DOI:
10.1039/D4MD00252K
(Research Article)
RSC Med. Chem., 2024,
15, 2351-2356
Discovery of first-in-class PROTACs targeting maternal embryonic leucine zipper kinase (MELK) for the treatment of Burkitt lymphoma†
Received
12th April 2024
, Accepted 4th June 2024
First published on 4th June 2024
Abstract
Maternal embryonic leucine zipper kinase (MELK) is a novel target for the treatment of various kinds of B-cell malignancies. However, the toxicity of inhibitors of MELK has led to clinical failures in cancer treatments. Moreover, inactivation of MELK catalytic domain is insufficient for achieving cancer cell apoptosis. To further confirm the role of MELK in Burkitt lymphoma treatment, we describe herein a structure-guided design of PROTACs targeting MELK. Through design, computer-assisted optimization and SAR studies, we developed the first-in-class MELK-targeting PROTAC MGP-39, which promoted a rapid and potent degradation of MELK in RAMOS cells. Additionally, the newly designed MELK degrader induced significant cell cycle arrest and apoptosis in cancer cells. Notably, compared to MELK inhibitors, MGP-39 has better anti-cancer activity and lower toxicity, indicating the practical role of PROTACs in avoiding the side effects of traditional inhibitors. More importantly, our results show that the use of a PROTAC can be adopted as a general and effective strategy for targeted cancer therapy.
Introduction
Burkitt's lymphoma (BL), a hematologic malignancy, is the most common non-Hodgkin's lymphoma (NHL) in children,1 with approximately 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people diagnosed worldwide each year.2 BL is a rapidly proliferating lymphoma derived from germinal-center B cells.3,4 BL typically has a dramatic clinical presentation, and rapidly spreads to intra-abdominal organs and the central nervous system (CNS).5 Therefore, immediate evaluation is warranted. Although intensive chemoimmunotherapy has appeared,6 relapse may occur shortly with dismal prognosis, and the survival rates are always lower than 30%.5,7
000 people diagnosed worldwide each year.2 BL is a rapidly proliferating lymphoma derived from germinal-center B cells.3,4 BL typically has a dramatic clinical presentation, and rapidly spreads to intra-abdominal organs and the central nervous system (CNS).5 Therefore, immediate evaluation is warranted. Although intensive chemoimmunotherapy has appeared,6 relapse may occur shortly with dismal prognosis, and the survival rates are always lower than 30%.5,7
Maternal embryonic leucine zipper kinase (MELK) is an enzyme encoded by the MELK gene. As an important member of the AMP-activated Ser/Thr kinase family, MELK plays a crucial role in cancer progression and drug resistance.8 MELK promotes cancer progression through the mTOR pathway.9,10 As mentioned, BL is a rapidly proliferating NHL. The MELK/mTOR pathway plays a crucial role in development, differentiation and signalling of BL and other NHLs.11–13 Meanwhile, MELK has proven to be a novel and practical target for treatment of B cell malignancies such as BL and MCL. Inhibition or degradation of MELK has potential benefits on the prognosis of BL.14,15 However, currently reported inhibitors of MELK display serious toxicity, which leads to clinical failures.16–18 Hence there is an urgent need to develop a new strategy to overcome these shortcomings.
Proteolysis-targeting chimeras (PROTACs) have recently garnered considerable research interest in academia and in the pharmaceutical industry.19,20 PROTACs constitute a series of bifunctional molecules each capable of bringing E3 ligase into the proximity of a target protein of interest (POI), inducing non-natural ubiquitin degradation of the target protein.21 Unlike traditional protein inhibitors, PROTACs achieve inactivation of the whole target protein, including its non-enzymatic functions, rather than inhibiting the kinase pocket only.22 Moreover, a typical advantage of PROTACs is their excellent selectivity.23 Based on the novel ubiquitination mechanism, selective degradation of a POI can be achieved without potential toxicity from protein inhibitors.24,25
Herein, for the first time, we describe the development of a novel degrader of MELK using the PROTAC strategy. The degrader, denoted as MGP-39, induced potent and rapid degradation of MELK in Burkitt lymphoma cells. In addition, our newly designed PROTAC also induced significant G2/M cell cycle arrest and apoptosis in RAMOS cells, indicating the therapeutic importance of MELK in B-cell malignancies. More importantly, compared to MELK inhibitors, MGP-39 showed better anti-cancer activity and lower toxicity (Fig. 1A and B). Therefore, the use of degradation, a novel method in medicinal chemistry, has been demonstrated to be a targeted and effective strategy in cancer therapy.
 |
| | Fig. 1 Development of novel PROTACs targeting the MELK protein. (A) Structural representation of MELK degrader design. (B) Mechanism of the newly developed PROTACs targeting MELK. | |
Results and discussion
The screening of novel MELK degraders
In order to develop MELK-targeting PROTAC-designed degraders, a MELK-targeting arm was conjugated to a MELK-degradation arm via linkers with variable lengths. As a result, a MELK ligand and pomalidomide were employed as corresponding MELK and E3 ligase binding partners. According to the design principles, PROTAC molecules in different combinations were prepared (Fig. 2A).
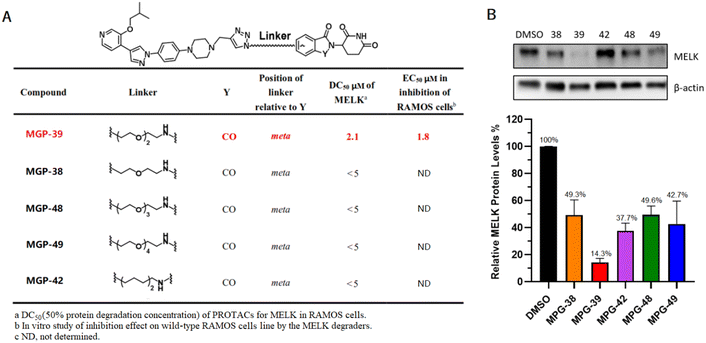 |
| | Fig. 2 Chemical structure and characterizations of novel MELK degraders. (A) SAR studies for MELK degrader evaluations. In a cell viability assay, 3000 cells per well in 96-well plates were incubated at 37 °C for 72–96 h. The final EC50 was calculated using CCK-8. (B) Immunoblotting analysis of novel PROTACs targeting MELK. RAMOS cells treated with compounds (5 μM) for 48 h; 2 × 105 cells per well in 12-well plates were incubated at 37 °C. Grayscale analysis data were generated using ImageJ. | |
The RAMOS cell line, being common for BL, was utilized for the evaluations. MGP-39, a bifunctional molecule with a triethylene glycol linker and pomalidomide ligand, was demonstrated from the results to have the strongest MELK-degrading ability and anti-cancer activity (Fig. 2A). Compound MPG-42, in which polyethylene glycol linker was replaced by an aliphatic chain with the same length, showed relatively low efficiency, indicating a significant impact of the identity of the linker on the bioactivity of the PROTAC (Fig. 2B). On the basis of a statistical analysis of western blots, MGP-39 was selected for further experiments.
The synthesis of MGP-39
The synthesis of the MELK degrader MGP-39 is shown in Scheme 1. Briefly, a substitution reaction was conducted with compound 13 and propargyl bromide as substrates to prepare intermediate 15. Compound 10 and amine derivatives were then utilized in substitution reactions to prepare intermediates B1. Finally, precursors 15 and B1 were transformed to MGP-39 and its analogues via a click reaction in the last synthetic step (for details of condition 1, please see ESI†).17 Note the relative simplicity of the MGP-39 synthetic route, convenient for any large-scale preparations to be considered in the future.
 |
| | Scheme 1 Synthesis of compound MGP-39 analogues. | |
Identification of MGP-39 as a MELK PROTAC degrader
PROTAC-induced degradation of MELK occurred rapidly and completely. The half-life of the MELK protein in RAMOS cells was <3 h in the presence of MGP-39 (Fig. 3A and ESI† Fig. S1). MLN4924 and MG132—as an NAE inhibitor and proteasome inhibitor, respectively—could effectively disable the function of MGP-39 in degrading MELK (Fig. 3B). These results confirmed that the MELK degradation was mediated by the ubiquitin-proteasome system (UPS), consistent with the PROTAC mechanism. To further demonstrate the degradation pathway used by MGP-39, relative MELK mRNA levels in RAMOS cells and DOHH2 cells treated with PROTACs were determined. Significantly increased levels of MELK mRNA were observed when MGP-39 was included (Fig. 3C), with the increase achieved through a negative feedback loop.
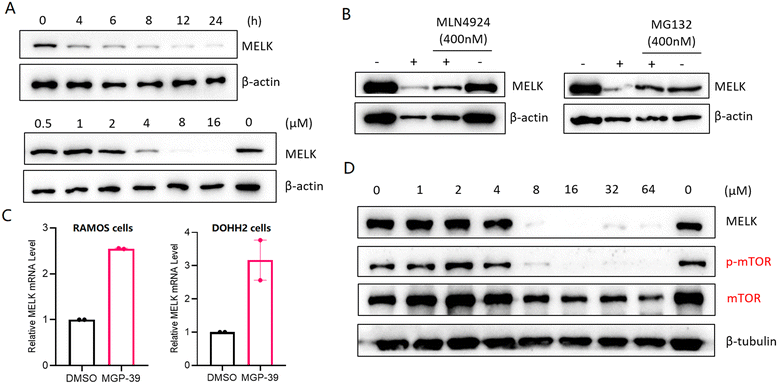 |
| | Fig. 3 Efficacy and mechanism studies of MGP-39 in degrading MELK. (A) Western blotting analysis for MELK in RAMOS cells with 20 μM MGP-39 for the indicated exposure times (top) and western blotting analysis for MELK in RAMOS cells with indicated concentrations of MGP-39 for 48 h. (B) Immunoblot for MELK and β-actin after a pretreatment for 4 h with MLN4924 or MG132 (400 nM), followed by a treatment with MGP-39 (20 μM) for 24 h in RAMOS cells. “+” refers to treatment with MGP-39, “−” refers to no treatment with MGP-39. (C) Relative MELK mRNA levels in RAMOS cells or DOHH2 cells treated with MGP-39 (20 μM) for 24 h. (D) Western blotting analysis for MELK, p-mTOR, mTOR, and β-tubulin proteins after 24 h of treatment of RAMOS cells with indicated concentrations of MGP-39. | |
To evaluate the detailed anti-cancer effects of MGP-39 in BL cells, we examined the activity of the downstream signaling molecule mTOR upon MELK degradation. Indeed, MGP-39 potently inhibited the phosphorylation of mTOR in accordance with MELK downregulation (Fig. 3D).
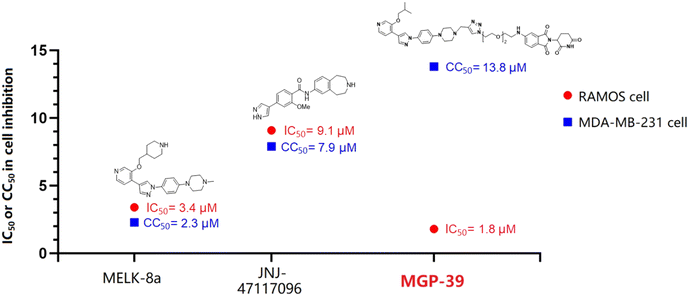
MGP-39 demonstrates higher efficacy and lower toxicity than do MELK inhibitors
As mentioned above, MELK is a novel and practical target for B-cell malignancy therapy. However, currently reported MELK inhibitors show serious toxicity, which leads to clinical failures. Although MELK is generally expressed in human cells, it is not necessary for the proliferation of basal-like breast cancer cells.26 Therefore, a cell viability assay was conducted using the MDA-MB-231 cell line to determine the toxicity of the novel MELK degrader MGP-39. As shown in Fig. 4A, MGP-39 exhibited better anti-cancer activity than did the two tested MELK inhibitors, and exhibited the lowest cytotoxicity against MDA-MB-231 cells (Fig. 4B). Furthermore, MGP-39 exhibited an obvious therapeutic window, while MELK inhibitors showed obvious general toxicity (Fig. 4D and 5), indicating the strong potential for developing a PROTAC-based therapeutic MELK regulator.
 |
| | Fig. 4 Cell viability assays of MELK regulators. (A) Anti-cancer effect of MELK degrader MGP-39 and MELK inhibitors JNJ-47117096/MELK-8a against the RAMOS cell line. (B) Cytotoxicity of MELK degrader MGP-39 and MELK inhibitors JNJ-47117096/MELK-8a, specifically toward the MDA-MB-231 cell line. (C) Relative protein levels of MELK and mTOR in RAMOS cells after being treated with MGP-39 for 24 h. (D) EC50s of MELK regulators in inhibiting RAMOS cells and MDA-MB-231 cells. | |
 |
| | Fig. 5 Therapeutic windows of MELK regulators. | |
MGP-39 induces cell cycle arrest and apoptosis in BL cells
To further investigate the anti-cancer mechanism of MGP-39, detections of apoptosis and cell cycle arrest were performed using flow cytometry. The results showed that MGP-39, as a MELK degrader, could effectively induce both early and total apoptosis of RAMOS cells (Fig. 6A). Meanwhile, the apoptosis function of compound MGP-39 was confirmed using a calcein AM/PI staining assay (Fig. 6B). The G2/M-phase checkpoint was established to prevent cells with damaged DNA from entering mitosis and allows for the repair of DNA. Therefore, cell cycle arrest at the G2/M phase is a promising therapeutic approach in the battle against cancer.27 Indeed, the MELK-targeting PROTAC MGP-39 can potently arrest the cell cycle at G2/M in the RAMOS cell line (Fig. 6C and ESI† Fig. S2). Moreover, a tube formation assay on HUVEC cells confirmed the lower general cytotoxicity of MGP-39 compared with MELK inhibitor JNJ-47117096 (Fig. 6D and ESI† Fig. S3), indicating that MGP-39, as a new type of MELK PROTAC, could be of clinical significance in the treatment of B-cell malignancies.
 |
| | Fig. 6 Apoptosis and cell cycle arrest triggered by MGP-39. (A) Flow cytometry quantification of early/late apoptotic cells (RAMOS) treated with 20 μM of MGP-39 for 24 h. **P < 0.01, ***P < 0.001. (B) Calcein AM/PI staining of RAMOS cells after being treated for 24 h with 20 μM MGP-39. ****P < 0.0001. (C) MGP-39 arrested the cell cycle at the G2/M phase in the RAMOS cell line. Cells were treated with 10 μM of the compound for 24 h and stained with propidium iodide. The cell cycle phases were determined using flow cytometry. (D) Tube formation assays for HUVEC cells treated with the indicated compounds (4 μM) for 24 h. | |
Conclusions
In summary, we have developed a first-in-class MELK-targeting PROTAC strategy for Burkitt lymphoma treatment. Remarkably, the representative PROTAC molecule MGP-39 could degrade the MELK protein with high efficacy. Furthermore, downstream signaling activities were strongly blocked by MGP-39, indicating a clear anti-cancer signaling pathway of the novel degrader. Moreover, MGP-39 arrested the cell cycle at G2/M and significantly induced early/late apoptosis in the RAMOS cell line. More importantly, compared with MELK inhibitors, the MELK degrader MGP-39 not only had better anti-cancer activity against Burkitt lymphoma, but also showed lower general cytotoxicity. Therefore, these findings indicated that MGP-39, a novel PROTAC molecule targeting MELK, may be used as part of a safe and effective method for cancer therapy and may have the potential to overcome the clinical failures of MELK inhibitors in treating NHL.
Author contributions
The project and experiments were designed by Y. S. and X. L. The chemistry was performed by Y. S., Q. H., X. L., Y. W. and N. Z. The biological research was carried out by Y. S., X. L. and W. Y. All authors contributed to the manuscript writing and review process.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2023-RC350-02).
Notes and references
- C. López, K. Kleinheinz, S. M. Aukema, M. Rohde, S. H. Bernhart, D. Hübschmann, R. Wagener, U. H. Toprak, F. Raimondi, M. Kreuz, S. M. Waszak, Z. Huang, L. Sieverling, N. Paramasivam, J. Seufert and S. Sungalee, Nat. Commun., 2019, 10, 1459 CrossRef.
- C. López, B. Burkhardt, J. K. C. Chan, L. Leoncini, S. M. Mbulaiteye, M. D. Ogwang, J. Orem, R. Rochford, M. Roschewski and R. Siebert, Nat. Rev. Dis. Primers, 2022, 8, 78 CrossRef.
- M. Méchali, Science, 2022, 377, 1259–1260 CrossRef PubMed.
- R. Wagener, J. Seufert, F. Raimondi, S. Bens, K. Kleinheinz, I. Nagel, J. Altmuller, H. Thiele, D. Hubschmann, C. W. Kohler, P. Nurnberg, R. Au-Yeung, B. Burkhardt, H. Horn, L. Leoncini, E. S. Jaffe, G. Ott, G. Rymkiewicz, M. Schlesner, R. B. Russell, W. Klapper and R. Siebert, Blood, 2019, 133, 962–966 CrossRef CAS PubMed.
- M. Roschewski, L. M. Staudt and W. H. Wilson, N. Engl. J. Med., 2022, 387, 1111–1122 CrossRef PubMed.
- K. Tomska, R. Kurilov, K. S. Lee, J. Hüllein, M. Lukas, L. Sellner, T. Walther, L. Wagner, M. Oleś, B. Brors, W. Huber and T. Zenz, Sci. Rep., 2018, 13, 12046 CrossRef.
- B. Burkhardt, U. Michgehl, J. Rohde, T. Erdmann, P. Berning, K. Reutter, M. Rohde, A. Borkhardt, T. Burmeister, S. Dave, A. Tzankov, M. Dugas, S. Sandmann, F. Fend, J. Finger and S. Mueller, Nat. Commun., 2022, 13, 3881 CrossRef CAS.
- Y. Zhang, X. Zhou, Y. Li, Y. Xu, K. Lu, P. Li and X. Wang, Oncogene, 2018, 37, 5520–5533 CrossRef CAS.
- H. A. Seong, R. Manoharan and H. Ha, Sci. Rep., 2017, 7, 42502 CrossRef CAS.
- Z. Li, H. Zhou, X. Zhai, L. Gao, M. Yang, B. An, T. Xia, G. Du, X. Li, W. Wang and B. Jin, Cell Death Dis., 2023, 14, 733 CrossRef CAS PubMed.
- M. Bhatti, T. Ippolito, C. Mavis, J. Gu, M. S. Cairo, M. S. Lim, F. Hernandez-Ilizaliturri and M. J. Barth, Onco Targets Ther, 2018, 9, 21820–21830 Search PubMed.
- F. Ni, X. Huang, Z. Chen, W. Qian and X. Tong, Sci. Rep., 2018, 8, 3317 CrossRef PubMed.
- W. Xu, P. Berning, T. Erdmann, M. Grau, N. Bettazová, M. Zapukhlyak, F. Frontzek, C. Kosnopfel, P. Lenz, M. Grondine, B. Willis, J. T. Lynch, P. Klener, S. Hailfinger, S. T. Barry and G. Lenz, Leukemia, 2023, 37, 178–189 CrossRef CAS PubMed.
- A. Maes, K. Maes, P. Vlummens, H. D. Raeve, J. Devin, V. Szablewski, K. D. Veirman, E. Menu, J. Moreaux, K. Vanderkerken and E. D. Bruyne, Blood Cancer J., 2019, 9, 87 CrossRef PubMed.
- C. Li, P. Xin, H. Xiao, Y. Zheng, Y. Huang and X. Zhu, Cancer Cell Int., 2015, 15, 1–9 CrossRef.
- B. J. Eisfelder, C. Saygin, J. Wynne, M. W. Colton, M. Fischietti, E. M. Beauchamp, J. X. Cheng, O. Odenike, G. Roboz, H. Alachkar and W. Stock, Blood Cancer J., 2021, 11, 48 CrossRef PubMed.
- B. B. Touré, J. Giraldes, T. Smith, E. R. Sprague, Y. Wang, S. Mathieu, Z. Chen, Y. Mishina, Y. Feng, Y. Yan-Neale, S. Shakya, D. Chen, M. Meyer, D. Puleo, J. T. Brazell, C. Straub, D. Sage, K. Wright, Y. Yuan, X. Chen, J. Duca, S. Kim, L. Tian, E. Martin, K. Hurov and W. Shao, J. Med. Chem., 2016, 59, 4711–4723 CrossRef PubMed.
- C. N. Johnson, V. Berdini, L. Beke, P. Bonnet, D. Brehmer, J. E. Coyle, P. J. Day, M. Frederickson, E. J. E. Freyne, R. A. H. J. Gilissen, C. C. F. Hamlett, S. Howard, L. Meerpoel, R. McMenamin, S. Patel, D. C. Rees, A. Sharff, F. Sommen, T. Wu and J. T. M. Linders, ACS Med. Chem. Lett., 2014, 23, 25–30 Search PubMed.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS.
- S. Montoya, J. Bourcier, M. Noviski, H. Lu, M. C. Thompson, A. Chirino, J. Jahn and A. K. Sondhi, Science, 2024, 383, eadi5798 CrossRef CAS PubMed.
- Y. Sun, X. Zhao, N. Ding, H. Gao, Y. Wu, Y. Yang, M. Zhao, J. Hwang, Y. Song, W. Liu and Y. Rao, Cell Res., 2018, 28, 779–781 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS.
- L. Wang, X. Shao, T. Zhong, Y. Wu, A. Xu, X. Sun, H. Gao, Y. Liu, T. Lan, Y. Tong, X. Tao, W. Du, W. Wang, Y. Chen, T. Li, X. Meng, H. Deng, B. Yang, Q. He, M. Ying and Y. Rao, Nat. Chem. Biol., 2021, 17, 567–575 CrossRef CAS.
- Y. Sun, X. Luo, Z. Yang, W. Lv, L. Chen, H. Li and Y. Rao, Chin. Chem. Lett., 2023, 34, 107924 CrossRef CAS.
- Z. Yang, Y. Sun, Z. Ni, C. Yang, Y. Tong, Y. Liu, H. Li and Y. Rao, Cell Res., 2021, 31, 1315–1318 CrossRef CAS PubMed.
- H. T. Huang, H. S. Seo, T. Zhang, Y. Wang, B. Jiang, Q. Li, D. L. Buckley, B. Nabet, J. M. Roberts, J. Paulk, S. Dastjerdi, G. E. Winter, H. McLauchlan, J. Moran, J. E. Bradner, M. J. Eck, S. Dhe-Paganon, J. J. Zhao and N. S. Gray, eLife, 2017, 6, e26693 CrossRef PubMed.
- W. J. Lu, W. Peng, Q. Sun, Y. Li, B. Chen, L. Yu, Y. Xu, S. Wang and Y. Zhao, Cell Death Dis., 2018, 4, 24 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Xiao
Liu
,
Qiyu
He
*,
Xiao
Liu
,
Qiyu
He






