
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeveraging bounded datapoints to classify molecular potency improvements†
Zachary
Fralish
 ,
Paul
Skaluba
and
Daniel
Reker
*
,
Paul
Skaluba
and
Daniel
Reker
*
Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA. E-mail: daniel.reker@duke.edu
First published on 31st May 2024
Abstract
Molecular machine learning algorithms are becoming increasingly powerful at predicting the potency of potential drug candidates to guide molecular discovery, lead series prioritization, and structural optimization. However, a substantial amount of inhibition data is bounded and inaccessible to traditional regression algorithms. Here, we develop a novel molecular pairing approach to process this data. This creates a new classification task of predicting which one of two paired molecules is more potent. This novel classification task can be accurately solved by various, established molecular machine learning algorithms, including XGBoost and Chemprop. Across 230 ChEMBL IC50 datasets, both tree-based and neural network-based “DeltaClassifiers” show improvements over traditional regression approaches in correctly classifying molecular potency improvements. The Chemprop-based deep DeltaClassifier outperformed all here evaluated regression approaches for paired molecules with shared and with distinct scaffolds, highlighting the promise of this approach for molecular optimization and scaffold-hopping.
Introduction
Major efforts are invested to optimize molecular potency during drug design. However, bottlenecks due to comparatively slow chemical syntheses during optimization often limit broader exploration of various chemical structures. To streamline synthesis and testing, molecular machine learning methods are increasingly employed to learn from historic data to prioritize the acquisition and characterization of new molecules.1However, during data generation, a substantial fraction of molecules is still incompletely characterized, leading to the reporting of bounded values in place of exact ones. Specifically, compound screening is often performed in a two-step process, where a large set of compounds is tested at a single concentration and only the most promising hits are further evaluated in full dose–response curves to determine IC50 values. This results in a substantial fraction of datapoints not being annotated with their exact IC50 values but instead with lower bounds. Conversely, upper bounds might be created through insufficient experimental resolution or solubility limits. In total, one fifth of the IC50 datapoints in ChEMBL datasets are bounded values (Fig. 1A).
 | ||
| Fig. 1 Schematic of classification approaches to handle bounded data (A) 230 IC50 datasets were analysed from ChEMBL33 that included molecules with exact and bounded values, the latter making up 20% of all datapoints. (B) Traditional regression models cannot incorporate bounded datapoints into training. (C) Using traditional regression models, classifications of predicted improvements can be calculated by first subtracting predictions for each molecule and then subsequently determining a class value by assessing the sign of the potency differences. (D) Pairwise model approaches can train on classified improvements from pairs of molecules, allowing for incorporation of bounded datapoints. (E) DeltaClassifiers can directly predict molecular improvements of molecular derivatizations from paired molecular representations. | ||
Furthermore, as the positive reporting bias imbalances available IC50 data towards the most potent compounds, incorporation of compounds with more mild activity could help counteract skewed class proportions and provide valuable chemical diversity during training (Table S1†).
Regression methods can be used to steer molecular optimization and discovery by predicting the potency of two molecules and comparing these predictions to select the molecule with higher predicted potency (Fig. 1C). However, as regression algorithms cannot train on bounded data, they only use a subset of the available training data with limited diversity (Fig. 1B, Table S1†).
We previously showed that leveraging pairwise molecular representations as training data for the established deep learning algorithm Chemprop can improve the prediction of the absorption, distribution, metabolism, excretion, and toxicity (ADMET) property differences between molecules compared to using state-of-the-art single molecule machine learning approaches.2 We hypothesized that we could extend this pairing approach into a novel classification task where the algorithm is tasked to predict which of the two paired molecules is more potent. This pairing would enable us to access bounded datapoints by pairing them with other molecules that are known to be more or known to be less potent (Fig. 1D). Providing this data to established classification algorithms can create a predictive tool that directly contrasts molecules to guide molecular optimization and discovery while incorporating all of the available training data (Fig. 1E).
Here, we evaluate the ability of established classification algorithms to learn from this paired data (which we call “DeltaClassifiers” when they are provided with our paired data) and compare their ability to correctly compare potencies of two molecules compared to simply using three established, state-of-the-art regression algorithms, namely the tree-based random forest,3 the gradient boosting method XGBoost,4 and the directed message passing neural network (D-MPNN) Chemprop.5,6 Across 230 ChEMBL IC50 datasets, both tree-based and neural network-based classification algorithms that train on paired representations exhibit improved performance over traditional regression approaches when classifying potency improvements. We believe that the DeltaClassifier concept and further extensions thereof will be able to access greater ranges of data to support drug design more accurately.
Results and discussion
Training deep models with bounded data
We hypothesized that using a paired approach to directly train on and classify molecular potency improvements would not only allow for the incorporation of bounded IC50 data into training, but also improve overall model performance. To evaluate this hypothesis, we created a novel machine learning task wherein molecular pairs function as the datapoints instead of individual molecules (Fig. 1D). The target variable is created by comparing the potency of the paired molecules and assigning class “1” when the second molecule is more potent and “0” otherwise (i.e., the first molecule is more potent or both molecules have equal potency). In other words, our classification tool answers the question “Is the second molecule more potent than the first molecule?” (Fig. 1D). If it is unknown if the potency is improved (e.g., both IC50 values in the pair are upper bounds), the pair is removed. Further, to account for experimental noise, we used a ‘demilitarized’ training approach where only molecular pairs with differences greater than 0.1 pIC50 were used for training and testing. This is to avoid training the model on potentially statistically insignificant potency differences as well as excluding data where the label could easily “flip” due to experimental uncertainty. Following filtering, any machine learning classification model capable of accepting two molecular inputs can be trained on this data to classify if the second molecule exhibits an improvement in potency over the first molecule.To evaluate the models, we used cross-validation to randomly split our ChEMBL benchmarking datasets into training and testing sets (Fig. S1†). Within each split, molecules were cross-merged to form all possible pairs and classified according to their ground-truth potency difference while filtering inconclusive and uncertain pairs as described above.
First, we tested the performance of the two-molecule implementation of the established D-MPNN Chemprop5 to solve this new classification task. For ease of readability, we call the predictive pipeline consisting of our molecular pair pre-processing approach and the established two-molecule Chemprop “DeepDeltaClassifier” (DΔC). Across 230 IC50 datasets, we found promising performance of this new approach for classifying molecular potency improvements with an average area under the receiver operating characteristic curve (ROCAUC) of 0.91 ± 0.04, ranging from 0.68–0.98, and average accuracy of 0.84 ± 0.04, ranging from 0.62–0.92, (Fig. 2, Tables 1 and S2, ESI† 2). With a ROCAUC never below 0.68 and reaching up to 0.98, it appears our approach is broadly applicable to various types of targets with very strong performance on select targets of clinical relevance. This encouraging performance highlights the ability of the Chemprop D-MPNN machine learning model to accurately solve our novel task, with high potential to serve as a guiding tool for molecular optimization.
 | ||
| Fig. 2 DeepDeltaClassifier performance following training with only exact values (DΔCOE), all data (DΔCAD), and demilitarized data (DΔC) tested on demilitarized data. (A) Violin plots of model performance following 1 × 10 cross-validation for 230 ChEMBL datasets in terms of accuracy, F1 score, and ROCAUC. (B) Pie charts showing percentage of datasets DΔC outcompeted DΔCOE and DΔCAD. | ||
| Model type | Model | Accuracy | F1 score | ROCAUC |
|---|---|---|---|---|
| Traditional (single molecule regression models) | Random forest | 0.80 ± 0.06 | 0.80 ± 0.06 | 0.87 ± 0.06 |
| XGBoost | 0.79 ± 0.06 | 0.79 ± 0.06 | 0.86 ± 0.07 | |
| Chemprop | 0.75 ± 0.07 | 0.75 ± 0.07 | 0.82 ± 0.08 | |
| DeltaClassifier (two molecule classification models) | ΔCL (XGBoost) | 0.82 ± 0.05 | 0.82 ± 0.05 | 0.90 ± 0.05 |
| DΔC (Chemprop) | 0.84 ± 0.04 | 0.84 ± 0.04 | 0.91 ± 0.04 |
To assess the impact of our demilitarization, we analogously implemented DΔC but trained on all data without filtering pairs with potency differences smaller than 0.1 pIC50 (DΔC all data, abbreviated as DΔCAD). DΔC and DΔCAD exhibited overall comparable performance with no significant difference between DΔC and DΔCAD for accuracy (p = 0.054), slight improvement for DΔC for F1 (p = 0.002), and slight improvement for DΔCAD for AUC (p = 0.003, Fig. 2, Table S2, ESI† 2) when evaluating on demilitarized test sets. Similar trends were observed for test sets with all pairs included (Table S3†) and all pairs except the same molecule pairs that are always classified as “0” (Table S4†). As DΔC accounts for experimental noise by training only on pairs with larger differences and shows no drop in performance with fewer training datapoints, we believe that our demilitarization is an appropriate pre-processing step to prepare datasets for training of algorithms to classify molecular potency improvements.
Since it is known that IC50 data has substantial variability,7,8 we also assessed whether stricter (i.e., larger) thresholds would provide further benefits to the model. To this end, we created additional models that were trained only on potency differences larger than 0.5 pIC50 and 1.0 pIC50. When evaluated on a test set that included all data to provide a uniform evaluation, these larger buffer zones led to a decrease in performance (p < 0.0001, Table S5†) compared to DΔC. This continued to be true when “trivial” same molecule pairs that are always classified as “0” were removed from the test set (p < 0.0001, Table S6†). This data suggests that our demilitarization of 0.1 pIC50 is sufficient to account for experimental error and potentially benefits from more data compared to stricter thresholds.
Finally, to determine if training on bounded datapoints improved performance compared to just training on the exact IC50 values, we analogously implemented the DΔC but trained only on molecular pairs with exact values (DΔC only equal, abbreviated as DΔCOE). DΔC significantly outperformed DΔCOE (p < 0.0001) across all metrics (Fig. 2, Table S2, ESI† 2). Similar trends were observed for test sets without filtering of low pIC50 differences (Table S3†) and without filtering but removal of same molecule pairs (Table S4†) highlighting how training on bounded datapoints can improve overall model performance. This suggests that our novel machine learning task can enable models to incorporate additional data into the model that significantly boosts performance. Adversarial Y-shuffling effectively disrupted chemical pattern and inhibitor relationships, leading to a collapse in DΔC performance as expected (Table S7†). We next set out to evaluate whether other algorithms beyond D-MPNN can solve this new DeltaClassifier task.
Tree-based DeltaClassifiers
In addition to implementing the Chemprop-based DeltaClassifier, we also implemented an XGBoost-based DeltaClassifier to evaluate how tree-based models would perform on this new task. XGBoost was selected due to its readily available GPU acceleration,4 which can speed up calculation on quadratically larger datasets created through our pairing. Due to their increased computational efficiency, we further refer to these XGBoost-based DeltaClassifiers as DeltaClassifierLite. Like the deep models, DeltaClassifierLite trained on demilitarized data (ΔCL) significantly outperformed training on only exact values (ΔCLOE, p < 0.0001, Fig. S2, Table S2, ESI† 2). Overall comparable performance was observed between ΔCL and an approach that used all data without filtering for small property differences (ΔCLAD) with no statistically significant difference for accuracy (p = 0.3), slight improvement for ΔCL for F1 (p = 0.002), and no significant difference for AUC (p = 0.7, Fig. S2, Table S2, ESI† 2). Analogously to DΔC, similar trends were observed for a test set with all pairs included (Table S3†) and all pairs except same molecule pairs (Table S4†). Adversarial Y-shuffling also collapsed ΔCL performance as expected (Table S7†). Altogether, these results support that our new classification task can be solved by both the deep D-MPNNs and the tree-based algorithms, although overall superior performance by the Chemprop implementation compared to the XGBoost version suggests the utility of deep neural networks to predict potency improvements between molecular derivatives.Comparisons with state-of-the-art regression approaches
Next, we investigated if either of the DeltaClassifiers would exhibit improved performance over using state-of-the-art regression approaches when predicting potency improvements between two molecules (Fig. 1C). We compared our DΔC and ΔCL approach against two tree-based machine learning algorithms, random forest and XGBoost, and the single-molecule regression version of Chemprop. The direct comparison between Chemprop and DΔC as well as between XGBoost and ΔCL allows for the direct quantification of the benefit of our pairing approach since they each use the same underlying predictive algorithms.The regression algorithms can only be trained on the training molecules with exact values and can then be used to predict absolute potency values of all test set molecule. Afterwards, potency improvements of pairs from the test set were inferred by subtracting the potency predictions to determine which compound was expected to be more potent (Fig. 1C). In addition to designating a positive difference as a positive class and a negative difference as a negative class, we also normalized the predicted differences between molecules to create a proxy for model confidence in potency differences (cf. methods).
In terms of accuracy, F1 Score, and ROCAUC, DΔC showed a statistically significant improvement over all regression methods (p < 0.0001, Table 1, Fig. 3A, ESI† 3). At the level of individual datasets, DΔC outcompeted all regression methods in at least 69% of datasets and was competitive in at least 96% of datasets for all metrics (p < 0.05, Fig. 3B). The largest benefit was seen over the regression version of Chemprop, wherein DΔC outcompeted in at least 222/230 datasets for all metrics and was not statistically significantly worse on any dataset, highlighting the particular benefit of combinatorial data expansion from molecular pairing for data-hungry deep models to boost their performance. ΔCL also outcompeted the regression methods in most datasets across all metrics, but at a consistently slightly lower percentage than DΔC (p < 0.05, Fig. S3†) across all metrics. When evaluating all five models on the level of each dataset DΔC showed the highest median Z-score followed by ΔCL across all metrics (Fig. 3C) and the same trends were observed for modified Z-scores (Fig. S4†). In terms of rank, DΔC showed the highest average rank for accuracy (1.29 ± 0.65), followed by ΔCL (2.13 ± 0.72), random forest (2.91 ± 0.80), XGBoost (3.84 ± 0.60), and Chemprop (4.84 ± 0.56) with similar trends for F1 score and AUC (Table S8†). DΔC also outcompeted all other approaches for test sets without filtering of low pIC50 differences (p < 0.0001, Table S3†) and without filtering of low pIC50 differences but removal of same molecule pairs (p < 0.0001, Table S4†). Additionally, we compared our approach against a simple k-nearest neighbours (k-NN) algorithm to ensure our approach outcompeted a standard non-parametric, supervised learning approach on this task. The parameter-free k-NN underperformed compared to our DeltaClassifiers models across all metrics (p < 0.0001, Table S9†). These results attest to the superior performance of the DeltaClassifier approach compared to simply following state-of-the-art regression methods or parameter free nearest neighbour models in classifying potency improvements between molecules.
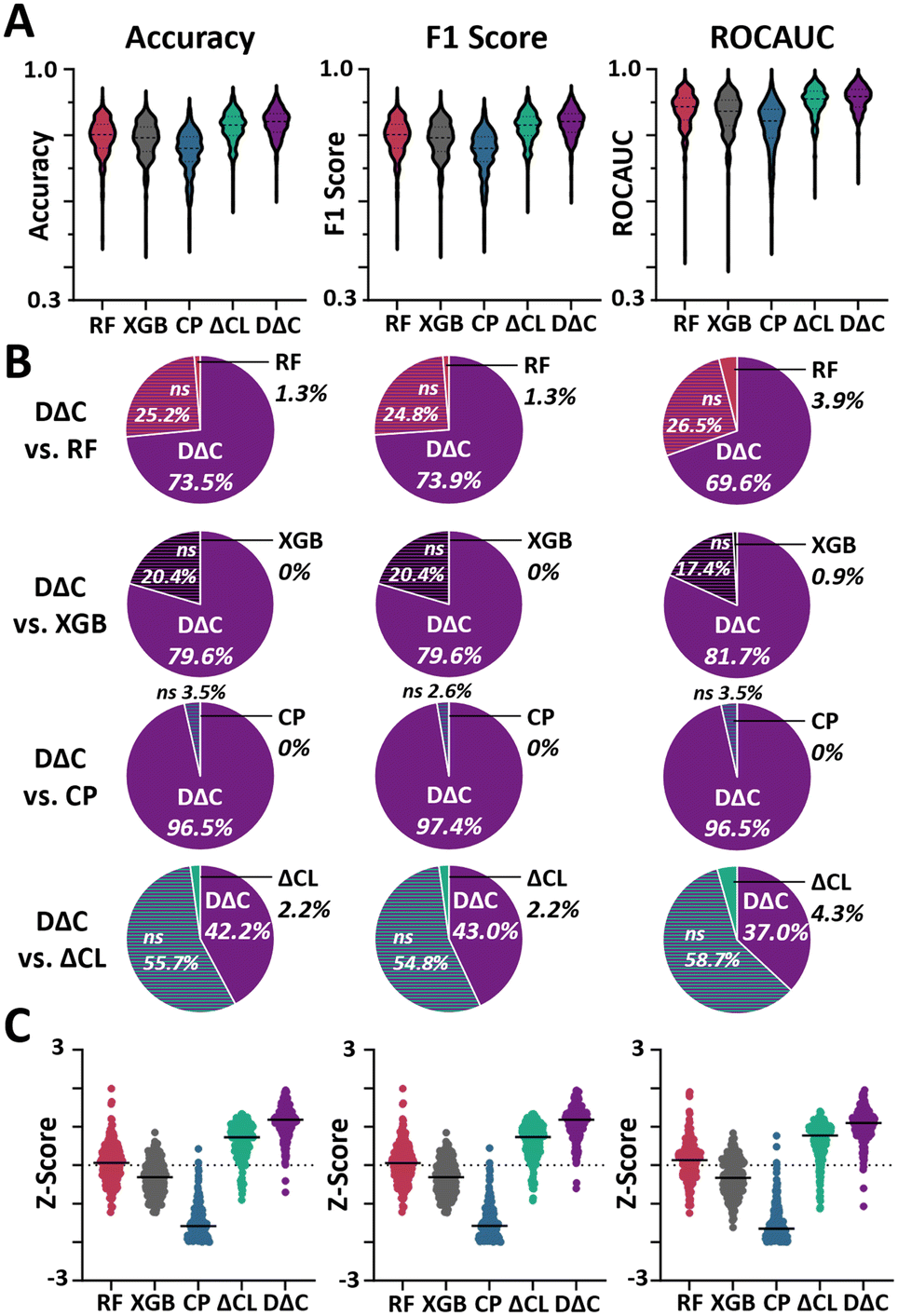 | ||
| Fig. 3 Comparison of DeltaClassifiers with regression approaches. Note that the DeepDeltaClassifier (DΔC) uses the neural network implementation of Chemprop (CP) and the DeltaClassifierLite (ΔCL) is based on XGBoost (XGB). The difference is that the DeltaClassifiers run these algorithms in classification mode after creating paired training data while the traditional implementations, including random forest (RF), run in regression mode. (A) Violin plots of average model performance following 3 × 10 cross-validation for 230 ChEMBL datasets in terms of accuracy, F1-score, and ROCAUC. (B) Pie charts showing percentage of datasets in which DΔC outcompeted (purple), exhibited no statistical difference (gradient), or underperformed compared to RF (red), XGB (black), CP (blue), and ΔCL (green) in terms of accuracy, F1-score, and ROCAUC. Statistical significance from paired t-test for three repeats (p < 0.05). (C) Z-scores for model performance in terms of accuracy, F1 score, and ROCAUC. | ||
To additionally evaluate how well DeltaClassifier approaches could predict molecules unlike those encountered during training, we performed another round of retrospective evaluation using a scaffold split to separate the training from the testing data. In alignment with our previous results, DΔC outcompeted all other approaches for scaffold-split test sets (p < 0.0001, Table S10†). DΔC also outcompeted all other approaches for test sets without filtering of low pIC50 differences (p < 0.0001, Table S11†) and without filtering of low pIC50 differences but removal of same molecule pairs (p < 0.0001, Table S12†). ΔCL also showed a statistically significant improvement over all regression methods across all metrics, but at a consistently slightly lower percentage than DΔC (p < 0.05, Tables S10–S12†). This outcome indicates that the DeltaClassifier approach also generalizes well to novel chemical classes, maintaining its superior performance over state-of-the-art regression methods.
When evaluated on a test set that was generated through pairing only molecules with exact values and no same molecular pairs, DΔC still outcompeted the regression version of Chemprop, XGBoost, and ΔCL (p < 0.0001, Table S13†), but exhibited similar performance compared to random forest in terms of accuracy (p = 0.3) and F1 score (p = 0.07), and lower performance in ROCAUC (p < 0.0001, Table S13†). This further attests to the strength of the DeltaClassifier approach to benefit from incorporating bounded potency values while the pairing alone might not inherently improve performance compared to robust tree-based models. This motivated us to investigate the impact of the amount of bounded data on DeltaClassifier performance.
Influence of bounded data on performance
Next, we sought to determine how the number of bounded datapoints in training data affects the improvement of DeltaClassifiers over regression methods and training DeltaClassifiers only on pairs made from exact IC50 values. The number of bounded datapoints in the training datasets correlated with the improvement of DΔC (Pearson's r = 0.58–0.75, Fig. 4) and ΔCL (Pearson's r = 0.56–0.70, Fig. S5†) over regression models and over training DeltaClassifiers with only pairs from exact IC50 values (DΔCOE and ΔCLOE). Therefore, our new pairing approach is most powerful if large amounts of bounded data are available that are not normally accessible to regression approaches. Importantly, these correlations are stronger (p = 0.0005) than the weaker correlations seen between dataset size and model performance (Pearson's r = 0.14–0.30, Fig. S6†), indicating that the benefit of the larger amounts of bounded data are not simply driven by larger dataset sizes. This evaluation suggests that the DeltaClassifier approach can be particularly helpful for datasets with large amounts of bounded datapoints. | ||
| Fig. 4 Percent of bounded data correlates with DΔC improvement over traditional models. Scatterplots showing correlation and Pearson's r values of DΔC performance improvement over random forest (RF, red), XGBoost (XGB, black) Chemprop (CP, blue), and DΔC trained only on exact values (DΔCOE, grey) following cross-validation for 230 ChEMBL datasets with the percent of bounded data within each dataset in terms of accuracy, F1 score, and ROCAUC. Note that DΔC uses the CP neural network and runs in classification mode after creating paired training data while CP runs in regression mode. | ||
Scaffold analysis
Next, we evaluated which model could most accurately predict potency improvements for pairs with either the same or with different scaffolds. After splitting test fold pairs into two separate groupings (shared or differing Murcko scaffolds, respectively), we evaluated all our models' performances on either test set after training the algorithms on the complete training folds containing pairs of both groupings. Gratifyingly, DΔC outperformed regression approaches both on predicting potency differences between molecules with different scaffolds (p < 0.0001, Tables S14/S15, Fig. S7A–F†) and between molecules with same scaffolds (p < 0.0001, Tables S16/S17, Fig. S7G–L†). DΔC achieved highest median Z scores across all datasets (Fig. S7C/I†) and showed highest average rank compared to all other investigated methods (Tables S15/S17†). This implies that a Chemprop-based DΔC could potentially be used both for fine-tuned compound optimization on the same scaffold while also enabling more drastic scaffold-hopping into new compound classes while optimizing potency. We next thought to further analyse whether DΔC would perform particularly well for certain target classes or datasets.Applicability domain
We analysed how dataset diversity and target type affected the improvement of DeltaClassifiers over regression methods. The percentage of unique scaffolds in the training datasets showed only a very a limited correlation with the improvement of DΔC (Pearson's r = 0.18–0.22, Fig. S8†) and ΔCL (Pearson's r = 0.20–0.26, Fig. S9†) over regression models. As such, our new pairing approach may exhibit enhanced predictive power with increasing numbers of differing training scaffolds but generally shows similar improvement over traditional methods regardless of dataset diversity. To determine whether certain target classes are most accessible to DeltaClassifier models, we analysed target performances grouped by their enzyme commission (EC) class. DΔC and ΔCL both significantly outperformed compared to all traditional regression approaches for all classes with greater than five targets represented (p < 0.0001, Tables S18–S20†) and outcompeted for all the remaining classes with less than five targets represented (Tables S21 and S22†) but due to the small number of represented targets there was insufficient resolution to guarantee statistical significance. This indicates that the DeltaClassifier approach is applicable regardless of enzyme class.Discussion
Here, we developed, validated, and characterized a new molecular pre-processing approach that enables established classification algorithms such as Chemprop and XGBoost to directly train on and classify potency improvements of molecular pairs. Across 230 datasets from ChEMBL, tree-based and deep DeltaClassifiers significantly improve performance over regression approaches to classify IC50 improvements between molecules. DeltaClassifiers showed greatest improvements for datasets with more bounded data, suggesting that this method could be particularly beneficial for targets with large amounts of bounded data, as can be expected for novel target classes and targets with poor druggability.DeltaClassifiers can benefit from increased training datapoints and cancellation of systematic errors within datasets through pairing9,10 while directly learning potency differences. This pairing approach benefits the neural network models even more than tree-based models, highlighting the particular advantage of combinatorial data expansion for data hungry deep models. This data augmentation also allows for expedited model convergence,2 leading to convergence of the Chemprop training on paired data after only 5 epochs compared to single-molecule Chemprop trained for 50 epochs (Table 1). Admittingly, paired methods are most efficiently applied to small or medium-sized datasets (<1000 datapoints) as their combinatorial expansion of training data leads to increased computational costs for each epoch. Altogether, the improved performance exhibited by DeltaClassifier over established methods across these benchmarks showcase its potential for potency classification with clear prospects for further improvements.
Related work
There are several related, powerful approaches to compare the properties of molecular pairs (Table S23†). Siamese neural networks consider two inputs and tandemly use the same parameters and weights to find similarities between inputs. As such, they use a distance function for locality-sensitive hashing as a contrastive learning approach. Siamese neural networks have been applied within the field of drug discovery to predict molecular similarity,11 bioactivity,12 toxicity,13 drug–drug interactions,14 relative free energy of binding,15 and transcriptional response similarity.16 These models have additionally shown particular promise when trained only on compounds with high similarity, highlighting how additional preprocessing steps, such as reducing exhaustive pairing to only the most similar pairs, can reduce computational costs while preserving predictive power for paired models.17 Although these models are similarly tailored to directly consider molecules as pairs, they are not inherently constructed to learn from bounded data and typically rely upon similarity metrics, such as cosine similarity, to determine distance between classes.There is also precedence of using bipartite ranking of chemical structures to additionally incorporate qualitative data alongside quantitative data for the prediction of molecular properties.18–20 For example, kernel-based ranking algorithms that minimize a ranking loss function in place of a classification or regression loss have been implemented for molecular ranking.20 More recently, a learning-to-rank framework has been implemented to rank candidates based on differences in IC50 for SARS-CoV-2 inhibition.18,19 Instead of incorporating bounded values, as we do for DeltaClassifiers, these approaches added labelled data (i.e., ‘inactive’) to regression data by considering all compounds with no measurable IC50 as less active than any active compound and discarding any molecular pair that contains two ‘inactive’ molecules. Compound rankings by quantitative structure–activity relationship (QSAR) models have also been shown to help integrate heterogeneous data from various assays to support IC50 prediction.9 These existing approaches for classifying molecular improvements (Table S23†) should be synergistic with our DeltaClassifier approach. Together, we believe that these methods show great promise to supplement or replace machine learning methods currently implemented for intricate molecular optimizations, chiefly when relying upon smaller datasets with bounded or noisy data.
Conclusions
As generating valuable data for drug discovery and development is expensive, there is a clear need for novel methods to integrate all available data into machine learning training. We here present DeltaClassifiers, a novel data pre-processing approach that enables classification models to access traditionally inaccessible bounded datapoints to guide potency optimizations and molecular discovery through directly contrasting molecular pairs. Given the Chemprop-based DeltaClassifiers' significant improvement in identifying potency improvements compared to traditional regression approaches, we believe that deep DeltaClassifiers and subsequent extensions stand to accurately guide potency optimizations in the future. This method is poised to prioritize the most promising next pharmaceutical candidates and could be directly incorporated into adaptive robotic platforms for automated discovery campaigns.21 Beyond its utility in drug development, we believe DeltaClassifier can be implemented for material selection and optimization, thereby to improving efficiency and quality for many important biological and chemical optimization tasks.Experimental
Datasets
ChEMBL3322 was filtered for single organism/protein IC50 of small molecules with molecular weights <1000 Da. To ensure sufficient data while preventing combinatorial explosion, we selected datasets containing 300–900 datapoints. Additionally, datasets were filtered to ensure no single IC50 value (e.g., “>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM”, e.g., ChEMBL target ID 4879459) accounted for more than half of all datapoints which occurred in 9 datasets. To further clean the data, datasets were screened for any invalid SMILES structure (RDKit) or molecule labelled with an IC50 value of ‘0’ or ‘N/A', and all such entries were removed. In addition, if a compound occurred multiple times in the database, this compound was also excluded. To naturally reduce dynamic range to preserve differences while not dramatically skewing scale, all IC50 values were then converted to pIC50 from nanomolar concentrations. This data curation workflow resulted in 230 benchmarking datasets (ESI† 4).
000 nM”, e.g., ChEMBL target ID 4879459) accounted for more than half of all datapoints which occurred in 9 datasets. To further clean the data, datasets were screened for any invalid SMILES structure (RDKit) or molecule labelled with an IC50 value of ‘0’ or ‘N/A', and all such entries were removed. In addition, if a compound occurred multiple times in the database, this compound was also excluded. To naturally reduce dynamic range to preserve differences while not dramatically skewing scale, all IC50 values were then converted to pIC50 from nanomolar concentrations. This data curation workflow resulted in 230 benchmarking datasets (ESI† 4).
Model architecture and implementation
For DΔC, we used the established, two-molecule version of the directed D-MPNN architecture implemented in Chemprop given its efficient computation and competitive performance for molecular data.6 By building on this architecture, results are directly comparable to the regression version of Chemprop and the benefits of our molecular pairing approach and integration of bounded data can be directly quantified. Two molecules formed an input pair for DΔC, while the regression version of Chemprop processed a single molecule to predict absolute potency values that were then subtracted to calculate potency differences between two molecules and used to classify IC50 improvements (Fig. 1C). In contrast, DΔC directly learned and classified IC50 improvements by training on input pairs and their classified potency differences (Fig. 1E). For all our D-MPNN models (regression Chemprop and two-molecule classification Chemprop used for DΔC), molecules were described using atom and bond features as previously described6 and were implemented with default parameters and using ‘sum’ aggregation. The Chemprop algorithm was set to ‘regression’ mode for the regression Chemprop implementation while the Chemprop algorithm was set to ‘classification’ mode for DΔC. As Chemprop was set for ‘regression’ mode for our regression implementation, it could only be trained on exact values within the training set. For the regression Chemprop implementation, “number of molecules” was set to 1 while for DΔC it was set to 2 to process molecular pairs.23 We previously optimized the number of epochs for molecular paired data2 and observed a convergence of performance by 5 epochs for paired deep models and a convergence by 50 epochs for singular deep models. Accordingly, we set epochs = 5 for DΔC and epochs = 50 for Chemprop.For random forest and XGBoost models, molecules were described using radial chemical fingerprints (Morgan circular fingerprint, radius 2, 2048 bits, rdkit.org). Random forest regression models were implemented with 500 trees and default parameters in scikit learn. XGBoost regression models were implemented with tree_method = ‘gpu_hist’ (to allow for gpu acceleration) and default parameters in scikit-learn. For random forest and XGBoost regression models, each molecule was processed individually such that predictions were made solely based on the fingerprint of a single molecule. Regression models were only able to be trained on exact values within the training set. For developing ΔCL, fingerprints for paired molecules were concatenated to form paired molecular representations to directly train on and classify potency improvements using the classification implementation of XGBoost.
For all traditional regression algorithms (Chemprop, random forest, and XGBoost), potency of the two molecules mi and mj were predicted separately as pred(mi) and pred(mj) and potency differences were calculated as di,j = pred(mi) − pred(mj) and using the sign of the prediction difference as the classification label yi,j = sign(di,j). In addition, each predicted difference was normalized as ni,j = [di,j − dmin]/[dmax − dmin], where dmin = mini,j(di,j) is the minimum predicted potency difference between all pairs of molecules mi and mj in the test dataset and dmax = maxi,j(di,j) is the maximum predicted potency differences between all pairs of molecules mi and mj in the test dataset. This normalization creates a normalized value ni,j ∈ [0,1] that is larger for molecule pairs with larger potency differences and therefore serves as a surrogate predictive confidence measure to enable ROCAUC calculations.
K-nearest neighbours (K-NNs) were trained on radial chemical fingerprints (Morgan circular fingerprint, radius 2, 2048 bits, rdkit.org) and implemented using the KNeighborsRegressor implementation in scikit learn with default parameters.
For standard approaches to classify potency improvements (Fig. S10†), machine learning models were trained on absolute IC50 values, and these models were used to predict absolute IC50 values for new molecules. These absolute IC50 values for two molecules were then subtracted and potency improvements were classified based on the sign of this difference. For DeltaClassifier models (Fig. S11†), the training data is first cross-merged into all possible molecule pairs and a ground-truth classification label is created using the sign of the subtracted ground-truth IC50 values. Additionally, during “demilitarization” (Fig. S12†), any pairs with a ground-truth IC50 difference below the demilitarization threshold value or with unknown potency differences were removed prior to training the model.
Model evaluation
To assess the impact of demilitarization on training of DeltaClassifiers and analyse modified test sets, models were evaluated using random 1 × 10-fold cross-validation (sklearn, ESI† 2). When comparing with traditional approaches, models were evaluated using random 3 × 10-fold cross-validation (ESI† 3). In all evaluations, each of the 230 datasets were individually modelled, and the resultant 230 separate machine learning models for each approach were evaluated using accuracy, F1 score, and ROCAUC. To prevent data leakage, data was first split into train and test sets during cross-validation prior to cross-merging to create molecule pairings (Fig. S1†).2 This ensured each molecule was only present in pairs made in the training or the test set but never both. If it was unknown if the potency was improved for a molecular pair (e.g., both molecules' potencies are denoted as upper bounds), the pair was removed (Fig. S12†). Additionally for demilitarized assessments and training, if the difference was less than 0.1 pIC50, the pair was removed to account for experimental noise and non-statistically significant potency differences (Fig. S12†). For assessments of training on only exact values, any datapoint denoted as ‘>’ or ‘<’ was removed. For scaffold-split assessments, 80–20 train-tests were implemented with a random state of 1 (deepchem). Scaffold analysis, analysis of the influence of bounded datapoints on model performance, and additional test sets (without filtering of low pIC50 differences or without filtering but removal of same molecule pairs) were made using cross-validation splits with a random state = 1. Z-scores were calculated using scipy and modified Z-scores (Mi) were calculated using the following equation:wherein
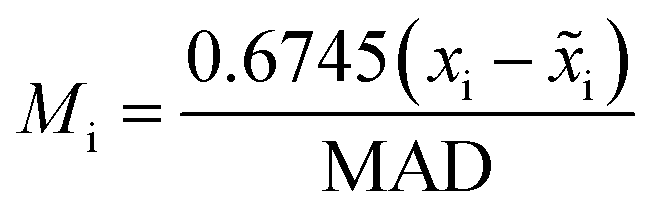
![[x with combining tilde]](https://www.rsc.org/images/entities/i_char_0078_0303.gif) i is the median and MAD is the median absolute deviation.24
i is the median and MAD is the median absolute deviation.24
Statistical comparisons were performed using the non-parametric Wilcoxon signed-rank test (p < 0.05) when comparing across the 230 datasets or across models and performed as paired t-tests (p < 0.05) for cross-validation repeats of a single dataset. Violin plots were made in GraphPad Prism 10.2.0 while scatterplots were made using matplotlib. The associated code and datasets are available in the GitHub repository, https://github.com/RekerLab/DeltaClassifier.
Data availability
The source code, datasets, and results supporting the conclusions of this article are available in the GitHub repository, https://github.com/RekerLab/DeltaClassifier.Author contributions
Z. F., P. S., and D. R. designed and implemented models. Z. F. and D. R. analysed data and generated figures. Z. F. and D. R. wrote the manuscript, with contributions from P. S. All authors reviewed and agreed on submitting the current version of the manuscript.Conflicts of interest
D. R. acts as a consultant to the pharmaceutical and biotechnology industry, as a mentor for Start2, and on the scientific advisory board of Areteia Therapeutics.Acknowledgements
We thank Pat Walters for insightful technical discussions about this work. We would like to thank the Chemprop and the Scikit-learn developers for making their machine learning algorithms publicly available. Z. F. is supported by the Department of Defense (DoD) through the National Defense Science & Engineering Graduate (NDSEG) Fellowship Program. This research was supported by the Duke Science & Technology Initiative and by the NIH NIGMS grant R35GM151255.References
- H. Van De Waterbeemd and E. Gifford, Nat. Rev. Drug Discovery, 2003, 2, 192–204 CrossRef CAS PubMed.
- Z. Fralish, A. Chen, P. Skaluba and D. Reker, Aust. J. Chem., 2023, 15, 101 Search PubMed.
- V. Svetnik, A. Liaw, C. Tong, J. C. Culberson, R. P. Sheridan and B. P. Feuston, J. Chem. Inf. Comput. Sci., 2003, 43, 1947–1958 CrossRef CAS PubMed.
- R. Mitchell and E. Frank, PeerJ Comput. Sci., 2017, 3, e127 CrossRef.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, J. Chem. Inf. Model., 2023, 64, 9–17 CrossRef PubMed.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley and M. Mathea, J. Chem. Inf. Model., 2019, 59, 3370–3388 CrossRef CAS PubMed.
- G. A. Landrum and S. Riniker, J. Chem. Inf. Model., 2024, 64(5), 1560–1567 CrossRef CAS PubMed.
- T. Kalliokoski, C. Kramer, A. Vulpetti and P. Gedeck, PLoS One, 2013, 8, e61007 CrossRef CAS PubMed.
- K. Matsumoto, T. Miyao and K. Funatsu, ACS Omega, 2021, 6, 11964–11973 CrossRef CAS PubMed.
- M. Tynes, W. Gao, D. J. Burrill, E. R. Batista, D. Perez, P. Yang and N. Lubbers, J. Chem. Inf. Model., 2021, 61, 3846–3857 CrossRef CAS PubMed.
- M. K. Altalib and N. Salim, ACS Omega, 2022, 7, 4769–4786 CrossRef CAS PubMed.
- D. Fernández-Llaneza, S. Ulander, D. Gogishvili, E. Nittinger, H. Zhao and C. Tyrchan, ACS Omega, 2021, 6, 11086–11094 CrossRef PubMed.
- H. Altae-Tran, B. Ramsundar, A. S. Pappu and V. Pande, ACS Cent. Sci., 2017, 3, 283–293 CrossRef CAS PubMed.
- K. Schwarz, A. Allam, N. A. Perez Gonzalez and M. Krauthammer, BMC Bioinf., 2021, 22, 1–19 CrossRef PubMed.
- A. T. McNutt and D. R. Koes, J. Chem. Inf. Model., 2022, 62, 1819–1829 CrossRef CAS PubMed.
- M. Jeon, D. Park, J. Lee, H. Jeon, M. Ko, S. Kim, Y. Choi, A.-C. Tan and J. Kang, Bioinformatics, 2019, 35, 5249–5256 CrossRef CAS PubMed.
- Y. Zhang, J. Menke, J. He, E. Nittinger, C. Tyrchan, O. Koch and H. Zhao, Aust. J. Chem., 2023, 15, 75 Search PubMed.
- K. L. Saar, W. McCorkindale, D. Fearon, M. Boby, H. Barr, A. Ben-Shmuel, C. M. Consortium, N. London, F. von Delft and J. D. Chodera, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2214168120 CrossRef CAS PubMed.
- A. Morris, W. McCorkindale, N. Drayman, J. D. Chodera, S. Tay, N. London and C. M. Consortium, Chem. Commun., 2021, 57, 5909–5912 RSC.
- S. Agarwal, D. Dugar and S. Sengupta, J. Chem. Inf. Model., 2010, 50, 716–731 CrossRef CAS PubMed.
- L. Bustillo, T. Laino and T. Rodrigues, Chem. Sci., 2023, 14, 10378–10384 RSC.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich and B. Al-Lazikani, Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS PubMed.
- F. H. Vermeire and W. H. Green, Chem. Eng. J., 2021, 418, 129307 CrossRef CAS.
- B. Iglewicz and D. C. Hoaglin, Volume 16: how to detect and handle outliers, Quality Press, 1993 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00325j |
| This journal is © The Royal Society of Chemistry 2024 |