
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel PROTAC probes targeting KDM3 degradation to eliminate colorectal cancer stem cells through inhibition of Wnt/β-catenin signaling†
Shadid U.
Zaman‡
a,
Piyusha P.
Pagare‡
a,
Hongguang
Ma
a,
Rosalie G.
Hoyle
a,
Yan
Zhang
 *a and
Jiong
Li
*abc
*a and
Jiong
Li
*abc
aDepartment of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, Virginia 23298-0540, USA. E-mail: yzhang2@vcu.edu; jli29@vcu.edu
bDepartment of Oral and Craniofacial Molecular Biology, Virginia Commonwealth University, Richmond, Virginia 23298-0540, USA
cMassey Cancer Center, Virginia Commonwealth University, Richmond, Virginia 23298-0540, USA
First published on 13th September 2024
Abstract
It has been demonstrated that the KDM3 family of histone demethylases (KDM3A and KDM3B) epigenetically control the functional properties of colorectal cancer stem cells (CSCs) through Wnt/β-catenin signaling. Meanwhile, a broad-spectrum histone demethylase inhibitor, IOX1, suppresses Wnt-induced colorectal tumorigenesis predominantly through inhibiting the enzymatic activity of KDM3. In this work, several cereblon (CRBN)-recruiting PROTACs with various linker lengths were designed and synthesized using IOX1 as a warhead to target KDM3 proteins for degradation. Two of the synthesized PROTACs demonstrated favorable degradation profile and selectivity towards KDM3A and KDM3B. Compound 4 demonstrated favorable in vitro metabolic profile in liver enzymes as well as no hERG-associated cardiotoxicity. Compound 4 also showed dramatic ability in suppressing oncogenic Wnt signaling to eliminate colorectal CSCs and inhibit tumor growth, with around 10- to 35-fold increased potency over IOX1. In summary, this study suggests that PROTACs provide a unique molecular tool for the development of novel small molecules from the IOX1 skeleton for selective degradation of KDM3 to eliminate colorectal CSCs via suppressing oncogenic Wnt signaling.
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed type of cancer and second leading cause of cancer-related deaths worldwide with about 935![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths reported in 2020.1 An increase in early detection has significantly improved the overall survival rate in CRC to 64%, but the 5 year survival rate for stage IV (metastatic) CRC remains unimproved at around 12% due to the lack of effective treatment options.1,2 Over 90% of all CRC cases are associated with hyperactivation of Wnt/β-catenin signaling, and Wnt/β-catenin signaling has been demonstrated to play an essential role in sustaining tumor CRC growth and development.3–7 Meanwhile, high Wnt activity has been shown to be a vital characteristic and a key driver of human colorectal cancer stem cells (CSCs) which are responsible for tumorigenesis, metastasis, and development of chemotherapy resistance. In fact, major functional markers of colorectal CSCs such as LGR5, ZNRF3, RNF43 and ASCL2 are Wnt direct target genes.7–12 The significance of Wnt signaling in CRC initiation and development, and validation of the Wnt signaling as a therapeutic target for CRC have already been well-recognized.13,14 Although substantial effort has been invested in therapeutically inhibiting Wnt/β-catenin signaling, no drug has reached clinics, indicating the urgent need for identifying new drug targets and developing more effective strategies to suppress oncogenic Wnt signaling.
000 deaths reported in 2020.1 An increase in early detection has significantly improved the overall survival rate in CRC to 64%, but the 5 year survival rate for stage IV (metastatic) CRC remains unimproved at around 12% due to the lack of effective treatment options.1,2 Over 90% of all CRC cases are associated with hyperactivation of Wnt/β-catenin signaling, and Wnt/β-catenin signaling has been demonstrated to play an essential role in sustaining tumor CRC growth and development.3–7 Meanwhile, high Wnt activity has been shown to be a vital characteristic and a key driver of human colorectal cancer stem cells (CSCs) which are responsible for tumorigenesis, metastasis, and development of chemotherapy resistance. In fact, major functional markers of colorectal CSCs such as LGR5, ZNRF3, RNF43 and ASCL2 are Wnt direct target genes.7–12 The significance of Wnt signaling in CRC initiation and development, and validation of the Wnt signaling as a therapeutic target for CRC have already been well-recognized.13,14 Although substantial effort has been invested in therapeutically inhibiting Wnt/β-catenin signaling, no drug has reached clinics, indicating the urgent need for identifying new drug targets and developing more effective strategies to suppress oncogenic Wnt signaling.
Our group has previously demonstrated that a KDM3 family of histone demethylases, specifically KDM3A and KDM3B, are significantly upregulated in colorectal CSCs and maintain the tumorigenic potential of colorectal CSCs through Wnt/β-catenin signaling. We also demonstrated that IOX1, a broad-spectrum inhibitor of histone demethylases, including KDM3, KDM4, KDM6B, and KDM2A, significantly suppresses Wnt/β-catenin signaling and the functional properties of colorectal CSCs primarily through inhibiting the enzymatic activity of KDM3.15,16 We showed that colorectal CSCs are more sensitive to IOX1 than non-CSCs, which makes IOX1 a promising structure to be optimized to develop new molecules that selectively target CSCs. However, IOX1 lacks selectivity between KDM3 and KDM4 family members and its potency for suppressing CRC tumorigenesis is relatively low with effective concentrations for suppressing CSC growth being 20–50 μM in most cell-based assays.16,17
In this work, we demonstrated that the shortcomings of IOX1 can be overcome by designing proteolysis targeting chimeras (PROTACs) carrying various linker lengths with the aim of improving selectivity and potency towards KDM3. PROTACs are being extensively studied as anti-cancer therapeutics.18,19 PROTACs can afford an advantage over the parent compound, IOX1, because they are expected to work catalytically to facilitate proteasomal degradation and thereby improve potency.20 Furthermore, it has been demonstrated that the linker length between the warhead and the E3 ligand can affect the selectivity towards the target protein.21–23 Thus, a series of PROTACs were synthesized utilizing IOX1 as the warhead and pomalidomide as the (cereblon-recruiting) E3 ligand, and their selectivity towards KDM3 and efficacy in suppressing CRC tumorigenesis were evaluated using in vitro and in vivo CRC models. We identified two potent IOX1-based PROTACs which selectively degraded KDM3 by optimizing the linker length, demonstrating the feasibility of improving the potency and selectivity toward KDM3 from the IOX1 skeleton via PROTAC modifications. Our study provides the proof-of-concept evidence that optimization of IOX1-based PROTACs creates opportunities for the selective degradation of KDM3 proteins, thereby effectively eliminating colorectal CSCs through inhibition of Wnt signaling, which can be tailored to develop novel Wnt-dependent targeted therapies to treat CRC.
Results and discussion
Molecular design and synthesis
A successful PROTAC is designed such that it brings together the protein of interest and E3-ligase to facilitate the ubiquitination of the protein of interest via the E3-ligase.18,24,25 Thus, the linker length, orientation, and point of attachment play an important role in its design. A structure-based approach was utilized to rationalize the molecular design of IOX1-based PROTACs. Employing molecular docking studies, previously it was demonstrated that IOX1 showed identical interactions with both KDM3A and KDM3B, and hence, in our current work we focused on the KDM3B–IOX1 protein–ligand complex.16 Our docking results showed that the hydroxy group was positioned to direct a linker chain towards the solvent-exposed area upon attachment. However, we recognized that the resulting phenolic esters would be suboptimal due to their chemical instability. Conversely, the carboxy group of IOX1, although bound within the binding pocket, was situated such that an ester or amide linkage would guide the linker chain out of the pocket and towards the solvent-exposed area (Fig. 1 and Hoyle 2021 (ref. 16). Thus, this carboxy group was defined as the point of attachment for the linker in our PROTAC design. | ||
| Fig. 1 (A) Binding pose of the optimal IOX1-PEG-polidomide (E3-ligand) in KDM3B (IOX1: magenta, PEG-linker: grey, pomalidomide: pink sticks, metal ion (Mn): green sphere, KDM3B: yellow surface; (B) chemical structure of synthesized PROTAC probes (IOX1: black, PEG-linker: red, pomalidomide: blue). | ||
As mentioned above, the optimal linker would facilitate the ubiquitination of the target protein, KDM3B, but not too long or too short to compromise the degradation efficiency.21,26–28 Polyethylene glycols (PEGs) with different chain lengths are the most commonly used PROTAC linkers accounting for almost 55% among published PROTAC molecules.28–30 Due to this and their commercial availability, PEG was chosen as the linker type. To determine the optimal linker length, IOX1 derivatives with various lengths of PEG chains were docked in the binding pocket of KDM3B (PDB ID: 4C8D) using GOLD2020.31,32 Following energy minimization of the highest scored protein–ligand complexes, it was observed that a minimum of 14-atoms were required for the linker to be out of the binding pocket of KDM3B and in the solvent exposed region (Fig. 1A). In addition, analyzing the X-ray structures of PROTAC bound ternary complexes of the CRBN-E3-ligase revealed that at least 4-atoms were required for the linker to be outside the binding pocket in the solvent exposed region from the E3-ligase.33 Thus, taken together along with the orientation of the KDM3B target protein, the starting point for exploring the linker length was determined to be 17 atoms (Fig. 1B). The flexible nature of PEG linkers makes it challenging to predict the optimal binding pose reliably using binary molecular docking studies. Nevertheless, molecular docking results were utilized to establish a reasonable starting point for exploring linker lengths. In order to calibrate the optimal linker length, PEG linkers with chain lengths 6 (PEG n = 1) – 24 (PEG n = 7) were explored. Lastly, thalidomide and its derivatives are one of the most reported CRBN selective E3-ligase ligands utilized for PROTACs including ARV-110 and ARV-471.33–39 Hence, thalidomide was selected as the E3-ligase ligand for PROTAC synthesis. Overall, this exploratory molecular design resulted in seven IOX1-based PROTAC analogs.
The synthetic route of the newly designed compounds 1–7 is outlined in Scheme 1. First, protection of the hydroxyl group on IOX1 was furnished by converting it to its methoxymethyl (MOM) ether. Second, pomalidomide, a derivative of thalidomide, was coupled with commercially available tert-butyloxycarbonyl protected polyethylene glycol (PEG) linkers of various lengths. After treatment with trifluoracetic acid to obtain the free amine, MOM-protected IOX1 and the linker–E3 ligase complex were coupled via the EDCI/HOBt coupling reaction. Finally, the hydroxyl group on IOX1 was deprotected under acidic conditions and the compounds were converted to their hydrochloride salt forms, fully characterized and submitted for subsequent in vitro and in vivo biological studies.
 | ||
| Scheme 1 Synthetic route for IOX1 PROTAC analogs: (a) MOM-Br, NAH, LiOH, THF, 0 °C; (b) DMA, DIPEA, 90 °C; (c) TFA, DCM, rt; (d) EDCI, HOBt, DMF, rt; (e) HCl, 1,4-dioxane, rt. | ||
Wnt luciferase report assay
To identify the most potent compounds in suppressing Wnt signaling effectively, we adopted our well-established Wnt luciferase reporter assays to assess their inhibitory activity in the 293T cells expressing a TCF-responsive luciferase reporter (293T-TCF-Luc).15 The cells were pre-treated with IOX1 or its respective PROTAC (compounds 1–7) for 4 hours, followed by 12 hour treatment of 20 mM LiCl, a GSK3 inhibitor, which induces the β-catenin/TCF-mediated transcription. Inhibition of the β-catenin-dependent transcription by the assayed compounds reduced expression of the luciferase activity which is indicated by reduction in luminescence. IOX1 and the seven PROTACs all showed dose-dependent reduction in luciferase activity upon 16 hour treatment but the PROTACs demonstrated lower IC50 values compared to the parent compound (Fig. 2A). Compound 4 was the most potent with a five-fold lower IC50 compared to IOX1 (8.9 μM vs. 48.6 μM, respectively). Furthermore, a clear relationship was observed between the IC50 and the spacer length between the warhead and the E3 ligand components of the PROTACs, with the linker length of 4 PEG units being the most optimal for activity (Fig. 2B). This corresponds to a linker length of 16 atoms which is very close to our proposed design. Our finding suggests that there is a “sweet-spot” of linker length for activity, a shorter linker length is insufficient, while a linker length longer than that also reduces activity.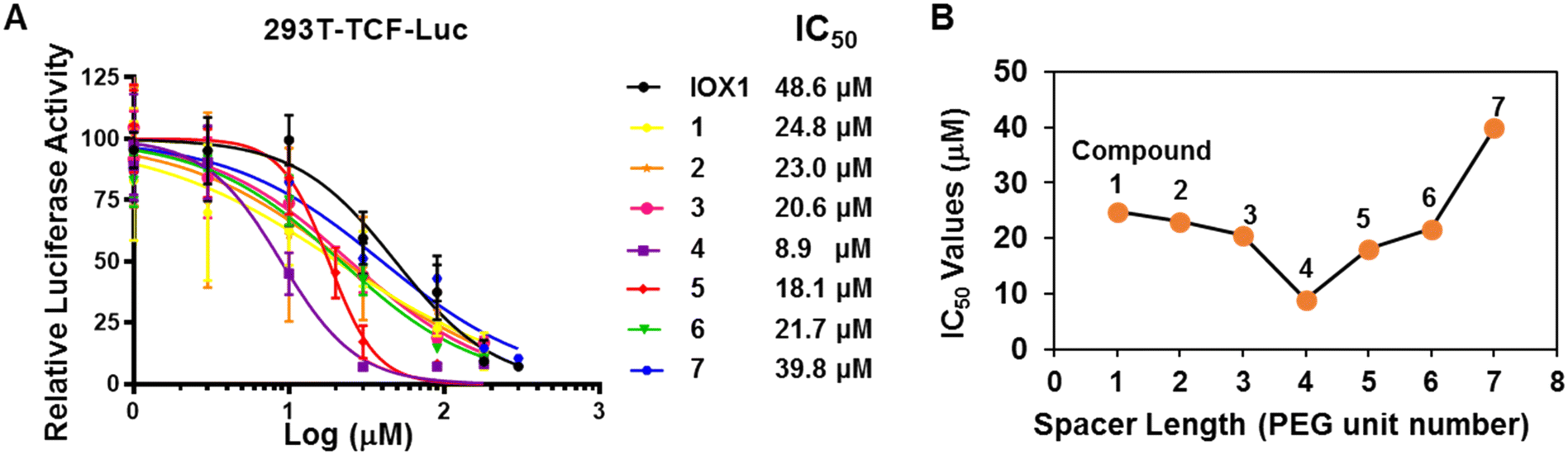 | ||
| Fig. 2 IOX1-PROTACs potently suppress Wnt signaling in CRC cells. (A) IOX1-PROTACs inhibited the Wnt luciferase reporter in 293T cells. (B) The relationship of the linker length with the IC50 of the IOX1-PROTACs. | ||
Degradation profiles of IOX1 PROTACs
Since IOX1 can inhibit both KDM3 (KDM3A and KDM3B) and KDM4 (KDM4A, KDM4B and KDM4C) families of histone demethylases, the IOX1-based PROTACs may degrade both KDM3 and KDM4 proteins.17 Based on the IC50 values obtained from the luciferase assay, compounds 3, 4, 5 and 6 were chosen for evaluation of the protein degradation profile in human CRC SW480 cells. Western blot analysis showed that compound 4 degrades both KDM3A and KDM3B at nanomolar concentrations, whereas compound 6 only has limited ability to degrade KDM3A and KDM3B in SW480 cells (Fig. 3A). In comparison, the parental compound, IOX1, did not induce KDM3A and KDM3B degradation in SW480 cells (Fig. S1†). Of note, the DC50 values of compound 4 are 13.73 nM and 172.6 nM for KDM3A and KDMB in SW480 cells, respectively (Fig. S2†). The Dmax values of compound 4 are 65.34% and 88.55% for KDM3A and KDM3B, respectively. Compounds 3 and 5 had little effect in degrading KDM3A and KDM3B (data not shown). A “hook effect” was observed with compounds 4 and 6 for KDM3A degradation at higher doses in SW480 cells. The “hook effect” is an intrinsic property of any PROTAC.40,41 This effect is a result of a saturation of PROTAC molecules that leads to increased binary complex formation and decreased ternary complex formation necessary for degradation and is correlated with the binding affinity of the warhead to the protein of interest. Since IOX1 has stronger binding affinity for KDM3A compared to other KDM proteins, this may explain why the “hook effect” is only observed for KDM3A degradation under the treatment conditions.17,42 It was also noted that KDM4 family proteins were not dramatically affected by the PROTACs (compounds 4 and 6). The KDM3 proteins' degradation profile was also evaluated in freshly isolated human CRC HCP-1 cells. As shown in Fig. S3,† compound 4 can also potently induce KDM3A and KDM3B degradation in HCP-1 cells. To validate the mechanism of action of PROTAC-induced degradation, a specific NEDD8-activating enzyme inhibitor, MLN4924, was adopted. As shown in Fig. 3B, the degradation of KDM3A or KDM3B induced by compound 4 could be restored by MLN4924 in SW480 cells. Furthermore, the degradation of KDM3A or KDM3B could also be suppressed by either pomalidomide or IOX1 in SW480 cells (Fig. 3B). Of note, pomalidomide treatment alone did not affect KDM3A or KDM3B expression in SW480 cells (Fig. S4A†). Finally, compounds 4 did not induce the known neo-substrate GSPT1 degradation in SW480 cells, which further confirmed the PROTAC-induced proteasomal degradation (Fig. S4B†). Taken together, our results indicated that the selectivity for KDM3 of the PROTAC molecules derived from the IOX1 can be achieved. | ||
| Fig. 3 IOX1-PROTACs induced KDM3A and KDM3B degradation in SW480 cells. (A) SW480 cells were treated with compounds 4 or 6 as indicated for 16 hours. (B) MLN4924, pomalidomide, or IOX1 restored PROTAC-induced KDM3A/B degradation in SW480 cells. MLN4924: 1 μM; pomalidomide: 100 μM; compound 4: 0.1 μM for KDM3A western blot and 1 μM for KDM3B western blot. | ||
RT-qPCR analysis of Wnt target gene expression
Based on the degradation profile of compounds 4 and 6, these were selected for further evaluation of the impact of KDM3 degradation on the Wnt signaling pathway. First, the basal toxicity of these two PROTACs was measured by CCK8 assay using non-malignant human colon epithelial cells, CRL-1790. Of note, KDM3 expression is undetected in CRL-1790. Both compounds had little impact on the cell growth in this cell line with TD50 values of 285.4 μM and 400.3 μM, respectively. (Fig. 4A). The cell growth was not dramatically affected at 60 μM for a two-day treatment. Therefore, concentrations below 60 μM were used for the following in vitro functional assays. | ||
| Fig. 4 IOX1-PROTAC suppresses Wnt target gene expression in CRC cells. (A) IOX1-PROTACs had little effect in suppressing the proliferation of CRL-1790 cells. The cells were treated with IOX1-PROTACs as indicated. CCK8 assay was performed 48 h post drug treatment. The TD50 values are listed in the table. (B) IOX1-PROTACs inhibited Wnt target gene expression in CRC cells. SW480 cells and HCP-1 were treated with compounds 4 or 6 as indicated for 16 hours. Data represent mean ± SD. **P < 0.01; unpaired two-tailed Student's t-test. | ||
To determine the impact on Wnt target gene transcription, RNA extracts obtained from SW480 and HCP-1 following 16-hour treatment with IOX1, 4 or 6 were subjected to RT-qPCR. The two PROTACs were able to profoundly suppress expression of Wnt target genes, including AXIN2, DKK1 and CCND1, by 50% or more in most cases at 50 μM concentration (Fig. 4B and S5†). The expression of colorectal CSC signature genes, such as ASCL2, RNF43, ZNRF3 and LGR5, was also strongly inhibited by the two PROTACs (Fig. 4B) in both SW480 and HCP-1. Notably, compound 4 showed a more dramatic inhibitory effect of these Wnt target genes' expression compared to IOX1, demonstrating the superiority to the parental compound.
Clonogenic assay
Clonogenic assay is an effective in vitro assay to determine whether compounds can inhibit growth of cells. Both 4 and 6 potently inhibited the colony formation of SW480 and HCP-1 cells in a dose-dependent manner and displayed advantageous doses as compared to IOX1 (Fig. S6A and B†).Tumorsphere formation assay
ALDH is a well-characterized marker of CSC-like activity in CRC. Previously, we found that ALDHHigh-CSCs have higher expression of KDM3A and KDM3B as compared to ALDHLow-non-CSC populations in CRC cells.16 To evaluate the inhibitory effect of IOX1-PROTACs on CSC-like behavior, tumor sphere formation assays were adopted. Both 4 and 6 displayed potency in suppressing the self-renewal ability of ALDHhigh-SW480 CSCs with around 5-fold higher potency than IOX1 based on their ED50 values (Fig. 5A and B). These molecules also potently inhibited the tumor sphere formation ability of ALDHhigh-HCP-1 CSCs. The ED50 values were 0.28 μM and 0.51 μM for 4 and 6, respectively, approximately 20- to 40-fold improvement compared to IOX1 (10.1 μM) (Fig. 5B). The therapeutic index (TI) values for each compound were calculated by normalizing the TD50 values of the CCK8 assay with the ED50 values. Compounds 4 and 6 showed 28-fold and 21-fold higher therapeutic index than IOX1, respectively (Fig. 5B). Because both KDM3A and KDM3B are significantly upregulated in ALDHHigh-CSCs as compared with ALDHLow-non-CSCs, CSCs are more sensitive to KDM3 inhibition, as found in our previous studies.16 Furthermore, CSCs only represent a small population in cultured CRC cells, which may explain the low IC50 values of IOX-PROTACs for CSC-based assays compared to the assays using the whole population of CRC cells. | ||
| Fig. 5 IOX1-PROTAC suppresses self-renewal of CSCs in vitro. (A and B) Tumor sphere formation assay showed that the self-renewal ability of CSCs was inhibited by IOX1-PROTACs. The represented images of tumor spheres (A) and dose response curve (B) of treatments in tumor sphere formation assays. The ED50 and TI values are listed in the table. | ||
In vitro metabolic stability and toxicity assessment
There are multiple factors influencing bioavailability, including not only permeability but also hepatic metabolic stability. Hepatic metabolism of small molecules primarily occurs through the cytochrome P450 (CYP) family of enzymes located in the hepatic endoplasmic reticulum, but non-CYP enzymes, such as phase II glucuronosyltransferases and sulfotransferases, also play a significant role.43,44 To evaluate overall liver metabolism in humans and rats, compound 4 was incubated in liver S9 fractions from both species. The clearance mechanism of compound 4 through phase II glutathione conjugation, glucuronidation and sulfation reactions was studied using glutathione S-transferases (GSTs), UDP-glucuronosyltransferase (UGT) and sulfotransferase (SULT) enzymes, with the addition of glutathione, uridine-5′-diphospho-α-D-glucuronic acid (UDPGA) and 3′-phosphoadenosine-5′-phosphosulfate (PAPS), respectively.45 Assuming first-order kinetics, the half-life of compound 4 was calculated to be 16 minutes in human liver S9 fractions and 20 minutes in rat liver S9 fractions (Table 1). This relatively high stability was also observed with the control compound terfenadine, tested in parallel. Furthermore, compound 4 exhibited apparent intrinsic clearance (CLint) values of 41.6 and 34.5 μL min−1 mg−1 in human and rat liver fractions, respectively (Table 1). According to the CLint classification bands for each species, these values indicate that compound 4 has moderate clearance in both humans and rats.46,47 This moderate clearance could be ideal for a PROTAC, as it needs to balance sufficient duration for effective protein degradation with timely clearance to avoid prolonged systemic exposure, which could lead to toxicity or off-target effects.| Compound | Human (liver, S9) | Rat (liver, S9) | ||
|---|---|---|---|---|
| t 1/2 (min) | CLint (μL min−1 mg−1) | t 1/2 (min) | CLint (μL min−1 mg−1) | |
| Compound 4 | 16.7 | 41.6 | 20.1 | 34.5 |
| Clozapine | >120 | <5.8 | 34.0 | 20.4 |
| Diclofenac | 18.5 | 37.5 | 101.8 | 6.8 |
| Imipramine | 102.7 | 22.8 | 20.3 | 113.7 |
| Propranolol | >120 | <19.3 | 14.1 | 164.2 |
| Terfenadine | 13.8 | 167.5 | 30.6 | 75.6 |
hERG-related cardiotoxicity is a critical safety evaluation parameter in early drug discovery campaigns. Compounds that exhibit hERG liability tend to block the inward rectifying voltage-gated K+ channel (IKr) in the heart, leading to QT interval prolongation and an increased risk of fatal arrhythmias.48 Consequently, the hERG inhibitory activity of compound 4 was assessed. Verapamil, an antiarrhythmic drug that selectively blocks the hERG potassium channel, was used as a positive control in the automated patch-clamp assay. The results showed that compound 4 had an IC50 value greater than 30 μM, indicating a low potential for causing hERG-related cardiotoxicity.49
In vivo anti-tumor activities
Compound 4 was selected to evaluate the efficacy in suppressing the tumorigenic potential of colorectal CSCs in vivo based on its highest potency in suppressing expression of colorectal CSC makers and the self-renewal ability of CSCs. ALDHHigh-HCP-1 CSCs were subcutaneously inoculated into the flanks of nude mice. The mice were then treated with IOX1 at 10 mg kg−1 or increasing doses of compound 4 at 3 mg kg−1, 10 mg kg−1, 35 mg kg−1 (equivalent to the same molar amount of IOX1 at 10 mg kg−1), or control vehicle for 16 days through IP injection. The treatment dose was selected based on Fig. 5 and our previous study, which suggested that the 10 mg kg−1 of IOX1 treatment daily can effectively suppress ALDHHigh-HCP-1 CSC-derived tumor growth in a similar xenograft mouse model of CRC.16 As shown in Fig. 6A–C, the CSC-derived tumor growth was significantly inhibited by administration of 4 at all three doses. There was no significant change in the tumor volume nor tumor weight between the IOX1 group and 4 treatment group at 3 mg kg−1, whereas both the tumor volume and weight were significantly decreased in the 10 and 35 mg kg−1 treatment groups with at least 10-fold improved potency over IOX1 (Fig. 6A–C). Of note, administration of 4 had little impact on the body weight of mice, suggesting that it is well-tolerated by mice (Fig. 6D). | ||
| Fig. 6 Compound 4 suppressed the tumorigenic potential of colorectal CSCs. (A and B) Compound 4 significantly inhibited tumorigenic potentials of ALDHhigh-HCP-1 cells in vivo. Data represent mean ± SD (n = 10). *P < 0.05 and **P < 0.01 by Student's t test. (C) Comparisons of tumor weights at the end of experiments (n = 10). *P < 0.05 and **P < 0.01 by one-way ANOVA. (D) 4 had little effect on the body weight change of mice. | ||
Immunohistochemical (IHC) staining of the tumor tissues confirmed the PROTAC-induced KDM3A and KDM3B degradation in vivo (Fig. 7). The Wnt target gene, CCND1, expression was also downregulated upon KDM3 degradation (Fig. 7). These results suggest that compound 4 has superiority in suppressing Wnt-induced tumorigenesis over IOX1. Interestingly, IHC showed a slight upregulation of KDM3A, but not for KDM3B, in tumors from mice that were treated with 35 mg kg−1 of 4 (Fig. 7). This is in line with the in vitro “hook effect” of KDM3A identified by western blot assays. Despite the upregulation of KDM3A, the Wnt-induced tumor growth was still drastically reduced in the 35 mg kg−1 treatment group, which may be due to the inhibition of the enzymatic activity of KDM3 by the IOX1 functional group in the PROTAC molecules.
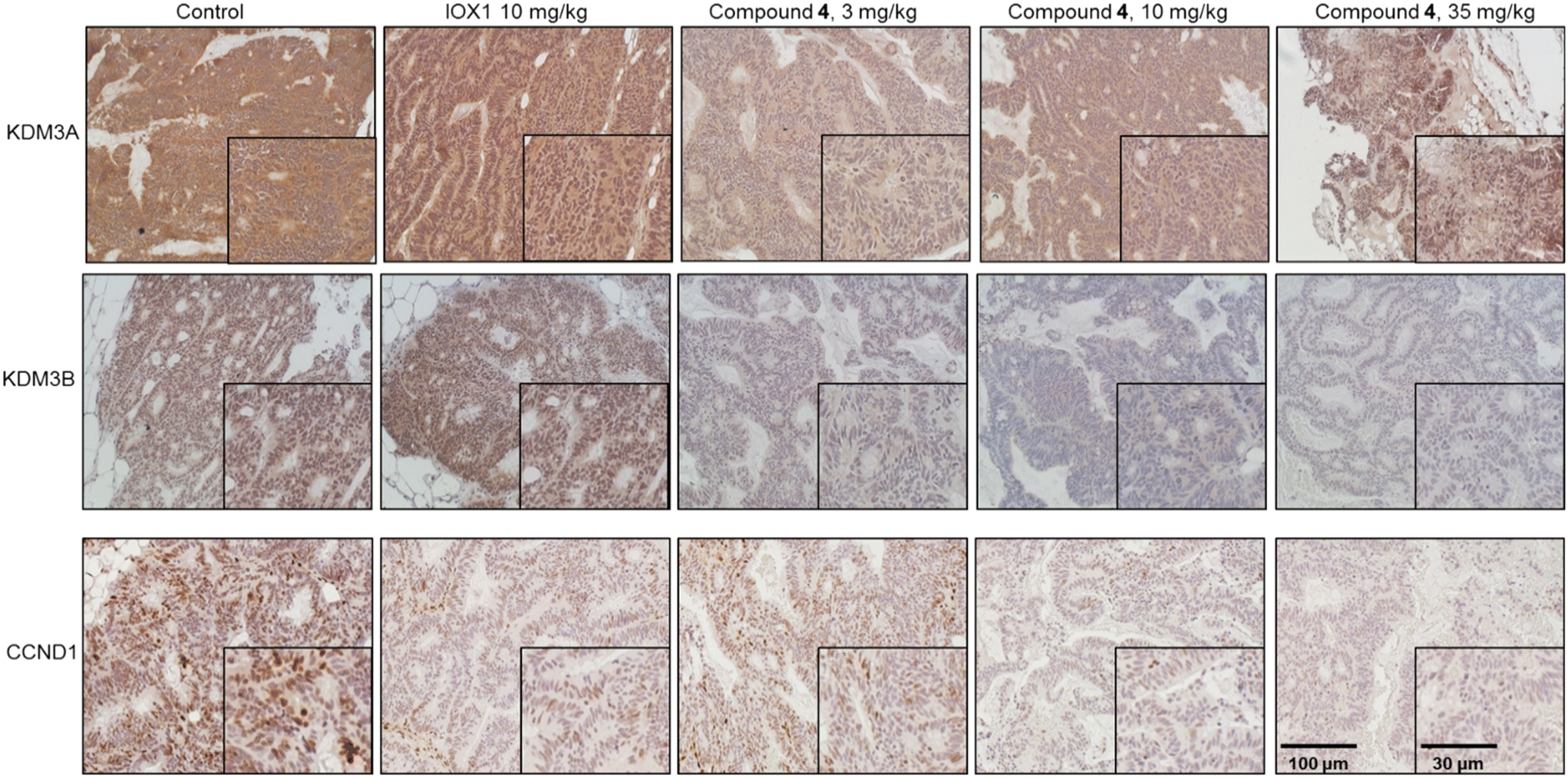 | ||
| Fig. 7 Administration of compound 4 degrades KDM3 and inhibited Wnt target gene expression in HCP-1 xenografts. Immunostaining of HCP1 xenografts using KDM3A, KDM3B, and CCND1 antibodies. | ||
Conclusion
Previously, our group demonstrated that KDM3 proteins are overexpressed in colorectal CSCs and control their tumorigenic potential through Wnt/β-catenin signaling. It was also demonstrated that IOX1 can suppress Wnt-induced colorectal tumorigenesis predominantly through enzymatic inhibition of the KDM3 proteins. This formed the basis for utilizing IOX1 to design molecules that target KDM3 proteins with high selectivity and potency, particularly for degrading these proteins using PROTAC technology. In this work, we optimized the linker length through a valid structure–activity relationship (SAR) strategy for a structure-based PROTAC design and developed two PROTACs that have higher selectivity towards KDM3 proteins over KDM4 proteins compared to IOX1. Both PROTACs had higher potency than IOX1, with one (compound 4) demonstrating up to 35-fold more potency in vitro and 10-fold potency in vivo than IOX1. Collectively, our investigation suggests that optimization of IOX1-based PROTACs may provide a promising strategy to develop molecules that can effectively suppress oncogenic Wnt signaling to eliminate colorectal CSCs through selective degradation of KDM3 proteins, which may lead to new therapeutics for the eradication of CRC, a life-threatening disease without an effective targeted therapeutic strategy at present.Experimental
Chemical syntheses
All non-aqueous reactions were carried out under a pre-dried nitrogen gas atmosphere. All solvents and reagents were purchased from either Combi-Blocks, Sigma-Aldrich, or Enamine LLC, and were used as received without further purification. Melting points (mp) were measured on an MPA100 OptiMelt automated melting point apparatus without correction. Analytical thin-layer chromatography (TLC) analyses were carried out on Analtech Uniplate F254 plates and flash column chromatography (FCC) was performed using silica gel (230–400 mesh, Merck). 1H (400 MHz) and 13C (100 MHz) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Ultrashield 400 Plus spectrometer. Chemical shifts were expressed in δ units (ppm), using TMS as an internal standard, and J values were reported in hertz (Hz). Mass spectra were obtained on an Applied BioSystems 3200 Q trap with a turbo V source for Turbolon Spray. Analytical reversed-phase high performance liquid chromatography (HPLC) was performed on a Varian ProStar 210 system using an Agilent Microsorb-MV 100-5 C18 column (250 × 4.6 mm). All analyses were conducted at ambient temperature with a flow rate of 0.5 mL min−1. HPLC eluent conditions: acetonitrile/water (with 0.1% trifluoroacetic acid), acetonitrile increased from 30% to 100% in gradient within 15 min of test. The UV detector was set up at 210 nm. The injection volume was 5 μL. The purities of the final compounds were calculated as the percentage peak area of the analyzed compound, and retention time (Rt) was presented in minutes. The purity of all newly synthesized compounds was identified as ≥95%.Step 1. 8-Hydroxyquinoline-5-carboxylic acid (1 eq.) was added portionwise to a stirring suspension of 60% NaH (2.5 eq.) in dry THF (30 mL) at 0 °C, and the resulting mixture was stirred under an inert atmosphere (N2) for 30 min. Then methoxymethyl bromide (MOM-Br, 2.5 eq.) dissolved in dry THF (20 mL) was added dropwise, and the suspension was allowed to warm to room temperature under an inert atmosphere (N2). After 4 h, a solution of LiOH (4.0 eq.) in water was added at 0 °C, and the resulting mixture was stirred at room temperature overnight. The reaction mixture was acidified with glacial acetic acid to pH = 4 and then extracted with dichloromethane (DCM; 5 × 90 mL); the organic phases were collected, dried over anhydrous Na2SO4, and filtered, and the solvent was removed under reduced pressure to give the target compound as a white solid.
Step 2. A mixture of 2-(2,6-dioxo-piperidin-3-yl)-4-fluoroisoindoline-1,3-dione (1 eq.), t-Boc-N-amido-PEG#-amine (1.5 eq.), and DIPEA (2 eq.) in DMF (5 mL) was heated to 90 °C and stirred overnight. Upon completion of the reaction via TLC, the reaction mixture was diluted with water and ethyl acetate and the organic layer was retrieved. The organic layer was then washed with brine and then dried with Na2SO4. The solvent was removed in vacuo and the crude product mixture was separated and purified via column chromatography (DCM/MeOH, 40:1 with 0.1% NH4OH).
Step 3. The pomalidomide-PEG-t-Boc-N-amine compound was dissolved in DCM (2 mL) and stirred for 5 minutes at room temperature. Then trifluoroacetic acid (2 mL) was added and the mixture was stirred until the reaction was complete via TLC (2 hours). The solvent was removed in vacuo and the resulting crude product was then taken to the next step without further purification. The deprotected crude product was dissolved in pre-dried DMF. Then EDCI, HOBt, triethylamine, and molecular sieves were added, and the solution was stirred in an ice bath for 1 hour. After 1 hour, MOM-protected IOX1 was added to the reaction mixture and was stirred overnight at room temperature. Once complete via TLC, the reaction was filtered over Celite, the solvent was removed in vacuo and column chromatography was run to separate and purify the product (DCM/MeOH, 30:1 with 0.1% NH4OH). The product was then dissolved in methanol (1 mL), followed by the addition of HCl solution in 1,4-dioxane (4 M). The mixture was stirred at room temperature overnight and then filtered. The target compound was obtained as a yellow solid.
Biological assays
000 cells per well density and were treated with the compounds upon attachment to the plate. The Wnt pathway was activated in the cells by treating with 20 mM lithium chloride 4 h post-treatment with the compounds. The cell lysates were collected 16 hours post-treatment with the compounds and the luciferase activity of the total cell lysate was measured using the Bright-Glo luciferase assay system (Promega). For each lysate, the obtained activity was normalized against the total protein concentration.
Abbreviations used
| ASCL2 | Achaete-scute family BHLH transcription factor 2 |
| ALDH | Aldehyde dehydrogenase |
| ATCC | American type culture collection |
| AXIN2 | Axis inhibition protein 2 |
| CCK8 | Cell counting kit 8 |
| CCND1 | Cyclin D1 |
| CSC | Cancer stem cell |
| CRBN | Cereblon |
| CRC | Colorectal cancer |
| DEAB | N,N-Diethylaminobenzaldehyde |
| DIPEA | N,N-Diisopropylethylamine |
| DKK1 | Dickkopf-1 |
| DMF | Dimethylformamide |
| DMEM | Dulbecco's modified Eagle medium |
| DMSO | Dimethyl sulfoxide |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| ED50 | Dose effective in 50% of test subjects |
| EGF | Epidermal growth factor |
| FACS | Fluorescence activated cell sorting |
| FBS | Fetal serum albumin |
| FCC | Flash column chromatography |
| FGF | Fibroblast growth factor |
| GSK3 | Glycogen synthase kinase 3 |
| HOBt | Hydroxybenzotriazole |
| HPLC | High performance liquid chromatography |
| HRMS | High-resolution mass spectrometry |
| IHC | Immunohistochemistry |
| KDM3 | Histone lysine demethylase 3 |
| KDM4 | Histone lysine demethylase 4 |
| LGR5 | Leucine rich repeat containing G protein coupled receptor 5 |
| MEBM | Mammary epithelial cell basal medium |
| MOM | Methoxymethyl |
| NMR | Nuclear magnetic resonance |
| PBS | Phosphate buffered saline |
| PEG | Polyethylene glycol |
| PROTAC | Proteolysis targeting chimera |
| PVDF | Polyvinylidene difluoride |
| qPCR | Quantitative polymerase chain reaction |
| RNF43 | Ring finger protein 43 |
| RT-qPCR | Reverse transcription quantitative polymerase chain reaction |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| THF | Tetrahydrofuran |
| TLC | Thin layer chromatography |
| TMS | Tetramethylsilane |
| ZNRF3 | Zinc and ring finger 3 |
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
The study was conceived and designed by J. L. and Y. Z. J. L. and Y. Z. developed the methodology. S. U. Z., P. P. P., H. M., and R. G. H. acquired data. S. U. Z., P. P. P., H. M., and R. G. H. analyzed and interpreted the data. The manuscript was written by S. U. Z. and P. P. P. with input from J. L. and Y. Z. J. L. and Y. Z. supervised the study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partially supported by an NIH/NIDCR grants (R03DE026822, R21DE033842, and R01DE033712) to J. L., NIH/NCATS grant (sub-award of KL2TR002648 from NIH/NCATS to VCU's CTSA) to J. L., VCU Presidential Research Quest Fund (J. L. and Y. Z.), and VCU Breakthroughs Fund (J. L.).References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- P. Rawla, T. Sunkara and A. Barsouk, Przegl. Gastroenterol., 2019, 14, 89–103 Search PubMed.
- F. A. Haggar and R. P. Boushey, Clin. Colon Rectal Surg., 2009, 22, 191–197 CrossRef PubMed.
- E. R. Fearon, Annu. Rev. Phytopathol., 2011, 6, 479–507 CrossRef.
- The Cancer Genome Atlas Network, Nature, 2012, 487, 330–337 CrossRef.
- M. Bienz and H. Clevers, Cell, 2000, 103, 311–320 CrossRef.
- R. Jackstadt, M. C. Hodder and O. J. Sansom, Annu. Rev. Cancer Biol., 2020, 4, 177–196 CrossRef.
- F. de Sousa e Melo, S. Colak, J. Buikhuisen, J. Koster, K. Cameron, J. H. de Jong, J. B. Tuynman, P. R. Prasetyanti, E. Fessler, S. P. van den Bergh, H. Rodermond, E. Dekker, C. M. van der Loos, S. T. Pals, M. J. van de Vijver, R. Versteeg, D. J. Richel, L. Vermeulen and J. P. Medema, Cell Stem Cell, 2011, 9, 476–485 CrossRef.
- A. G. Schepers, H. J. Snippert, D. E. Stange, M. van den Born, J. H. van Es, M. van de Wetering and H. Clevers, Science, 2012, 337, 730–735 CrossRef CAS.
- F. de Sousa e Melo, A. V. Kurtova, J. M. Harnoss, N. Kljavin, J. D. Hoeck, J. Hung, J. E. Anderson, E. E. Storm, Z. Modrusan, H. Koeppen, G. J. P. Dijkgraaf, R. Piskol and F. J. de Sauvage, Nature, 2017, 543, 676–680 CrossRef CAS.
- N. Takahashi, K. Yamaguchi, T. Ikenoue, T. Fujii and Y. Furukawa, PLoS One, 2014, 9, e86582 CrossRef.
- M. Shimokawa, Y. Ohta, S. Nishikori, M. Matano, A. Takano, M. Fujii, S. Date, S. Sugimoto, T. Kanai and T. Sato, Nature, 2017, 545, 187–192 CrossRef CAS PubMed.
- L. E. Dow, K. P. O'Rourke, J. Simon, D. F. Tschaharganeh, J. H. van Es, H. Clevers and S. W. Lowe, Cell, 2015, 161, 1539–1552 CrossRef CAS.
- M. Sawa, M. Masuda and T. Yamada, Expert Opin. Ther. Targets, 2016, 20, 419–429 CrossRef CAS PubMed.
- J. Li, B. Yu, P. Deng, Y. Cheng, Y. Yu, K. Kevork, S. Ramadoss, X. Ding, X. Li and C.-Y. Wang, Nat. Commun., 2017, 8, 15146 CrossRef PubMed.
- R. G. Hoyle, H. Wang, Y. Cen, Y. Zhang and J. Li, Mol. Cancer Ther., 2021, 20, 191–202 CrossRef PubMed.
- R. Schiller, G. Scozzafava, A. Tumber, J. R. Wickens, J. T. Bush, G. Rai, C. Lejeune, H. Choi, T.-L. Yeh, M. C. Chan, B. T. Mott, J. S. O. McCullagh, D. J. Maloney, C. J. Schofield and A. Kawamura, ChemMedChem, 2014, 9, 566–571 CrossRef PubMed.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef.
- X. Wang, Z.-L. Qin, N. Li, M.-Q. Jia, Q.-G. Liu, Y.-R. Bai, J. Song, S. Yuan and S.-Y. Zhang, Eur. J. Med. Chem., 2024, 267, 116166 CrossRef PubMed.
- A. Mares, A. H. Miah, I. E. D. Smith, M. Rackham, A. R. Thawani, J. Cryan, P. A. Haile, B. J. Votta, A. M. Beal, C. Capriotti, M. A. Reilly, D. T. Fisher, N. Zinn, M. Bantscheff, T. T. MacDonald, A. Vossenkamper, P. Dace, I. Churcher, A. B. Benowitz, G. Watt, J. Denyer, P. Scott-Stevens and J. D. Harling, Commun. Biol., 2020, 3, 1–13 CrossRef PubMed.
- B. E. Smith, S. L. Wang, S. Jaime-Figueroa, A. Harbin, J. Wang, B. D. Hamman and C. M. Crews, Nat. Commun., 2019, 10, 131 CrossRef.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2018, 25, 78–87.e5 CrossRef PubMed.
- C. Donoghue, M. Cubillos-Rojas, N. Gutierrez-Prat, C. Sanchez-Zarzalejo, X. Verdaguer, A. Riera and A. R. Nebreda, Eur. J. Med. Chem., 2020, 201, 112451 CrossRef.
- S.-L. Paiva and C. M. Crews, Curr. Opin. Chem. Biol., 2019, 50, 111–119 CrossRef PubMed.
- S.-M. Qi, J. Dong, Z.-Y. Xu, X.-D. Cheng, W.-D. Zhang and J.-J. Qin, Front. Pharmacol., 2021, 12, 692574 CrossRef.
- P. M. Cromm and C. M. Crews, Cell Chem. Biol., 2017, 24, 1181–1190 CrossRef.
- K. Cyrus, M. Wehenkel, E.-Y. Choi, H.-J. Han, H. Lee, H. Swanson and K.-B. Kim, Mol. BioSyst., 2011, 7, 359–364 RSC.
- R. I. Troup, C. Fallan and M. G. J. Baud, Explor. Targeted Anti-Tumor Ther., 2020, 1, 273–312 Search PubMed.
- M. Tanaka, J. M. Roberts, H.-S. Seo, A. Souza, J. Paulk, T. G. Scott, S. L. DeAngelo, S. Dhe-Paganon and J. E. Bradner, Nat. Chem. Biol., 2016, 12, 1089–1096 CrossRef PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K.-H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef.
- M. Vollmar, C. Johansson, C. Gileadi, S. Goubin, A. Szykowska, T. Krojer, L. Crawley, F. von Delft, C. H. Arrowsmith, C. Bountra, A. Edwards and U. Oppermann, Crystal structure of JmjC domain of human histone 3 Lysine-specific demethylase 3B (KDM3B), PDB crystal structure, 2013, DOI:10.2210/pdb4C8D/pdb.
- R. P. Nowak, S. L. DeAngelo, D. Buckley, Z. He, K. A. Donovan, J. An, N. Safaee, M. P. Jedrychowski, C. M. Ponthier, M. Ishoey, T. Zhang, J. D. Mancias, N. S. Gray, J. E. Bradner and E. S. Fischer, Nat. Chem. Biol., 2018, 14, 706–714 CrossRef.
- G. Lu, R. E. Middleton, H. Sun, M. Naniong, C. J. Ott, C. S. Mitsiades, K.-K. Wong, J. E. Bradner and W. G. Kaelin, Science, 2014, 343, 305–309 CrossRef PubMed.
- J. Krönke, N. D. Udeshi, A. Narla, P. Grauman, S. N. Hurst, M. McConkey, T. Svinkina, D. Heckl, E. Comer, X. Li, C. Ciarlo, E. Hartman, N. Munshi, M. Schenone, S. L. Schreiber, S. A. Carr and B. L. Ebert, Science, 2014, 343, 301–305 CrossRef PubMed.
- L. B. Snyder, J. J. Flanagan, Y. Qian, S. M. Gough, M. Andreoli, M. Bookbinder, G. Cadelina, J. Bradley, E. Rousseau, J. Chandler, R. Willard, J. Pizzano, C. M. Crews, A. P. Crew, J. Houston, M. D. Moore, R. Peck and I. Taylor, Cancer Res., 2021, 81, 44 CrossRef.
- L. B. Snyder, T. K. Neklesa, X. Chen, H. Dong, C. Ferraro, D. A. Gordon, J. Macaluso, J. Pizzano, J. Wang, R. R. Willard, N. Vitale, R. Peck, M. D. Moore, C. M. Crews, J. Houston, A. P. Crew and I. Taylor, Cancer Res., 2021, 81, 43 CrossRef.
- P. P. Chamberlain and B. E. Cathers, Drug Discovery Today: Technol., 2019, 31, 29–34 CrossRef PubMed.
- M. E. Matyskiela, W. Zhang, H.-W. Man, G. Muller, G. Khambatta, F. Baculi, M. Hickman, L. LeBrun, B. Pagarigan, G. Carmel, C.-C. Lu, G. Lu, M. Riley, Y. Satoh, P. Schafer, T. O. Daniel, J. Carmichael, B. E. Cathers and P. P. Chamberlain, J. Med. Chem., 2018, 61, 535–542 CrossRef PubMed.
- M. Pettersson and C. M. Crews, Drug Discovery Today: Technol., 2019, 31, 15–27 CrossRef.
- X. Li, W. Pu, Q. Zheng, M. Ai, S. Chen and Y. Peng, Mol. Cancer, 2022, 21, 99 CrossRef PubMed.
- S. B. Hatch, C. Yapp, R. C. Montenegro, P. Savitsky, V. Gamble, A. Tumber, G. F. Ruda, V. Bavetsias, O. Fedorov, B. Atrash, F. Raynaud, R. Lanigan, L. Carmichael, K. Tomlin, R. Burke, S. M. Westaway, J. A. Brown, R. K. Prinjha, E. D. Martinez, U. Oppermann, C. J. Schofield, C. Bountra, A. Kawamura, J. Blagg, P. E. Brennan, O. Rossanese and S. Müller, Epigenet. Chromatin, 2017, 10, 9 CrossRef.
- F. Esteves, J. Rueff and M. Kranendonk, J. Xenobiot., 2021, 11, 94–114 CrossRef CAS PubMed.
- M. Zhao, J. Ma, M. Li, Y. Zhang, B. Jiang, X. Zhao, C. Huai, L. Shen, N. Zhang, L. He and S. Qin, Int. J. Mol. Sci., 2021, 22, 12808 CrossRef CAS PubMed.
- L. Di and R. S. Obach, AAPS J., 2015, 17, 352–357 CrossRef CAS.
- J. B. Houston, Biochem. Pharmacol., 1994, 47, 1469–1479 CrossRef CAS PubMed.
- A.-K. Sohlenius-Sternbeck, L. Afzelius, P. Prusis, J. Neelissen, J. Hoogstraate, J. Johansson, E. Floby, A. Bengtsson, O. Gissberg, J. Sternbeck and C. Petersson, Xenobiotica, 2010, 40, 637–649 CrossRef CAS PubMed.
- A. Garrido, A. Lepailleur, S. M. Mignani, P. Dallemagne and C. Rochais, Eur. J. Med. Chem., 2020, 195, 112290 CrossRef CAS.
- H. Yu, B. Zou, X. Wang and M. Li, Acta Pharmacol. Sin., 2016, 37, 111–123 CrossRef CAS PubMed.
- S. J. Richardson, A. Bai, A. A. Kulkarni and M. F. Moghaddam, Drug Metab. Lett., 2016, 10, 83–90 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00122b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |