
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new class of 7-deazaguanine agents targeting autoimmune diseases: dramatic reduction of synovial fibroblast IL-6 production from human rheumatoid arthritis patients and improved performance against murine experimental autoimmune encephalomyelitis†
Michelle
Cotter
a,
Shauna M.
Quinn
b,
Ursula
Fearon
c,
Sharon
Ansboro
c,
Tatsiana
Rakovic
c,
John M.
Southern
 *a,
Vincent P.
Kelly
*b and
Stephen J.
Connon
*a
*a,
Vincent P.
Kelly
*b and
Stephen J.
Connon
*a
aSchool of Chemistry, Trinity College, Trinity Biomedical Sciences Institute, 152-160 Pearse Street, Dublin, Ireland. E-mail: southerj@tcd.ie; connons@tcd.ie
bSchool of Biochemistry & Immunology, Trinity College, Trinity Biomedical Sciences Institute, 152-160 Pearse Street, Dublin, Ireland. E-mail: kellyvp@tcd.ie
cSchool of Medicine, Trinity College, Trinity Biomedical Sciences Institute, 152-160 Pearse Street, Dublin, Ireland
First published on 28th March 2024
Abstract
A simple in vitro assay involving the measurement of IL-6 production in human synovial fibroblasts from rheumatoid arthritis patients has been utilised to select candidates from a targeted library of queuine tRNA ribosyltransferase (QTRT) substrates for subsequent in vivo screening in murine experimental autoimmune encephalomyelitis (EAE – a model of multiple sclerosis). The in vitro activity assay discriminated between poor and excellent 7-deazaguanine QTRT substrates and allowed the identification of several structures which subsequently outperformed the previous lead in EAE. Two molecules were of significant promise: one rigidified analogue of the lead, and another considerably simpler structure incorporating an oxime motif which differs structurally from the lead to a considerable extent. These studies provide data from human cells for the first time and have expanded both the chemical space and current understanding of the structure–activity relationship underpinning the remarkable potential of 7-deazguanines in a Multiple Sclerosis disease model.
Introduction
Multiple sclerosis (MS) is the most common chronic autoimmune disease of the central nervous system1 affecting ca. 3 million patients worldwide; with rising prevalance.2 Considerable therapeutic strides have been made through the development of disease-modifying drugs – however a curative treatment remains elusive.1,3 The search for new targets for the treatment of this multifaceted disease is hence both broadening and intensifying.4Recently it has been shown that one such target is tRNA (1, Fig. 1). Queuine (2) is a hypermodified 7-deazaguanine nucleobase which is irreversibly incorporated (in place of an expelled existing guanine (3) base) into eukaryotic tRNAs 1 associated with histidine, tyrosine, aspartic acid and asparagine at the wobble position to give new queuosine-modified tRNA 4.5 This highly unusual reaction is catalysed by the heterodimeric enzyme queuine tRNA ribosyltransferase (QTRT, 5).6–10 Queuine is exclusively synthesised by eubacteria and must be obtained from either dietary sources or gut fauna by mammals,11,12 it is not an essential nutrient in mice13 however queuine-deprived mice exhibit a pathological sensitivity to tyrosine deficiency.5 A comprehensive picture of queuine's biological raison d'etre has not emerged.
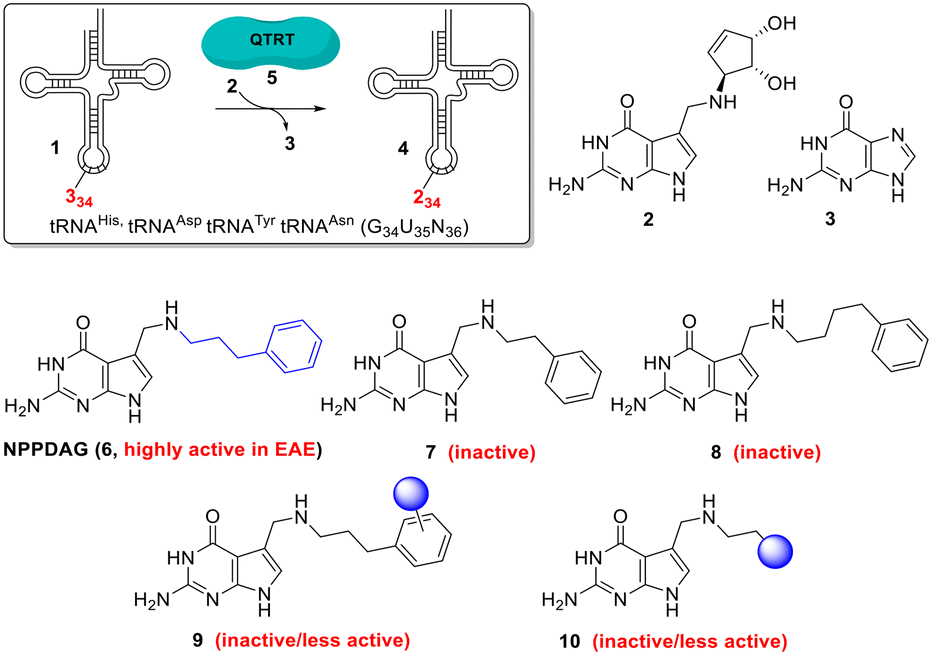 | ||
| Fig. 1 The QTRT-catalysed wobble-base exchange reaction involving tRNAHis,Asp,Tyr,Asn, the structures of queuine, guanine, NPPDAG and related analogues. | ||
Modification of tRNA by queuine has been shown to play a role in the control of protein translational speed/folding and codon usage bias.14,15 Influence over cell proliferation5 and metabolism16 has been demonstrated; levels of queuine modification have been correlated with age/cellular differentiation stage17–19 and negatively correlated with the progression of several cancers.20–22 These modalities of influence associated with queuine are interesting in the context of the growing attention being directed at potential treatments for MS based on promoting differentiation to regulatory T- (Treg) cells.4,23,24
While QTRT will catalyse the base exchange exclusively involving the 4 aforementioned tRNAs and eschew the modification of the myriad of other RNA molecules in the cell,25 it exhibits a surprisingly wide substrate scope with respect to the deazaguanine component.25,26 This allows the possibility of exploiting QTRT to rapidly, conveniently and dramatically alter the properties of tRNA at the anticodon loop, provided that it has not already been elaborated with queuine. Hypomodification by queuine is a characteristic of undifferentiated, rapidly proliferating cells20–22,27,28 and thus by extension we hypothesised that tRNA in clonally expanding T-cells could be susceptible to modification with artificial nucleobases in a fashion that mature, terminally differentiated cells would not. This would allow the selective targeting of a subset of the proliferative T-cells driving autoimmune disease in a way to which the majority of cell populations are inert. We previously reported the synthesis and evaluation of the deazaguanine NPPDAG (6, Fig. 1) in murine experimental autoimmune encephalomyelitis (EAE).28 The artificial nucleobase is a substrate for QTRT and is incorporated into tRNA while avoiding genotoxicity through the action of the hypoxanthine–guanine phosphoribosyltransferase (HPRT) enzyme. NPPDAG possessed unprecedented activity: administration of 5 daily doses (30 mg Kg−1) led to complete symptom remission, while QTRT-deficient mice were refractory to therapy. Levels of effector and cytotoxic T-cells in both the periphery and the brain of treated animals were markedly reduced without overt immunosuppression via general lymphocyte population diminution. Spleen cells from NPPDAG-treated animals remained functional – they proliferated normally in the presence of a pan-stimulant yet remained unresponsive when re-exposed to EAE-inducing antigen.
NPPDAG represents a queuine analogue where the aminomethyl-7-deazaguanine moiety has been retained and the cyclopentene diol unit has been exchanged for a substituent comprising a 3-carbon chain and an aromatic ring (highlighted in blue in structure 6). Subsequent studies29 aimed at optimising the structure of this substituent revealed an exceptionally narrow structure–activity relationship in the context of EAE treatment (i.e. through the modification of the behaviour of tRNA by the covalently incorporated artificial nucleobase). For instance, despite demonstrably serving as efficient QTRT substrates; both 2- and 4-carbon chain analogues (7 and 8 respectively, Fig. 1) were inactive as immunomodulatory agents, as were the vast majority of variants incorporating substituted benzene rings of general type 9 (electron withdrawing/electron donating substituents, ring extensions, heterocyclic analogues (save a 3-thiophenyl isostere), protic substituents, all substitution patterns). Removal of the aromatic ring and replacement with either aliphatic chains or queuine-fragments (i.e.10, Fig. 1) were also not advantageous.
Results and discussion
Compound efficacy was initially determined using two in vitro assays – a screen monitoring the secretion of IFN-γ by spleen cells from EAE-diseased mice and a CD3+ T cell proliferation assay involving spleen cells. Both assays used cells from wildtype and QTRT-deficient mice. If sufficiently active, screening progressed to the murine EAE model.29 While this regime allowed the identification of potentially efficacious candidate molecules, we wished to develop a more streamlined initial screening methodology – preferably not reliant on a two-assay system and the availability of cells from EAE-diseased animals.In addition, we were interested in the activity of NPPDAG and variants as potential treatments of rheumatoid arthritis (RA) – a painful and mobility-attenuating chronic condition among the most prevalent autoimmune diseases which affects ca. 1% of the global population.30 While RA and MS are distinct and complex multi-faceted autoimmune conditions, the contributions from activated macrophages, Th1 and Th17 cells (which are key in MS) towards driving RA are significant,31 and we had shown that deazaguanine analogues could significantly reduce the populations of these (inter alia) pathological cells in EAE diseased mice.28 Given that the mode of action of NPPDAG is not confined exclusively to direct effects on T-cells, we arrived at an unusual hypothesis: that evaluation of the influence of NPPDAG analogues on synovial fibroblasts from human RA patients (based on changes in concentrations of the signature cytokine IL-6 (ref. 32)) could not only serve as a simple screen to establish if deazaguanines could be active in RA, it could also possess predictive utility in EAE and additionally provide useful information regarding the behavior of these compounds in human cells. In this regard we note that dimethyl fumarate – an FDA-approved drug for use in MS and psoriasis which influences the proinflammatory Th17–Th1 response – has very recently shown promise in reducing IL-6 levels from fibroblast-like synoviocytes from human RA patients.33,34
In preliminary experiments, synovial fibroblasts from RA patients were activated with TNF-α and treated with NPPDAG (6) at either 100 μM or 200 μM and the levels of the pro-inflammatory cytokine IL-6 measured using an ELISA assay. DMSO (4% in PBS buffer) was utilised as a control. Gratifyingly, a dose-dependent reduction in the cytokine concentration was observed after 72 h (Fig. 2).
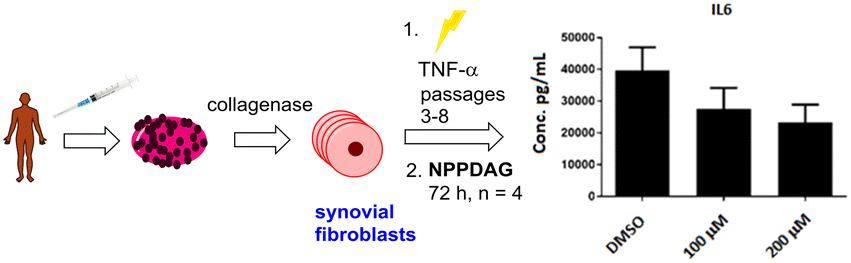 | ||
| Fig. 2 Reduction in production of IL-6 by synovial fibroblasts from RA patients in the presence of NPPDAG. | ||
With proof of concept in human RA synovial fibroblasts in hand, attention now switched to the activity of NPPDAG analogues. Earlier studies29 had shown that changes to the length of the linker chain associated with NPPDAG led to compounds inactive in EAE screening this constraint, coupled with the requirement for a simple aromatic moiety at the chain terminus, pointed to a requirement for the NPPDAG side chain to attain a particular conformation to alter the properties of the tRNA anticodon loop. We therefore began with retention of the NPPDAG structural core and a focus on mild rigidification of the linker chain via substitution (Fig. 3). All compounds were evaluated at 200 μM concentrations and unless stated, were excellent substrates for QTRT and could be incorporated into tRNA.25 DMSO (4% in PBS buffer) was again used as a control, alongside IgG and positive controls (Tofacitinib – Pfizer, Janus kinase inhibitor, 1 μM) and Adalimumab (Humira – Abbvie, anti-TNF-α monoclonal antibody, 1 μM).
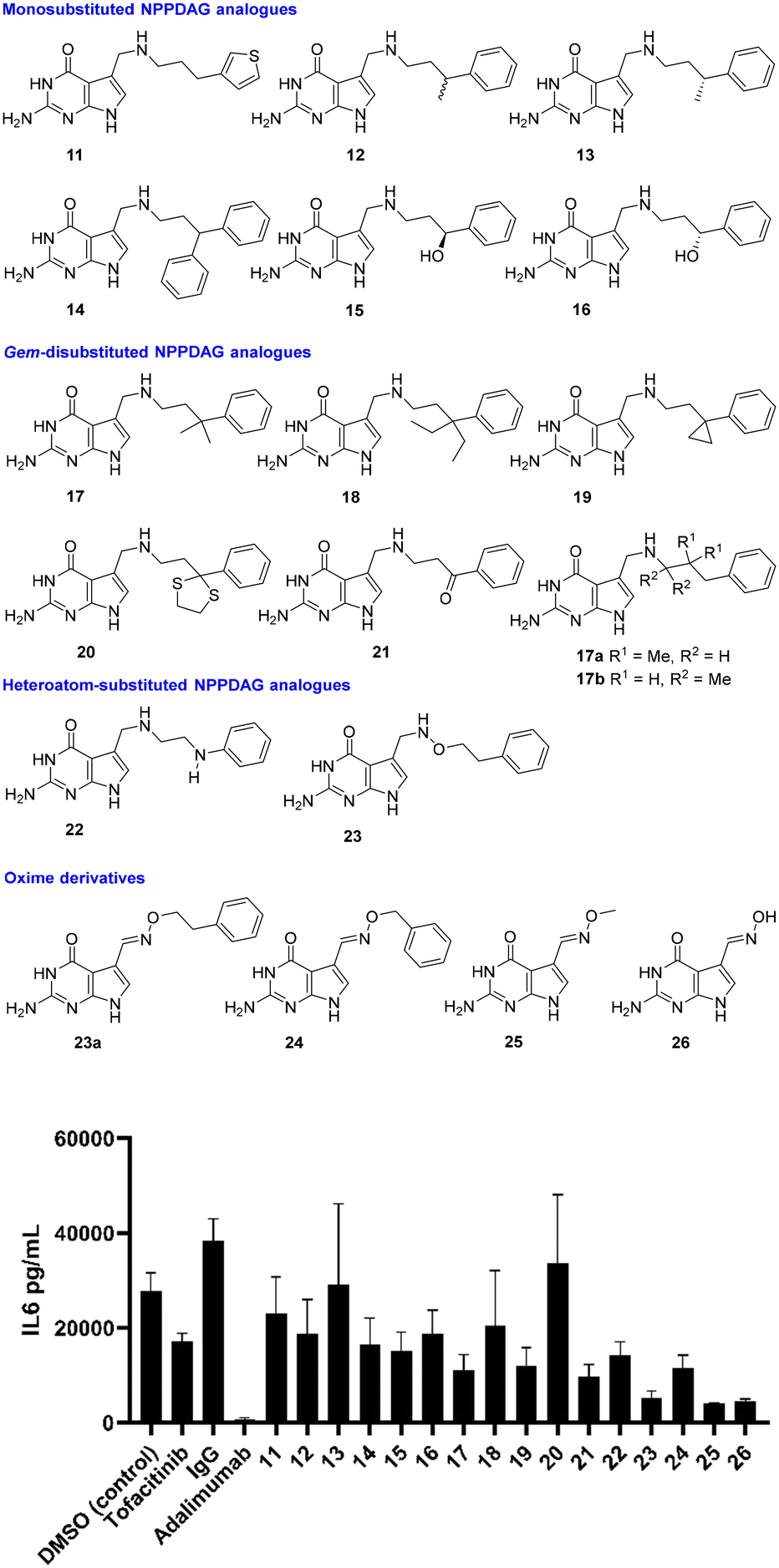 | ||
| Fig. 3 Reduction in production of IL-6 by synovial fibroblasts from RA patients in the presence of a library of NPPDAG derivatives. | ||
The 3-thiophene derivative 11 was previously29 shown to possess potent in vitro activity in cells from EAE-diseased mice but was not evaluated in EAE itself – here it proved capable of lowering IL-6 production relative to vehicle control, with activity levels close to those obtained using 6. Methyl substitution at the benzylic position (i.e. racemate 12) resulted in modest improvement, while the (R)-enantiomer of the same compound (i.e.13) was essentially inactive. This would signal significant activity associated with the (S)-antipode, which was not synthesised due to improved activity obtained from simpler materials in the study. Installation of a second phenyl unit at the same position gave rise to 14, which possessed activity levels akin to 11; while hydroxyl substitution at the benzylic position (i.e. enantiomers 15 and 16) seemed only marginally advantageous where the stereogenic center had the (S)-configuration.
With little achieved from mono-substitution at the benzylic position, gem-disubstituted analogues were next evaluated. These, it was envisaged, would be significantly less conformationally flexible than NPPDAG itself, without being considered rigid materials. The dimethyl derivative 17 exhibited markedly improved activity – considerably superior to either NPPDAG or any of the mono-substituted derivatives 11–16. Augmentation of the steric demand at the quaternary carbon atom (i.e.18) resulted in a less dramatic fall in IL-6 concentration, however it should be noted that the substrate competency of 18 toward QTRT is <20% of that associated with 6.25 The smaller gem-disubstituted cyclopropyl analogue 19 also exhibited impressive activity – albeit to a lesser extent than observed using 17.
Consistent with this trend was the poor performance of the dithiolane 20 (bulky benzylic position, mediocre QTRT compatibility) and the excellent activity detected in experiments involving the less sterically demanding precursor ketone 21 – which could be readily manipulated by QTRT.25 While it is tempting to consider the electronic effect of the ketone on the aromatic ring, we have previously shown (multiple examples) that the installation of electron withdrawing substituents either on or in the ring was not advantageous29 – thus, at this juncture we would suggest that activity here could be primarily due to rigidification. Given the superiority of 17 over 6 (Fig. 1 and 3), isomers 17a–b were synthesised where the gem-dimethyl unit was relocated to either the middle of the chain or adjacent to the secondary amine group. The former material was highly insoluble and the latter a poor QTRT substrate,25 so neither were carried forward for screening.
The final module of screening in RA was intended to be a short examination of the influence of heteroatoms in the chain itself, as exemplified by the aniline derivative 22 and the alkoxyamine 23. Interestingly, both proved capable of ameliorating the production of IL-6 to a greater degree than 6, with 23 proving the most potent compound screened thus far. The factors underpinning the striking increase in potency associated with 23 were not immediately obvious. With an initial view to investigating the effects of further rigidification on in vitro compound behaviour, we prepared 23a – the (E)-oxime variant of 23. This compound was a completely incompetent substrate for QTRT25 and was not subsequently evaluated in the synovial fibroblast assay. The homologue with a chain shorter by one methylene unit however (i.e.24) proved approximately as active as 17 – which is extraordinary in light of the fact that it does not possess a chain of sufficient length between the exocyclic N-atom and the aromatic ring to be compatible with the established SAR (see Fig. 1).29 Accordingly, simple O-methyl-substituted and unsubstituted oxime derivatives of 7-deazaguanine (i.e.25 and 26) were evaluated. These are excellent QTRT substrates25 which led to the lowest IL-6 concentrations detected in the study.
The synovial fibroblast screen identified a number of analogues of 6 which served as excellent QTRT substrates and possessed significantly superior activity to the lead structure (e.g.17, 21 and 23). In addition, 2 simple oximes with structures not anticipated to be consistent with activity showed particular promise. Returning to our initial hypothesis regarding the predictive power of the synovial fibroblast assay in identifying candidates for screening in EAE, a selection of these compounds were evaluated in vivo in chronic, monophasic, myelin oligodendrocyte glycoprotein (MOG33–35)-mediated murine EAE (Fig. 4). Mice were treated daily via intraperitoneal injection when a disease score 2 (limp tail, wobbly gait – usually 9 days post immunisation) was attained. A lower dose equimolar to 15 mg Kg−1 of NPPDAG (30 mg Kg−1 of NPPDAG leads to full remission) was utilised to facilitate efficacy comparisons.
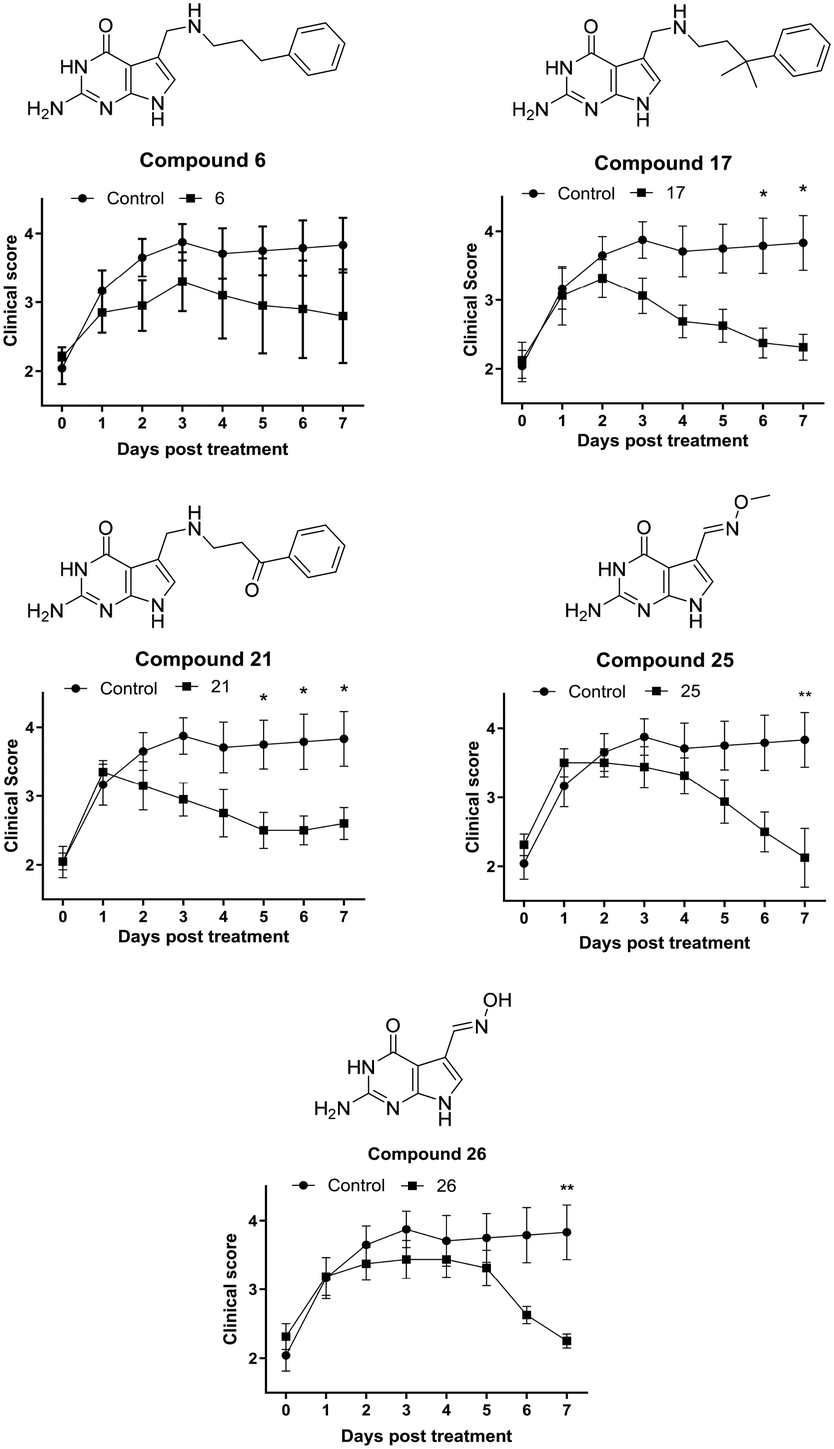 | ||
| Fig. 4 Evaluation of selected compounds in EAE. Upon reaching a clinical score of approximately 2, EAE diseased female mice were administered PBS as control or equimolar concentrations of 6 (15 mg Kg−1), 17 (16 mg Kg−1), 21 (16 mg Kg−1) 25 (9 mg Kg−1) or 26 (9 mg kg−1) i.p. daily for 7 days, n = 5. Animals were monitored daily for clinical score. Data represent the mean ± S.E.M. *P < 0.05, **P < 0.01, by two-way Anova with post hoc pair-wise comparison. | ||
On treatment with 6, the disease course diverged from the controls after one day: progression peaked at day 3 post treatment and the animals condition gradually improved thereafter to a final disease score just under 3 after 7 days – consistent with earlier studies using 6 at this lower dose.28 Gratifyingly, administration of compound 17 – the gem-dimethyl analogue of 6 – resulted in a peak at day 2, a more rapid disease remission and an improved final score. The ketone 21 also proved capable of arresting disease progression after one day of treatment, but does not appear as efficacious overall as 17. The more difficult-to-synthesise 23 was not selected for full evaluation, however in a 4 day treatment study it performed similarly to 17 (data not shown). The methyl-oxime 25 exhibited a small deviation in behavior – again disease progression was arrested after one day of treatment, followed by 3 days of modest improvement. Thereafter remission was more rapid – with the animals returning to a disease score of 2 after 7 days. Its O-unsubstituted analogue 26 displayed a similar profile – albeit with a longer plateau phase post treatment and a marginally higher final disease score.
Experimental
In vitro rheumatoid arthritis synovial fibroblast (RASFC) assays
PBMC toxicity screen
To ensure that the reductions seen in IL-6 were not the result of compound toxicity, tests were performed on matched PBMCs. PBMCs were isolated from whole blood by Ficoll density gradient centrifugation (Lymphoprep™; STEMCELL technologies, Canada) and were incubated for 24 h with the selected compounds at four different concentrations (5, 50, 100 and 200 μM) as well as DMSO controls as shown. Cells were then washed in PBS and stained with a Near IR viability dye for 30 min. Cells were washed and immediately analysed on a flow cytometer. PBMC cell viability assays for selected compounds can be found in the ESI† (Fig. S1).In vivo experimental autoimmune encephalomyelitis (EAE) experiments35
For EAE studies, mice were maintained under specific pathogen-free conditions. Experiments were conducted under licence from the Health Products Regulatory Authority (Ref: A19136/P086), with the approval of the Trinity College Dublin Animal Research Ethics Committee.• Complete Freund's Adjuvant (CFA) was prepared by diluting complete Freund's adjuvant (4 mg mL−1M. tuberculosis; Chondrex) to a concentration of 2 mg mL−1 in incomplete Freund's adjuvant.
• MOG solution was prepared by re-suspending an appropriate amount of mouse MOG(35–55) peptide (GenScript; supplied frozen down as a powder) in PBS (Sigma) to give a stock solution of 5 mg mL−1.
• CFA/MOG Emulsion comprised a 50% suspension of CFA, 10% MOG solution in 40% PBS solution (i.e. 10% of the solution above). It was prepared by the following method:
a. PBS solution and MOG solution were first placed into a 50 ml plastic tube with CFA subsequently placed on top.
b. The mixture was homogenised using a laboratory bench homogeniser at a speed of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm, until the tube can be turned upside down without the emulsion spilling. The correct consistency of the MOG emulsion is necessary to ensure dispersion does not occur in the animal after administration. The consistency was tested as follows: PBS solution was placed in a petri dish. MOG emulsion was withdrawn from the 50 mL tube with a syringe using a blue pipette tip. A drop of MOG emulsion was added to the PBS. A correct preparation should maintain its consistency and not disperse into the PBS.
500 rpm, until the tube can be turned upside down without the emulsion spilling. The correct consistency of the MOG emulsion is necessary to ensure dispersion does not occur in the animal after administration. The consistency was tested as follows: PBS solution was placed in a petri dish. MOG emulsion was withdrawn from the 50 mL tube with a syringe using a blue pipette tip. A drop of MOG emulsion was added to the PBS. A correct preparation should maintain its consistency and not disperse into the PBS.
c. Once prepared, the emulsion was loaded into a 1 mL Tuberculin syringe fitted with a 23 Gauge needle.
• Pertussis toxin (PT; Katetsuken) was made up in the morning of disease induction, with sufficient volume prepared for both day 0 and day 2 injections. PT stock was 200 μg ml−1 which equals 200 ng μL−1. Each mouse was injected with 200 ng (equating to 1 μL of PT stock) in 200 μL PBS.
Induction of disease. • Day 0
a. The mice were weighed and marked with a permanent marker on the tail.
b. Each mouse was injected intraperitoneally with 200 μL of PT (tuberculin syringe and a 27 Gauge needle).
c. Subsequently, each mouse was subcutaneously injected on the back with 200 μL of CFA/MOG.
• Day 2
a. Each mouse was weighed again.
b. Animals were injected intraperitoneally with 200 μL of PT prepared previously.
• Day 5-onwards
a. The mice were weighed every day and the data recorded.
b. The first day of overt disease occurs approximately from day 9 onwards.
0.25 little or no weight loss, but loss of power in the tip of the tail.
0.5 small amount of weight loss, but loss of power to half of the tail.
1.0 small amount of weight loss, but loss of power to entire tail.
1.25/1.5 increased body weight loss and some loss in tail power.
2.0 characteristic cowboy gait (animal rear extended).
2.5 characteristic cowboy gait with maintained ability to support rear.
3.0 movement of rear legs, but unable to support rear.
3.5 loss of movement in only one rear leg and unable to support rear.
3.75 loss of rear leg movement and unable to support rear.
4.0 complete loss of movement in rear legs with locomotion using only front legs.
4.25/4.5 weak front leg movement and complete paralysis of rear.
4.5 animal locomotion considered severe enough to euthanise the animal.
5.0 any animal discovered dead overnight.
- A 100 mM solution of dry, powdered compound was prepared in DMSO.
- The 100 mM stock was diluted 25 fold in PBS to give a compound concentration of 4 mM.
- Each mouse was injected intraperitoneally with 200 μL of diluted compound everyday for 7 days.
- This approach allowed the assessment of compounds on an equimolar basis to alleviate effects influenced by the different molecular weight of the compounds.
- Control animals administered PBS solution containing 4% DMSO (n = 6).
- Compound 6 treated animals, 15 mg Kg−1 (n = 5).
- Compound 7 treated animals, 16 mg Kg−1 (n = 4).
- Compound 21 treated animals, 16 mg Kg−1 (n = 5).
- Compound 23 treated animals, 15 mg Kg−1 (n = 5, 4 day study).
- Compound 25 treated animals, 9 mg Kg−1 (n = 4).
- Compound 26 treated animals, 9 mg Kg−1 (n = 4).
Animals that failed to succumb to disease or whose disease had progressed to a score of 3 or beyond were not included in the study.
Deazaguanine substrates
Compounds 6, 11–26 were prepared previously to evaluate their QTRT compatibility and their characterisation – melting points, HRMS and copies of the associated 1H NMR and 13C NMR spectra which attest to >95% purity have been reported.25,28,29 The synthesis of key compounds 6, 17, 21, 25 and 26 evaluated in EAE are described below – associated 1H- and 13C NMR spectra have been provided in the ESI.†General
Proton nuclear magnetic resonance spectra were recorded on a Bruker 400 MHz or 600 MHz (as specified) or Agilent 400 MHz spectrometer in DMSO-d6 relative to residual DMSO (δ = 2.50 ppm). Chemical shifts are reported in ppm and coupling constants in Hertz. Carbon (100 MHz and 150 MHz) spectra were recorded on the same instruments with total proton decoupling. All melting points are uncorrected. Infrared spectra were obtained on a Perkin Elmer spectrophotometer. Flash chromatography was carried out using silica gel, particle size 0.04–0.063 mm. TLC analysis was performed on precoated 60F254 slides, and visualised by either UV irradiation, KMnO4 staining, or phosphomolybdic acid staining as appropriate. All chemicals were obtained from commercial sources and used as received unless otherwise stated.General procedure A: synthesis amine hydrochloride salts
:1 dichloromethane:methanol) to yield the desired compound as a white solid (210 mg, 81%), m.p. > 300 °C (decomp.).
A solution of the trityl-protected compound (210.0 mg, 0.39 mmol) in 1.25 M methanolic HCl (3 mL) was prepared and stirred at room temperature for 16 h. The precipitated product was removed by filtration and washed with dichloromethane to yield the title compound as a white powder, (84 mg, 65%, 53% over two steps), m.p. >300 °C (decomp.). 1H NMR (400 MHz, DMSO-d6) δ 1.84–1.98 (2H, m), 2.63 (2H, t, J 7.6), 2.85–2.95 (2H, m), 4.13 (2H, t, J 4.8), 6.51 (2H, bs, NH), 6.80 (1H, s), 7.14–7.21 (3H, m), 7.23–7.30 (2H, m), 9.10 (2H, bs, NH), 11.02 (1H, bs, NH), 11.29 (1H, bs, NH). 13C NMR (150 MHz, DMSO-d6): δ 27.7, 32.3, 43.0, 45.6, 98.7, 108.9, 117.9, 126.5, 128.7, 128.9, 141.1, 152.6, 153.4, 160.7. HRMS (m/z ESI+): found: 298.1662 ([M + H]+; C16H20N5O requires: 298.1668). νmax (film)/cm−1: 1456, 1625, 2443, 2713, 2756, 2873, 2933, 3184.
:3 hexane ethyl acetate) to yield the desired compound as a white solid (137 mg, 0.30 mmol, 43%), m.p. 186–190 °C.
A cooled (0 °C) solution of the trityl-protected compound (58 mg, 0.13 mmol) in 1 M HCl in dioxane (4.0 mL) was prepared and stirred for 10 minutes before the ice bath was removed and the reaction mixture stirred for 2 h at room temperature. The precipitated product was removed by filtration and washed with diethyl ether to yield the title compound as a white powder, (10 mg, 0.041 mmol, 32%), m.p. >250 °C (decomp.). 1H NMR (400 MHz, DMSO-d6): δ 3.87 (1H, s), 6.18 (2H, bs), 7.38 (1H, s), 7.84 (1H, s), 10.46 (1H, bs), 11.47 (1H, bs). 13C NMR (100 MHz, DMSO-d6): δ 61.7, 97.4, 108.7, 123.4, 138.8, 148.5, 152.6, 158.7. HRMS (m/z – ESI): found: 230.0668 ([M + Na]+ C8H9N5NaO2; requires: 230.0654). νmax (film)/cm−1: 1053, 1593, 1672, 2854, 3132.
:3 hexane ethyl acetate) to yield the desired compound as a white solid (220 mg, 0.40 mmol, 56%), m.p. >300 °C.
A cooled (0 °C) solution of the trityl-protected compound (203 mg, 0.37 mmol) in 1 M HCl in dioxane (4.0 mL) and methanol (1.0 mL) was prepared and stirred for 10 minutes at 0 °C before the ice bath was removed and the reaction mixture stirred for 2 h at room temperature. The precipitated product was removed by filtration and washed with diethyl ether to yield the title compound as an off-white powder, (70 mg, 0.30 mmol, 81%), m.p. >250 °C (decomp.). 1H NMR (400 MHz, DMSO-d6): δ 7.52 (1H, d, J 2.4), 7.84 (1H, s), 10.87 (1H, bs), 11.60 (1H, bs). 13C NMR (100 MHz, DMSO-d6): δ 97.9, 109.8, 124.0, 138.5, 144.3, 152.5, 158.4. HRMS (m/z – APCI): found: 192.0525 ([M − H]− C7H6N5O2; requires: 192.0521). νmax (film)/cm−1: 1578, 1671, 2625, 2971, 3088, 3676.
Conclusions
In summary, it has been found that a simple in vitro screen involving the measurement of IL-6 production by synovial fibroblasts from human RA patients could be used to select compounds for in vivo evaluation in murine EAE. The in vitro assay revealed several deazaguanine QTRT substrates – from a library targeting side-chain rigidification – as being capable of reducing the levels of the key pro-inflammatory cytokine beyond that (relative to DMSO vehicle control) associated with the use of lead compound NPPDAG (6). In cases where the introduction of chain-rigidifying functionality also brought about (previously determined25) poorer QTRT substrate competency (e.g.18 and 20); commensurately weaker suppression of IL-6 production resulted. The somewhat unexpected superiority of the oxy-derivative of 6 (i.e.23) prompted the evaluation of simple oxime derivatives, which resulted in the identification of the considerably more potent derivatives 25 and 26. Five deazaguanines were selected for full evaluation in murine EAE at concentrations equivalent to 15 mg Kg−1 of NPPDAG, all of which resulted in superior outcomes in terms of early arrest of disease progression and lower final clinical scores than when 6 was utilised. The gem-dimethyl analogue of 6 (i.e.17) and the methyl oxime 25 appear particularly promising and warrant further investigation. The emergence of the oximes as active candidates is fascinating given that they are excellent QTRT substrates yet are located outside the chemical space outlined in extensive previous SAR studies focused on molecules related to 6. In this regard it is interesting that the genotoxic QTRT substrate 6-thioguanine (which also represents a considerably truncated pharmacophore relative to 6) has also been shown to be active against EAE in mice without the HPRT enzyme (responsible for its toxicity).28 At this juncture, it appears that a small O-substituent in the oxime series (i.e. –H or Me) and an O-atom replacing the C-atom adjacent to the methylene amine in derivatives of NPPDAG are most beneficial from an activity standpoint. Investigations to determine (inter alia) if the oximes operate in a similar fashion to 6-thioguanine without the attendant genotoxicity are underway.Author contributions
S. J. C., J. M. S., V. P. K. and U. F. designed the experimental protocol and drafted the manuscript. M. C. performed the synthesis of the candidate molecules. S. M. Q. performed the EAE experiments and S. A. and T. R. performed the in vitro assays. All authors read and approved the final manuscript.Conflicts of interest
S. J. C., J. M. S. and V. P. K. are founders of a company – Azadyne Ltd. – involved in developing drug candidates for the treatment of autoimmune disease. No data or savoir-faire generated by Azadyne is incorporated into this manuscript.Acknowledgements
Financial support from the School of Chemistry, Trinity College Dublin for a studentship for M. C. is gratefully acknowledged. The work described herein also emanated from research conducted with the financial support of Science Foundation Ireland under Grant number 17/TIDA/5029 and Enterprise Ireland Grant number CF/2015//0029. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.References
- D. S. Reich, C. F. Lucchinetti and P. A. Calabresi, N. Engl. J. Med., 2018, 378, 169–180 CrossRef CAS PubMed.
- C. Walton, R. King, L. Rechtman, W. Kaye, E. Leray, R. A. Marrie, N. Robertson, N. La Rocca, B. Uitdehaag, I. van der Mei, M. Wallin, A. Helme, C. A. Napier, N. Rijke and P. Baneke, Mult. Scler., 2020, 26, 1816–1821 CrossRef PubMed.
- J. H. Yang, T. Rempe, N. Whitmire, A. Dunn-Pirio and J. S. Graves, Front. Neurol., 2022, 13, 824926 CrossRef PubMed.
- L. Bierhansl, H.-P. Hartung, O. Aktas, T. Ruck, M. Roden and S. G. Meuth, Nat. Rev. Drug Discovery, 2022, 21, 578 CrossRef CAS PubMed.
- C. Fergus, D. Barnes, M. A. Alqasem and V. P. Kelly, Nutrients, 2015, 7, 2897–2929 CrossRef CAS PubMed.
- N. Okada, F. Harada and S. Nishimura, Nucleic Acids Res., 1976, 3, 2593–2603 CrossRef CAS PubMed.
- N. Shindo-Okada, N. Okada, T. Ohgi, T. Goto and S. Nishimura, Biochemistry, 1980, 19, 395–400 CrossRef CAS PubMed.
- Y.-C. Chen, V. P. Kelly, S. V. Stachura and G. A. Garcia, RNA, 2010, 16, 958–968 CrossRef CAS PubMed.
- M. A. Alqasem, C. Fergus, J. M. Southern, S. J. Connon and V. P. Kelly, Chem. Commun., 2020, 56, 3915–3918 RSC.
- M. Sebastiani, C. Behrens, S. Dörr, H.-D. Gerber, R. Benazza, O. Hernandez-Alba, S. Cianférani, G. Klebe, A. Heine and K. Reuter, ACS Chem. Biol., 2022, 17, 2229–2247 CrossRef CAS PubMed.
- W. R. Farkas, J. Biol. Chem., 1980, 255, 6832–6835 CrossRef CAS PubMed.
- J. R. Katze, B. Basile and J. A. McCloskey, Science, 1982, 216, 55–56 CrossRef CAS PubMed.
- T. Rakovich, C. Boland, I. Bernstein, V. M. Chikwana, D. Iwata-Reuyl and V. P. Kelly, J. Biol. Chem., 2011, 286, 19354–19363 CrossRef CAS PubMed.
- T. Tuorto, C. Legrand, C. Cirzi, G. Federico, R. Liebers, M. Müller, A. E. Ehrenhofer-Murray, G. Dittmar, H.-J. Gröne and F. Lyko, EMBO J., 2018, 37, e99777 CrossRef PubMed.
- M. Müller, C. Legrand, F. Tuorto, V. P. Kelly, Y. Atlasi, F. Lyko and A. E. Ehrenhofer-Murray, Nucleic Acids Res., 2019, 47, 3711–3727 CrossRef PubMed.
- P. Hayes, C. Fergus, M. Ghanim, C. Cirzi, L. Burtnyak, C. J. McGrenaghan, F. Tuorto, D. P. Nolan and V. P. Kelly, Nutrients, 2020, 12, 871 CrossRef CAS PubMed.
- Y. L. Chen and R. T. Wu, Cancer Res., 1994, 54, 2192–2198 CAS.
- C. J. Morgan, F. L. Merrill and R. W. Trewyn, Cancer Res., 1996, 56, 594–598 CAS.
- P. Thumbs, T. T. Ensfelder, M. Hillmeier, M. Wagner, M. Heiss, C. Scheel, A. Schön, M. Müller, S. Michalakis, S. Kellner and T. Carell, Angew. Chem., Int. Ed., 2020, 59, 12352–12356 CrossRef CAS PubMed.
- B. S. Huang, R. T. Wu and K. Y. Chien, Cancer Res., 1992, 52, 4696–4700 CAS.
- W. Baranowski, G. Dirheimer, J. A. Jakowicki and G. Keith, Cancer Res., 1994, 54, 4468–4471 CAS.
- G. Dirheimer, W. Baranowski and G. Keith, Biochimie, 1995, 77, 99–103 CrossRef CAS PubMed.
- P. J. Eggenhuizen, B. H. Ng and J. D. Ooi, Int. J. Mol. Sci., 2020, 21, 7015 CrossRef CAS PubMed.
- T. K. Goswami, M. Singh, M. Dhawan, S. Mitra, T. Bin Emran, A. A. Rabaan, A. Al Mutair, Z. Al Alawi, S. Alhumaid and K. Dhama, Hum. Vaccines Immunother., 2022, 18, 2035117 CrossRef PubMed.
- C. Fergus, M. Al-qasem, M. Cotter, C. M. McDonnell, E. Sorrentino, F. Chevot, K. Hokamp, M. O. Senge, J. M. Southern, S. J. Connon and V. P. Kelly, Nucleic Acids Res., 2021, 49, 4877–4890 CrossRef CAS PubMed.
- W. R. Farkas, K. B. Jacobson and J. R. Katze, Biochim. Biophys. Acta, 1984, 781, 64–75 CrossRef CAS PubMed.
- R. P. Singhal, R. A. Kopper, S. Nishimura and N. Shindo-Okada, Biochem. Biophys. Res. Commun., 1981, 99, 120–126 CrossRef CAS PubMed.
- S. Varghese, M. Cotter, F. Chevot, C. Fergus, C. Cunningham, K. H. Mills, S. J. Connon, J. M. Southern and V. P. Kelly, Nucleic Acids Res., 2017, 45, 2029–2039 CAS.
- M. Cotter, S. Varghese, F. Chevot, C. Fergus, V. P. Kelly, S. J. Connon and J. M. Southern, ChemMedChem, 2023, 18, e202300207 CrossRef CAS PubMed.
- S. Shams, J. M. Martinez, J. R. D. Dawson, J. Flores, M. Gabriel, G. Garcia, A. Guevara, K. Murray, N. Pacifici, M. V. Vargas, T. Voelker, J. W. Hell and J. F. Ashouri, Front. Pharmacol., 2021, 12, 680043 CrossRef CAS PubMed.
- P. Luo, P. Wang, J. Xu, W. Hou, P. Xu and K. Xu, Bone Joint Res., 2022, 11, 426–438 CrossRef PubMed.
- F. Pandolfi, L. Franza, V. Carusi, S. Altamura, G. Andriollo and E. Nucera, Int. J. Mol. Sci., 2020, 21, 5238 CrossRef PubMed.
- P. Zafari, M. Taghadosi, F. Faramarzi, M. Rajabinejad and A. Rafiei, Inflammation, 2023, 46, 612–622 CrossRef CAS PubMed.
- G. Montes Diaz, R. Hupperts, J. Fraussen and V. Somers, Autoimmun. Rev., 2018, 17, 1240–1250 CrossRef CAS PubMed.
- I. M. Stromnes and J. M. Goverman, Nat. Protoc., 2006, 1, 1810–1819 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra for active compounds, figures related to PBMC cell viability and animal weights during treatment. See DOI: https://doi.org/10.1039/d4md00028e |
| This journal is © The Royal Society of Chemistry 2024 |