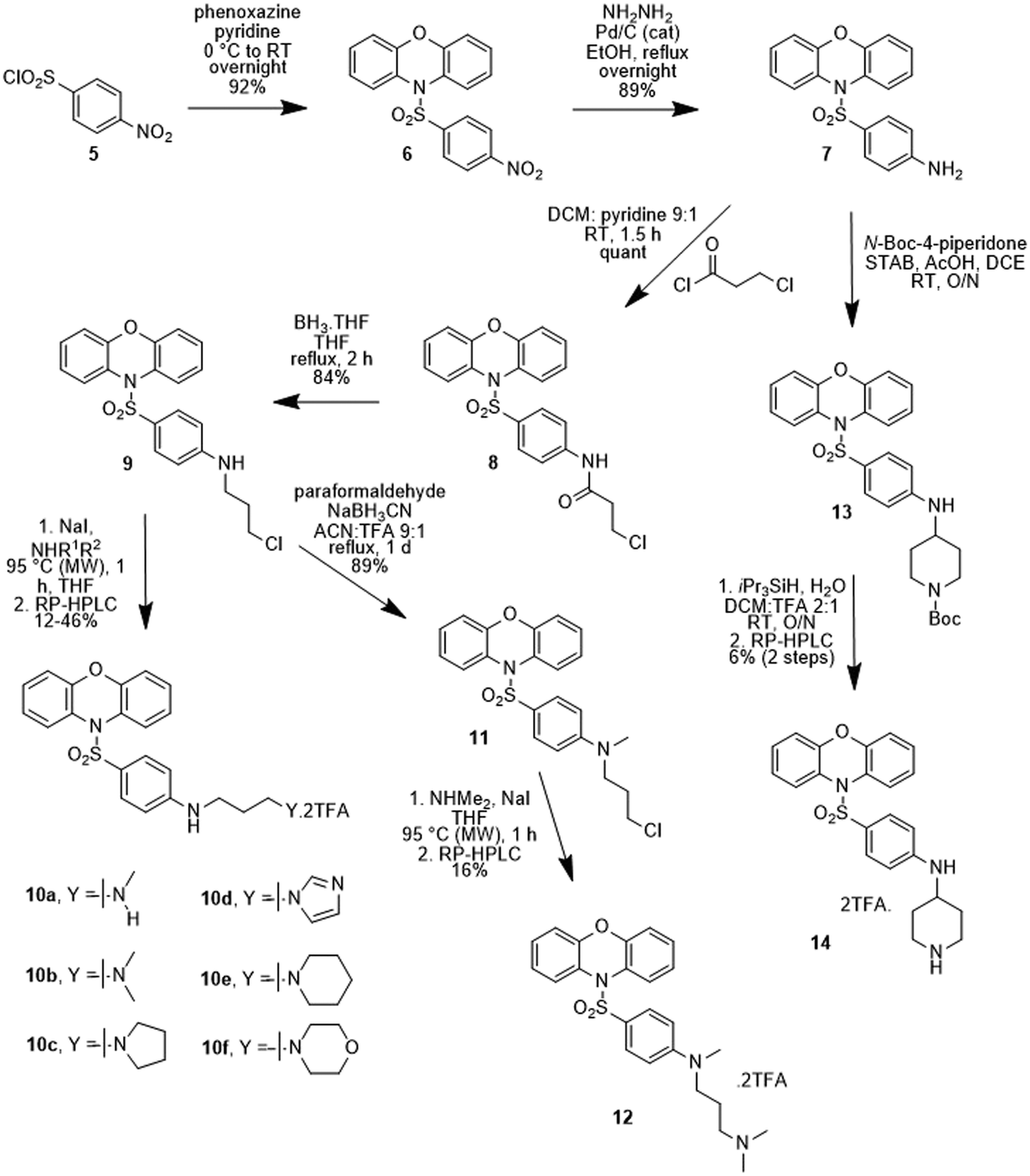 | ||
| Scheme 2 Synthesis of sulfonylphenoxazine analogues 10a–10f, 12 and 14. | ||
A methoxy analogue of the amines 4a–4f was also prepared in order to gauge the importance of the terminal amine on CaV2.2 binding affinity. The methoxy compound (15) was prepared from 3via a one-pot Finkelstein reaction followed a substitution reaction with methoxide (Scheme 3). Under the conditions of the reaction 15 was produced as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with the corresponding allyl ether, which resulted from an elimination reaction, with the two products readily separated using a combination of normal and reversed phase chromatography.
:1 mixture with the corresponding allyl ether, which resulted from an elimination reaction, with the two products readily separated using a combination of normal and reversed phase chromatography.
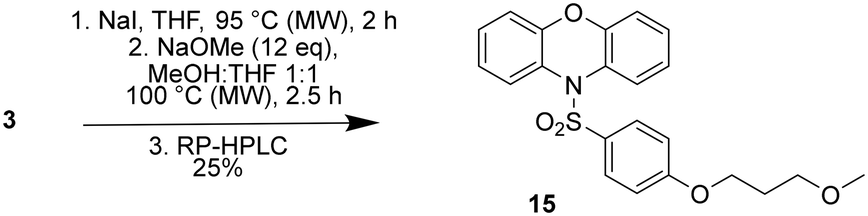 | ||
| Scheme 3 Synthesis of sulfonylphenoxazine analogue 15. | ||
Calcium ion channel inhibition studies
The ability of the sulfonylphenoxazines (4a–4f, 10a–10f, 12, 14 and 15) to inhibit functional hCaV2.2 channels was assessed with a calcium flux imaging assay with human neuroblastoma SH-SY5Y cells, as previously described.5,14,17 The assays were performed in the presence of the CaV1 blocker nifedipine and cilnidipine was used as a positive control. The determined IC50 values for 4a–4f, 10a–10f, 12 and 14, together with those previously reported for compounds 1a–1d and 16 (Fig. 2), are shown in Table 1. Good quality data was obtained for the new compounds, with the measured inhibition for the most active compounds showing narrow errors and confidence intervals. Noteworthy is the observation that the activity for compounds 4a, 4b and 10c is equivalent to the best so far recorded for this class of compound in this hCaV2.2 assay. As was observed with the previously reported open chain sulfonylamides (e.g. compound 16),17 substitution of the acyl functionality for a sulfonyl link has led to a marked improvement in activity (for example the previously reported results for acyl compounds 1b–d with those of the corresponding sulfonyl compounds 4b, 4c and 4f), with dimethylamine 4b showing >10-fold improvement cf.1b. It was hoped that the replacement of the ether linker with an amine in compounds 10a–10f, 12 and 14 would provide further hydrogen bonding opportunities in the CaV2.2 binding site occupied by these compounds (see below) and hence even stronger inhibition, but in most cases no great improvement in activity was seen, with only the imidazole 10d showing just less than a 4-fold improvement (cf.4d). The importance of the terminal amine was, however, demonstrated by the marked drop off in affinity shown by the methoxy compound 15 compared to all the corresponding sulfonylphenoxazines 4a–4f. The majority the sulfonylphenoxazines were also tested for their ability to inhibit CaV3.2 channels recombinantly expressed in HEK293T cells using a similar calcium flux imaging assay,14,17 but no strong inhibition was observed, with their measured IC50 values all falling in the 40–70 μM range (data not shown), and revealing only weak structure–activity correlations.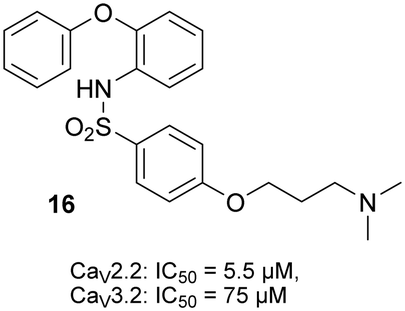 | ||
| Fig. 2 Chemical structure and ion channel inhibition activity, determined by calcium influx fluorescence imaging assays, of recently reported17 open chain sulfonylamide 16. | ||
| Compound | IC50 (μM) | SEM | 95% CI (μM) |
|---|---|---|---|
| a From ref. 14. b From ref 17. | |||
| Cilnidipine | 26 | 4 | 9–43 |
| 1a | 404 | 39 | 250–650 |
| 1b | 44 | 2 | 35–55 |
| 1c | 36 | 2 | 28–44 |
| 1d | 129 | 6 | 103–161 |
| 4a | 4.6 | 0.6 | 3.8–5.3 |
| 4b | 4.3 | 0.6 | 3.6–5.0 |
| 4c | 8.0 | 1.0 | 6.7–9.3 |
| 4d | 48.7 | 3.2 | 44.4–53.0 |
| 4e | 7.6 | 1.1 | 6.1–9.1 |
| 4f | 29.0 | 2.5 | 25.7–32.3 |
| 10a | 10.9 | 1.6 | 7.4–14.4 |
| 10b | 9.1 | 1.0 | 6.5–11.6 |
| 10c | 4.4 | 0.5 | 3.2–5.6 |
| 10d | 13.7 | 1.7 | 8.3–19.1 |
| 10e | 15.2 | 2.1 | 10.4–20.0 |
| 10f | 20.8 | 1.6 | 15.4–26.2 |
| 12 | 18.7 | 3.6 | 13.8–23.6 |
| 14 | 19.9 | 2.4 | 16.6–23.2 |
| 15 | 172 | 21 | 144–199 |
| 16 | 5.5 | 0.8 | 4.9-6.1 |
Molecular modelling
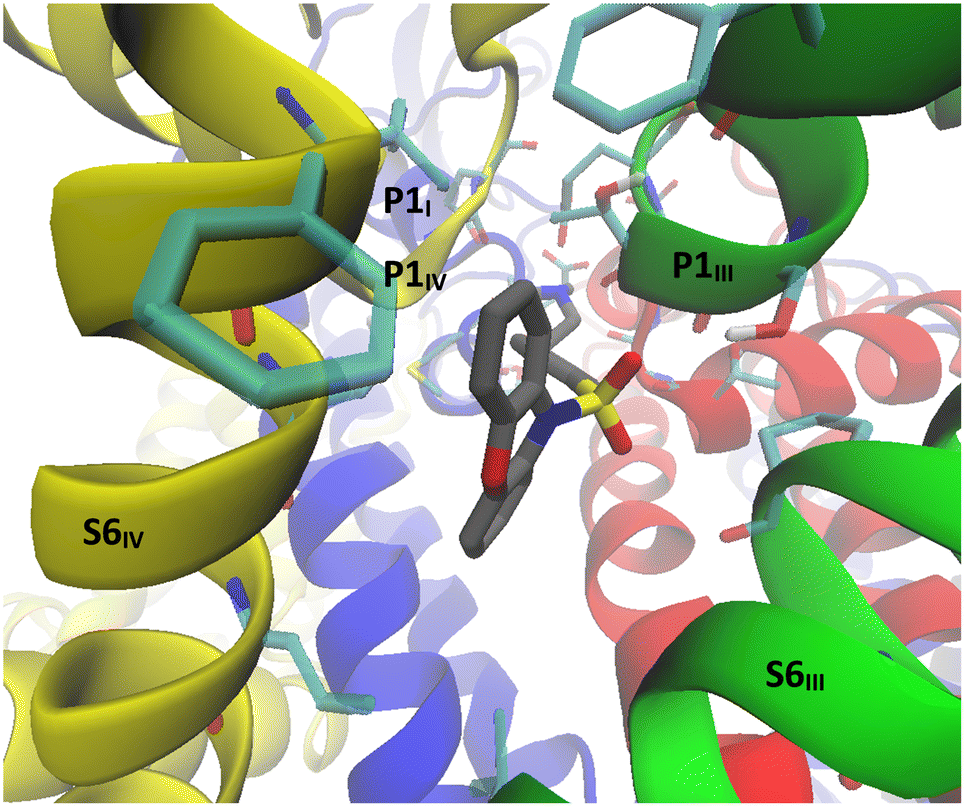
In order to gain a better understanding of how this class of CaV2.2 channel inhibitors exert their blocking effect, a computational study was undertaken. Previously, three main binding sites for calcium channel inhibitors that occur above the channel's selectivity filter were identified.5 In Fig. 3 these are designated as binding sites 1, 2 and 3 on a representation of the deactivated cryo-EM structure of CaV2.2 reported by Gao et al. (PDB:7MIY).18 Site 1, formed around the S5-P1III, P2III and P2IV segments,§ is where ω-conotoxin MVIIA was determined to bind by Gao et al.18 Site 2, consisting of the pocket between the S5III, S6III and S6IV segments, is analogous to the dihydropyridine binding site in CaV1.1 (ref. 19) and is where amitriptyline, maprotiline and other tricyclic antidepressants were predicted to bind in CaV2.2.5 Site 3, located above the S6I, S6II, S6III and S6IV segments, is analogous to the diltiazem binding site in CaV1.1 (ref. 19) and is where desipramine and opipramol were predicted to bind in CaV2.2.5 | ||
| Fig. 3 Side view of the α1 subunit of the CaV2.2 Cryo-EM structure.18 Domains I to IV are represented in blue, red, green and yellow respectively. The approximate boundaries of docking sites 1, 2 and 3 are designated in light blue, purple and orange rectangles respectively. | ||
A rigid docking study was undertaken in an attempt to identify the most likely binding sites for the N-sulfonylphenoxazines in the CaV2.2 channel and to understand better the origins of the observed trends in inhibition values. The cryo-EM structure of the channel, determined by Gao et al.,18 was imported into Schrödinger Maestro®20 and the compounds prepared in this study were then rigidly docked into the three sites designated in Fig. 3. There was no observed correlation between the docking scores obtained and experimentally determined inhibition values, but the docking scores obtained for sites 2 and 3 were consistently more favourable than those for site 1.
Images of the results obtained with the dimethylamine (4b) docked into the three binding sites are shown in Fig. 4 and S1.† Consistent with the results of the modelling studies of the binding of tricyclic antidepressants (TCAs) to CaV2.2,5 all three poses show salt-bridge and hydrogen bond interactions between the ammonium tail and the glutamate residues of the selectivity filter. Similar interactions are not possible with the methoxy analogue (15), which experimentally was found to be a two orders of magnitude weaker inhibitor than its amine analogue (4a), emphasising that the ability of the ammonium tails of these compounds to bind to the CaV2.2 selectivity filter appears to be a critical feature of their inhibition effect. The phenoxazine head group of the compounds in this study was found to be coordinated to S6 segments, which together form the internal gate of the channel. It has previously been hypothesised that close coordination of small molecule inhibitors with the S6 segments of the protein structure prevents these segments from efficiently moving away from each other when the channel opens, as having the drug desorb would be thermodynamically unfavourable.5,19 This may also explain why MONIRO-1, an earlier analogue of the compounds studied here, was found to be a state-dependent inhibitor of CaV2.2 with a higher affinity for the inactivated state,21 in which the internal gate is closed. It is not clear why MONIRO-1 would be state-dependent if it bound to CaV2.2 in docking site 1, as the P2 segments are not known to undergo conformational shifts during channel activation, unlike the S6 segments.22,23 Coordination of the phenoxazine head group of the compounds in this study to S6 segments may also explain the relatively low but significant residual activity of 15 (IC50: 172 μM). While this compound lacks the ammonium tail group to disrupt the selectivity filter, it would be able interact with the S6 segments in the same way as 4b and the other phenoxazine analogues appear to do. Therefore, most factors seem to indicate that docking sites 2 and 3 are more likely than docking site 1 to be the actual binding site of the phenoxazine compounds developed.
 | ||
| Fig. 4 Ligand interaction diagrams of protonated 4b in docking site 1 (A), docking site 2 (B), and docking site 3 (C). | ||
The substitution of the acyl link to the phenoxazine unit with a sulfonyl attachment resulted in an order of magnitude improvement in potency (compare IC50s for 1b–1d with 4b, 4c and 4f) and when 4b was docked into binding site 2 an interaction with the sulfonamide oxygens was observed. In this case Ser1362 was seen to form a hydrogen bond with one of the sulfonamide oxygens. While this was not evident with all of the sulfonylphenoxazines examined, when docked into binding site 2 (Fig. 5), the sulfonamide group was consistently found to be within 3 Å of hydrogen donor residues – either Ser1362, Thr1363 and/or Ser1696. No such association was found when the phenoxazines were docked into site 3. Based on these findings, it appears that the binding of the sulfonylphenoxazines into site 2 is primarily responsible for their inhibition of CaV2.2 ion channels. Qualitatively, docking site 1 is a larger pocket, rich in polar and charged residues, and in contact with water. It appears less suited for binding lipophilic molecules like the sulfonylphenoxazines, compared to docking site 2 and 3, which are located in the transmembrane and are rich with lipophilic residues.
 | ||
| Fig. 5 Diagram of protonated 4b docked into docking site 2 of the CaV2.2 Cryo-EM structure. | ||
The series in which the ether linkage was replaced with an amine (10a–10f, 12 and 14) was prepared in the hope that additional hydrogen bond and/or salt bridge associations would improve the affinity to the CaV2.2 channel. In most cases, this change had a limited or even slightly detrimental effect on channel affinities, with only the imidazole 10d showing a marked improvement. Consistent with this, no interactions with either the aryl ether oxygen of 4a–4f or the aniline nitrogen of 10a–10f could be detected in the structures docked into the cryo-EM structure of the channel.
In vitro metabolic stability assessments
Six sulfonylphenoxazines (4a–4f) were assessed for their stability in male rat plasma using diazepam as a positive control. In stark contrast to that previously reported for the acylphenoxazine 1b, which was found to hydrolyse with a t1/2 = 13.9 min,14 only the monomethylamine 4a showed any significant degradation, with an average of 73 ± 5% remaining after 24 hours. Greater than 90% of the other five sulfonylphenoxazines (4b–4f) remained intact after the same period. The same six sulfonylphenoxazines were also subjected to an in vitro liver microsome stability test, this time using diazepam as the control. The results are presented in Table 2 both in terms of an intrinsic clearance (CLint), which is equivalent to kcat/Km for a single enzyme reaction, and a ratio of a compound's CLint to that measured for diazepam. LCMS analysis of the assay mixtures showed that hydroxylation was a major metabolic route for all six compounds, together with demethylation for the methylamines 4a and 4b. Given that diazepam is considered to be a long-acting drug in humans, with a t1/2 = 20–35 h in the body, the methylamines 4a and 4b in particular showed promising stability.| Compound | 4a | 4b | 4c | 4d | 4e | 4f |
|---|---|---|---|---|---|---|
| CLint (μL min−1 mg−1 protein) | 34 | 105 | 83 | 132 | 97 | 239 |
| Cpd CLint/diazepam CLint | 1.8 | 2.0 | 4.4 | 6.9 | 5.1 | 12.5 |
Potential to penetrate the blood–brain barrier
An obvious consideration when attempting to develop central nervous system (CNS)-active drugs is their potential to cross the blood–brain barrier and enter the CNS. A widely used predicter developed by Pfizer scientists is the CNS MPO desirability tool,15,16 which uses a set of six physico-chemical properties to rank compounds on a scale of 0–6 in terms of their likelihood to penetrate the CNS. A score ≥4 suggests that a compound is likely to be able to enter the CNS. The CNS MPO scores calculated for the sulfonylphenoxazines prepared in this study are shown in Table 3. With the exception of the piperidine derivatives 4e and 10e, the sulfonylphenoxazines yielded highly favourable CNS MPO scores (4.0–4.5), with 4b in particular showing an improved MPO score (4.5) over the previously described open chain sulfonamide 16 (4.3).17 An MPO score of 4.5 is well within the range of many currently prescribed CNS-active drugs.16 Importantly, 4b has a similar CNS MPO score to tramadol (4.6) and is superior to the two TCAs clomipramine (3.1) and nortriptyline (3.3), and the two neuropathic pain medications gabapentin (4.2) and pregabalin (4.2).16Conclusions
As was recently reported for open chain phenoxy-substituted CaV2.2 blockers,17 the substitution of an acyl link for a sulfonyl functionality in the phenoxazine series has not only led to significantly improved stability in rat plasma, but has also seen a 4–10-fold enhancement in affinity for the target neuronal calcium ion channel, leading to compounds with equivalent effectiveness to the best reported for this class of compound. For the first time, molecular modelling based on a published CaV2.2 cryo-EM structure, has provided insight on how this class of channel blockers inhibit the passage of calcium ions through the channel. These results suggest that the ammonium side chains of these inhibitors associate with the channel's selectivity filter and amino acid residues above the selectivity filter engage with the aromatic head structure. This is a region where tricyclic antidepressants like amitriptyline have been predicted to bind, and in so doing, limit the channel's ability to transition from a closed to an open state. Hydrogen bonding to the sulfonyl functionality also appears to be important here, which would explain why the sulfonyl compounds tend to show stronger inhibition than the corresponding acyl derivatives. It was hoped that the replacement of the ether link for an amine would pick up additional interactions however in most cases improved binding strength was not observed. As indicated, most models show the ammonium side chain strongly associating with the carboxylate residues that make up the selectivity filter and consistent with this, the affinity of the methoxy analogue (15) was found to be greatly diminished relative to the corresponding methylamine (4a).Again, for the first time, this class of compound was tested for metabolic stability in an in vitro liver microsome assay. Hydroxylation and demethylation were found to be the main metabolic routes, with the rate of degradation of the methylamines 4a and 4b comparing favourably with the positive control diazepam, a drug that is considered long acting. Finally, the likelihood of the dimethylamine 4b to enter the CNS, as judged by its calculated MPO score, appears to be high, yielding a value well within the range of many CNS-active drugs and superior to the neuropathic pain medications gabapentin and pregabalin, and the related open chain analogue 16.
In summary, the replacement of the acyl link in previously reported phenoxazine-based CaV2.2 inhibitors has led to compounds with improved metabolic stability and affinity for the channel that is equivalent to the best reported for this class of compound. One of the most active compounds (4b) is also more likely to penetrate the blood brain barrier, based on its CNS MPO score. In addition, molecular modelling using a recently reported cryo-EM structure of the channel has been employed to predict where in the channel this class of inhibitor binds and how they exert their inhibitory effect. Based on our previous findings, future optimisation of the lead structure will involve exploring substitution of the aromatic rings. Additionally, future studies will investigate the state-dependency of the most active compounds using electrophysiology assays, and will test the most potent lead compound for blood–brain barrier penetration.
Experimental
Chemistry
Thin-layer chromatography (TLC) was performed using TLC Silica Gel 60 F254 and visualised under UV lamp or through the use of an appropriate stain such as potassium permanganate or ninhydrin. Melting points were recorded in an ISG melting point apparatus.
Proton nuclear magnetic resonance (1H NMR) and Fluorine nuclear magnetic resonance (19F NMR) spectra were recorded on a Bruker AV400 or AV600 as specified. The resonance shifts were assigned based on the chemical shift (δ – measured in ppm), multiplicity (s – singlet, d – doublet, t – triplet, q – quartet, etc.), number of protons, observed coupling constant (J – measured in Hz). Carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded on Bruker AV400 or AV600 at 100 or 150 MHz respectively. All chemical shifts referenced to either TMS or the residual solvent peak unless otherwise stated.
High-resolution mass spectrometry (APCI, ESI) was conducted on a Thermo Scientific QExactive FT-MS. Positive ion EI mass spectra were performed using a Thermo Scientific DFS mass spectrometer using an ionisation energy of 70 eV. Accurate mass measurements were obtained with a resolution of 5000–10000 using PFK (perfluorokerosene) as the reference compound.
High Performance Liquid Chromatography (HPLC) methods (Methods 1–5) are detailed in the Supplementary Information.
1H NMR (CDCl3, 400 MHz) δ (ppm) = 7.69 (dd, J = 7.8, 1.8 Hz, 2H), 7.26–7.12 (m, 4H), 7.07–6.98 (m, 2H), 6.84 (dd, J = 7.9, 1.6 Hz, 2H), 6.76–6.68 (m, 2H), 4.12 (t, J = 5.9 Hz, 2H), 3.74 (t, J = 6.2 Hz, 2H), 2.25 (p, J = 6.1 Hz, 2H).
13C NMR (CDCl3, 100 MHz) δ(ppm) = 162.70, 151.20, 129.93, 128.32, 128.16, 127.43, 126.45, 123.78, 116.28, 114.10, 64.67, 41.14, 31.98.
1H NMR (MeOD, 600 MHz) δ (ppm) = 7.62 (dd, J = 8.0, 1.6 Hz, 2H), 7.28 (td, J = 7.8, 1.6 Hz, 2H), 7.21 (td, J = 7.7, 1.4 Hz, 2H), 7.01–6.95 (m, 2H), 6.86 (td, J = 8.0, 1.8 Hz, 4H), 4.13 (t, J = 5.7 Hz, 2H), 3.21 (t, J = 7.3 Hz, 2H), 2.75 (s, 3H), 2.22–2.14 (m, 2H).
13C NMR (MeOD, 150 MHz) δ (ppm) = 162.75, 151.19, 129.68, 128.39, 127.72, 127.41, 126.33, 123.52, 116.01, 114.00, 65.24, 46.67, 32.45, 25.53.
HRMS (APCI): m/z calculated for [M + H]+: 411.1373, found 411.1374.
1H NMR (MeOD, 600 MHz) δ (ppm) = 7.62 (dd, J = 8.0, 1.6 Hz, 2H), 7.28 (td, J = 7.8, 1.6 Hz, 2H), 7.21 (td, J = 7.7, 1.4 Hz, 2H), 7.01–6.95 (m, 2H), 6.89–6.83 (m, 4H), 4.13 (t, J = 5.8 Hz, 2H), 3.36–3.33 (m, 2H), 2.95 (s, 6H), 2.27–2.19 (m, 2H).
13C NMR (MeOD, 150 MHz) δ (ppm) = 162.73, 151.20, 129.69, 128.39, 127.73, 127.41, 126.33, 123.52, 116.02, 113.99, 65.00, 55.10, 42.20, 24.08.
HRMS (APCI): m/z calculated for [M + H]+: 425.1530, found 425.1526.
1H NMR (MeOD, 600 MHz) δ (ppm) = 7.61 (dd, J = 8.0, 1.6 Hz, 2H), 7.28 (td, J = 7.8, 1.7 Hz, 2H), 7.20 (td, J = 7.7, 1.4 Hz, 2H), 7.00–6.95 (m, 2H), 6.87–6.81 (m, 4H), 4.11 (t, J = 5.8 Hz, 2H), 3.63–3.56 (m, 2H), 3.32–3.26 (m, 2H), 2.97 (td, J = 12.6, 3.0 Hz, 2H), 2.28–2.20 (m, 2H), 2.02–1.95 (m, 2H), 1.91–1.73 (m, 3H), 1.60–1.49 (m, 1H).
13C NMR (MeOD, 150 MHz) δ (ppm) = 162.76, 151.19, 129.70, 128.41, 127.71, 127.34, 126.31, 123.52, 116.03, 113.96, 65.06, 54.18, 53.05, 23.58, 22.90, 21.27.
HRMS (APCI): m/z calculated for [M + H]+: 451.1686, found 451.1685.
1H NMR (MeOD, 600 MHz) δ (ppm) = 8.99 (s, 1H), 7.69 (t, J = 1.8 Hz, 1H), 7.62 (dd, J = 8.0, 1.6 Hz, 2H), 7.59 (t, J = 1.7 Hz, 1H), 7.28 (td, J = 7.7, 1.8 Hz, 2H), 7.21 (td, J = 7.5, 1.3 Hz, 2H), 7.00–6.94 (m, 2H), 6.86 (dd, J = 8.1, 1.4 Hz, 2H), 6.83–6.77 (m, 2H), 4.48 (t, J = 7.0 Hz, 2H), 4.10 (t, J = 5.7 Hz, 2H), 2.40 (p, J = 6.2 Hz, 2H).
13C NMR (MeOD, 150 MHz) δ (ppm) = 162.70, 151.21, 135.23, 129.70, 128.39, 127.73, 127.37, 126.34, 123.52, 122.05, 119.84, 116.01, 113.90, 64.93, 46.53, 29.11.
HRMS (APCI): m/z calculated for [M + H]+: 448.1326, found 448.1327.
1H NMR (MeOD, 600 MHz) δ (ppm) = 7.61 (dd, J = 8.0, 1.6 Hz, 2H), 7.28 (td, J = 7.8, 1.6 Hz, 2H), 7.20 (td, J = 7.7, 1.4 Hz, 2H), 7.00–6.95 (m, 2H), 6.88–6.81 (m, 4H), 4.12 (t, J = 5.8 Hz, 2H), 3.83–3.55 (m, 2H), 3.43–3.37 (m, 2H), 3.13 (s, 2H), 2.30–1.98 (m, 6H).
13C NMR (MeOD, 150 MHz) δ (ppm) = 162.76, 151.20, 129.70, 128.40, 127.72, 127.36, 126.32, 123.52, 116.02, 113.98, 64.98, 53.96, 52.17, 48.19, 25.43, 22.57.
HRMS (APCI): m/z calculated for [M + H]+: 465.1843, found 465.1845.
1H NMR (MeOD, 600 MHz) δ (ppm) = 7.62 (dd, J = 8.0, 1.6 Hz, 2H), 7.28 (td, J = 7.9, 1.6 Hz, 2H), 7.21 (td, J = 7.7, 1.4 Hz, 2H), 7.02–6.94 (m, 2H), 6.89–6.83 (m, 4H), 4.17–4.04 (m, 4H), 3.83–3.74 (m, 2H), 3.60–3.52 (m, 2H), 3.41–3.32 (m, 2H), 3.25–3.15 (m, 2H), 2.30–2.22 (m, 2H).
13C NMR (MeOD, 150 MHz) δ (ppm) = 162.71, 151.20, 129.72, 128.39, 127.73, 127.47, 126.34, 123.53, 116.00, 113.96, 64.93, 63.69, 54.46, 51.92, 23.27.
HRMS (APCI): m/z calculated for [M + H]+: 467.1635, found 467.1636.
MP: 170–172 °C.
1H NMR (CDCl3, 400 MHz) δ (ppm) = 8.15–8.07 (m, 2H), 7.70 (dd, J = 7.8, 1.8 Hz, 2H), 7.33–7.18 (m, 6H), 6.86 (dd, J = 7.9, 1.6 Hz, 2H).
13C NMR (CDCl3, 100 MHz) δ (ppm) = 151.09, 141.00 (HMBC), 129.02, 128.98, 127.96, 125.67, 124.24, 123.56, 116.65.
MP: 110–112 °C.
1H NMR (CDCl3, 400 MHz) δ (ppm) = 7.67 (dd, J = 7.8, 1.8 Hz, 2H), 7.24–7.12 (m, 5H), 6.90–6.79 (m, 4H), 6.49 (d, J = 8.5 Hz, 2H).
13C NMR (CDCl3, 100 MHz) δ 151.24, 150.93, 129.87, 128.22, 128.14, 126.65, 123.95, 123.66, 116.19, 113.58.
1H NMR (CDCl3, 400 MHz) δ (ppm) = 7.58 (dd, J = 7.8, 1.8 Hz, 2H), 7.41–7.35 (m, 2H), 7.16–7.05 (m, 5H), 6.98–6.91 (m, 2H), 6.74 (dd, J = 7.9, 1.6 Hz, 2H), 3.78 (t, J = 6.3 Hz, 2H), 2.76 (t, J = 6.3 Hz, 2H).
13C NMR (CDCl3, 100 MHz) δ (ppm) = 168.01, 151.17, 142.28, 130.48, 129.11, 128.50, 128.05, 126.22, 123.87, 118.64, 116.43, 40.52, 39.41.
1H NMR (CDCl3, 400 MHz) δ (ppm) = 8.55 (s, 1H), 7.58 (dd, J = 7.9, 1.8 Hz, 2H), 7.15–7.00 (m, 4H), 6.80–6.68 (m, 4H), 6.30–6.16 (m, 2H), 3.55 (t, J = 6.4 Hz, 2H), 3.25 (t, J = 6.7 Hz, 2H), 2.02–1.87 (m, 2H).
13C NMR (CDCl3, 100 MHz) δ (ppm) = 151.88, 151.25, 147.56, 139.00, 129.86, 128.24, 128.11, 126.72, 125.30, 123.64, 122.49, 116.16, 111.14, 42.25, 40.38, 31.54.
1H NMR (MeOD, 400 MHz) δ (ppm) = 7.49 (dd, J = 7.9, 1.6 Hz, 2H), 7.17 (td, J = 7.8, 1.6 Hz, 2H), 7.08 (td, J = 7.7, 1.5 Hz, 2H), 6.77 (dd, J = 8.1, 1.4 Hz, 2H), 6.70–6.61 (m, 2H), 6.37–6.29 (m, 2H), 3.13 (td, J = 8.2, 7.5, 4.2 Hz, 4H), 2.62 (s, 3H), 1.97–1.85 (m, 2H).
13C NMR (MeOD, 100 MHz) δ (ppm) = 152.94 (HMBC), 151.29, 129.48, 128.14, 127.78, 126.62, 123.30, 120.93, 115.93, 110.51, 55.55, 42.13, 39.26, 23.69.
HRMS (APCI): m/z calculated for [M + H]+: 410.1533, found 410.1533.
1H NMR (MeOD, 400 MHz) δ 7.59 (dd, J = 7.9, 1.6 Hz, 2H), 7.22 (dtd, J = 32.4, 7.6, 1.6 Hz, 4H), 6.89–6.72 (m, 4H), 6.49–6.34 (m, 2H), 3.26–3.17 (m, 4H), 2.90 (s, 6H), 2.06–1.93 (m, 2H).
13C NMR (MeOD, 100 MHz) δ 152.97 (HMBC), 151.28, 129.49, 128.11, 127.80, 126.64, 123.30, 121.03, 115.90, 110.48, 55.56, 42.12, 39.24, 23.71.
HRMS (APCI): m/z calculated for [M + H]+: 424.1689, found 424.1690.
1H NMR (MeOD, 400 MHz) δ (ppm) = 7.59 (dd, J = 7.9, 1.7 Hz, 2H), 7.31–7.13 (m, 4H), 6.86 (dd, J = 8.1, 1.5 Hz, 2H), 6.78–6.71 (m, 2H), 6.46–6.37 (m, 2H), 3.71–3.62 (m, 2H), 3.30–3.20 (m, 6H), 3.14–3.03 (m, 2H), 2.23–1.95 (m, 6H).
13C NMR (MeOD, 100 MHz) δ (ppm) = 152.92 (HMBC), 151.29, 129.49, 128.10, 127.79, 126.66, 123.29, 121.00 (HMBC), 115.90, 110.48, 53.91, 52.73, 39.33, 25.12, 22.57.
HRMS (APCI): m/z calculated for [M + H]+: 450.1846, found 450.1843.
1H NMR (MeOD, 600 MHz) δ (ppm) = 8.77 (s, 1H), 7.62–7.57 (m, 3H), 7.51 (s, 1H), 7.26 (td, J = 7.7, 1.7 Hz, 2H), 7.18 (td, J = 7.7, 1.5 Hz, 2H), 6.86 (dd, J = 8.1, 1.4 Hz, 2H), 6.78–6.72 (m, 2H), 6.41–6.35 (m, 2H), 4.34 (t, J = 7.2 Hz, 2H), 3.17 (t, J = 6.7 Hz, 2H), 2.18 (p, J = 6.9 Hz, 2H).
13C NMR (MeOD, 100 MHz) δ 153.00, 151.29, 135.07, 129.46, 128.14, 127.78, 126.62, 123.30, 121.87, 120.22, 115.93, 110.46, 104.98, 46.88, 39.06, 28.87.
HRMS (APCI): m/z calculated for [M + H]+: 447.1485, found 447.1485.
1H NMR (CD3CN, 600 MHz) δ 7.47 (dd, J = 8.0, 1.6 Hz, 2H), 7.17 (td, J = 7.8, 1.6 Hz, 2H), 7.08 (td, J = 7.7, 1.4 Hz, 2H), 6.77 (dd, J = 8.1, 1.4 Hz, 2H), 6.62 (d, J = 8.9 Hz, 2H), 6.31–6.24 (m, 2H), 3.33 (d, J = 12.2 Hz, 2H), 3.02 (t, J = 6.6 Hz, 2H), 2.98–2.90 (m, 2H), 2.66 (d, J = 14.1 Hz, 2H), 1.87 (dd, J = 9.9, 4.9 Hz, 2H), 1.79–1.58 (m, 6H).
13C NMR (CD3CN, 150 MHz) δ 153.66, 151.87, 130.35, 129.28, 128.66, 127.32, 124.46, 121.63, 116.97, 111.55, 55.22, 53.70, 40.40, 23.61, 23.37, 22.07.
HRMS (ESI): m/z calculated for [M + H]+: 464.2002, found: 464.2004.
1H NMR (MeOD, 600 MHz) δ (ppm) = 7.59 (dd, J = 8.0, 1.6 Hz, 2H), 7.25 (td, J = 7.7, 1.6 Hz, 2H), 7.18 (td, J = 7.7, 1.4 Hz, 2H), 6.86 (dd, J = 8.1, 1.4 Hz, 2H), 6.79–6.73 (m, 2H), 6.45–6.39 (m, 2H), 4.12–4.02 (m, 2H), 3.81–3.71 (m, 2H), 3.50 (d, J = 12.5 Hz, 2H), 3.28–3.21 (m, 4H), 3.19–3.09 (m, 2H), 2.07–1.98 (m, 2H).
13C NMR (MeOD, 150 MHz) δ (ppm) = 152.89, 151.28, 129.49, 128.10, 127.78, 126.64, 123.28, 121.06, 115.91, 110.48, 63.64, 55.01, 51.86, 39.30, 22.89.
HRMS (APCI): m/z calculated for [M + H]+: 466.1795, found 466.1794.
1H NMR (CD3CN, 600 MHz) δ 7.53 (dd, J = 8.0, 1.6 Hz, 2H), 7.21 (td, J = 7.7, 1.6 Hz, 2H), 7.13 (td, J = 7.7, 1.4 Hz, 2H), 6.81 (dd, J = 8.1, 1.4 Hz, 2H), 6.74–6.69 (m, 2H), 6.47–6.41 (m, 2H), 3.34 (t, J = 7.4 Hz, 2H), 2.97–2.89 (m, 2H), 2.86 (s, 3H), 2.67 (s, 6H), 1.88–1.83 (m, 2H + CD3CN).
13C NMR (CD3CN, 150 MHz,) δ 153.27, 151.86, 130.16, 129.25, 128.65, 127.34, 124.43, 121.18, 116.98, 111.16, 55.40, 49.31, 43.06, 38.24, 22.28.
HRMS (ESI): m/z calculated for [M + H]+: 438.1846, found: 438.1849.
1H NMR (MeOD, 600 MHz) δ (ppm) = 7.59 (dd, J = 8.0, 1.6 Hz, 2H), 7.25 (td, J = 7.9, 1.4 Hz, 2H), 7.18 (td, J = 7.6, 1.4 Hz, 2H), 6.86 (dd, J = 8.1, 1.4 Hz, 2H), 6.79–6.73 (m, 2H), 6.49–6.43 (m, 2H), 3.67–3.61 (m, 1H), 3.48–3.41 (m, 2H), 3.14 (td, J = 12.6, 3.1 Hz, 2H), 2.20 (dd, J = 14.6, 3.8 Hz, 2H), 1.67 (dtd, J = 14.2, 10.6, 3.9 Hz, 2H).
13C NMR (MeOD, 150 MHz) δ (ppm) = 151.76, 151.31, 129.54, 128.09, 127.80, 126.66, 123.28, 121.11, 115.90, 110.81, 46.20, 42.67, 28.30.
HRMS (APCI): m/z calculated for [M + H]+: 421.1455, found 421.1456.
1H NMR (600 MHz, Methanol-d4) δ 7.61 (dd, J = 7.9, 1.6 Hz, 2H), 7.31–7.25 (m, 2H), 7.20 (td, J = 7.7, 1.4 Hz, 2H), 6.97–6.92 (m, 2H), 6.88 (dd, J = 8.1, 1.4 Hz, 2H), 6.84–6.79 (m, 2H), 4.60 (s, 3H), 4.08 (t, J = 6.3 Hz, 2H), 3.56 (t, J = 6.2 Hz, 2H), 2.03 (p, J = 6.2 Hz, 2H).
13C NMR (150 MHz, MeOD) δ 163.44, 151.25, 129.64, 128.37, 127.70, 126.63, 126.36, 123.43, 116.05, 113.91, 68.54, 65.04, 57.48, 28.90.
HRMS (ESI): m/z calculated for [M + H]+: 412.1213, found: 412.1211.
Biological evaluations
000 and 10000 cells per well, respectively, in 384 well flat clear-bottom black plates (Corning, NY, USA) and cultured at 37 °C in a humidified 5% CO2 incubator 48 h before assay. Cells were loaded with 20 μL per well of Calcium 6 dye (Molecular Devices) reconstituted in assay buffer containing (in mM) 140 NaCl, 11.5 glucose, 5.9 KCl, 1.4 MgCl2, 1.2 NaH2PO4, 5 NaHCO3, 1.8 CaCl2,10 HEPES pH 7.4 and 0.1% bovine serum albumin (BSA, Sigma), and incubated for 30 min at 37 °C in a humidified 5% CO2 incubator. Nifedipine 10 μM (CaV1 blocker) was added to the dye solution for CaV2.2 assay. The Ca2+ fluorescence responses were recorded at excitation 470–495 nm and emission 515–575 nm for 10 s to set the baseline, 300 s after addition of compound and for further 300 s after channel activation induced by the addition of 90 mM KCl and 5 mM CaCl2 for CaV2.2, or 40 mM KCl and 5 mM CaCl2 for CaV3.2. Compound stock solutions were prepared at 100 mM in 100% DMSO and diluted further in the assay buffer to 1% DMSO, for the highest tested concentration of 100 μM of compound, and serial-diluted 3-fold in assay buffer.
Rat plasma stability assessments
Six sulfonylphenoxazines (4a–4f) were assessed for their stability in male rat plasma using diazepam as a positive control, using a protocol adapted from that previously reported.14 Male rat plasma sourced from Sprague Dawley was purchased from Monash Animal Research Platform (MARP) and stored at −80 °C. Samples were defrosted at room temperature prior to usage. Male rat blood plasma (597 μL) was incubated at 37 °C for thirty minutes, then 3 μL of a 30 mM solution of the compound of interest (final concentration: 150 μM) and diazepam (internal standard, final concentration: 150 μM) in DMSO were added. The mixture was left incubating at 37 °C for 24 h; aliquots of 40 μL were removed at t = 0, 5, 15, 30, 45, 60, 75, 90, 105, 120, 180 and 3600 min, and immediately quenched by adding 40 μL of ACN. The samples were vortexed, diluted with 120 μL of deionized water, vortexed again, cooled on ice for 30 min and finally centrifuged at 10000 rcf for 15 min. The supernatant solution was then collected and stored at 4 °C until analysis by HPLC following Method 4.
For each compound of interest, the experiment was repeated in triplicate alongside one positive control experiment with diltiazem. The amount of compound of interest remaining at the various time points was determined from the peak area ratio of compound to diazepam. The data was then plotted and, where appropriate, fitted to a one-phase decay model using GraphPad Prism 8.0.2. Representative chromatograms are included in ESI.†
Liver microsome stability assessments
Phosphate-buffered saline (277 μL, pH = 7.4) was incubated at 37 °C for twenty minutes, then 32 μL of a freshly prepared 10 mM NADPH solution (final concentration: 1 mM), 3.2 μL of a 100 μM stock solution of the compound of interest (ACN 1:1 water, final concentration 1 μM) and 8 μL of a solution of a 20 mg mL−1 male rat liver microsomes (Sigma-Aldrich: reference number M9066; stored at −80 °C until used; final concentration 0.5 mg mL−1) were added. The mixture was left incubating at 37 °C for one hour; aliquots of 30 μL were removed at t = 0, 5, 10, 15, 20, 25, 30, 40, and 60 min, and immediately quenched by adding 60 μL of a solution of 0.5% TFA in methanol. The samples were vortexed, cooled on ice for 30 min and finally centrifugated at 10000 rcf for 15 min. The supernatant solution was then collected and analysed by LC/HRMS following Method 5.
For each compound of interest, the experiment was repeated in triplicate alongside one positive control experiment with diazepam. For each sample, the chromatogram of the ion corresponding to the compound of interested was extracted. A calibration curve was made in order to determine the amount of compound of interest remaining at each time point. The data was then plotted and fitted to a one-phase decay model using GraphPad Prism 8.0.2. Representative chromatograms are attached in ESI.†
Computational methods
The cryo-EM structure of the α1 subunit of the N-type calcium channel determined by Gao et al. (PDB: 7MIY)18 was imported into Schrödinger Maestro® (version 2021-3).20 The Small-Molecule Drug Discovery Suite was used to perform docking studies. Using the receptor grid generation utility, docking grids for the three sites of interest were generated by defining a 20 × 20 × 20 Å3 box around the centroid of the following segments (approximately represented on Fig. 3):• Docking site 1: P2III and P2IV segments.
• Docking site 2: S5III, S6III and S6IV segments.
• Docking site 3: lower parts of the S6I, S6II, S6III and S6IV segments.
The structures of the compounds of interest were then imported and their possible ionization states at pH = 7.4 ± 2.0 were generated using the LigPrep utility. Rigid docking of the compounds of interest was performed with the docking grid corresponding to sites 1, 2 and 3 using the Glide Ligand Docking utility. Docking scores were exported and plotted in GraphPad 8.0.2 for analysis. Fig. 3 and 5 were generated using the Visual Molecular Dynamics (VMD) 1.9.3 package. VMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign.26
Author contributions
Synthesis: MS. Calcium influx bioassays: MH, YD, FCC. Rat plasma stability and liver microsomal stability: MS. Data analysis: MS, FCC, MH, YD, RJL. Computational methods: MS, MK. Manuscript preparation: PJD, KLT. Project conception and supervision: PJD, KLT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Monash University and CSIRO are acknowledged for funding. This work was also supported by a NHMRC Program Grant (APP1072113, RJL) and NHMRC Fellowship (1119056, RJL). We gratefully acknowledge Dr David Chalmers (MIPS, Monash University, Australia) for providing access to Schrödinger software.Notes and references
- D. Bouhassira, M. Lantéri-Minet, N. Attal, B. Laurent and C. Touboul, Pain, 2008, 136, 380 CrossRef PubMed.
- A. S. Fisher, M. T. Lanigan, N. Upton and L. A. Lione, Front. Pharmacol., 2021, 11, 614990 CrossRef PubMed.
- S. Y. Yoon and J. Oh, Korean J. Intern. Med., 2018, 33, 1058 CrossRef CAS PubMed.
- S. Sindrup, M. Otto, N. B. Finnerup and T. S. Jensen, Basic Clin. Pharmacol. Toxicol., 2005, 96, 399 CrossRef CAS PubMed.
- F. C. Cardoso, M. Schmit, M. J. Kuiper, R. J. Lewis, K. L. Tuck and P. J. Duggan, RSC Med. Chem., 2022, 13, 183 RSC.
- K. E. Galluzzi, J. Am. Osteopath. Assoc., 2005, 105, S12 Search PubMed.
- G. Cruccu and A. Truini, Pain and Therapy, 2017, 6, 35 CrossRef PubMed.
- T. P. Snutch, NeuroRx, 2005, 2, 662 CrossRef PubMed.
- P. Beswick, 7.03 – Progress in the Discovery of Ca Channel Blockers for the Treatment of Pain A2 – Chackalamannil, Samuel, in Comprehensive Medicinal Chemistry III, ed. D. Rotella and S. E. Ward, Elsevier, Oxford, 2017, pp. 65–130 Search PubMed.
- R. Patel, C. Montagut-Bordas and A. H. Dickenson, Br. J. Pharmacol., 2018, 175, 2173 CrossRef CAS PubMed.
- https://www.biospace.com/article/releases/persica-pharmaceuticals-completes-recruitment-into-modic-trial-assessing-efficacy-of-pp353-to-treat-chronic-lower-back-pain/, viewed 5th June 2024.
- https://staybletherapeutics.com/research/, viewed 5th June 2024.
- P. J. Duggan and K. L. Tuck, Toxins, 2015, 7, 4175 CrossRef CAS PubMed.
- F. C. Cardoso, M.-A. Marliac, C. Geoffroy, M. Schmit, A. Bispat, R. J. Lewis, K. L. Tuck and P. J. Duggan, Bioorg. Med. Chem., 2020, 28, 115655 CrossRef CAS PubMed.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2010, 1, 435 CrossRef CAS PubMed.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2016, 7, 767 CrossRef CAS PubMed.
- A. S. Bispat, F. C. Cardoso, M. M. Hasan, Y. Dongol, R. Wilcox, R. J. Lewis, P. J. Duggan and K. L. Tuck, RSC Med. Chem., 2024, 15, 916 RSC.
- S. Gao, X. Yao and N. Yan, Nature, 2021, 596, 143 CrossRef CAS PubMed.
- Y. Zhao, G. Huang, J. Wu, Q. Wu, S. Gao, Z. Yan, J. Lei and N. Yan, Cell, 2019, 177, 1495 CrossRef CAS PubMed.
- Schrödinger Release 2020–3: Maestro, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- J. R. McArthur, L. Motin, E. C. Gleeson, S. Spiller, R. J. Lewis, P. J. Duggan, K. L. Tuck and D. J. Adams, Br. J. Pharmacol., 2018, 175, 2284 CrossRef CAS PubMed.
- S. Hering, E. M. Zangerl-Plessl, S. Beyl, A. Hohaus, S. Andranovits and E. N. Timin, Pflugers Arch., 2018, 470, 1291 CrossRef CAS PubMed.
- S. Beyl, P. Kügler, M. Kudrnac, A. Hohaus, S. Hering and E. Timin, J. Gen. Physiol., 2009, 134, 231 CrossRef CAS PubMed.
- D. Pedersen and C. Rosenbohm, Synthesis, 2001, 16, 2431 Search PubMed.
- H. Fukuda, F. Karaki, K. Dodo, T. Noguchi-Yachide, M. Ishikawa, Y. Hashimoto and K. Ohgane, Bioorg. Med. Chem. Lett., 2017, 27, 2781 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of 2, NMR spectra, rat plasma stability and liver microsomal stability studies. See DOI: https://doi.org/10.1039/d4md00336e |
| ‡ Current address: Pharmacy Discipline, Life Science School, Khulna University, Khulna, 9208, Bangladesh. |
| § The abbreviations used to identify the various segments and loops in the CaV2.2 protein structure are defined in a previous publication.5 |
| This journal is © The Royal Society of Chemistry 2024 |