
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrating a quinone substructure into histone deacetylase inhibitors to cope with Alzheimer's disease and cancer†
Melissa
Guardigni‡
a,
Giulia
Greco‡
b,
Eleonora
Poeta‡
c,
Alan
Santini
a,
Elisa
Tassinari
a,
Christian
Bergamini
c,
Chiara
Zalambani
c,
Angela
De Simone
 d,
Vincenza
Andrisano
a,
Elisa
Uliassi
c,
Barbara
Monti
c,
Maria Laura
Bolognesi
*c,
Carmela
Fimognari
a and
Andrea
Milelli
*a
d,
Vincenza
Andrisano
a,
Elisa
Uliassi
c,
Barbara
Monti
c,
Maria Laura
Bolognesi
*c,
Carmela
Fimognari
a and
Andrea
Milelli
*a
aDepartment for Life Quality Studies, Alma Mater Studiorum – University of Bologna, 47921 Rimini, Italy. E-mail: andrea.milelli3@unibo.it
bDepartment of Chemistry “Giacomo Ciamician”, Alma Mater Studiorum – University of Bologna, 40129, Bologna, Italy
cDepartment of Pharmacy and Biotechnology, Alma Mater Studiorum – University of Bologna, 40126 Bologna, Italy. E-mail: marialaura.bolognesi@unibo.it
dDepartment of Drug Science and Technology, University of Turin, 10125 Turin, Italy
First published on 2nd May 2024
Abstract
Alzheimer's disease (AD) and cancer are among the most devastating diseases of the 21st century. Although the clinical manifestations are different and the cellular mechanisms underlying the pathologies are opposite, there are different classes of molecules that are effective in both diseases, such as quinone-based compounds and histone deacetylase inhibitors (HDACIs). Herein, we investigate the biological effects of a series of compounds built to exploit the beneficial effects of quinones and histone deacetylase inhibition (compounds 1–8). Among the different compounds, compound 6 turned out to be a potent cytotoxic agent in SH-SY5Y cancer cell line, with a half maximal inhibitory concentration (IC50) value lower than vorinostat and a pro-apoptotic activity. On the other hand, compound 8 was nontoxic up to the concentration of 100 μM and was highly effective in stimulating the proliferation of neural precursor cells (NPCs), as well as inducing differentiation into neurons, at low micromolar concentrations. In particular, it was able to induce NPC differentiation solely towards a neuronal-specific phenotype, without affecting glial cells commitment.
Introduction
Histone deacetylases (HDACs) are transcriptional regulators that have attracted considerable attention in both chemical biology and medicinal chemistry in the last thirty years.1 From a drug-discovery perspective, they are one of the most druggable of the epigenetic family, from which the identification of histone deacetylase inhibitors (HDACIs) has raised great therapeutic hopes. Although HDAC aberrant activity is associated with multiple pathological conditions, to date HDACs have been mainly related with tumor progression, and HDACIs have been approved for treatments of T-cell lymphoma, multiple myeloma, and breast cancer in combinatorial therapy.2,3 However, a recent great deal of attention has been paid to the role of HDACs in regulating brain function, development, and degeneration.4 Particularly, HDAC2 overexpression has been shown to decrease the dendritic spine density, synapse number, synaptic plasticity, and memory formation;5 for this reason, this isoform has been proposed as a promising target for both cancer and neurodegenerative disorders.6 Similarly, while the expression of HDAC6 has been initially linked to tumorigenesis and the metastasis of cancer cells, increased HDAC6 expression has been also found in postmortem brain samples from Alzheimer's disease (AD) patients.7Furthermore, HDAC6 has been shown to facilitate tau-mediated toxicity8 and HDAC6 inhibition has proven to be effective also in in vivo models of AD.9 As a consequence, vorinostat, the first US Food and Drug Administration (FDA)-approved HDAC drug, as well as AMX0035, a fixed-dose combination of HDACI sodium phenylbutyrate and tauroursodeoxycholic acid, are being evaluated in clinical trials for AD (NCT03056495 and NCT03533257). Thus, it appears that HDACIs have potential against both AD and cancer, which are among the most pressing therapeutic challenges worldwide.
In an intriguing and similar manner, quinone-based molecules have been proven to exert beneficial effects as both antiproliferative10,11 and neuroprotective agents.12 Quinones have traditionally represented a major source of anticancer compounds. These agents are unique among cancer therapeutics because of their capability to generate toxic free radical species following catalyzed bioreduction of the quinone moiety. For instance, daunomycin, doxorubicin, mitomycin C, and mitoxantrone have been approved by the FDA while many others are currently in clinical trials (e.g., lapachol, Fig. 1). More recently, quinone-based compounds have shown interesting activities also in the context of neurodegenerative diseases (NDs).12 For instance, Bolognesi and coworkers reported that quinone-based compounds (e.g., memoquin in Fig. 1), being reduced to hydroquinones, can act as radical scavengers and exhibit neuroprotective effects.13–15 Furthermore, vitamin K and its analogs bearing the menadione moiety have shown neural differentiation-inducing activity.16,17 To note, these properties can be modulated through proper medicinal chemistry.
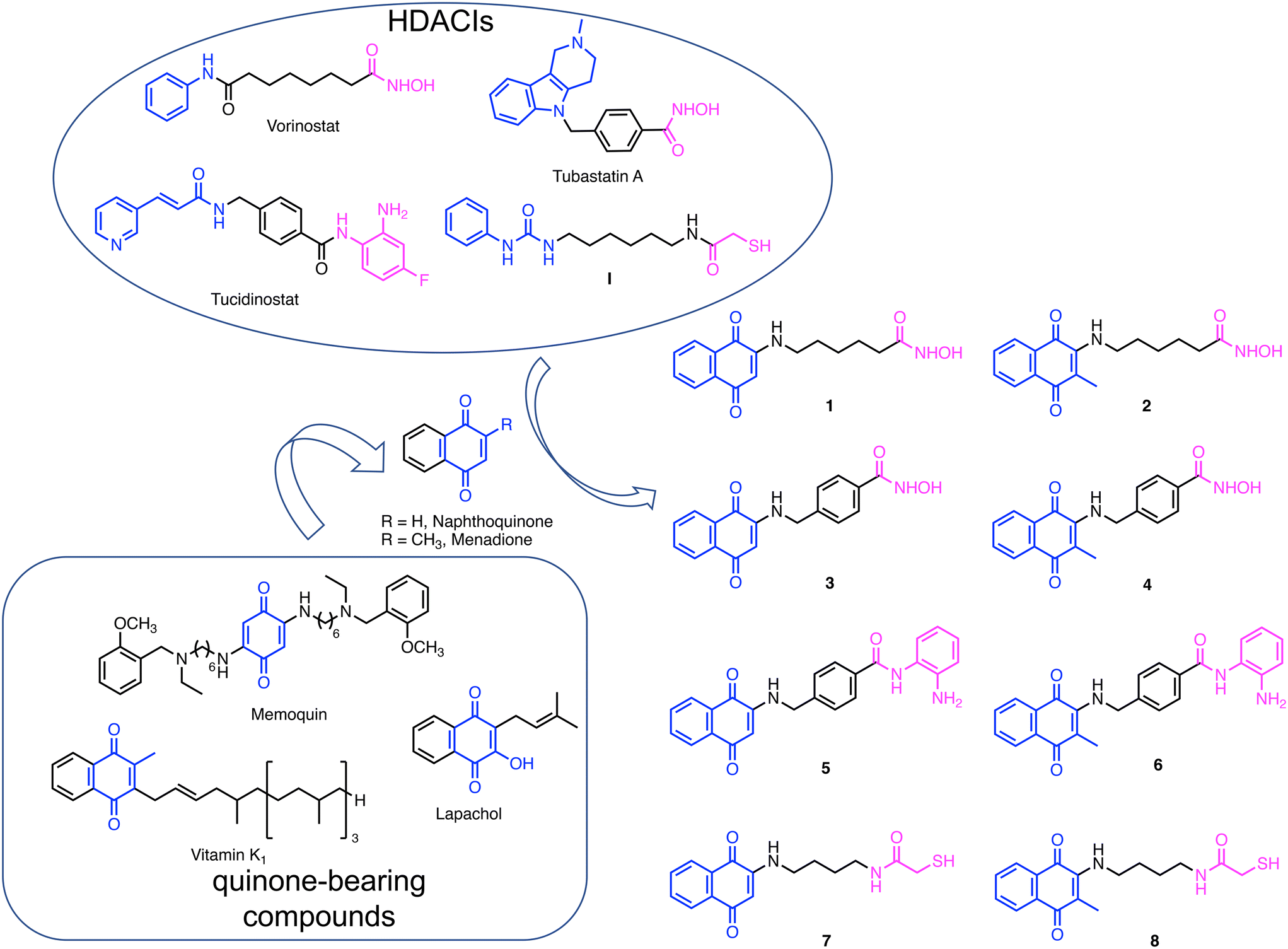 | ||
| Fig. 1 Design strategy leading to the target compounds 1–8. Referring to the HDACs' pharmacophore features, the CAP group is colored in blue, the linker in black, and the zinc-binding group in magenta. | ||
In the present investigation, we sought to generate a set of hybrid molecules by amalgamating HDACIs and quinone molecular frameworks into new single molecules. We aimed to develop molecules comprising the beneficial properties of these chemical substructures and to explore their potential against two of the most common chronic-degenerative conditions, i.e., cancer and NDs.
Results and discussions
Drug design
To design this new class of compounds, we explored the pharmacophoric model of HDACIs that comprises a zinc binding group (ZBG), responsible for binding the Zn2+ ion located at the bottom of the catalytic site, a linker, and a CAP group, usually an aromatic surface that can establish additional interactions with the external surface of the enzyme and strengthen target engagement. The CAP group is highly permissive and, therefore, a vast array of chemotypes have been reported. Due to the CAP modification tolerance, moieties endowed with additional biochemical properties have been employed, giving rise to dual inhibitors. Thanks to this approach, several different HDAC-based multiple agents have been developed as both antiproliferative and neuroprotective agents.18,19 In this investigation, naphthoquinone was selected as the CAP group. We were inspired by the work of Chou et al.,20 who reported a class of novel HDACIs bearing a naphthoquinone structure and displaying a selective inhibition profile against HDAC6. Indeed, naphthoquinone meets the structural requirements for a CAP group, namely a large aromatic surface capable of establishing π–π and cation–π interactions and the presence of groups able to establish hydrogen bonds with the biological counterpart(s). In addition to the naphthoquinone, we also considered menadione, which is present in several vitamin K analogs, as it can also exert interesting biological properties (Fig. 1).21,22 Moreover, having a methyl group in the α-position relative to the side substituent, it is not subject to attack by the nucleophilic groups of proteins.As for ZBGs, given the large availability of molecular frameworks and their distinctive properties, we decided to explore three different ones: hydroxamic acid, benzamide, and mercaptoacetic groups. Hydroxamic acid, due to its high affinity for Zn2+, is found in the majority of reported HDACIs. Other notable ZBGs include benzamide and its derivatives, as in tucidinostat,23 and mercaptoacetamide (as in I reported by Kozikowski's group), which confer superior neuroprotective effects in cortical neurons compared to hydroxamates.24,25
Based on these considerations, we designed a new series of hybrid compounds by combining naphthoquinone/menadione with different ZBGs, specifically (Fig. 1):
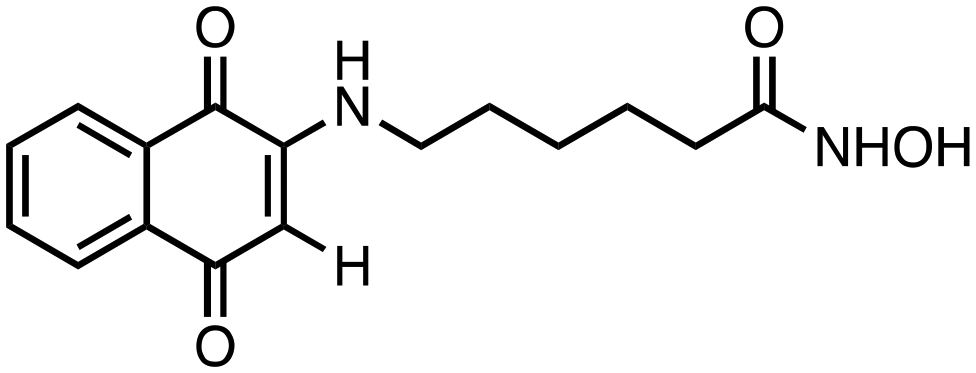
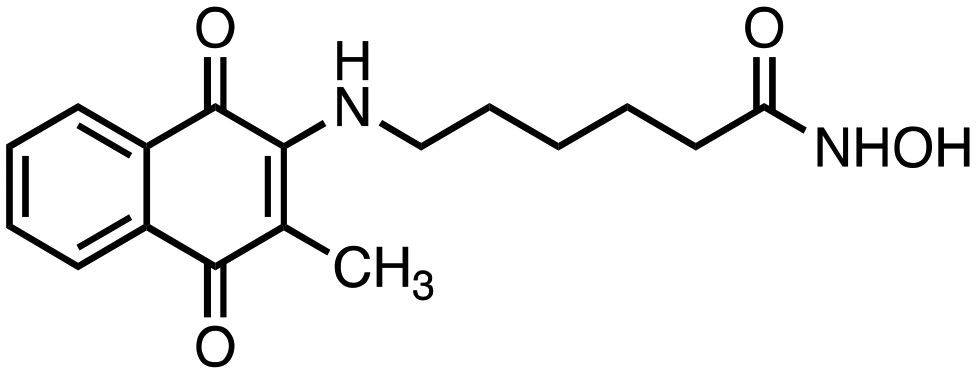
1) compounds 1 and 2 bearing an alkylhydroxamic acid functionality, as in vorinostat;
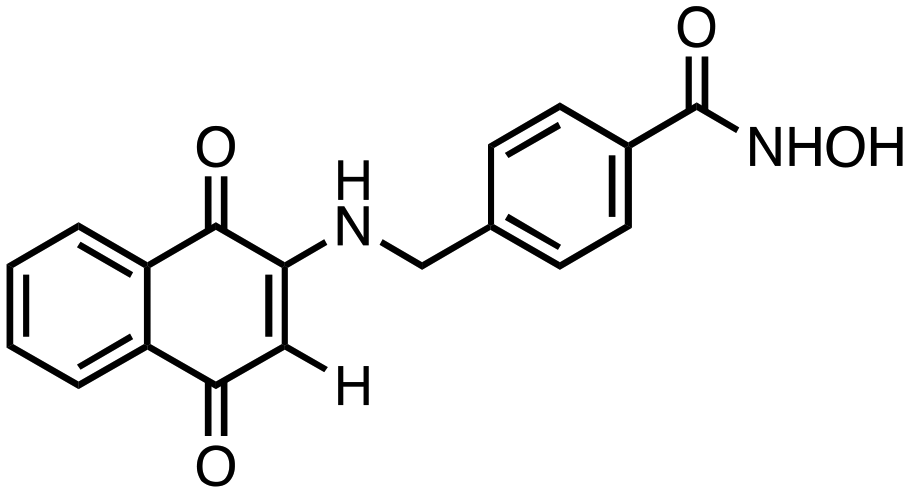
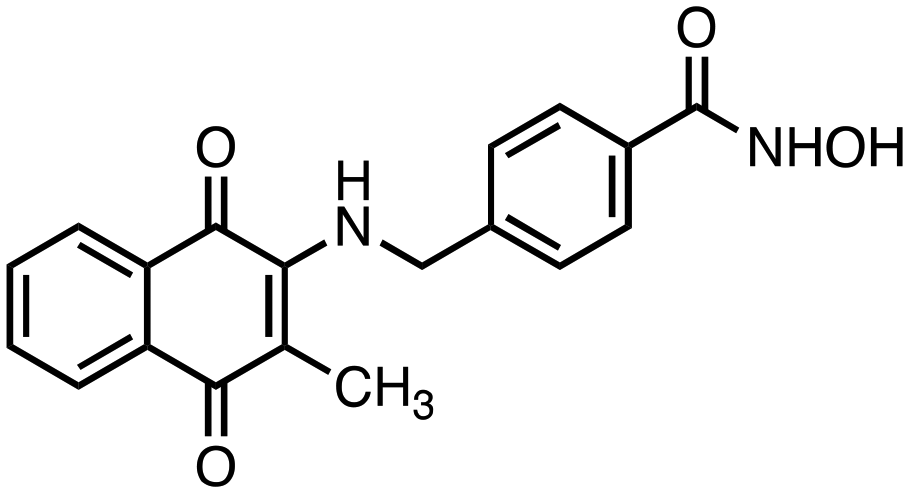
2) compounds 3 and 4 bearing a benzylhydroxamic acid functionality, as in tubastatin;
3) compounds 5 and 6 bearing a benzamide functionality, as in tucidinostat;
4) compounds 7 and 8 bearing a mercaptoacetamide functionality, as in I.
Synthesis
Target compounds 1–8 were synthesized following the procedure reported in Schemes 1 and 2. First, we synthesized the side chains, which were sequentially coupled with the appropriate quinone. 6-Amino-N-(trityloxy)hexanamide was synthesized following the procedure we have previously developed,26 while for compounds 11, 13, and 16, the synthetic procedure is reported in Scheme 1. For the synthesis of 11, we started from the trifluoroacetamide derivative 9,27 which was activated with EDCI/HOBT and then reacted with Tetrahydropyranyl-protected hydroxylamine to furnish the adduct 10. The latter was then subjected to basic hydrolysis to give the primary amine 11. Similarly, 13 was synthesized starting from the common intermediate 9, which underwent activation with iso-butylchloroformate, followed by reaction with tert-butyl (2-aminophenyl)carbamate28 to give 12. Basic hydrolysis furnished the primary amine 13. For the synthesis of 16, we started from the carboxylic acid 14,29 which was activated with EDCI/HOBT and reacted with Boc-protected diaminobutane30 to give 15, which was then deprotected through acidic hydrolysis.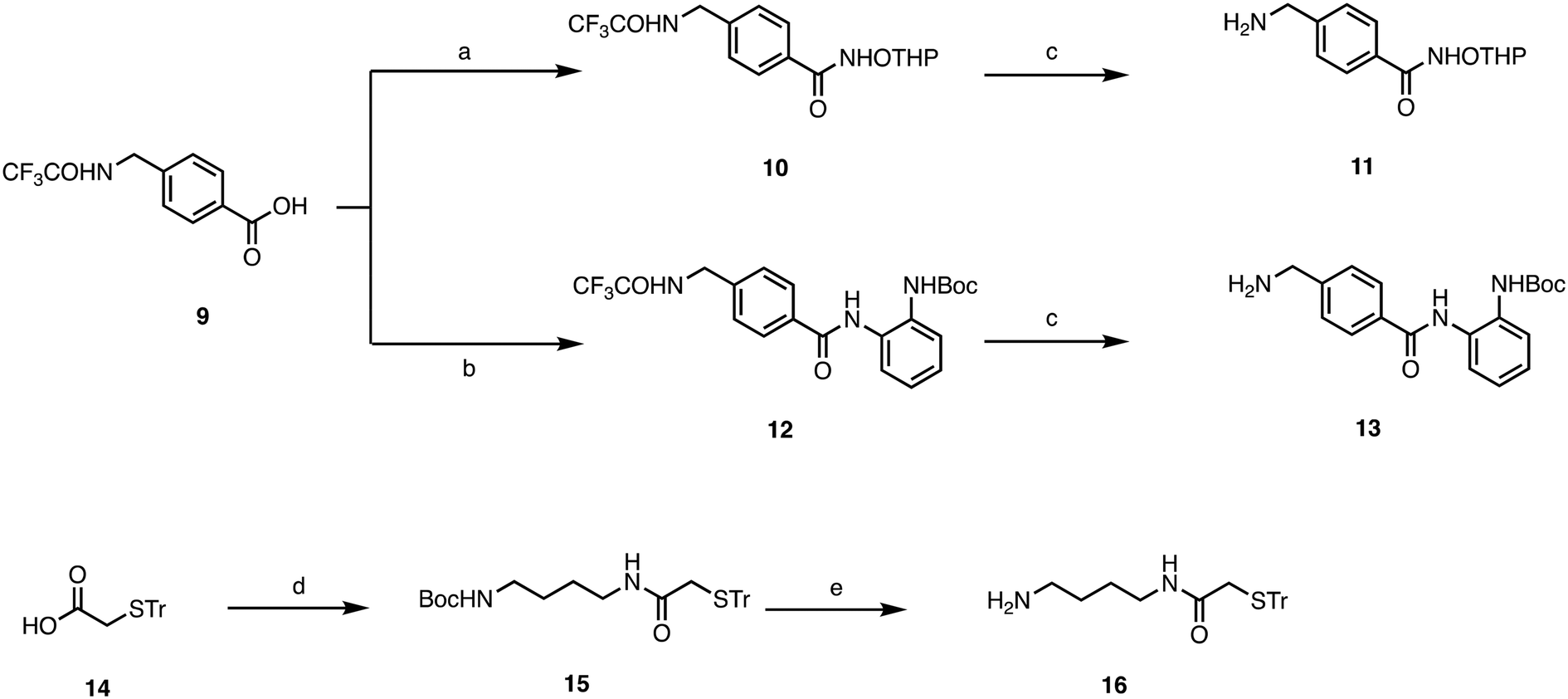 | ||
| Scheme 1 a) 1. EDCI, HOBT, DMF, 0 °C, 30 min; 2. NH2OTHP, DMF, rt, 12 h; b) 1. IBCF, NMM, DMF, 0 °C, 30 min; 2. tert-butyl (2-aminophenyl)carbamate, DMF, 12 h, rt; c) K2CO3, MeOH/H2O, rt, 12 h; d) 1. EDCI, HOBT, DMF, 0 °C, 30 min; 2. NH2(CH2)4NHBoc, DMF, 12 h, rt; e) HCl 6N, MeOH, rt, 12 h. | ||
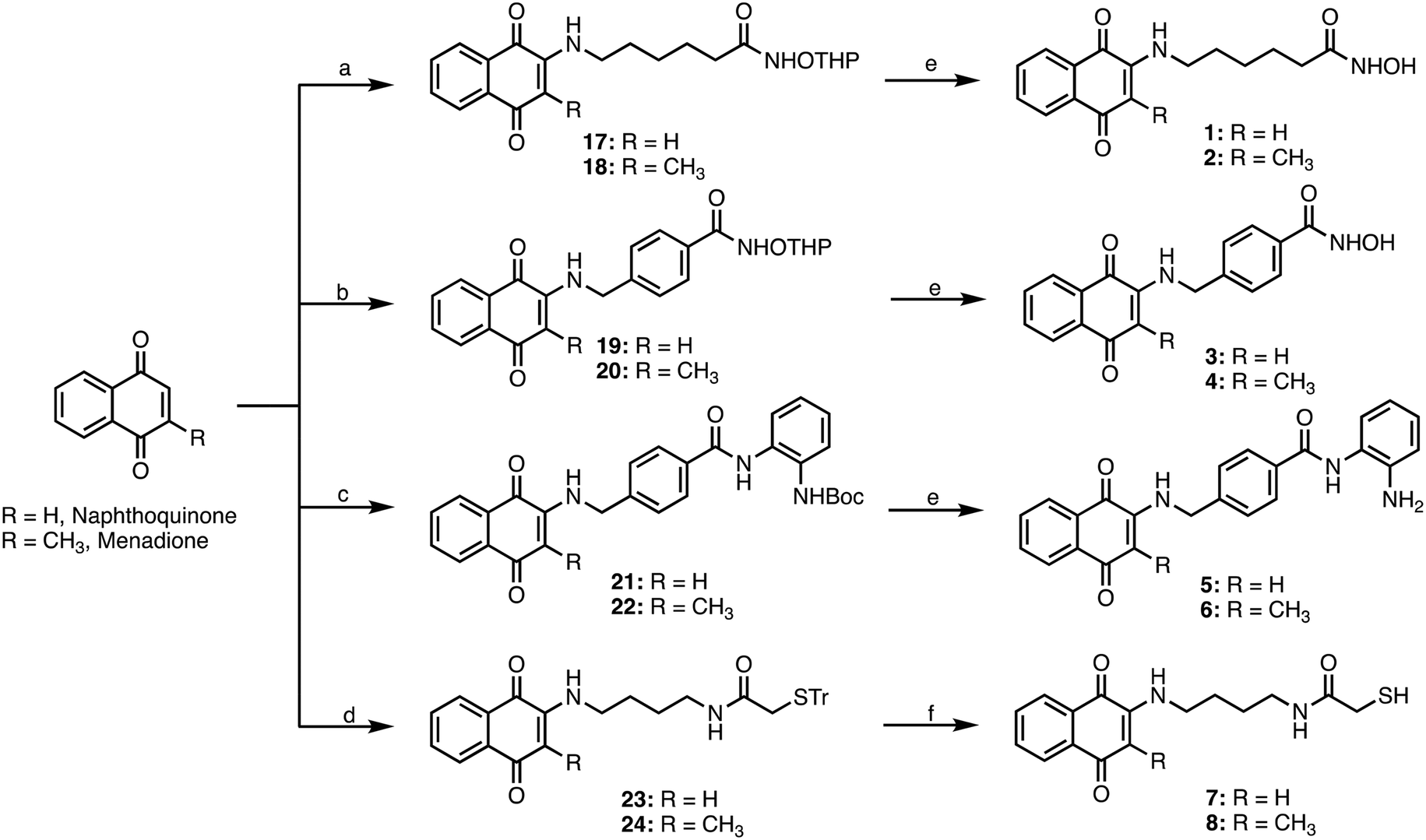 | ||
| Scheme 2 a) 6-Amino-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide, EtOH, rt, 12 h; b) 11, EtOH, rt, 12 h; c) 13, SnCl2, EtOH, rt, 12 h; d) 16, EtOH, rt, 12 h; e) TFA, DCM, rt, 12 h; f) TFA, Et3SiH, DCM, rt, 12 h. | ||
The four different side chains 6-amino-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide,2611, 13, and 16 were reacted with the appropriate 1,4-naphthoquinone or menadione to give the adducts 17–24, which were then deprotected with TFA to give the target compounds 1–8 (Scheme 2).
Being aware that the assessment of the cytotoxic effects could offer the opportunity to classify our compounds as antineurodegenerative or anticancer agents, we first evaluated the cytotoxicity of the designed compounds. Then, based on the obtained results, different in vitro and in cell studies were carried out to elucidate the mechanisms of action of the most promising compounds.
Cytotoxic effects in SH-SY5Y cells
As a first step, the target compounds 1–8 were evaluated for their potential cytotoxic effects on the neuroblastoma SH-SY5Y cell line after 24, 48, or 72 h of treatment at concentrations ranging from 0.1 to 100 μM. The HDACI vorinostat was used as a positive control. Among all the compounds, compound 6, characterized by menadione and benzamide as ZBGs, exhibited the highest cytotoxicity. It significantly decreased cell viability in a time- and concentration-dependent manner (Fig. 2), displaying a half maximal inhibitory concentration (IC50) value of 1.56 μM after 24 h, which was 32 times lower than that obtained for vorinostat (Table 1). Compared to compound 6, its desmethyl analog 5 was significantly less active, and its cytotoxicity was comparable to that of vorinostat. Compounds 1–4, characterized by the hydroxamic acid group as ZBGs, showed moderate cytotoxic activity at all three times points. In contrast, compounds 7 and 8, bearing a mercaptoacetamide as the ZBG, were found to have the lowest cytotoxicity. As for compound 8, in fact, at the highest concentration tested (100 μM) the cell viability was about 70% at all treatment timings. Regarding its demethylated form 7, similar results were observed, although its IC50 value at 24 h of treatment was significantly higher (Fig. 2 and Table 1).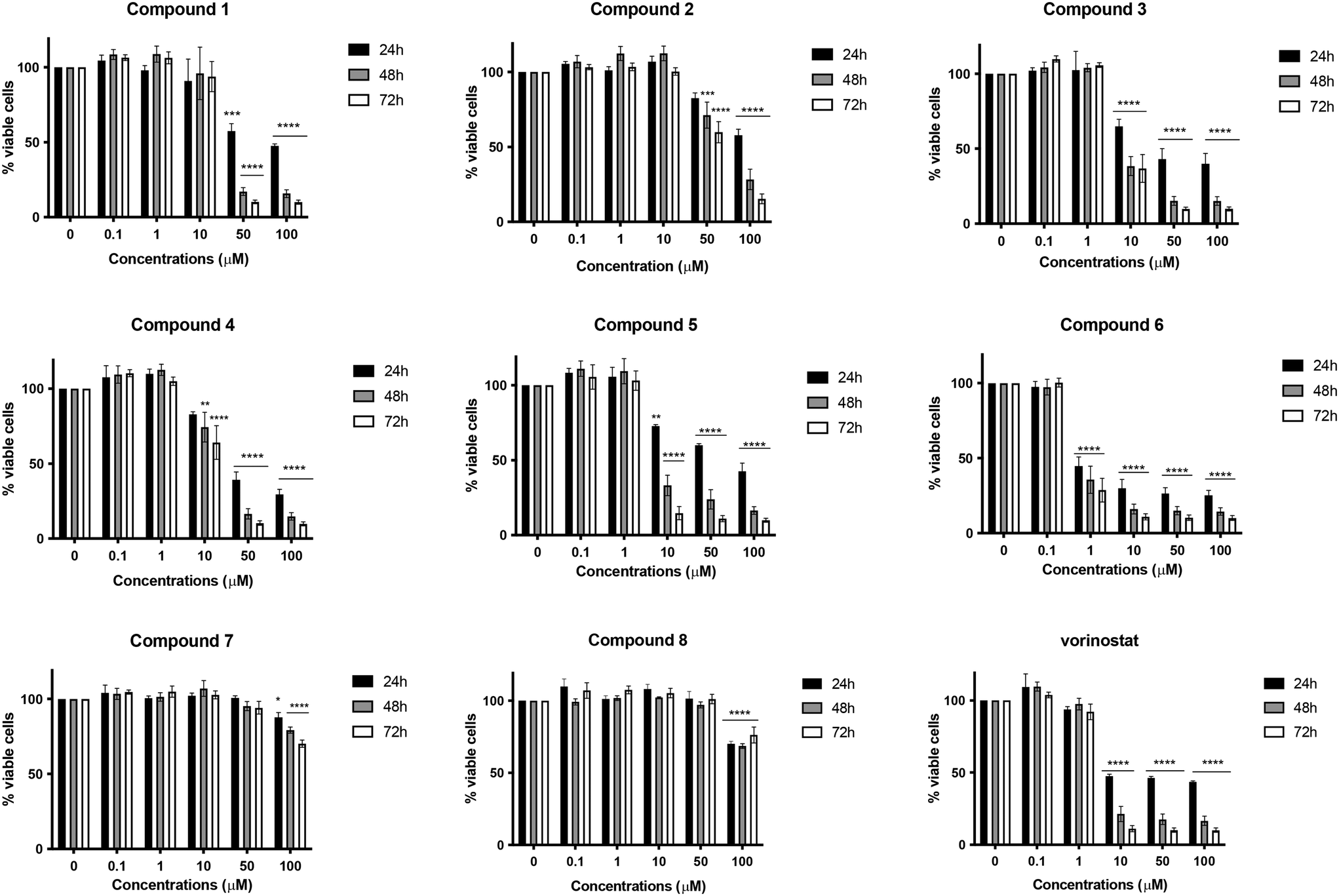 | ||
| Fig. 2 Percentage (%) of SH-SY5Y viable cells after treatment with increasing concentrations of the tested compounds or vorinostat for 24, 48 or 72 h. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 versus untreated cells, two-way ANOVA (Dunnett's post hoc comparison test). | ||
| Compound | Structure | IC50 (μM) ± SEM | ||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| 1 |
|
79.99 ± 13.01 | 26.93 ± 8.07 | 22.47 ± 6.25 |
| 2 |
|
177.7 ± 27.29 | 84.40 ± 22.46 | 52.97 ± 10.60 |
| 3 |
|
38.96 ± 8.01 | 8.67 ± 1.56 | 7.74 ± 1.64 |
| 4 |
|
39.03 ± 5.74 | 19.78 ± 4.37 | 13.97 ± 2.81 |
| 5 |
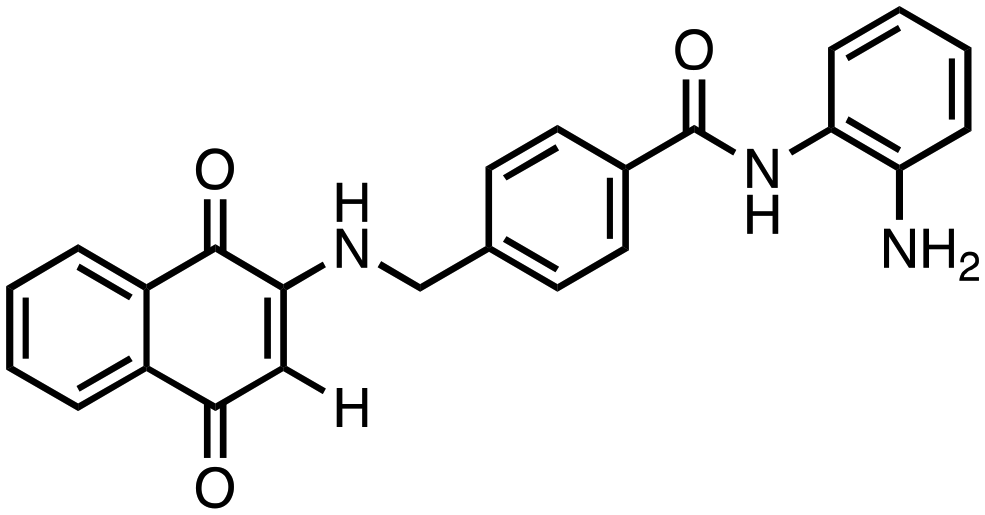
|
59.50 ± 8.10 | 8.53 ± 2.06 | 4.42 ± 1.15 |
| 6 |
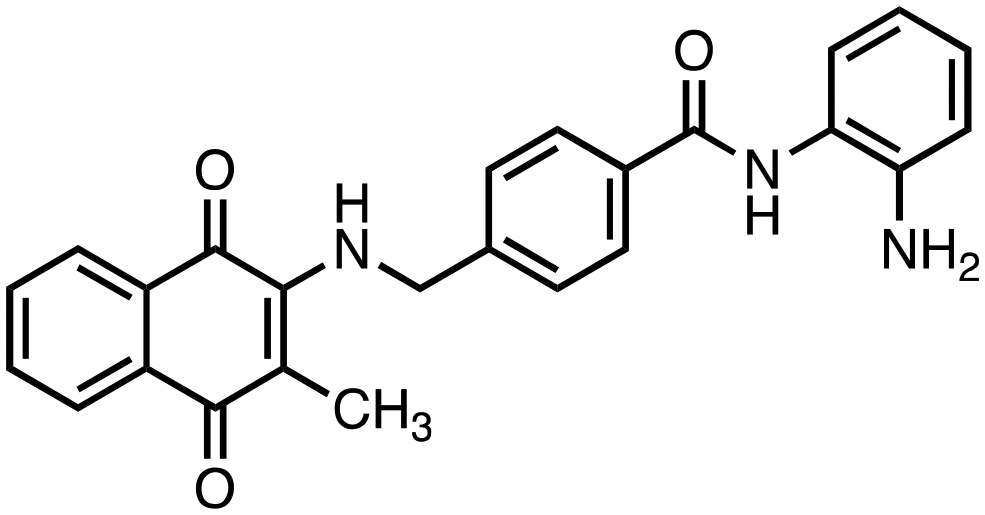
|
1.56 ± 0.60 | 0.78 ± 0.20 | 0.62 ± 0.14 |
| 7 |
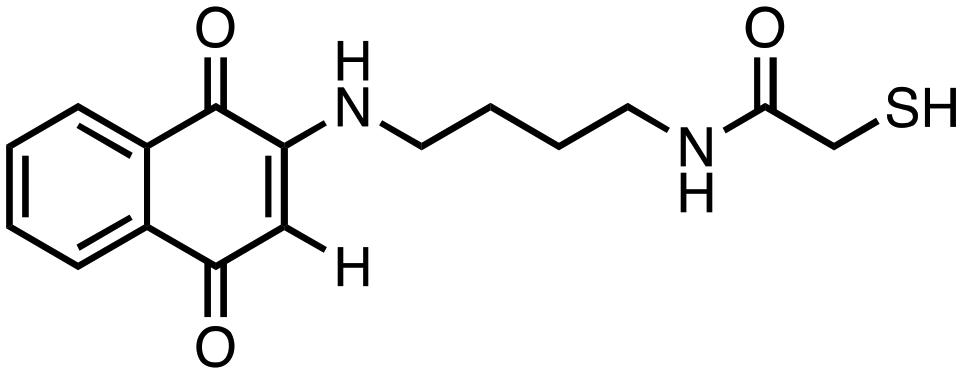
|
>100 | >100 | >100 |
| 8 |
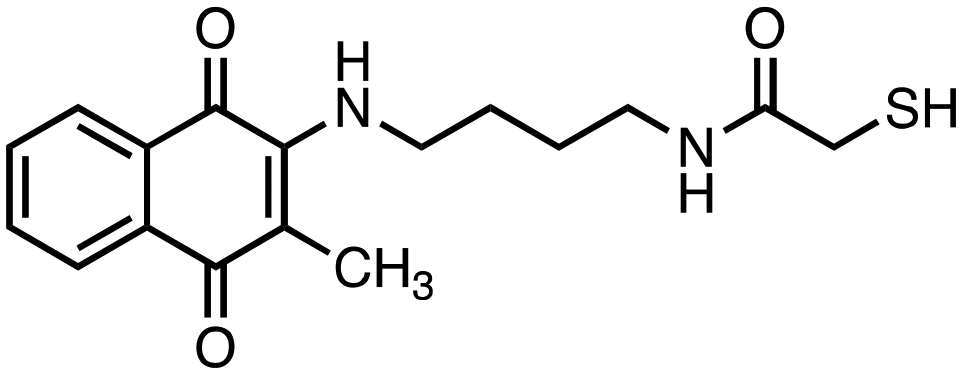
|
>100 | >100 | >100 |
| Vorinostat |
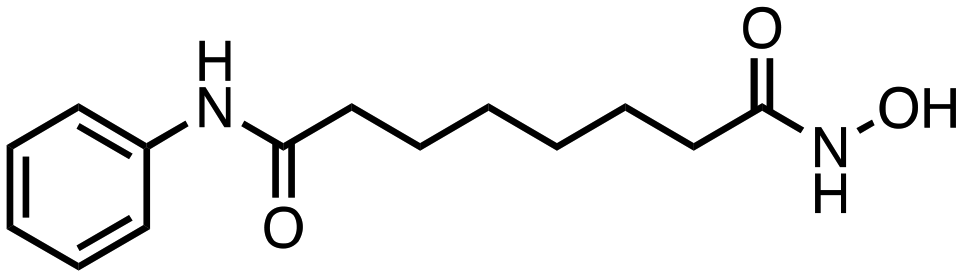
|
32.20 ± 8.68 | 5.43 ± 1.35 | 3.59 ± 0.80 |
Based on the toxicity data, it was interesting to note that the cytotoxic effect did not significantly depend on the substitution pattern on the quinone moiety, i.e., methyl versus desmethyl compounds, but rather it had a strong dependence on the nature of the ZBGs. In fact, although the most toxic compound in the series carried the benzamide group, compounds characterized by the presence of hydroxamic acid, either alkyl or aryl, also showed some level of toxicity, albeit modest in some cases. The compounds characterized by mercaptoacetamide ZBGs were essentially nontoxic, showing a slight decrease in cell viability only at the 100 μM concentration. These findings are perfectly in line with what has been reported in the literature; indeed, Kozikowski et al. outlined that mercaptoacetamide derivatives are generally less toxic in cortical neurons than the corresponding hydroxamic acids.24 Furthermore, the same study reported that mercaptoacetamide-based inhibitors showed greater neuroprotective activity in cortical neurons subjected to toxic stimuli than hydroxamic acid-based inhibitors.24
Based on these results, we selected compounds 6 and 8 for further biochemical evaluations. In particular, compound 6 was evaluated for its ability to act as a potential anticancer agent, while compound 8, due to its lack of cytotoxicity, was evaluated for its potential use in the context of NDs. For both compounds, we determined HDAC2 and HDAC6 in vitro inhibitions, the ability to block HDAC activity in cells, and the effects on oxidative stress.
HDAC2 and HDAC6 inhibitions
We evaluated compounds 6 and 8 for their ability to inhibit HDAC2 and HDAC6. Indeed, despite these two isoforms belonging to two different HDAC classes, and having different cellular localization, both of them have implications in cancer and AD.5,6,31–33 As reported in Table 2, both compounds were able to inhibit the activity of HDAC2 and HDAC6 with an IC50 in the low micromolar range of concentrations (compound 6: HDAC2: 2.10 ± 0.02 μM, HDAC6: 3.76 ± 0.16 μM; compound 8: HDAC2: 25.19 ± 1.89 μM, HDAC6: 30.05 ± 0.16 μM). However, both compounds showed no selectivity between the two isoforms. This was partially in contrast with the literature reporting that mercaptoacetamide-based HDACIs display higher HDAC6 potency and selectivity over class I HDAC isozymes.25To evaluate whether the in vitro inhibitory activities translate into the intracellular inhibition of HDACs, further in-cell-based assays for the selected compound 6 were carried out. Western blotting analysis was performed using the human neuroblastoma SH-SY5Y cell line to determine the effects of compound 6 on the lysines acetylation levels of histones H2/H3. Cells were treated with compound 6 at a 5 μM concentration for 6 h. As can be observed in Fig. S1,† compound 6 was able to induce a massive hyperacetylation of histone H2/H3, indicating a clear in-cell enzymatic inhibition.
Effects on oxidative stress
Quinone-based compounds, such as compounds 6 and 8, can act as anti- or pro-oxidants depending on the specific set of conditions. To evaluate the effect of the compounds on cellular redox homeostasis, we treated human neuroblastoma SH-SY5Y cells with 2 μM of compounds 6 and 8 for 24 h in the presence or absence of the exogenous oxidative stress inducer tert-butyl hydroperoxide (TBH). The effect of the target compounds on both endogenous and induced oxidative stress was assessed using the fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFDA). After cleavage of the acetate groups by intracellular esterases and oxidation by intracellular reactive oxygen species (ROS), the non-fluorescent H2DCF is oxidized by ROS to the highly fluorescent 2′,7′-dichlorofluorescein (DCF). Compound 6 showed a significant induction of oxidative stress compared to the control, whereas compound 8 showed only a tendency to increase ROS production. Furthermore, both molecules did not show any protective effect against THB-induced oxidative stress (Fig. 3).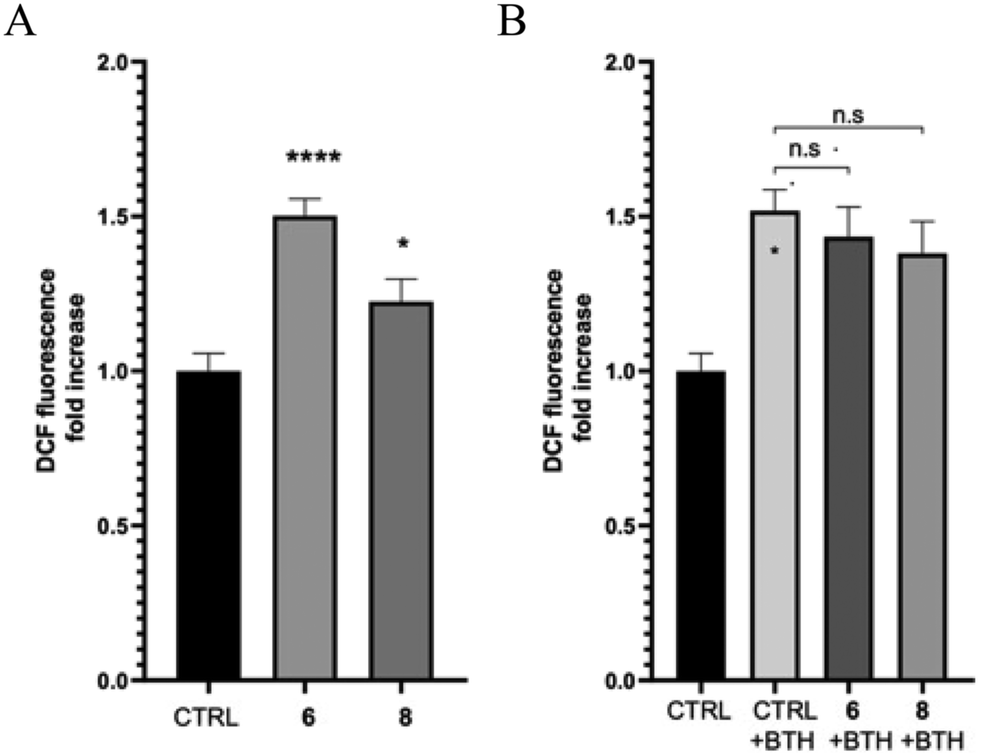 | ||
| Fig. 3 Determination of effects on oxidative stress. A) Determination of the ROS in SH-SY5Y cells treated with 2 μM of compounds 6 and 8 or vehicle (CTRL) using the fluorogenic probe DCFDA (n = 5). B) Determination of the ROS in SH-SY5Y cells pretreated with compounds 6 and 8 or vehicle for 24 h. Oxidative stress was induced with 100 μM tert-butyl hydroperoxide for 30 min (n = 5). Data are presented as the mean ± s.e.m. (*p ≤ 0.05, ****p ≤ 0.0001 versus ctrl, unpaired t-test with Welch's correction). | ||
The potential neuroprotective activity of compound 8 was also evaluated by analyzing its ability to protect SH-SY5Y cells from the cytotoxic effect caused by hydrogen peroxide (H2O2). Pre-treatment with compound 8 at all concentrations (1–50 μM) did not rescue SH-SY5Y cell viability (Fig. 4). In addition, the viability of cells pretreated with compound 8 at 50 μM and exposed to H2O2 was even lower than that of cells treated with H2O2 alone (28.2% versus 50.8%) (Fig. 4). This behavior might be ascribed to the fact that the capacity of quinones to exert their antioxidant activity resides in the hydroquinone form. NAD(P)H quinone dehydrogenase 1 (NQO1) is an inducible enzyme responsible for the reduction of natural quinones to antioxidant hydroquinones.34 Interestingly, NQO1 levels are increased in AD patients.35
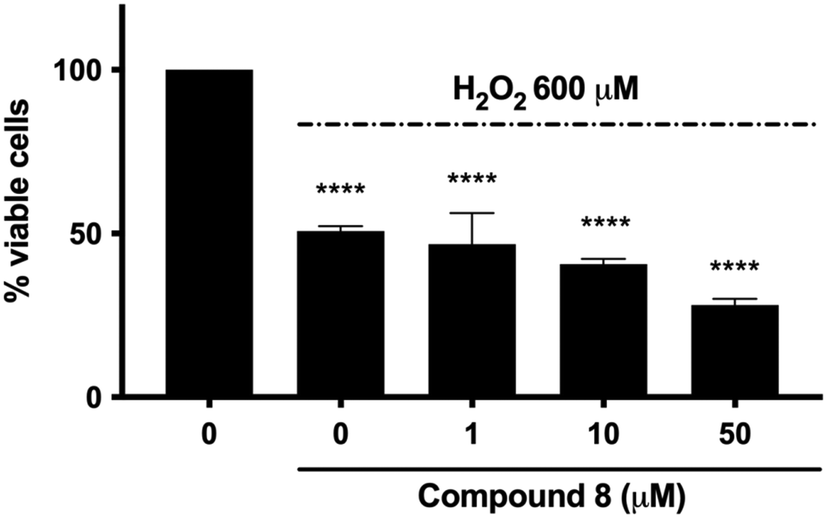 | ||
| Fig. 4 Percentage (%) of viable SH-SY5Y cells after pre-treatment with compound 8 (1, 10, or 50 μM) for 24 h, treatment with H2O2 (600 μM) for 1 h, and 22 h recovery in drug-free complete medium. ****p < 0.0001 versus untreated cells, one-way ANOVA (Tukey's post hoc comparison test). | ||
To verify the antioxidant potential of 8 in H2O2-stressed SH-SY5Y cells mimicking NQO1 overexpression, which is typical of AD, they were treated with sulforaphane (SFN), one of the most potent NQO1 inducers known to date.36 Surprisingly, no increase in the protective activity was observed (data not shown). Similarly, we did not observe any change in the toxic activity of compound 6 in SH-SY6Y cells pretreated with SFN (data not shown).
Overall, these findings suggest that compound 8 cannot counteract the cytotoxic effect of the pro-oxidant agent H2O2 and, at higher concentrations, increases H2O2-induced cytotoxicity.
Modulation of cell death by compound 6
To better explore the pharmacological potential of compound 6 in cancer cells, we first analyzed its cytostatic activity. After 72 h of treatment, no cell-cycle block was recorded in the G0/G1, S, or G2/M phases, indicating that compound 6 did not exert any cytostatic effect. Of note, compound 6 treatment increased the percentage of cells in the sub-G1-phase in a concentration-dependent manner: from 28.03% at 1 μM concentration to 42.32% at 10 μM, compared with 5.56% of untreated cells (Fig. 5a). The sub-G1 population identifies cells with fragmented DNA. Although DNA fragmentation is a crucial morphological feature of apoptotic cell death, even necrotic cells could have a fractional DNA content.37,38 Therefore, the increase in cells in the sub-G1 phase indicates that compound 6 causes cell death, but this cannot prove apoptosis involvement. Hence, to evaluate the pro-apoptotic activity of compound 6, we examined caspase-3 activation and poly(ADP-ribose) polymerase (PARP) cleavage, being two crucial events in the final stage of apoptotic cell death.39 Treatment with compound 6 for 72 h increased caspase-3 activity 1.2-fold at 1 μM and 1.7-fold at 10 μM compared to the untreated cells (Fig. 5b). Flow cytometry analysis also indicated that compound 6 significantly decreased whole PARP expression in a concentration-dependent manner, thus indicating an increase in the expression of the cleaved counterpart. In particular, whole PARP1 expression was reduced by 44.81% and 56.81% at 1 and 10 μM, respectively, after 72 h treatment (Fig. 5c). Taken together, these results indicate that compound 6 induces apoptosis in SH-SY5Y cells.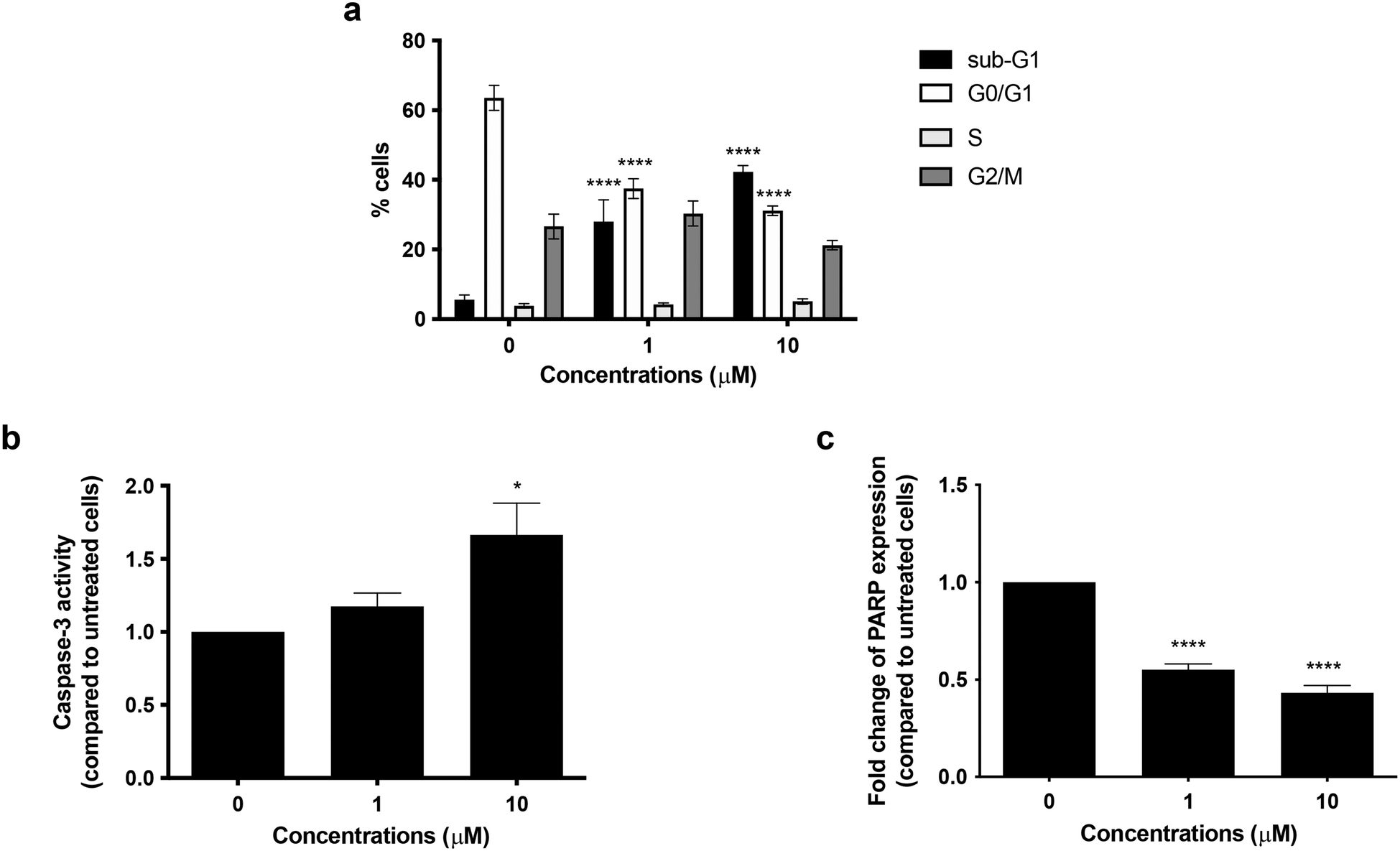 | ||
| Fig. 5 Cell-cycle distribution (a), caspase-3 activity (b), and PARP protein expression (c) in SH-SY5Y cells treated with compound 6 at 1 or 10 μM for 72 h. *p < 0.05; ****p < 0.0001 versus untreated cells, one-way ANOVA (Bonferroni's post hoc comparison test). | ||
Role of ROS generation in the cytotoxic effect of compound 6
Since compound 6 increased ROS intracellular levels in SH-SY5Y cells, we investigated whether the pro-oxidant behavior of compound 6 may contribute to its cytotoxicity. Pre-treatment with the antioxidant N-acetyl-L-cysteine (NAC) increased the viability of SH-SY5Y cells treated with compound 6 at 1 μM by 22.3% (Fig. 6), counteracting the cytotoxic effect elicited by the hybrid derivative. In contrast, the cytotoxic effect of compound 6 at 10 μM was not mitigated, most likely due to the compound's higher toxicity.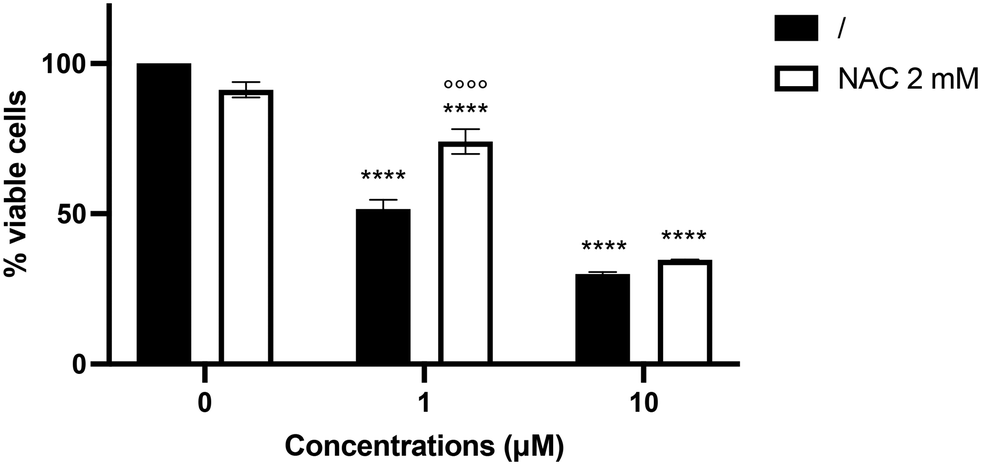 | ||
| Fig. 6 Percentage (%) viable SH-SY5Y cells after pre-treatment with NAC 2 mM for 1 h, and treatment with compound 6 at 1 or 10 μM for 24 h. ****p < 0.0001 versus untreated cells. °°°°p < 0.0001 versus compound 6-treated cells, two-way ANOVA (Tukey's post hoc comparison test). | ||
Study of compound 8's cytotoxicity in human hepatoma cell line (HepG2)
Hepatotoxicity would be of importance for the overall drug-likeness of potential drugs, and particularly for compound 8, as quinones are known for their hepatotoxicity.14 Thus, experiments were performed in HepG2 cells (Fig. S2†). After 24 h incubation at 1–100 μM, compound 8 showed no significant toxicity up to 10 μM with a calculated IC50 of 25.52 μM.Effects of compound 8 on progenitor cells differentiation into neuronal cells
In AD, neurons are the brain's cells that are largely damaged and the connections within neuronal networks are also interrupted. A potential strategy for modifying the course of AD involves the utilization of small molecules able to induce the proliferation and/or differentiation of neural progenitor cells (NPCs), a potential source of new neurons, to mitigate cognitive impairments and counteract neurodegeneration.40,41 In fact, NPCs are multipotent cells that can generate neurons, astrocytes, and oligodendrocytes, and quinone-based compounds, such as vitamin K derivatives, as well as HDACIs, are known to induce the differentiation of NPCs into neuronal cells.16,42Therefore, to investigate the effect of compound 8 on NPCs proliferation and differentiation, experiments were performed on an in vitro model of neurospheres (i.e., floating cultures of NSCs and neuronal, astrocytic, and oligodendrocytic progenitors at different stages of maturation) derived from the sub-ventricular zone (SVZ) of 8-months-old C57BL/6N wild-type male mice (Mus musculus). For proliferation analysis, neurospheres were plated in suspension as single cells in the presence of compound 8 (2.5, 5, 10, 25, 50 μM) or dimethyl sulfoxide (DMSO) as the control. After 7 days in vitro (DIV), while higher concentrations (25, 50 μM) were revealed to be toxic, the lower 2.5 and 5 μM concentrations of compound 8 significantly increased the neurospheres number per well (Fig. 7a and b) without affecting the neurospheres size (Fig. 7a and c) compared to the control, suggesting a positive effect in stimulating NPCs proliferation.
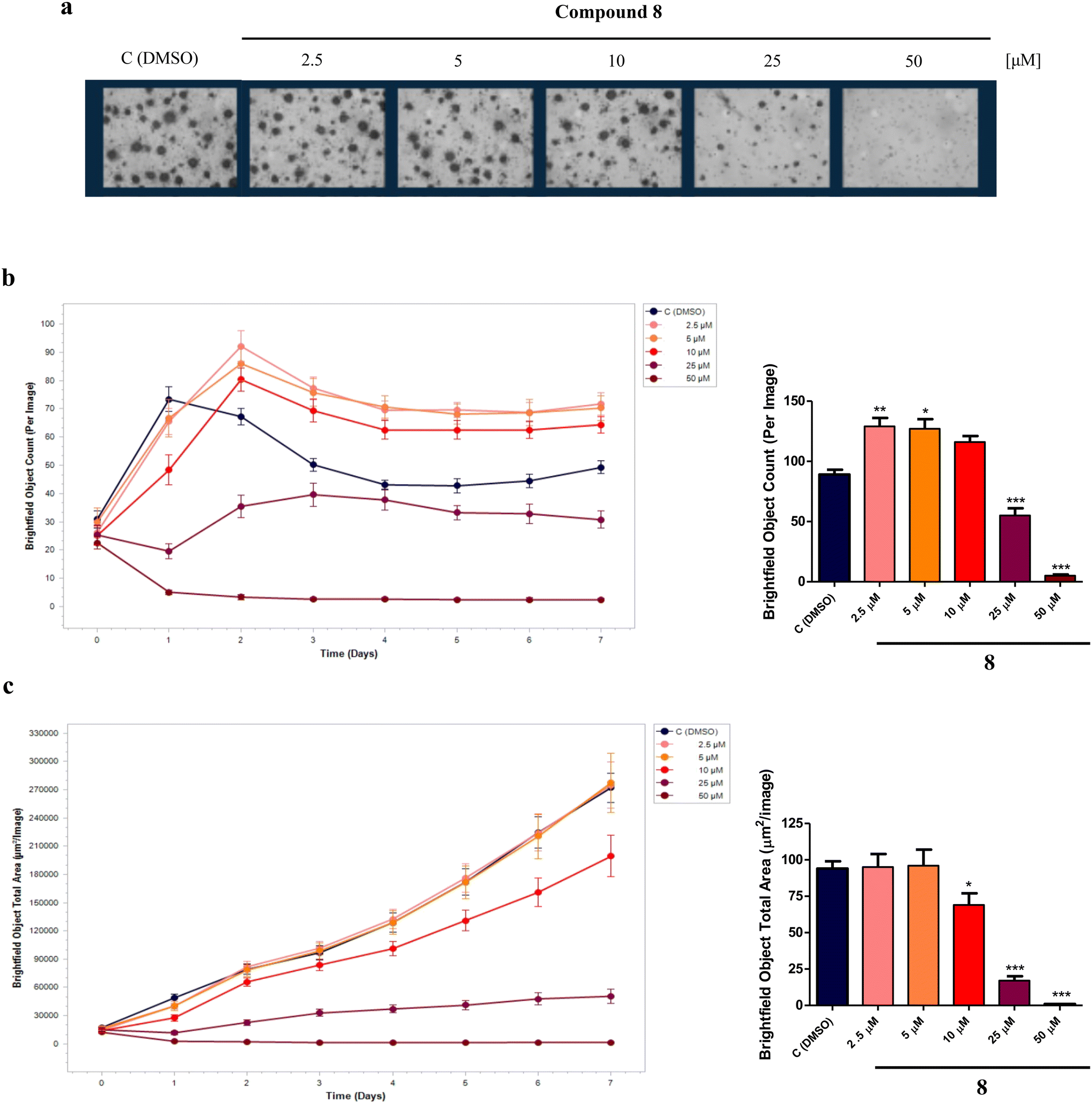 | ||
| Fig. 7 (a) Brightfield images per well of neurospheres after 7DIV in the presence of compound 8 (2.5, 5, 10, 25, 50 μM) or DMSO (control); 10× objective. (b) Neurospheres number per image (brightfield object count) and (c) neurospheres size per image (brightfield object total area) with relative densitometries. Values are the mean ± SD of 4 independent experiments. *pp < 0.05, **pp < 0.01, ***pp < 0.001, compared to control; one-way ANOVA (Bonferroni's post hoc comparison test). | ||
The effective concentrations of compound 8 in neurospheres proliferation (2.5 and 5 μM) were then selected to assess the effect on the NPCs spontaneous differentiation. To this aim, entire 7 days formed-neurospheres were plated on a Matrigel matrix to allow adhesion, spreading, and consequently differentiation, in the presence or absence of compound 8. After 7DIV, cells were fixed and stained with Nestin (NPCs specific marker),43 doublecortin (DCX, immature neurons marker),43 DCX (immature neurons marker),44 Olig2 (oligodendrocytes progenitor oligodendrocyte precursor cells [OPCs] marker),45 and GFAP (astrocytes marker)46 for immunofluorescence analysis. As shown, compound 8 at both 2.5 and 5 μM decreased Nestin+ and significantly increased DCX+ cells compared to the DMSO control (Fig. 8a–c) without affecting Olig2 and glial fibrillary acidic protein (GFAP) expression (Fig. 8a, d and e), indicating an induction of NPCs differentiation toward a neuron-specific phenotype without affecting the OPCs and astrocytes specification.
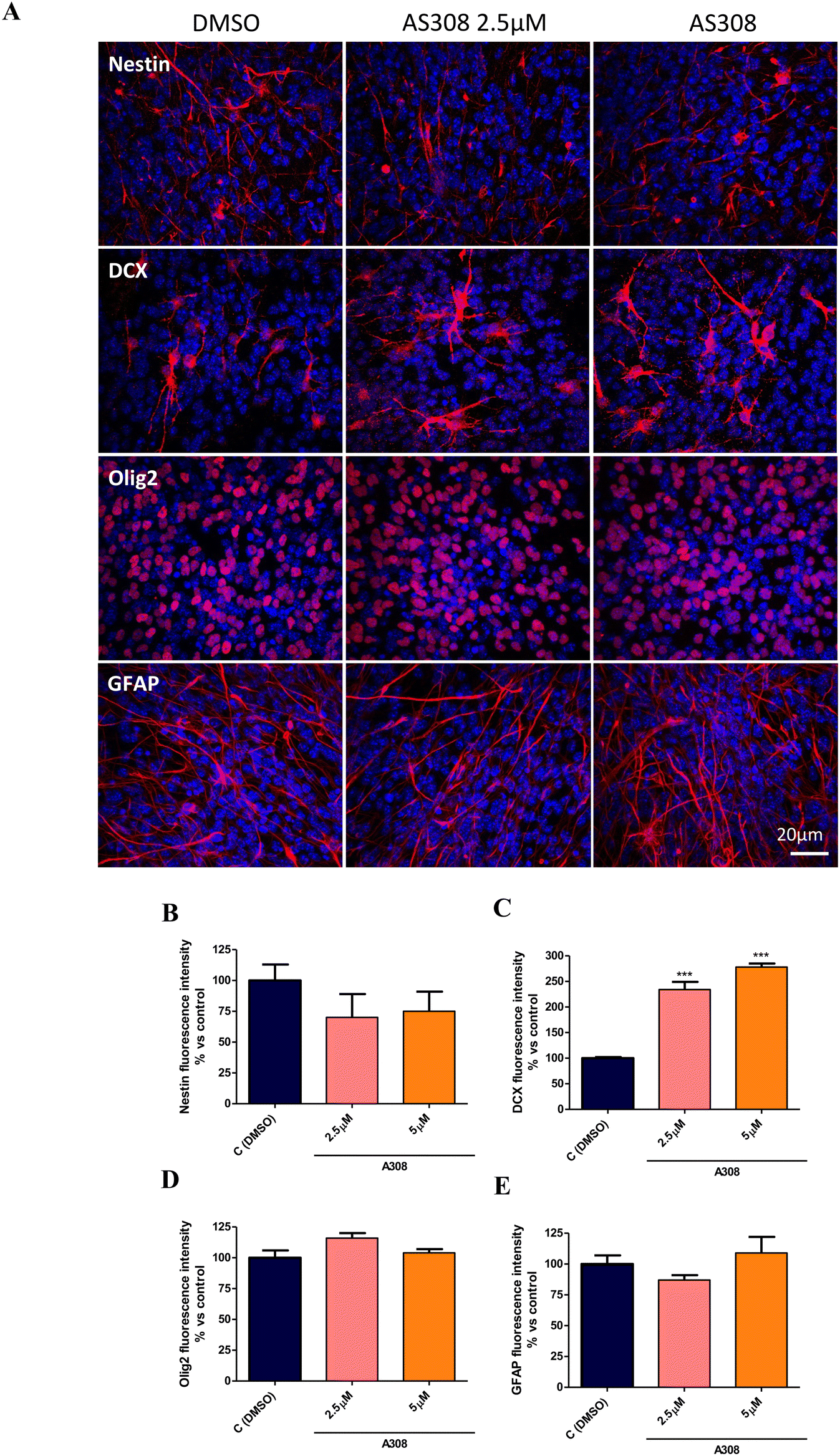 | ||
| Fig. 8 (a) Immunostaining and fluorescence intensity analysis with differentiation markers on 7DIV spontaneously differentiated neurospheres in the presence of compound 8 (2.5 μM, 5 μM) or DMSO (control). Values are the mean ± SD of 3 different fields acquired for each condition. 60× objective; 20 μm bar scale. ***p < 0.001, compared to DMSO control; one-way ANOVA (Bonferroni's post hoc comparison test). | ||
Discussion and conclusions
Since vorinostat FDA's approval in 2009, HDACIs have been considered as key players in several drug-discovery programs by both companies and academia. Indeed, due to their mechanism of actions, i.e., modulation of gene expressions, they can be successfully used to treat many and various pathological conditions. Actually, although vorinostat has been approved for the treatment of cutaneous T-cell lymphoma, it has also entered clinical trials for AD (NCT03056495) and drug-resistant epilepsy (NCT03894826).In the present investigation, we developed new hybrid HDACIs by using quinones as a CAP group and employing different available ZBGs. In particular, as a CAP group we focused our attention on naphthoquinone and its closely related analog menadione. The choice of quinones as a CAP group is based on their different pharmacological activities, including anti-AD and HDAC inhibitory properties.10,20 Indeed, the aim of this work was to depict the activity profile of new hybrids obtained by merging selected quinones moieties to different ZBGs.
As ZBGs, we employed hydroxamic acid, benzamide, and mercaptoacetic groups since they are present in some of the most interesting HDACIs developed so far. The preliminary results, based on cytotoxicity studies, demonstrated that, in addition to the nature of the CAP group, the biological activity could be modulated by varying the nature of the ZBGs. This aspect was evident when considering the opposite behavior showed by compounds 6 and 8, which shared the same CAP group, i.e., menadione. However, these two compounds differed by the ZBG: compound 6 featured a benzamide, while compound 8 carried a mercaptoacetic group. Their opposite effects in the cytotoxic assays were not surprising considering the broad spectrum of activities that both quinones and HDACIs can have. Compound 6 showed a marked cytotoxic activity in neuronal SH-SY5Y cells, being more potent than the reference compound vorinostat. Conversely, compound 8 demonstrated no neurotoxicity up to the highest concentration tested (100 μM). Both compounds were then characterized in terms of inhibitory activity toward HDAC2 and HDAC6 isoforms in vitro and in cells showing comparable inhibitory activities. Conversely, despite having the same quinone CAP group, compound 6 showed pro-oxidant activity, while compound 8 showed no effect in modulating ROS homeostasis. Considering the different behaviors, both compounds were subjected to different biological evaluations with the aim of disclosing their anticancer and neuroprotective profile, respectively. We indeed demonstrated that compound 6 exerted apoptotic cell death and that its cytotoxic activity was partially mediated by an increase in ROS production. On the other hand, compound 8 was highly effective in inducing the differentiation of NPCs into neurons, at concentrations in the low micromolar range. In particular, it was able to induce NPCs differentiation solely toward a neuronal-specific phenotype, without affecting glial cells commitment, i.e., OPCs and astrocytes. This is extremely relevant as, at early stages, NDs are characterized by the degeneration and death of neurons in specific brain areas, often related to NPCs proliferation/differentiation dysfunctions, making the replacement of degenerated neurons difficult. However, compound 8 showed potential hepatotoxicity, as its IC50 against HepG2 cells (25.52 μM) was quite close to the concentrations at which it exerts its neuroregenerative effects (2.5 and 5 μM). This is an issue to be considered in the further optimization stage.
To sum up, we demonstrated that the combination of a quinone moiety with a specific ZBG may produce HDACIs with different biological properties. This finding was fully demonstrated by depicting the activity profiles for two selected compounds, namely 6 and 8. The obtained results demonstrated that compound 6, characterized by benzamide as the ZBG, was more effective than vorinostat in inducing apoptotic death in neuroblastoma SH-SY5Y cell lines. On the other hand, the conjugation of the quinone moiety with the mercaptoacetamide group led to compound 8, which was nontoxic up to a concentration of 100 μM and was characterized by its ability to induce NPCs differentiation into neurons. The latter characteristic is extremely interesting since the discovery of small molecules able to affect the neural stem cells pool could provide a source of new neurons able to replace degenerated ones, counteracting cognitive impairment and AD progression.47
Therefore, compounds 6 and 8, albeit characterized by opposite behaviors, emerged as interesting hit compounds and are currently under in-depth biological characterization in our laboratories.
Experimental section
Chemistry
All chemicals were purchased from Aldrich Chemistry (Milan, Italy), Alfa Aesar (Milan, Italy), and FluoroChem (Cambridge, UK) and were of the highest purity. The solvents were analytical grade. Reaction progress was followed by thin-layer chromatography on precoated silica gel 60 F254 plates (Merck, Darmstadt, Germany). Chromatographic separations were performed on 0.040–0.063 mm silica gel 40 columns via the flash method (Merck). The 1H-NMR and 13C-NMR spectra were recorded on a Varian Gemini spectrometer at 400 and 100 MHz, respectively. Chemical shifts (δ) were reported as parts per million relative to tetramethylsilane, used as the internal standard; coupling constants (J) are reported in Hertz (Hz). Standard abbreviations indicating spin multiplicities are given as follows: s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Missing proton signals in some spectra refer to NH or OH, which, in some deuterated solvents, could be not detected. Low-resolution and high-resolution mass spectra were recorded on a VG707EH-F or a Xevo G2-XS QTof system, and electrospray ionization (ESI), both in the positive and negative modes. All the final compounds showed ≥95% purity by analytical HPLC. The compounds were named following IUPAC rules as applied by ChemBioDraw Ultra (version 19.0).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 9:1 ratio) to give 1–6 as red-orange oils.
:0 to 9:1 ratio) to give 1–6 as red-orange oils.
6-((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)-N-hydroxyhexanamide (1). Synthesized from compound 17 (0.170 g, 0.4 mmol). Obtained 0.093 g (75% yield). 1H-NMR (400 MHz, DMSO) δ 1.26–1.32 (m, 2H), 1.48–1.59 (m, 4H), 1.95 (t, 2H, J = 7.4 Hz), 3.13–3.18 (m, 2H), 5.66 (s, 1H), 7.58 (brs, 1H), 7.69–7.73 (m, 1H), 7.80–7.83 (m, 1H), 7.92–7.98 (m, 2H), 8.65 3 (brs, 1H), 10.34 (brs, 1H); 13C-NMR (100 MHz, DMSO) δ 24.85, 26.10, 27.03, 32.10, 41.77, 99.18, 125.30, 125.86, 130.39, 132.10, 133.21, 134.82, 148.50, 169.00, 181.18, 181.50; HRMS (ESI): C16H19N2O4 [M + H]+: calcd 303.1345, found 303.1289.
N-Hydroxy-6-((3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)hexanamide (2). Synthesized from compound 18 (0.054 g, 0.1 mmol). Obtained 0.024 g (58% yield). 1H-NMR (400 MHz, DMSO) δ 1.20–1.29 (m, 2H), 1.45–1.58 (m, 4H), 1.98 (t, 2H, J = 7.4 Hz), 2.41 (s, 3H) 3.35–3.39 (m, 2H), 7.23 (brs, 1H), 7.71–7.75 (m, 1H), 7.81–7.85 (m, 1H), 7.92–7.99 (m, 2H), 8.51 (brs, 1H), 10.38 (brs, 1H); 13C-NMR (100 MHz, DMSO) δ 12.20, 24.81, 26.50, 27.33, 31.98, 42.00, 101.12, 125.10, 125.77, 130.56, 132.19, 134.01, 134.75, 149.40, 169.32, 181.08, 181.43; HRMS (ESI): C17H21N2O4 [M + H]+: calcd 317.1501, found 317.1489.
4-(((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)-N-hydroxybenzamide (3). Synthesized from compound 19 (0.050 g, 0.1 mmol). Obtained 0.026 g (67% yield). 1H-NMR (400 MHz, DMSO) δ 4.49 (d, 2H, J = 8.0 Hz), 5.53 (s, 1H), 7.40–7.42 (m, 3H), 7.69–7.73 (m, 2H), 7.81–7.82 (m, 1H), 7.88–8.89 (m, 1H), 8.01–8.02 (m, 1H), 8.25 (brs, 1H), 9.01 (brs, 1H), 11.17 (brs, 1H); 13C-NMR (100 MHz, DMSO) δ 46.21, 104.67, 125.22, 125.89, 126.95, 128.13, 131.32, 132.54, 132.89, 133.43, 136.32, 144.01, 147.12, 163.15, 182.17, 182.42; HRMS (ESI): C18H15N2O4 [M + H]+: calcd 323.1032, found 323.1055.
N-Hydroxy-4-(((3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)benzamide (4). Synthesized from compound 20 (0.070 g, 0.1 mmol). Obtained 0.027 g (48% yield). 1H-NMR (400 MHz, DMSO) δ 2.00 (s, 3H), 4.80–4.82 (d, 2H, J = 8.0 Hz), 7.28–7.36 (m, 3H), 7.67–7.77 (m, 3H), 7.90–7.99 (m, 3H), 8.90 (brs, 1H), 11.15 (brs, 1H); 13C-NMR (100 MHz, DMSO) δ 10.38, 47.19, 111.70, 125.37, 125.65, 126.24, 127.12, 130.35, 131.36, 132.22, 132.55, 134.39, 143.93, 146.55, 164.11, 181.80, 182.05; HRMS (ESI): C19H17N2O4 [M + H]+: calcd 337.1188, found 337.1151.
N-(2-Aminophenyl)-4-(((1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)benzamide (5). Synthesized from compound 21 (0.068 g, 0.1 mmol). Obtained 0.039 g (72% yield). 1H-NMR (400 MHz, CDCl3) δ 4.48–4.50 (d, 2H, J = 8.0 Hz), 5.80 (s, 1H), 7.06–7.08 (m, 2H), 7.19–7.23 (m, 3H), 7.42–7.44 (m, 2H), 7.62–7.66 (m, 1H), 7.72–7.76 (m, 1H), 7.80–7.82 (m, 2H), 7.86–7.93 (m, 3H), 8.07–8.10 (m, 2H); 13C-NMR (100 MHz, DMSO) δ 44.12, 102.98, 116.32, 120.03, 123.22, 126.78, 126.01, 126.55, 126.89, 127.12, 128.89, 130.26, 131.73, 132.88, 133.14, 133.64, 135.64, 145.17, 148.22, 161.24, 182.22, 182.56; HRMS (ESI): C24H20N3O3 [M + H]+: calcd 398.1505, found 398.1527.
N-(2-Aminophenyl)-4-(((3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)benzamide (6). Synthesized from compound 22 (0.070 g, 0.1 mmol). Obtained 0.020 g (34% yield). 1H-NMR (400 MHz, DMSO) δ 2.02 (s, 3H), 4.84–4.86 (d, 2H, J = 8.0 Hz), 6.65–6.69 (m, 1H), 6.82–6.84 (m, 1H), 6.98–7.02 (m, 1H), 7.17–7.19 (m, 1H), 7.33–7.37 (m, 1H), 7.41–7.43 (m, 2H), 7.68–7.72 (m, 1H), 7.76–7.80 (m, 1H), 7.86–7.95 (m, 4H), 9.69 (s, 1H); 13C-NMR (100 MHz, DMSO) δ 10.41, 47.24, 111.84, 116.86, 117.38, 119.30, 123.28, 125.39, 125.66, 126.14, 126.48, 126.66, 128.07, 130.34, 132.24, 132.56, 133.07, 134.41, 144.31, 146.57, 165.18, 181.84, 182.06; HRMS (ESI): C25H22N3O3 [M + H]+: calcd 412.1661, found 412.1649.
N-(4-((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)butyl)-2-mercaptoacetamide (7). Synthesized from compound 23 (0.190 g, 0.3 mmol). Obtained 0.059 g (55% yield). 1H-NMR (400 MHz, DMSO) δ 1.42–1.49 (m, 2H), 1.55–1.62 (m, 2H), 3.06–3.11 (m, 4H), 3.19 (s, 2H), 5.69 (s, 1H), 7.52–7.58 (m, 1H), 7.70–7.74 (m, 1H), 7.80–7.84 (m, 1H), 7.93–7.99 (m, 1H); 13C-NMR (100 MHz, DMSO) δ 26.44, 29.88, 38.75, 42.55, 44.98, 101.45, 125.67, 125.88, 131.32, 132.44, 132.97, 134.77, 146.89, 167.88, 181.76, 182.21; HRMS (ESI): C16H19N2O3S [M + H]+: calcd 319.1116, found 319.1089.
2-Mercapto-N-(4-((3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)butyl)acetamide (8). Synthesized from compound 24 (0.190 g, 0.3 mmol). Obtained 0.048 g (44% yield). 1H-NMR (400 MHz, DMSO) δ 1.41–1.48 (m, 2H), 1.53–1.59 (m, 2H), 2.08 (s, 3H), 3.07–3.12 (m, 2H), 3.43 (s, 2H) 3.51–3.53 (m, 2H), 6.56–6.59 (m, 1H), 7.65–7.69 (m, 1H), 7.74–7.78 (m, 1H), 7.89–7.92 (m, 1H), 8.07–8.10 (m, 1H); 13C-NMR (100 MHz, DMSO) δ 10.61, 26.19, 28.08, 38.60, 41.95, 44.06, 110.74, 125.33, 125.57, 130.23, 132.02, 132.72, 134.37, 146.56, 167.57, 181.56, 182.18; HRMS (ESI): C17H21N2O3S [M + H]+: calcd 333.1273, found 333.1244.
N-((Tetrahydro-2H-pyran-2-yl)oxy)-4-((2,2,2-trifluoroacetamido)methyl)benzamide (10). To a solution of acid 9 (0.879 g, 3.55 mmol) in anhydrous DMF (10 ml), EDCI (1.36 g, 7.1 mmol) and HOBT (0.480 g, 3.55 mmol) were added and the mixture was stirred at 0 °C for 30 min. O-(Tetrahydro-2H-pyran-2-yl)hydroxylamine (0.500 g, 4.27 mmol) was added and the resulting mixture was stirred at room temperature for 12 h. Water was added and the mixture was extracted with ethyl acetate (3 × 20 ml). The organic layers were dried and evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (from 8
:2 to 6:4). Obtained 0.790 g (64% yield). 1H-NMR (400 MHz, DMSO) δ 1.52–1.59 (m, 2H), 1.67–1.72 (m, 2H), 2.10–2.14 (m, 1H), 3.50–3.54 (m, 1H), 3.84–3.86 (m, 1H), 4.02–4.07 (m, 1H), 4.44 (d, 2H, J = 6.4 Hz), 4.99 (m, 1H), 7.36 (d, 2H, J = 8.4 Hz), 7.75 (d, 2H, J = 8.4 Hz), 10.05 (brs, 1H), 11.61 (brs, 1H).
4-(Aminomethyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (11). To a solution of 10 (0.790 g, 2.29 mmol) in a mixture of methanol/water in a 2
:1 ratio, potassium carbonate was added (0.946 g, 6.8 mmol) and the mixture was stirred at room temperature for 12 h. The solvents were removed under vacuum to give a residue that was purified through flash chromatography using as eluent a mixture of dichloromethane/methanol/aqueous 33% ammonia (9:1:0.1) to give 11 as a colorless oil. Obtained 0.515 g (64% yield). 1H-NMR (400 MHz, DMSO) δ 1.53–1.58 (m, 3H), 1.72–1.74 (m, 3H), 3.50–3.54 (m, 1H), 3.83 (s, 2H), 4.03–4.08 (m, 1H); 4.99 (m, 1H), 7.42 (d, 2H, J = 8.4 Hz), 7.74 (d, 2H, J = 8.0 Hz); 13C-NMR (150 MHz, DMSO) δ 18.77, 25.18, 28.37, 44.49, 49.03, 61.83, 101.42, 127.58, 127.72, 131.12, 146.02.
Tert-Butyl (2-(4-((2,2,2-trifluoroacetamido)methyl)benzamido)phenyl)carbamate (12). To a solution of acid 9 (1.20 g, 4.80 mmol) in anhydrous DMF (10 ml) under a stream of nitrogen, IBCF (0.68 ml, 0.716 g, 5.26 mmol) and NMM (0.58 ml, 0.533 g, 5.26 mmol) were added and the mixture was stirred at 0 °C for 30 min. Tert-Butyl (2-aminophenyl)carbamate (1 g, 4.80 mmol) was added and the resulting mixture was stirred at room temperature for 12 h. Water was added and the mixture was extracted with dichloromethane (3 × 20 ml). The organic layers were washed with brine, then dried and evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (8
:2). Obtained 0.785 g (37% yield). 1H-NMR (400 MHz, DMSO) δ 1.13 (s, 9H), 4.85 (d, 2H, J = 6.2 Hz), 7.17–7.19 (m, 1H), 7.32–7.35 (m, 2H), 7.38 (d, 2H, J = 8.4 Hz), 7.81–7.92 (m, 3H).
Tert-Butyl (2-(4-(aminomethyl)benzamido)phenyl)carbamate (13). To a solution of 12 (0.785 g, 2.3 mmol) in 12 ml of methanol/water (2
:1 ratio), potassium carbonate was added (0.953 g, 6.9 mmol) and the resulting mixture was stirred at room temperature overnight. The solvent was removed in vacuo and the resulting residue was taken up with CH2Cl2 (20 ml) and washed with brine (20 ml × 4) to furnish 13 as a yellow oil. Obtained 0.500 g (64% yield). 1H-NMR (400 MHz, CDCl3) δ 1.53 (s, 9H), 4.31 (s, 2H), 6.85 (brs, 1H), 7.14–7.16 (m, 1H), 7.19–7.23 (m, 2H), 7.41 (d, 2H, J = 8.8 Hz), 7.77–7.80 (m, 1H), 7.99 (d, 2H, J = 8.0 Hz), 9.34 (brs, 1H).
Tert-Butyl (4-(2-(tritylthio)acetamido)butyl)carbamate (15). To a solution of 14 (0.26 g, 4.48 mmol) in 10 ml of anhydrous DMF under a stream of nitrogen at 0 °C, EDCI (0.791 g, 4.13 mmol) and HOBT (0.658 g, 4.88 mmol) were added and the mixture was stirred for 10 min. After this, tert-butyl (4-aminobutyl)carbamate (0.652 g, 3.75 mmol) was added and the mixture was stirred at room temperature for 12 h. Ethyl acetate and water were added and the organic layer was washed with brine, then dried and evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (9
:1 to 7:3). Obtained 0.536 g (28% yield). 1H-NMR (400 MHz, DMSO) δ 1.42 (s, 9H), 1.45–1.52 (m, 4H), 2.62–2.77 (m, 2H), 2.84 (s, 2H), 2.95–2.99 (m, 2H), 7.07–7.32 (m, 15H).
N-(4-Aminobutyl)-2-(tritylthio)acetamide (16). To a solution of 15 (0.536 g, 1 mmol) in methanol 10 ml, HClaq 6N (6 ml) was added and the mixture was stirred at room temperature for 12 h. Potassium carbonate was added until pH 8 and then the solvent was removed under vacuum. The residue was taken up with water and extracted with ethyl acetate (3 × 20 ml), the organic layers were washed with brine, then dried and concentrated to give 16 as a yellow oil. Obtained 0.400 g (quantitative yield). 1H-NMR (400 MHz, DMSO) δ 1.29–1.33 (m, 4H), 2.54–2.73 (m, 2H), 2.79 (s, 2H), 2.89–2.93 (m, 2H), 7.21–7.34 (m, 15H), 7.85 (brs, 1H).
6-((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide (17). A solution of 6-amino-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide (0.3 g, 1.3 mmol) and naphthoquinone (0.126 g, 0.8 mmol) in 15 ml of ethanol was stirred at room temperature for 12 h. The solvent was evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (8
:2 to 5:5). Obtained 0.170 g (55% yield). 1H-NMR (400 MHz, DMSO) δ 1.27–1.33 (m, 2H), 1.49–1.57 (m, 7H), 1.70–1.76 (m, 3H), 1.98 (t, 2H, J = 7.6 Hz), 3.11–3.19 (m, 2H), 3.50–3.54 (m, 1H), 3.99–4.03 (m, 1H), 4.97–5.01 (m, 1H), 5.69 (s, 1H), 7.68–7.76 (m, 1H), 7.83–7.87 (m, 1H), 7.80–7.96 (m, 2H).
6-((3-Methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide (18). A solution of 6-amino-N-((tetrahydro-2H-pyran-2-yl)oxy)hexanamide (0.35 g, 1.5 mmol) and menadione (0.156 g, 0.91 mmol) in 15 ml of ethanol was stirred at room temperature for 12 h. The solvent was evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (8
:2 to 5:5). Obtained 0.210 g (57% yield). 1H-NMR (400 MHz, DMSO) δ 0.87–0.93 (m, 6H) 1.53–1.58 (m, 3H), 1.72–1.74 (m, 5H), 2.20 (s, 3H), 3.43 (t, 2H, J = 8.0 Hz), 3.50–3.54 (m, 1H), 4.03–4.08 (m, 1H); 4.97–5.00 (m, 1H), 7.53–7.58 (m, 1H), 7.64–7.68 (m, 1H), 7.97–7.99 (m, 1H), 8.06–8.08 (m, 1H).
4-(((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (19). A solution of compound 11 (0.100 g, 0.4 mmol) and naphtoquinone (0.095 g, 0.6 mmol) in 15 ml of ethanol was stirred at room temperature for 12 h. The solvent was evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (8
:2). Obtained 0.05 g (31% yield). 1H-NMR (400 MHz, DMSO) δ 1.55–1.59 (m, 3H), 1.61–1.68 (m, 3H), 3.45–3.51 (m, 1H), 3.89 (s, 2H), 3.92–3.99 (m, 1H), 4.05–4.08 (m, 1H), 5.55 (s, 1H), 7.44 (d, 2H, J = 8.4 Hz), 7.67 (d, 2H, J = 8.0 Hz), 7.71–7.73 (m, 1H), 7.77–7.81 (m, 1H), 7.87–7.89 (m, 1H), 7.97–7.99 (m, 1H).
4-(((3-Methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (20). A solution of compound 11 (0.350 g, 1.4 mmol) and menadione (0.240 g, 1.4 mmol) in 15 ml of ethanol was stirred at room temperature for 12 h. The solvent was evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (8
:2 to 5:5). Obtained 0.19 g (31% yield). 1H-NMR (400 MHz, DMSO) δ 1.53–1.61 (m, 3H), 1.78–1.86 (m, 3H), 2.12 (s, 3H), 3.59–3.63 (m, 1H), 3.95–3.98 (m, 1H), 4.73 (s, 2H), 5.6 (m, 1H), 7.26–7.32 (m, 2H), 7.56–7.58 (m, 1H), 7.63–7.67 (m, 1H), 7.73–7.75 (m, 2H), 7.96–7.99 (m, 1H), 8.02–8.05 (m, 1H).
Tert-Butyl (2-(4-(((1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)benzamido)phenyl)carbamate (21). A solution of compound 13 (0.230 g, 0.67 mmol), naphthoquinone (0.071 g, 0.45 mmol), and SnCl2 (0.006 g, 0.07 mmol) in 10 ml of ethanol was stirred at room temperature for 12 h. The solvent was evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (8
:2). Obtained 0.10 g (45% yield). 1H-NMR (400 MHz, CDCl3) δ 1.50 (s, 9H), 4.47 (s, 2H), 5.77 (s, 1H), 7.18 (d, 1H, J = 6.8 Hz), 7.22–7.25 (m, 2H), 7.40–7.43 (m, 2H), 7.62–7.66 (m, 1H), 7.72–7.76 (m, 1H), 7.83 (d, 1H, J = 8.4 Hz), 7.98 (d, 2H, J = 8.0 Hz), 8.04–8.10 (m, 2H), 9.22 (brs, 1H).
Tert-Butyl (2-(4-(((3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)benzamido)phenyl)carbamate (22). A solution of compound 13 (0.100 g, 0.29 mmol), menadione (0.050 g, 0.29 mmol), and SnCl2 (0.006 g, 0.03 mmol) in 10 ml of ethanol was stirred at room temperature for 12 h. The solvent was evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (8
:2 to 7:3). Obtained 0.07 g (49% yield). 1H-NMR (400 MHz, DMSO) δ 1.56 (s, 9H), 1.98 (s, 3H), 3.85 (s, 2H), 6.66–6.69 (m, 1H), 6.79–6.82 (m, 1H), 6.99–7.03 (m, 1H), 7.20–7.23 (m, 1H), 7.30–7.36 (m, 1H), 7.65–7.69 (m, 2H), 7.76–7.84 (m, 2H), 7.90–7.99 (m, 3H), 9.75 (brs, 1H).
N-(4-((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)butyl)-2-(tritylthio)acetamide (23). A solution of compound 16 (0.200 g, 0.49 mmol) and naphtoquinone (0.078 g, 0.49 mmol) in 15 ml of ethanol was stirred at room temperature for 12 h. The solvent was evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (8
:2 to 5:5). Obtained 0.160 g (58% yield). 1H-NMR (400 MHz, DMSO) δ 1.36–1.42 (m, 2H), 1.49–1.56 (m, 2H), 2.75 (s, 2H), 2.96–3.00 (m, 2H), 3.13–3.16 (m, 2H), 5.68 (s, 1H), 7.22–7.35 (m, 15H), 7.53–7.56 (m, 1H), 7.70–7.74 (m, 1H), 7.80–7.84 (m, 2H), 7.95–7.99 (m, 2H).
N-(4-((3-Methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)butyl)-2-(tritylthio)acetamide (24). A solution of compound 16 (0.280 g, 0.69 mmol) and menadione (0.119 g, 0.69 mmol) in 15 ml of ethanol was stirred at room temperature for 12 h. The solvent was evaporated to give a residue that was purified through flash chromatography using as eluent a mixture of petroleum ether/ethyl acetate (8
:2 to 6:4). Obtained 0.070 g (18% yield). 1H-NMR (400 MHz, DMSO) δ 1.32–1.39 (m, 2H), 1.44–1.53 (m, 2H), 2.15 (s, 3H), 2.93–2.98 (m, 2H), 3.32 (s, 2H), 3.33–3.41 (m, 2H), 7.11–7.27 (m, 15H), 7.57–7.62 (m, 1H), 7.61–7.69 (m, 1H), 7.78–7.85 (m, 1H), 7.89–7.96 (m, 2H).
Biology
Cell culture
The SH-SY5Y human neuroblastoma cell line was cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 UI ml−1 penicillin, 100 μg ml−1 streptomycin, and 40 μg ml−1 gentamicin in a 5% CO2 atmosphere at 37 °C with saturating humidity. The human hepatoma cell line (HepG2) was cultured in modified Eagle's medium (MEM) supplemented with 10% fetal bovine serum, 100 UI ml−1 penicillin, and 100 μg ml−1 streptomycin in a 5% CO2 atmosphere at 37 °C with saturating humidity. The cell viability and number were measured by the trypan blue exclusion method.Cell viability
SH-SY5Y cells were seeded in 96-well plates at a density of 0.75 × 104 cells per well. After 24 h of incubation, the cells were treated with increasing concentrations (0.1–100 μM) of all the tested compounds for 24, 48, or 72 h. SH-SY5Y cell viability was assessed using a spectrophotometric assay based on 4-methylumbelliferyl heptanoate (MUH, Sigma Aldrich, Merck, St. Luis, MO, USA) reagent. MUH generates the highly fluorescent 4-methylumbelliferone due to its hydrolysis by the esterases and lipases of viable cells. Thus, fluorescence is proportional to cell viability. After treatment, the cells were washed in phosphate buffer saline (PBS) 1× and resuspended in PBS 1× containing MUH 0.01 mg mL−1. After incubation in the dark, for 30 min at 37 °C and 5% CO2, fluorescence (330 nm excitation; 450 nm emission) was detected using a Victor X3 microplate reader (Perkin Elmer, Waltham, MA, USA). Cell viability in percentage (%) was calculated normalizing the fluorescence of the treated cells to that of the untreated samples. To examine the ability of compound 8 to prevent oxidative stress-induced cell death by H2O2, the cells were pretreated with the tested compound (1, 10, or 50 μM) for 24 h, exposed for 1 h to H2O2 (300 μM), and then incubated for 22 h in drug-free complete medium. In the other experiments, the cells were pretreated with NAC (2 mM, 1 h or SFN 2.5 μM, 24 h) and then treated with compound 6 (0.1, 1, or 10 μM) or 8 (1, 10, or 50 μM) for an additional 24 h.HepG2 cells were plated at 1.5 × 104 cells per well in a 96-well plate (Sarstedt, Milan, Italy). After 24 h, the cells were exposed to different concentrations of compound 8 (ranging from 0.010 μM to 100 μM) dissolved in complete DMEM medium or vehicle. After a 24 h treatment period, the culture medium was replaced with 0.1 mL of 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) solution (Sigma-Aldrich, Milan, Italy) prepared in PBS at a concentration of 0.2 mg mL−1, and the cells were incubated for 2 h at 37 °C in 5% CO2 and a humidified atmosphere. After this time, the MTT solution was removed and the formazan crystals were dissolved in 100 μL DMSO and the plate was stirred for 30 min. Absorbance was measured at 570 nm using a multi-well plate reader (Tecan, Männedorf, CH). Data analysis was performed using Prism GraphPad software (GraphPad Software, version 6.0, San Diego, CA, USA). The results are reported as the percentage of live cells compared to controls treated with vehicle [mean ± standard deviation (SD), n = 4].
Analysis of cell cycle
SH-SY5Y cells treated with compound 6 for 72 h were collected, fixed with 70% ice-cold ethanol, and then incubated with Guava Cell Cycle Reagent (Merck Millipore, Burlington, MA, USA), containing propidium iodide. After incubation for 30 min in the dark at room temperature, the cells were analyzed by flow cytometry using a Guava EasyCyte 6-2L flow cytometer (Guava Technologies, Merck Millipore, Darmstadt, Germany).Detection of apoptosis
:100, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), washed, and incubated with Alexa Fluor 488-labeled secondary antibody (1:200, Invitrogen). After incubation, the cells were washed and analyzed via flow cytometry with recording the mean fluorescence intensity (MFI) values. PARP expression was indicated as the fold change of MFI of the treated samples compared to that of the control cells.
ROS determination
SH-SY5Y cells were seeded in 96-well plates at a density of 1 × 104 cells per well. After an incubation period of 24 h to allow adhesion, the cells were exposed to vehicle or 2 μM compounds for a further 24 h. The cells were then washed with PBS and exposed to 10 μM DCFDA (2′,7′-dichlorofluorescein diacetate, DCFH-DA) in DMEM for 30 min. After washing again with PBS, fluorescence was measured in each well using a plate reader (EnSpire, PerkinElmer) with excitation and emission wavelengths of 485 nm and 535 nm, respectively. Oxidative stress was induced in the cells by incubation with 100 μM TBH in PBS for 30 min after incubation with DCFDA. Data shown are the mean ± standard deviation (SD) of the fluorescence signal normalized to the protein content, determined by the Lowry method. All experiments were performed in triplicate.Histone post-translational modification
SH-SY5Y cells were seeded in a 10 cm dish at a density of 20 × 104 cells per cm2. After 48 h, cells were incubated for 6 h with 5 μM compound or vehicle. Then, the cells were harvested by trypsinization, washed with 10 mM sodium butyrate in PBS, and the nuclei were isolated according to Micheletti et al.49 In brief, a nuclear pellet was suspended in 0.1 mL ice-cold H2O, and concentrated H2SO4 was added to the suspension to give a final concentration of 0.4 N. After incubation at 4 °C for 1 h, the suspension was centrifuged for 5 min at 14000g, and the supernatant was taken and mixed with 1 mL of acetone. After overnight incubation, the coagulate material was collected by microcentrifugation and air-dried. This histone fraction was dissolved in 20 μL of H2O. Proteins were quantified using a protein assay kit (Bio-Rad, Hercules, CA, USA). Histones were detected resolving samples on a 15% gel in Tris/glycine buffer at 150 V for 90 min. Western blotting was performed in transfer buffer at 100 V for 1 h. The nitrocellulose membrane was incubated with mouse anti-acetylated lysines (Millipore, Billerica, MA, USA) primary antibodies for 1 h. After washing with PBS-polyoxyethylene sorbitan monolaurate 0.1% (TWEEN 20) (St. Louis, MO, USA), the membrane was incubated as before with mouse secondary HRP-conjugated antibodies (GE Healthcare, Chicago, Il, USA). After washing with PBS-TWEEN 20 0.1%, antibody binding was detected by an Amersham ECL Plus Western Blotting Detection System (GE Healthcare, Chicago, Il, USA). Densitometry analysis was performed using a Fluor-S Max MultiImager (Bio-Rad, Hercules, CA, US), and relative quantification of the histone acetylation signals was done by using a densitometric approach and normalized on the H1 signal as a control.
Neurospheres cultures
NPCs were initially obtained by the SVZ microdissection of 8-months-old C57BL/6N wild-type male mice (Mus musculus), as previously published.50 To perform the experiments, neurospheres were cultured in suspension and passed every week (5/7 days of growth). Cells were collected and pelleted for 5 min at 1000 rpm, washed in PBS (0.9% NaCl in 50 mM phosphate buffer pH 7.4), and centrifugated again for 5 min at 1000 rpm. Spheres were dissociated through 5 min incubation in Accutase (Aurogene; Roma, Italy) at 37 °C, and basal DMEM F-12 was added to stop the reaction. Following centrifugation for 5 min at 1000 rpm, single cells were plated with a cell density of 5 × 103 cells per cm2 in 35 mm dishes in neurospheres culture medium: DMEM-F12 (Gibco; ThermoFisher Scientific, Waltham, MA, USA) supplemented with 2 mM glutamine, 10 μg ml−1 insulin from bovine pancreas (Sigma-Aldrich; St. Louis, MO, USA), 20 ng ml−1 epidermal growth factor (EGF; PeproTech EC, London, UK), 20 ng ml−1 fibroblast growth factor-2 (FGF2; PeproTech), 1% N2 (ThermoFisher Scientific), 1% B27 (ThermoFisher Scientific), 10 units per ml penicillin, and 10 μg streptomycin.Neurospheres proliferation
To investigate the effect of compound 8 on neurospheres growth rate, neural precursor cells were plated 7DIV in suspension in 96-well plates (5 × 103 cells per well) in the presence of compound 8 (2.5, 5, 10, 25, 50 μM) in neurospheres culture medium; the same volume of DMSO was used as the control. One image per well was acquired every day by using the Sartorius Incucyte® Live-Cell Analysis System and evaluated through the Brightfield Spheroid Analysis Software Module. Only aggregates with areas larger than 400 μm2 were considered for statistical analysis.Neurospheres differentiation analysis
To assess the effect of compound 8 on NPCs differentiation, 30 neurospheres were plated on 13 mm glass coverslips in complete DMEM F-12 medium in the presence of different concentrations of the compound; DMSO was used as the control. To allow stem cells adhesion, the coverslips were previously incubated with Matrigel matrix (Corning; New York, USA) at 37 °C for at least 3 h. After 7DIV, the neurospheres were fixed 20 min with 4% PFA in PBS 0.1% pH 7.4 and left in PBS until use. To perform the immunostainings, the membranes were permeabilized in 0.1% Triton/PBS and aspecific sites were blocked for 1 h with 0.1% Triton/PBS and 5% normal goat serum (Sigma-Aldrich; St. Louis, MO, USA). Cells were then incubated overnight at 4 °C with primary antibodies: anti-DCX (Abcam, Cambridge, UK; Cat# ab18723, RRID: AB_732011), GFAP (Dakopatts; Cat# sc-33673, RRID: AB_627673), Olig2 (Santa Cruz Biotechnology; Cat# sc-48817, RRID: AB_2157550), and Nestin (Abcam; Cat# ab22035, RRID: AB_446723). The next day, after 3 washes in 0.1% Triton/PBS, specific secondary antibodies were added for 2 h at rt away from light: Donkey anti-Mouse IgG Alexa Fluor 555 (Abcam; Cat# ab150106, RRID: AB_2857373), Goat anti-Rabbit IgG Alexa Fluor 555 (Abcam; Cat# ab150078, RRID: AB_2722519). The primary antibodies were diluted 1:500 and Alexa secondary antibodies 1:1000 in 0.1% Triton/PBS with 2% normal goat serum. Following 3 washes in 0.1% Triton/PBS and one with PBS, the nuclei were stained with Hoechst 33258 (2 μg ml−1, Sigma-Aldrich) for 5 min. Glass coverslips in PBS were mounted by using Ultracruz Aqueous Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI, Santa Cruz Biotechnology, cat. no. sc-24941) and stored at 4 °C in the dark until acquisition. Neurospheres confocal images were obtained with Nikon EZ-C1 microscope (60× objective) and the z-stack function (1024 steps and 1 μm thickness layers; 40 total stacks). 3D image reconstruction was performed using Fiji ImageJ2 software, with the z-project plugin and selecting the sum stacks function. The fluorescence intensity index was estimated as the ratio of markers' positive cells intensity/total cells fluorescence intensity stained with DAPI.
Statistical analysis
All results were expressed as the mean ± SEM or SD of at least 3 independent experiments. Statistical analyses were performed using one- or two-way ANOVA and Dunnett, Tukey, or Bonferroni tests as the post hoc comparison tests. IC50 values (concentrations that inhibit 50% cell viability) were calculated from the dose–response curve using the nonlinear regression [log(inhibitor) versus normalized response]. The statistical software GraphPad InStat 8.0 version (GraphPad Prism, San Diego, CA, USA) was used, and p-values < 0.05 were considered statistically significant.Abbreviations
| AD | Alzheimer's disease |
| DCF | 2′,7′-Dichlorofluorescein |
| DCFDA | 2′,7′-Dichlorofluorescin diacetate |
| FDA | Food and Drug Administration |
| HDACIs | Histone deacetylase inhibitors |
| HDACs | Histone deacetylases |
| NAC | N-Acetyl-L-cysteine |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| NDs | Neurodegenerative diseases |
| NPCs | Neural precursor cells |
| NSC | Neural stem cells |
| PARP | Poly(ADP-ribose) polymerase |
| SFN | Sulforaphane |
| SVZ | Sub-ventricular zone |
| TBH | Tert-Butyl hydroperoxide |
| ZBG | Zinc binding group |
Author contributions
M. G., A. S. and E. U. performed chemical synthesis and compounds characterization; E. T., V. A. and A. D. S. performed enzymatic in vitro assay; G. G., E. P., C. B., C. Z., B. M. and C. F. designed and performed in cell assays; M. L. B. and A. M. conceived the idea, supervised the work, analyzed data and wrote the manuscript with contributions of all co-authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the University of Bologna (RFO) and University of Turin (SPY_RILO_21_01, SPY_RILO_22_01). Thanks are expressed to Luca Pincigher for technical assistance. “The Sartorius Incucyte® Live-Cell Analysis System was provided by Centro di Ricerca Biomedica Applicata (CRBA)”.References
- T. C. S. Ho, A. H. Y. Chan and A. Ganesan, J. Med. Chem., 2020, 63, 12460–12484 CrossRef CAS PubMed.
- K. J. Falkenberg and R. W. Johnstone, Nat. Rev. Drug Discovery, 2014, 13, 673–691 CrossRef CAS PubMed.
- A. D. Bondarev, M. M. Attwood, J. Jonsson, V. N. Chubarev, V. V. Tarasov and H. B. Schiöth, Br. J. Clin. Pharmacol., 2021, 87, 4577–4597 CrossRef PubMed.
- V. Kumar, S. Kundu, A. Singh and S. Singh, Curr. Neuropharmacol., 2022, 20, 158–178 CrossRef CAS PubMed.
- J. S. Guan, S. J. Haggarty, E. Giacometti, J. H. Dannenberg, N. Joseph, J. Gao, T. J. Nieland, Y. Zhou, X. Wang, R. Mazitschek, J. E. Bradner, R. A. DePinho, R. Jaenisch and L. H. Tsai, Nature, 2009, 459, 55–60 CrossRef CAS PubMed.
- P. Gediya, P. K. Parikh, V. K. Vyas and M. D. Ghate, Eur. J. Med. Chem., 2021, 216, 113332 CrossRef CAS PubMed.
- R. W. Sabnis, ACS Med. Chem. Lett., 2021, 12, 1202–1203 CrossRef CAS PubMed.
- N. Govindarajan, P. Rao, S. Burkhardt, F. Sananbenesi, O. M. Schlüter, F. Bradke, J. Lu and A. Fischer, EMBO Mol. Med., 2013, 5, 52–63 CrossRef CAS PubMed.
- Y. Li, S. Sang, W. Ren, Y. Pei, Y. Bian, Y. Chen and H. Sun, Eur. J. Med. Chem., 2021, 226, 113874 CrossRef CAS PubMed.
- L. Zhang, G. Zhang, S. Xu and Y. Song, Eur. J. Med. Chem., 2021, 223, 113632 CrossRef CAS PubMed.
- E. N. da Silva Júnior, G. A. M. Jardim, C. Jacob, U. Dhawa, L. Ackermann and S. L. de Castro, Eur. J. Med. Chem., 2019, 179, 863–915 CrossRef PubMed.
- Á. Cores, N. Carmona-Zafra, J. Clerigué, M. Villacampa and J. C. Menéndez, Antioxidants, 2023, 12, 1464 CrossRef PubMed.
- V. Capurro, P. Busquet, J. P. Lopes, R. Bertorelli, G. Tarozzo, M. L. Bolognesi, D. Piomelli, A. Reggiani and A. Cavalli, PLoS One, 2013, 8, e56870 CrossRef CAS PubMed.
- E. Nepovimova, E. Uliassi, J. Korabecny, L. E. Peña-Altamira, S. Samez, A. Pesaresi, G. E. Garcia, M. Bartolini, V. Andrisano, C. Bergamini, R. Fato, D. Lamba, M. Roberti, K. Kuca, B. Monti and M. L. Bolognesi, J. Med. Chem., 2014, 57, 8576–8589 CrossRef CAS PubMed.
- R. Perone, C. Albertini, E. Uliassi, F. Di Pietri, P. de Sena Murteira Pinheiro, S. Petralla, N. Rizzardi, R. Fato, L. Pulkrabkova, O. Soukup, A. Tramarin, M. Bartolini and M. L. Bolognesi, ChemMedChem, 2021, 16, 187–198 CrossRef CAS PubMed.
- H. Yoshimura, Y. Hirota, S. Soda, M. Okazeri, Y. Takagi, A. Takeuchi, C. Tode, M. Kamao, N. Osakabe and Y. Suhara, Bioorg. Med. Chem. Lett., 2020, 30, 127059 CrossRef CAS PubMed.
- Y. Hirota and Y. Suhara, Int. J. Mol. Sci., 2019, 20, 3006 CrossRef CAS PubMed.
- A. De Simone and A. Milelli, ChemMedChem, 2019, 14, 1067–1073 CrossRef CAS PubMed.
- V. K. Patel, E. Shirbhate, P. Tiwari, R. Kore, R. Veerasamy, A. Mishra and H. Rajak, Curr. Med. Chem., 2023, 30, 2762–2795 CrossRef CAS PubMed.
- E. S. Inks, B. J. Josey, S. R. Jesinkey and C. J. Chou, ACS Chem. Biol., 2012, 7, 331–339 CrossRef CAS PubMed.
- K. D. Badave, A. A. Khan and S. Y. Rane, Anti-Cancer Agents Med. Chem., 2016, 16, 1017–1030 CrossRef CAS PubMed.
- K. Kimura, Y. Hirota, S. Kuwahara, A. Takeuchi, C. Tode, A. Wada, N. Osakabe and Y. Suhara, J. Med. Chem., 2017, 60, 2591–2596 CrossRef CAS PubMed.
- L. Zhang, J. Zhang, Q. Jiang and W. Song, J. Enzyme Inhib. Med. Chem., 2018, 33, 714–721 CrossRef CAS PubMed.
- A. P. Kozikowski, Y. Chen, A. Gaysin, B. Chen, M. A. D'Annibale, C. M. Suto and B. C. Langley, J. Med. Chem., 2007, 50, 3054–3061 CrossRef CAS PubMed.
- M. T. Tavares, A. P. Kozikowski and S. Shen, Eur. J. Med. Chem., 2021, 209, 112887 CrossRef CAS PubMed.
- A. De Simone, V. La Pietra, N. Betari, N. Petragnani, M. Conte, S. Daniele, D. Pietrobono, C. Martini, S. Petralla, R. Casadei, L. Davani, F. Frabetti, P. Russomanno, E. Novellino, S. Montanari, V. Tumiatti, P. Ballerini, F. Sarno, A. Nebbioso, L. Altucci, B. Monti, V. Andrisano and A. Milelli, ACS Med. Chem. Lett., 2019, 10, 469–474 CrossRef CAS PubMed.
- S. Gao, J. Zang, Q. Gao, X. Liang, Q. Ding, X. Li, W. Xu, C. J. Chou and Y. Zhang, Bioorg. Med. Chem., 2017, 25, 2981–2994 CrossRef CAS PubMed.
- S. Mehndiratta, M. H. Lin, Y. W. Wu, C. H. Chen, T. Y. Wu, K. H. Chuang, M. W. Chao, Y. Y. Chen, S. L. Pan, M. C. Chen and J. P. Liou, Eur. J. Med. Chem., 2020, 185, 111725 CrossRef CAS PubMed.
- T. M. Vishwanatha, E. Bergamaschi and A. Dömling, Org. Lett., 2017, 19, 3195–3198 CrossRef CAS PubMed.
- A. Milelli, C. Marchetti, M. L. Greco, F. Moraca, G. Costa, E. Turrini, E. Catanzaro, N. Betari, C. Calcabrini, C. Sissi, S. Alcaro, C. Fimognari, V. Tumiatti and A. Minarini, Eur. J. Med. Chem., 2017, 128, 107–122 CrossRef CAS PubMed.
- G. Li, Y. Tian and W. G. Zhu, Front. Cell Dev. Biol., 2020, 8, 576946 CrossRef PubMed.
- P. LoPresti, Cells, 2020, 10, 12 CrossRef PubMed.
- G. I. Aldana-Masangkay and K. M. Sakamoto, J. Biomed. Biotechnol., 2011, 2011, 875824 Search PubMed.
- M. L. Bolognesi, R. Banzi, M. Bartolini, A. Cavalli, A. Tarozzi, V. Andrisano, A. Minarini, M. Rosini, V. Tumiatti, C. Bergamini, R. Fato, G. Lenaz, P. Hrelia, A. Cattaneo, M. Recanatini and C. Melchiorre, J. Med. Chem., 2007, 50, 4882–4897 CrossRef CAS PubMed.
- K. S. SantaCruz, E. Yazlovitskaya, J. Collins, J. Johnson and C. DeCarli, Neurobiol. Aging, 2004, 25, 63–69 CrossRef CAS PubMed.
- J. M. Han, Y. J. Lee, S. Y. Lee, E. M. Kim, Y. Moon, H. W. Kim and O. Hwang, J. Pharmacol. Exp. Ther., 2007, 321, 249–256 CrossRef CAS PubMed.
- P. Pozarowski and Z. Darzynkiewicz, Methods Mol. Biol., 2004, 281, 301–311 CAS.
- M. Kajstura, H. D. Halicka, J. Pryjma and Z. Darzynkiewicz, Cytometry, Part A, 2007, 71, 125–131 CrossRef PubMed.
- L. Galluzzi, I. Vitale, S. A. Aaronson, J. M. Abrams, D. Adam, P. Agostinis, E. S. Alnemri, L. Altucci, I. Amelio, D. W. Andrews, M. Annicchiarico-Petruzzelli, A. V. Antonov, E. Arama, E. H. Baehrecke, N. A. Barlev, N. G. Bazan, F. Bernassola, M. J. M. Bertrand, K. Bianchi, M. V. Blagosklonny, K. Blomgren, C. Borner, P. Boya, C. Brenner, M. Campanella, E. Candi, D. Carmona-Gutierrez, F. Cecconi, F. K. Chan, N. S. Chandel, E. H. Cheng, J. E. Chipuk, J. A. Cidlowski, A. Ciechanover, G. M. Cohen, M. Conrad, J. R. Cubillos-Ruiz, P. E. Czabotar, V. D'Angiolella, T. M. Dawson, V. L. Dawson, V. De Laurenzi, R. De Maria, K. M. Debatin, R. J. DeBerardinis, M. Deshmukh, N. Di Daniele, F. Di Virgilio, V. M. Dixit, S. J. Dixon, C. S. Duckett, B. D. Dynlacht, W. S. El-Deiry, J. W. Elrod, G. M. Fimia, S. Fulda, A. J. García-Sáez, A. D. Garg, C. Garrido, E. Gavathiotis, P. Golstein, E. Gottlieb, D. R. Green, L. A. Greene, H. Gronemeyer, A. Gross, G. Hajnoczky, J. M. Hardwick, I. S. Harris, M. O. Hengartner, C. Hetz, H. Ichijo, M. Jäättelä, B. Joseph, P. J. Jost, P. P. Juin, W. J. Kaiser, M. Karin, T. Kaufmann, O. Kepp, A. Kimchi, R. N. Kitsis, D. J. Klionsky, R. A. Knight, S. Kumar, S. W. Lee, J. J. Lemasters, B. Levine, A. Linkermann, S. A. Lipton, R. A. Lockshin, C. López-Otín, S. W. Lowe, T. Luedde, E. Lugli, M. MacFarlane, F. Madeo, M. Malewicz, W. Malorni, G. Manic, J. C. Marine, S. J. Martin, J. C. Martinou, J. P. Medema, P. Mehlen, P. Meier, S. Melino, E. A. Miao, J. D. Molkentin, U. M. Moll, C. Muñoz-Pinedo, S. Nagata, G. Nuñez, A. Oberst, M. Oren, M. Overholtzer, M. Pagano, T. Panaretakis, M. Pasparakis, J. M. Penninger, D. M. Pereira, S. Pervaiz, M. E. Peter, M. Piacentini, P. Pinton, J. H. M. Prehn, H. Puthalakath, G. A. Rabinovich, M. Rehm, R. Rizzuto, C. M. P. Rodrigues, D. C. Rubinsztein, T. Rudel, K. M. Ryan, E. Sayan, L. Scorrano, F. Shao, Y. Shi, J. Silke, H. U. Simon, A. Sistigu, B. R. Stockwell, A. Strasser, G. Szabadkai, S. W. G. Tait, D. Tang, N. Tavernarakis, A. Thorburn, Y. Tsujimoto, B. Turk, T. Vanden Berghe, P. Vandenabeele, M. G. Vander Heiden, A. Villunger, H. W. Virgin, K. H. Vousden, D. Vucic, E. F. Wagner, H. Walczak, D. Wallach, Y. Wang, J. A. Wells, W. Wood, J. Yuan, Z. Zakeri, B. Zhivotovsky, L. Zitvogel, G. Melino and G. Kroemer, Cell Death Differ., 2018, 25, 486–541 CrossRef PubMed.
- C. Zhou, W. Ni, T. Zhu, S. Dong, P. Sun and F. Hua, Front. Neurosci., 2022, 16, 884667 CrossRef PubMed.
- E. Uliassi, A. Gandini, R. C. Perone and M. L. Bolognesi, Future Med. Chem., 2017, 9, 995–1013 CrossRef CAS PubMed.
- J. Yang, Y. Tang, H. Liu, F. Guo, J. Ni and W. Le, BMC Biol., 2014, 12, 95 CrossRef PubMed.
- D. Park, A. P. Xiang, F. F. Mao, L. Zhang, C. G. Di, X. M. Liu, Y. Shao, B. F. Ma, J. H. Lee, K. S. Ha, N. Walton and B. T. Lahn, Stem Cells, 2010, 28, 2162–2171 CrossRef CAS PubMed.
- F. Francis, A. Koulakoff, D. Boucher, P. Chafey, B. Schaar, M. C. Vinet, G. Friocourt, N. McDonnell, O. Reiner, A. Kahn, S. K. McConnell, Y. Berwald-Netter, P. Denoulet and J. Chelly, Neuron, 1999, 23, 247–256 CrossRef CAS PubMed.
- D. H. Meijer, M. F. Kane, S. Mehta, H. Liu, E. Harrington, C. M. Taylor, C. D. Stiles and D. H. Rowitch, Nat. Rev. Neurosci., 2012, 13, 819–831 CrossRef CAS PubMed.
- C. Nolte, M. Matyash, T. Pivneva, C. G. Schipke, C. Ohlemeyer, U. K. Hanisch, F. Kirchhoff and H. Kettenmann, Glia, 2001, 33, 72–86 CrossRef CAS.
- S. G. Davies, P. D. Kennewell, A. J. Russell, P. T. Seden, R. Westwood and G. M. Wynne, J. Med. Chem., 2015, 58, 2863–2894 CrossRef CAS PubMed.
- F. Halley, J. Reinshagen, B. Ellinger, M. Wolf, A. L. Niles, N. J. Evans, T. A. Kirkland, J. M. Wagner, M. Jung, P. Gribbon and S. Gul, J. Biomol. Screening, 2011, 16, 1227–1235 CrossRef CAS PubMed.
- G. Micheletti, C. Boga, G. Drius, S. Bordoni and N. Calonghi, Molecules, 2024, 29, 238 CrossRef CAS PubMed.
- S. Petralla, L. E. Peña-Altamira, E. Poeta, F. Massenzio, M. Virgili, S. N. Barile, L. Sbano, E. Profilo, M. Corricelli, A. Danese, C. Giorgi, R. Ostan, M. Capri, P. Pinton, F. Palmieri, F. M. Lasorsa and B. Monti, Int. J. Mol. Sci., 2019, 20, 4486 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00175c |
| ‡ M. G., G. G. and E. P. contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |