
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibition of N-type calcium channels by phenoxyaniline and sulfonamide analogues†
Anjie S.
Bispat
 ab,
Fernanda C.
Cardoso
c,
Md. Mahadhi
Hasan‡
c,
Yashad
Dongol
c,
Ricki
Wilcox
a,
Richard J.
Lewis
c,
Peter J.
Duggan
*bd and
Kellie L.
Tuck
*a
ab,
Fernanda C.
Cardoso
c,
Md. Mahadhi
Hasan‡
c,
Yashad
Dongol
c,
Ricki
Wilcox
a,
Richard J.
Lewis
c,
Peter J.
Duggan
*bd and
Kellie L.
Tuck
*a
aSchool of Chemistry, Monash University, Victoria 3800, Australia. E-mail: Kellie.Tuck@monash.edu
bCSIRO Manufacturing, Research Way, Clayton, Victoria 3168, Australia. E-mail: Peter.Duggan@csiro.au
cInstitute for Molecular Bioscience, The University of Queensland, St Lucia, QLD 4072, Australia
dCollege of Science and Engineering, Flinders University, Adelaide, South Australia 5042, Australia
First published on 31st January 2024
Abstract
Building on previous investigations, structural modifications to the neuronal calcium ion channel blocker MONIRO-1 and related compounds were conducted that included replacement of the amide linker with an aniline and isosteric sulfonamide moiety, and the previously used strategy of substitution of the guanidinium group with less hydrophilic amine functionalities. A comprehensive SAR study revealed a number of phenoxyaniline and sulfonamide compounds that were more potent or had similar potency for the CaV2.2 and CaV3.2 channel compared to MONIRO-1 when evaluated in a FLIPR-based intracellular calcium response assay. Cytotoxicity investigations indicated that the sulfonamide analogues were well tolerated by Cos-7 cells at dosages required to inhibit both calcium ion channels. The sulfonamide derivatives were the most promising CaV2.2 inhibitors developed by us to date due, possessing high stability in plasma, low toxicity (estimated therapeutic index > 10), favourable CNS MPO scores (4.0–4.4) and high potency and selectivity, thereby, making this class of compounds suitable candidates for future in vivo studies.
Introduction
The sensation of pain is an intrinsic self-preservation mechanism that alerts the body to possible dangers. Nociceptive pain, the body's natural response to mechanical, thermal or chemical trauma, can be managed by removal of the noxious stimuli and may also necessitate the usage of analgesics and/or other clinical interventions depending on the severity of the injury.1,2 However, chronic pain, especially neuropathic pain, which affects approximately 7–8% of the population, does not respond favourably to traditional therapies even when there is no apparent stimuli acting on the body.3,4 Currently, the approved treatment for neuropathic pain includes three potent N-type calcium channel blockers; namely gabapentin 1, pregabalin 2 and ziconotide (Prialt®), a synthetic version of ω-conotoxin MVIIA 3 found naturally in the venom of the marine snail Conus Magus (Fig. 1).5,7 Unfortunately, the use of ziconotide is severely limited due to the intrathecal mode of administration and narrow therapeutic window. Gabapentin 1 and pregabalin 2, provide only minimal relief and also cause serious unwanted side-effects such as depression, blurred vision and decreased motor coordination.6,8 Consequently, several research groups, including our own, have engaged in research programs to develop alternative small molecule CaV2.2 (N-type) calcium ion channel blockers for the treatment of neuropathic pain.9–27 Likewise, various research studies have shown that the CaV3.2 (T-type) voltage-gated calcium channel is also a promising therapeutic target for alleviating neuropathic pain.24–26,28–33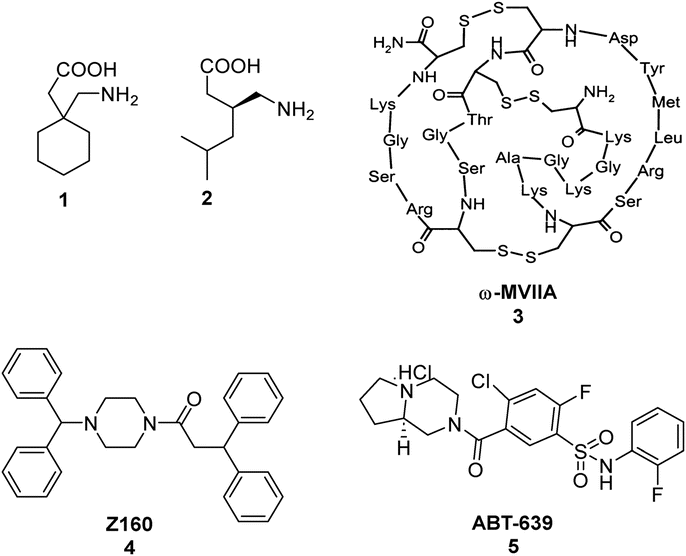 | ||
| Fig. 1 Structures of gabapentin 1, pregabalin 2, ω-conotoxin MVIIA 3,7 marketed as ziconotide (Prialt®), Z160 4 and ABT-639 5. | ||
Over the years, a small number of N- and T- type calcium channel blockers were evaluated in clinical trials for the treatment of neuropathic pain arising from various diseases. Unfortunately, the majority of drug candidates failed to meet the primary outcomes and were subsequently terminated. For instance, the drug Z160 4 (Fig. 1), a state-dependent CaV2.2 inhibitor originally developed by Zalicus Pharmaceuticals, a subsidiary of Epirus Pharmaceuticals, was well tolerated in Phase I clinical trials, however, Z160 4 failed to demonstrate efficacy in patients with lumbosacral radiculopathy and post-herpetic neuralgia during the relevant phase II clinical evaluations.34 Similarly, the drug ABT-639 5 (Fig. 1), a potent CaV3.2 blocker developed by AbbVie, failed to demonstrate analgesic efficacy in patients with diabetic neuropathy and was eventually terminated during phase II clinical trials.34–37 Due to the limited availability of effective medications for the management of neuropathic pain, our research investigations have focused on the discovery of small molecule CaV2.2 and CaV3.2 inhibitors with a number of phenoxyanilides being evaluated for their activity at the two calcium ion channels (representative structures are shown in Fig. 2).15,18,20 Our initial studies using whole-cell patch clamp electrophysiology experiments showed that MONIRO-1 6a demonstrated good selectivity for CaV2.2 (IC50 = 34 μM) and CaV3.2 (IC50 = 1.7 μM) channels over other neuronal calcium channels.20 Moreover, the research outcomes of follow-up SAR studies indicated that the removal of the fluoro substituent in MONIRO-1 6a resulted in the analogous compound 7a exhibiting a 5-fold decrease in potency for the CaV2.2 channel, indicating that the fluoro group was essential for enhanced binding to the channel.22 Encouragingly, the replacement of the guanidinium group with a tertiary amine moiety, which was performed in an effort to improve drug-like characteristics and increase transport across the blood–brain barrier (BBB), resulted in compounds 6b, the tertiary amine variant of MONIRO-1, and 7b displaying comparable potency to that observed for MONIRO-1 6a.22 However, MONIRO-1 6a and related analogues 6b, 7a and 7b tend to be prone to rapid metabolism into toxic phenoxyaniline and as a result they are non-ideal drug leads. Therefore, we explored constrained tertiary amine functionalised analogues, with the aim of enhancing activity and decreasing metabolic degradation.22 While, the activity of the constrained analogues 8–10 for the CaV2.2 channel was similar to MONIRO-1 6a and analogues, the restriction of rotation in most cases led to a decrease in activity for the CaV3.2 channel.22 The acylated phenoxazine 8 was observed to rapidly hydrolyse in rat plasma (t½ = 13.9 min) to the corresponding carboxylic acid and phenoxazine, the dihydrodibenzoazepine 9 and dibenzoazepine 10 analogues were more stable, with both having a half-life greater than 60 h.22 Noting the structural similarity of these latter compounds to tricyclic antidepressants (TCAs), we recently reported CaV2.2 inhibition results that support the off-label use of TCAs with neuropathic pain.23
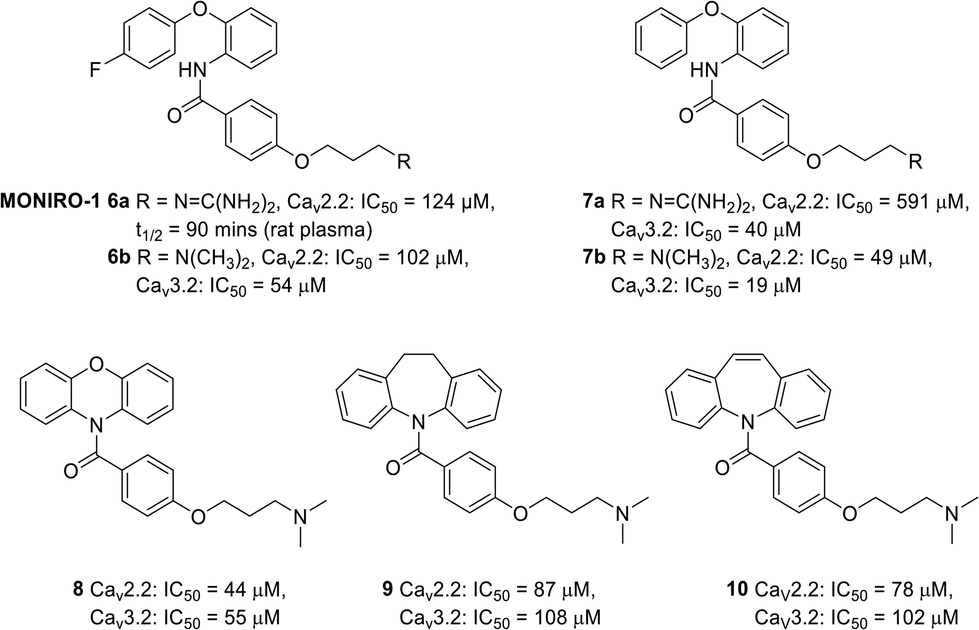 | ||
| Fig. 2 Chemical structures and their activity, determined by calcium influx fluorescence-imaging assays, of MONIRO-1 6a, and related analogues 6b, 7a and 7b, and the constrained analogues 8–10. | ||
The research findings highlighted above provided valuable insights for further optimisation of compounds to give improved activity at the CaV2.2 and CaV3.2 channels, superior pharmacokinetic profiles when compared to MONIRO-1 6a, and more favourable drug-like properties. To overcome the stability challenges observed in several of the phenoxyanilides analogues, two libraries of compounds, replacing the labile amide bond with either a secondary aniline or an isosteric sulfonamide functional group, were prepared. Additionally, the previously used strategy of replacing the guanidinium group of MONIRO-1 6a with less hydrophilic amine functionalities could lead to the development of more drug-like compounds with greater potential to cross the BBB. In this paper we report the synthesis and activity at the CaV2.2 and CaV3.2 channels of 54 compounds (Fig. 3), and for a representative subset, their plasma stability and an evaluation of their cytotoxicity.
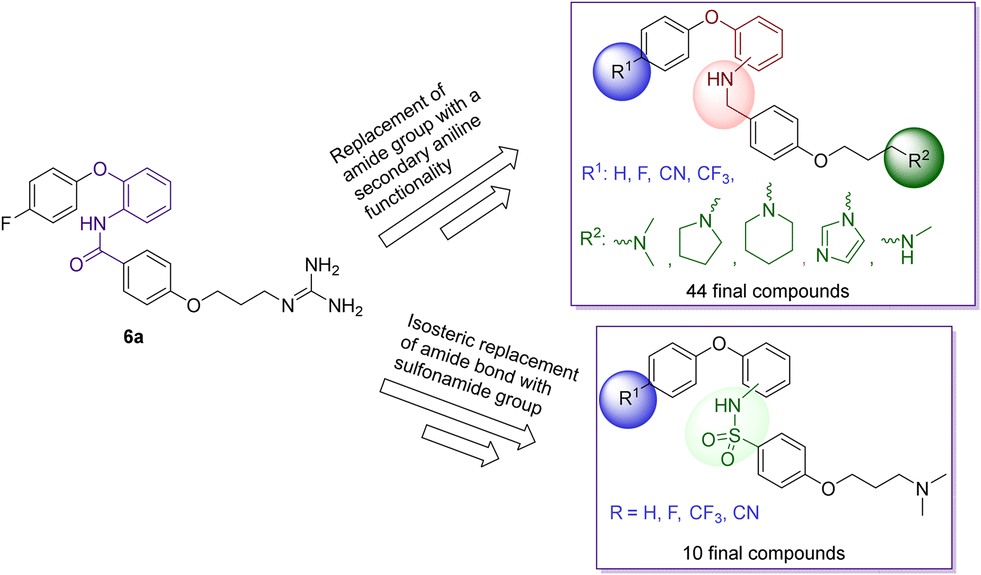 | ||
| Fig. 3 Schematic depiction of the library of compounds synthesised and tested; replacement of the labile amide bond of MONIRO-1 6a with either a secondary aniline or an isosteric sulfonamide functional group, and replacement of the guanidinium group with less hydrophilic amines. | ||
Results and discussion
Chemistry – compound design and synthesis
For all compounds, substituents on the aromatic phenoxy ring (R1) were positioned para to the phenoxy linkage, consistent with the location of this group in the MONIRO-1 analogues. The substituents, –F, –CF3 and –CN, selected for their pharmacological relevance, were used to investigate the influence of electronic factors on the activity for the CaV2.2 and CaV3.2 channels with the unsubstituted analogue (R1 = H) included for comparison purposes. For the phenoxyaniline analogues the position of the secondary aniline functionality was varied to evaluate how structural differences would affect binding to the calcium ion channels. The choice of amines was based on our previous findings and the various amine groups present in CNS drugs or drug candidates. The terminal amine groups investigated were dimethylamine, pyrrolidine, piperidine, imidazole and methylamine (Fig. 3). This allowed us to assess the effect of pKa, the number of hydrogen bond donors (HBD) and ring size on potency. For the sulfonamide analogues a smaller library was synthesised, which was guided by the biological findings with the phenoxyanilines. All compounds were assessed for their inhibition of the CaV2.2 and CaV3.2 channels, with representative compounds analysed for plasma stability and cytotoxicity.A library of 44 phenoxyaniline analogues that vary in substitution of the terminal aromatic ring, regiochemistry of the central ring and the terminal amine moiety were synthesised according to Scheme 1. A reductive amination reaction between the corresponding anilines 11–20 (see ESI†) and the aromatic aldehyde 21 (ref. 27) provided the required chloro species 22–31, from which the desired phenoxyaniline analogues 32(a–e)–41(a–c) could be derived. The dimethylamine sulfonamide–based analogues 42–51 were synthesised from the sulfonyl acid 52 (see ESI†) and the corresponding aniline 11–20 (Scheme 2). Prior to biological testing, all final compounds were purified by preparative RP–HPLC using a mobile phase containing TFA. The TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amine stoichiometry in the purified products was determined by 19F NMR spectroscopic analysis as described in detail in the ESI.†
:amine stoichiometry in the purified products was determined by 19F NMR spectroscopic analysis as described in detail in the ESI.†
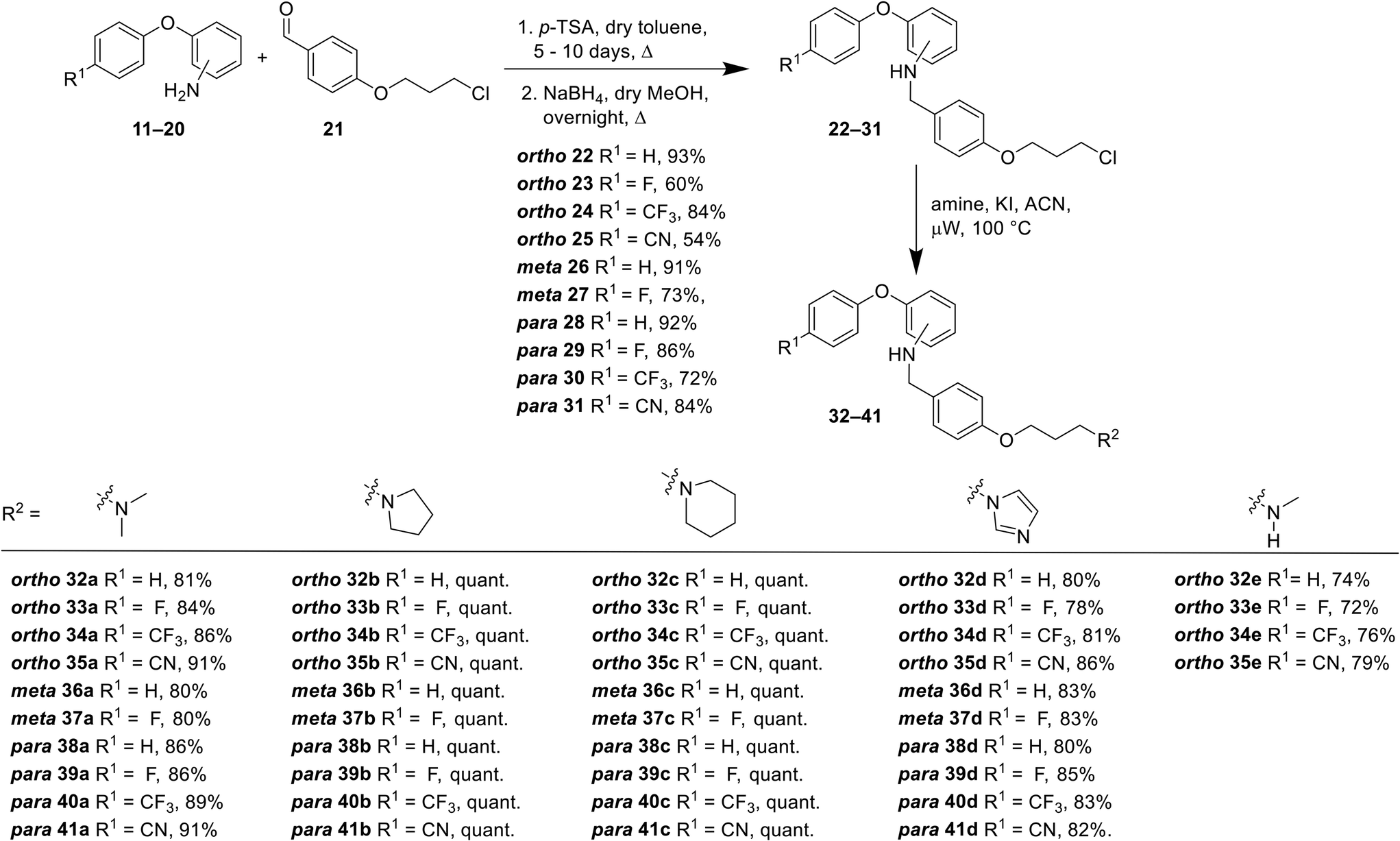 | ||
| Scheme 1 Synthesis of the phenoxyaniline analogues 32–41. | ||
 | ||
| Scheme 2 Synthesis of the sulfonamide analogues 42–51. | ||
Biology – CaV2.2 and CaV3.2 activity, cytotoxicity studies
The activity of all analogues for the CaV2.2 and CaV3.2 channels was evaluated using the previously reported FLIPR calcium imaging assay.22,23 CaV2.2 inhibition was evaluated with the neuroblastoma SH-SY5Y cell line, in the presence of the CaV1 blocker nifedipine, and HEK 293T cells expressing recombinant human CaV3.2 α1 subunit were used to evaluate CaV3.2 inhibition. The positive control for the CaV2.2 channel and CaV3.2 channel assay was NNC 55-0396. Representative dose response curves are shown in Fig. 4, and IC50 values are summarised in Tables 1–3.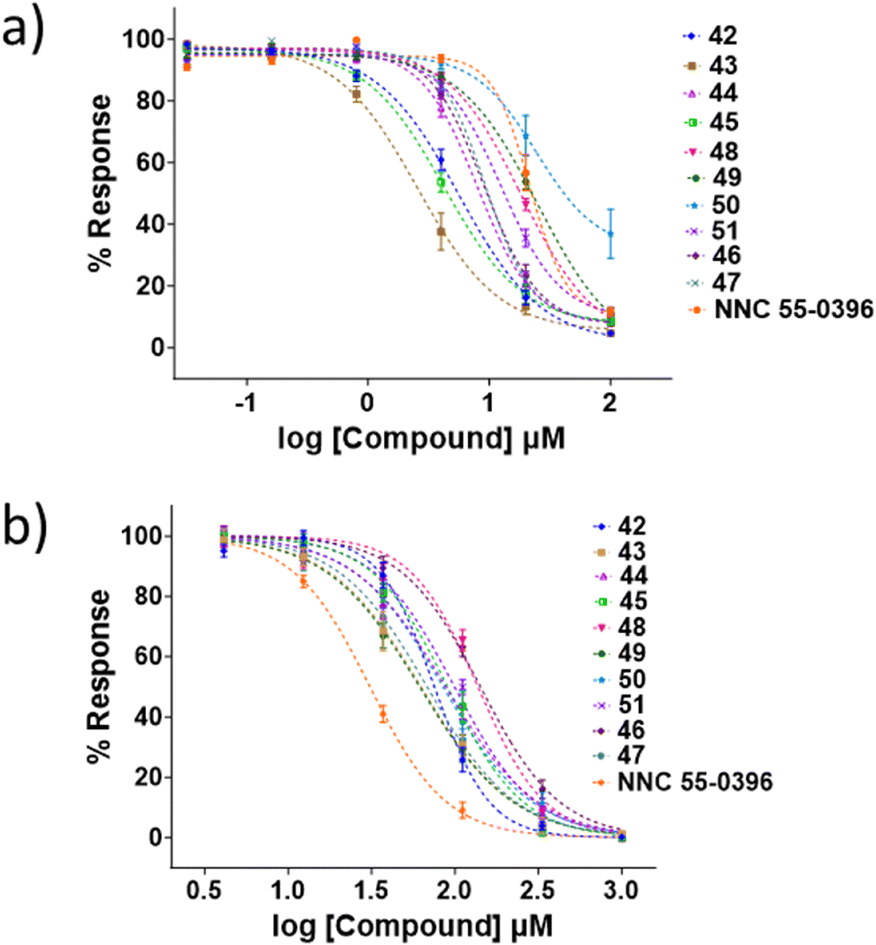 | ||
| Fig. 4 Dose response curves, determined by calcium influx fluorescence-imaging assays of the sulfonamide compounds 42–51 in a) hCaV2.2 and b) hCaV3.2 channels. Data are presented as mean ± SEM from n = 3–5 independent experiments. The calculated IC50 values are shown in the relevant tables. | ||
| Compd. | CaV2.2 | CaV3.2 | Compd. | CaV2.2 | CaV3.2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | SEM | 95% CI (μM) | IC50 (μM) | SEM | 95% CI (μM) | IC50 (μM) | SEM | 95% CI (μM) | IC50 (μM) | SEM | 95% CI (μM) | ||
| 32a | 49 | 10.9 | 45–55 | 379 | 13.8 | 346–416 | 35e | 65 | 6.2 | 61–70 | 157 | 8.2 | 146–168 |
| 33a | 55 | 8.4 | 50–61 | 267 | 19.0 | 240–294 | 42 | 5.5 | 0.8 | 4.9–6.1 | 75 | 9.6 | 68–83 |
| 34a | 30 | 5.7 | 27–32 | 238 | 25.5 | 216–261 | 43 | 2.8 | 1.0 | 2.4–3.4 | 63 | 19.0 | 54–73 |
| 35a | 47 | 3.7 | 43–51 | 319 | 57.6 | 290–348 | 44 | 8.8 | 2.3 | 7.7–9.9 | 88 | 22.6 | 76–101 |
| 36a | 54 | 10.6 | 49–60 | 258 | 37.2 | 228–288 | 45 | 4.8 | 1.4 | 4.0–5.4 | 90 | 14.2 | 81–100 |
| 37a | 40 | 6.2 | 36–45 | 228 | 15.7 | 205–252 | 46 | 10 | 1.8 | 9.0–11 | 142 | 14.1 | 126–160 |
| 38a | 81 | 27.4 | 69–95 | 308 | 32.3 | 272–344 | 47 | 9.9 | 2.0 | 9.0–11 | 68 | 8.9 | 60–77 |
| 39a | 39 | 12.4 | 33–46 | 227 | 22.0 | 207–248 | 48 | 18 | 0.6 | 16–19 | 137 | 7.2 | 121–154 |
| 40a | 60 | 10.7 | 54–67 | 271 | 47.5 | 244–300 | 49 | 21 | 0.7 | 19–24 | 61 | 15.9 | 54–69 |
| 41a | 59 | 4.5 | 55–63 | 216 | 67.3 | 192–244 | 50 | 53 | 1.0 | 39–76 | 85 | 18.1 | 75–96 |
| 32e | 84 | 13.1 | 77–92 | 178 | 24.8 | 163–194 | 51 | 14 | 0.5 | 12–15 | 99 | 3.2 | 88–112 |
| 33e | 63 | 10.5 | 57–68 | 167 | 5.2 | 151–184 | NNC55-0396 | 24.3 | 2.8 | 21–28 | 30.9 | 3.0 | 28–34 |
| 34e | 58 | 12.5 | 53–64 | 204 | 17.2 | 189–219 | |||||||
| Compd. | CaV2.2 | CaV3.2 | Compd. | CaV2.2 | CaV3.2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | SEM | 95% CI (μM) | IC50 (μM) | SEM | 95% CI (μM) | IC50 (μM) | SEM | 95% CI (μM) | IC50 (μM) | SEM | 95% CI (μM) | ||
| 32b | 34 | 8.8 | 30–38 | 111 | 11.2 | 102–121 | 32c | 62 | 17.5 | 55–69 | 188 | 15.0 | 171–206 |
| 33b | 33 | 8.4 | 30–36 | 154 | 10.7 | 142–166 | 33c | 18 | 2.2 | 16–20 | 126 | 3.5 | 114–141 |
| 34b | 51 | 11.6 | 46–58 | 110 | 15.5 | 101–120 | 34c | 27 | 3.4 | 23–30 | 146 | 18 | 118–175 |
| 35b | 23 | 2.0 | 21–25 | 123 | 3.1 | 110–138 | 35c | 8.0 | 0.6 | 7–9 | 67 | 8.0 | 36–127 |
| 36b | 29 | 3.4 | 27–31 | 158 | 34.2 | 142–176 | 36c | 63 | 59–67 | 200 | 14.0 | 182–219 | |
| 37b | 43 | 4.0 | 40–45 | 196 | 54.3 | 176–218 | 37c | 20 | 0.8 | 18–23 | 292 | 20 | 245–327 |
| 38b | 36 | 4.0 | 34–39 | 201 | 30.6 | 184–219 | 38c | 54 | 49–60 | 237 | 13.7 | 218–258 | |
| 39b | 48 | 3.1 | 44–51 | 208 | 39.8 | 191–226 | 39c | 30 | 1.7 | 27–36 | 67 | 5.9 | 36–128 (∼50% inhibition at 500 μM) |
| 40b | 50 | 2.6 | 46–55 | 182 | 9.2 | 165–202 | 40c | 37 | 3.9 | 33–40 | 26.0 | 4.5 | 18–41 (∼50% inhibition at 500 μM) |
| 41b | 28 | 1.6 | 26–31 | 146 | 24.3 | 134–160 | 41c | 19 | 1.3 | 17–22 | 22 | 5.6 | 14–38 (∼60% inhibition at 500 μM) |
| Compd. | CaV2.2 | CaV3.2 | Compd. | CaV2.2 | CaV3.2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | SEM | 95% CI (μM) | IC50 (μM) | SEM | 95% CI (μM) | IC50 (μM) | SEM | 95% CI (μM) | IC50 (μM) | SEM | 95% CI (μM) | ||
| 32d | 78 | 27.3 | 55–111 | 316 | 52.1 | 283–349 | 37d | 70 | 15.5 | 51–96 | 385 | 27.3 | 353–426 |
| 33d | 33 | 8.8 | 28–38 | 196 | 28.0 | 174–220 | 38d | 70 | 24.4 | 58–86 | 234 | 26.1 | 206–265 |
| 34d | 125 | 8.9 | 116–136 | 287 | 29.4 | 262–312 | 39d | 68 | 20.2 | 59–79 | 337 | 51.1 | 308–366 |
| 35d | 39 | 16.1 | 32–48 | 238 | 50.2 | 215–262 | 40d | 283 | 31.1 | 257–309 | 170 | 40.6 | 150–192 |
| 36d | 68 | 32.6 | 55–84 | 258 | 26.1 | 236–281 | 41d | 75 | 35.2 | 55–100 | 371 | 38.0 | 344–407 |
As the ortho-phenoxyanilines generally exhibited superior CaV2.2 activity, representative ortho-phenoxyanilines (32e R1 = H, R2 = NHMe; 33a R1 = F, R2 = NMe2; 33c R1 = F, R2 = piperidine; 34b R1 = CF3, R2 = pyrrolidine and 35d R1 = CN, R2 = imidazole), were evaluated for their stability in rat plasma,21,38 in addition to the sulfonamides 45, 47 and 48. For these experiments the internal standard, diazepam, was added directly to the plasma along with the test compound. Diazepam was selected as the reference compound due to the stability of the compound in rat plasma over the period assessed and also because it had a different retention time on RP-HPLC analysis than the test compounds. Additionally, the activity of plasma enzymes in each batch of plasma utilized in this study was assessed using the positive control, diltiazem, and this experiment was performed in parallel with the test compounds. The phenoxyanilines 32e, 33a, 33c, 34b and 35d, and sulfonamides 45, 47 and 48 tested were all highly stable in rat plasma, each with a half-life greater than 60 hours. Comparing these findings to that obtained for MONIRO-1 6a and 6b,22 a vast increase in stability is apparent. Both MONIRO-1 6a and the corresponding dimethylamine 6b were determined to have a half-life in rat plasma of only ∼90 min, whereas all the analogues tested in this study showed considerably greater stability with half-lives of >60 hours. These research findings indicate that replacement of the amide functional group with either a secondary amine or sulfonamide had the expected effect of significantly reducing the compound's susceptibility to plasma hydrolysis.
The probability of the phenoxyaniline and sulfonamide analogues permeating the BBB and the CNS was assessed using the central nervous system multiparameter optimisation (CNS MPO) desirability tool.39,40 This algorithm, developed by Pfizer, assesses six physicochemical parameters; MW, clogP, clogD at a pH of 7.4, topological polar surface area (TPSA); number of hydrogen-bond donors (HBDs), and the pKa of the most basic centre, to identify lead compounds with an increased chance of penetrating the CNS. A CNS MPO score of ≥4, on a scale of 0–6, is indicative that the molecule has a high likelihood of entering the CNS. The phenoxyanilines prepared in this study typically had scores in the order of <3.5 and the sulfonamides were approximately 4 (see ESI† for the calculated CNS MPO scores).
For the first time we have also evaluated representative compounds, consisting of phenoxyanilide 7b,22 the phenoxyanilines 32e, 33a, 33c, 34b and 35d, and the sulfonamides 42, 44, 45, 47 and 48, for their cytotoxicity to Cos-7 cells. The Cos-7 cell line was chosen as it is an immortalised cell line with fibroblast-like morphology and originates from the African green monkey (Cercopithecus aethiops) kidney cells,41 that has previously been used for toxicity studies.42,43 A resazurin reduction assay, Promega CellTitre-Blue® viability assay, was utilized for this study.44,45 Metabolically viable cells were detected by measuring the reduction of resazurin to resorufin, a fluorescent molecule, by oxidoreductases in the mitochondrial electron transport chain. The number of viable cells were determined by fluorimetry and the 50% cytotoxic concentration (CC50) values were determined from the fitted dose–response curve (Table 4). Additionally, the molecules' therapeutic index (TI), which is the ratio between the dosage of a drug that elicits an adverse effect (CC50) and the dosage required to produce a therapeutic response (IC50), was estimated.46 Generally, a TI value >10 is indicative of a drug with a good safety profile as in clinical settings drugs with a narrow TI usually require constant monitoring of the levels in plasma to prevent toxic effects in the patient.47 The amide 7b was found to be well tolerated by Cos-7 cells up to concentrations as high as 100 μM as were the sulfonamides 42, 44 and 45. Overall, the sulfonamide analogues had lower cytotoxicity than the phenoxyanilines.
| Compound | CC50 (μM) | CaV2.2 | CaV3.2 | ||
|---|---|---|---|---|---|
| IC50 (μM) | TI | IC50 (μM) | TI | ||
| 7b | >100 | 49 | >2.0 | 19 | >5.3 |
| 32e | 33 | 84 | 0.4 | 178 | 0.2 |
| 33a | 93 | 55 | 1.7 | 267 | 0.3 |
| 33c | 37 | 18 | 2.1 | 126 | 0.3 |
| 34b | 30 | 51 | 0.6 | 110 | 0.3 |
| 35d | 211 | 39 | 5.4 | 238 | 0.9 |
| 42 | >100 | 5.5 | >18 | 75 | >1.3 |
| 44 | 109 | 8.8 | 12 | 88 | 1.2 |
| 45 | 100 | 4.8 | 21 | 90 | 1.1 |
| 47 | 92 | 9.9 | 9.3 | 68 | 1.4 |
| 48 | 54 | 21 | 2.6 | 137 | 0.4 |
Discussion
The large number of phenoxyaniline compounds 32–35a–e and 36–41a–d (44 compounds) synthesised and tested allowed us to further explore the structure–activity relationship by evaluating the influence of the substitution pattern of the aniline functional group, the substituent at the R1 position, and the effect of varying the guanidinium group with less hydrophilic amine functionalities. For a given series of the phenoxyaniline analogues, those containing the same substituent at both R1 and R2, the position of the aniline substituent on the central aromatic ring (ortho, meta or para) generally did not have a significant effect on the potency of the compounds for the CaV2.2 channel. However, in some cases, for instance with the –CN analogues, the ortho-substituted compounds 35a,c,d were found to be 2-fold more active than the analogous para-substituted compounds 41a,c,d for the CaV2.2 channel. Similarly, for the –F imidazole-based analogues, the ortho-derivative 33d was 2-fold more active at the CaV2.2 channel compared to the corresponding para- (39d) and meta-substituted (37d) compounds. Similarly, the substituent at R1 for a particular series of compounds did not have a substantial effect on potency for the CaV2.2 channel, except for the piperidine and imidazole-based analogues. There was a more than 3-fold increase in affinity for the CaV2.2 for both the –F (33c, 37c, 39c) and –CF3 (34c and 40c) piperidine-based analogues regardless of the substitution of the aniline group when compared to the corresponding unsubstituted compound (R1 = H) 32c, 36c, 38c. For the –CN derivatives, there was about a 3-fold in potency for 41c and more importantly an 8-fold increase in potency for 35cversus38c and 32c respectively. Based on the biological results obtained for the piperidine-based phenoxyanilines, the affinity for the CaV2.2 channel generally increased as the electron-withdrawing nature of R1 increased suggesting that an electron-deficient aromatic ring and was critical for binding whilst an increase in the hydrogen bond acceptor (HBA) properties of the substituent led to reduction in affinity for the channel. For the ortho substituted imidazole-based anilines, a similar trend was observed where there was a 2-fold increase in potency for the CaV2.2 channel for both the –F 33d and –CN 35d compounds versus the unsubstituted derivative 32c. However, the additional HBA properties of the –CF3 imidazole-based analogues, 34d and 40d, potentially resulted in this compound having decreased affinity for the receptor when assessed against the analogous unsubstituted derivatives, 32d and 38d respectively. By modifying the tail amine functionality, it was observed that the tertiary amines are preferred with the order of activity following the trend of piperidine > pyrrolidine > N,N-dimethylated > imidazole when evaluated against the CaV2.2 channel. The mono-methylated analogues 32e–35e were generally less active than the respective dimethylated derivatives (32a–35a) suggesting that the incorporation of the HBD at the tail group was not essential for binding to the receptor. The incorporation of the piperidine and pyrrolidine rings at R2 resulted in compounds (32b/c–41b/c) with improved binding affinity for the CaV2.2 channel compared to MONIRO-1 6a with the 6-membered heterocycle generally being preferred. The imidazole moiety has a pKa value of approximately 6.5, therefore, at physiological pH this group would only be partially ionised whereas the pyrrolidine and piperidine groups, which both have a pKa of about 9.9, would be completely protonated. As the imidazole derivatives 32d–41d were less active than the corresponding pyrrolidine 32b–41b and piperidine 32c–41c analogues, it can be concluded that a fully ionised tertiary amine group, R2 was critical for binding to the CaV2.2 channel. With regards to the CaV3.2 channel, the phenoxyanilines were overall inactive at this channel, however, the results obtained for the –CF340c and –CN 41c piperidine-based derivatives were promising.Unlike the analogous phenoxyaniline 32a–41a, the position of the sulfonamide group on the central aromatic ring did affect the affinity of the sulfonamide analogues 42–51 for the CaV2.2 channel where the activity followed the trend such that ortho- 42–45 > meta- 46 and 47 > para-substituted 48–51 sulfonamide derivatives. Overall, the ortho- 42–45 and meta-substituted 46 and 47 compounds were highly active for the CaV2.2 channel, with IC50 values less than 10 μM. Among them the ortho-substituted fluorinated compound 43 exhibited the highest potency of 2.8 μM. This is the highest activity that we have observed in our research program focused on the rationally designed small-molecule inhibitors targeting the CaV2.2 channel. Within the para-substituted series the –CN analogue was the most active, which mirrors that observed in the aniline series. In contrast, the substitution of the sulfonamide on the aromatic ring did not influence the binding to the CaV3.2 channel. Whilst the sulfonamide analogues showed higher potency against the CaV3.2 channel compared to the corresponding phenoxyaniline derivatives; they were still deemed to be essentially inactive against this channel.
The current research study also allowed us to do a preliminary evaluation of how the substitution of the amide linker with the aniline and sulfonamide functionalities affected binding to the two calcium ion channels of interest. For the unsubstituted compounds, R1 = H, the replacement of the amide group 7b with the aniline moiety did not affect the binding affinity for the CaV2.2 channel whereas there was a 2-fold increase in potency for the fluorinated phenoxyaniline analogue 33aversus the analogous amide compound 6b. On the other hand, a 9-fold and 36-fold increase in activity at the CaV2.2 channel was observed for the –H 42 and –F 43 substituted sulfonamide derivatives respectively versus the analogous amide compounds, 7b and 6b. Conversely, the replacement of the amide moiety (7b and 6b), with the aniline (32a and 33a), and sulfonamide (42 and 43), groups led to decreased binding affinity for the CaV3.2 channel.
The preliminary results obtained from the cytotoxicity studies were encouraging. The previously synthesised compound 7b (ref. 22) was well tolerated by Cos-7 cells up to concentrations as high as 100 μM, however, cleavage of the amide bond (t1/2 = 89 min) is still a major bioavailability and toxicity concern. Cytotoxicity studies with the evaluated phenoxyaniline compounds revealed unacceptable levels of toxicity, and the majority of compounds had low estimated TI values, which were less than 2. The phenoxyanilines typically had CNS MPO scores in the order of <3.5, and, combined with their high toxicity, means that replacement of the amide functionality of MONIRO-1 6a with an amine will not result in an acceptable drug-lead. However, the comprehensive SAR of this functionality has provided useful information that can be utilised in the rational design of future molecules. In contrast, the sulfonamide compounds tested were well tolerated by Cos-7 cells, and compounds 42 and 45 had estimated TI values greater than 18. From this study it can be concluded that the sulfonamide analogues 42–47 were the most promising drug leads, particularly 42, 43 and 45, due to their safe toxicological profile (estimated TI > 10), high CNS MPO scores (4.0–4.4) and, most importantly, high potency and selectivity for the CaV2.2 channel.
Conclusions
Although it was recognised that the phenoxyaniline series were likely to be toxic and this was confirmed by the results of the cytotoxicity studies, which showed that the majority of the phenoxyaniline compounds evaluated had low estimated TI values < 2 at both receptors, removing anilines completely from the drug design space can severely limit the discovery process. Therefore, it was of interest to study the SAR of these compounds as they can be synthesised with relative ease. Overall, it was found that a tertiary amine functionality that can fully ionise at physiological pH was critical for binding to both calcium ion channels. Additionally, an electron-withdrawing substituent particularly the –CN group can potentially improve the potency for the CaV2.2 channel, whilst, an increase in the HBA properties may result in a decrease in activity for the receptor as seen in the imidazole-based –CF3 derivatives. The affinity of the piperidine-based phenoxyaniline compounds particularly the –CN derivatives 35c and 41c for both the CaV2.2 and CaV3.2 channel respectively was promising as they provide valuable insights into the factors that affect binding to channel. Additionally, there are opportunities to make modifications to 35c and 41c to produce compounds that are more potent for both receptors and have more favourable drug-like properties.The activity for the sulfonamide compounds for the CaV3.2 channel was somewhat encouraging, however, optimisation studies and further modifications is still required to obtain improved activity at this receptor. Fortunately, the replacement of the amide linker of MONIRO-1 6a with an isosteric sulfonamide group combined with substituting the guanidinium group with a dimethylated amine moiety led to the discovery of the most potent CaV2.2 blockers developed in our research group to date. Considering the CaV2.2 activity of the sulfonamide derivatives, their plasma stability and cytotoxicity, as well as their ability to cross the BBB, it can be concluded that 42 and 45 are the most promising compounds to take forward in future in vivo studies.
Experimental
Chemistry
000 using perfluorokerosene (PFK) as the reference compound. Infrared spectra (IR) were recorded on an Agilent Technologies Cary 630 FTIR as thin films. Infrared band frequencies are reported in wavenumbers (cm−1). Analytical grade reagents and solvents were used as purchased without further purification unless otherwise stated. Aldehyde 21 was synthesised according to the procedure noted in ref. 27. Air-sensitive reactions were performed under an atmosphere of N2 using a standard Schlenk line and glassware for moisture-sensitive reactions were dried overnight in the oven at 110 °C. Microwave reactions were conducted on a CEM Discover SP microwave synthesiser at 150 W. Compounds were purified by preparative RP–HPLC using a 1260 infinity quaternary LC system with a Phenomenex Luna C8 column (150 × 21.5 mm, 5 μm), buffer A – 100% MilliQ H2O/0.1% TFA and buffer B – 80% ACN/20% MilliQ H2O /0.1% TFA, flow rate: 10 mL min−1, elution method: 90% buffer A and 10% buffer B for 2 min, gradient run 90% buffer A and 10% buffer B to 10% buffer A and 90% buffer B from 2 to 30 min, 10% buffer A and 90% buffer B from 30 to 40 min, 90% buffer A and 10% buffer B from 40 to 42 min.
:40% CHCl3:57% petroleum benzine). A portion was purified by preparative RP–HPLC to obtain an analytically pure sample for biological studies and the 1H,13C and 19F NMR spectra of the respective compound was recorded.
:30% CHCl3:67% petroleum benzine). A portion was purified by preparative RP–HPLC to obtain an analytically pure sample for biological studies. The 1H and 13C NMR spectra of the compound was recorded while the 19F NMR spectrum of the TFA salt was recorded.
:50% CHCl3:47% petroleum benzine). A portion was purified by preparative RP–HPLC to obtain an analytically pure sample biological studies and the 1H,13C and 19F NMR spectra of the respective TFA salt was recorded.
:93% petroleum benzine); IR (neat, cm−1): 3445, 2969, 2927, 1607, 1511, 1490, 1243, 1215, 1174, 1051, 745; 1H NMR (400 MHz, CDCl3-d) δ 7.35–7.27 (m, 2H), 7.25–7.18 (m, 2H), 7.10–6.94 (m, 4H), 6.88–6.81 (m, 3H), 6.72 (dd, J = 8.0, 1.5 Hz, 1H), 6.64 (td, J = 7.7, 1.5 Hz, 1H), 4.54 (t, J = 5.6 Hz, 1H), 4.30 (d, J = 5.7 Hz, 2H), 4.10 (t, J = 6.1 Hz, 2H), 3.74 (t, J = 6.1 Hz, 2H), 2.23 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 158.05, 157.67, 143.16, 140.49, 131.72, 129.80, 128.67, 125.02, 122.85, 119.34, 117.57, 116.99, 114.75, 111.87, 64.47, 47.37, 41.65, 32.44; HRMS (APCI): m/z calcd C22H23ClNO2 [M + H]+ 368.1417, found, 368.1413.
:90% petroleum benzine); IR (neat, cm−1): 3403, 2872, 1602, 1498, 1435, 1243, 1201, 1115, 1044, 849, 809, 735; 1H NMR (600 MHz, CDCl3-d) δ 7.26–7.22 (m, 2H), 7.03–6.96 (m, 3H), 6.96–6.90 (m, 2H), 6.88–6.84 (m, 2H), 6.79 (dd, J = 7.7, 1.5 Hz, 1H), 6.72 (dd, J = 8.1, 1.5 Hz, 1H), 6.63 (td, J = 7.7, 1.5 Hz, 1H), 4.54 (d, J = 5.4 Hz, 1H), 4.30 (d, J = 5.1 Hz, 2H), 4.10 (t, J = 6.1 Hz, 2H), 3.74 (t, J = 6.1 Hz, 2H), 2.23 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3-d) δ 158.56 (d, J = 240.8 Hz), 158.06, 153.37 (d, J = 2.4 Hz), 143.67, 140.18, 128.68, 124.91, 119.05 (d, J = 8.1 Hz), 118.57, 116.98, 116.24 (d, J = 23.1 Hz), 114.73, 111.85, 64.42, 47.34, 41.61, 32.38; 19F NMR (376 MHz, CDCl3-d) δ −121.56; HRMS (APCI): m/z calcd C22H22O2NClF [M + H]+ 386.1318, found, 386.1319.
:93% petroleum benzine); IR (neat, cm−1): 3444, 3419, 2927, 1608, 1511, 1330, 1238, 1165, 1101, 1068, 836, 740; 1H NMR (400 MHz, CDCl3-d) δ 7.57 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 7.09 (t, J = 7.9 Hz, 1H), 7.03 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 7.9 Hz, 1H), 6.86 (d, J = 8.1 Hz, 2H), 6.77 (d, J = 7.9 Hz, 1H), 6.70–6.64 (t, J = 7.9 Hz, 1H), 4.43 (s, 1H), 4.30 (s, 2H), 4.10 (t, J = 6.1 Hz, 2H), 3.75 (t, J = 6.1 Hz, 2H), 2.24 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3-d) δ 160.55, 158.04, 141.63, 140.58, 131.31, 128.48, 127.11 (q, J = 4.5 Hz), 126.10, 124.48 (q, J = 32.7 Hz), 124.40 (q, J = 271.5 Hz), 120.32 (d, J = 1.9 Hz), 117.12, 116.73, 114.60, 112.29, 64.24, 47.04, 41.48, 32.21; 19F NMR (377 MHz, CDCl3) δ −62.17; HRMS (APCI): m/z calcd C23H21O2NClF3 [M]+ 435.1207, found, 435.1208.
:85% petroleum benzine); IR (CDCl3, cm−1): 3411, 2931, 2225, 1602, 1501, 1230, 1163, 1036, 907, 834, 797; 1H NMR (600 MHz, CDCl3-d) δ 7.58 (d, J = 8.8 Hz, 2H), 7.18 (d, J = 8.5 Hz, 2H), 7.10 (ddd, J = 8.1, 7.6, 1.5 Hz, 1H), 6.99 (d, J = 8.8 Hz, 2H), 6.91 (dd, J = 7.6, 1.5 Hz, 1H), 6.84 (d, J = 8.5 Hz, 2H), 6.77 (d, J = 8.1 Hz, 1H), 6.70 (t, J = 7.6 Hz, 1H), 4.33 (s, 1H), 4.28 (s, 2H), 4.09 (t, J = 6.1 Hz, 2H), 3.74 (t, J = 6.1 Hz, 2H), 2.23 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 161.08, 157.73, 140.71, 140.00, 133.84, 130.77, 128.27, 126.26, 120.37, 118.60, 117.13, 116.82, 114.35, 112.46, 105.35, 64.04, 46.82, 41.36, 31.94; HRMS (APCI): m/z calcd C23H22O2N2Cl [M]+ 392.1286, found, 392.1285.
:85% petroleum benzine); IR (neat, cm−1): 3400, 3061, 2881, 1615, 1578, 1505, 1487, 1240, 1215, 1145, 818, 752; 1H NMR (400 MHz, CDCl3-d) δ 7.39–7.28 (m, 2H), 7.30–7.19 (m, 2H), 7.16–7.06 (m, 2H), 7.06–6.97 (m, 2H), 6.94–6.84 (m, 2H), 6.42–6.32 (m, 2H), 6.31 (t, J = 2.3 Hz, 1H), 4.21 (d, J = 5.3 Hz, 2H), 4.11 (t, J = 6.1 Hz, 2H), 3.99 (s, 1H), 3.75 (t, J = 6.1 Hz, 2H), 2.24 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 158.52, 158.14, 157.33, 149.83, 131.45, 130.30, 129.69, 128.95, 123.11, 119.07, 114.79, 108.17, 107.88, 103.42, 64.47, 47.82, 41.63, 32.39; HRMS (APCI): m/z calcd C22H22ClNO2 [M]+ 367.1339, found, 367.1332.
:85% petroleum benzine); IR (neat, cm−1): 3398, 2884, 1603, 1579, 1495, 1446, 1232, 1197, 1047, 828; 1H NMR (400 MHz, CDCl3-d) δ 7.25 (d, J = 8.7 Hz, 2H), 7.09 (t, J = 8.1 Hz, 1H), 7.04–6.93 (m, 4H), 6.87 (d, J = 8.7 Hz, 2H), 6.36 (ddd, J = 8.1, 2.3, 0.9 Hz, 1H), 6.29 (ddd, J = 8.1, 2.3, 0.9 Hz, 1H), 6.23 (t, J = 2.3 Hz, 1H), 4.21 (d, J = 5.3 Hz, 2H), 4.11 (t, J = 5.8 Hz, 2H), 4.01 (t, J = 6.1 Hz, 1H), 3.75 (t, J = 6.1 Hz, 2H), 2.24 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3-d) δ 159.02, 158.82 (d, J = 241.3 Hz), 158.17, 152.96, 149.85, 130.36, 128.94, 120.73 (d, J = 8.2 Hz), 116.22 (d, J = 23.3 Hz), 114.80, 108.10, 107.20, 102.79, 64.46, 47.83, 41.65, 32.40; 19F NMR (376 MHz, CDCl3-d) δ −120.99; HRMS (APCI): m/z calcd C22H22O2NClF [M + H]+ 386.1318, found, 386.1313.
:85% petroleum benzine); mp: 54.3–56.1 °C; IR (neat, cm−1): 3390, 2970, 2902, 1610, 1587. 1508,1490, 1230, 1047, 818, 750; 1H NMR (400 MHz, CDCl3-d) δ 7.36–7.30 (m, 2H), 7.30–7.24 (m, 2H), 7.03 (ddt, J = 7.4, 5.9, 1.1 Hz, 1H), 6.98–6.87 (m, 6H), 6.70–6.60 (m, 2H), 4.27 (s, 2H), 4.15 (t, J = 6.1 Hz, 2H), 3.90 (s, 1H), 3.78 (t, J = 6.1 Hz, 2H), 2.27 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 159.17, 158.15, 147.87, 144.96, 131.74, 129.60, 128.97, 122.05, 121.31, 117.25, 114.80, 113.94, 64.48, 48.45, 41.64, 32.40; HRMS (APCI): m/z calcd C22H22ClNO2 [M]+ 367.1339, found, 367.1331.
:85% petroleum benzine); mp: 72.4–74.8 °C; IR (neat, cm−1): 3386, 2936, 1609, 1496, 1243, 1208, 1171, 1048, 827, 807; 1H NMR (400 MHz, CDCl3-d) δ 7.34–7.27 (m, 2H), 7.02–6.92 (m, 2H), 6.92–6.82 (m, 6H), 6.66–6.57 (m, 2H), 4.24 (s, 2H), 4.12 (t, J = 6.1 Hz, 2H), 3.88 (s, 1H), 3.75 (t, J = 6.1 Hz, 2H), 2.24 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3-d) δ 158.12, 158.09 (d, J = 240.0 Hz), 154.93 (d, J = 2.3 Hz), 148.33, 144.89, 128.90, 120.77, 118.62 (d, J = 8.1 Hz), 115.99 (d, J = 23.2 Hz), 114.76, 113.91, 64.56, 48.35, 41.61, 32.35; 19F NMR (376 MHz, CDCl3-d) δ −122.70; HRMS (APCI): m/z calcd C22H22O2NClF [M + H]+ 386.1318, found, 386.1315.
:93% petroleum benzine); mp: 69.5–72.1 °C; IR (neat, cm−1): 3409, 2946, 1611, 1504, 1323, 1239, 1154, 1104, 1063, 840, 823; 1H NMR (400 MHz, CDCl3-d) δ 7.55–7.48 (m, 2H), 7.34–7.27 (m, 2H), 7.00–6.93 (m, 2H), 6.93–6.88 (m, 4H), 6.68–6.62 (m, 2H), 4.26 (s, 2H), 4.12 (t, J = 6.1 Hz, 2H), 4.10–3.84 (m, 1H), 3.76 (t, J = 6.1 Hz, 2H), 2.24 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3-d) δ 162.03, 158.10, 146.23, 145.62, 131.44, 128.83, 126.92 (q, J = 3.7 Hz), 124.45 (q, J = 272.9 Hz), 123.65 (q, J = 32.6 Hz), 121.70, 116.50, 114.68, 113.85, 64.32, 48.08, 41.54, 32.23; 19F NMR (377 MHz, CDCl3) δ −62.12; HRMS (APCI): m/z calcd C23H21O2NClF3 [M]+ 435.1207, found, 435.1208.
:85% petroleum benzine); mp: 74.3–76.7 °C; IR (neat, cm−1): 3368, 2873, 2226, 1600, 1497, 1238, 1165, 1108, 829; 1H NMR (600 MHz, CDCl3-d) δ 7.55(d, J = 8.9 Hz, 2H), 7.30 (d, J = 8.6 Hz, 2H), 6.94 (d, J = 8.9 Hz, 2H), 6.92–6.87 (m, 4H), 6.66 (d, J = 8.6 Hz, 2H), 4.26 (s, 2H), 4.12 (t, J = 6.1 Hz, 2H), 3.75 (t, J = 6.1 Hz, 2H), 2.24 (p, J = 6.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 162.83, 157.99, 145.71, 145.42, 133.90, 131.17, 128.74, 121.70, 119.01, 116.84, 114.61, 113.89, 104.65, 64.28, 48.01, 41.50, 32.16; HRMS (APCI): m/z calcd C23H22O2N2Cl [M]+ 392.1286, found, 392.1285.
:90% DCM); IR (neat, cm−1): 3425, 2942, 2765, 1608, 1509, 1215, 1173, 1035, 822, 739, 691; 1H NMR (400 MHz, MeOD-d4) δ 7.31–7.21 (m, 2H), 7.19–7.10 (m, 2H), 7.04–6.97 (m, 1H), 6.96–6.85 (m, 3H), 6.81–6.73 (m, 3H), 6.67 (dd, J = 8.1, 1.4 Hz, 1H), 6.57 (td, J = 7.7, 1.5 Hz, 1H), 4.22 (s, 2H), 3.92 (t, J = 6.2 Hz, 2H), 2.54–2.42 (m, 2H), 2.24 (s, 6H), 1.95–1.85 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 159.34, 159.25, 144.21, 141.87, 133.10, 130.67, 129.36, 126.04, 123.54, 120.67, 118.06, 117.76, 115.47, 113.43, 67.00, 57.37, 47.75, 45.39, 28.18; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C24H29N2O2 [M + H]+ 377.2224, found, 377.2214.
:90% DCM); IR (neat, cm−1): 3424, 1672, 1499, 1195, 1173, 1123, 829, 720; 1H NMR (400 MHz, MeOD-d4) δ 7.21–7.12 (m, 2H), 7.04–6.95 (m, 2H), 6.94–6.84 (m, 3H), 6.84–6.77 (m, 2H), 6.73 (dd, J = 7.9, 1.4 Hz, 1H), 6.67 (dd, J = 8.1, 1.4 Hz, 1H), 6.56 (td, J = 7.7, 1.5 Hz, 1H), 4.24 (s, 2H), 3.94 (t, J = 6.1 Hz, 2H), 2.55–2.43 (m, 2H), 2.25 (s, 6H), 1.97–1.85 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 159.65 (d, J = 239.2 Hz), 159.37, 155.21 (d, J = 2.4 Hz), 144.67, 141.70, 133.13, 129.38, 126.02, 120.18, 119.62 (d, J = 8.2 Hz), 117.77, 116.96 (d, J = 23.5 Hz), 115.48, 113.47, 67.00, 57.37, 47.74, 45.38, 28.17; 19F NMR (377 MHz, MeOD-d4) δ −123.70, −76.55; HRMS (APCI): m/z calcd C24H28N2O2F [M + H]+ 395.2129, found, 395.2118.
:90% DCM); IR (neat, cm−1): 3434. 2966, 1673, 1609, 1510, 1324, 1166, 1105, 1064829, 799, 720; 1H NMR (400 MHz, MeOD-d4) δ 7.59 (d, J = 8.6 Hz, 2H), 7.19 (d, J = 8.6 Hz, 2H), 7.02 (d, J = 8.6 Hz, 2H), 7.00 (dd, J = 7.8, 1.5 Hz, 1H), 6.88 (dd, J = 7.8, 1.5 Hz, 1H), 6.85 (d, J = 8.6 Hz, 2H), 6.71 (dd, J = 7.8, 1.5 Hz, 1H), 6.64 (td, J = 7.8, 1.5 Hz, 1H), 4.29 (s, 2H), 4.07 (t, J = 5.7 Hz, 2H), 3.37–3.32 (m, 2H), 2.93 (s, 6H), 2.26–2.11 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 162.52, 158.87, 142.79, 142.12, 133.87, 129.42, 128.06 (q, J = 3.8 Hz), 127.15, 125.85 (q, J = 270.2 Hz), 125.22 (q, J = 32.5 Hz), 121.87, 117.92, 117.59, 115.51, 113.87, 65.92, 56.93, 47.46, 43.65, 25.77; 19F NMR (376 MHz, MeOD-d4) δ −62.87; HRMS (APCI): m/z calcd C25H28N2O2F3 [M + H]+ 445.2097, found, 405.2085.
:90% DCM); IR (neat, cm−1): 3420, 2968, 2226, 1678, 1610, 1512, 1236, 1171, 1130, 1056, 832, 721; 1H NMR (400 MHz, MeOD-d4) δ 7.63 (d, J = 9.0 Hz, 2H), 7.17 (d, J = 8.7 Hz, 2H), 7.06–7.01 (m, 1H), 6.98 (d, J = 9.0 Hz, 2H), 6.88 (dd, J = 7.9, 1.5 Hz, 1H), 6.84 (d, J = 8.7 Hz, 2H), 6.73 (dd, J = 7.9, 1.5 Hz, 1H), 6.65 (td, J = 7.9, 1.5 Hz, 1H), 4.26 (s, 2H), 4.06 (t, J = 5.7 Hz, 2H), 3.37–3.31 (m, 2H), 2.92 (s, 6H), 2.26–2.13 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 163.33, 158.90, 142.23, 142.03, 135.24, 133.70, 129.40, 127.53, 122.08, 119.85, 118.05, 118.02, 115.51, 114.12, 106.23, 65.92, 56.84, 47.48, 43.60, 25.74; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C25H28N3O2 [M + H]+ 402.2176, found, 402.2166.
:90% DCM); IR (neat, cm−1): 3409, 2941, 2765, 1581, 1486, 1216, 1148, 1005, 823, 754, 688; 1H NMR (400 MHz, MeOD-d4) δ 7.31–7.22 (m, 2H), 7.22–7.15 (m, 2H), 7.06–6.96 (m, 2H), 6.93–6.85 (m, 2H), 6.85–6.79 (m, 2H), 6.37 (ddd, J = 8.2, 2.2, 0.9 Hz, 1H), 6.22 (t, J = 2.2 Hz, 1H), 6.18 (ddd, J = 8.2, 2.2, 0.9 Hz, 1H), 4.14 (s, 2H), 3.95 (t, J = 6.1 Hz, 2H), 2.50 (t, J = 6.1 Hz, 2H), 2.26 (s, 6H), 1.99–1.86 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 159.46, 159.30, 158.85, 151.73, 133.12, 130.92, 130.59, 129.54, 123.81, 119.64, 115.45, 109.52, 107.99, 104.46, 67.01, 57.39, 45.39, 28.18; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C24H29N2O2 [M + H]+ 377.2224, found, 377.2215.
:90% DCM); IR (MeOH, cm−1): 3405, 2969, 1672, 1608, 1498, 1195, 1128, 1027, 830, 799, 721; 1H NMR (400 MHz, MeOD-d4) δ 7.23–7.13 (m, 2H), 7.03–6.93 (m, 3H), 6.93–6.85 (m, 2H), 6.84–6.79 (m, 2H), 6.40–6.33 (m, 1H), 6.21–6.12 (m, 2H), 4.15 (s, 2H), 3.97 (t, J = 6.2 Hz, 2H), 2.55–2.47 (m, 2H), 2.27 (s, 6H), 1.98–1.88 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 159.88, 159.87 (d, J = 239.6 Hz), 159.32, 154.74 (d, J = 2.5 Hz), 151.77, 133.12, 130.95, 129.47, 121.36 (d, J = 1.6 Hz), 121.28, 116.93 (d, J = 23.5 Hz), 115.44, 109.53, 107.45, 103.95, 67.02, 57.42, 48.00, 45.40, 28.23; 19F NMR (377 MHz, MeOD-d4) δ −76.55, −122.36; HRMS (APCI): m/z calcd C24H28N2O2F [M + H]+ 395.2129, found, 395.2118.
:90% DCM); IR (neat, cm−1): 3250, 2957, 2825, 1612, 1510, 1498, 1227, 1041, 1005, 831, 746, 689; 1H NMR (400 MHz, CDCl3-d) δ 7.27 (ddd, J = 8.4, 6.8, 2.6 Hz, 4H), 7.03–6.97 (m, 1H), 6.97–6.85 (m, 6H), 6.67–6.59 (m, 2H), 4.23 (s, 2H), 4.02 (t, J = 7.2 Hz, 2H), 3.87 (s, 1H), 2.47 (t, J = 7.2 Hz, 2H), 2.27 (s, 6H), 1.98 (p, J = 7.2 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 159.20, 158.47, 147.81, 145.02, 131.34, 129.61, 128.96, 122.04, 121.36, 117.22, 114.78, 113.92, 66.39, 56.55, 48.51, 45.64, 27.68; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C24H29N2O2 [M + H]+ 377.2224, found, 377.2213.
:90% DCM); IR (neat, cm−1): 3247, 2959, 2777, 1609, 1495, 1239, 1205, 1177, 1007, 822, 722, 670; 1H NMR (400 MHz, CDCl3-d) δ 7.32–7.26 (m, 2H), 7.00–6.93 (m, 2H), 6.93–6.80 (m, 6H), 6.66–6.56 (m, 2H), 4.23 (d, J = 4.2 Hz, 2H), 4.02 (t, J = 6.6 Hz, 2H), 3.87 (s, 1H), 2.47 (t, J = 6.6 Hz, 2H), 2.27 (s, 6H), 1.97 (p, J = 6.6 Hz, 2H); 13C NMR (101 MHz, CDCl3-d) δ 158.48, 158.16 (d, J = 240.0 Hz), 154.99 (d, J = 2.4 Hz), 148.37, 144.97, 131.31, 128.95, 120.86, 118.65 (d, J = 8.0 Hz), 116.05 (d, J = 23.1 Hz), 114.79, 113.96, 66.38, 56.55, 48.50, 45.62, 27.65; 19F NMR (376 MHz, CDCl3) δ −122.20; HRMS (APCI): m/z calcd C24H28N2O2F [M + H]+ 395.2129, found, 395.2116.
:90% DCM); IR (neat, cm−1): 3251, 2958, 2826, 1677, 1504, 1326, 1237, 1156, 1104, 1065, 1008, 831, 721; 1H NMR (400 MHz, CDCl3-d) δ 7.51 (d, J = 8.6 Hz, 2H), 7.29 (d, J = 6.5 Hz, 3H), 6.96 (d, J = 8.6 Hz, 2H), 6.93–6.86 (m, 4H), 6.65 (d, J = 8.9 Hz, 2H), 4.25 (d, J = 5.0 Hz, 2H), 4.02 (t, J = 6.4 Hz, 2H), 3.96 (t, J = 5.0 Hz, 1H), 2.46 (t, J = 6.4 Hz, 2H), 2.26 (s, 6H), 1.97 (p, J = 6.4 Hz, 2H); 13C NMR (151 MHz, CDCl3-d) δ 162.14, 158.55, 146.42, 145.73, 131.13, 128.94, 127.03 (q, J = 3.7 Hz), 124.47 (q, J = 270.9 Hz), 123.90 (q, J = 32.8 Hz), 121.84, 116.60, 114.83, 113.97, 66.43, 56.55, 48.39, 45.67, 27.72; 19F NMR (377 MHz, MeOD-d4) δ −76.55, −62.99; HRMS (APCI): m/z calcd C25H28N2O2F3 [M + H]+ 445.2097, found, 405.2085.
:90% DCM); IR (neat, cm−1): 3348, 2963, 2225, 1666, 1610, 1494, 1240, 1167, 1124, 1056, 828, 708, 720; 1H NMR (400 MHz, MeOD-d4) δ 7.68 (d, J = 8.8 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H), 7.12–6.99 (m, 6H), 6.95 (d, J = 8.6 Hz, 2H), 4.40 (s, 2H), 4.10 (t, J = 5.7 Hz, 2H), 3.40–3.32 (m, 2H), 2.91 (s, 6H), 2.29–2.16 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 163.30, 159.99, 152.07, 140.09, 135.45, 131.43, 128.93, 122.46, 120.92, 119.66, 118.99, 115.77, 106.87, 65.99, 56.75, 52.61, 43.58, 25.71;19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C25H28N3O2 [M + H]+ 402.2176, found, 402.2161.
:95% DCM); IR (MeOH, cm−1): 3423, 2926, 1672, 1608, 1510, 1198, 1125, 829, 746, 721; 1H NMR (400 MHz, MeOD-d4) δ 7.38–7.29 (m, 2H), 7.27–7.19 (m, 2H), 7.16–7.07 (m, 1H), 7.06–6.98 (m, 1H), 6.98–6.90 (m, 3H), 6.90–6.82 (m, 3H), 6.80 (dd, J = 8.1, 1.5 Hz, 1H), 4.38 (s, 2H), 4.06 (t, J = 5.8 Hz, 2H), 3.77–3.59 (m, 2H), 3.42–3.34 (m, 2H), 3.18–2.98 (m, 2H), 2.29–2.09 (m, 4H), 2.09–1.88 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 159.64, 158.08, 147.12, 136.15, 130.91, 130.40, 125.50, 124.59, 122.77, 119.87, 119.13, 117.82, 115.64, 65.88, 55.27, 53.77, 50.40, 27.03, 23.95; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C26H31N2O2 [M + H]+ 403.238, found, 403.2372.
:95% DCM); IR (neat, cm−1): 3422, 2881, 1673, 1609, 1500, 1243, 1195, 1124, 829, 742, 720; 1H NMR (400 MHz, MeOD-d4) δ 7.29–7.19 (m, 2H), 7.13–7.04 (m, 2H), 7.00 (ddd, J = 8.1, 7.2, 1.5 Hz, 1H), 6.98–6.93 (m, 2H), 6.93–6.87 (m, 3H), 6.84 (ddd, J = 8.1, 7.2, 1.5 Hz, 1H), 6.78 (dd, J = 8.1, 1.5 Hz, 1H), 4.39 (s, 2H), 4.08 (t, J = 5.7 Hz, 2H), 3.78–3.62 (m, 2H), 3.50–3.35 (m, 2H), 3.18–3.03 (m, 2H), 2.26–2.10 (m, 4H), 2.10–1.93 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 59.71 (d, J = 238.8 Hz), 158.87, 155.33 (d, J = 2.5 Hz), 144.71, 141.71, 133.93, 129.50, 126.01, 120.31, 119.57 (d, J = 8.3 Hz), 117.83, 116.96 (d, J = 23.7 Hz), 115.50, 113.52, 65.85, 55.42, 53.95, 47.66, 27.13, 23.96; 19F NMR (376 MHz, MeOD-d4) δ −121.99; −76.55; HRMS (APCI): m/z calcd C26H30N2O2F [M + H]+ 421.2286, found, 421.2279.
:95% DCM); IR (neat, cm−1): 3421, 1673, 1609, 1510, 1324, 1230, 1167, 1105, 1064, 829, 743, 720; 1H NMR (600 MHz, MeOD-d4) δ 7.59 (d, J = 8.7 Hz, 2H), 7.23–7.13 (m, 2H), 7.06–6.97 (m, 3H), 6.88 (dd, J = 7.9, 1.5 Hz, 1H), 6.86–6.82 (m, 2H), 6.71 (dd, J = 7.9, 1.5 Hz, 1H), 6.64 (td, J = 7.9, 1.5 Hz, 1H), 4.28 (s, 2H), 4.07 (t, J = 5.7 Hz, 2H), 3.69 (s, 2H), 3.42–3.35 (m, 2H), 3.10 (s, 2H), 2.26–2.09 (m, 4H), 2.03 (s, 2H); 13C NMR (151 MHz, MeOD-d4) δ 162.50, 158.89, 142.80, 142.09, 133.79, 129.42, 127.26, 125.84 (q, J = 270.4 Hz), 125.22 (q, J = 32.5 Hz), 121.85, 117.93, 117.59, 115.48, 113.88, 65.85, 55.37, 53.92, 47.47, 27.11, 23.96; 19F NMR (376 MHz, MeOD-d4) δ −62.54, −76.55; HRMS (APCI): m/z calcd C27H30N2O2F3 [M + H]+ 471.2254, found, 471.2250.
:95% DCM); IR (neat, cm−1): 3387, 2933, 2222, 1672, 1603, 1499, 1231, 1163, 1125, 833, 759, 718; 1H NMR (400 MHz, MeOD-d4) δ 7.68–7.59 (m, 2H), 7.22–7.14 (m, 2H), 7.07–6.95 (m, 3H), 6.89 (dd, J = 7.9, 1.5 Hz, 1H), 6.86–6.80 (m, 2H), 6.73 (dd, J = 7.9, 1.5 Hz, 1H), 6.65 (td, J = 7.9, 1.5 Hz, 1H), 4.27 (s, 2H), 4.07 (t, J = 5.7 Hz, 2H), 3.57–3.32 (m, 6H), 2.28–2.14 (m, 2H), 2.14–2.00 (m, 4H); 13C NMR (101 MHz, MeOD-d4) δ 163.38, 158.90, 142.18, 142.15, 135.25, 133.76, 129.39, 127.54, 122.13, 119.85, 118.03, 117.93, 115.47, 114.05, 106.22, 65.86, 55.36, 53.91, 47.42, 27.14, 23.96; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C27H30N3O2 [M + H]+ 428.2333, found 428.2327.
:95% DCM); IR (neat, cm−1): 3429, 2964, 1664, 1611, 1486, 1228, 1125, 828, 720, 688; 1H NMR (400 MHz, MeOD-d4) δ 7.39–7.17 (m, 4H), 7.009–6.96 (m, 2H), 6.95–6.81 (m, 4H), 6.39 (ddd, J = 8.2, 2.2, 0.9 Hz, 1H), 6.23 (t, J = 2.3 Hz, 1H), 6.20 (ddd, J = 8.2, 2.2, 0.9 Hz, 1H), 4.20 (s, 2H), 4.08 (t, J = 5.7 Hz, 2H), 3.69 (s, 2H), 3.45–3.34 (m, 2H), 3.17–3.01 (m, 2H), 2.28–2.09 (m, 4H), 2.03 (s, 2H); 13C NMR (101 MHz, MeOD-d4) δ 159.52, 158.85, 151.41, 133.64, 130.94, 130.62, 129.66, 123.88, 119.65, 115.47, 109.76, 108.26, 104.69, 65.85, 55.36, 53.91, 48.13, 27.13, 23.96; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C26H31N2O2 [M + H]+ 403.238, found, 403.2375.
:95% DCM); IR (MeOH, cm−1):3353, 1672, 1605, 1497, 1240, 1195, 1126, 830, 721; 1H NMR (400 MHz, MeOD-d4) δ 7.30–7.20 (m, 2H), 7.20–7.11 (m, 1H), 7.10–6.99 (m, 2H), 6.98–6.84 (m, 4H), 6.58 (ddd, J = 8.1, 2.2, 0.9 Hz, 1H), 6.41 (ddd, J = 8.1, 2.2, 0.9 Hz, 1H), 6.33 (t, J = 2.3 Hz, 1H), 4.28 (s, 2H), 4.10 (t, J = 5.7 Hz, 2H), 3.76–3.64 (m, 2H), 3.46–3.37 (m, 2H), 3.19–3.04 (m, 2H), 2.29–2.11 (m, 4H), 2.11–1.94 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 159.94 (d, J = 239.6 Hz), 159.91, 158.88, 154.71 (d, J = 2.5 Hz), 151.25, 133.51, 131.00, 129.64, 121.36 (d, J = 8.3 Hz), 116.96 (d, J = 23.6 Hz), 115.48, 109.88, 107.89, 104.30, 65.86, 55.36, 53.91, 48.19, 27.13, 23.96; 19F NMR (376 MHz, MeOD-d4) δ −76.55, −121.78; HRMS (APCI): m/z calcd C26H30N2O2F [M + H]+ 421.2286, found, 421.2278.
:95% DCM); IR (neat, cm−1): 3389, 2952, 1656, 1610, 1510, 1227, 1178, 1129, 830, 755, 720, 688; 1H NMR (400 MHz, MeOD-d4) δ 7.36–7.28 (m, 2H), 7.28–7.20 (m, 2H), 6.97 (tt, J = 7.4, 1.1 Hz, 1H), 6.94–6.88 (m, 2H), 6.88–6.81 (m, 2H), 6.81–6.75 (m, 2H), 6.69–6.62 (m, 2H), 4.24 (s, 2H), 4.09 (t, J = 5.7 Hz, 2H), 3.69 (s, 2H), 3.44–3.37 (m, 2H), 3.27–2.85 (m, 2H), 2.32–1.89 (m, 6H); 13C NMR (101 MHz, MeOD-d4) δ 160.56, 158.92, 148.97, 146.40, 133.78, 130.52, 129.82, 122.96, 121.85, 117.98, 115.49, 65.86, 55.39, 53.94, 48.98, 27.14, 23.96; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C26H31N2O2 [M + H]+ 403.238, found, 403.2373.
:95% DCM); IR (MeOH, cm−1): 3375, 1674, 1606, 1496, 1244, 1201, 1132, 832, 721; 1H NMR (400 MHz, MeOD-d4, 50 °C) δ 7.36–7.26 (m, 2H), 7.17–7.11 (m, 2H), 7.11–7.03 (m, 2H), 7.02–6.89 (m, 6H), 4.42 (s, 2H), 4.11 (t, J = 5.8 Hz, 2H), 3.71 (s, 2H), 3.44–3.36 (m, 2H), 3.11 (s, 2H), 2.38–2.19 (m, 2H), 2.19–1.84 (m, 4H); 13C NMR (101 MHz, MeOD-d4, 50 °C) δ 160.42 (d, J = 240.9 Hz), 160.39, 156.86, 154.30, 135.61, 132.09, 127.20, 122.83, 121.56 (d, J = 8.4 Hz), 120.38, 117.35 (d, J = 23.7 Hz), 115.93, 66.13, 55.38, 54.24, 53.84, 27.01, 23.98; 19F NMR (377 MHz, MeOD-d4) δ −121.01, −76.55; HRMS (APCI): m/z calcd C26H30N2O2F [M + H]+ 421.2286, found, 421.2278.
:95% DCM); IR (neat, cm−1): 3357, 1665, 1613, 1506, 1324, 1235, 1160, 1106, 1064, 830, 799, 720; 1H NMR (400 MHz, MeOD-d4) δ δ 7.61 (d, J = 8.4 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H), 7.04 (d, J = 8.4 Hz, 2H), 6.99–6.89 (m, 6H), 4.36 (s, 2H), 4.10 (t, J = 5.7 Hz, 2H), 3.79–3.62 (m, 2H), 3.46–3.36 (m, 2H), 3.17–3.04 (m, 2H), 2.26–2.10 (m, 4H), 2.10–1.95 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 163.87, 158.91, 147.70, 147.05, 133.98, 129.68, 127.98 (q, J = 3.8 Hz), 125.88 (q, J = 270.2 Hz), 124.61 (q, J = 32.5 Hz), 122.48, 117.46, 115.50, 115.11, 65.93, 55.40, 53.98, 48.45, 27.26, 23.98; 19F NMR (376 MHz, MeOD-d4) δ −62.72; HRMS (APCI): m/z calcd C27H30N2O2F3 [M + H]+ 471.2254, found, 471.2250.
:95% DCM); IR (MeOH, cm−1): 3358, 2954, 2223, 1656, 1601, 1493, 1242, 1171, 1130, 811, 799, 720; 1H NMR (400 MHz, MeOD-d4) δ 7.62 (d, J = 8.9 Hz, 2H), 7.31 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.9 Hz, 3H), 6.91 (d, J = 8.6 Hz, 3H), 6.84 (d, J = 8.9 Hz, 2H), 6.69 (d, J = 8.9 Hz, 2H), 4.26 (s, 2H), 4.09 (t, J = 5.7 Hz, 2H), 3.70 (s, 2H), 3.44–3.37 (m, 2H), 3.11 (s, 2H), 2.26–2.10 (m, 4H), 2.04 (s, 2H); 13C NMR (101 MHz, MeOD-d4) δ 164.66, 158.96, 147.54, 146.69, 135.24, 133.63, 129.76, 122.56, 119.87, 118.01, 115.51, 115.38, 105.68, 65.87, 55.39, 53.93, 48.67, 27.14, 23.97; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C27H30N3O2 [M + H]+ 428.2333, found 428.2327.
:95% DCM); IR (neat, cm−1): 3425, 2959, 1671, 1608, 1510, 1242, 1196, 1173, 1121, 798, 719, 692; 1H NMR (400 MHz, MeOD-d4) δ 7.37–7.25 (m, 2H), 7.25–7.17 (m, 2H), 7.05 (ddt, J = 8.5, 7.9, 1.5 Hz, 1H), 7.00–6.88 (m, 3H), 6.88–6.82 (m, 2H), 6.78 (dd, J = 7.9, 1.5 Hz, 1H), 6.72 (dd, J = 7.9, 1.5 Hz, 1H), 6.63 (td, J = 7.9, 1.5 Hz, 1H), 4.30 (s, 2H), 4.06 (t, J = 5.7 Hz, 2H), 3.58 (d, J = 12.5 Hz, 2H), 3.30–3.22 (m, 2H), 2.94 (td, J = 12.5, 3.0 Hz, 2H), 2.26–2.13 (m, 2H), 1.96 (d, J = 12.5 Hz, 2H), 1.90–1.80 (m, 1H), 1.81–1.67 (m, 2H), 1.60–1.44 (m, 1H); 13C NMR (101 MHz, MeOD-d4) δ 159.12, 158.98, 144.71, 140.98, 133.30, 130.71, 129.69, 125.94, 123.72, 120.59, 118.57, 118.21, 115.50, 114.13, 65.94, 55.96, 54.44, 48.09, 25.22, 24.31, 22.67; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C27H33N2O2 [M + H]+ 417.2537, found, 417.2533.
:95% DCM); IR (neat, cm−1): 3422, 1671, 1608, 1499, 1241, 1195, 1121, 827, 742, 719; 1H NMR (501 MHz, MeOD-d4) δ 7.23 (d, J = 8.6, 2H), 7.07–6.98 (m, 2H), 6.97–6.89 (m, 3H), 6.86 (d, J = 8.6, 2H), 6.76 (dd, J = 7.9, 1.5 Hz, 1H), 6.68 (dd, J = 8.1, 1.5 Hz, 1H), 6.59 (td, J = 7.7, 1.5 Hz, 1H), 4.30 (s, 2H), 4.08 (t, J = 5.7 Hz, 2H), 3.58 (d, J = 12.7 Hz, 2H), 3.30–3.24 (m, 2H), 2.95 (t, J = 12.7 Hz, 2H), 2.27–2.14 (m, 2H), 2.04–1.66 (m, 5H), 1.62–1.45 (m, 1H); 13C NMR (126 MHz, MeOD-d4) δ 159.72 (d, J = 239.0 Hz), 158.88, 155.33 (d, J = 2.3 Hz), 144.72, 141.71, 133.91, 129.51, 126.01, 120.30, 119.58 (d, J = 8.1 Hz), 117.83, 116.96 (d, J = 23.6 Hz), 115.48, 113.52, 65.95, 56.02, 54.49, 47.67, 25.27, 24.37, 22.69; 19F NMR (376 MHz, MeOD-d4) δ −76.55, −123.54; HRMS (APCI): m/z calcd C27H32N2O2F [M + H]+ 435.2442, found, 435.2429.
:95% DCM); IR (neat, cm−1): 3425, 2951, 1671, 1609, 1510, 1325, 1230, 1165, 1105, 1064, 828, 742, 719; 1H NMR (501 MHz, MeOD-d4, −15 °C) δ 7.62 (d, J = 8.6 Hz, 2H), 7.21 (d, J = 8.6 Hz, 2H), 7.03 (d, J = 8.6 Hz, 2H), 6.99 (td, J = 7.8, 1.5 Hz, 1H), 6.89 (dd, J = 7.8, 1.5 Hz, 1H), 6.85 (d, J = 8.6 Hz, 2H), 6.69–6.59 (m, 2H), 4.29 (s, 2H), 4.05 (t, J = 5.6 Hz, 2H), 3.87–3.37 (m, 2H), 3.29–3.21 (m, 2H), 2.92 (s, 2H), 2.29–2.11 (m, 2H), 2.11–1.36 (m, 6H); 13C NMR (126 MHz, MeOD-d4) δ 162.52, 158.89, 142.78, 142.14, 133.82, 129.43, 128.07 (q, J = 3.8 Hz), 127.16, 125.86 (q, J = 269.7 Hz), 125.21 (q, J = 32.5 Hz), 121.88, 117.90, 117.59, 115.46, 113.86, 65.99, 56.06, 54.52, 47.45, 25.34, 24.43, 22.77; 19F NMR (376 MHz, MeOD-d4) δ −62.73, −76.55; HRMS (APCI): m/z calcd C28H32N2O2F3 [M + H]+ 485.2410, found, 485.2406.
:95% DCM); IR (neat, cm−1): 3388, 2951, 2225, 1672, 1610, 1501, 1231, 1165, 1123, 828, 744, 719; 1H NMR (400 MHz, MeOD-d4) δ 7.64 (d, J = 9.0 Hz, 2H), 7.17 (d, J = 8.8 Hz, 2H), 7.07–7.01 (m, 1H), 6.99 (d, J = 9.0 Hz, 2H), 6.89 (dd, J = 7.9, 1.5 Hz, 1H), 6.83 (d, J = 8.8 Hz, 2H), 6.73 (dd, J = 7.9, 1.5 Hz, 1H), 6.65 (td, J = 7.9, 1.5 Hz, 1H), 4.27 (s, 2H), 4.06 (t, J = 5.7 Hz, 2H), 3.72–3.45 (m, 2H), 3.30–3.24 (m, 2H), 3.11–2.78 (m, 2H), 2.29–2.13 (m, 2H), 2.07–1.63 (m, 5H), 1.63–1.40 (m, 1H); 13C NMR (101 MHz, MeOD-d4) δ 163.38, 158.88, 142.19, 142.13, 135.25, 133.77, 129.40, 127.54, 122.12, 119.85, 118.03, 117.94, 115.45, 114.06, 106.22, 65.95, 55.98, 54.46, 47.43, 25.25, 24.33, 22.68; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C28H32N3O2 [M + H]+ 442.2489, found 442.2486.
:95% DCM); IR (neat, cm−1): 3353, 2949, 1671, 1609, 1488, 1219, 1198, 1126, 830, 721, 691; 1H NMR (400 MHz, MeOD-d4) δ 77.36–7.17 (m, 4H), 7.16–7.02 (m, 2H), 6.96–6.81 (m, 4H), 6.53 (ddd, J = 8.1, 2.2, 0.9 Hz, 1H), 6.41–6.32 (m, 2H), 4.26 (s, 2H), 4.09 (t, J = 5.7 Hz, 2H), 3.59 (d, J = 12.5 Hz, 2H), 3.30–3.26 (m, 2H), 2.95 (td, J = 12.5, 3.0 Hz, 2H), 2.28–2.16 (m, 2H), 1.97 (d, J = 12.5 Hz, 2H), 1.90–1.68 (m, 3H), 1.62–1.44 (m, 1H); 13C NMR (101 MHz, MeOD-d4) δ 159.52, 158.87, 158.83, 151.25, 133.56, 130.95, 130.62, 129.71, 123.90, 119.66, 115.46, 109.86, 108.39, 104.80, 65.95, 56.00, 54.47, 48.20, 25.26, 24.34, 22.68; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C27H33N2O2 [M + H]+ 417.2537, found, 417.2536.
:95% DCM); IR (neat, cm−1): 3343, 2950, 1671, 1605, 1494, 1240, 1195, 1125, 828, 720, 689; 1H NMR (501 MHz, MeOD-d4) δ 7.24 (d, J = 8.6 Hz, 2H), 7.05–6.95 (m, 3H), 6.93–6.81 (m, 4H), 6.37 (ddd, J = 8.1, 2.1, 1.0 Hz, 1H), 6.20–6.10 (m, 2H), 4.19 (s, 2H), 4.08 (t, J = 5.7 Hz, 2H), 3.86–3.36 (m, 2H), 3.30–3.27 (m, 2H), 3.21–2.71 (m, 2H), 2.28–2.16 (m, 2H), 1.85 (s, 6H); 13C NMR (126 MHz, MeOD-d4) δ 159.92 (d, J = 239.4 Hz), 159.91, 158.80, 154.79 (d, J = 2.5 Hz), 151.77, 133.88, 130.94, 129.53, 121.32 (d, J = 8.4 Hz), 116.94 (d, J = 23.6 Hz), 115.44, 109.58, 107.47, 103.96, 65.96, 56.04, 54.50, 47.91, 25.31, 24.38, 22.71; 19F NMR (376 MHz, MeOD-d4) δ −76.55, −122.77; HRMS (APCI): m/z calcd C27H32N2O2F [M + H]+ 435.2442, found, 435.2436.
:95% DCM); IR (neat, cm−1): 3378, 2958, 1664, 1610, 1511, 1228, 1179, 1130, 830, 720, 689; 1H NMR (400 MHz, MeOD-d4) δ 7.35–7.28 (m, 2H), 7.28–7.18 (m, 2H), 6.97 (tt, J = 7.4, 1.1 Hz, 1H), 6.93–6.86 (m, 2H), 6.86–6.81 (m, 2H), 6.81–6.73 (m, 2H), 6.70–6.59 (m, 2H), 4.23 (s, 2H), 4.08 (t, J = 5.7 Hz, 2H), 3.57 (s, 2H), 3.30–3.23 (m, 2H), 2.95 (s, 2H), 2.31–2.13 (m, 2H), 2.10–1.36 (m, 6H); 13C NMR (101 MHz, MeOD-d4) δ 160.57, 158.89, 148.83, 146.56, 133.83, 130.52, 129.79, 122.93, 121.87, 117.95, 115.46, 115.37, 65.95, 55.99, 54.45, 48.75, 25.25, 24.33, 22.68; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C27H33N2O2 [M + H]+ 417.2537, found, 417.2532.
:95% DCM); IR (MeOH, cm−1): 3347, 2951, 1672, 1608, 1495, 1242, 1198, 1130, 830, 800, 721; 1H NMR (400 MHz, MeOD-d4) δ 7.35–7.26 (m, 2H), 7.04–6.93 (m, 2H), 6.88–6.82 (m, 2H), 6.82–6.74 (m, 2H), 6.73–6.64 (m, 2H), 4.25 (s, 2H), 4.09 (t, J = 5.7 Hz, 2H), 3.59 (d, J = 12.2 Hz, 2H), 3.30–3.26 (m, 2H), 2.95 (td, J = 12.2, 3.0 Hz, 2H), 2.30–2.13 (m, 2H), 1.97 (d, J = 12.2 Hz, 2H), 1.91–1.67 (m, 3H), 1.63–1.44 (m, 1H); 13C NMR (101 MHz, MeOD-d4) δ 159.45 (d, J = 238.6 Hz), 159.01, 156.40 (d, J = 2.4 Hz), 149.90, 145.65, 133.31, 129.97, 121.43, 119.61 (d, J = 8.2 Hz), 116.86 (d, J = 23.6 Hz), 115.99, 115.48, 65.96, 56.00, 54.48, 49.24, 25.27, 24.36, 22.69; 19F NMR (376 MHz, MeOD-d4) δ −123.98; HRMS (APCI): m/z calcd C27H32N2O2F [M + H]+ 435.2442, found, 435.2440.
:95% DCM); IR (neat, cm−1): 3356, 2953, 1665, 1606, 1505, 1324, 1236, 1160, 1105, 830, 800, 719; 1H NMR (600 MHz, MeOD-d4) δ 7.55 (d, J = 8.6 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H), 6.96 (d, J = 8.6 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 8.9 Hz, 2H), 6.67 (d, J = 8.9 Hz, 2H), 4.25 (s, 2H), 4.09 (t, J = 5.7 Hz, 2H), 3.59 (d, J = 11.9 Hz, 2H), 3.31–3.27 (m, 2H), 2.96 (s, 2H), 1.96 (s, 2H), 1.90–1.69 (m, 3H), 1.53 (s, 1H); 13C NMR (151 MHz, MeOD-d4) δ 162.45, 157.48, 146.23, 145.68, 132.56, 128.29, 126.57 (q, J = 3.8 Hz), 124.47 (q), 123.20 (q, J = 32.6 Hz), 121.06, 116.06, 114.08, 113.74, 64.57, 54.62, 53.09, 47.06, 23.88, 22.96, 21.28; 19F NMR (376 MHz, MeOD-d4) δ −62.76, −76.55; HRMS (APCI): m/z calcd C28H32N2O2F3 [M + H]+ 485.2410, found, 485.2401.
:95% DCM); IR (neat, cm−1): 3371, 2950, 2223, 1656, 1602, 1493, 1242, 1170, 1132, 822, 770, 721; 1H NMR (400 MHz, MeOD-d4, −15 °C) δ 7.64 (d, J = 8.9 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H), 6.94 (d, J = 8.9 Hz, 3H), 6.90 (d, J = 8.6 Hz, 3H), 6.83 (d, J = 8.9 Hz, 2H), 6.66 (d, J = 8.9 Hz, 2H), 4.25 (s, 2H), 4.07 (t, J = 5.6 Hz, 2H), 3.52 (s, 2H), 3.28 (dd, J = 6.8, 3.9 Hz, 2H), 2.94 (s, 2H), 2.30–2.12 (m, 2H), 2.10–1.36 (m, 6H); 13C NMR (126 MHz, MeOD-d4) δ 164.75, 158.90, 147.99, 146.38, 135.23, 133.93, 129.67, 122.57, 119.88, 117.96, 115.48, 115.06, 105.62, 66.00, 56.05, 54.52, 48.36, 25.34, 24.41, 22.73; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C28H32N3O2 [M + H]+ 442.2489, found 442.2484.
:95% DCM); IR (MeOH, cm−1): 3418, 3148, 2509, 1671, 1607, 1509, 1201, 1130, 801, 743, 692; 1H NMR (501 MHz, MeOD-d4, − 15 °C) δ 9.01 (s, 1H), 7.70 (s, 1H), 7.58 (s, 1H), 7.38–7.26 (m, 2H), 7.21 (d, J = 8.4 Hz, 2H), 7.05 (td, J = 7.7, 1.5 Hz, 1H), 6.98–6.87 (m, 3H), 6.79 (td, J = 7.7, 1.5 Hz, 3H), 6.65 (dd, J = 7.7, 1.5 Hz, 1H), 6.61 (td, J = 7.7, 1.5 Hz, 1H), 4.47 (t, J = 6.8 Hz, 2H), 4.29 (s, 2H), 3.99 (t, J = 5.5 Hz, 2H), 2.35 (p, J = 6.3 Hz, 2H); 13C NMR (126 MHz, MeOD-d4, − 15 °C) δ 159.27, 158.79, 144.22, 141.31, 136.73, 133.34, 130.72, 129.47, 126.01, 123.56, 123.43, 121.14, 120.72, 118.02, 115.11, 113.68, 65.33, 48.01, 47.51, 30.69; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C25H26N3O2 [M + H]+ 400.202, found, 400.2017.
:95% DCM); IR (neat, cm−1): 3420, 2933, 2877, 1607, 1498, 1231, 1195, 1108, 818, 738, 663; 1H NMR (600 MHz, acetone-d6) δ 7.58 (s, 1H), 7.27 (d, J = 8.8 Hz, 2H), 7.18 (s, 1H), 7.13–7.06 (m, 2H), 7.04–6.90 (m, 4H), 6.87 (d, J = 8.8 Hz, 2H), 6.79 (dd, J = 7.9, 1.5 Hz, 1H), 6.72 (dd, J = 7.9, 1.5 Hz, 1H), 6.59 (td, J = 7.9, 1.5 Hz, 1H), 4.35 (d, J = 5.7 Hz, 2H), 4.25 (t, J = 6.9 Hz, 2H), 3.94 (t, J = 6.9 Hz, 2H), 2.24 (p, J = 6.9 Hz, 2H); 13C NMR (101 MHz, acetone-d6) δ 159.07 (d, J = 238.5 Hz), 158.76, 154.91 (d, J = 2.3 Hz), 143.93, 141.49, 138.35, 133.08, 129.71, 129.18, 125.90, 120.21, 120.09, 119.37 (d, J = 8.3 Hz), 117.12, 116.82 (d, J = 23.5 Hz), 115.27, 112.78, 65.16, 47.05, 44.11, 31.58; 19F NMR (376 MHz, MeOD-d4) δ −76.55, −122.20; HRMS (APCI): m/z calcd C25H25N3O2F [M + H]+ 418.1925, found, 418.1921.
:95% DCM); IR (neat, cm−1): 3419, 2933, 1608, 1509, 1322, 1226, 1158, 1105, 1063, 819, 739, 663; 1H NMR (400 MHz, acetone-d6) δ 77.68 (d, J = 8.6 Hz, 2H), 7.55 (s, 1H), 7.24 (d, J = 8.6 Hz, 2H), 7.13 (s, 1H), 7.09–6.99 (m, 3H), 6.98–6.89 (m, 2H), 6.85 (d, J = 8.6 Hz, 2H), 6.76 (dd, J = 8.2, 1.5 Hz, 1H), 6.65 (td, J = 8.2, 1.5 Hz, 1H), 4.34 (d, J = 4.5 Hz, 2H), 4.23 (t, J = 6.9 Hz, 2H), 3.93 (t, J = 6.9 Hz, 2H), 2.23 (p, J = 6.9 Hz, 2H); 13C NMR (101 MHz, acetone-d6) δ 162.10, 158.75, 142.00, 141.85, 138.32, 132.95, 129.69, 129.11, 127.92 (q, J = 3.8 Hz), 127.04, 125.49 (q, J = 270.6 Hz), 124.41 (q, J = 32.5 Hz), 121.67, 120.04, 117.40, 117.30, 115.25, 113.22, 65.14, 46.90, 44.04, 31.58; 19F NMR (376 MHz, MeOD-d4) δ −62.46, −76.55; HRMS (APCI): m/z calcd C26H24N3O2F3 [M + H]+ 468.1890, found, 468.1890.
:95% DCM); IR (neat, cm−1): 3400, 2928, 2522, 2221, 1673, 1602, 1499, 1232, 1175, 1115, 872, 831, 741, 720; 1H NMR (501 MHz, MeOD-d4, − 15 °C) δ 9.00 (s, 1H), 7.82–7.63 (m, 3H), 7.58 (s, 1H), 7.17 (d, J = 8.2 Hz, 2H), 7.07–6.96 (m, 3H), 6.90 (dd, J = 7.9, 1.4 Hz, 1H), 6.79 (d, J = 8.2 Hz, 2H), 6.70–6.59 (m, 2H), 4.47 (t, J = 6.7 Hz, 2H), 4.27 (s, 2H), 3.99 (t, J = 6.7 Hz, 2H), 2.48–2.20 (m, 2H); 13C NMR (126 MHz, MeOD-d4, − 15 °C) δ 163.43, 158.73, 142.11, 141.76, 136.76, 135.27, 133.56, 129.22, 127.60, 123.40, 122.27, 121.25, 119.86, 117.87, 117.65, 115.09, 113.78, 106.06, 65.32, 47.96, 46.94, 30.71; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C26H25N4O2 [M + H]+ 425.1969, found 425.1968.
:95% DCM); IR (neat, cm−1): 3242, 2948, 2873, 1614, 1512, 1488, 1244, 1212, 1145, 817, 754, 692; 1H NMR (400 MHz, acetone-d6) δ 7.55 (s, 1H), 7.38–7.30 (m, 2H), 7.30–7.23 (m, 2H), 7.13 (s, 1H), 7.10–7.02 (m, 2H), 7.01–6.90 (m, 3H), 6.90–6.84 (m, 2H), 6.45 (ddd, J = 8.1, 2.2, 0.8 Hz, 1H), 6.31 (t, J = 2.2 Hz, 1H), 6.20 (ddd, J = 8.1, 2.3, 0.8 Hz, 1H), 4.30–4.16 (m, 4H), 3.94 (t, J = 6.5 Hz, 2H), 2.24 (p, J = 6.5 Hz, 2H); 13C NMR (101 MHz, acetone) δ 159.02, 158.73, 158.39, 151.46, 138.37, 132.94, 130.79, 130.48, 129.77, 129.38, 123.64, 120.05, 119.37, 115.24, 108.97, 107.41, 103.91, 65.14, 47.47, 44.01, 31.57; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C25H26N3O2 [M + H]+ 400.202, found, 400.2011.
:95% DCM); IR (neat, cm−1): 3225, 2944, 2835, 1609, 1497, 1234, 1195, 1109, 1034, 824, 728, 661; 1H NMR (600 MHz, acetone-d6) δ 7.52 (s, 1H), 7.27 (d, J = 8.6 Hz, 2H), 7.15–7.06 (m, 3H), 7.04 (t, J = 8.2 Hz, 1H), 7.01–6.95 (m, 2H), 6.91 (s, 1H), 6.90–6.85 (m, 2H), 6.44 (ddd, J = 8.2, 2.3, 0.8 Hz, 1H), 6.27 (t, J = 2.3 Hz, 1H), 6.17 (ddd, J = 8.2, 2.3, 0.8 Hz, 1H), 4.27–4.21 (m, 4H), 3.94 (t, J = 6.0 Hz, 2H), 2.24 (p, J = 6.4 Hz, 2H); 13C NMR (101 MHz, acetone-d6) δ 159.47, 159.35 (d, J = 239.0 Hz), 158.74, 154.26 (d, J = 2.4 Hz), 151.47, 138.40, 132.92, 130.83, 129.82, 129.37, 121.24 (d, J = 8.3 Hz), 120.04, 116.87 (d, J = 23.4 Hz), 115.24, 108.90, 106.85, 103.38, 65.14, 47.49, 43.99, 31.58; 19F NMR (376 MHz, MeOD-d4) δ −121.54; HRMS (APCI): m/z calcd C25H25N3O2F [M + H]+ 418.1925, found, 418.1931.
:95% DCM); IR (neat, cm−1): 3247, 2939, 2821, 1608, 1509, 1489, 1228, 1064, 824, 741, 662; 1H NMR (600 MHz, acetone-d6) δ 7.55 (s, 1H), 7.33 (d, J = 8.8 Hz, 2H), 7.31–7.26 (m, 2H), 7.13 (s, 1H), 6.99 (tt, J = 7.3, 1.1 Hz, 1H), 6.93 (s, 1H), 6.92–6.89 (m, 2H), 6.88–6.85 (m, 2H), 6.85–6.80 (m, 2H), 6.73–6.67 (m, 2H), 4.27 (s, 2H), 4.25 (t, J = 6.9 Hz, 2H), 3.95 (t, J = 6.9 Hz, 2H), 2.25 (p, J = 6.9 Hz, 2H); 13C NMR (151 MHz, acetone) δ 160.33, 158.78, 147.63, 146.72, 138.43, 133.24, 130.37, 129.77, 129.46, 122.57, 121.84, 120.13, 117.56, 115.28, 114.37, 65.18, 48.06, 44.06, 31.59; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C25H26N3O2 [M + H]+ 400.202, found, 400.2015.
:95% DCM); IR (neat, cm−1): 3236, 2943, 2835, 1606, 1494, 1241, 1207, 1035, 821, 729, 659; 1H NMR (400 MHz, acetone-d6) δ 7.56 (s, 1H), 7.32(d, J = 8.6 Hz, 2H), 7.13 (s, 1H), 7.09–6.99 (m, 2H), 6.94 (s, 1H), 6.92–6.84 (m, 4H), 6.85–6.76 (m, 2H), 6.73–6.64 (m, 2H), 4.33–4.17 (m, 4H), 3.94 (t, J = 6.5 Hz, 2H), 2.23 (p, J = 6.5 Hz, 2H); 13C NMR (101 MHz, acetone-d6) δ 158.74, 158.66 (d, J = 237.7 Hz), 156.33 (d, J = 2.2 Hz), 148.04, 146.68, 138.36, 133.19, 129.73, 129.44, 121.47, 119.99, 119.10 (d, J = 8.4 Hz), 116.68 (d, J = 23.4 Hz), 115.25, 114.36, 65.13, 48.01, 44.01, 31.55; 19F NMR (376 MHz, acetone) δ −124.75, −76.55; HRMS (APCI): m/z calcd C25H25N3O2F [M + H]+ 418.1925, found, 418.1922.
:95% DCM); IR (neat, cm−1): 3243, 2939, 1614, 1508, 1324, 1236, 1104, 1065, 830, 742, 662; 1H NMR (400 MHz, acetone-d6) δ 7.63 (d, J = 8.8 Hz, 2H), 7.57 (s, 1H), 7.34 (d, J = 8.6 Hz, 2H), 7.28–7.06 (m, 1H), 7.02 (d, J = 8.8 Hz, 2H), 6.96 (s, 1H), 6.93–6.85 (m, 4H), 6.74 (d, J = 8.9 Hz, 2H), 4.29 (s, 2H), 4.25 (t, J = 6.9 Hz, 2H), 3.95 (t, J = 6.9 Hz, 2H), 2.25 (p, J = 6.9 Hz, 2H); 13C NMR (101 MHz, acetone-d6) δ 163.50, 158.80, 147.47, 146.13, 138.49, 133.08, 129.80, 129.47, 127.87 (q, J = 3.8 Hz), 125.52 (q, J = 270.0 Hz), 123.81 (q, J = 32.4 Hz), 122.35, 120.21, 117.25, 115.28, 114.42, 65.15, 47.93, 44.04, 31.57; 19F NMR (376 MHz, MeOD-d4) δ −62.72, −76.55; HRMS (APCI): m/z calcd C26H24N3O2F3 [M + H]+ 468.1890, found, 468.1890.
:95% DCM); IR (MeOH, cm−1): 3389, 2926, 2225, 1610, 1496, 1239, 1168, 1110, 830, 743; 1H NMR (501 MHz, acetone-d6) δ 7.70 (d, J = 9.0 Hz, 2H), 7.55 (s, 1H), 7.33 (d, J = 8.6 Hz, 2H), 7.15 (s, 1H), 7.00 (d, J = 9.0 Hz, 2H), 6.94 (s, 1H), 6.93–6.87 (m, 4H), 6.74 (d, J = 8.9 Hz, 2H), 4.29 (s, 2H), 4.25 (t, J = 6.9 Hz, 2H), 3.95 (t, J = 6.9 Hz, 2H), 2.25 (p, J = 6.9 Hz, 2H); 13C NMR (101 MHz, acetone) δ 164.08, 158.81, 147.68, 145.62, 138.18134.98, 133.04, 129.74, 129.46, 122.41, 120.86, 119.41, 117.68, 115.29, 114.42, 105.54, 65.16, 47.90, 44.09, 31.55; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C26H25N4O2 [M + H]+ 425.1969, found 425.1966.
:90% DCM); IR (neat, cm−1): 3401, 2951, 1667, 1607, 1490, 1242, 1174, 1047, 838, 746, 724, 691; 1H NMR (501 MHz, MeOD-d4) δ 7.34–7.25 (m, 2H), 7.22 (d, J = 8.6 Hz, 2H), 7.08–7.00 (m, 1H), 6.97–6.89 (m, 3H), 6.87 (d, J = 8.6 Hz, 2H), 6.78 (dd, J = 7.9, 1.5 Hz, 1H), 6.68 (dd, J = 7.9, 1.5 Hz, 1H), 6.59 (td, J = 7.9, 1.5 Hz, 1H), 4.29 (s, 2H), 4.08 (t, J = 6.1 Hz, 2H), 3.20 (t, J = 6.1 Hz, 2H), 2.72 (s, 3H), 2.14 (p, J = 6.1 Hz, 2H); 13C NMR (126 MHz, MeOD-d4) δ 159.34, 158.88, 144.29, 141.88, 133.88, 130.67, 129.47, 126.01, 123.55, 120.73, 118.04, 117.81, 115.53, 113.47, 66.20, 47.69, 33.88, 27.18; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C23H27N2O2 [M + H]+ 363.2067, found, 363.2066.
:90% DCM); IR (neat, cm−1): 3421, 2933, 1607, 1498, 1240, 1196, 1110, 1035, 824, 738; 1H NMR (400 MHz, MeOD-d4) 7.28–7.20 (m, 2H), 7.13–7.04 (m, 2H), 7.04–6.93 (m, 3H), 6.93–6.73 (m, 5H), 4.39 (d, J = 5.7 Hz, 2H), 4.09 (t, J = 6.3 Hz, 2H), 3.21 (t, J = 6.3 Hz, 2H), 2.73 (s, 3H), 2.16 (p, J = 6.3 Hz, 2H); 13C NMR (101 MHz, MeOD-d4) δ 160.12 (d, J = 239.9 Hz), 159.43, 154.45, 146.63, 137.79, 131.55, 130.38, 125.67, 121.28, 120.42 (d, J = 8.3 Hz), 119.72, 117.18 (d, J = 23.7 Hz), 116.52, 115.63, 66.21, 49.57, 48.36, 33.85, 27.16; 19F NMR (377 MHz, MeOD-d4) δ −76.55, −122.35; HRMS (APCI): m/z calcd C23H26N2O2F [M + H]+ 381.1973, found, 381.1963.
:90% DCM); IR (neat, cm−1): 3420, 2761, 1666, 1609, 1510, 1230, 1165, 1105, 1064, 836, 799, 721; 1H NMR (600 MHz, MeOD-d4) δ 7.59 (d, J = 8.7 Hz, 2H), 7.19 (d, J = 8.6 Hz, 2H), 7.02 (d, J = 8.7 Hz, 2H), 6.99 (dd, J = 8.0, 1.5 Hz, 1H), 6.88 (dd, J = 8.0, 1.5 Hz, 1H), 6.86 (d, J = 8.6 Hz, 2H), 6.71 (dd, J = 8.0, 1.5 Hz, 1H), 6.64 (td, J = 8.0, 1.5 Hz, 1H), 4.28 (s, 2H), 4.08 (t, J = 5.7 Hz, 2H), 3.20 (t, J = 5.7 Hz, 2H), 2.72 (s, 3H), 2.18–2.10 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 162.52, 158.88, 142.78, 142.14, 133.83, 129.40, 128.07 (q, J = 3.8 Hz), 127.15, 125.85 (q, J = 271.3 Hz), 125.22 (q, J = 32.6 Hz), 121.87, 117.91, 117.59, 115.51, 113.86, 66.19, 48.47, 47.46, 33.87, 27.18; 19F NMR (376 MHz, MeOD-d4) δ −62.83, −76.55; HRMS (APCI): m/z calcd C24H26N2O2F3 [M + H]+ 431.1941, found, 431.1929.
:90% DCM); IR (neat, cm−1): 3411, 2940, 2222, 1677, 1603, 1499, 1235, 1176, 1133, 1051, 837, 762, 720; 1H NMR (501 MHz, MeOD-d4) δ 7.64 (d, J = 8.9 Hz, 2H), 7.18 (d, J = 8.6 Hz, 2H), 7.03 (dd, J = 7.6, 1.5 Hz, 1H), 6.99 (d, J = 8.9 Hz, 2H), 6.89 (dd, J = 7.9, 1.5 Hz, 1H), 6.85 (d, J = 8.6 Hz, 2H), 6.72 (dd, J = 7.6, 1.5 Hz, 1H), 6.65 (td, J = 7.6, 1.5 Hz, 1H), 4.27 (s, 2H), 4.08 (t, J = 6.1 Hz, 2H), 3.20 (t, J = 6.1 Hz, 2H), 2.73 (s, 3H), 2.14 (p, J = 6.1 Hz, 2H); 13C NMR (126 MHz, MeOD-d4) δ 163.38, 158.90, 142.20, 142.12, 135.25, 133.76, 129.37, 127.53, 122.11, 119.83, 118.04, 117.94, 115.51, 114.04, 106.24, 66.18, 48.43, 47.43, 33.85, 27.17; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C24H26N3O2 [M + H]+ 388.2020, found, 388.2009.
:90% DCM); IR (neat, cm−1): 3044, 1672, 1490, 1333, 1252, 1152, 751; 1H NMR (600 MHz, MeOD-d4) δ 7.65–7.60 (m, 2H), 7.60–7.55 (m, 1H), 7.27–7.19 (m, 2H), 7.11–7.06 (m, 1H), 7.04 (dd, J = 6.1, 3.6 Hz, 2H), 6.90–6.84 (m, 2H), 6.68–6.63 (m, 1H), 6.63–6.58 (m, 2H), 4.10 (t, J = 5.8 Hz, 2H), 3.37–3.33 (m, 2H), 2.94 (s, 6H), 2.28–2.20 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 163.24, 157.69, 150.93, 133.43, 130.70, 130.46, 129.21, 127.45, 126.68, 124.65, 124.49, 119.75, 119.10, 115.52, 66.30, 56.51, 43.60, 25.56; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C23H26O4N2S 426.1608 [M]+, found, 426.1609; C23H27O4N2S 427.1686 [M + H]+, found, 427.1684.
:90% DCM); IR (neat, cm−1): 3072, 1672, 1493, 1333, 1153, 830; 1H NMR (600 MHz, MeOD-d4) δ 7.64–7.60 (m, 2H), 7.60–7.56 (m, 1H), 7.09–7.04 (m, 2H), 6.99–6.93 (m, 2H), 6.89–6.85 (m, 2H), 6.69–6.64 (m, 1H), 6.62–6.56 (m, 2H), 4.10 (t, J = 5.8 Hz, 2H), 3.37–3.33 (m, 2H), 2.95 (s, 6H), 2.28–2.20 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 163.22, 160.11 (d, J = 240.4 Hz), 153.82 (d, J = 2.6 Hz), 151.07, 133.59, 130.42, 129.21, 127.63, 127.14, 124.71, 121.02 (d, J = 8.4 Hz), 119.08, 117.04 (d, J = 23.7 Hz), 115.50, 66.29, 56.55, 43.61, 25.59; 19F NMR (376 MHz, MeOD-d4) δ −76.55, −121.81; HRMS (APCI): m/z calcd [M + H]+, found, HRMS (APCI): m/z calcd C23H25O4N2FS 444.1514 [M]+, found, 444.1516; C23H27O4N2FS 445.1592 [M + H]+, found, 445.159.
:90% DCM); IR (neat, cm−1): 3079, 1671, 1595, 1494, 1324, 1255, 1106, 831; 1H NMR (600 MHz, MeOD-d4) δ 7.67–7.63 (m, 1H), 7.59–7.54 (m, 2H), 7.50–7.45 (m, 2H), 7.16 (pd, J = 7.5, 1.8 Hz, 2H), 6.88–6.84 (m, 1H), 6.83–6.78 (m, 2H), 6.75–6.69 (m, 2H), 4.08 (t, J = 5.8 Hz, 2H), 3.35–3.32 (m, 2H), 2.94 (s, 6H), 2.30–2.16 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 163.21, 161.33, 149.09, 133.54, 130.32, 130.29, 127.97 (q, J = 3.8 Hz), 127.75, 127.17, 126.15, 125.74 (q, J = 270.6 Hz), 125.66 (q, J = 32.5 Hz), 121.24, 118.54, 115.51, 66.25, 56.49, 43.59, 25.52; 19F NMR (376 MHz, MeOD-d4) δ −62.63, −76.55; HRMS (APCI): m/z calcd C24H25O4N2F3S 494.1482 [M]+, found, 494.1482; C24H26O4N2F3S 495.156 [M + H]+, found, 495.1555.
:90% DCM); IR (neat, cm−1): 3385, 3069, 2214, 1672, 1595, 1496, 1367, 1331, 1161, 829; 1H NMR (600 MHz, MeOD-d4) δ 7.58–7.53 (m, 2H), 7.47 (dd, J = 8.2, 1.4 Hz, 1H), 7.38–7.32 (m, 2H), 7.31–7.24 (m, 2H), 7.15 (ddd, J = 8.6, 6.9, 2.1 Hz, 1H), 6.71–6.67 (m, 2H), 6.67–6.62 (m, 2H), 3.93 (t, J = 5.9 Hz, 2H), 3.38–3.33 (m, 2H), 2.96 (s, 6H), 2.28–2.20 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 164.16, 149.43, 143.17, 134.48, 134.25, 131.66, 128.82, 128.33, 126.10, 125.31, 124.32, 121.48, 115.66, 115.52, 101.02, 66.26, 56.46, 43.69, 25.62; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C24H25O4N3S 451.156 [M]+, found, 451.1562; C24H25O4N3S 452.1639 [M + H]+, found, 452.1628.
:90% DCM); IR (neat, cm−1): 3078, 1676, 1592, 1489, 1339, 1202, 1162, 1120, 771; 1H NMR (600 MHz, MeOD-d4) δ 7.67 (d, J = 8.9 Hz, 2H), 7.37–7.31 (m, 2H), 7.16 (t, J = 8.1 Hz, 1H), 7.13 (tt, J = 8.1, 1.1 Hz, 1H), 7.01 (d, J = 8.9 Hz, 2H), 6.90–6.86 (m, 2H), 6.81 (ddd, J = 8.1, 2.1, 0.9 Hz, 1H), 6.70 (t, J = 2.2 Hz, 1H), 6.64 (ddd, J = 8.1, 2.1, 0.9 Hz, 1H), 4.15 (t, J = 5.7 Hz, 2H), 3.36–3.32 (m, 2H), 2.93 (s, 6H), 2.28–2.19 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 163.29, 159.43, 158.07, 140.63, 132.91, 131.28, 130.91, 130.48, 124.74, 120.13, 116.35, 115.70, 115.39, 111.66, 66.37, 56.54, 43.61, 25.54; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C23H26O4N2S 426.1608 [M]+, found, 426.161; C23H27O4N2S 427.1683 [M + H]+, found, 427.1684.
:90% DCM); IR (neat, cm−1): 3077, 2888, 1672, 1594, 1496, 1339, 1200, 1120, 1086, 773; 1H NMR (501 MHz, MeOD-d4) δ 7.67 (d, J = 8.9 Hz, 2H), 7.16 (t, J = 8.1 Hz, 1H), 7.11–7.03 (m, 2H), 7.01 (d, J = 8.9 Hz, 2H), 6.92–6.85 (m, 2H), 6.81 (ddd, J = 8.1, 2.1, 0.9 Hz, 1H), 6.67 (t, J = 2.1 Hz, 1H), 6.64 (ddd, J = 8.1, 2.1, 0.9 Hz, 1H), 4.15 (t, J = 5.8 Hz, 2H), 3.37–3.32 (m, 2H), 2.93 (s, 6H), 2.29–2.17 (m, 2H); 13C NMR (126 MHz, MeOD-d4) δ 163.31, 160.39 (d, J = 240.7 Hz), 159.75, 154.00 (d, J = 2.5 Hz), 140.65, 133.01, 131.33, 130.45, 121.89 (d, J = 8.3 Hz), 117.29 (d, J = 23.6 Hz), 116.48, 115.74, 115.06, 111.37, 66.41, 56.57, 43.65, 25.54; 19F NMR (377 MHz, MeOD-d4) δ −76.55, −121.33; HRMS (APCI): m/z calcd C23H25O4N2FS 444.1514 [M]+, found, 444.1516; C23H27O4N2FS 445.1592 [M + H]+, found, 445.159.
:90% DCM); IR (neat, cm−1): 3047, 1670, 1488, 1331, 1245, 1150, 831; 1H NMR (501 MHz, MeOD-d4) δ 7.77–7.71 (m, 2H), 7.41–7.35 (m, 2H), 7.18–7.10 (m, 3H), 7.10–7.05 (m, 2H), 7.00–6.94 (m, 2H), 6.91–6.86 (m, 2H), 4.21 (t, J = 5.8 Hz, 2H), 3.45–3.36 (m, 2H), 3.00 (s, 6H), 2.39–2.27 (m, 2H); 13C NMR (126 MHz, MeOD-d4) δ 161.88, 157.19, 154.58, 132.78, 131.53, 129.47, 129.04, 123.56, 123.03, 118.89, 118.25, 114.30, 65.06, 55.09, 42.25, 24.09; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C23H26O4N2S 426.1608 [M]+, found, 426.1609; C23H27O4N2S 427.1686 [M + H]+, found, 427.1683.
:90% DCM); IR (neat, cm−1): 3072, 1671, 1494, 1331, 1255, 1190, 1151, 831; 1H NMR (501 MHz, MeOD-d4) δ 7.70 (d, J = 8.8 Hz, 2H), 7.15–7.04 (m, 4H), 7.02 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.9 Hz, 2H), 6.86–6.77 (m, 2H), 4.16 (t, J = 5.8 Hz, 2H), 3.43–3.30 (m, 2H), 2.97 (s, 6H), 2.25–2.22 (m, 2H); 13C NMR (126 MHz, MeOD-d4) δ 163.22, 160.15 (d, J = 240.7 Hz), 156.27, 154.35 (d, J = 2.8 Hz), 134.08, 132.81, 130.38, 124.93, 121.42 (d, J = 8.4 Hz), 119.80, 117.22 (d, J = 23.6 Hz), 115.68, 66.38, 56.48, 43.63, 25.46; 19F NMR (376 MHz, MeOD-d4) δ −76.55, −121.84; HRMS (APCI): m/z calcd C23H25O4N2FS 444.1514 [M]+, found, 444.1515; C23H27O4N2FS 445.1592 [M + H]+, found, 445.159.
:90% DCM); IR (neat, cm−1): 3054, 1671, 1595, 1499, 1324, 1249 1151, 1105, 832; 1H NMR (600 MHz, MeOD-d4) δ 7.70 (d, J = 8.9 Hz, 2H), 7.61 (d, J = 9.0 Hz, 2H), 7.12 (d, J = 8.9 Hz, 2H), 7.05–6.99 (m, 4H), 6.93 (d, J = 8.9 Hz, 2H), 4.15 (t, J = 5.8 Hz, 2H), 3.36–3.32 (m, 2H), 2.93 (s, 6H), 2.27–2.19 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 163.28, 162.22, 153.96, 135.69, 133.04, 130.47, 128.27 (q, J = 3.8 Hz), 125.82 (q, J = 32.6 Hz), 125.70 (q, J = 270.5 Hz), 124.62, 121.72, 118.72, 115.68, 66.37, 56.55, 43.61, 25.54; 19F NMR (376 MHz, MeOD-d4) δ −63.14, −76.55; HRMS (APCI): m/z calcd C24H25O4N2F3S 494.1482 [M]+, found, 494.1482; C24H26O4N2F3S 495.156 [M + H]+, found, 495.155.
:90% DCM); IR (neat, cm−1): 3047, 2227, 1670 1596, 1494, 1334, 1245, 1155, 1094, 832; 1H NMR (600 MHz, MeOD-d4) δ 7.73 (d, J = 8.7 Hz, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.17 (d, J = 8.9 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.9 Hz, 2H), 4.17 (t, J = 5.8 Hz, 2H), 3.39–3.34 (m, 2H), 2.96 (s, 6H), 2.31–2.21 (m, 2H); 13C NMR (151 MHz, MeOD-d4) δ 163.30, 163.16, 153.15, 136.16, 135.43, 132.96, 130.46, 124.52, 122.12, 119.63, 118.90, 115.70, 106.84, 66.38, 56.53, 43.61, 25.53; 19F NMR (376 MHz, MeOD-d4) δ −76.55; HRMS (APCI): m/z calcd C24H25O4N3S 452.1639 [M + H]+, found, 452.1629.
Biology
000 and 10000 cells per well, respectively, in 384-well flat clear-bottom black plates (Corning, NY, USA) and cultured at 37 °C in a humidified 5% CO2 incubator 48 h before assay. Cells were loaded with 20 μL per well of calcium 6 dye (Molecular Devices) reconstituted in assay buffer containing (in mM) 140 NaCl, 11.5 glucose, 5.9 KCl, 1.4 MgCl2, 1.2 NaH2PO4, 5 NaHCO3, 1.8 CaCl2,10 HEPES pH 7.4 and 0.1% bovine serum albumin (BSA, Sigma), and incubated for 30 min at 37 °C in a humidified 5% CO2 incubator. For CaV2.2 assays, nifedipine 10 μM (CaV1 blocker) was added to the dye solution. The Ca2+ fluorescence responses were recorded at excitation 470–495 nm and emission 515–575 nm for 10 s to set the baseline, 300 s after addition of compound and for further 300 s after channel activation induced by the addition of 90 mM KCl and 5 mM CaCl2 for CaV2.2, or 40 mM KCl and 5 mM CaCl2 for CaV3.2. Compound stock solutions were prepared at 100 mM in 100% DMSO and diluted further in the assay buffer to 1% DMSO, for the highest tested concentration of 100 μM of compound, and serial-diluted 3-fold in assay buffer.
The maximum or maximum–minimum fluorescence signals were used to calculate the half-maximum inhibition effect (IC50). Curve fitting was achieved using GraphPad Prism Version 10 (GraphPad Software Inc, San Diego, CA, USA) with non-linear regression with log[inhibitor] versus normalized response and variable Hill slope for dose–responses. Data were represented as mean ± 95% CI and SEM from n = 3–5 independent experiments.
Rat plasma (4 mL) was transferred into an Eppendorf tube and incubated at 37 °C for 15 min. An aliquot of the incubated rat plasma (995 μL) was transferred to an Eppendorf tube containing a solution of the test compound and internal standard (5 μL of solution: 30 mM test compound and 25 mM diazepam in DMSO). The final concentration of the test compound and diazepam in plasma was 150 and 125 μM respectively (n = 3). The compound–plasma mixtures were incubated at 37 °C. At timed intervals of 0, 5, 15, 30, 45, 60, 75, 90, 105, 120, 180 and 3600 min, aliquots (50 μL) of each incubate were removed and acetonitrile (50 μL) was added immediately. For the sulfonamide compounds, the samples were vortexed, diluted with H2O (150 μL), vortexed again and cooled on ice for 30 min whilst the phenoxyaniline compounds were vortex and placed immediately on ice for 30 min. The samples were centrifuged for 15 min at 14000 rpm and the supernatant collected and analysed by RP–HPLC. The ratio of the test compound to diazepam was calculated and the data normalised. The stability curves were plotted using GraphPad Prism 8.0.2. Curve fitting was done using “one-phase decay – non-linear regression” to calculate the half-life (t1/2).
Author contributions
Synthesis: AB, RW. Calcium influx bioassays: AB, FCC, MH, YD. Rat plasma stability and cytotoxicity: AB. Data analysis: AB, FCC, MH, YD, RJL. Manuscript preparation: AB, PJD, KLT. Project conception and supervision: PJD, KLT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Monash University and CSIRO are acknowledged for funding. This work was also supported by a NHMRC Program Grant (APP1072113, RJL) and NHMRC Fellowship (1119056, RJL). The assistance of the Andrews group at Monash University, especially Sarmi Munuganti, with the cytotoxicity studies is gratefully acknowledged.References
- R. Planells-Cases, E. Perez-Paya, A. Messeguer, C. Carreno and A. Ferrer-Montiel, Mini-Rev. Med. Chem., 2003, 3, 749–756 CrossRef CAS PubMed.
- S. M. Todorovic and V. Jevtovic-Todorovic, Pflügers Arch., 2013, 465, 921–927 CrossRef CAS PubMed.
- R. H. Dworkin, A. B. O'Connor, M. Backonja, J. T. Farrar, N. B. Finnerup, T. S. Jensen, E. A. Kalso, J. D. Loeser, C. Miaskowski, T. J. Nurmikko, R. K. Portenoy, A. S. C. Rice, B. R. Stacey, R.-D. Treede, D. C. Turk and M. S. Wallace, Pain, 2007, 132, 237–251 CrossRef CAS PubMed.
- G. Cruccu and A. Truini, Pain Ther., 2017, 6, 35–42 CrossRef PubMed.
- H. Safavi-Hemami, S. E. Brogan and B. M. Olivera, J. Proteomics, 2019, 190, 12–20 CrossRef CAS PubMed.
- S. Lee, Curr. Neuropharmacol., 2013, 11, 606–620 CrossRef CAS.
- S. Menzler, J. A. Bikker, N. Suman-Chauhan and D. C. Horwell, Bioorg. Med. Chem. Lett., 2000, 10, 345–347 CrossRef CAS PubMed.
- C. Toth, Ther. Adv. Drug Saf., 2014, 5, 38–56 CrossRef CAS PubMed.
- G. W. Zamponi, Z.-P. Feng, L. Zhang, H. Pajouhesh, Y. Ding, F. Belardetti, H. Pajouhesh, D. Dolphin, L. A. Mitscher and T. P. Snutch, Bioorg. Med. Chem. Lett., 2009, 19, 6467–6472 CrossRef PubMed.
- C. Abbadie, O. B. McManus, S.-Y. Sun, R. M. Bugianesi, G. Dai, R. J. Haedo, J. B. Herrington, G. J. Kaczorowski, M. M. Smith, A. M. Swensen, V. A. Warren, B. Williams, S. P. Arneric, C. Eduljee, T. P. Snutch, E. W. Tringham, N. Jochnowitz, A. Liang, D. Euan MacIntyre, E. McGowan, S. Mistry, V. V. White, S. B. Hoyt, C. London, K. A. Lyons, P. B. Bunting, S. Volksdorf and J. L. Duffy, J. Pharmacol. Exp. Ther., 2010, 334, 545–555 CrossRef CAS PubMed.
- X. Beebe, D. Darczak, R. F. Henry, T. Vortherms, R. Janis, M. Namovic, D. Donnelly-Roberts, K. L. Kage, C. Surowy, I. Milicic, W. Niforatos, A. Swensen, K. C. Marsh, J. M. Wetter, P. Franklin, S. Baker, C. Zhong, G. Simler, E. Gomez, J. M. Boyce-Rustay, C. Z. Zhu, A. O. Stewart, M. F. Jarvis and V. E. Scott, Bioorg. Med. Chem., 2012, 20, 4128–4139 CrossRef CAS PubMed.
- V. E. Scott, T. A. Vortherms, W. Niforatos, A. M. Swensen, T. Neelands, I. Milicic, P. N. Banfor, A. King, C. Zhong, G. Simler, C. Zhan, N. Bratcher, J. M. Boyce-Rustay, C. Z. Zhu, P. Bhatia, G. Doherty, H. Mack, A. O. Stewart and M. F. Jarvis, Biochem. Pharmacol., 2012, 83, 406–418 CrossRef CAS PubMed.
- P. P. Shao, F. Ye, P. K. Chakravarty, D. J. Varughese, J. B. Herrington, G. Dai, R. M. Bugianesi, R. J. Haedo, A. M. Swensen, V. A. Warren, M. M. Smith, M. L. Garcia, O. B. McManus, K. A. Lyons, X. Li, M. Green, N. Jochnowitz, E. McGowan, S. Mistry, S.-Y. Sun, C. Abbadie, G. J. Kaczorowski and J. L. Duffy, J. Med. Chem., 2012, 55, 9847–9855 CrossRef CAS PubMed.
- N. L. Subasinghe, M. J. Wall, M. P. Winters, N. Qin, M. L. Lubin, M. F. A. Finley, M. R. Brandt, M. P. Neeper, C. R. Schneider, R. W. Colburn, C. M. Flores and Z. Sui, Bioorg. Med. Chem. Lett., 2012, 22, 4080–4083 CrossRef CAS.
- C. E. Tranberg, A. Yang, I. Vetter, J. R. McArthur, J. B. Baell, R. J. Lewis, K. L. Tuck and P. J. Duggan, Mar. Drugs, 2012, 10, 2349–2368 CrossRef CAS PubMed.
- P. P. Shao, F. Ye, P. K. Chakravarty, J. B. Herrington, G. Dai, R. M. Bugianesi, R. J. Haedo, A. M. Swensen, V. A. Warren, M. M. Smith, M. L. Garcia, O. B. McManus, K. A. Lyons, X. Li, M. Green, N. Jochnowitz, E. McGowan, S. Mistry, S.-Y. Sun, C. Abbadie, G. J. Kaczorowski and J. L. Duffy, ACS Med. Chem. Lett., 2013, 4, 1064–1068 CrossRef CAS PubMed.
- P. J. Duggan and K. L. Tuck, Toxins, 2015, 7, 4175–4198 CrossRef CAS PubMed.
- E. C. Gleeson, J. E. Graham, S. Spiller, I. Vetter, R. J. Lewis, P. J. Duggan and K. L. Tuck, Mar. Drugs, 2015, 13, 2030–2045 CrossRef CAS PubMed.
- A. Mollica, R. Costante, E. Novellino, A. Stefanucci, S. Pieretti, F. Zador, R. Samavati, A. Borsodi, S. Benyhe, I. Vetter and R. J. Lewis, Chem. Biol. Drug Des., 2015, 86, 156–162 CrossRef CAS PubMed.
- J. R. McArthur, L. Motin, E. C. Gleeson, S. Spiller, R. J. Lewis, P. J. Duggan, K. L. Tuck and D. J. Adams, Br. J. Pharmacol., 2018, 175, 2284–2295 CrossRef CAS PubMed.
- A. Sairaman, F. C. Cardoso, A. Bispat, R. J. Lewis, P. J. Duggan and K. L. Tuck, Bioorg. Med. Chem., 2018, 26, 3046–3059 CrossRef CAS PubMed.
- F. C. Cardoso, M.-A. Marliac, C. Geoffroy, M. Schmit, A. Bispat, R. J. Lewis, K. L. Tuck and P. J. Duggan, Bioorg. Med. Chem., 2020, 28, 115655 CrossRef CAS PubMed.
- F. C. Cardoso, M. Schmit, M. J. Kuiper, R. J. Lewis, K. L. Tuck and P. J. Duggan, RSC Med. Chem., 2022, 13, 183–195 RSC.
- D. Perret and Z. D. Luo, Neurotherapeutics, 2009, 6, 679–692 CrossRef CAS PubMed.
- T. P. Snutch, NeuroRx, 2005, 2, 662–670 CrossRef PubMed.
- A. Calderon-Rivera, K. Gomez, S. Loya-López, E. M. K. Wijeratne, H. Stratton, C. Tang, P. Duran, K. Masterson, O. Alsbiei, A. A. L. Gunatilaka and R. Khanna, Neurobiol. Pain, 2023, 13, 100116 CrossRef CAS PubMed.
- P. J. Duggan, R. J. Lewis, L. Y. Phei, N. G. Lumsden, K. L. Tuck and A. Yang, Bioorg. Med. Chem. Lett., 2009, 19, 2763–2765 CrossRef CAS PubMed.
- K.-H. Choi, Expert Opin. Drug Discovery, 2013, 8, 919–931 CrossRef CAS PubMed.
- G. W. Zamponi, Nat. Rev. Drug Discovery, 2016, 15, 19–34 CrossRef CAS PubMed.
- S. Cai, K. Gomez, A. Moutal and R. Khanna, Transl. Res., 2021, 234, 20–30 CrossRef CAS PubMed.
- Y. Nam, K. D. Ryu, C. Jang, Y. H. Moon, M. Kim, D. Ko, K.-S. Chung, M. A. Gandini, K.-T. Lee, G. W. Zamponi and J. Y. Lee, Bioorg. Med. Chem., 2020, 28, 115491 CrossRef CAS PubMed.
- I. J. Amaye, P. L. Jackson-Ayotunde and M. Martin-Caraballo, Bioorg. Med. Chem., 2022, 65, 116766 CrossRef CAS PubMed.
- A. Garcia-Caballero, V. M. Gadotti, M. Y. Ali, C. Bladen, E. Gambeta, J. F. Van Humbeck, J. L. MacCallum and G. W. Zamponi, ACS Chem. Neurosci., 2022, 13, 524–536 CrossRef CAS PubMed.
- P. Beswick, 7.03 – Progress in the Discovery of Ca Channel Blockers for the Treatment of Pain A2 – Chackalamannil, Samuel, in Comprehensive Medicinal Chemistry III, ed. D. Rotella and S. E. Ward, Elsevier, Oxford, 2017, pp. 65–130 Search PubMed.
- M. F. Jarvis, V. E. Scott, S. McGaraughty, K. L. Chu, J. Xu, W. Niforatos, I. Milicic, S. Joshi, Q. Zhang and Z. Xia, Biochem. Pharmacol., 2014, 89, 536–544 CrossRef CAS PubMed.
- D. Ziegler, W. R. Duan, G. An, J. W. Thomas and W. Nothaft, Pain, 2015, 156, 2013–2020 CrossRef CAS PubMed.
- Q. Zhang, Z. Xia, S. Joshi, V. E. Scott and M. F. Jarvis, ACS Med. Chem. Lett., 2015, 6, 641–644 CrossRef CAS PubMed.
- L. Di, E. H. Kerns, Y. Hong and H. Chen, Int. J. Pharm., 2005, 297, 110–119 CrossRef CAS PubMed.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2010, 1, 435–449 CrossRef CAS PubMed.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2016, 7, 767–775 CrossRef CAS PubMed.
- M. Nozaki, S. Haraguchi, T. Miyazaki, D. Shigeta, N. Kano, X.-F. Lei, J.-R. Kim-Kaneyama, H. Minakata, A. Miyazaki and K. Tsutsui, Sci. Rep., 2018, 8, 2167 CrossRef PubMed.
- K. J. Burke, L. J. Stephens, M. V. Werrett and P. C. Andrews, Chem. – Eur. J., 2020, 26, 7657–7671 CrossRef CAS PubMed.
- R. N. Duffin, V. L. Blair, L. Kedzierski and P. C. Andrews, Eur. J. Med. Chem., 2020, 186, 111895 CrossRef CAS PubMed.
- K. Präbst, H. Engelhardt, S. Ringgeler and H. Hübner, in Cell Viability Assays: Methods and Protocols, ed. D. F. Gilbert and O. Friedrich, Springer New York, New York, 2017, pp. 1–17 Search PubMed.
- J. Á. Rodríguez-Corrales and J. S. Josan, in Proteomics for Drug Discovery: Methods and Protocols, ed. I. M. Lazar, M. Kontoyianni and A. C. Lazar, Springer New York, New York, 2017, pp. 207–219 Search PubMed.
- E. S. Lowe and J. J. L. Lertora, in Principles of Clinical Pharmacology (Third Edition), ed. A. J. Atkinson, S.-M. Huang, J. J. L. Lertora and S. P. Markey, Academic Press, 2012, pp. 343–356 Search PubMed.
- L. McCallum, S. Lip and S. Padmanabhan, in Handbook of Pharmacogenomics and Stratified Medicine, ed. S. Padmanabhan, Academic Press, San Diego, 2014, pp. 365–383 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00714f |
| ‡ Current address: Pharmacy Discipline, Life Science School, Khulna University, Khulna, 9208, Bangladesh. |
| This journal is © The Royal Society of Chemistry 2024 |