
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The importance of tyrosines in multimers of cyclic RGD nonapeptides: towards αvβ6-integrin targeted radiotherapeutics†
Neil Gerard
Quigley‡
a,
Maximilian Alexander
Zierke‡
a,
Beatrice Stefanie
Ludwig
b,
Frauke
Richter
a,
Nghia Trong
Nguyen
b,
Falco
Reissig
c,
Jakub
Šimeček
c,
Susanne
Kossatz
*b and
Johannes
Notni
 *ac
*ac
aInstitute of Pathology, School of Medicine and Health, Technische Universität München, Munich, Germany
bDepartment of Nuclear Medicine, University Hospital Klinikum Rechts der Isar and Central Institute for Translational Cancer Research, (TranslaTUM), School of Medicine and Health, Technische Universität München, Munich, Germany
cTRIMT GmbH, Carl-Eschebach-Str. 7, D-01454 Radeberg, Germany. E-mail: jn@trimt.de
First published on 19th April 2024
Abstract
In a recent paper in this journal (RSC Med. Chem., 2023, 14, 2429), we described an unusually strong impact of regiospecific exchange of phenylalanines by tyrosines in 10 gallium-68-labeled trimers of certain cyclic RGD peptides, c[XRGDLAXp(NMe)K] (X = F or Y), on non-specific organ uptakes. We found that there was, in part, no correlation of liver uptake with established polarity proxies, such as the octanol–water distribution coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D). Since this observation could not be explained straightforwardly, we suggested that the symmetry of the compounds had resulted in a synergistic interaction of certain components of the macromolecules. In the present work, we investigated whether a comparable effect also occurred for a series of 5 tetramers labeled with lutetium-177. We found that in contrast to the trimers, liver uptake of the tetramers was well correlated to their polarity, indicating that the unusual observations along the trimer series indeed was a unique feature, probably related to their particular symmetry. Since the Lu-177 labeled tetramers are also potential agents for treatment of a variety of αvβ6-integrin expressing cancers, these were evaluated in mice bearing human lung adenocarcinoma xenografts. Due to their tumor-specific uptake and retention in biodistribution and SPECT imaging experiments, these compounds are considered a step forward on the way to αvβ6-integrin-targeted anticancer agents. Furthermore, we noticed that the presence of tyrosines in general had a positive impact on the in vivo performance of our peptide multimers. In view of the fact that a corresponding rule was already proposed in the context of protein engineering, we argue in favor of considering peptide multimers as a special class of small or medium-sized proteins. In summary, we contend that the performance of peptide multimers is less determined by the in vitro characteristics (particularly, affinity and selectivity) of monomers, but rather by the peptides' suitability for the overall macromolecular design concept, and peptides containing tyrosines are preferred.
D). Since this observation could not be explained straightforwardly, we suggested that the symmetry of the compounds had resulted in a synergistic interaction of certain components of the macromolecules. In the present work, we investigated whether a comparable effect also occurred for a series of 5 tetramers labeled with lutetium-177. We found that in contrast to the trimers, liver uptake of the tetramers was well correlated to their polarity, indicating that the unusual observations along the trimer series indeed was a unique feature, probably related to their particular symmetry. Since the Lu-177 labeled tetramers are also potential agents for treatment of a variety of αvβ6-integrin expressing cancers, these were evaluated in mice bearing human lung adenocarcinoma xenografts. Due to their tumor-specific uptake and retention in biodistribution and SPECT imaging experiments, these compounds are considered a step forward on the way to αvβ6-integrin-targeted anticancer agents. Furthermore, we noticed that the presence of tyrosines in general had a positive impact on the in vivo performance of our peptide multimers. In view of the fact that a corresponding rule was already proposed in the context of protein engineering, we argue in favor of considering peptide multimers as a special class of small or medium-sized proteins. In summary, we contend that the performance of peptide multimers is less determined by the in vitro characteristics (particularly, affinity and selectivity) of monomers, but rather by the peptides' suitability for the overall macromolecular design concept, and peptides containing tyrosines are preferred.
Introduction
Peptide multimers have gained a firm place in the canon of compound classes used in radiopharmacy.1 This is because the combination of multiple copies of the same binding motif for a biological target in a single molecule increases avidity for that target—a principle that nature has always exploited in the form of regular antibodies, which are nothing else than dimers of targeting proteins.2 A particularly important component of a synthetic multimer is the scaffold, because its valency, geometry, and symmetry determine multiplicity, distance, and interaction between the conjugated functional monomers.3,4 The spatial distribution and motility of the comprised monomeric ligands can furthermore be adjusted using linkers of different lengths and rigidity to optimize avidity,5,6 the latter being considered a cumulative effect of individual target binding events.7 This strategy has been extensively exploited in connection with perhaps the most widely used class of ligands for αvβ3-integrin, the cyclic pentapeptides c(RGDxX) (x = f, y; X = K, E).8 Clinical data have suggested that radiolabeled c(RGDxK) dimers9 outperform the respective monomers for αvβ3-integrin targeted positron emission tomography (PET) diagnostics in patients.10 Comparative studies revealed that multimers with more than two c(RGDxK) units possess even higher avidities than monomers and dimers,11–13 which frequently also resulted in higher target-specific uptakes in vivo.14–16 This design principle has been applied to various targeting moieties with high relevance in radiopharmacy, such as fibroblast activation protein inhibitors (FAPi)17 or prostate-specific membrane antigen (PSMA) inhibitors,18–20 although it did not always lead to improved in vivo properties.21 However, the concept generally works well for radiopharmaceuticals based on peptides and peptidomimetics targeting integrins,22 such as αvβ3,14–16 α5β1,23,24 αvβ8,25,26 or αvβ6.27,28 The approach certainly is a valuable addition to the radiopharmaceutical toolbox,1 as it has been shown that even peptides whose affinities are too low for the radiolabeled monomers to be used as radiotracers can be useful for effective in vivo addressing when their target-specific binding is boosted by multimerization.26,28,29However, multimerization does not only enhance avidity to the desired target but may also amplify unknown weak interactions.30 Peptide multimers therefore can exhibit altered target specificity or unexpectedly bind to sites not recognized by the respective monomers, which makes the successful design of multimeric probes and therapeutics a challenging endeavour. In addition, they differ from oligopeptides in that they have several times the molecular weight, which puts them on the borderline of proteins. Together with their sometimes altered polarity, they can therefore exhibit significant differences in pharmacokinetic parameters such as tissue penetration, elimination pathways and kinetics, and plasma half-life.31 Boehmer et al. have furthermore pointed out that multimers are characterized by a higher complexity that needs to be better understood to enable a rational design of clinically applicable agents.32 The increased complexity of multimers apparently leads to a higher degree of indeterminancy in the structure–activity relationships, because the components of complex systems can interact in unpredictable ways and thus produce completely new properties, which is known as emergence.33 Concerning multimers, this means that such emergent properties cannot in principle be calculated or otherwise derived straightforwardly from data for the respective monomers. Comprehensive data collection for libraries of multimers seems to be the only way to close this gap in understanding.34
One aspect of multimerization that is particularly difficult to predict is nonspecific accumulation in non-target organs and tissues, which can emerge unexpectedly and render an agent with otherwise promising properties, such as high avidity and target-specific uptake, virtually useless.35 Although most binding motifs are well characterized in terms of their specificity for their respective pharmacological target, usually only a small fraction of the multitude of possible interactions with other cell types, surface proteins, transporters, enzymes, or other biological contact sites in a living organism is known, which of course applies all the more the larger the respective molecule and thus the greater the number of possible contact sites.
To this end, we recently investigated the influence of small structural variations on the biodistribution of the αvβ6-integrin targeted positron emission tomography (PET) radiopharmaceutical 68Ga-Trivehexin (Fig. 1).36 This molecule comprises three copies of the cyclic nonapeptide c[YRGDLAYp(NMe)K], which are attached to a central triazacyclononane-triphosphinate (TRAP, see Fig. 1)37 structure that serves as both a highly efficient chelator for 68Ga38 and a C3-symmetrical trimeric scaffold.3968Ga-Trivehexin thus comprises 6 tyrosines, which we regioselectively and consecutively substituted by phenylalanines.34 The influence of these substitutions on the biodistribution proved to be counterintuitive to some extent. For example, although a high non-specific liver uptake of the all-Phe derivative was reduced by 50% upon introduction of a single Tyr, and by almost three quarters by two Tyr's, there was no concordant change in polarity proxies, such as logD. We concluded that non-specific tissue uptake is most likely an emergent phenomenon arising from synergistic interaction of phenyl groups in these highly symmetric molecules.34 One question that arose in the course of this study was whether the observed correlations are primarily caused by the highly ordered triangular symmetry of the compounds, or whether comparable effects can also be reproduced within a similar set of structures that exhibit a different symmetry.
 | ||
| Fig. 1 Structures of compounds discussed in the text. | ||
In order to shed more light on this question, the present study takes a closer look at comparable structures based on the DOTPI framework (Fig. 1).40 This tetrameric scaffold is a larger congener of TRAP and can be functionalized in a similar manner, i.e., by virtue of the copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) using the respective azide-functionalized derivative,41,42 which provides access to symmetrically and asymmetrically substituted peptide tetramers in practical amounts.43 DOTPI rapidly forms kinetically inert complexes with many radiometal ions that emit particle radiation, such as the alpha emitter 213BiIII,44 or trivalent radiolanthanides and especially the popular beta emitter 177LuIII.40 We herein report synthesis and preclinical data (ex vivo biodistribution and SPECT imaging in tumor-bearing mice) for tetrameric conjugates of αvβ6-integrin binding cyclic nonapeptides with the sequence cyclo[XRGDLAXp(NMe)K] (X = F or Y), which also constitutes the next step towards αvβ6-integrin targeted radiotherapeutics on the basis of these peptides. Such agents are currently considered of high clinical relevance45,46 because αvβ6-integrin is upregulated in various malignant cancers,47 especially in pancreatic ductal adenocarcinoma (PDAC),48 oral squamous cell carcinoma (OSCC),49 ovarian50 and cervical cancer,51 as well as in primaries and brain metastases52 of non-small cell lung cancer (NSCLC).53
Results
Peptide conjugation via CuAAC
Monomeric peptide building blocks FF,29FY,34 and YY36 (Fig. 2) were synthesized as described previously by coupling 4-pentynoic acid to the Lys side chains of the three cyclopeptides cyclo[FR(Pbf)GD(tBu)LAFp(NMe)K], cyclo[FR(Pbf)GD(tBu)LAY(tBu)p(NMe)K], or cyclo[Y(tBu)R(Pbf)GD(tBu)LAY(tBu)p(NMe)K], followed by acidic deprotection. With regard to our preceding study on comparable trimeric peptides,34 we found that the increase in possible Phe → Tyr exchange positions and the change in symmetry leads to an even greater number of possible isomers, rendering a full-scale in vivo evaluation unfeasible. The imperative to reduce animal experiments to a reasonable minimum forced us to limit this investigation to the most relevant examples. We therefore refrained from including the fourth possible monomer YF (in which RA is OH and RB is H, see Fig. 2) into this investigation because in the preceding study,34 we observed that replacement of FF by YF invariantly had the same influence on biodistribution as FY, albeit to a slightly lower extent. In addition, FY showed higher affinity and selectivity for αvβ6-integrin than YF,34 which is why FY seemed to be generally more relevant for this type of study. | ||
| Fig. 2 Synthesis of tetrameric conjugates of αvβ6-integrin binding peptides. Synthesis of Y0, Y4, and Y8 was done by reacting DOTPI-tetraazide with the peptides FF, YF, and YY, respectively. The asymetrical conjugate Y3 was isolated from the CuAAC reaction of DOTPI-tetraazide with a mixture of FF and YF by means of preparative HPLC. Functional groups related to the CuAAC are highlighted in red. | ||
The synthesis of the homo-tetramers Y0, Y4, and Y8 was done by CuAAC reactions as shown in Fig. 2, using FF, FY, and YY, respectively, as building blocks. Y3 was synthesized in the same way, applying a combinatorial reaction scheme using a 1:3 mixture of FF and FY, after which the conjugate Y3 was isolated by preparative HPLC. Other conjugates were not isolated. Since reaction yields were fairly low, we tested auxiliary ligands during CuAAC or alternative scavengers for demetallation for the example of the synthesis of Y0, and found that the yields can generally be substantially increased through optimisation if necessary. Furthermore, we were concerned about a high kidney uptake that was observed for all TRAP-trimers irrespective of the presence or absence of tyrosines.34 The same situation was expected for the DOTPI-tetramers, potentially leading to issues caused by a high renal radiation burden during translation. We therefore also elaborated a congener of Y4 with tripeptide linkers between chelator and peptides, referred to as GFK-Y4 (Fig. 3). The linker motif, Gly-Phe-Lys (GFK), has been described as stable in blood plasma, and readily cleavable by enzymes present on the brush border membrane of renal tubules.54 The conjugate was also assembled via CuAAC using a respective alkyne-functionalized building block, GFK-FY (Fig. 3).
 | ||
| Fig. 3 Structure of the tetramer GFK-Y4 obtained by CuAAC of DOTPI-tetraazide with GFK-FY, a peptide building block comprising a mono-tyrosine cyclic peptide as featured in FY and the brush-border cleavable linker GFK (for a full synthesis scheme, see Experimental section). Functional groups related to the CuAAC are highlighted in red. | ||
In vivo biodistribution and PET imaging
Radiolabeling of all conjugates was done in a straightforward manner, using n.c.a. 177Lu in sodium acetate buffer (pH 5.5–6), affording the radiolabeled compounds in high purities (>98% by radio-HPLC and radio-TLC; for details see Experimental section) without a separate purification step. Octanol–water distribution coefficients were determined by shake-flask and are summarized in Table 1.DOW), determined by shake-flask method (n = 8)
| Compound | LogDOW |
|---|---|
| Y0 | −0.37 ± 0.07 |
| Y3 | −1.16 ± 0.23 |
| Y4 | −1.48 ± 0.25 |
| Y8 | −1.91 ± 0.20 |
| GFK-Y4 | −1.64 ± 0.28 |
The biodistribution of the 177Lu-labeled peptide conjugates was investigated in severe combined immunodeficiency (SCID) mice bearing subcutaneous xenografts of the αvβ6-integrin expressing human lung adenocarcinoma cell line H2009 on their right shoulders. Two representative time points after i.v. administration (p.i.) were evaluated. Uptake data were acquired 90 min p.i. to facilitate direct comparison with the respective 68Ga-labeled trimers,34 and additionally 3 d p.i. to demonstrate long-term tumor retention, which is relevant for radiotherapeutic application (Fig. 4).
 | ||
| Fig. 4 Biodistribution data for 177Lu-labeled peptide tetramers in H2009 (human lung adenocarcinoma) xenografted mice. For details and numerical data in tabular form, see ESI.† | ||
For the 90 min time point, a uniform pattern for the influence of Phe → Tyr exchange on organ uptakes could not be discerned. For example, lung, stomach, intestine, and tumor uptakes were highest for 177Lu-Y4, but this compound showed the lowest uptake in the liver (Fig. 4a). Similar observations were made for the other compounds. An exception was 177Lu-GFK-Y4, whose uptakes were by far the lowest in almost all organs except the kidneys. The same situation was found 3 d p.i., with the difference that kidney activity was also lowest here (Fig. 4b). Tumor uptake of the other investigated compounds was comparable, albeit slightly in favor of 177Lu-Y3 and 177Lu-Y4. The same pattern was observed for the stomach, which makes sense insofar as a physiological expression of β6-integrin in murine stomach has been confirmed earlier.29 The fact that tumor uptakes for 177Lu-Y3 and 177Lu-Y4 after 3 d were somewhat higher than those of 177Lu-Y0 and 177Lu-Y8 could also be caused by different administered total peptide amounts (averagely, 42 and 64 pmol for Y3 and Y4, respectively, vs. 92 and 200 pmol for Y0 and Y8, respectively; see ESI,† Tables S2, S4, S6, and S11). Although the target-specific tissue uptake of integrin ligands is usually not severely influenced by moderate changes (i.e., ±50%) of injected peptide mass,23 a factor of approx. 4 between 177Lu-Y3/Y4 and 177Lu-Y8 might nonetheless have resulted in a relative underestimation of the tumor uptake of the latter in this study.
The tumor-to-organ ratios deserve particular attention for potential radiotherapeutics because off-target nonspecific uptakes are determining radiotoxicity and thus, clinical applicability. Again, no consistent pattern could be identified (Fig. 4c). The tumor-to-blood ratio was lowest for 177Lu-Y8 among the tetramers without GFK linkers, but the same compound performed best in terms of tumor-to-pancreas and particularly tumor-to-intestines ratios. Since unwanted uptake in the intestines has proven the most critical issue hampering translation of various αvβ6-integrin targeted radiopharmaceuticals,55 we considered 177Lu-Y8 to be the most promising compound in the series, despite the overall ambiguous data situation.
We determined the biodistribution for 177Lu-Y8 for another time point, 24 h p.i., which revealed that the compound rapidly cleared from the blood stream but was retained in many organs (Fig. 5). We noted that uptake and retention was somewhat higher for the αvβ6-integrin positive H2009 tumor and stomach and furthermore observed a similar clearance pattern, which substantiates the interpretation that the prolonged tissue accumulation was target-specific. The long-term tumor retention of 177Lu-Y8 was further illustrated by single-photon emission computed tomography (SPECT) imaging (Fig. 6). The images showed an uneven tumor uptake, which corresponds well to the generally heterogeneous αvβ6-integrin expression in this model.28,29 Volume-of-interest (VOI) based quantification of tumor uptakes did not quite correspond to biodistribution, but nevertheless corroborated a tumor retention of 177Lu-Y8 over days. However, the images also showed a high uptake in the kidneys, which did not decrease over time, consistent with the biodistribution data shown in Fig. 5a.
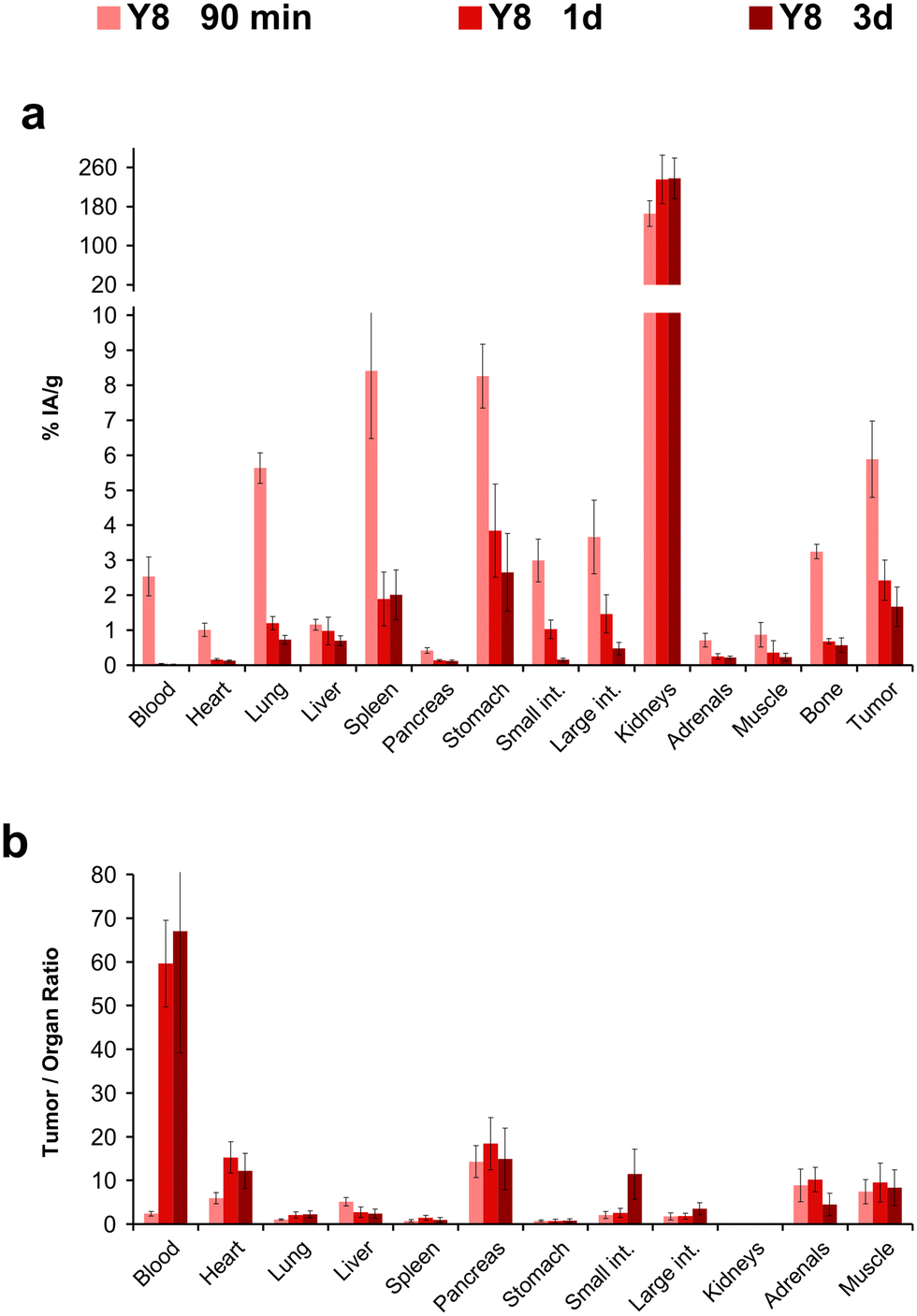 | ||
| Fig. 5 Biodistribution in H2009 tumor mice, and resulting tumor-to-organ ratios for the 177Lu-labeled tetramer Y8 at different time points after injection. For details and numerical data in tabular form, see ESI.† | ||
 | ||
| Fig. 6 Single-photon emission computed tomography (SPECT) of a SCID mouse bearing a human H2009 tumor xenograft on the right shoulder, recorded at 4 time points (1 h, 3 h, 24 h, and 72 h) after i.v. injection of 36.5 MBq of 177Lu-Y8, scan time 1 h. Tumor position is indicated by white dotted circle. Adjacent figures denote ROI-based tumor uptakes in % IA mL−1. | ||
Discussion
Towards αvβ6-integrin targeted radiotherapeutics
In a series of 10 different 68Ga-labeled peptide trimers featuring different combinations of αvβ6-integrin targeting cyclic nonapeptides monomers of the generic formula cyclo[XRGDLAXp(NMe)K] (X = Phe or Tyr), the trimer comprising only tyrosines showed the lowest degree of nonspecific organ accumulation in preclinical models.34 This radiotracer, referred to as 68Ga-Trivehexin (see Fig. 1),36 has convincingly demonstrated its utility for PET imaging of cancers and metastases,45 particularly of pancreatic carcinoma.56 Its low intestinal uptake has proven beneficial in a clinical setting, because it resulted in a better delineation of metastases than radiotracers based on other αvβ6-integrin binding peptides.46,55 The next step towards improved clinical treatment of cancer patients was, of course, the development of corresponding therapeutic αvβ6-integrin radioligands to enable a theranostic approach.46,57We previously evaluated the potential of various 177Lu labeled 1:1 chelator–peptide conjugates of cyclo[FRGDLAFp(NMe)K] but found that such monomers are generally not suitable for radioligand therapy due to low uptake and retention in tumors (<0.8% IA g−1, 90 min p.i.).58 The novel peptide tetramers 177Lu-Y0, 177Lu-Y3, 177Lu-Y4, and 177Lu-Y8 showed a markedly improved tumor uptake (range 3.2 to 6.9% IA g−1, 90 min p.i.) and a retention over days (range 1.7 to 2.1% IA g−1, 3 d p.i.; see Fig. 4, Fig. 6, and ESI† Tables S1–S11). However, one cannot fail to notice a high and persistent kidney uptake of these agents, which is expected to limit their clinical applicability because of a high risk of radiation-induced renal damage.59 This problem has been observed for many radiolabeled peptides and proteins, and is most likely caused by a long residence time of the radiometabolites generated by lysosomal proteolysis of the radiopeptides in renal tubular cells.60
Different approaches have been pursued to solve this issue. One strategy aims at selective breakdown of radiopharmaceuticals that have accumulated in the kidneys. This can be achieved by introducing peptidic linkers that are stable in blood plasma but are cleaved by enzymes present on the brush border membrane of renal tubules, to selectively generate small radiometabolites in the kidneys that are rapidly excreted via the urine.61 One such linker is the amino acid sequence GFK that is recognized by neutral endopeptidase (NEP) located in the renal brush border.54 We consequently equipped one of the tetramers, Y4, with this sequence in all peptide–chelator linkages, resulting in the conjugate GFK-Y4 (Fig. 3). Unfortunately, the approach did not result in the expected substantial improvement of the tumor-to-kidney ratio. The renal retention of 177Lu-GFK-Y4 was still high after 3 d, and the compound furthermore showed lower tumor uptake and faster elimination from the blood stream than the other tetramers (Fig. 4). Such an in vivo behavior suggests that the GFK sequence is actually cleaved before reaching the brush border, leading to premature decomposition and excretion of a large fraction of the radiopharmaceutical before it can bind to its target on the tumor cells. The desired reduction in renal uptake while maintaining tumor uptake could therefore not be achieved in this explorative experiment. Further linker variations or other established methods to reduce the radiation dose of the kidneys, such as the simultaneous infusion of renal protection agents,62 shall therefore be evaluated in the future.
Structure, polarity, and biodistribution revisited
The other aim of this study was to figure out whether multimers of cyclo[XRGDLAXp(NMe)K] (X = F or Y) that are based on another scaffold than TRAP show the same surprising trends of polarity measures, especially logD, and non-specific organ uptakes, especially in the liver. We recall that for the trimer series, replacement of less than 50% of phenylalanines by tyrosines did not change the logD but reduced liver uptake by up to 72%.34 Such an observation was not made for the tetramers of this work (Fig. 4). We found a consistent decrease of both parameters with gradual Phe → Tyr exchange. The first 3 Phe → Tyr substitutions (Y0 → Y3) still had a larger influence on both parameters than the last 4 exchanges (Y4 → Y8), but the overall picture is well in line with the textbook scheme, i.e., a higher degree of polarity reduces liver absorption (Fig. 7).
 | ||
| Fig. 7 Comparison of trends in polarity proxies and liver uptakes, 3 d p.i. Connecting lines between data points are intended only to visualize trends and do not indicate a functional correlation. | ||
Fig. 8 further illustrates that the tetramer series did not show the anomaly that was previously observed for the corresponding library of TRAP-based trimers, namely, that polarity proxies such as logD had no predictive value for certain in vivo properties, such as liver uptake.34 For the tetramers, an increase of the logD always corresponded to an increase of liver uptake (Fig. 8a). It thus appears possible to derive a bijective correlation function, which could be used to approximate the liver uptake of a novel compound in that series based on its logD (exemplified by the dotted line in Fig. 8a). In lack of a suitable theory, we are however not able to define which type of regression could be appropriate to obtain a fit function for a correlation between the logarithm of the octanol–PBS distribution coefficient and the liver uptake expressed as % IA g−1. In contrast, a fit was not meaningful at all for the trimers,34 which is particularly evident from the fact that in the corresponding plot (see Fig. 8b), five out of ten data points for the trimer series are more or less vertically aligned. A hypothetical correlation function would therefore have to comprise non-bijective sections (indicated by a vertical dotted line in Fig. 8b), which means it does not exist.
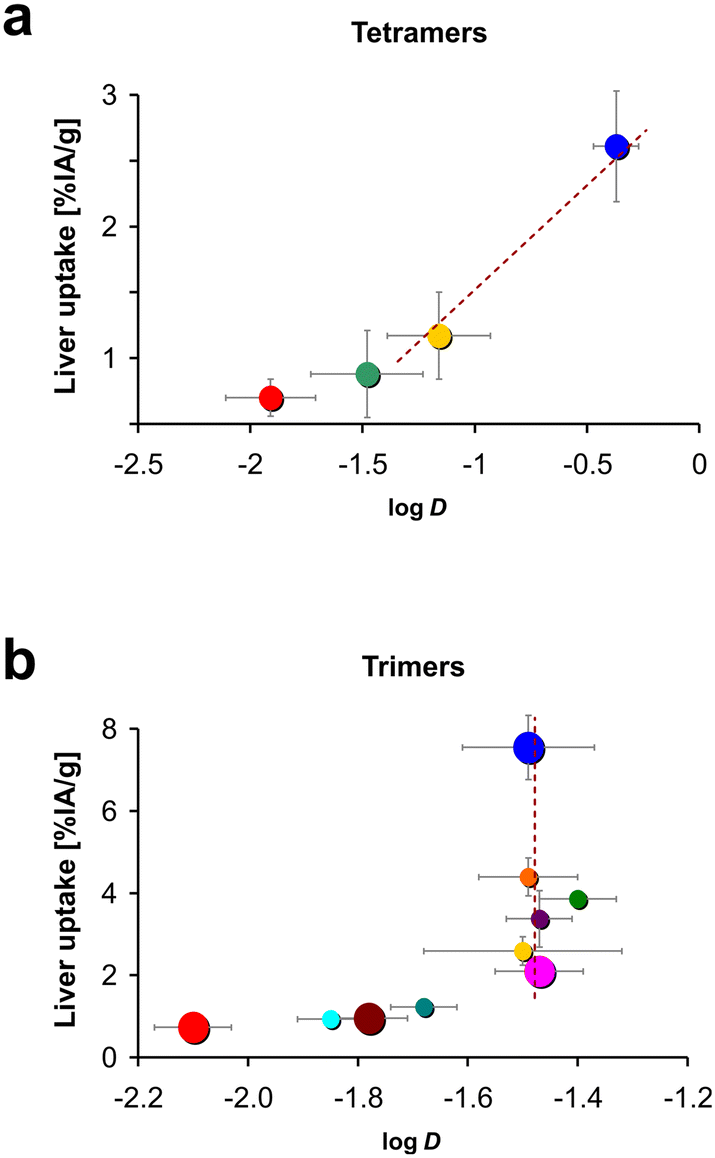 | ||
| Fig. 8 Plots of liver uptakes against octanol–water distribution coefficients (logD) for a) peptide tetramers (this work) and b) Trivehexin-related trimers reported earlier (RSC Med. Chem. 2023, 14, 2429).34 Large-size circles in b highlight the conjugates of the trimer series (Y0, Y2F, Y3F, and Y6, see ref. 34, Fig. 1) which, in terms of percentage and distribution of Phe/Tyr units, are most equivalent to the four tetramers in this work. Color codes of circles correspond to the ones used for the respective compound series; for trimers see ref. 34, Fig. 2a; for the tetramers, see this work, Fig. 4. Note that similar colors in a and b do not necessarily indicate a structural match. | ||
Taken together, we found that in contrast to αvβ6-integrin binding cyclic RGD peptide trimers,34 the tetramers behaved “normally”, i.e., the relationship between polarity and liver absorption adhered to the textbook pattern. Their conformational structure apparently does not promote a synergistic interaction between certain structural elements or the peptide monomers that would lead to non-linear effects in this regard. Therefore, it seems plausible to assume that for the DOTPI conjugates, in vitro polarity proxies such as logD may be more useful to guide early-stage drug development than in case of TRAP trimers.
The importance of tyrosines in peptide multimers
Regardless of whether or not a synergistic interaction of phenylalanines caused a disproportionately high non-specific organ uptake, one fundamental observation made for both groups of cyclopeptide multimers studied (68Ga-TRAP trimers or 177Lu-DOTPI tetramers) was the same: more tyrosines improved the in vivo performance. In particular, undesired non-target uptake was reduced, while target-specific binding in vivo (i.e. tumor uptake) of the conjugates remained more or less unchanged or was even improved. Similar findings have already been forged into a golden rule for the development of synthetic binding proteins. Koide and Sidhu pointed out in 2009 that non-natural engineered proteins showed better binding properties if they consisted predominantly of Tyr, Ser, and Gly.63 These authors described the unique role of Tyr as follows: “The physicochemical properties of tyrosine make it the amino acid that is most effective for mediating molecular recognition, and protein engineers have taken advantage of these characteristics to build tyrosine-rich protein binding sites that outperform natural proteins in terms of affinity and specificity.” However, the same concept does not seem to play a major role for peptides, as there is apparently no systematic tendency to favor tyrosine-containing sequences over comparable peptides without Tyr. To stay with integrin ligands, even a glance into the wealth of RGD peptide literature8,57 quickly reverals that there is apparently no clear preference for either of the two most popular αvβ3-integrin binding motifs, c(RGDyK) and c(RGDfK), although the former contains a D-Tyr and the latter does not.With regard to the present work, it should be mentioned that during the development of the nonapeptides used herein, the sequence cyclo[FRGDLAFp(NMe)K] showed a slightly better αvβ6-integrin affinity and subtype selectivity than its two siblings with one Phe replaced by a Tyr.64 Therefore, it was initially chosen as a lead structure for radiopharmaceutical development.29,58 However, as long as such peptide monomers are not directly used as drugs (either non-functionalized or equipped with simple tags) but as components of larger constructs, it seems reasonable not to rely exclusively on the “best” peptide, but also to consider their “second best” congeners as long as they contain more tyrosines. Our data convincingly demonstrated that it can be advantageous to follow this rationale in the development of radiopharmaceuticals, as our multimers comprising only cyclo[FRGDLAFp(NMe)K] were outperformed by those with tyrosines, although all corresponding Tyr-containing monomers exhibited less favorable in vitro affinities and selectivities.34
Conclusion
The overarching message of our two multimerization studies (RSC Med. Chem., 2023, 14, 2429,34 and this work) is that the development of peptide multimers should preferably be guided by the concepts and rules known from protein engineering. This is because peptide multimers are not simply somewhat larger molecules whose properties are the sum of the characteristics of the monomeric peptide components of which they are composed. Rather, they should be perceived as a special class of small or medium-sized proteins, which have their own special design rules that need to be systematically explored. We conclude that the performance of multimers is not primarily determined by the in vitro properties (especially affinity and selectivity) of the peptide monomers, but rather on the peptides' suitability for the overall macromolecular design concept. One lesson we have learned is that tyrosines in the peptide sequence can contribute a great deal to this suitability. This particular aspect has proven to be crucial for the development of αvβ6 integrin-targeted radiopharmaceuticals.Experimental Section
Materials & methods
Analytical HPLC was performed on a Shimadzu (Kyoto, Japan) Prominence HPLC system equipped with either a Dr. Maisch ReproSil Pur C18 AQ 150 × 4.6 mm, 5 μm, 120 Å (AC1) or a Phenomenex Kinetex C18 150 × 4.6 mm 5 μm column (AC2). Semipreparative HPLC was performed on a Shimadzu HPLC system equipped with either a Dr. Maisch ReproSil Pur C18 AQ 250 × 10 mm, 5 μm, 120 Å column with 5 mL min−1 flow (PC1) or a Dr. Maisch ReproSil Pur C18 150 × 30 mm, 5 μm, 120 Å column with 20 mL min−1 flow (PC2). Solvents for both systems were water (A) and acetonitrile (B) each supplemented with 0.1% (vol) trifluoroacetic acid (TFA). ESI-MS spectra (positive mode) were recorded on a Shimadzu LC-MS 2020 single quadrupol mass spectrometer. Solvents for MS applications were water (A) and acetonitrile (B) each supplemented with 0.1% (vol) formic acid. The other experimental procedures (determination of logD7.4, cultivation of H2009 human lung adenocarcinoma cells, generation of tumor xenografts in CB17 SCID mice, and biodistribution) were performed as described before.28 All animal experiments have been carried out according to applicable law and institutional guidelines of Technical University of Munich, and were approved by the responsible local authority (Regierung von Oberbayern). The H2009 cells were regularly authenticated and tested for mycoplasma contamination. DOTPI-tetraazide,43FF (previously termed AvB6),29FY,34 and YY (also referred to as Tyr2-alkyne)36 were synthesized as described previously.
Syntheses
The synthesis of the Y4 analog GFK-linker functionalized peptide building block GFK-Y4 is outlined in Fig. 9. Corresponding chromatograms and spectra are given in the ESI.† | ||
| Fig. 9 Synthesis of the building block GFK-FY, a derivative of FY featuring the brush border cleavable linker GFK. | ||
:2.5:2.5) (3 × 4 mL). After removal of all volatiles in vacuo, the residue was purified via semipreparative HPLC (10–40% B in 15 min, PC2) and subsequently lyophylized (50%, 430 mg, 1 mmol). m/z: 431.3 [M + H+]+, 453.3 [M + Na+]+. GFK-alkyne RP-HPLC (10–90% B in 15 min, AC2): 6.0 min.
:2.5:2.5) (3 mL) for final deprotection. TFA was removed under a stream of air and the peptide was purified via semipreparative HPLC (20–60% B in 15–min, PC2). After lyophylization GFK-FY was obtained as a colorless solid (85%, 44.1 mg, 28 μmol). m/z: 1576.1 [M + H+]+, 788.5 [M + 2H+]2+. GFK-FY RP-HPLC (10–90% B in 15 min, AC2): 6.2 min.
:1). Copper(II) acetate hydrate (1.49 mg, 7.5 μmol), sodium ascorbate (17.8 mg, 90 μmol) and tris(3-hydroxypropyltriazolylmethyl)amine (THPTA, 0.33 mg, 0.75 μmol) in 500 μL H2O/tBuOH (1:1). Catalyst solution was added to azide/alkyne solution and the mixture was stirred for 10 min at room temperature. Cu species were demetallated as described above with NOTA (6.82 mg, 22.5 μmol) and bispidol (2.68 mg, 4.5 μmol) in 1 mL H2O/ACN pH 2.4 for 1 h at 60 °C. Y0 was obtained as the TFA salt in form of a colorless solid (46.7%, 4.0 mg, 0.70 μmol). m/z: 1403.1 [M + 4H+]4+, 1122.5 [M + 5H+]5+, 935.3 [M + 6H+]6+. 177Lu-Y0 RP-HPLC (0–99% B in 15 min, AC1): tR = 9.4 min.
:2), CuII acetate hydrate (2.0 mg, 10 μmol, 2.2 eq.), Na ascorbate (36.1 mg, 182 μmol, 40.0 eq.). A CuII complex of Y3 was isolated from the CuAAC reaction mixture by preparative HPLC (32–48% B in 20 min, PC1). Afterwards, Cu species were demetallated as described above, followed by final purification by HPLC. (35% B isocratic, PC1). After lyophilization Y3 was obtained as the TFA salt in form of a colorless solid (1.7%, 450 μg, 78 nmol). m/z: 1886.0 [M + 3H+]3+, 1414.8 [M + 4H+]4+, 1132.1 [M + 5H+]5+, 943.6 [M + 6H+]6+. 177Lu-Y3 RP-HPLC (0–99% B in 15 min, AC1): tR = 8.6 min.
:1.7). Cu species were demetallated as described above with NOTA (7.02 mg, 23.15 μmol) in 0.5 mL H2O. GFK-Y4 was obtained as the TFA salt in form of a colorless solid (22.4%, 1.28 mg, 0.17 μmol). m/z: 1480.52 [M + 5H+]5+, 1234.00 [M + 6H+]6+, 1057.71 [M + 7H+]7+. 177Lu-GFK-Y4 RP-HPLC (0–99% B in 15 min, AC1): tR = 8.1 min.
Radiolabeling
Labeling of the tetramers with 177Lu was performed manually in 300 μL of a 1 M NaOAc/HOAc-buffer (pH 5.9). 1–7 nmol (1–7 μL of an 1 mM stock solution) of labeling precursor were incubated with 5–106 μL (32–106 MBq) of 177LuCl3 in 0.04 M HCl (Endolucin Beta, ITM, Germany) for 15–60 min at 95 °C. Completion of the reaction was monitored with radio-TLC or radio-HPLC (for chromatograms see ESI†).SPECT imaging
SPECT imaging was performed for 60 min per animal on a nanoScan SPECT/CT (Mediso, Budapest, Hungary) at different timepoints (1 h, 3 h, 24 h, 72 h) after intravenous injection of approx. 40 MBq [177Lu]Lu-Y8 per animal (n = 2). Reconstruction, image analysis and quantification of SPECT data were performed using Nucline and Interview fusion software (both Mediso).Conflicts of interest
N. G. Q. and J. N. are inventors on patent applications related to αvβ6-integrin binding peptide conjugates and 68Ga-Trivehexin. J. N. and J. Š. are CSO and CEO, respectively, and co-founders of TRIMT GmbH (Radeberg, Germany) who has licensed IP from TU Munich. J. N. is furthermore a member of the Scientific Advisory Board of Radiopharm Theranostics LLC (Carlton, Australia) who has licensed IP from TRIMT GmbH. S. K. receives research support from TRIMT GmbH.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (SFB 824, project A10) is gratefully acknowledged by M. A. Z., N. G. Q., Fr. R., and J. N. This work was supported by the Sächsische Aufbaubank (project INTRA, #100685203), co-financed by the European Union.References
- G. Carlucci, H. J. K. Ananias, Z. Yu, C. Van de Wiele, R. A. Dierckx, I. J. de Jong and P. H. Elsinga, Curr. Pharm. Des., 2012, 18, 2501–2516 CrossRef CAS PubMed.
- S. I. Rudnick and G. P. Adams, Cancer Biother. Radiopharm., 2009, 24, 155–161 CrossRef CAS PubMed.
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef CAS PubMed.
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef CAS PubMed.
- K. Abstiens, M. Gregoritza and A. M. Goepferich, ACS Appl. Mater. Interfaces, 2018, 11, 1311–1320 CrossRef PubMed.
- Y. Mizuno, K. Kimura, S. Onoe, M. Shukuri, Y. Kuge and H. Akizawa, J. Med. Chem., 2021, 64, 16008–16019 CrossRef CAS PubMed.
- P. I. Kitov and D. R. Bundle, J. Am. Chem. Soc., 2003, 125, 16271–16284 CrossRef CAS PubMed.
- C. Liolios, C. Sachpekidis, A. Kolocouris, A. Dimitrakopoulou-Strauss and P. Bouziotis, Molecules, 2021, 26, 1792 CrossRef CAS PubMed.
- S. Liu, Bioconjugate Chem., 2009, 20, 2199–2213 CrossRef CAS PubMed.
- H. Chen, G. Niu, H. Wu and X. Chen, Theranostics, 2016, 6, 78–92 CrossRef CAS PubMed.
- G. Thumshirn, U. Hersel, S. L. Goodman and H. Kessler, Chem. – Eur. J., 2003, 9, 2717–2725 CrossRef CAS PubMed.
- C. Wängler, S. Maschauer, O. Prante, M. Schäfer, R. Schirrmacher, P. Bartenstein, M. Eisenhut and B. Wängler, ChemBioChem, 2010, 11, 2168–2181 CrossRef PubMed.
- P. Kaeopookum, M. Petrik, D. Summer, M. Klinger, C. Zhai, C. Rangger, R. Haubner, H. Haas, M. Hajduch and C. Decristoforo, Nucl. Med. Biol., 2019, 78–79, 1–10 CrossRef CAS PubMed.
- I. Dijkgraaf, J. A. W. Kruijtzer, S. Liu, A. C. Soede, W. K. G. Oyden, F. H. M. Corstens, R. M. J. Liskamp and O. C. Boerman, Eur. J. Nucl. Med. Mol. Imaging, 2007, 34, 267–273 CrossRef CAS PubMed.
- J. Notni, K. Pohle and H. J. Wester, Nucl. Med. Biol., 2013, 40, 33–41 CrossRef CAS PubMed.
- A. M. Singh, W. Liu, G. Hao, A. Kumar, A. Gupta, O. K. Öz, J.-T. Hsieh and X. Sun, Bioconjugate Chem., 2011, 22, 1650–1662 CrossRef CAS PubMed.
- M. Martin, S. Ballal, M. P. Yadav, C. Bal, Y. Van Rymenant, J. De Loose, E. Verhulst, I. De Meester, P. Van Der Veken and F. Roesch, Cancers, 2023, 15, 1889 CrossRef CAS PubMed.
- M. Schäfer, U. Bauder-Wüst, K. Leotta, F. Zoller, W. Mier, U. Haberkorn, M. Eisenhut and M. Eder, EJNMMI Res., 2012, 2, 23 CrossRef PubMed.
- A. Wurzer, J. Pollmann, A. Schmidt, D. Reich, H. J. Wester and J. Notni, Mol. Pharmaceutics, 2018, 15, 4296–4302 CrossRef CAS PubMed.
- D. Reich, A. Wurzer, M. Wirtz, V. Stiegler, P. Spatz, J. Pollmann, H. J. Wester and J. Notni, Chem. Commun., 2017, 53, 2586–2589 RSC.
- N. Böhnke, B. Indrevoll, S. Hammer, A. Papple, A. Kristian, H. Briem, A. Celik, D. Mumberg, A. Cuthbertson and S. Zitzmann-Kolbe, Eur. J. Nucl. Med. Mol. Imaging, 2023, 51, 669–680 CrossRef PubMed.
- K. Steiger, N. G. Quigley, T. Groll, F. Richter, M. A. Zierke, A. J. Beer, W. Weichert, M. Schwaiger, S. Kossatz and J. Notni, EJNMMI Res., 2021, 11, 106 CrossRef CAS PubMed.
- J. Notni, K. Steiger, F. Hoffmann, D. Reich, M. Schwaiger, H. Kessler and H. J. Wester, J. Nucl. Med., 2016, 57, 1618–1624 CrossRef CAS PubMed.
- J. Notni, K. Steiger, F. Hoffmann, D. Reich, T. G. Kapp, F. Rechenmacher, S. Neubauer, H. Kessler and H. J. Wester, J. Nucl. Med., 2016, 57, 460–466 CrossRef CAS PubMed.
- F. Reichart, O. V. Maltsev, T. G. Kapp, A. F. B. Räder, M. Weinmüller, U. K. Marelli, J. Notni, A. Wurzer, R. Beck, H. J. Wester, K. Steiger, S. Di Maro, F. S. Di Leva, L. Marinelli, M. Nieberler, U. Reuning, M. Schwaiger and H. Kessler, J. Med. Chem., 2019, 62, 2024–2037 CrossRef CAS PubMed.
- N. G. Quigley, K. Steiger, F. Richter, W. Weichert, S. Hoberück, J. Kotzerke and J. Notni, EJNMMI Res., 2020, 10, 133 CrossRef CAS PubMed.
- F. S. Di Leva, S. Tomassi, S. Di Maro, F. Reichart, J. Notni, A. Dangi, U. K. Marelli, D. Brancaccio, F. Merlino, H. J. Wester, E. Novellino, H. Kessler and L. Marinelli, Angew. Chem., Int. Ed., 2018, 57, 14645–14649 CrossRef CAS PubMed.
- N. G. Quigley, S. Tomassi, F. S. Di Leva, S. Di Maro, F. Richter, K. Steiger, S. Kossatz, L. Marinelli and J. Notni, ChemBioChem, 2020, 21, 2836–2843 CrossRef CAS PubMed.
- J. Notni, D. Reich, O. V. Maltsev, T. G. Kapp, K. Steiger, F. Hoffmann, I. Esposito, W. Weichert, H. Kessler and H. J. Wester, J. Nucl. Med., 2017, 58, 671–677 CrossRef CAS PubMed.
- T. Curk, J. Dobnikar and D. Frenkel, Design principles for super selectivity using multivalent interactions in Multivalency: Concepts, Research & Applications ed. J. Huskens, L. J. Prins, R. Haag and B. J. Ravoo, John Wiley & Sons Ltd., 2018, pp. 75–101 Search PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- V. I. Böhmer, V. Szymanski, B. L. Feringa and P. H. Elsinga, Trends Mol. Med., 2021, 27, 379–393 CrossRef PubMed.
- P. A. Corning, Complexity, 2002, 7, 18–30 CrossRef.
- N. G. Quigley, F. Richter, S. Kossatz and J. Notni, RSC Med. Chem., 2023, 14, 2564–2573 RSC.
- S. Maschauer, J. Einsiedel, D. Reich, H. Hübner, P. Gmeiner, H. J. Wester, O. Prante and J. Notni, Pharmaceuticals, 2017, 10, 29 CrossRef PubMed.
- N. G. Quigley, K. Steiger, S. Hoberück, N. Czech, M. A. Zierke, S. Kossatz, M. Pretze, F. Richter, W. Weichert, C. Pox, J. Kotzerke and J. Notni, Eur. J. Nucl. Med. Mol. Imaging, 2022, 49, 1136–1147 CrossRef CAS PubMed.
- J. Notni, P. Hermann, J. Havlíčková, J. Kotek, V. Kubíček, J. Plutnar, N. Loktionova, P. J. Riss, F. Rösch and I. Lukeš, Chem. – Eur. J., 2010, 16, 7174–7185 CrossRef CAS PubMed.
- J. Notni, K. Pohle and H. J. Wester, EJNMMI Res., 2012, 2, 28 CrossRef PubMed.
- J. Notni, J. Šimeček and H. J. Wester, ChemMedChem, 2014, 9, 1107–1115 CrossRef CAS PubMed.
- J. Šimeček, P. Hermann, J. Havlíčková, E. Herdtweck, T. G. Kapp, N. Engelbogen, H. Kessler, H. J. Wester and J. Notni, Chem. – Eur. J., 2013, 19, 7748–7757 CrossRef PubMed.
- Z. Baranyai, D. Reich, A. Vágner, M. Weineisen, I. Tóth, H. J. Wester and J. Notni, Dalton Trans., 2015, 44, 11137–11146 RSC.
- J. Notni and H. J. Wester, Chem. – Eur. J., 2016, 22, 11500–11508 CrossRef CAS PubMed.
- A. Wurzer, A. Vágner, D. Horváth, F. Fellegi, H. J. Wester, F. K. Kálmán and J. Notni, Front. Chem., 2018, 6, 107 CrossRef PubMed.
- J. Šimeček, P. Hermann, C. Seidl, F. Bruchertseifer, A. Morgenstern, H. J. Wester and J. Notni, EJNMMI Res., 2018, 8, 78 CrossRef PubMed.
- P. Thakral, S. S. Das, S. Dhiman, D. Manda, C. B. Virupakshappa, D. Malik and I. Sen, Cancer Biother. Radiopharm., 2023, 38, 468–474 CrossRef CAS PubMed.
- S. Kossatz, A. J. Beer and J. Notni, Cancers, 2021, 13, 5958 CrossRef CAS PubMed.
- J. Niu and Z. Li, Cancer Lett., 2017, 403, 128–137 CrossRef CAS PubMed.
- K. Steiger, A. M. Schlitter, W. Weichert, I. Esposito, H. J. Wester and J. Notni, Mol. Imaging, 2017, 16, 1536012117709384 CrossRef PubMed.
- M. Lawaetz, A. Christensen, K. Juhl, K. Karnov, G. Lelkaitis, A. M. Kanstrup Fiehn, A. Kjaer and C. von Buchwald, Int. J. Mol. Sci., 2023, 24, 3853 CrossRef CAS PubMed.
- N. Ahmed, C. Riley, G. E. Rice, M. A. Quinn and M. S. Baker, J. Histochem. Cytochem., 2002, 50, 1371–1379 CrossRef CAS PubMed.
- S. Hazelbag, G. G. Kenter, A. Gorter, E. J. Dreef, L. A. Koopman, S. M. Violette, P. H. Weinreb and G. J. Fleuren, J. Pathol., 2007, 212, 316–324 CrossRef CAS PubMed.
- A. S. Berghoff, A. K. Kovande, T. Melchardt, R. Bartsch, J. A. Hainfellner, B. Sipos, J. Schittenhelm, C. C. Zielinski, G. Widhalm, K. Dieckmann, M. Weller, S. L. Goodman, P. Birner and M. Preusser, Clin. Exp. Metastasis, 2014, 31, 841–851 CrossRef CAS PubMed.
- A. N. Elayadi, S. N. Samli, L. Prudkin, Y. H. Liu, A. Bian, X. J. Xie, I. I. Wistuba, J. A. Roth, M. J. McGuire and K. C. Brown, Cancer Res., 2007, 67, 5889–5895 CrossRef CAS PubMed.
- C. Suzuki, T. Uehara, N. Kanazawa, S. Wada, H. Suzuki and Y. Arano, J. Med. Chem., 2018, 61, 5257–5268 CrossRef CAS PubMed.
- R. H. Kimura, A. Iagaru and H. H. Guo, Front. Nucl. Med., 2023, 3, 1271208 CrossRef.
- N. G. Quigley, N. Czech, W. Sendt and J. Notni, Eur. J. Nucl. Med. Mol. Imaging, 2021, 48, 4107–4108 CrossRef CAS PubMed.
- J. Notni, Pharmaceuticals, 2022, 16, 56 CrossRef PubMed.
- S. F. Färber, A. Wurzer, F. Reichart, R. Beck, H. Kessler, H. J. Wester and J. Notni, ACS Omega, 2018, 3, 2428–2436 CrossRef PubMed.
- A. S. Parihar, S. Chopra and V. Prasad, Transl. Oncol., 2022, 15, 101295 CrossRef CAS PubMed.
- H. Akizawa, T. Uehara and Y. Arano, Adv. Drug Delivery Rev., 2008, 60, 1319–1328 CrossRef CAS PubMed.
- Y. Arano, Nucl. Med. Biol., 2021, 92, 149–155 CrossRef CAS PubMed.
- E. J. Rolleman, M. Melis, R. Valkema, O. C. Boerman, E. P. Krenning and M. de Jong, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 1018–1031 CrossRef PubMed.
- S. Koide and S. S. Sidhu, ACS Chem. Biol., 2009, 4, 325–334 CrossRef CAS PubMed.
- O. V. Maltsev, U. K. Marelli, T. G. Kapp, F. S. Di Leva, S. Di Maro, M. Nieberler, U. Reuning, M. Schwaiger, E. Novellino, L. Marinelli and H. Kessler, Angew. Chem., Int. Ed., 2016, 55, 1535–1539 CrossRef CAS PubMed.
- T. K. Heath, M. R. Lutz, C. T. Reidl, E. R. Guzman, C. A. Herbert, B. P. Nocek, R. C. Holz, K. W. Olsen, M. A. Ballicora and D. P. Becker, PLoS One, 2018, 13, e0196010 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00073k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |