
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral hydroxymethyl-1H,3H-pyrrolo[1,2-c]thiazoles: the search for selective p53-activating agents for colorectal cancer therapy†
Mees M.
Hendrikx
a,
Adelino M. R.
Pereira
 a,
Ana B.
Pereira
a,
Carla S. C.
Carvalho
b,
João L. P.
Ribeiro
a,
Maria I. L.
Soares
a,
Lucília
Saraiva
*b and
Teresa M. V. D.
Pinho e Melo
*a
a,
Ana B.
Pereira
a,
Carla S. C.
Carvalho
b,
João L. P.
Ribeiro
a,
Maria I. L.
Soares
a,
Lucília
Saraiva
*b and
Teresa M. V. D.
Pinho e Melo
*a
aUniversity of Coimbra, Coimbra Chemistry Centre – Institute of Molecular Sciences and Department of Chemistry, 3004-535 Coimbra, Portugal. E-mail: tmelo@ci.uc.pt
bLAQV/REQUIMTE, Laboratório de Microbiologia, Departamento de Ciências Biológicas, Faculdade de Farmácia, Universidade do Porto, Porto, Portugal
First published on 12th April 2024
Abstract
MANIO is an efficient p53-activating anticancer agent with remarkable selectivity to the p53 pathway and promising antitumor activity against colorectal cancer (CRC). Herein, a library of novel MANIO derivatives, including hydroxymethyl- and bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazoles, was synthesized by rational structural modulation. The antiproliferative activity of twenty derivatives was evaluated in a panel of human CRC cells with different p53 status. From this library, five compounds with R- and S-configuration and with aromatic or heteroaromatic groups at position 3, including the enantiomer of MANIO, were identified as selective towards p53-expressing cancer cells. On the other hand, two compounds with S-configuration, 6-hydroxymethyl- and 7-hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazoles, showed high cytotoxicity against WTp53-expressing HCT116 colon cells but, unlike MANIO, exhibited p53-independent inhibitory activity in CRC. The results described provide relevant structural and pharmacophoric data for the design of new p53-activating agents for precision therapy of CRC or other p53-related cancers harboring both wild-type or mutated p53 forms.
Introduction
Cancer is currently a major public health concern, with both incidence and mortality rates expected to increase in the coming decades.1,2 Among the various types of cancer, colorectal cancer (CRC) is the third most common cancer worldwide and the second leading cause of cancer-related deaths.3,4 The high mortality rates, often associated with treatment resistance and metastasization, highlight the limited effectiveness of existing therapies in treating CRC.4 Progress in understanding the development of CRC has revealed that p53 dysfunction is a key event in both localized and advanced cases of CRC. Impairment of the p53 pathway, either by TP53 mutation or inhibition of p53 by its negative regulators, is a significant event in both localized and advanced CRC. Thus, restoration of p53 activity has emerged as one of the most attractive anticancer therapeutic strategies.Recently, our research team disclosed a new p53-activating anticancer drug, (3S)-6,7-bis(hydroxymethyl)-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (MANIO) (Fig. 1).5 MANIO represents a privileged anticancer drug compared to other currently available p53-activating agents regarding its antitumor activity in several cancer types, including CRC. MANIO showed a remarkable selectivity to the p53 pathway, activating wild-type (WT)p53 and restoring WT-like function to mutant (mut)p53 in human cancer cells. The half maximal inhibitory concentration (IC50) value of MANIO in WTp53-expressing HCT116 colon cells, HCT116 p53+/+ (0.97 μM), was approximately 50-fold lower than that obtained in HCT116 p53−/−cells (48.25 μM), indicating a significant p53-dependent growth inhibitory effect. MANIO has been shown to directly interact with the p53 DNA-binding domain (DBD), leading to activation of the p53 pathway. In fact, MANIO works as a bridging molecule between p53 and DNA. Ramos et al. have demonstrated that MANIO binds to a pocket formed between one dimer of the WTp53 DBD protein and the minor groove of the DNA molecule.5 MANIO interacts with the WTp53 DBD protein backbone through hydrogen bonds between its hydroxyl groups and the amide backbone groups of methionine 243 of chain A (M243A) and methionine 243 of chain B (M243B) (each belong to a different monomer). The remaining interactions of MANIO are stacking interactions made with the bases of DNA (DG8, DG12). In the specific case of mutp53 R248W, a highly frequent p53 mutation, in most clusters, the ligand makes a hydrogen bond between one of its hydroxyl oxygen atoms and the W248 side-chain nitrogen. The ligand phenyl group is close to the DNA bases adenine 6 and thymine 7. These interactions may eventually compensate for the loss of direct contacts between lysine 248 residue and the DNA. The latter interaction results in increased p53 stability, enhanced DNA-binding capacity and increased transcriptional activity. MANIO synergizes with conventional chemotherapeutics (e.g., doxorubicin (DOXO), cisplatin (CISP) and 5-fluorouracil (5-FU)) in patient-derived and immortalized CRC cells expressing WTp53 or mutp53. In addition, MANIO demonstrated in vivo p53-dependent anti-tumor activity in xenograft mouse models of CRC with no adverse side effects. It also demonstrated favorable drug-likeness and pharmacokinetic (PK) properties for a clinical candidate.5
 | ||
| Fig. 1 Chemical structure and key features of (3S)-6,7-bis(hydroxymethyl)-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (MANIO) (1). | ||
In this study, structural modulation of MANIO, a lead p53-activating anticancer molecule, was performed with two main goals: to synthesize new bioactive 1H,3H-pyrrolo[1,2-c]thiazoles and to gain in-depth knowledge of structure–activity relationships (SAR).
Results and discussion
Synthesis of new MANIO-like derivatives
The structural modulation strategy focused on structural changes of MANIO at position 3 of the 1H,3H-pyrrolo[1,2-c]thiazole core, namely the introduction of new aromatic, heteroaromatic, and alkyl substituents and the modification of the absolute configuration. These changes included the introduction of p-fluorophenyl, p-(trifluoromethoxy)phenyl, p-methoxyphenyl, bulky naphthyl and quinolinyl groups, and alkyl groups such as benzyl and methyl. Modifications in other positions of the bicyclic system involved the removal of one hydroxymethyl group or the oxidation of the sulphur atom.Chiral 1H,3H-pyrrolo[1,2-c]thiazoles with R-configuration 3 were synthesized in moderate yields (35–69%) according to a known synthetic procedure (Scheme 1).6 Thiazolidines 2 were obtained from the reaction of L-cysteine with the corresponding aromatic aldehydes as mixtures of 2R,4R- and 2S,4R-diastereoisomers. The synthetic sequence proceeds via 1,3-dipolar cycloaddition of dimethyl acetylenedicarboxylate (DMAD) with the bicyclic münchnone generated in situ from thiazolidine 2, followed by elimination of carbon dioxide. The acylation of the diastereoisomeric mixtures of thiazolidines with acetic anhydride or acyl chlorides can lead the to the selective synthesis of N-acetyl-thiazolidine-4-carboxylic acids as pure stereoisomers with 2S,4R- or 2R,4R-configuration, depending on the reaction conditions.7–9 Thiazolidine 2 was heated in a solution of acetic anhydride in the presence of the dipolarophile. Under these reaction conditions the N-acylation occurs in situ giving selectively N-acetyl thiazolidine-4-carboxylic acids with 2R,4R-configuration. Thus, starting from thiazolidine-4-carboxylic acids 2 as a mixture of 2R,4R- and 2S,4R-diastereoisomers, chiral 1H-pyrrolo[1,2-c]thiazoles 3 were obtained as single enantiomer with R-configuration. In this process the chirality at C-4 of the thiazolidine is lost and the chirality at C-2 (C-3 in the product) is retained. The reduction of 3 was carried out with lithium aluminium hydride to afford the target 6,7-bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazoles 4 in moderate to good yields (34–75% for alcohols 4a, 4d and 4e). The formation of alcohols 4b and 4c bearing an OCF3 or CF3 group in the para position of the phenyl group was confirmed by proton NMR spectroscopy. However, we decided not to proceed with the biological evaluation studies due to the lack of stability of these compounds.
 | ||
| Scheme 1 Synthesis of chiral 6,7-bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazoles 4. | ||
Chiral 6,7-bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazole 7 with S-configuration was also synthesized (Scheme 2). In order to obtain N-acetyl-thiazolidine-4-carboxylic acid with (2S,4R) stereochemistry we used an experimental procedure previously described in the literature.10 The reaction of thiazolidine 2f with acetyl chloride in dry pyridine carried out at 0 °C allowed the selective formation of (2S,4R)-N-acetyl-thiazolidine-4-carboxylic acid 5. Reaction of 5 with DMAD afforded the 1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylate 6 with S-configuration in 69% yield. Further reduction with lithium aluminium hydride afforded 6,7-bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazole 7 in good yield (74%).
 | ||
| Scheme 2 Synthesis of (3S)-6,7-bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazole 7. | ||
(3S)-7-Hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (11) and (3S)-6-hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (12), which differ in the position of the hydroxymethyl group, were also synthesized (Scheme 3). Chiral 1H,3H-pyrrolo[1,2-c]thiazole carboxylates 9 and 10 were synthesized in moderate yield (67%) as a regioisomeric mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) following a known synthetic procedure.6 Diastereoisomerically pure (2S,4R)-N-acetyl-thiazolidine-4-carboxylic acid 810 was used to generate the corresponding bicyclic münchnone in situ, which reacted with methyl propiolate via 1,3-dipolar cycloaddition. The mixture of products could not be separated by chromatography. However, 1H,3H-pyrrolo[1,2-c]thiazole-6-carboxylate 10 was isolated in pure form by selective crystallization from diethyl ether/hexane. The structural assignment of compound 10, made by comparison with the characterization data of the corresponding R-enantiomer,11 which differed only in the optical rotation, allowed us to conclude that we were in the presence of a 1H,3H-pyrrolo[1,2-c]thiazole-6-carboxylate with S-configuration.
:1) following a known synthetic procedure.6 Diastereoisomerically pure (2S,4R)-N-acetyl-thiazolidine-4-carboxylic acid 810 was used to generate the corresponding bicyclic münchnone in situ, which reacted with methyl propiolate via 1,3-dipolar cycloaddition. The mixture of products could not be separated by chromatography. However, 1H,3H-pyrrolo[1,2-c]thiazole-6-carboxylate 10 was isolated in pure form by selective crystallization from diethyl ether/hexane. The structural assignment of compound 10, made by comparison with the characterization data of the corresponding R-enantiomer,11 which differed only in the optical rotation, allowed us to conclude that we were in the presence of a 1H,3H-pyrrolo[1,2-c]thiazole-6-carboxylate with S-configuration.
 | ||
| Scheme 3 Synthesis of hydroxymethyl-1H,3H-pyrrolo[1,2-c]thiazoles 11 and 12. ayield over two steps. | ||
The reduction of (3S)-1H,3H-pyrrolo[1,2-c]thiazole-6-carboxylate 10 was carried out with lithium aluminium hydride to afford the corresponding alcohol 12 in 55% yield. A mixture of the regioisomeric alcohols 7-hydroxymethyl-1H,3H-pyrrolo[1,2-c]thiazole 11 and 6-hydroxymethyl-1H,3H-pyrrolo[1,2-c]thiazole 12 was obtained by reduction of the crude mixture of the corresponding carboxylate derivatives 9 and 10 with lithium aluminium hydride. Compounds 11 and 12 were successfully separated by column chromatography. This two-step synthetic approach afforded alcohols 11 and 12 in 10.5% and 16% overall yields, respectively.
The sulfone functional group is known to impart polarity to molecules, reducing their lipophilicity and improving their solubility in aqueous media, and the cyclic sulfone nucleus in particular is commonly found in biologically active compounds.12 Thus, the sulfone of the enantiomer of MANIO was prepared starting from dimethyl (3R)-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylate (13), whose synthesis was previously described by our group.13 In this case, the reduction was carried out with a sodium borohydride-methanol system instead of LiAlH4, as the latter was found to reduce both the ester and the sulfone groups. The reaction of sulfone 13 with the NaBH4-methanol system in refluxing THF for 24 h allowed the synthesis of the target sulfone 15 in 52% yield, which was obtained together with the mono-reduced derivative, compound 14, in 31% yield (Scheme 4). Attempts to favor the exclusive formation of compound 15, by prolonging the reaction time, were unsuccessful.
 | ||
| Scheme 4 Reduction of dimethyl (3R)-5-methyl-2,2-dioxo-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylate (13). | ||
6,7-Bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazoles 17 bearing methyl or benzyl groups at position 3 were also prepared from the corresponding 1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylates 1614,15 as outlined in Scheme 5. The target chiral molecules 17 were obtained in yields ranging from 57 to 75%.
 | ||
| Scheme 5 Synthesis of chiral 3-methyl- and 3-benzyl-6,7-bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazoles. | ||
Antiproliferative effect on CRC cells of MANIO-like derivatives
A small library of hydroxymethyl- and bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazoles and a bis(hydroxymethyl)-1H,3H-pyrazolo[1,5-c]thiazole were screened for p53-dependent anticancer activity. These included 1H,3H-pyrrolo[1,2-c]thiazoles, the synthesis of which has been described above, as well as hydroxymethyl- and bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazoles 18 and 6,7-bis(hydroxymethyl)-1H,3H-pyrazolo[1,5-c]thiazole 19 previously described by our research group (Fig. 2).10,11,16 | ||
| Fig. 2 Chemical structures of previously synthesized 1H,3H-pyrrolo[1,2-c]thiazoles and 1H,3H-pyrazolo[1,5-c]thiazole.10,11,16 | ||
In vitro studies regarding the antiproliferative activity of compounds 18 and 19 have been carried out in a panel of human colorectal cancer cells with different p53 status, WTp53-expressing HCT116 colon cells (HCT116 p53+/+) and p53 null isogenic derivative (HCT116 p53−/−). The comparison of the activity of the compounds was made by analyzing the corresponding IC50 values (Table 1). 1H,3H-Pyrrolo[1,2-c]thiazole 18a, the enantiomer of MANIO, was characterized by its high selectivity for HCT116 colon cells expressing p53 HCT116 p53+/+ with IC50 = 5.11 μM, whereas the IC50 value obtained for the p53 null isogenic derivative (HCT116 p53−/−) was higher than 50 μM. Nevertheless, the IC50 value for 18a against HCT116 p53+/+ was 5-fold higher than that obtained for MANIO in the same cells. Compound 18b, containing a hydroxymethyl group at C-6, and 6,7-bis(hydroxymethyl) derivative 18g, containing a p-methoxyphenyl group at position 3, both with R-configuration, also showed a marked selectivity to p53-expressing cancer cells, with IC50 values of 6.68 and 4.61 μM, respectively, compared to p53-null cells (>50 μM). Among the other compounds tested, none of them showed relevant selectivity for the WTp53-expressing HCT116 colon cells. Compound 18c, which differs from 18b only in the position of the hydroxymethyl group, shows a marked loss of selectivity for HCT116 colon cells expressing p53 HCT116 p53+/+. A similar loss of selectivity was observed for compound 18d, which differs from 18b only in having no substituents at C-3. Compound 18e, without substituents at the 5-position, showed a loss of selectivity of the same order of magnitude compared to compound 18a. Although the introduction of a p-methoxyphenyl group at position 3 resulted in good performance, as observed for compound 18g, the introduction of a bulkier trimethoxyphenyl group (compound 18f) resulted in a complete loss of activity against HCT116 colon cells. On the other hand, compound 18h, with a p-hydroxyphenyl group at C-3, showed lower selectivity and an IC50 against HCT116 p53+/+ 3-fold higher than that observed for 18a. Finally, compound 19 with a 1H,3H-pyrazolo[1,5-c]thiazole core showed no activity against HCT116 colon cells.
| Compound | IC50 (μM) | |
|---|---|---|
| HCT116 p53+/+ | HCT116 p53−/− | |
| 1 (MANIO) | 0.97 ± 0.04 | 48.25 ± 1.97 |
| 18a (MANIO enantiomer) | 5.11 ± 0.42 | 51.83 ± 2.73 |
| 18b | 6.68 ± 1.22 | >50 |
| 18c | 29.34 ± 2.08 | >50 |
| 18d | 28.32 ± 3.51 | >50 |
| 18e | 20.30 ± 1.53 | >50 |
| 18f | >50 | >50 |
| 18g | 4.61 ± 0.59 | >50 |
| 18h | 16.65 ± 2.31 | >50 |
| 19 | >50 | >50 |
SAR data allowed the identification of potential pharmacophores in the molecule and the definition of new MANIO-like derivatives to be studied (Fig. 3). It has been shown that the presence of an aryl group at C-3, a methyl group at C-5 and the presence of an hydroxymethyl group at position 6 may be crucial to ensure good antiproliferative activity in HCT116 colon cells expressing WTp53. Thus, the design of new 1H,3H-pyrazolo[1,5-c]thiazoles involved the structural modulation at position 3 through the introduction of fluorine containing aromatic substituents, naphthyl or quinolinyl groups. Among the new synthesized compounds, enantiomers of the most promising compounds studied (compounds 18b and 18g) were also prepared, as well as the sulfone of the enantiomer of MANIO. To confirm the importance of an aryl group at C-3 in ensuring good anticancer activity, the replacement of this type of functional group by alkyl groups was also carried out, resulting in novel 3-alkylated-6,7-bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazole derivatives.
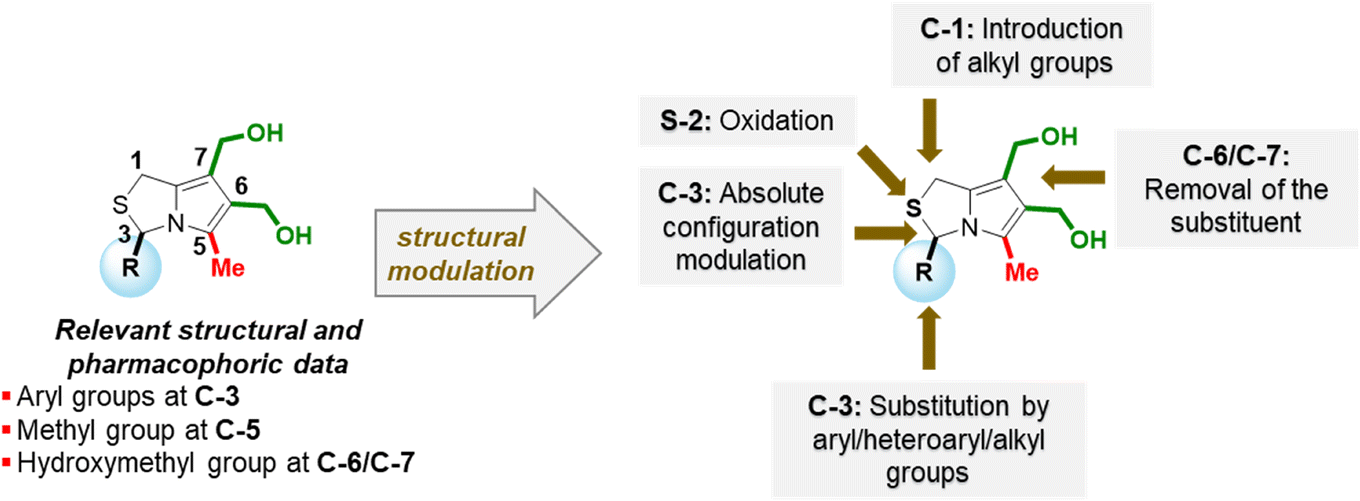 | ||
| Fig. 3 Structural modulation approaches based on SAR data. | ||
The antiproliferative activity of the newly synthesized 1H,3H-pyrrolo[1,2-c]thiazoles 4, 7, 11, 12, 15 and 17 as well as compound 20, whose synthesis was previously reported by our group,10 was evaluated in a panel of human colorectal cancer cells with different p53 status (Fig. 4, Table 2). (3R)-6,7-Bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazole 4a with a p-fluorophenyl substituent at position 3 showed good selectivity for HCT116 p53+/+ with an IC50 of 4.43 μM in contrast to the IC50 of 32.50 μM against HCT116 p53−/− cells. Compound 4e, containing a quinolinyl group at C-3, had a slightly higher IC50 value in the HCT116 p53+/+ cells (7.50 μM) than 4a, however it showed higher selectivity for the p53 pathway, with the IC50 value obtained in HCT116 p53−/− cells higher than 50 μM. Compound 15, the sulfone of the enantiomer of MANIO, showed higher IC50 values than MANIO enantiomer (compound 18a, Table 1), indicating that sulfur oxidation did not lead to an improvement of the anticancer activity. Compound 20, which differs from MANIO only by the presence of two methyl groups at position 1, showed moderate anticancer activity in HCT116 p53+/+ cells (IC50 = 24.34 μM) and no dependence on the p53 pathway (HCT116 p53−/−; IC50 = 22.15 μM).
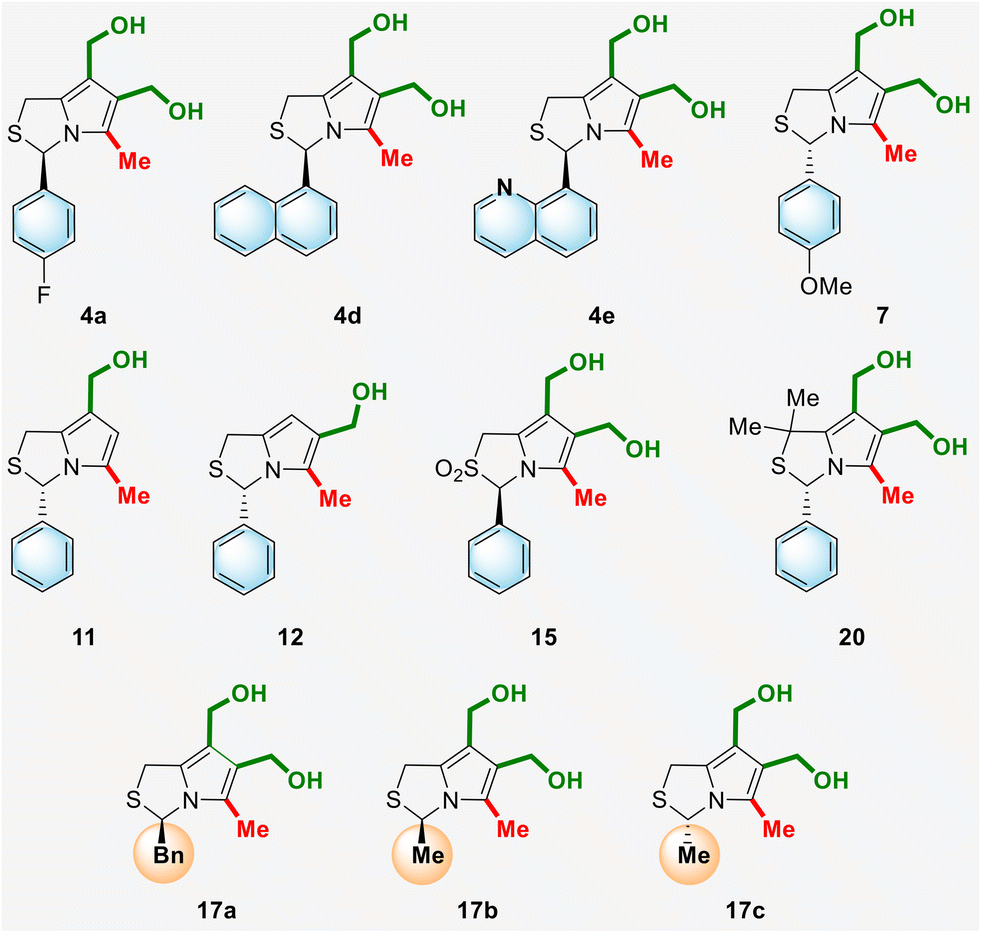 | ||
| Fig. 4 Chemical structures of new chiral hydroxymethyl- and bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazoles. | ||
| Compound | IC50 (μM) | |
|---|---|---|
| HCT116 p53+/+ | HCT116 p53−/− | |
| ND: not determined due to insolubility in the medium used to grow the cells. | ||
| 4a | 4.43 ± 2.13 | 32.50 ± 1.30 |
| 4d | ND | ND |
| 4e | 7.50 ± 1.10 | >50 |
| 7 | 1.47 ± 0.66 | 23.50 ± 2.50 |
| 11 | 0.14 ± 0.07 | 18.11 ± 1.62 |
| 12 | 0.99 ± 0.05 | 4.33 ± 1.10 |
| 15 | >15 | >50 |
| 17a | 16.27 ± 1.15 | 29.11 ± 1.00 |
| 17b | 33.28 ± 1.73 | 46.19 ± 2.31 |
| 17c | 31.40 ± 3.51 | 42.73 ± 3.61 |
| 20 | 24.34 ± 2.86 | 22.15 ± 4.17 |
3-Alkyl-1H,3H-pyrrolo[1,2-c]thiazoles 17 showed moderate p53 dependence, with IC50 values in HCT116 p53+/+ (16–33 μM) only about 10 μM lower than those in p53 null isogenic cells (29–46 μM). As mentioned above, MANIO interacts with the WTp53 DBD protein backbone through hydrogen bonds between its hydroxyl groups and the amide backbone groups of the methionine residues of two different monomers, and through stacking interactions made with the bases of the DNA.5 This is in agreement with the results obtained for the alkylated derivatives, in particular for compounds 17b and 17c, where these aromatic stacking interactions are not possible.
The IC50 value of compound 7 in HCT116 p53+/+ (1.47 μM) was 3-fold lower than that obtained for its enantiomer, compound 18g (see Table 1). However, in contrast to its enantiomer, it showed lower selectivity for HCT116 p53−/− cells (IC50 = 23.50 μM).
Mono-hydroxymethyl derivatives, regioisomers 11 and 12, showed excellent activity for HCT116 p53+/+ cells, with IC50 values of 0.11 and 0.99 μM, respectively. Remarkably, the IC50 value of 11 is about 200-fold lower than that obtained for its enantiomer (compound 18c, Table 1) but 7-fold lower than that obtained for MANIO. However, compounds 11 and 12 showed a low p53-dependent anti-proliferative effect with IC50 values of 18.11 and 4.33 μM for HCT116 p53−/− cells, respectively.
Antiproliferative effect on MDA-MB-231, PANC-1 and A375 cells of MANIO-like derivatives
The antiproliferative activity of the most active compounds against HCT116 colon cells (HCT116 p53+/+) was then evaluated in a panel of different human cancer cell lines, triple-negative breast cancer (MDA-MB-231), pancreatic adenocarcinoma (PANC-1) and melanoma (A375) (Table 3). However, none of the compounds showed relevant activity in any of the cell lines tested. Of note is the moderate activity of MANIO, its enantiomer and compound 12 in the human melanoma cell line (IC50 < 15 μM).| Compound | IC50a (μM) | ||
|---|---|---|---|
| MDA-MB-231 | PANC-1 | A375 | |
| a Values correspond to the media ± standard deviation of 3 independent experiments. | |||
| 1 (MANIO) | >50 | 42.15 ± 2.32 | 13.20 ± 0.71 |
| 18a (MANIO enantiomer) | >50 | 33.10 ± 1.27 | 14.49 ± 1.85 |
| 18b | >50 | >50 | 39.50 ± 1.41 |
| 18g | >50 | >50 | >50 |
| 4a | >50 | 20.15 ± 2.76 | 36.14 ± 1.32 |
| 4e | >50 | 17.35 ± 2.48 | 24.35 ± 2.62 |
| 7 | 16.57 ± 1.86 | >50 | 22.80 ± 0.91 |
| 12 | 24.11 ± 2.09 | 35.00 ± 2.57 | 11.78 ± 1.84 |
| 11 | >50 | >50 | >50 |
Conclusion
Structural modulation of the lead compound MANIO was performed resulting in the synthesis of thirteen new 1H,3H-pyrrolo[1,2-c]thiazoles with R and S configuration. The antiproliferative activity of a library of twenty compounds was assessed in a panel of human colorectal cancer cells with different p53 status, providing relevant structural and pharmacophoric information.Among the compounds tested, five, including the enantiomer of MANIO, all with aromatic or heteroaromatic substituents at C-3, showed selectivity towards p53-expressing cancer cells, with IC50 values in cancer cells expressing WTp53 (HCT116 p53+/+) ranging between 1.47 and 7.50 μM and IC50 > 23.50 μM in p53-null isogenic derivatives (HCT116 p53−/−). It was observed that the p-methoxyphenyl-substituted derivative with S-configuration showed significantly higher activity against HCT116 p53+/+ cells than its enantiomer, 1.47 vs. 4.61 μM.
Mono-hydroxymethyl derivatives with S-configuration stood out for their high antiproliferative activity in colon cancer cells expressing WTp53, with IC50 < 1 μM, higher activity than the corresponding enantiomers and MANIO, but no p53-dependent growth-inhibitory effect.
Collectively, these results indicate that the presence of aromatic substituents at C-3 and a methyl group at C-5 are essential to ensure effective p53-dependent growth inhibitory activity. Furthermore, the absolute configuration at the chiral center was shown to play an important role in the antiproliferative activity in WTp53-expressing colon cancer cells. It was also observed that the position of the hydroxymethyl group in mono-hydroxymethyl derivatives affects also the inhibitory activity. Interestingly, while for 1H,3H-pyrrolo[1,2-c]thiazoles with R configuration the 6-hydroxymethyl derivative was significantly more active than the 7-hydroxymethyl derivative, in the case of 1H,3H-pyrrolo[1,2-c]thiazoles with S configuration, both 6-hydroxymethyl and 7-hydroxymethyl derivative show outstanding antiproliferative activity in WTp53-expressing HCT116 colon cells, but p53-independent inhibitory activity.
Overall, the present work represents a successful strategy of modulation and rational structural optimization of a lead molecule, which allowed to gather pertinent structural and pharmacophoric data for the design of new p53-activating agents for precision therapy of CRC or other cancers harboring WT or mutp53.
Experimental
Chemistry
Thin-layer chromatography (TLC) analyses were performed using precoated silica gel plates. Column chromatography was performed with silica gel 60 as the stationary phase. 1H Nuclear magnetic resonance (NMR) spectra (400 MHz), 13C NMR spectra (100 MHz) and 19F spectra (376 MHz) were recorded in CDCl3, CD3OD or hexadeuterated dimethylsulfoxide (DMSO-d6). Chemical shifts are expressed in parts per million (ppm) relatively to internal tetramethylsilane (TMS), and coupling constants (J) are in hertz. Infrared (IR) spectra were recorded in a Fourier transform spectrometer using either a KBr matrix or diamond attenuated total reflectance (ATR) mode. Elemental analyses were carried out with an Elemental Vario Micro Cube analyser. High-resolution mass spectra (HRMS) were obtained on a TOF VG Autospect M spectrometer with electron impact (EI), on a Thermo Orbitrap Q-Exactive Focus spectrometer with electrospray ionization (ESI) or on a Bruker MicroTOF (APCI-FIA-TOF). Melting points were determined in open glass capillaries and are uncorrected. Optical rotations were measured on an Optical Activity AA-5 electrical polarimeter.Thiazolidine-4-carboxylic acids 2a,82c,172d,182f,16N-acetyl-thiazolidine 8,10 1H,3H-pyrrolo[1,2-c]thiazoles 16,14,15 hydroxymethyl- and bis(hydroxymethyl)-1H,3H-pyrrolo[1,2-c]thiazoles 18,10,11,16 bis(hydroxymethyl)-1H,3H-pyrazolo[1,5-c]thiazole 19,10 and 1H,3H-pyrrolo[1,2-c]thiazole-2,2-dioxide 1313 were prepared as described in the literature.
2-(4-(Trifluoromethoxy)phenyl)-1,3-thiazolidine-4-carboxylic acid (2b). Obtained from 4-(trifluoromethoxy)benzaldehyde (5.51 g, 29.0 mmol) as described in the general procedure. Yield: 75% (6.39 g), white solid, mp 146.6–147.6 °C. The 1H NMR spectrum obtained at 25 °C showed the presence of the two diastereoisomers (2R,4R) and (2S,4R) (ratio 40
:60); 1H NMR (400 MHz, DMSO-d6): δ = 3.08–3.14 (2 × m, 2H), 3.32 and 3.38 (2 × dd, J = 10.2, 7.0 Hz and J = 10.0, 7.0 Hz, 2H), 3.92 and 4.18 (2 × dd, J = 8.8, 7.0 Hz and J = 6.8, 5.1 Hz, 2H), 5.56 and 5.74 (2 × s, 2H), 7.32 and 7.36 (2 × d, J = 8.1 Hz and J = 8.1 Hz, ArH, 4H), 7.56–7.58 and 7.66–7.68 (2 × m, ArH, 4H). 13C NMR (100 MHz, DMSO-d6): δ = 38.0 and 38.3, 64.8 and 65.6, 69.9 and 70.6, 120.0 (q,1JC–F = 254 Hz), 120.7 and 120.9, 128.8 and 129.4, 138.6 and 141.2, 147.5 and 148.0, 172.0 and 172.8. 19F NMR (376 MHz, DMSO-d6): δ = −56.79 and −56.78. IR (ATR): ν = 1572, 1509, 1437, 1381, 1251, 1208, 1163 cm−1. HRMS (ESI): calcd for C11H11NO3F3S [M + H]+ 294.0406; found 294.0401.
2-(Quinolin-8-yl)thiazolidine-4-carboxylic acid (2e). Obtained from quinoline-8-carbaldehyde (2.50 g, 15.9 mmol) as described in the general procedure. Yield: 95% (3.96 g), white solid, mp 129.8–131.1 °C. The 1H NMR spectrum obtained at 25 °C showed the presence of the two diastereoisomers (2R,4R) and (2S,4R) (ratio 50
:50); 1H NMR (400 MHz, CD3OD): δ = 3.44–3.49 (2 × m, 2H), 3.59–3.68 (2 × m, 2H), 4.32 and 4.57 (2 × pseudo-t, J = 7.6 Hz and J = 6.5 Hz, 2H), 6.52 and 6.72 (2 × s, 2H), 7.58–7.66 (m, ArH, 4H), 7.95–8.02 (m, ArH, 4H), 8.39–8.42 (m, ArH, 2H), 8.96–8.98 (m, ArH, 2H). 13C NMR (100 MHz, DMSO-d6): δ = 37.3, 38.4, 65.3, 65.8, 66.2, 67.9, 121.4, 121.6, 124.6, 126.2, 126.3, 127.1, 127.4, 127.9, 130.0, 128.2, 136.3, 136.7, 140.2, 144.9, 145.2, 149.2, 149.6, 172.4, 172.9. IR (ATR): ν = 1606, 547, 1500, 1397, 1359, 1314 cm−1. HRMS (ESI): calcd for C13H13N2O2S [M + H]+ 261.0692; found 261.0688.
(2S,4R)-3-Acetyl-2-(4-methoxyphenyl)thiazolidine-4-carboxylic acid (5). The compound was prepared according to a procedure described in the literature for the synthesis of (2S,4R)-3-acetyl-2-phenylthiazolidine-4-carboxylic acid.10 Yield: 61%, white solid, mp 148.4–149.9 °C (from ethanol). The 1H NMR spectrum obtained at 25 °C showed the presence of the two rotational isomers; 1H NMR (400 MHz, CD3OD): δ = 1.76 and 2.01 (2 × s, 3H), 3.15–3.47 (2 × m, 2H), 3.73 and 3.76 (2 × s, 3H), 5.10 and 5.32 (2 × d, J = 6.9 Hz and J = 6.0 Hz, 1H), 6.12 and 6.33 (2 × s, 1H), 6.83 and 6.92 (d, J = 8.7 Hz, 2H), 7.19 and 7.24 (d, J = 8.7 Hz, 2H). IR (ATR): ν = 1706, 1597, 1513, 1403, 1243, 1182, 1030 cm−1. HRMS (ESI): calcd for C13H16NO4S [M + H]+ 282.0795; found 282.0792. [α]20D = −270 (c 0.5, DCM).
Dimethyl (3R)-3-(4-fluorophenyl)-5-methyl-1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylate (3a). Obtained from thiazolidine 2a (2.02 g, 8.8 mmol) and DMAD (1.88 g, 13.2 mmol, 1.5 equiv.) as described in the general procedure. Reaction time: 12 h. Purification by column chromatography [hexane/EtOAc (1
:2), (1:1), then (2:1)] gave compound 3a as solid (2.30 g, 69%). Recrystallization from EtOAc/hexane gave the compound as a pale-yellow solid, mp 101.8–102.3 °C. 1H NMR (400 MHz, CDCl3): δ = 2.00 (s, 3H), 3.83 (s, 1H), 3.83 (s, 1H), 4.32 (d, J = 15.0 Hz, 1H), 4.47 (dd, J = 15.0, 1.6 Hz, 1H), 6.28 (d, J = 1.6 Hz, 1H), 7.03–7.08 (m, 4H). 13C NMR (100 MHz, CDCl3): δ = 11.4, 30.0, 51.4, 51.6, 64.4, 107.0, 116.3 (d,2JC–F = 22.9 Hz), 117.6, 127.6 (d,3JC–F = 8.4 Hz), 130.5, 136.0 (d,4JC–F = 3.2 Hz), 140.3, 162.8 (d,1JC–F = 247 Hz), 164.0, 165.2. 19F NMR (376 MHz, CDCl3): δ = −114.47. IR (ATR): ν = 2952, 1773, 1699, 1508, 1436, 1291, 1219, 1150, 1090 cm−1. HRMS (ESI): calcd for C17H17NO4FS [M + H]+ 350.0857; found 350.0853. [α]20D = +185 (c 1, CH2Cl2).
Dimethyl (3R)-5-methyl-3-(4-(trifluoromethoxy)phenyl)-1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylate (3b). Obtained from thiazolidine 2b (2.00 g, 6.8 mmol) and DMAD (1.45 g, 10.2 mmol, 1.5 equiv.) as described in the general procedure. Reaction time: 12 h. Purification by column chromatography [hexane/EtOAc (1
:1)] gave compound 3b as a colorless oil (1.85 g, 65%). 1H NMR (400 MHz, CDCl3): δ = 2.02 (s, 3H), 3.83 (s, 6H), 4.33 (d, J = 15.0 Hz, 1H), 4.47 (dd, J = 15.0, 1.6 Hz, 1H), 6.29 (d, J = 1.5 Hz, 1H), 7.06–7.12 (m, 2H), 7.20 (d, J = 8.2 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 11.5, 30.0, 51.5, 51.6, 64.1, 107.1, 116.5, 120.3 (d,1JC–F = 256 Hz), 121.7, 127.2, 130.5, 138.9, 140.3, 149.4 (d,3JC–F = 1.5 Hz), 163.9, 165.2. 19F NMR (376 MHz, CDCl3): δ = −57.88. IR (ATR): ν = 2952, 1702, 1509, 1439, 1252, 1208, 1155, 1092 cm−1. HRMS (ESI): calcd for C18H17NO5F3S [M + H]+ 416.0774; found 416.0770. [α]20D = +107 (c 0.5, CH2Cl2).
Dimethyl (3R)-5-methyl-3-(4-(trifluoromethyl)phenyl)-1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylate (3c). Obtained from thiazolidine 2c (2.00 g, 7.2 mmol) and DMAD (1.54 g, 10.8 mmol, 1.5 equiv.) as described in the general procedure. Reaction time: 4 h. Purification by column chromatography [hexane/EtOAc (3
:1)] gave compound 3c as an oil (1.44 g, 50%). Recrystallization from EtOAc/hexane gave the compound as white solid, mp 105.6–106.6 °C. 1H NMR (400 MHz, CDCl3): δ = 2.03 (s, 3H), 3.83 (s, 3H), 3.84 (s, 3H), 4.34 (d, J = 14.9 Hz, 1H), 4.47 (dd, J = 1.5 and 14.9, 1H), 6.32 (d, J = 1.2 Hz, 1H), 7.16 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 8.2 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 11.5, 30.1, 51.5, 51.6, 64.1, 107.3, 117.8, 123.7 (q,1JC–F = 272 Hz), 125.9, 126.4 (q,3JC–F = 3.7 Hz), 130.4, 131.2 (q,2JC–F = 32.7 Hz), 140.3, 144.2, 163.9, 165.1. 19F NMR (376 MHz, CDCl3): δ = −62.79. IR (ATR): ν = 2950, 1705, 1437, 1323, 1204, 1126, 1090, 1065 cm−1. HRMS (ESI): calcd for C18H17NO4F3S [M + H]+ 400.0825; found 400.0822. [α]20D = +180 (c 0.5, CH2Cl2).
Dimethyl (3R)-5-methyl-3-(naphthalen-1-yl)-1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylate (3d). Obtained from thiazolidine 2d (2.00 g, 7.7 mmol) and DMAD (1.64 g, 11.6 mmol, 1.5 equiv.) as described in the general procedure. Reaction time: 4 h. Purification by column chromatography [hexane/EtOAc (3
:1), then hexane/EtOAc (2:1)] gave compound 3d as a solid (1.03 g, 35%). Recrystallization from EtOAc/hexane gave the compound as pale-yellow solid, mp 192.9–194.8 °C. 1H NMR (400 MHz, CDCl3): δ = 2.08 (s, 3H), 3.85 (s, 6H), 4.36 (s, 2H), 6.51 (bs, 1H), 6.98 (s, 1H), 7.34 (pseudo-t, J = 7.7 Hz, 1H), 7.55–7.62 (m, 2H), 7.82 (d, J = 8.3 Hz, 1H), 7.88–7.93 (m, 2H). 13C NMR (100 MHz, CDCl3): δ = 11.5, 29.8, 51.4, 51.6, 61.1, 107.5, 117.2, 121.0, 122.1, 125.5, 126.4, 127.1, 129.3, 129.3, 131.1, 134.0, 135.4, 141.1, 164.0, 165.3. IR (ATR): ν = 2947, 1726, 1698, 1534, 1433, 1204, 1154, 1093 cm−1. HRMS (ESI): calcd for C21H20NO4S [M + H]+ 382.1108; found 382.1103. [α]20D = +396 (c 0.5, CH2Cl2).
Dimethyl (3R)-5-methyl-3-(quinolin-8-yl)-1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylate (3e). Obtained from thiazolidine 2e (2.01 g, 7.7 mmol) and DMAD (1.64 g, 11.6 mmol, 1.5 equiv.) as described in the general procedure. Reaction time: 4 h. Purification by column chromatography [hexane/EtOAc (1
:1)] gave compound 3e as a solid (1.35 g, 46%). Recrystallization from EtOAc/hexane gave the compound as white solid, mp 203.9–205.8 °C (with decomposition). 1H NMR (400 MHz, CDCl3): δ = 2.10 (s, 3H), 3.85 (s, 3H), 3.86 (s, 3H), 4.31 (d, J = 15.1 Hz, 1H), 4.36 (dd, J = 0.8 and 15.1 Hz, 1H), 6.79 (d, J = 7.0 Hz, 1H), 7.43 (pseudo-t, J = 7.7 Hz, 1H), 7.50 (dd, J = 4.2 and 8.3 Hz, 1H), 7.54 (br s, 1H), 7.80 (dd, J = 1.1 and 8.2 Hz, 1H), 8.20 (dd, J = 1.7 and 8.3 Hz, 1H), 8.97 (dd, J = 1.7 and 4.2 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ = 11.5, 29.5, 51.4, 51.6, 60.1, 107.3, 117.0, 121.8, 123.8, 126.4, 128.5, 131.0, 136.3, 138.2, 141.5, 144.5, 150.0, 164.1, 165.4. IR (ATR): ν = 2946, 1725, 1696, 1534, 1444, 1385, 1303, 1204, 1156, 1092 cm−1. HRMS (ESI): calcd for C20H19N2O4S [M + H]+ 383.1060; found 383.1057. [α]20D = +434 (c 0.5, CH2Cl2).
Dimethyl (3S)-3-(4-methoxyphenyl)-5-methyl-1H,3H-pyrrolo[1,2-c]thiazole-6,7-dicarboxylate (6). Obtained from N-acetyl-thiazolidine 5 (0.81 g, 2.9 mmol) and DMAD (0.61 g, 4.3 mmol, 1.5 equiv.) as described in the general procedure. Reaction time: 12 h. Purification by column chromatography [hexane/EtOAc (1
:1)] gave compound 6 as a solid (0.72 g, 69%). Recrystallization from EtOAc/hexane gave the compound as pale-yellow solid, mp 86.3–88.1 °C. 1H NMR (400 MHz, CDCl3): δ = 2.00 (s, 3H), 3.80 (s, 3H), 3.82 (s, 3H), 3.83 (s, 3H), 4.30 (d, J = 14.9 Hz, 1H), 4.47 (dd, J = 1.7 and 14.9, 1H), 6.27 (d, J = 1.5 Hz, 1H), 6.85–6.87 (m, 2H), 7.01–7.03 (m, 2H). 13C NMR (100 MHz, CDCl3): δ = 11.4, 30.0, 51.4, 51.6, 55.4, 64.8, 106.7, 114.6, 117.4, 127.2, 130.8, 131.9, 140.4, 160.1, 164.1, 165.5. IR (ATR): ν = 2958, 1687, 1513, 1447, 1230, 1169, 1091, 1027 cm−1. HRMS (ESI): calcd for C18H20NO5S [M + H]+ 362.1057; found 362.1053. [α]20D = −183 (c 0.5, CH2Cl2).
Methyl (3S)-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-7-carboxylate (9) and methyl (3S)-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-6-carboxylate (10). Obtained from N-acetyl-thiazolidine 8 (2.00 g, 7.96 mmol) and methyl propiolate (1.20 g, 14.33 mmol, 1.8 equiv.) as described in the general procedure. Reaction time: 16 h. Purification by column chromatography [hexane/EtOAc (2
:1)] gave a mixture of 9 and 10 (50:50) (1.46 g, 67%). Compound 10 could be separated by selective crystallization with diethyl ether–hexane. Methyl (3S)-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-6-carboxylate (10): pale-yellow solid, mp 89.7–91.7 °C. 1H NMR (400 MHz, CDCl3): δ = 2.18 (s, 3H), 3.78 (s, 3H), 4.02 (d, J = 13.1 Hz, 1H), 4.27 (d, J = 13.1 Hz, 1H), 6.25 (s, 1H), 6.33 (s, 1H), 7.01 (dd, J = 1.7 and 7.7 Hz, 2H), 7.29–7.35 (m, 3H). 13C NMR (100 MHz, CDCl3): δ = 11.8, 28.2, 50.8, 64.0, 101.7, 116.6, 125.4, 128.6, 129.1, 132.0, 133.2, 141.1, 165.8. IR (ATR): ν = 2923, 1688, 1523, 1363, 1222, 1169, 1068 cm−1. HRMS (ESI): calcd for C15H16NO2S [M + H]+ 274.0896; found 274.0892. [α]20D = −320 (c 0.5, CH2Cl2). Methyl (3S)-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-7-carboxylate (9): 1H NMR (400 MHz, CDCl3): δ = 1.82 (s, 3H), 3.81 (s, 3H), 4.34 (d, J = 14.7 Hz, 1H), 4.51 (dd, J = 1.8 and 14.7 Hz), 6.27 (d, J = 1.8 Hz, 1H), 6.36 (s, 1H), 7.06 (dd, J = 1.7 and 7.6 Hz, 2H), 7.30–7.35 (m, 3H). 13C NMR (100 MHz, CDCl3): δ = 12.1, 30.4, 51.0, 65.0, 106.9, 112.2, 125.7, 125.8, 128.8, 129.1, 140.3, 140.4, 165.2.
(3R)-6,7-Bis(hydroxymethyl)-3-(4-fluorophenyl)-5-methyl-1H,3H-pyrrolo[1,2-c]thiazole (4a). Obtained from compound 3a (1.06 g, 2.80 mmol) as described in the general procedure. Purification by column chromatography [hexane/EtOAc (1
:2), then hexane/EtOAc (1:3)] followed by trituration with diethyl ether gave compound 4a as a pale yellow solid (290 mg, 34%). mp ≥ 85 °C, with decomposition. 1H NMR (400 MHz, CDCl3): δ = 1.83 (s, 3H), 2.99 (bs, 2H), 4.07 (d, J = 12.8 Hz, 1H), 4.27 (d, J = 12.8 Hz, 1H), 4.45 (d, J = 12.3 Hz, 1H), 4.49 (d, J = 12.3 Hz, 1H), 4.55 (s, 2H), 6.21 (d, J = 0.8 Hz, 1H), 6.97–7.06 (m, 4H). 13C NMR (100 MHz, CDCl3): δ = 10.0, 27.6, 56.2, 56.5, 63.7, 113.5, 116.0 (d,2JC–F = 21.9 Hz), 122.9, 123.7, 127.6 (d,3JC–F = 8.4 Hz), 131.4, 137.5 (d,4JC–F = 3.1 Hz), 162.6 (d,1JC–F = 248 Hz). 19F NMR (376 MHz, CDCl3): δ = −113.04. IR (ATR): ν = 3326, 2863, 1604, 1508, 1354, 1226, 997 cm−1. HRMS (APCI): calcd for C15H15FNOS [M − OH]+ 276.0853; found 276.0855. [α]20D = +221 (c 0.5, CH2Cl2).
(3R)-6,7-Bis(hydroxymethyl)-5-methyl-3-(4-(trifluoromethoxy)phenyl)-1H,3H-pyrrolo[1,2-c]thiazole (4b). Obtained from compound 3b (1.72 g, 4.15 mmol) as described in the general procedure. Purification by column chromatography [hexane/EtOAc (1
:2), then hexane/EtOAc (1:3)] gave compound 4b (60 mg, 4%) in a very impure form.
(3R)-6,7-Bis(hydroxymethyl)-5-methyl-3-(4-(trifluoromethyl)phenyl)-1H,3H-pyrrolo[1,2-c]thiazole (4c). Obtained from compound 3c (0.93 g, 2.34 mmol) as described in the general procedure. Purification by column chromatography [hexane/EtOAc (1
:2), hexane/EtOAc (1:5), then EtOAc] gave compound 4c in trace amount and in a very impure form.
(3R)-6,7-Bis(hydroxymethyl)-5-methyl-3-(naphthalen-1-yl)-1H,3H-pyrrolo[1,2-c]thiazole (4d). Obtained from compound 3d (0.47 g, 1.15 mmol) as described in the general procedure. Purification by column chromatography [hexane/EtOAc (1
:2), hexane/EtOAc (1:5), then EtOAc] gave compound 4d as a white solid (255 mg, 53%). mp ≥ 180 °C, with decomposition (triturated from diethyl ether). 1H NMR (400 MHz, CDCl3): δ = 1.92 (s, 3H), 2.68 (bs, 2H), 4.08 (d, J = 12.5 Hz, 1H), 4.22 (d, J = 12.8 Hz, 1H), 4.55 (d, J = 12.0 Hz, 1H), 4.60 (d, J = 12.2 Hz, 1H), 4.63 (s, 2H), 6.51 (bs, 1H), 6.91 (s, 1H), 7.33 (pseudo-t, J = 7.7 Hz, 1H), 7.53–7.60 (m, 2H), 7.79 (d, J = 8.2 Hz, 1H), 7.90 (d, J = 7.9 Hz, 2H). IR (ATR): ν = 3297, 2869, 1541, 1431, 1353, 979, 777 cm−1. HRMS (APCI): calcd for C19H18NOS [M − OH]+ 308.1104; found 308.1106. [α]20D = +540 (c 0.5, CH2Cl2).
(3R)-6,7-Bis(hydroxymethyl)-5-methyl-3-(quinolin-8-yl)-1H,3H-pyrrolo[1,2-c]thiazole (4e). Obtained from compound 3e (1.24 g, 3.24 mmol) as described in the general procedure. Recrystallization of the crude product from EtOAc/hexane followed by trituration with diethyl ether gave the compound 4e as a white solid (0.80 g, 75%). mp 148.3–150.1 °C. 1H NMR (400 MHz, CDCl3): δ = 1.95 (s, 3H), 2.52 (bs, 2H), 4.03 (d, J = 13.0 Hz, 1H), 4.20 (d, J = 13.0 Hz, 1H), 4.57 (d, J = 12.4 Hz, 1H), 4.61 (d, J = 12.4 Hz, 1H), 4.63 (d, J = 12.8 Hz, 1H), 4.66 (d, J = 12.8 Hz, 1H), 6.76 (d, J = 7.1 Hz, 1H), 7.39–7.43 (m, 1H), 7.44 (s, 1H), 7.48 (dd, J = 4.2 and 8.3, Hz, 1H), 7.76 (dd, J = 0.9 and 8.2 Hz, 1H), 8.18 (dd, J = 1.7 and 8.3 Hz, 1H), 8.98 (dd, J = 1.7 and 4.2 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ = 9.9, 27.1, 56.6, 56.9, 59.6, 113.8, 121.6, 123.0, 123.2, 124.0, 126.5, 128.0, 128.4, 132.3, 136.2, 139.6, 144.7, 149.8. IR (ATR): ν = 3207, 2917, 2850, 1498, 1422, 1361, 984, 798 cm−1. HRMS (ESI): calcd for C18H19N2O2S [M − H]+ 327.1162; found 327.1156. [α]20D = +570 (c 1, CH2Cl2).
(3S)-6,7-Bis(hydroxymethyl)-3-(4-methoxyphenyl)-5-methyl-1H,3H-pyrrolo[1,2-c]thiazole (7). Obtained from compound 6 (0.51 g, 1.42 mmol) as described in the general procedure. Purification by trituration with diethyl ether yields compound 7 as a white solid (323 mg, 74%). mp 113–115 °C. 1H NMR (400 MHz, CDCl3): δ = 1.83 (s, 3H), 2.43 (bs, 1H), 2.57 (bs, 1H), 3.79 (s, 3H), 4.07 (d, J = 12.8 Hz, 1H), 4.28 (d, J = 12.8 Hz, 1H), 4.48 (d, J = 12.3 Hz, 1H), 4.52 (d, J = 12.2 Hz, 1H), 4.58 (s, 2H), 6.21 (d, J = 0.9 Hz, 1H), 6.82–6.86 (m, 2H), 7.00–7.04 (m, 2H). 13C NMR (100 MHz, CDCl3): δ = 10.0, 27.6, 55.3, 56.4, 56.7, 64.2, 113.3, 114.3, 123.1, 123.5, 127.2, 131.5, 133.4, 159.7. IR (ATR): ν = 3361, 2866, 1613, 1541, 1255, 1171, 1008, 819 cm−1. Anal. Calcd for C16H19NO3S: C, 62.93; H, 6.27; N, 4.59; S, 10.50. Found: C, 62.80; H, 6.11; N, 4.44; S, 10.81. [α]20D = −280 (c 0.5, CH2Cl2).
(3S)-7-Hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (11) and (3S)-6-hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (12). Obtained from the crude mixture of 9 and 10 [ratio (1
:1), from the reaction of N-acetyl-thiazolidine 8 (6.16 mmol) and methyl propiolate, as described previously] and LiAlH4 (6.16 mmol), as described in the general procedure. In this case the reaction mixture was refluxed for 4 h after the addition was completed. Purification by column chromatography [hexane/EtOAc (2:1), then hexane/EtOAc (1:1)] gave in order of elution (3S)-7-hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (11) (159 mg, 10.5%) and (3S)-6-hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (12) (247 mg, 16%). (3S)-7-Hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (11). White solid. mp 91–93 °C (triturated with diethyl ether). 1H NMR (400 MHz, CDCl3): δ = 1.36 (bs, 1H),1.83 (s, 3H), 4.09 (d, J = 12.8 Hz, 1H), 4.31 (d, J = 12.8 Hz, 1H), 4.52 (pseudo-t, J = 12.2 Hz, 2H), 5.98 (s, 1H), 6.22 (s, 1H), 6.90–7.14 (m, 2H), 7.28–7.35 (m, 3H). 13C NMR (100 MHz, CDCl3): δ = 12.1, 27.9, 58.3, 64.3, 111.3, 114.1, 125.1, 125.7, 128.4, 129.0, 131.8, 141.7. IR (ATR): ν = 3376, 2907, 1438, 1377, 1344, 1004, 990, 708 cm−1. HRMS (ESI): calcd for C14H15NOSNa [M + Na]+ 268.0767; found 268.0761. [α]20D = −280 (c 0.5, CH2Cl2). (3S)-6-Hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (12). Pale pink solid. mp 61–63 °C (triturated with diethyl ether). 1H NMR (400 MHz, CDCl3): δ = 1.26 (bs, 1H), 1.87 (s, 3H,), 4.05 (d, J = 12.8 Hz, 1H), 4.30 (d, J = 12.8 Hz, 1H), 4.45 (s, 2H), 5.93 (s, 1H), 6.22 (d, J = 1.4 Hz, 1H), 7.03–7.05 (m, 2H), 7.28–7.32 (m, 3H). 13C NMR (100 MHz, CDCl3): δ = 10.0, 28.8, 58.2, 64.2, 100.1, 122.9, 124.9, 125.7, 128.4, 129.0, 133.0, 141.9. IR (ATR): ν = 3363, 2899, 1337, 1110, 986, 695 cm−1. HRMS (APCI): calcd for C14H14NS [M − OH]+ 228.0841; found 228.0843. [α]20D = −300 (c 0.5, CH2Cl2).
(3S)-6-Hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole (12). Obtained from compound 10 (0.45 g, 1.64 mmol) and LiAlH4 (3.94 mmol) as described in the general procedure. In this case the reaction mixture was refluxed for 4 h after the addition was completed. Purification by column chromatography [hexane/EtOAc (1
:1)] followed by trituration with diethyl ether yields compound 12 as a pale pink solid (221 mg, 55%). Compound 12 was identified by comparison with the specimen previously prepared (see above).
(3R)-6,7-Bis(hydroxymethyl)-3-benzyl-5-methyl-1H,3H-pyrrolo[1,2-c]thiazole (17a). Obtained from compound 16a (0.63 g, 1.83 mmol) as described in the general procedure. Purification by column chromatography [hexane/EtOAc (1
:3), then hexane/EtOAc (1:5)] gave compound 17a as a colorless oil (363 mg, 68%). 1H NMR (400 MHz, CDCl3): δ = 2.27 (s, 3H), 2.62 (bs, 2H), 3.15–3.21 (m, 2H), 3.52 (d, J = 12.9 Hz, 1H), 3.65 (d, J = 12.9 Hz, 1H), 4.40 (s, 2H), 4.50 (d, J = 12.6 Hz, 1H), 4.54 (d, J = 12.7 Hz, 1H), 5.41 (t, J = 4.9 Hz, 1H), 7.03–7.05 (m, 2H), 7.23–7.25 (m, 3H). 13C NMR (100 MHz, CDCl3): δ = 10.1, 26.8, 44.5, 56.3, 56.5, 63.5, 113.2, 121.9, 123.2, 127.2, 128.2, 130.0, 131.5, 135.5. IR (ATR): ν = 3306, 2913, 2861,1429, 1355, 1237, 1030, 978, 696 cm−1. HRMS (APCI): calcd for C16H18NOS [M − OH]+ 272.1104; found 272.1107. [α]20D = +200 (c 1, CH2Cl2).
(3R)-6,7-Bis(hydroxymethyl)-3,5-dimethyl-1H,3H-pyrrolo[1,2-c]thiazole (17b). Obtained from compound 16b (1.21 g, 4.50 mmol) as described in the general procedure. Purification by trituration with diethyl ether yields compound 17b as a white solid (549 mg, 57%). mp 65–66 °C. 1H NMR (400 MHz, CDCl3): δ = 1.70 (d, J = 6.1 Hz, 1H), 2.23 (s, 3H), 2.44 (bs, 2H), 3.92 (d, J = 12.9 Hz, 1H), 4.21 (d, J = 12.9 Hz, 1H), 4.48–4.55 (m, 4H), 5.33 (q, J = 5.9 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ = 10.0, 25.7, 26.7, 56.3, 56.6, 58.1, 113.2, 122.0, 123.0, 130.6. IR (ATR): ν = 3272, 2913, 2858, 1528, 1437, 1367, 1325, 1241, 971 cm−1. HRMS (EI): calcd for C10H15NO2S [M]+ 213.0824; found 213.0828. [α]20D = +110 (c 1, CH2Cl2).
(3S)-6,7-Bis(hydroxymethyl)-3,5-dimethyl-1H,3H-pyrrolo[1,2-c]thiazole (17c). Obtained from compound 16c (1.12 g, 4.17 mmol) as described in the general procedure. Purification by trituration with diethyl ether yields compound 17c as a white solid (669 mg, 75%). mp 65–66 °C. 1H NMR (400 MHz, CDCl3): δ = 1.70 (d, J = 6.2 Hz, 1H), 2.23 (s, 3H), 2.69 (bs, 2H), 3.91 (d, J = 12.9 Hz, 1H), 4.20 (d, J = 12.9 Hz, 1H), 4.46–4.52 (m, 4H), 5.33 (q, J = 5.9 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ = 10.0, 25.7, 26.7, 56.3, 56.6, 58.1, 113.2, 122.0, 123.0, 130.6. IR (KBr): ν = 3286, 2913, 2877, 1529, 1442, 1327, 985 cm−1. HRMS (APCI): calcd for C10H14NOS [M − OH]+ 196.0791; found 196.0791. [α]20D = −110 (c 1, CH2Cl2).
:3), hexane/EtOAc (1:5), then EtOAc] gave in order of elution methyl (3R)-7-hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-6-carboxylate-2,2-dioxide (14) (107 mg, 31%) and (3R)-6,7-bis(hydroxymethyl)-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-2,2-dioxide (15) (166 mg, 53%).
Methyl (3R)-7-hydroxymethyl-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-6-carboxylate-2,2-dioxide (14). mp 112–114 °C. 1H NMR (400 MHz, CDCl3): δ = 2.27 (s, 3H), 3.87 (s, 3H), 4.22 (d, J = 15.2 Hz, 1H), 4.48 (d, J = 15.2 Hz, 1H), 4.61 (d, J = 13.2 Hz, 1H), 4.69 (d, J = 13.2 Hz, 1H), 5.91 (s, 1H), 7.02–7.04 (m, 2H), 7.41–7.47 (m, 3H). 13C NMR (100 MHz, CDCl3): δ = 12.6, 48.2, 51.5, 57.1, 77.8, 113.6, 118.9, 122.2, 126.8, 129.6, 130.62. 130.64, 135.7, 166.2. IR (KBr): ν = 3132, 1712, 1672, 1454, 1404, 1317, 1138 cm−1. HRMS (EI): calcd for C16H17NO5S [M]+ 335.0827; found 335.0827. [α]20D = 0.0 (c 1, CH2Cl2).
(3R)-6,7-Bis(hydroxymethyl)-5-methyl-3-phenyl-1H,3H-pyrrolo[1,2-c]thiazole-2,2-dioxide (15). mp > 170 °C (with decomposition). 1H NMR (400 MHz, CDCl3): δ = 2.00 (s, 3H), 2.50 (bs, 1H), 2.77 (bs, 1H), 4.22 (d, J = 15.0 Hz, 1H), 4.43 (d, J = 15.0 Hz, 1H), 4.59–4.65 (m, 4H), 5.88 (s, 1H), 7.05–7.07 (m, 2H), 7.41–7.45 (m, 3H). 13C NMR (100 MHz, CDCl3): δ = 10.1, 48.4, 55.8, 56.6, 78.2, 118.2, 119.2, 121.6, 126.2, 127.1, 129.4, 130.4, 131.3. IR (KBr): ν = 3532, 3454, 2981, 2923, 2873, 1311, 1130, 987, 700 cm−1. HRMS (ESI): calcd for C15H17NO4SNa [M + Na]+ 330.0771; found 330.0764. [α]20D = +20.0 (c 0.25, CH2Cl2).
Biological evaluation
Author contributions
MH, AMP, ABP, MS: chemical synthesis; CC, JR: cell biology studies; MS, LS, TPM: project conception and supervision; MS, LS, TPM: manuscript preparation.Conflicts of interest
One patent application protecting the compounds disclosed in this manuscript has been filed by the following authors M. I. L. S., L. S., and T. M. V. D. P. M.Acknowledgements
The authors thank Coimbra Chemistry Centre (CQC), supported by the Portuguese Agency for Scientific Research “Fundação para a Ciência e a Tecnologia” (FCT), through project UIDB/00313/2020 (https://doi.org/10.54499/UIDB/00313/2020) and UIDP/00313/2020 (https://doi.org/10.54499/UIDP/00313/2020), co-funded by COMPETE2020-UE. This research was also funded by FCT/MCTES, through LAQV/REQUIMTE (UIDP/QUI/50006/2020). M. I. L. S. (DL57/2016/CP1370/CT0041, https://doi.org/10.54499/DL57/2016/CP1370/CT0041), J. L. P. R. (PhD fellowship PD/BD/143160/2019 “MedChemTrain programme”) and C. S. C. C. (PhD fellowship 2021.06305.BD) thank FCT for financial support. The authors also acknowledge the UC-NMR facility for obtaining the NMR data (https://www.uc.pt/uc-nmr/).References
- G. W. Prager, S. Braga, B. Bystricky, C. Qvortrup, C. Criscitiello, E. Esin, G. S. Sonke, G. A. Martínez, J. S. Frenel, M. Karamouzis, M. Strijbos, O. Yazici, P. Bossi, S. Banerjee, T. Troiani, A. Eniu, F. Ciardiello, J. Tabernero, C. C. Zielinski, P. G. Casali, F. Cardoso, J. Y. Douillard, S. Jezdic, K. McGregor, G. Bricalli, M. Vyas and A. Ilbawi, ESMO Open, 2018, 3, e000285 CrossRef PubMed.
- WHO report on cancer: setting priorities, investing wisely and providing care for all, World Health Organization, Geneva, 2020 Search PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- E. Dekker, P. J. Tanis, J. L. A. Vleugels, P. M. Kasi and M. B. Wallace, Lancet, 2019, 394, 1467–1480 CrossRef PubMed.
- H. Ramos, M. I. L. Soares, J. Silva, L. Raimundo, J. Calheiros, C. Gomes, F. Reis, F. A. Monteiro, C. Nunes, S. Reis, B. Bosco, S. Piazza, L. Domingues, P. Chlapek, P. Vlcek, P. Fabian, A. T. Rajado, A. T. P. Carvalho, R. Veselska, A. Inga, T. M. V. D. Pinho e Melo and L. Saraiva, Cell Rep., 2021, 35, 108982 Search PubMed.
- T. M. V. D. Pinho e Melo, M. I. L. Soares, D. M. Barbosa, A. M. d'A. R. Gonsalves, A. M. Beja, J. A. Paixão, M. R. Silva and L. A. da Veiga, Tetrahedron, 2000, 56, 3419–3424 Search PubMed.
- L. Szilágyi and Z. Györgydeák, J. Am. Chem. Soc., 1979, 101, 427–432 CrossRef.
- Z. Györgydeák, M. Kajtár-Peredy, J. Kajtár and M. Kajtár, Liebigs Ann. Chem., 1987, 927–934 CrossRef.
- T. M. V. D. Pinho e Melo, C. S. B. Gomes, A. M. d'A. R. Gonsalves, J. A. Paixão, A. M. Beja, M. R. Silva and L. A. da Veiga, Tetrahedron, 2002, 58, 5093–5102 Search PubMed.
- M. I. L. Soares, A. F. Brito, M. Laranjo, J. A. Paixão, M. F. Botelho and T. M. V. D. Pinho e Melo, Eur. J. Med. Chem., 2013, 60, 254–262 CrossRef CAS PubMed.
- M. I. L. Soares, A. F. Brito, M. Laranjo, A. M. Abrantes, M. F. Botelho, J. A. Paixão, A. M. Beja, M. R. Silva and T. M. V. D. Pinho e Melo, Eur. J. Med. Chem., 2010, 45, 4676–4681 CrossRef CAS PubMed.
- A. Regueiro-Ren, in Applications of Heterocycles in the Design of Drugs and Agricultural Products, ed. N. A. Meanwell and M. L. Lolli, 2021, vol. 134, pp. 1–30 Search PubMed.
- T. M. V. D. Pinho e Melo, M. I. L. Soares, A. M. D. R. Gonsalves and H. McNab, Tetrahedron Lett., 2004, 45, 3889–3893 Search PubMed.
- T. M. V. D. Pinho e Melo, M. I. L. Soares, A. M. d'A. R. Gonsalves, J. A. Paixão, A. M. Beja, M. R. Silva, L. A. da Veiga and J. C. Pessoa, J. Org. Chem., 2002, 67, 4045–4054 Search PubMed.
- T. M. V. D. Pinho e Melo, M. I. L. Soares, A. M. d'A. R. Gonsalves, J. A. Paixão, A. M. Beja and M. R. Silva, J. Org. Chem., 2005, 70, 6629–6638 CrossRef CAS PubMed.
- K. Santos, M. Laranjo, A. M. Abrantes, A. F. Brito, C. Goncalves, A. B. S. Ribeiro, M. F. Botelho, M. I. L. Soares, A. S. R. Oliveira and T. M. V. D. Pinho e Melo, Eur. J. Med. Chem., 2014, 79, 273–281 CrossRef CAS PubMed.
- S. Yang, T. N. Wang, Y. A. Zhou, L. Shi, A. D. Lu and Z. W. Wang, Molecules, 2021, 26, 383 CrossRef CAS PubMed.
- I. R. Schmolka and P. E. Spoerri, J. Org. Chem., 1957, 22, 943–946 CrossRef CAS.
- N. Boechat, J. C. S. da Costa, J. D. Mendonça, P. S. M. de Oliveira and M. V. N. de Souza, Tetrahedron Lett., 2004, 45, 6021–6022 CrossRef CAS.
- L. Raimundo, A. Paterna, J. Calheiros, J. Ribeiro, D. S. P. Cardoso, I. Piga, S. J. Neto, D. Hegan, P. M. Glazer, S. Indraccolo, S. Mulhovo, J. L. Costa, M.-J. U. Ferreira and L. Saraiva, Br. J. Pharmacol., 2021, 178, 3627–3647 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00076e |
| This journal is © The Royal Society of Chemistry 2024 |