
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and cytotoxic activity of madecassic acid–silybin conjugate compounds in liver cancer cells†
Chien Van
Tran
ab,
Thao Thi Phuong
Tran
ab,
Anh The
Nguyen
a,
Loc Van
Tran
a,
Ninh Thi
Pham
a,
Luu Thi
Nguyen
a,
Dung Thi
Nguyen
a,
Michelle D.
Garrett
c,
Nga Thi
Nguyen
d,
Thao Thi
Do
 d,
Christopher J.
Serpell
*e and
Sung Van
Tran
*a
d,
Christopher J.
Serpell
*e and
Sung Van
Tran
*a
aInstitute of Chemistry, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet Road, Cau Giay, Hanoi, Vietnam. E-mail: tranvansungvhh@gmail.com
bGraduate University of Science and Technology, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet Road, Cau Giay, Hanoi, Vietnam
cSchool of Biosciences, University of Kent, Stacey Building, Canterbury, Kent CT2 7NJ, UK
dInstitute of Biotechnology, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet Road, Cau Giay, Hanoi, Vietnam
eDepartment of Pharmaceutical and Biological Chemistry, School of Pharmacy, University College London, 29-39 Brunswick Square, London, WC1N 1AX, UK. E-mail: chris.serpell@ucl.ac.uk
First published on 2nd August 2024
Abstract
A series of 14 conjugates of 2α,3β,23-triacetyl-madecassic acid and silybin were designed and synthesized. The madecassic acid unit was linked to silybin either directly at position C-7 or C-3; or through an amino acid linker (glycine, β-alanine, or 11-aminoundecanoic acid) at position C-3. The conjugates were tested in vitro for their cytotoxic effect on HepG2 cells using the MTT assay. The results confirmed that the conjugated compounds demonstrated a stronger cytotoxic effect compared to the parent compounds. Of these compounds, the most promising conjugate, compound 8, was evaluated for cytotoxic activity in the additional Hep3B, Huh7, and Huh7R human hepatocellular carcinoma cell lines and also for cell cycle changes and induction of apoptosis in HepG2 cells. This compound caused a rapid and significant induction of caspase 3 activity and induced cell cycle arrest in the S phase – effects distinct from the activity of madecassic acid. This is the first study on the synthesis and cytotoxicity of madecassic acid–silybin conjugates, and of their testing against liver cancer cell lines and provides evidence for a distinct biological profile versus madecassic acid alone.
1. Introduction
Liver cancer was one of the top five causes of cancer death in 185 countries in 2020, with an estimated 906![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases and 830000 deaths globally. The number of liver cancer deaths is predicted to reach approximately 1.3 million people by 2040, representing a 56.4% increase upon 2020.1 Liver cancer is caused by viral infections including hepatitis B virus (HBV), hepatitis C virus (HCV), and hepatitis D virus (HDV), and is becoming one of the most challenging and urgent problems particularly in Viet Nam today. An estimated 25000 new cases of liver cancer were reported in 2020, making Viet Nam the fifth highest country in the world for incidence of this disease.1–3 Since 2018, liver cancer has risen above lung cancer to become the leading cause of cancer death in Viet Nam.2,3
000 new cases and 830000 deaths globally. The number of liver cancer deaths is predicted to reach approximately 1.3 million people by 2040, representing a 56.4% increase upon 2020.1 Liver cancer is caused by viral infections including hepatitis B virus (HBV), hepatitis C virus (HCV), and hepatitis D virus (HDV), and is becoming one of the most challenging and urgent problems particularly in Viet Nam today. An estimated 25000 new cases of liver cancer were reported in 2020, making Viet Nam the fifth highest country in the world for incidence of this disease.1–3 Since 2018, liver cancer has risen above lung cancer to become the leading cause of cancer death in Viet Nam.2,3
The herb Centella asiatica (L.) Urb. (Apiaceae) is extensively grown throughout Viet Nam. This medicinal plant has a wide range of biological properties, including antioxidant, anticancer, and wound healing activity.4–6 Madecassic acid is one of the three main pentacyclic triterpenoid acids produced by the plant, together with asiatic acid and terminolic acid.7 Madecassic acid has been reported to have valuable pharmacological activities, such as wound healing,8 antioxidant,9 anti-inflammatory,10 and antidiabetic effects.11 Recently, madecassic acid has been shown to induce apoptosis in the CT26 mouse colon cancer cell line and its anticancer activity can be enhanced through chemical modification. Indeed, new madecassic acid derivatives have been reported to have significantly increased cytotoxicity towards cancer cell lines, including hepatocellular carcinoma models.12,13
Silybins (also known as silibinins) A and B are flavonolignan framework compounds isolated from the seeds of Silybum marianum (L.) Gaertn. (Asteraceae), and are well-known for their hepatoprotective activity,14 strong antioxidant potential and anti-lipid peroxidation action.15,16 Recent studies have found that silybins possess cytotoxic activity in a mouse model of prostate cancer,17 in the human non-small-cell lung carcinoma H1299, H322 and H460 cell lines,18 and the SW480 human colorectal cancer cell line.19 In addition, they have proven cytotoxic potential in bladder, skin, prostate, colon and lung cancers20,21 in which they regulate the cancer cell cycle, apoptosis, and autophagy, as well as inhibiting tumor-inducing factors, potentially through the HGF/c-Met, Wnt/β-catenin, and PI3K/Akt/mTOR signaling pathways.22,23 Structural modifications of silybin could provide further interesting information on the mechanism of action of this molecule and hence further potential applications.
Synthesis of hybrid molecules composed of multiple bioactive compounds into a single molecule could be an effective strategy to produce novel and better active substances for the treatment of cancer.24 Bio-conjugated molecules could have superior efficacy compared to a single drug due to the minimization of unwanted side-effects and synergism of two or more active moieties in one molecule, which enables both to be delivered at the same time and to the same place.25–27 Although this will necessarily result in larger molecules, which are classically expected to have poorer pharmacokinetics, there is an increasing awareness that molecules beyond the usual rules for drug design are worth exploring.28
Inspired by the potential of hybrid natural product compounds, we here report the design and synthesis of a new set of madecassic acid-silybin conjugates which were anticipated to be able to both kill liver cancer cells and provide protection to healthy hepatocytes, potentially reducing side effects. These hybrids are linked through an ester bond between a hydroxy group at C-7 or C-3 of silybin and the carboxyl group of madecassic acid, or via an amino acid as a spacer (Fig. 1). The synthesized compounds were evaluated for their antiproliferative potency in vitro against the HepG2 human liver cancer cell line. The most potent compound, 8, was further evaluated for cytotoxic activity in Hep3B, Huh7, and Huh7R liver cancer cell lines together with cell cycle analysis.
 | ||
| Fig. 1 A) Chemical structures of silybin (1, A/B designation depends on variable stereochemistry) and madecassic acid (2); B) rational design of MA–SB conjugates. | ||
2. Results and discussion
2.1. Chemistry
Silybin (1) is a flavonoligan containing five functional hydroxyl groups, therefore regioselective esterification between the carboxylic acid group of madecassic acid (MA, 2) and a single alcohol on the silybin moiety may be somewhat problematic (Fig. 1). Initially, our aim was to conjugate madecassic acid with the hydroxy group at C-23 of silybin through an ester. Accordingly, silybin was converted into 3,5,7,20-O-tetraacetyl silybin as described by Armando,29 followed by reaction with the carboxyl of 2α,3β,23-triacetyl-madecassic acid (3) or its glycine derivatives (Scheme 1). Various esterification conditions were applied, such as Staab's reagent (1,1′-carbonyldiimidazole/4-dimethylaminopyridine), Steglich esterification (DCC/DMAP/THF),30 or the Mitsunobu reaction (DEAD/Ph3P),31,32 but these attempts failed to obtain the desired ester. | ||
| Scheme 1 Selective esterification of 2α,3β,23-triacetyl-madecassic acid with silybin at position 23. | ||
Due to the failure of 3,5,7,20-O-tetraacetylsilybin to participate in selective esterification, free silybin, without any protection, was used as the starting material. As reported by Li33 and Decker34 regioselective 23-OH esterification of the silybin moiety is possible under Mitsunobu conditions. However, in our case the esterification did not occur as planned with MA or its derivatives. Further alternative routes were therefore pursued.
As illustrated in Scheme 2, treatment of acid 3 with either thionyl chloride or oxalyl chloride to produce an intermediate MA–chloride acid followed by exposure to silybin (1) in the presence of triethylamine (TEA) led to the formation of esters 4 and 5, in which NMR analysis of compound 5 showed a dehydration of the hydroxyl group at C-3 on silybin by the appearance of a signal of a singlet proton resonance for H-3 at δH 6.56, and carbon resonance at C-3 at δC 104.38 (3-CH).31 The selective attachment to the 7-OH group of silybin was confirmed based on NMR analysis, in agreement with the report by Křen and Decker.34,35 On the other hand, under Steglich conditions using DCC/DMAP, esterification of acid 3 with silybin 1 provided ester 6 with a selective attachment to the 3-OH group of silybin. The NMR analysis of ester 6 confirmed an elimination of protons H-2 and H-3 on the silybin moiety, which took part in an oxidation under mild basic conditions to create 2,3-dehydrosilybin derivatives. The conclusion was based on the disappearance of proton signals in the NMR at δH 5.0 (d, J = 11.5 Hz, H-2) and 4.54 (dd, J = 3.5; 11.5 Hz, H-3), together with carbon signals at δC 84.7 (C-2) and at δC 73.7 (C-3),35 accompanied by the appearance of two new carbon signals upfield at δC 156.7 (C-2) and 132.5 (C-3).36,37
 | ||
| Scheme 2 Synthesis of madecassic acid conjugated with silybin. Reagents and conditions: a) Ac2O, pyridine, rt, 12 h 84%; b) i) oxalyl chloride, DCM, 18 h, rt, ii) TEA, DCM, silybin, rt, 4 (40%), 5 (20%); c) Silybin, Dicyclohexylcarbodiimide (DCC), DMAP, 0 °C, rt, 34 h, 42%; d) i) oxalyl chloride, DCM, rt, 14 h, ii) amino acid (glycine; or β-alanine; or 11-aminoundecanoic acid (11-AUDA)), TEA, DCM, rt, 18 h; iii) Silybin, DCC, DMAP, THF, 0 °C, rt, 18 h. | ||
The conjugation of 2α,3β,23-triacetyl-madecassic acid with silybin via an amino acid as a spacer (glycine, β-alanine, or 11-aminoundecanoic acid (11-AUDA)), was carried out in a two-step reaction sequence. Treatment of triacetyl madecassic acid (3) with oxalyl chloride to furnish the intermediate acyl chloride acid was followed by reaction with one of the three amino acids and then subsequent coupling with silybin 1 under Steglich conditions, using DCC/DMAP in dry tetrahydrofuran (THF) at room temperature. The corresponding conjugates 8–10 were obtained in overall yields of 20–26%. Analysis of NMR spectroscopy of these conjugates 8–10 confirmed regioselective esterification to the 3-OH group of the silybin moiety. A comparison of the 1H NMR spectra of the silybin (1) with its conjugated ester revealed that the chemical shift of H-3 proton multiplet in the silybin moiety had shifted downfield from a range of 4.65–4.64 ppm38 up to 5.82–5.84 ppm. To date, reported silybin ester derivatives at position C-3 are rare: Antoszczak et al. reported the synthesis of conjugates of silybin with the antibiotics salinomucin and monensin. The authors obtained the conjugates through an ester linkage in the 23-OH group with yields of 43% and 35%, respectively, and no conjugates at the 3-OH group of silybin.39 However, our results are in good agreement with the results by Křen30 who reported a selective attachment to the 3-OH group of the silybin moiety under Steglich esterification conditions to form 3-O-galloylsilybin. Acetylation of compounds 6, 8–10 with acetic anhydride in pyridine at room temperature provided the corresponding acetylated products (7, 11–13). Their structures were confirmed by the analysis of the NMR and MS spectroscopic data.
In order to evaluate the role of the 6-OH group in madecassic acid in cytotoxicity to HepG2 cells, a series of conjugates 15–18 were synthesized (Scheme 3). Triacetyl madecassic acid (3) was first treated with thionyl chloride in the presence of pyridine to give the dehydrated compound 14 (ref. 12) in 47% yield after a silica gel column chromatography. In a similar two-step sequence reaction, conversion of acid 14 into the intermediate chloride acid followed by coupling with amino acids (glycine; or β-alanine), and subsequently esterified with silybin (1) under Steglich conditions provided esters 15–16 (21–23%). Treatment of these esters with acetic anhydride provided products 17 and 18 in 68% and 58% yields, respectively, after silica gel column chromatography (Scheme 3).
 | ||
| Scheme 3 Synthesis of dehydrated madecassic acid conjugated with silybin. Reagents and conditions: a) pyridine, SOCl2, pyridine, 2 h, rt, 47%. b) i) Oxalyl chloride, DCM, rt, 14 h, ii) amino acid (glycine; or β-alanine), TEA, DCM, rt, 18 h; iii) silybin, dicyclohexylcarbodiimide (DCC), DMAP, THF, 0 °C, rt, 18 h; c) Ac2O, pyridine, rt, 12 h. | ||
2.2. Biological activities
| Compound | Conjugation positiona | Linkage | Modificationb | GI50 (μM) |
|---|---|---|---|---|
| a Alcohol on the silybin unit through which conjugation is achieved. b +Ac = acetylation, −H2O = dehydration on 2,3 positions on silybin (S) or 5,6 positions on madecassic acid (M). Ellipticine was used as a positive control and all GI50 values were generated using a 96 hour MTT assay (n ≥ 3 assay repeats). | ||||
| Silybin (1) | — | — | — | 285.40 ± 1.62 |
| Madecassic acid (2) | — | — | — | 161.0 ± 1.28 |
| 3 | — | — | +Ac | 44.12 ± 2.08 |
| 4 | 7 | Direct ester | — | >500 |
| 5 | 7 | Direct ester | −H2O(S) | >500 |
| 6 | 3 | Direct ester | — | 217.37 ± 10.76 |
| 7 | 3 | Direct ester | +Ac | 471.13 ± 17.82 |
| 8 | 3 | Glycine | — | 32.5 ± 3.59 |
| 9 | 3 | β-Alanine | — | 141.37 ± 39.22 |
| 10 | 3 | 11-AUDA | — | 383.12 ± 40.69 |
| 11 | 3 | Glycine | +Ac | 122.48 ± 4.66 |
| 12 | 3 | β-Alanine | +Ac | 359.62 ± 12.35 |
| 13 | 3 | 11-AUDA | +Ac | 198.93 ± 48.55 |
| 14 | — | — | −H2O(M) | 38.47 ± 5.78 |
| 15 | 3 | Glycine | −H2O(M) | 173.50 ± 96.94 |
| 16 | 3 | β-Alanine | −H2O(M) | 343.62 ± 19.12 |
| 17 | 3 | Glycine | −H2O(M) +Ac | 243.41 ± 21.96 |
| 18 | 3 | β-Alanine | −H2O(M) +Ac | 352.08 ± 33.11 |
| Ellipticine | — | — | — | 0.42 ± 0.01 |
Patterns in the data were identified: acetylation of the hydroxyl groups at positions 2α,3β,23 of both madecassic acid and its dehydrated product (3 and 14) significantly increased antiproliferative activity on the HepG2 cell line. The conjugates between silybin or tetra-acetyl silybin with triacetyl madecassic acid (3) using glycine as a spacer also enhanced this activity (compounds 8 and 11), and the compound with silybin displaying free hydroxyls (8) had greater activity versus the equivalent acetylated version (11).
| Compound | GI50 (μM) | ||
|---|---|---|---|
| Hep3B | Huh7 | Huh7R | |
| Madecassic acid (2) | 177.80 ± 7.36 | 197.90 ± 9.80 | 215.43 ± 4.14 |
| 8 | 9.61 ± 0.81 | 13.78 ± 0.45 | 14.23 ± 1.74 |
| Compound | Live | Cells (%) | ||
|---|---|---|---|---|
| Early apoptosis | Late apoptosis | Necrosis | ||
| Untreated | 88.62 | 2.98 | 3.06 | 5.34 |
| Camptothecin | 79.08 | 14.78 | 2.30 | 3.85 |
| 2 (1 × IC50) | 80.48 | 9.00 | 4.29 | 6.23 |
| 2 (3 × IC50) | 72.03 | 13.37 | 6.16 | 8.44 |
| 8 (1 × IC50) | 71.11 | 10.65 | 4.92 | 13.32 |
| 8 (3 × IC50) | 84.00 | 1.28 | 0.91 | 13.81 |
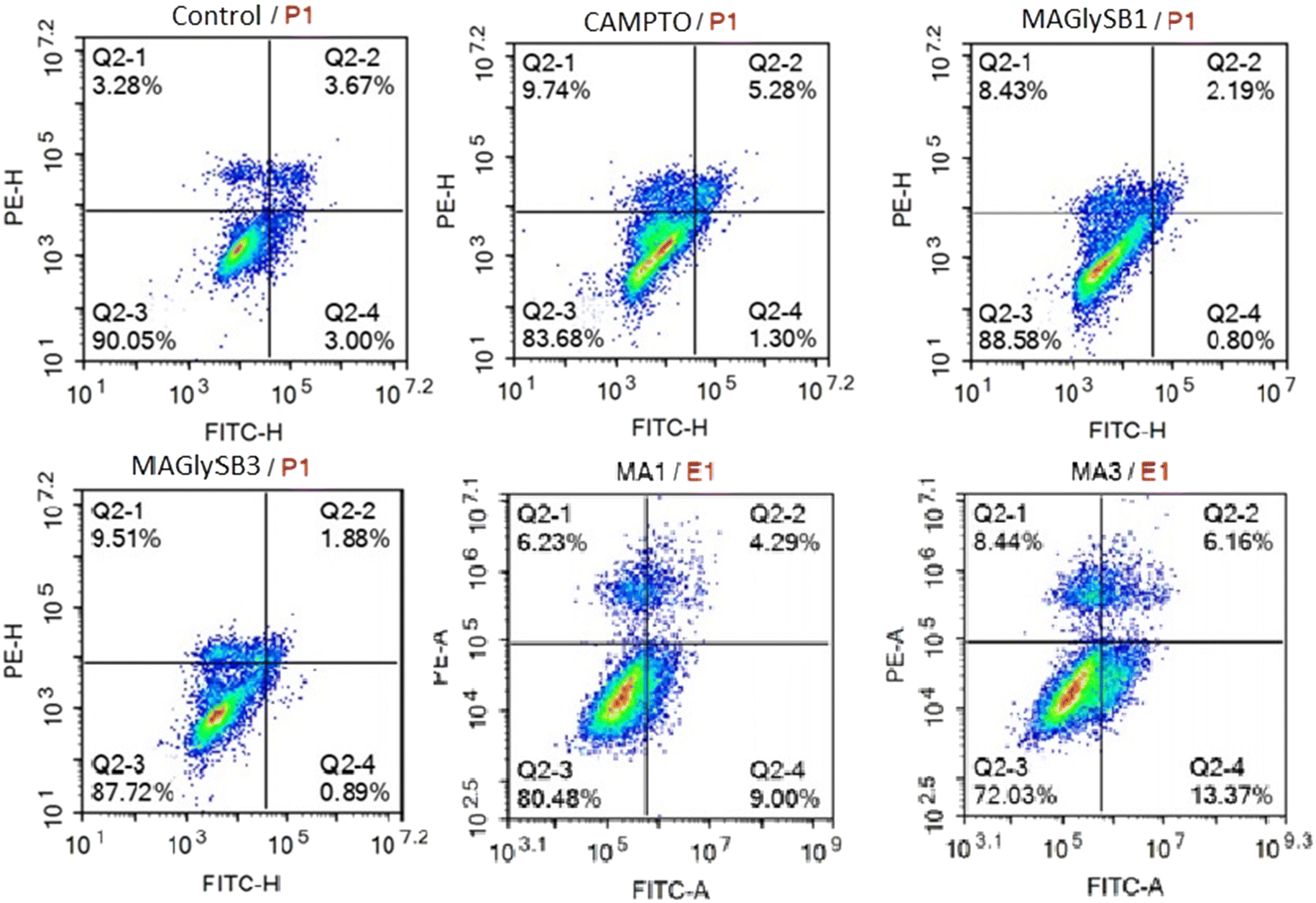 | ||
| Fig. 2 The effect of MA (2) and compound 8 on early and late apoptosis and necrosis. HepG2 cells were incubated with compounds at 1 × GI50 and 3 × GI50 for each compound for 24 hours followed by annexin V-FITC and PI staining and analysis by flow cytometry. Camptothecin treated cells were used a positive control with untreated cells as the negative control. | ||
Because cysteinyl aspartate specific proteinase-3 (caspase-3) plays an important role in the apoptotic pathway, the capacity of madecassic acid (2) and compound 8 to induce caspase 3 activity was measured at 1, 6, and 24 hours post treatment. The results in Table 4 show that compound 2 caused a significant increase in caspase 3 activity in HepG2 cells at 6 h post treatment, and at 3 × GI50, with a fold change vs. the control of 1.71. Interestingly, compound 8 significantly enhanced caspase 3 activity at 1, 6, and 24 h post treatment with 3 × GI50, and at 24 hours post treatment with both 0.3 × and 1 × GI50 concentrations.
| Compound | Conc. | Fold change of activities | |||||
|---|---|---|---|---|---|---|---|
| 1 h | 6 h | 24 h | |||||
| Mean | SE | Mean | SE | Mean | SE | ||
| a P < 0.05. b P < 0.01. c P < 0.001. | |||||||
| 2 | 0.3 × GI50 | 0.74 | 0.02 | 0.81 | 0.02 | 0.99 | 0.10 |
| 1.0 × GI50 | 0.94 | 0.10 | 1.16 | 0.05 | 0.68 | 0.04 | |
| 3.0 × GI50 | 0.97 | 0.02 | 1.71 | 0.09 | 0.54 | 0.01 | |
| 8 | 0.3 × GI50 | 0.59 | 0.03 | 0.76 | 0.04 | 1.62 | 0.01 |
| 1.0 × GI50 | 0.76 | 0.09 | 1.09 | 0.06 | 1.86 | 0.05 | |
| 3.0 × GI50 | 1.69 | 0.14 | 1.27 | 0.13 | 1.37 | 0.01 | |
| Camptothecin | 0.5 μM | 1.94 | 0.05 | 2.21 | 0.05 | 2.19 | 0.02 |
| Untreated cells | 1.00 | 0.05 | 1.00 | 0.03 | 1.00 | 0.01 | |
| Compound | % G0/G1 | % S | % G2/M |
|---|---|---|---|
| Control (DMSO 0.5%) | 24.63 | 46.83 | 27.69 |
| 2 (1 × IC50) | 19.29 | 58.52 | 21.51 |
| 2 (3 × IC50) | 54.22 | 24.25 | 20.71 |
| 8 (1 × IC50) | 26.72 | 61.09 | 9.78 |
| 8 (3 × IC50) | 27.07 | 60.41 | 11.80 |
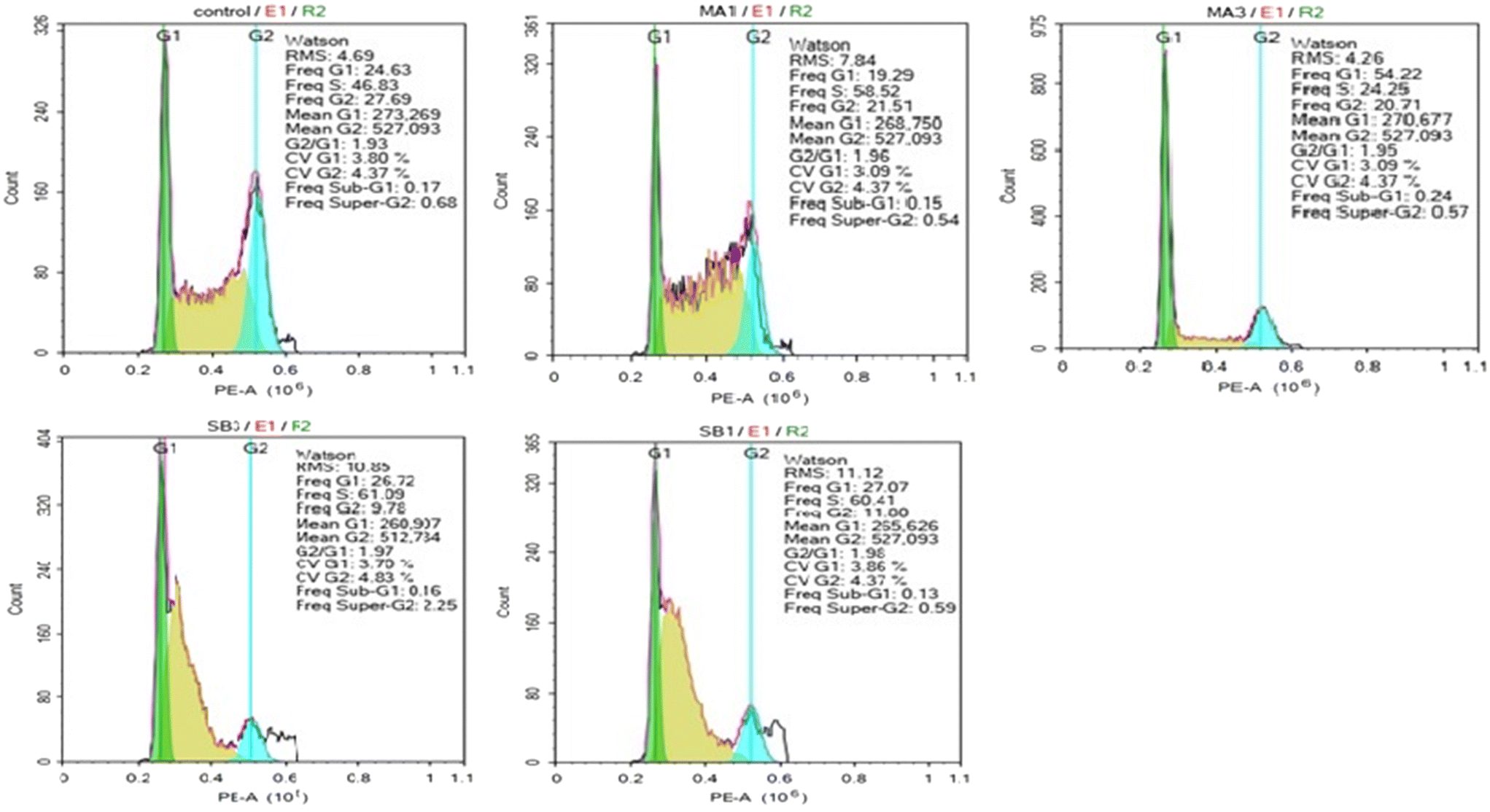 | ||
| Fig. 3 The cell cycle effects of madecassic acid (2) and compound 8. HepG2 cells were incubated with compounds at concentrations of 1 and 3 × GI50 for 24 hours followed by PI staining and analysis by flow cytometry. Untreated cells served as a negative control. | ||
3. Conclusion
Fourteen novel conjugates of madecassic acid and silybin have been synthesized through esterification or using amino acids as linkers. The formation of esters at silybin positions 3 or 7 could be controlled depending on the reaction conditions. The new conjugates were evaluated for their cytotoxicity on the HepG2 hepatocellular carcinoma line. Nine compounds showed higher cytotoxic activity than silybin and five compounds exhibited greater cytotoxic activity than madecassic acid itself. Alongside its hepatoprotective activity, silybin has been reported to have cytotoxic activity on HepG2 cells,41 a finding supported by our work. Among them, three compounds were very strongly active, and of these, compound 8 was taken forward for further evaluation. Compound 8 demonstrated strong cytotoxic activity against an additional three liver cancer cell lines (HepG3, Huh7 and Huh7R) along with S phase cell cycle arrest and rapid induction of caspase 3 activity in HepG2 cells, which is distinct from madecassic acid (2). These results provide the first evidence of a distinct biological profile of a madecassic acid–silybin conjugate versus madecassic acid (2) alone, and could enable the development of treatments that combine cytotoxic effects on liver cancer cells with broader hepatoprotection. The activity of the synthesized hybrid compounds on healthy hepatocytes and other cell types is currently unknown and should be further investigated ideally through in vivo testing.4. Experimental section
4.1. Chemistry
Chemicals were purchased from Merck and Sigma-Aldrich and used without further purification. Madecassic acid was isolated and purified from Centella asiatica cultivated from Hue province by our laboratory. Solvents were redistilled before being used. NMR spectra (1H, 13C, HMBC, HSQC) were recorded on a Bruker AVANCE 500 MHz with tetramethylsilane (TMS) as the internal standard for 1H and solvent signals for 13C NMR. Chemical shifts are reported in parts per million (δ ppm). J coupling constants in Hz. Proton spectra multiplicities are abbreviated as: MA madecassic acid, SB silybin, s singlet, brs broad single, d doublet, t triplet, q quartet, quin quintet, m multiplet, dd doublet of doublets, dt doublet of triplets, td triplet of doublets. Electrospray ionization (ESI) mass spectra were measured on a 1100 Agilent LC/MS ion trap. Reactions were monitored by thin-layer chromatography using silica gel G60 F254 (Merck). Silica gel 300–400 mesh (Merk) was used for column chromatography.Hepatocellular carcinoma cell lines (HepG2, Hep3B, Huh and Huh7R) were the kindly gift of Prof. Chi-Ying Huang, Taiwan. DMEM (Gibco, USA) consisted of 10% fetal bovine serum (FBS, Gibco), 2 mM L-glutamine (Gibco, USA) and 1% anti–anti was used for growth cancer cells. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), ribonuclease (RNase), and propidium iodide (PI) were original from Sigma Chemical (St. Louis, MO, USA). The eBioessence™ Annexin V Apoptosis Detection Kit was purchased from Invitrogen (Carlsbad, CA, USA). Caspase-3 assay kit (colorimetric) sourced from Abcam (Cambridge, UK).
4.1.1.1. 2α,3β,23-Triacetyloxy-6β-hydroxyurs-12-en-28-oic acid (3). A stirred solution of madecassica cid 2 (1 g, 2.0 mmol) in pyridine (10 mL) was treated with acetic anhydride (600 mg, 6.0 mmol) and stirred for 12 h at room temperature. Reaction mixture was concentrated under reduced pressure and ethyl acetate (200 mL) was added. The solution was washed with 1 N HCl, brine solution, and dried over Na2SO4. Removal of solvent and purification over SiO2 column (n-hexane/EtOAc; 2/1) gave 3 (1.60 g, 84%) as a white powder. Rf = 0.25 (n-hexane/EtOAc; 2/1). ESI-MS m/z: 629.4 [M − H]−. 1H NMR (500 MHz, CDCl3) δ 5.28 (t, 1H, J = 3.5 Hz, H-12), 5.23 (td, 1H, J = 11.0, 4.5 Hz, H-2), 5.01 (d, 1H, J = 10.5 Hz, H-3), 4.34 (brs, 1H, H-6), 3.94 (d, 1H, J = 12.0 Hz, H-23), 2.12 (d, 1H, J = 11.0 Hz, H-18), 2.09 (overlap, 1H, H-11), 2.09 (s, 3H, Ac), 2.06–2.04 (m, 1H, H-1), 2.05 (s, 3H, Ac), 1.98 (s, 3H, Ac), 1.85–1.83 (m, 1H, H-11), 1.75–1.65 (m, 5H), 1.55–1.51 (m, 1H, H-9), 1.49 (s, 3H, H-25), 1.36–1.29 (m, 3H), 1.28 (s, 3H, H-24), 1.14–1.11 (m, 1H, H-1), 1.05 (s, 3H, H-27), 1.05 (s, 3H, H-26), 0.94 (d, 3H, J = 6.5 Hz, H-30), 0.86 (d, 3H, J = 6.5 Hz, H-29); 13C NMR (125 MHz, CDCl3) δ 183.6 (C-28), 170.8 (C23-OAc), 170.4 (2× OAc), 137.2 (C-13), 125.6 (C-12), 74.9 (C-3), 69.9 (C-2), 67.9 (C-6), 65.4 (C-23), 52.4 (C-18), 48.2 (C-17), 47.9 (C-9), 47.8, 45.8 (C-1), 42.4 (C-4; C-14), 40.7, 39.1, 38.8, 38.6, 37.3, 36.6, 30.6, 27.9, 24.1, 23.5, 23.3 (C-27), 21.1 (C-30), 21.0 (AcO), 21.0 (AcO), 20.8 (AcO), 18.6 (C-25), 18.4 (C-26), 16.9 (C-29), 15.3 (C-24).
4.1.1.2. Synthesis of esters 4 and 5. A stirred solution of acid 3 (50 mg, 0.079 mmol) in dry DCM (5 mL) was added oxalyl chloride (0.2 mL). After being stirred at room temperature for 18 h, the reaction mixture was concentrated under reduced pressure to dryness, which was then treated with silybin (57 mg, 0.119 mmol) and triethyl amine (0.03 mL, 0.237 mmol) in dry DCM (5 mL). Reaction mixture was stirred further overnight and purified over a silica gel column chromatography (DCM/MeOH, 25/1) to obtain esters 4 (30 mg, 40%) and 5 (16 mg, 20%).
7-O-(2α,3β,23-Triacetyloxy-6β-hydroxyurs-12-en-28-oyl)silybin (4). White amorphous powder, Rf = 0.15 (DCM/MeOH; 25/1), ESI-MS m/z: 1095.4 [M + H]+. 1H NMR (500 MHz, CDCl3) δ 11.05 (s, 1H, OH), 7.18 (dd, 1H, J = 1.0 Hz, 9.0 Hz), 7.05–7.01 (m, 2H), 6.95 (s, 1H), 6.93 (overlap, 2H), 6.26–6.23 (m, 1H), 6.21–6.18 (m, 1H), 5.76 (brs, 1H, OH), 5.58–5.55 (m, 1H), 5.48–5.45 (m, 1H), 5.29 (td, 1H, J = 11.0, 4.0 Hz, H-2-SB), 5.16–5.12 (m, 1H), 5.04 (d, 1H, J = 12.0 Hz, H-3-SB), 4.96 (d, 1H, J = 8.5 Hz), 4.58 (d, 1H, J = 5.0 Hz), 4.26 (s, 1H, H-6-MA), 4.07–4.05 (m, 1H), 3.92 (overlap, 1H, H-23-SB), 3.92 (s, 3H, OMe-SB), 3.79 (d, 1H, J = 12.6 Hz), 3.69 (d, 1H, J = 12.0 Hz, H-23-SB), 3.62 (d, 1H, J = 12.6 Hz), 3.55 (dd, 1H, J = 3.5 Hz, 12.5 Hz), 3.47 (brs, 1H, OH), 2.21 (d, 1H, J = 12.0 Hz), 2.11 (overlap, 2H, H-18, H-11-MA), 2.07 (s, 3H, Ac), 2.04 (s, 3H, Ac), 1.98 (s, 3H, Ac), 1.27 (s, 3H, H-25-MA), 1.21 (overlap, 3H, H-24-MA), 1.12 (s, 3H, H-27-MA), 1.04 (s, 3H, H-26-MA), 0.90 (d, 3H, J = 6.0 Hz, H-30-MA), 0.80 (d, 3H, J = 6.0 Hz, H-29-MA). 13C NMR (125 MHz, CDCl3) δ 197.3 (CO-4-SB), 174.7 (C-28), 171.0 (OAc), 170.8/170.5/170.4 (OAc), 170.3 (OAc), 162.8, 159.9, 146.9, 146.9, 144.3, 143.9, 135.1, 129.1, 127.8, 127.1, 123.8, 122.6, 121.2, 120.8, 117.4/117.4, 116.5/116.2, 114.7, 109.6, 103.9, 103.6, 102.2, 83.1 (C-2-SB), 78.3 (C-10-SB), 76.4 (C-11-SB), 74.8 (C-3-MA), 72.6 (C-3-SB), 69.1 (C-2-MA), 68.2 (C-6-MA), 64.9 (C-23-MA), 61.6 (C-23-SB), 56.1 (OMe-SB), 54.0, 53.4 (C-18-MA), 49.6 (C-17-MA), 49.2 (C-9-MA), 47.6, 46.5/46.3 45.0 (C-1-MA), 44.2, 43.7, 43.3, 42.3 (C-4; C-14), 41.4, 40.9, 38.8, 38.3, 36.6/36.1, 32.2, 30.5, 29.7, 24.0, 23.4 (C-27), 21.1 (C-30), 21.0 (Ac), 20.8 (Ac), 19.1 (C-25-MA), 18.6 (C-26-MA), 17.1/17.0 (C-29-MA), 14.1 (C-24-MA).
7-O-(2α,3β,23-Triacetyloxy-6β-hydroxyurs-12-en-28-oyl)hydnocarpin D (5). White amorphous powder, Rf = 0.20 (DCM/MeOH; 25/1), ESI-MS m/z: 1077.2 [M + H]+. 1H NMR (500 MHz, CDCl3) δ 11.75 (s, 1H, OH), 7.86–7.82 (2H, overlap), 7.09 (s, 1H), 6.98–6.96 (m, 3H), 6.72 (s, 2H), 6.45 (s, 1H), 5.92 (brs, 1H), 5.59–5.55 (m, 1H), 5.47 (d, 1H, J = 12.5 Hz, H-12-MA), 5.30–5.26 (m, 1H), 5.14 (dd, 1H, J = 11.0, 4.5 Hz, H-2-SB), 4.99 (brs, 1H), 4.36 (s, 1H, H-6-MA), 4.26 (d, 1H, J = 12.0 Hz), 4.13 (brs, 1H), 3.93 (s, 3H, overlap, OMe-SB), 3.86–3.81 (m, 1H), 3.70 (d, 1H, J = 12.0 Hz, H-23-SB), 3.60 (d, 1H, J = 10.5 Hz), 2.38 (d, 1H, J = 11.0 Hz), 2.19–2.13 (m, 2H, overlap, H-18, H-11-MA), 2.08 (s, 3H, Ac), 2.04 (s, 3H, Ac), 1.98 (s, 3H, Ac), 1.47 (s, 3H, H-25-MA), 1.26 (3H, overlap, H-24-MA), 1.25 (s, 3H, H-27-MA), 1.11 (3H, H-26-MA), 0.99 (3H, H-30-MA), 0.90 (d, 3H, J = 6.5 Hz, H-29-MA). 13C NMR (125 MHz, CDCl3) δ 175.6 (C-28), 172.9 (CO), 170.9 (OAc), 170.4 (2× OAc), 160.5, 156.6, 155.6, 147.1, 146.6, 145.5, 143.9, 136.9, 127.6, 126.2, 123.9, 121.9, 120.9, 117.3, 117.0, 114.8, 109.6, 104.3, 101.1, 78.7, 76.4, 74.9, 69.9, 67.8, 65.3 (C-23-SB), 61.6, 56.1 (MeO-SB), 52.9, 48.9 (C-17-MA), 48.2 (C-9), 47.8, 45.9 (C-1-MA), 42.8, 42.4 (C-4; C-14-MA), 41.1, 39.2, 38.9, 38.8, 37.3, 36.5, 30.6, 28.0, 24.2, 23.4 (C-27-MA), 21.1 (C-30-MA), 20.9 (Ac), 20.8 (2× Ac), 19.1 (C-25-MA), 18.6 (C-26-MA), 16.9 (C-29-MA), 15.3 (C-24-MA).
4.1.1.3. 3-O-(2α,3β,23-Triacetyloxy-6β-hydroxyurs-12-en-28-oyl)-2,3-dehydrosilybin (6). Acid 3 (50 mg, 0.079 mmol) was treated with DCC (49 mg, 0.237 mmol), silybin (76 mg, 0.158 mmol) and DMAP (10 mg, 0.079 mmol) in dry THF (5 mL) at room temperature for 34 h. Afterward, an amount of oxalic acid (15 mg, 0.12 mmol) was added to destroy the excess of DCC. The reaction mixture was cooled to 0 °C for 30 min, then precipitate was removed by filtration. The filtrate was concentrated under reduced pressure to dryness which was subjected over a silica gel column chromatography eluting with DCM/acetone (5/1) to obtain ester 6 (36 mg, 42%). White amorphous powder, Rf = 0.2 (DCM/acetone; 5/1), ESI-MS m/z: 1094.1 [M + H]+. 1H NMR (600 MHz, CDCl3) δ 11.75 (brs, 1H, OH), 7.87 (s, 1H), 7.81 (d, 1H, J = 8.4 Hz), 7.10 (d, 1H, J = 8.4 Hz), 6.98 (s, 2H), 6.96 (1H, overlap), 6.75 (1H, s), 6.48 (s, 1H), 5.40 (brs, 1H, H-12-MA), 5.23 (t, 1H, J = 4.5 Hz, H-2-MA), 5.02 (1H, overlap, H-3-MA), 5.06–5.00 (m, 2H), 4.36 (brs, 1H, H-6-MA), 4.16–4.12 (m, 1H), 3.95 (1H, overlap, H-23-SB), 3.94 (s, 3H, OMe-SB), 3.85 (d, 1H, J = 12.6 Hz), 3.72 (d, 1H, J = 12.0 Hz, H-23-SB), 3.61 (d, 1H, J = 12.6 Hz), 2.12 (1H, overlap, H-18-MA), 2.13–2.09 (m, 1H, H-11-MA), 2.07 (s, 3H, Ac), 2.06–2.02 (m, 1H, H-1-MA), 2.05 (s, 3H, Ac), 1.99 (s, 3H, Ac), 1.94–1.86 (m, 10H), 1.47/1.46 (s, 3H, H-25-MA), 1.28 (3H, overlap, H-24-MA), 1.15/1.14 (s, 3H, H-27-MA), 1.11 (s, 3H, H-26), 0.99 (d, 3H, J = 6.0 Hz, H-30-MA), 0.90 (d, 3H, J = 6.0 Hz, H-29-MA). 13C NMR (150 MHz, CDCl3) δ 175.6 (C-28), 170.9 (OAc), 170.5 (2× OAc), 160.5, 156.7 (C-2-SB), 147.1/146.1, 146.6/145.5, 143.9, 136.9/136.4, 132.5 (C-3-SB), 130.9, 128.8, 127.6, 126.2, 123.9, 121.9, 120.8, 117.3, 117.0, 114.8, 109.6, 106.9, 104.3, 101.1, 78.8 (C-10-SB), 76.4 (C-11-SB), 74.9 (C-3-MA), 69.9 (C-2-MA), 67.9 (C-6-MA), 65.3 (C-23-SB), 56.1 (OMe-SB), 52.9 (C-18-MA), 48.9 (C-17-MA), 48.1 (C-9-MA), 47.8, 45.9 (C-1-MA), 42.9, 42.4 (C-4; C-14-MA), 41.1, 39.2, 38.9, 38.8, 37.3, 36.5, 30.6, 28.0, 24.2, 23.8, 23.4 (C-27-MA), 21.1 (C-30), 20.9 (Ac), 20.8 (Ac), 19.1 (C-25-MA), 18.6 (C-26-MA), 16.9 (C-29-MA), 15.3 (C-24-MA).
4.1.1.4. 3-O-(2α,3β,23-Triacetyloxy-6β-hydroxyurs-12-en-28-oyl)-5,7,20,23-O-tetraacetyl-2,3-dehydrosilybin (7). A stirred solution of 6 (15 mg, 0.023 mmol) in pyridine (1 mL) was treated with acetic anhydride (0.1 mL) and stirred further 12 h at room temperature. The reaction mixture was concentrated under reduced pressure and ethyl acetate (20 mL) was added. The organic phase was washed brine solution (2 × 10 mL) and dried over Na2SO4. Removal of solvent and purification over SiO2 column (n-hexane–EtOAc; 2/1) gave 7 (14 g, 78%) as a grey foam. Rf = 0.15 (n-hexane/EtOAc; 3/1), ESI-MS m/z: 1262.1 [M + H]+. 1H NMR (600 MHz, CDCl3) δ 7.50 (s, 1H), 7.47–7.44 (m, 1H), 7.25 (1H, overlap), 7.12–7.09 (m, 2H), 6.99 (2H, overlap), 6.77 (s, 1H), 6.48 (s, 1H), 5.41 (brs, 1H, H-12-MA), 5.22 (td, 1H, J = 11.4, 5.4 Hz, H-2-MA), 5.01 (2H, overlap, H-3-MA, H-11-SB), 4.41 (d, 1H, J = 12.4 Hz), 4.37–4.32 (m, 1H, H-6-MA), 4.35–4.30 (m, 1H), 4.05–4.02 (m, 1H, H-23-SB), 3.95 (1H, overlap, H-23-SB), 3.93 (s, 3H, OMe-SB), 3.72 (d, 1H, J = 12.0 Hz, H-23-MA), 2.42 (s, 3H, OAc), 2.35 (1H, overlap), 2.33 (s, 3H, AcO-SB), 2.33 (s, 3H, AcO-SB) 2.32 (s, 3H, AcO-SB), 2.12 (2H, overlap, H-18, H-11-MA), 2.07 (s, 3H, Ac), 2.05 (s, 3H, Ac), 2.03 (s, 3H, Ac), 1.98 (s, 3H, AcO-SB), 1.99 (s, 3H, Ac), 1.94–1.82 (m, 10H), 1.47/1.46 (s, 3H, H-25-MA), 1.28 (3H, overlap, H-24-MA), 1.11 (s, 3H, H-27-MA), 1.10 (s, 3H, H-26-MA), 0.99 (d, 3H, J = 6.0 Hz, H-30), 0.88 (d, 3H, J = 6.0 Hz, H-29-MA). 13C NMR (150 MHz, CDCl3) δ 174.5 (C-28), 170.9 (OAc), 170.5 (2× OAc), 170.4, 170.2, 169.4, 168.8, 167.9, 156.7 (C-2-SB), 154.8, 154.7, 151.8, 150.3, 145.7, 143.5, 143.5, 140.7, 136.9, 134.1, 133.5 (C-3-SB), 130.9, 128.8, 126.2, 123.4, 119.8, 117.6, 117.4, 114.5, 113.6, 111.1, 108.7, 76.4 (C-10-SB), 75.9 (C-11-SB), 74.9 (C-3-MA), 69.9 (C-2-MA), 68.2, 67.8 (C-6-MA), 65.3 (C-23-SB), 56.1 (OMe-SB), 52.9 (C-18-MA), 49.3 (C-17-MA), 48.0 (C-9), 47.8, 45.9 (C-1), 42.7, 42.4 (C-4; C-14-MA), 41.0, 39.1, 38.9, 38.8, 37.3, 36.4, 30.6, 28.9, 24.2, 23.8, 23.4 (C-27), 21.1 (2× OAc), 20.9 (AcO), 20.8 (AcO), 20.7 (2× OAc), 18.9 (C-25-MA), 18.6 (C-26-MA), 16.9 (C-29-MA), 15.3 (C-24-MA).
4.1.2.1. Silybin-3-yl(N-(2α,3β,23-triacetyloxy-6β-hydroxyurs-12-en-28-oyl))aminoacetate (8). Yield 25% (46 mg), white amorphous powder, Rf = 0.18 (DCM/acetone (4/1), ESI-MS m/z: 1175.4 [M + Na]+. 1H NMR (600 MHz, CD3OD) δ 7.11–7.08 (m, 1H), 7.04 (d, 1H, J = 4.8 Hz), 7.05–7.02 (m, 2H), 6.95–6.92 (m, 1H), 6.88–6.85 (m, 1H), 5.98 (s, 1H, H-6-SB), 5.96 (s, 1H, H-8-SB), 5.84 (dd, 1H, J = 11.4, 14.4. Hz, H-3-SB), 5.39 (2H, overlap, H-12, H-2-SB), 5.28–5.23 (m, 1H, H-2-MA), 4.97 (2H, overlap, H-10-SB, H-3-MA), 4.29 (brs, 1H, H-6-MA), 4.13–4.09 (m, 1H, H-11-SB), 3.97 and 3.92 (each 1H, overlap, CH2-Gly), 3.92 and 3.77 (d, each 1H, J = 12.6 Hz, H-23-MA), 3.90 (s, 3H, OMe-SB), 3.73 (d, 1H, J = 10.8 Hz, H-23-SB), 3.51 (dd, 1H, J = 12.6, 4.8 Hz, H-23-SB), 2.13–2.09 (m, 2H, overlap, H-11, H-18), 2.05 (s, 3H, Ac), 2.04 (s, 3H, Ac), 1.99 (s, 3H, Ac), 1.89–1.85 (m, 1H), 1.76–1.71 (m, 1H, H-11), 1.68–1.63 (m, 4H), 1.48 (s, 3H, H-25), 1.36–1.29 (m, 3H), 1.30 (s, 3H, H-24), 1.12 (s, 3H, H-27), 1.05 (s, 3H, H-26), 0.98 (d, 3H, J = 7.2 Hz, H-30), 0.91 (d, 3H, J = 7.2 Hz, H-29). 13C NMR (150 MHz, CD3OD) δ 192.2 (C4-SB), 180.3 (C-28-MA), 172.6 (C23-OAc), 172.3 (OAc), 172.2 (OAc), 170.5, 165.5, 164.0, 149.3, 148.4, 145.9, 145.4, 139.1 (C-13-MA), 129.9, 129.4, 127.3 (C-12-MA), 121.8, 118.3, 117.4, 116.3, 112.2, 102.1, 97.8 (C-6-SB), 96.7 (C-8-SB), 82.0 (C-2-SB), 80.1 (C-11-SB), 77.8 (C-3-SB), 76.7 (C-10-SB), 74.5 (C-3-MA), 71.2 (C-2-MA), 67.9 (C-6-MA), 66.3 (C-23-MA), 62.1 (C-23-SB), 56.6 (OMe-SB), 54.3 (C-18), 48.1 (C-17), 47.0 (C-9), 43.7, 41.9 (CH2-Gly), 41.2, 40.8, 40.2, 40.1, 38.6, 38.4, 31.9, 28.9, 25.3, 24.4, 24.1, 21.5 (C-30), 20.9 (Ac), 20.7 (2× Ac), 19.1 (C-25), 18.7 (C-26), 17.4 (C-29), 15.7 (C-24).
4.1.2.2. Silybin-3-yl(N-(2α,3β,23-triacetyloxy-6β-hydroxyurs-12-en-28-oyl))-3-aminopropanoate (9). Yield 20% (36 mg), white amorphous powder, Rf = 0.15 (DCM/acetone (3/1), ESI-MS m/z: 1167.2 [M + H]+. 1H NMR (600 MHz, CD3OD) δ 7.16–7.08 (m, 2H), 7.05–7.01 (m, 2H), 6.93–6.90 (m, 1H), 6.87–6.84 (m, 1H), 5.97/5.95 (s, 1H, H-6-SB), 5.92/5.90 (s, 1H, H-8-SB), 5.86 (dd, 1H, J = 12.6 Hz, H-3-SB), 5.36 (2H, overlap, H-12-MA, H-2-SB), 5.27–5.21 (m, 1H, H-2-MA), 4.97 (d, 1H, J = 10.2 Hz, H-10-SB), 4.94–4.90 (m, 1H, H-3), 4.29 (brs, 1H, H-6-MA), 4.15–4.11 (m, 1H, H-11-SB), 3.90 and 3.75 (each 1H, overlap, H-23), 3.89/3.87 (s, 3H, OMe-SB), 3.76–3.72 (m, 1H, H-23-SB), 3.58–3.53 (m, 1H, β-CH2-Ala), 3.51 (1H, overlap, H-23-SB), 3.47–3.42 (m, 1H, β-CH2-Ala), 2.78 (t, 1H, J = 6.0 Hz, α-CH2-Ala), 2.49–2.44 (m, 1H, α-CH2-Ala), 2.15–2.10 (m, 2H, overlap, H-11, H-18), 2.09 (s, 3H, Ac), 2.07 (s, 3H, Ac), 1.99 (s, 3H, Ac), 1.85–1.80 (m, 2H), 1.72–1.65 (m, 4H), 1.44 (s, 3H, H-25), 1.29 (s, 3H, H-24), 1.11 (s, 3H, H-27), 1.07 (s, 3H, H-26), 0.97 (d, 3H, J = 7.2 Hz, H-30), 0.91 (d, 3H, J = 7.2 Hz, H-29).
4.1.2.3. Silybin-3-yl(N-(2α,3β,23-triacetyloxy-6β-hydroxyurs-12-en-28-oyl))-11-amino-undecanoate (10). Yield 26% (50 mg), white amorphous powder, Rf = 0.2 (DCM/acetone (4/1), ESI-MS m/z: 1279.4 [M + H]+. 1H NMR (500 MHz, CDCl3) δ 7.07–7.03 (m, 3H), 6.95–6.89 (m, 2H), 5.98 (brs, 1H, H-6-SB), 5.95 (brs, 1H, H-8-SB), 5.72 (dd, 1H, J = 12.5 Hz, H-3-SB), 5.32 (brs, 1H, H-12-MA), 5.21 (td, 1H, J = 11.0, 4.5 Hz, H-2-MA), 5.05–5.01 (m, 1H, overlap, H-3-MA), 4.98 (1H, overlap, H-2-SB), 4.50–4.46 (m, 1H), 4.26 (brs, 1H, H-6-MA), 4.18 (t, 1H, J = 7.0 Hz, H-10), 4.01 (brs, 1H, H-23-MA), 3.88/3.81 (s, 3H, OMe-SB), 3.73 (1H, overlap, H-23-MA), 3.72 (d, 1H, J = 12.0 Hz, H-23-SB), 3.58–3.54 (m, 1H, H-23-SB), 3.23–3.19 (m, 2H, overlap, CH2-11-AUDA), 2.56 (t, 2H, J = 7.0 Hz, CH2-11-AUDA), 2.03 (s, 3H, AcO), 2.00 (s, 3H, AcO), 1.95 (s, 3H, OAc), 1.96–1.92 (m, 1H), 1.85–1.81 (m, 2H), 1.74–1.57 (m, 7H), 1.48–1.37 (m, 10H), 1.26–1.12 (m, 26H, CH2-11-AUDA), 1.03 (s, 3H, H-27), 1.02 (s, 3H, H-26), 0.92 (s, 3H, H-30), 0.85 (d, 3H, J = 6.5 Hz, H-29). 13C NMR (125 MHz, CDCl3) δ 185.6 (C-4-SB), 176.7 (C-28), 172.2 (COO-3-SB), 171.4, 170.9 (OAc), 139.3, 135.2, 134.3 (C-13-MA), 132.5, 131.1, 128.9, 125.6 (C-12-MA), 120.8, 119.9, 116.5, 110.0, 97.2 (C-6-SB), 96.1 (C-8-SB), 83.0 (C-2-SB), 78.5 (C-10-MA), 75.1 (C-11-SB), 73.9 (C-3-MA), 73.2 (C-3-SB), 70.2 (C-2-MA), 67.2 (C-6-MA), 65.3 (C-23-MA), 61.4 (C-23-SB), 56.1 (OMe-SB), 54.2, 53.5 (C-18), 48.1 (C-17), 47.8 (C-9), 45.9 (C-1), 43.0, 42.5 (C-4), 39.9, 39.8 (CH2-11-AUDA), 38.8, 37.4, 34.1 (CH2-11-AUDA), 29.8–29.1 (8× CH2), 28.8, 27.1, 24.8, 23.8, 23.3 (C-27), 21.2 (C-30), 21.1 (Ac), 20.9 (Ac), 20.8 (Ac), 18.6 (C-25), 18.2 (C-26), 17.2 (C-29), 15.3 (C-24).
4.1.3.1. (5,7,20,23-O-Tetraacetyl)silybin-3-yl(N-(2α,3β,23-triacetyloxy-6β-hydroxyurs-12-en-28-oyl))aminoacetate (11). Yield 74% (16 mg), white amorphous powder, Rf = 0.22 (n-hexane/EtOAc; 2/1), ESI-MS m/z: 1321.3 [M + H]+. 1H NMR (500 MHz, CDCl3) δ 7.09–7.06 (m, 2H), 7.04–6.80 (m, 5H), 6.78 (d, 1H, J = 2.0 Hz, H-6-SB), 6.60 (d, 1H, J = 2.0 Hz, H-8-SB), 5.64 (dd, 1H, J = 12.5 Hz, H-3-SB), 5.42 (brs, 1H, H-12-MA), 5.38 (dd, 1H, J = 12.5, 3.5 Hz, H-2-SB), 5.23 (td, 1H, J = 11.0, 4.5 Hz, H-2-MA), 5.01 (dd, 1H, J = 10.5, 3.0 Hz, H-2-SB), 4.96 (d, 1H, J = 10.5 Hz, H-3-MA), 4.36 and 3.99 (dd, each 1H, J = 13.5, 9.0 Hz, H-23-SB), 4.29 (brs, 1H, H-6), 4.09 and 3.98 (dd, 1H, J = 19.0, 5.5 Hz, CH2-Gly), 3.97 (1H, overlap, H-23-MA), 3.87/3.86 (s, 3H, OMe-SB), 3.72 (d, 1H, J = 12.0 Hz, H-23-MA), 2.35 (s, 3H, AcO-SB), 2.32 (s, 3H, AcO-SB), 2.30 (s, 3H, AcO-SB), 2.14–2.11 (m, 1H, overlap, H-11), 2.06 (s, 3H, Ac), 2.05 (s, 3H, Ac), 2.03 (s, 3H, Ac), 1.96 (s, 3H, AcO-SB), 1.70–1.65 (m, 9H, H-11), 1.48 (s, 3H, H-25), 1.38–1.29 (m, 6H), 1.26 (s, 3H, H-24), 1.05 (s, 3H, H-27), 1.02 (s, 3H, H-26), 0.92 (s, 3H, H-30), 0.87 (d, 3H, J = 6.5 Hz, H-29). 13C NMR (125 MHz, CDCl3) δ 184.4 (C4-SB), 177.9 (C-28), 170.8 (C-23-OAc), 170.4 (2× OAc), 170.3 (OAc), 169.1 (COO-3-SB), 168.8 (CO-Gly), 167.8 (2× OAc), 162.5, 156.6, 151.7, 151.5, 144.1, 143.7, 140.6, 134.4 (C-13), 130.8, 128.8, 128.1, 126.3 (C-12), 123.2, 120.7, 119.9, 117.6, 116.4, 111.3, 109.1, 80.8 (C-2-SB), 76.4 (C-10-SB), 75.7 (C-11-SB), 74.9 (C-3-MA), 74.5 (C-3-SB), 69.9 (C-2-MA), 67.6 (C-6-MA), 65.3 (C-23-MA), 62.6 (C-23-SB), 56.1 (OMe-SB), 53.6 (C-18-MA), 48.1 (C-17-MA), 47.9 (C-9-MA), 47.8, 45.9 (C-1-MA), 42.8 (C4-MA), 41.1 (CH2-Glycine), 39.7, 38.9, 38.8, 37.3, 36.9, 30.9, 28.9, 27.7, 24.8, 23.8, 23.4 (C-27), 21.2 (C-30), 21.7 (Ac), 20.9 (2× Ac), 20.8 (Ac), 20.7 (2× Ac), 20.6 (2× Ac), 18.6 (C-25), 18.0 (C-26), 17.1 (C-29), 15.3 (C-24).
4.1.3.2. (5,7,20,23-O-Tetraacetyl)silybin-3-yl(N-(2α,3β,23-triacetyloxy-6β-hydroxyurs-12-en-28-oyl))-3-aminopropanoate (12). Yield 61% (13 mg), white amorphous powder, Rf = 0.25 (n-hexane/EtOAc; 2/1), ESI-MS m/z: 1335.1 [M + H]+. 1H NMR (500 MHz, CDCl3) δ 7.14–7.09 (m, 2H), 7.05–6.80 (m, 4H, overlap), 6.80 (d, 1H, J = 2.0 Hz), 6.60 (d, 1H, J = 2.0 Hz), 6.32 (t, 1H, NH), 5.74/5.73 (d, 1H, J = 12.5 Hz, H-3-SB), 5.40 (dd, 1H, J = 12.5, 3.5 Hz, H-2-SB), 5.36/5.31 (brs, 1H, H-12-MA), 5.21 (td, 1H, J = 11.0, 4.5 Hz, H-2-MA), 5.08–5.00 (m, 1H, overlap, H-10-SB), 4.98 (d, 1H, J = 10.5 Hz, H-3-MA), 4.36 and 4.03 (dt, each 1H, J = 13.5, 9.0 Hz, H-23-SB), 4.29 (1H, overlap, H-6-MA), 4.27 (1H, overlap), 3.92 (dd, 1H, J = 12.0, 6.0 Hz, H-23-MA), 3.87/3.86 (s, 3H, OMe-SB), 3.70 (d, 1H, J = 12.0 Hz, H-23-MA), 3.59–3.53 (m, 2H, β-CH2-Ala), 2.55–2.51 (m, 2H, α-CH2- Ala), 2.35 (s, 3H, Ac), 2.32 (s, 3H, Ac), 2.30 (s, 3H, Ac), 2.10 (s, 3H, Ac), 2.04 (s, 3H, Ac), 2.03 (s, 3H, Ac), 2.0 (s, 3H, Ac), 1.97 (s, 3H, Ac), 1.69–1.63 (m, 9H, H-11-MA), 1.45 (s, 3H, H-25-MA), 1.27 (s, 3H, H-24-MA), 1.04 (s, 3H, H-27-MA), 1.03 (s, 3H, H-26-MA), 0.94 (s, 3H, H-30-MA), 0.86 (s, 3H, H-29-MA). 13C NMR (125 MHz, CDCl3) δ 185.9 (C4-SB), 174.5 (C-28), 171.5 (COO-3-SB), 171.2, 170.9 (OAc), 170.4 (OAc), 170.1 (OAc), 168.9, 168.8 (OAc), 167.8 (OAc), 162.5, 156.8, 151.7, 151.4, 144.1, 143.9, 140.6, 134.3 (C-13-MA), 130.8, 128.3, 126.0 (C-12-MA), 123.3, 121.2, 119.8, 117.5/117.4, 116.5/116.4, 111.4/111.3, 109.1, 81.0/80.8 (C-2-SB), 78.3 (C-10-MA), 76.4 (C-3-MA), 75.7 (C-11-MA), 74.9 (C-3-MA), 73.6 (C-3-SB), 69.9 (C-2-MA), 67.7 (C-6-MA), 65.3 (C-23-MA), 62.7/62.6 (C-23-SB), 56.1 (OMe-SB), 53.4 (C-18-MA), 48.1 (C-17-MA), 47.8 (C-9-MA), 47.7, 45.9 (C-1-MA), 42.8 (C-4-MA), 39.7, 38.9, 38.7, 37.3, 35.3 (β-CH2- Ala), 34.3 (α-CH2- Ala), 30.9, 29.7, 27.7, 24.6, 23.3 (C-27), 21.2 (C-30), 21.2 (Ac), 21.1 (2× Ac), 20.9 (2× Ac), 20.8(2× Ac), 20.7 (Ac), 18.6 (C-25-MA), 18.4 (C-26-MA), 17.1 (C-29-MA), 15.3/15.2 (C-24-MA).
4.1.3.3. (5,7,20,23-O-Tetraacetyl)silybin-3-yl(N-(2α,3β,23-triacetyloxy-6β-hydroxyurs-12-en-28-oyl))-11-amino-undecanoate (13). Yield 68% (15 mg), white amorphous powder, Rf = 0.30 (n-hexane/EtOAc; 2/1), ESI-MS m/z: 1446.8 [M + H]+. 1H NMR (600 MHz, CDCl3) δ 7.12 (brs, 1H), 7.11–7.03 (m, 1H), 7.02–6.90 (m, 4H), 6.79–6.74 (m, 1H), 6.59–6.54 (m, 1H), 5.84 (brs, 1H, NH), 5.70/5.68 (td, 1H, J = 15 Hz, H-3-SB), 5.38–5.34 (m, 2H, overlap, H-2-SB, H-12-MA), 5.23 (td, 1H, J = 13.2, 5.4 Hz, H-2), 5.02 (dd, 1H, J = 12.6, 1.8 Hz, H-3-MA), 4.96–4.95 (m, 1H, H-2-SB), 4.37 and 4.00 (dd, each 1H, J = 16.2, 10.8 Hz, H-23-SB), 4.36–3.32 (1H, overlap, H-6-MA), 3.94 (d, 1H, J = 14.4 Hz, H-23-MA), 3.87/3.86 (s, 3H, OMe-SB), 3.72 (d, 1H, J = 14.4 Hz, H-23), 3.27 (quin, 1H, J = 6.6 Hz, CH2-11-AUDA), 3.06–3.01 (m, 1H, CH2-11-AUDA), 2.58 (t, 1H, J = 7.8 Hz, CH2-11-AUDA), 2.37 (s, 3H, Ac), 2.34 (s, 3H, Ac), 2.27 (s, 3H, Ac), 2.10–2.07 (m, 1H, overlap, H-11), 2.06–2.03 (13H, overlap, 4× Ac), 1.94 (s, 3H, OAc), 1.76–1.64 (m, 9H, H-11-MA), 1.49 (s, 3H, H-25-MA), 1.29–1.21 (m, 28H), 1.08 (s, 3H, H-27-MA), 1.05 (s, 3H, H-26-MA), 0.95 (s, 3H, H-30-MA), 0.88 (d, 3H, J = 6.5 Hz, H-29-MA). 13C NMR (150 MHz, CDCl3) δ 185.4 (C4-SB), 177.7 (C-28), 171.9 (COO-3-SB), 171.6, 170.8 (OAc), 170.4 (2× OAc), 169.1 (OAc), 168.8 (OAc), 167.8 (2× OAc), 162.6, 156.4, 151.7, 151.7/151.4, 143.9, 143.6, 140.6, 139.2, 134.3 (C-13), 130.9, 128.8, 125.3 (C-12), 123.3, 121.3, 119.8, 117.3, 116.5, 111.3, 111.0, 108.9, 81.1/81.0 (C-2-SB), 78.3 (C-10-SB), 76.4, 75.6 (C-11-MA), 74.9 (C-3-MA), 73.2 (C-3-SB), 69.9 (C-2-MA), 67.6 (C-6-MA), 65.3 (C-23-MA), 62.7 (C-23-SB), 56.0 (OMe-SB), 54.0, 53.4 (C-18-MA), 48.1 (C-17-MA), 47.8 (C-9), 47.7, 45.9 (C-1-MA), 42.4 (C-4), 40.8, 39.8, 39.5 (CH2-11-AUDA), 38.8, 37.3, 34.0 (CH2-11-AUDA), 30.9, 29.7–29.1 (8× CH2-11-AUDA), 28.9, 27.8, 24.9, 23.8, 23.4 (C-27-MA), 21.2 (C-30), 21.7 (Ac), 21.1 (2× Ac), 20.9 (2× Ac), 20.8 (AcO), 20.7 (Ac), 20.6 (Ac), 18.6 (C-25-MA), 18.3 (C-26-MA), 17.2 (C-29-MA), 15.3 (C-24-MA).
4.1.5.1. Silybin-3-yl(N-(2α,3β,23-triacetyloxyursa-5,12-dien-28-oyl))aminoacetate (15). Yield 23% (40 mg), white amorphous powder, Rf = 0.20 (DCM/acetone (4/1), ESI-MS m/z: 1135.2 [M + H]+. 1H NMR (500 MHz, CDCl3) δ 11.34 (s, 1H, OH), 7.11–7.06 (m, 2H), 6.95–6.91 (m, 4H), 6.57 (brs, 1H, NH), 6.06 (brs, 1H, H-8-SB), 5.99 (brs, 1H, H-6-SB), 5.71–6.98 (m, 1H, H-3-SB), 5.56–5.51 (m, 1H, H-6-MA), 5.48–5.43 (m, 1H, H-12-MA), 5.31–5.25 (m, 1H), 5.16–5.12 (m, 1H, H-2-SB), 4.93 (d, 1H, J = 10.5 Hz, H-3-MA), 4.22 and 3.69 (d, each 1H, J = 12.0 Hz, H-23-MA), 4.10–4.07 and 3.96–3.93 (m, 1H, overlap, CH2-Gly), 3.96/3.95 (3H, OMe-SB), 3.79 (1H, overlap, H-23-SB), 3.52 (1H, overlap, H-23-SB), 2.33 (dd, 1H, J = 19.0, 6.5 Hz, H-7), 2.18 (d, 1H, J = 11.5 Hz, H-18), 2.08 (s, 3H, Ac), 2.06 (s, 3H, Ac), 1.98 (s, 3H, Ac), 1.85–1.80 (m, 4H), 1.76–1.60 (m, 7H), 1.35–1.28 (m, 5H), 1.21 (s, 3H, H-25), 1.15 (s, 3H, H-24), 0.98 (s, 3H, H-27), 0.92 (s, 3H, H-30), 0.89 (s, 3H, H-26), 0.84 (3H, overlap, H-29). 13C NMR (125 MHz, CDCl3) δ 190.2 (C-4-SB), 178.8 (C-28), 171.1 (OAc), 170.8 (OAc), 170.4 (OAc), 168.8 (COO-3-SB), 166.9, 164.3, 162.3, 147.0, 146.5, 144.3 (C-5-MA), 140.1 (C-13-MA), 137.2, 130.9, 128.8, 127.7, 127.1 (C-12-MA), 123.7, 122.6 (C-6-MA), 120.8, 120.5, 117.3, 116.2, 114.8, 109.7, 97.6 (C-6-SB), 96.2 (C-8-SB), 82.9 (C-2-SB), 80.5 (C-10-SB), 78.3 (C-11-SB), 76.3 (C-3-SB), 73.5 (C-3-MA), 69.1 (C-2-MA), 65.0 (C-23-MA), 61.6 (C-23-SB), 56.1 (OMe-SB), 54.7 (C-18-MA), 48.1, 46.6 (C-9-MA), 45.1, 43.5, 42.3 (C-1-MA), 41.4 (CH2-Gly), 38.9, 38.8, 38.2 (C-8, C-10-MA), 36.6, 32.3 (C-7-MA), 30.9, 27.0, 23.8 (C-27), 22.9 (C-25), 22.8 (C-29), 21.8 (C-26), 21.1 (Ac), 21.0 (Ac), 20.8 (Ac), 17.3 (C-24-MA), 16.8.
4.1.5.2. Silybin-3-yl(N-(2α,3β,23-triacetyloxyursa-5,12-dien-28-oyl))-3-aminopropanoate (16). Yield 21% (38 mg), white amorphous powder, Rf = 0.15 (DCM/acetone (4/1), ESI-MS m/z: 1149.2 [M + H]+. 1H NMR (500 MHz, CDCl3) δ 11.42 (s, 1H, OH), 7.09–7.05 (m, 2Hm), 6.97–6.91 (m, 4H), 6.08 (brs, 1H, H-6-SB), 6.03 (brs, 1H, H-8-SB), 5.71 (dt, 1H, J = 12.5 Hz, H-3-SB), 5.57–5.52 (m, 1H, H-6-MA), 5.41–5.37 (m, 1H, H-12-MA), 5.29–5.24 (m, 1H, H-2-MA), 5.13 (d, 1H, J = 10.5 Hz, H-3-MA), 4.91 (d, 1H, J = 6.0 Hz, H-2-SB), 4.23 and 3.68 (d, each 1H, J = 12.0 Hz, H-23-MA), 4.09–4.04 (m, 1H, H-10-SB), 3.91 (s, 3H, OMe-SB), 3.84–3.80 and 3.56–3.51 (m, each 1H, H-23-SB), 3.59–3.53 (m, 1H, β-CH2-Ala), 2.53–2.48 (m, 1H, α-CH2-Ala), 2.36–2.31 (m, 1H, H-7), 2.20–2.15 (m, 1H, H-18), 2.08 (s, 3H, Ac), 2.04 (s, 3H, Ac), 1.99 (s, 3H, Ac), 1.79–1.73 (m, 4H), 1.67–1.60 (m, 5H), 1.35–1.28 (m, 6H), 1.18 (3H, overlap, H-25), 1.12 (s, 3H, H-24), 0.96 (3H, overlap, H-27), 0.92 (s, 3H, H-30), 0.89 (s, 3H, H-26), 0.86 (3H, overlap, H-29). 13C NMR (125 MHz, CDCl3) δ 184.5 (C-4-SB), 178.7 (C-28), 171.3 (OAc), 170.9 (OAc), 170.4 (OAc), 167.2 (COO-3-SB), 164.3, 162.5, 147.0, 146.5, 144.3 (C-5-MA), 144.2, 143.7, 139.9 (C-13-MA), 137.2, 130.9, 128.8, 128.5, 127.7, 126.6 (C-12-MA), 123.1, 122.7 (C-6-MA), 120.8, 120.5, 117.2, 116.4, 114.8, 109.7, 101.4, 97.6 (C-6-SB), 96.3 (C-8-SB), 80.5 (C-2-SB), 78.4 (C-10-SB), 76.4 (C-3-SB), 75.9 (C-11-SB), 73.6 (C-3-MA), 69.4 (C-2-MA), 68.2, 65.0 (C-23-MA), 61.6 (C-23-SB), 56.1 (OMe-SB), 53.8 (C-18), 48.1, 46.7 (C-9-MA), 45.1, 43.5, 42.3 (C-1-MA), 38.9, 38.8, 38.5 (C-8, C-10-MA), 36.9, 35.3 (β-CH2-Ala), 33.9 (α-CH2- Ala), 32.5 (C-7), 30.9, 27.0, 23.8, 22.9 (C-25-MA), 22.8 (C-29), 21.8 (C-26-MA), 20.9 (Ac), 20.8 (Ac), 20.6 (Ac), 17.3 (C-24), 14.0.
4.1.6.1. (5,7,20,23-O-Tetraacetyl)silybin-3-yl(N-(2α,3β,23-triacetyloxyursa-5,12-dien-28-oyl))aminoacetate (17). Yield 61% (10 mg), white amorphous powder, Rf = 0.25 (n-hexane/EtOAc; 2/1), ESI-MS m/z: 1303.4 [M + H]+. 1H NMR (600 MHz, CDCl3) δ 7.11–7.08 (m, 2H), 7.02–6.96 (m, 4H), 6.79 (d, 1H, J = 1.2 Hz, H-8-SB), 6.59 (d, 1H, J = 1.2 Hz, H-6-SB), 6.46 (brs, 1H, NH), 5.66 (td, 1H, J = 12.6 Hz, H-3-SB), 5.58–5.52 (m, 1H, H-6-MA), 5.50–5.46 (m, 1H, H-12-MA), 5.41–5.36 (m, 1H, H-2-SB), 5.30–5.25 (m, 1H, H-2-MA), 5.13 (d, 1H, J = 11.4 Hz, H-3-MA), 4.96 (d, 1H, J = 8.4 Hz, H-11-SB), 4.37 (dt, 1H, J = 10.8, 2.4 Hz, CH2-23-SB), 4.29–4.24 (m, 1H, H-10-SB), 4.22–4.20 (m, 1H, H-23-MA), 4.14–4.09 (m, 1H, CH2-Gly), 3.99 (dd, 1H, J = 16.2, 10.8 Hz, H-23-SB), 3.95–3.90 (m, 1H, CH2-Gly), 3.86 (s, 3H, OMe-SB), 3.72 (d, 1H, J = 12.0 Hz, H-23-MA), 2.37 (s, 3H, Ac), 2.32 (s, 3H, Ac), 2.29 (s, 3H, Ac), 2.08 (s, 3H, Ac), 2.06 (s, 3H, Ac), 2.04 (s, 3H, Ac), 1.98 (s, 3H, Ac), 1.78–1.63 (m, 11H), 1.35–1.28 (m, 5H), 1.22 (s, 3H, H-25-MA), 1.12 (s, 3H, H-24-MA), 0.98 (s, 3H, H-27-MA), 0.94 (s, 3H, H-30), 0.88 (s, 3H, H-26-MA), 0.85 (3H, overlap, H-29-MA). 13C NMR (150 MHz, CDCl3) δ 184.5 (C-4-SB), 178.0 (C-28), 171.0 (OAc), 170.8 (OAc), 170.4 (OAc), 170.3, 169.1, 169.0 (COO-3-SB), 168.7, 167.8, 162.3, 156.6, 151.7, 151.5, 144.2 (C-5-MA), 143.8, 143.6, 140.6, 140.2 (C-13-MA), 135.1, 134.3, 132.5, 130.9, 128.8, 127.9, 126.9 (C-12-MA), 123.7, 123.3, 122.6 (C-6-MA), 120.9, 120.7, 119.9, 117.6, 116.4, 111.4, 111.2, 110.4, 109.1, 80.8 (C-2-SB), 77.8 (C-10-SB), 76.4 (C-11-SB), 75.7 (C-3-MA), 73.5 (C-3-SB), 70.1, 69.1 (C-2-MA), 68.2, 64.9 (C-23-MA), 62.6 (C-23-SB), 56.1 (OMe-SB), 53.9 (C-18-MA), 48.3, 46.0 (C-9-MA), 45.1, 43.7, 42.4 (C-1-MA), 41.0 (CH2-Gly), 38.9, 38.8, 38.8 (C-8, C-10-MA), 38.4, 36.6, 32.3 (C-7-MA), 30.8, 30.6, 30.4, 28.9, 27.6, 23.8, 22.9 (C-25-MA), 22.8 (C-29), 22.4, 21.2 (Ac), 21.1 (OAc), 20.9 (Ac), 20.8 (2× Ac), 20.7 (Ac), 20.6 (Ac), 17.3 (C-24-MA), 16.8.
4.1.6.2. (5,7,20,23-O-Tetraacetyl)silybin-3-yl(N-(2α,3β,23-triacetyloxyursa-5,12-dien-28-oyl))-3-amino-propanoate (18). Yield 54% (9 mg), white amorphous powder, Rf = 0.18 (n-hexane/EtOAc; 2/1), ESI-MS m/z: 1317.3 [M + H]+. 1H NMR (600 MHz, CDCl3) δ 7.12–7.08 (m, 2H), 7.00–6.94 (m, 4H), 6.80 (s, 1H, H-8-SB), 6.41 (s, 1H, H-6-SB), 5.74 (dd, 1H, J = 12.6 Hz, H-3-SB), 5.57–5.55 (m, 1H, H-6-MA), 5.41–5.38 (m, 1H, H-12-MA), 5.34–5.32 (m, 2H), 5.15–5.11 (m, 1H, H-3-MA), 4.93 (d, 1H, J = 12.6 Hz, H-2-SB), 4.36 and 4.00 (d, each 1H, J = 7.5 Hz, H-23-SB), 4.26–4.23 and 3.75–3.71 (m, each 1H, overlap, H-23-MA), 4.22 (1H, overlap, H-10-SB), 3.86 (s, 3H, OMe-SB), 3.45–3.42 and 3.36–3.31 (m, each 1H, β-CH2-Ala), 2.47 (2H, overlap, α-CH2-Ala), 2.37 (s, 3H, Ac), 2.32 (s, 3H, Ac), 2.29 (s, 3H, Ac), 2.08 (s, 3H, Ac), 2.03 (s, 3H, Ac), 2.0 (s, 3H, Ac), 1.97 (s, 3H, Ac), 1.95–1.90 (m, 5H), 1.76–1.61 (m, 15H), 1.25 (3H, overlap, H-25-MA), 1.12 (s, 3H, H-24-MA), 0.96 (3H, overlap, H-27-MA), 0.92 (s, 3H, H-30-MA), 0.89 (3H, overlap, H-26-MA), 0.88 (3H, overlap, H-29-MA). 13C NMR (150 MHz, CDCl3) δ 184.5 (C-4-SB), 178.6 (C-28), 171.1 (OAc), 170.4 (2× OAc), 168.7, 167.8 (COO-3-SB), 162.6, 157.1/156.6, 151.7/151.4, 143.9, 143.5 (C-5-MA), 140.6 (C-13-MA), 134.3, 132.5, 130.9, 128.8, 126.5 (C-12-MA), 123.6, 123.3, 122.7 (C-6-MA), 119.9, 111.2, 109.1, 81.2 (C-2-SB), 78.5 (C-10-SB), 76.5 (C-11-SB), 75.6 (C-3-MA), 73.6 (C-3-SB), 69.1 (C-2-MA), 64.9 (C-23-MA), 62.6 (C-23-SB), 56.1 (OMe-SB), 49.3, 47.9 (C-9-MA), 46.5, 45.1, 43.7, 42.4 (C-1-MA), 40.9, 38.8, 38.5, 38.2 (C-8, C-10-MA), 37.1, 36.6 (β-CH2-Ala), 34.9 (α-CH2-Ala), 32.3 (C-7), 30.9, 30.4, 29.4, 29.1, 28.9, 23.8, 23.5, 22.9 (C-25-MA), 22.7 (C-29-MA), 22.3 (C-26-MA), 21.2 (OAc), 21.1 (Ac), 20.9 (2× Ac), 20.8 (2× Ac), 20.7 (OAc), 17.3 (C-24), 14.0.
4.2. Growth inhibition assay
Liver ancer cell lines HepG2, Hep3B, Huh7 and Huh7R were grown in DMEM containing 10% FBS and 1% antibiotic (anti–anti, Gibco, ThermoFisher Scienctific) in a humidified atmosphere at 37 °C and 5% CO2. In order to test the cytotoxic effect of compounds on different cell lines, cells were detached from the adherent culture surface by trypsin–EDTA (0.05%) and then seeded into 96 wells-plate at the density of 3 × 104 cell per mL and treated with an eight titration of each compound. Ellipticine was used as a reference (positive) control. After 96 hours of incubation, all cell culture media was removed from wells followed by addition of 100 μL MTT solution (0.5 mg mL−1 solute in fresh medium) per well and incubation at 37 °C for 4 hours. The MTT solution was then removed and DMSO (200 μL) to each well to solubilise the formazan product and mix each sample again using a pipette before determining the absorbance at 570 nm. GI50 values (compound concentration that reduces the MTT assay value by 50% versus untreated control) were calculated using GraphPad Prism 5.0 software.4.3. Cell cycle determination
To evaluate cell cycle effects, HepG2 cells were seeded in T25 flasks at a density of 1 × 105 cell per mL at 37 °C in 5% CO2 and treated with compound at concentrations of 1 × IC50 and 3 × IC50 from the result of the cytotoxic assay. After 24 hours of treatment, cells were gently harvested with trypsin and then washed with PBS (pH 7,4). In the next step, 70% ethanol was slowly added to the cells after which they were kept in a fridge (4 °C) for at least 2 h to fix the cells. After fixation the cells were pelleted by centrifugation and the ethanol completely removed resuspension with RNase A (1 mg mL−1) and incubation at 37 °C for 15 min, followed by PI staining for 45 min. Finally, at least 10000 cells were analysed using a NovoCyte flow cytometer system with NovoExpress software (ACEA Bioscience Inc.) to generate cell cycle profiles.
4.4. Caspase 3 inducible assay
A Caspase-3 Colorimetric Assay Kit (Biovision Inc.) was used for establishing the caspase 3 activity of tested samples. Treated cells were lysed in lysis buffer for 10 minutes in ice and centrifuged at 10000 × g for 1 minute to remove residual cellular debris (cell pellet). After determination of the protein concentration of each sample (Bradford assay), 80 μg of each sample in 50 μL assay buffer was added to 50 μL DTT (10 mM) and 5 μL of DEVD-pNA (200 μM) in each well of a 96-well plate in triplicate. The plate was incubated at 37 °C for 1 hour. The absorbance was read at 405 nm using a microplate reader (BioTek, ELx800).
4.5. Detection of apoptotic inducible activities by flow cytometry
The eBioessence™ Annexin V Apoptosis Detection Kit was used to measure the percentage of apoptotic cells after treatment with each compound for 24 hours according to the manufacturer's instructions. Briefly, after harvesting by trypsin–EDTA, cells were washed with cold PBS to completely remove trace medium. Cell pellets were resuspended in binding buffer and then stained with annexin V-FITC for 15 minutes at room temperature (protected from light) before being washed with the binding buffer to remove the unstained dye, resuspension in 190 μL of binding buffer and addition of 10 μL of PI solution (20 μg mL−1) in a binding buffer. Analysis was performed using a NovoCyte flow cytometer system and NovoExpress software (ACEA Bioscience Inc.) to identify the proportion of apoptotic cells.4.6. Statistical analysis
Results were analysed using Excel and GraphPad Prism 5.0 software and reported as mean ± standard error (SE). GraphPad Prism 5 software using unpaired t-test and one-way analysis of variance was used for data analysis. A value of P < 0.05 was considered to indicate statistical significance.Data availability
Processed data for this paper is included in the main manuscript. Raw data for this paper (molecular characterisation, assay readout) is available from the authors upon request.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We thank the UK's Engineering and Physical Science Research Council and Global Challenges Research Fund (EP/T020164/1) and the University of Kent, United Kingdom and Vietnam Academy of Science and Technology (VAST) for financial support under the project.References
- H. Rumgay, M. Arnold, J. Ferlay, O. Lesi, C. J. Cabasag, J. Vignat, M. Laversanne, K. A. McGlynn and I. Soerjomataram, Global burden of primary liver cancer in 2020 and predictions to 2040, J. Hepatol., 2022, 77, 1598–1606, DOI:10.1016/j.jhep.2022.08.021.
- International Agency for Research on Cancer (IARC), https://gco.iarc.fr/today/data/factsheets/populations/704-viet-nam-fact-sheets.pdf Search PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2021, 71, 209–249, DOI:10.3322/caac.21660.
- V. V. Chi, Dictionary of medicinal plants in Vietnam, Vietnam Publisher of Medicine, 2012, vol. 2 Search PubMed.
- I. E. Orhan, Centella asiatica (L.) urban: from traditional medicine to modern medicine with neuroprotective potential, Evid. Based Complement. Alternat. Med., 2012, 2012, 946259, DOI:10.1155/2012/946259.
- R. Tabassum, K. Vaibhav, P. Shrivastava, A. Khan, M. E. Amed, H. Javed, F. Islam, S. Amad, M. S. Siddiqui, M. M. Salhi and F. Islam, Centella asiatica attenuates the neuro behavioral, neuro chemical and histological changes in transient focal middle cerebral artery occlusion rats, Neurol. Sci., 2013, 34, 925–933, DOI:10.1007/s10072-012-1163-1.
- B. Brinkhaus, M. Lindner, D. Schuppan and E. G. Hahn, Chemical, pharmacological and clinical profile of the East Asian medical plant Centella asiatica, Phytomedicine, 2000, 7, 427–448, DOI:10.1016/S0944-7113(00)80065-3.
- F. Bonte, M. Dumas, C. Chaudagne and A. Meybeck, Influence of asiatic acid, madecassic acid, and asiaticoside on human collagen I synthesis, Planta Med., 1994, 60, 133–135, DOI:10.1055/s-2006-959434.
- M. C. Yin, M. C. Lin, M. C. Mong and C. Y. Lin, Bioavailability, Distribution, and Antioxidative Effects of Selected Triterpenes in Mice, J. Agric. Food Chem., 2012, 60, 7697–7701, DOI:10.1021/jf302529x.
- J. H. Won, J. S. Shin, H. J. Park, H. J. Jung, D. J. Koh, B. G. Jo, J. Y. Lee, K. Yun and K. T. Lee, Anti-inflammatory effects of madecassic acid via the suppression of NF-kappaB pathway in LPS-induced RAW 264.7 macrophage cells, Planta Med., 2010, 76, 251–257, DOI:10.1055/s-0029-1186142.
- Y. M. Hsu, Y. C. Hung, L. Hu, Y. J. Lee and M. C. Yin, Anti-Diabetic Effects of Madecassic Acid and Rotundic Acid, Nutrients, 2015, 7, 10065–10075, DOI:10.3390/nu7125512.
- A. S. C. Valdeira, E. Darvishi, G. M. Woldemichael, J. A. Beutler, K. R. Gustafson and J. A. R. Salvador, Madecassic Acid Derivatives as Potential Anticancer Agents: Synthesis and Cytotoxic Evaluation, J. Nat. Prod., 2019, 82, 2094–2105, DOI:10.1021/acs.jnatprod.8b00864.
- T. V. Loc, V. T. Q. Nhu, T. V. Chien, T. T. T. Ha, T. T. P. Thao and T. V. Sung, Synthesis of madecassic acid derivatives and their cytotoxic activity, Z. Naturforsch., 2018, 73B, 91–98, DOI:10.1515/znb-2017-0172.
- Z. Svagera, N. Skottová, P. Vána, R. Vecera, K. Urbánek, M. Belejová, P. Kosina and V. Simánek, Plasma lipoproteins in transport of silibinin, an antioxidant flavonolignan from Silybum marianum, Phytother. Res., 2003, 17, 524–530, DOI:10.1002/ptr.1187.
- I. György, S. Antus, A. Blázovics and G. Földiák, Substituent effects in the free radical reactions of silybin: radiation-induced oxidation of the flavonoid at neutral pH, Int. J. Radiat. Biol., 1992, 61, 603–609, DOI:10.1080/09553009214551411.
- H. Basaga, G. Poli, C. Tekkaya and I. Aras, Free radical scavenging and antioxidative properties of 'silibin' complexes on microsomal lipid peroxidation, Cell Biochem. Funct., 1997, 15, 27–33, DOI:10.1002/(SICI)1099-0844(199703)15:1<27::AID-CBF714>3.0.CO;2-W.
- R. P. Singh, K. Raina, G. Sharma and R. Agarwal, Silybin inhibits established prostate tumor growth, progression, invasion, and metastasis and suppresses tumor angiogenesis and epithelial mesenchymal transition in transgenic adenocarcinoma of the mouse prostate model mice, Clin. Cancer Res., 2008, 14, 7773–7780, DOI:10.1158/1078-0432.CCR-08-1309.
- S. Mateen, A. Tyagi, C. Agarwal, R. P. Singh and R. Agarwal, Silibinin inhibits human nonsmall cell lung cancer cell growth through cell-cycle arrest by modulating expression and function of key cell-cycle regulators, Mol. Carcinog., 2010, 49, 247–258, DOI:10.1002/mc.20595.
- B. Velmurugan, S. C. Gangar, M. Kaur, A. Tyagi, G. Deep and R. Agarwal, Silibinin exerts sustained growth suppressive effect against human colon carcinoma SW480 xenograft by targeting multiple signaling molecules, Pharm. Res., 2010, 27, 2085–2097, DOI:10.1007/s11095-010-0207-6.
- G. Deep and R. Agarwal, Chemopreventive efficacy of silymarin in skin and prostate cancer, Integr. Cancer Ther., 2007, 6, 130–145, DOI:10.1177/1534735407301441.
- G. Deep and R. Agarwal, Anti-metastatic Efficacy of Silibinin: Molecular Mechanisms and Therapeutic Potential against Cancer, Cancer Metastasis Rev., 2010, 29, 447–463, DOI:10.1007/s10555-010-9237-0.
- Z. Jahanafrooz, N. Motamed, B. Rinner, A. Mokhtarzadeh and B. Baradaran, Silibinin to improve cancer therapeutic, as an apoptotic inducer, autophagy modulator, cell cycle inhibitor, and microRNAs regulator, Life Sci., 2018, 213, 236–247, DOI:10.1016/j.lfs.2018.10.009.
- N. Y. S. Yassin, S. F. AbouZid, A. M. El-Kalaawy, T. M. Ali, M. M. Almehmadi and O. M. Ahmed, Silybum marianum total extract, silymarin and silibinin abate hepatocarcinogenesis and hepatocellular carcinoma growth via modulation of the HGF/c-Met, Wnt/β-catenin, and PI3K/Akt/mTOR signaling pathways, Biomed. Pharmacother., 2022, 145, 112409, DOI:10.1016/j.biopha.2021.112409.
- N. Godarzi, R. Varshochian, G. Kamalinia and F. Atyabi, A review of polysaccharide cytotoxic drug conjugates for cancer therapy, Carbohydr. Polym., 2013, 92, 1280–1293, DOI:10.1016/j.carbpol.2012.10.036.
- A. Patrelli and S. Giordano, From single- to multitarget drugs in cancer therapy: when aspecificity becomes an advantage, Curr. Med. Chem., 2008, 15, 422–432, DOI:10.2174/092986708783503212.
- V. Pokrovskaya, V. Belakhov, S. Mariana Hainrichson and T. B. Yaron, Design, synthesis, and evaluation of novel fluoroquinolone-aminoglycoside hybrid antibiotics, J. Med. Chem., 2009, 52, 2243–2254, DOI:10.1021/jm900028n.
- M. Decker, Hybrid molecules incorporating natural products: applications in cancer therapy, neurodegenerative disorders and beyond, Curr. Med. Chem., 2011, 18, 1464–1475, DOI:10.2174/092986711795328355.
- E. Price, M. Weinheimer, A. Rivkin, G. Jenkins, M. Nijsen, P. B. Cox and D. DeGoey, Beyond Rule of Five and PROTACs in Modern Drug Discovery: Polarity Reducers, Chameleonicity, and the Evolving Physicochemical Landscape, J. Med. Chem., 2024, 67, 5683–5698, DOI:10.1021/acs.jmedchem.3c02332.
- A. Zarrelli, A. Sgambato, V. Petito, L. De Napoli, L. Previtera and G. Di Fabio, New C-23 modified of silybin and 2,3-dehydrosilybin: synthesis and preliminary evaluation of antioxidant properties, Bioorg. Med. Chem. Lett., 2011, 21, 4389–4392, DOI:10.1016/j.bmcl.2011.06.049.
- R. Gažák, K. Valentová, K. Fuksová, P. Marhol, M. Kuzma, M. A. Medina, I. Oborná, J. Ulrichová and V. Křen, Synthesis and Antiangiogenic Activity of New Silybin Galloyl Esters, J. Med. Chem., 2011, 54, 7397–7407, DOI:10.1021/jm201034h.
- G. Huang, S. Schramm, J. Heilmann, D. Biedermann, V. Křen and M. Decker, Unconventional application of the Mitsunobu reaction: Selective flavonolignan dehydration yielding hydnocarpins, Beilstein J. Org. Chem., 2016, 12, 662–669, DOI:10.3762/bjoc.12.66.
- F. Wang, K. Huang, L. Yang, J. Gong, Q. Tao, H. Li, Y. Zhao, S. Zeng, X. Wu, J. Stöckigt, X. Li and J. Qu, Preparation of C-23 esterified silybin derivatives and evaluation of their lipid peroxidation inhibitory and DNA protective properties, Bioorg. Med. Chem., 2009, 17, 6380–6389, DOI:10.1016/j.bmc.2009.07.023.
- J. P. Dai, L. Q. Wu, R. Li, Z. F. Zhao, Q. Y. Wan, X. X. Chen, W. Z. Li, G. F. Wang and K. S. Li, Identification of 23-(S)-2-Amino-3-Phenylpropanoyl-Silybin as an Antiviral Agent for Influenza A Virus Infection In Vitro and In Vivo, Antimicrob. Agents Chemother., 2013, 57, 4433–4443, DOI:10.1128/AAC.00759-13.
- S. Schramm, G. Huang, S. Gunesch, F. Lang, J. Roa, P. Hogger, R. Sabate, P. Maher and M. Decker, Regioselective synthesis of 7-O-esters of the flavonolignan silibinin and SARs lead to compounds with overadditive neuroprotective effects, Eur. J. Med. Chem., 2018, 146, 93–107, DOI:10.1016/j.ejmech.2018.01.036.
- R. Gazák, K. Purchartová, P. Marhol, L. Zivná, P. Sedmera, K. Valentová, N. Kato, H. Matsumura, K. Kaihatsu and V. Křen, Antioxidant and antiviral activities of silybin fatty acid conjugates, Eur. J. Med. Chem., 2010, 45, 1059–1067, DOI:10.1016/j.ejmech.2009.11.056.
- A. Zanarotti, Stereochemistry of Silychristin, Mild Dehydrogenation of Flavononols, Heterocycles, 1982, 19, 1585–1586, DOI:10.3987/R-1982-09-1585.
- R. Gazák, A. Svobodová, J. Psotová, P. Sedmera, V. Prikrylová, D. Walterová and V. Kren, Oxidised derivatives of silybin and their antiradical and antioxidant activity, Bioorg. Med. Chem., 2004, 12, 5677–5687, DOI:10.1016/j.bmc.2004.07.064.
- D. Y. W. Lee and Y. Liu, Molecular Structure and Stereochemistry of Silybin A, Silybin B, Isosilybin A,and Isosilybin B, Isolated from Silybum marianum (Milk Thistle), J. Nat. Prod., 2003, 66, 1171–1174, DOI:10.1021/np030163b.
- M. Antoszczak, G. Klejborowska, M. Kruszyk, E. Maj, J. Wietrzyk and A. Huczyński, Synthesis and Antiproliferative Activity of Silybin Conjugates with Salinomycin and Monensin, Chem. Biol. Drug Des., 2015, 86, 1378–1386, DOI:10.1111/cbdd.12602.
- B. Pucci, M. Kasten and A. Giordano, Cell cycle and apoptosis, Neoplasia, 2000, 2, 291–299, DOI:10.1038/sj.neo.7900101.
- L. Varghese, C. Agarwal, A. Tyagi, R. P. Singh and R. Agarwal, Silibinin Efficacy against Human Hepatocellular Carcinoma, Clin. Cancer Res., 2005, 11, 8441–8448, DOI:10.1158/1078-0432.CCR-05-1646.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00170b |
| This journal is © The Royal Society of Chemistry 2024 |