
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective synthesis and antiproliferative activity of allo-gibberic acid-based 1,3-aminoalcohol regioisomers†
Zein Alabdeen
Khdar
a,
Tam Minh
Le
ab,
Zsuzsanna
Schelz
 c,
István
Zupkó
c and
Zsolt
Szakonyi
*a
c,
István
Zupkó
c and
Zsolt
Szakonyi
*a
aInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös utca 6, H-6720 Szeged, Hungary. E-mail: szakonyi.zsolt@szte.hu; Fax: +36 62 545705; Tel: +36 62 546809
bHUN-REN–SZTE Stereochemistry Research Group, University of Szeged, Eötvös u. 6, H-6720 Szeged, Hungary
cInstitute of Pharmacodynamics and Biopharmacy, University of Szeged, H-6720 Szeged, Hungary
First published on 12th February 2024
Abstract
A new library of allo-gibberic acid-based aminoalcohol regioisomers was synthesised stereoselectively starting from commercially available gibberellic acid, which yields allo-gibberic acid under mild acidic conditions. The successful formation of hydroxymethyl ketone derivative 5, by acid-mediated rearrangement of previously prepared epoxide, paved the way to obtain the desired 1,3-aminoalcohols through Schiff base formation. To obtain the desired regioisomers, the primary alcohol functionality of 5 was subjected to mesylation, then replaced with either primary amine or sodium azide. The formed azide derivative was subjected to either CuAAC reaction to obtain 1,2,3-triazoles or underwent Pd-catalysed hydrogenolysis to obtain primary aminoalcohol, which was further transformed into 1,3-aminoalcohols by reductive alkylation. All prepared aminoalcohols were identified in a satisfactory manner using modern spectroscopic techniques and assessed for their antiproliferative activity against a panel of human cancer cell lines. The antiproliferative effects of the prepared compounds were assayed by in vitro MTT method against a panel of human cancer cell lines (HeLa, SiHa, A2780, MCF-7 and MDA-MB-231). A significant difference was observed in the antiproliferative activity between the regioisomers. Some compounds exerted outstanding activities against the malignant cells with limited action on fibroblasts, indicating considerable cancer selectivity.
1. Introduction
Based on the variety of research that has been carried out in drug discovery and medicinal chemistry, it is a well-known reality that nature, including plants, animals, marine organisms and microorganisms, remains the richest, most varied and sustainable source of structurally diverse compounds with critical biological activity.1,2 The ent-kaurane tetracyclic diterpenes represent a large group of natural products widely found in different plant families and endowed with remarkable biological activities.3 Structurally, the majority of ent-kauranes discovered so far have a tetracyclic core structure formed by combining a perhydrophenanthrene skeleton with a cyclopentane ring.4,5 Almost 600 of the known ent-kaurane diterpenes have been identified and characterised within the Isodon (Lamiaceae) plants, which have been employed to treat cancer and other inflammatory diseases for centuries.6 The antiproliferative activity of different ent-kaurane diterpenoids has received considerable attention and has been deeply studied. Fujita et al. determined the structure–activity relationship of enmein and oridonin, which suggests the role of α,β-unsaturated ketone as a Michael receptor in easing the addition of nucleophiles such as alkanethiols resulting in inactivation of SH enzymes.7 However, various pathways can be included in the anticancer activity of ent-kaurane diterpenes, such as telomerase activity inhibition (eriocalyxin B),6,8 p53 activation (oridonin)9,10 and AKT kinase inhibition (excisanin A).11 Gibberellins (GAs), a class of ent-kaurane diterpenes, are phytohormones responsible for seed germination, plant growth and development. Gibberellic acid (GA3) 1, a well-known member of the GA family, attracted the interest of many researchers as a scaffold for antitumor studies because of its diverse functionalities, which are accessible for chemical modifications as well as its commercial availability at tonne quantities by fermentation of Gibberella fujikuroi.12,13 Subsequently, a variety of derivatives with different functionalities has been reported and showed promising antiproliferative activity with different possible mechanisms. Chen et al. recorded the complete inhibition of topoisomerase-I activity by gibberellic acid methyl ester bearing two α,β-unsaturated ketone moieties.14 Wu et al. reported that S phase arrest in the cell cycle and apoptosis could be induced by allo-gibberic-based 1,2,3-triazoles with an α,β-unsaturated ketone moiety.15 Zhu et al. prepared a series of allo-gibberic acid benzyl esters endowed with strong antitumor activity. This activity could be linked to inhibiting FGFR1 and KDR activation.16 Moreover, Goel et al. demonstrated that the expression of PARP-1 and caspase 3 can be inhibited by ipomone, a novel gibberic acid-derived diterpene.17On the other hand, 1,3-aminoalcohols and their N-substituted analogues have been studied extensively for their application as chiral auxiliaries for different reactions, such as ring opening, aldol condensation and catalytic reduction18 as well as for their diverse biological activities. A library of isosteviol-based N-substituted 1,3-aminoalcohols was recently prepared and showed remarkable antiproliferative activity with a preference for benzyl substituents.19 Moreover, 1,3-aminoalcohols have proven to be promising building blocks for new biologically active heterocyclic derivatives such as oxazines, thiazines and pyrimidines. 2,4-Diaminopyrimidine derivatives with potent antiproliferative activity were prepared starting from pinane- and (−)-isopulegol-based 1,3-aminoalcohols.20,21
In our previous work, we designed and evaluated the antiproliferative activity of a series of N-substituted allo-gibberic acid-based aminodiols. We found that N-naphthylethyl-substituted aminodiol derivatives showed the most promising antiproliferative activity. In particular, compound A presented in Fig. 1 exhibited a modest cancer selectivity (IC50 = 4.38–7.49 μM) over NIH/3 T3 fibroblast cells (IC50 = 10.88 μM).22 Furthermore, a group of allo-gibberic acid-based aminoalcohols B shown in Fig. 1 has been recently considered for their antimicrobial activity against a panel of multidrug-resistant Gram-negative bacteria.23 Combining these results with our continuous effort to obtain promising, more potent and selective anticancer agents has led us to the field of diterpene-based aminoalcohols. Herein, we report the synthesis of new gibberellic acid-based 1,3-aminoalcohol derivatives and their in vitro antiproliferative evaluation against different human cancer cell lines.
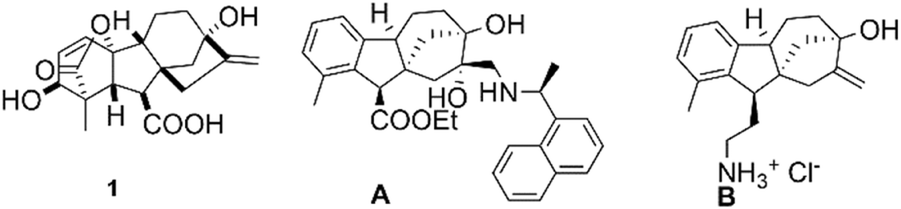 | ||
| Fig. 1 The structure of gibberellic acid 1 and its bioactive derivatives. | ||
2. Results and discussion
2.1. Synthetic procedure
2.1.1.1. Synthesis of key intermediates (hydroxymethyl ketone 5 and primary aminoalcohol 7). The allylic alcohol derivative, allo-gibberic acid 2, was obtained by HCl-mediated hydrolysis of the commercially available gibberellic acid 1 following literature methods.12,16 As mentioned in our previous work, cis-epoxy alcohol 4 was prepared in a stereospecific manner by m-CPBA-mediated epoxidation of allo-gibberic acid ethyl ester 3 (Scheme 1).22
 | ||
| Scheme 1 Synthesis of epoxy alcohol 4. (i) HCl 1.2 M, 65 °C, 3 h, 70%; (ii) C2H5I (2 eq.), TBAF 1 M in THF (2.5 eq.), dry THF, Ar atm., 25 °C, 4 h, 95%; (iii) C2H5I (2.5 eq.), Cs2CO3 (2 eq.), MeCN, reflux, 1 h, 99%; (iv) m-CPBA 75% (2.5 eq.), dry DCM, 25 °C, 2 h, 62%. | ||
The key intermediate hydroxymethyl ketone 5 was obtained by HCl-mediated rearrangement of epoxy alcohol 4. In this process, a six-membered transition state is formed and the carbonium ion is stabilised by Wagner–Meerwein rearrangement with the inversion of ring C/D configuration.24 Moreover, compound 5 could also be prepared more efficiently on a gram scale, with higher yield and shorter reaction, by BF3·Et2O-catalysed rearrangement, which can be explained by the coordination between the Lewis acid and the epoxide oxygen.25
Primary amines have attracted substantial interest as building blocks for the biosynthesis of bioactive molecules and they can be obtained simply by hydrogenation of unsaturated ligands, mainly oximes, containing the C–N bond. However, the heterogeneous catalytic hydrogenation of aldoximes and ketoximes remains the most resourceful selective method to obtain the corresponding amines.26 Similarly, to obtain primary 1,3-aminoalcohol 7, hydroxymethyl ketone 5 was first subjected to an oximation reaction by hydroxylamine hydrochloride in the presence of NaHCO3. The formed oxime 6 underwent further RANEY® Ni-catalysed hydrogenation in THF and led to compound 7 with good yield and in a stereospecific manner as shown by NMR experiments (Scheme 2).
 | ||
| Scheme 2 Synthesis of primary 1,3-aminoalcohol 7. (i) HCl (3 N), 25 °C, 72 h, 80%; (ii) cat. BF3·Et2O, toluene, 25 °C, 12 h, 95%; (iii) H2NOH·HCl (2 eq.), NaHCO3 (1 eq.), EtOH, reflux, 12 h, 93%; (iv) RANEY® Ni, H2 (10 atm), THF, 25 °C, 24 h, 72%. | ||
2.1.1.2. Synthesis of allo-gibberic acid-based 1,3-aminoalcohols via Schiff base formation. A series of N-substituted 1,3-aminoalcohols was prepared by either reductive amination of hydroxymethyl ketone 5 with different primary amines or reductive alkylation of primary aminoalcohol 7 with several aldehydes via the formation of Schiff bases followed by NaBH4-mediated reduction. However, the efficiency of reductive amination of compound 5 was enhanced by using a catalytic amount of BF3·Et2O, as it promotes the nucleophilic attack of the primary amine on the carbonyl carbon.27 A variety of nitrogen substituents were employed to study the steric and electronic effects on the antiproliferative activity of the designed aminoalcohols (Scheme 3 and Table 1).
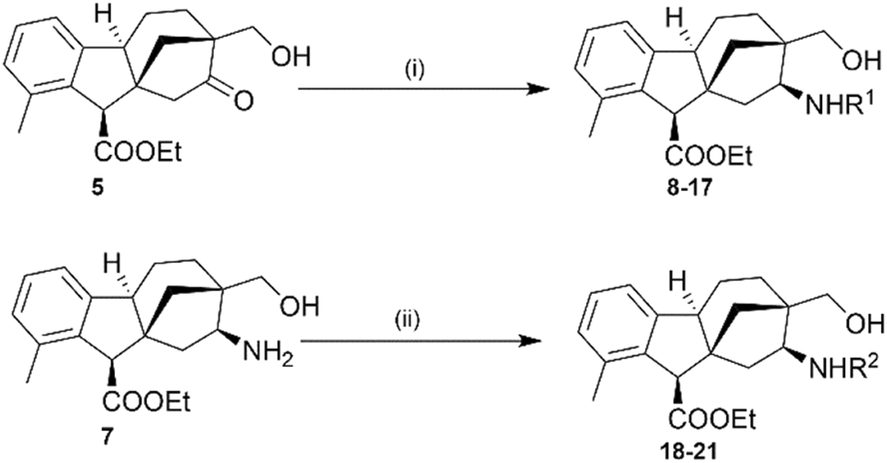 | ||
| Scheme 3 Preparation of secondary 1,3-aminoalcohols 8–21. (i) (1) R1NH2 (2 eq.), BF3·Et2O (5 μL), dry toluene, reflux (Dean–Stark), 24–48 h; (2) NaBH4 (2 eq.), dry EtOH, 25 °C, 3 h, 54–70%; (ii) (1) aldehydes (1.2 eq.), dry EtOH, 25 °C, 3 h; (2) NaBH4 (2 eq.), dry EtOH, 25 °C, 3 h, 46–55%. | ||
| Entry | Compound | R1/R2 | Yield (%) |
|---|---|---|---|
| 1 | 8 | Benzyl | 65 |
| 2 | 9 | 1-Naphthylmethyl | 62 |
| 3 | 10 | (S)-(−)-1-(2-Naphthyl)ethyl | 60 |
| 4 | 11 | (R)-(−)-1-(2-Naphthyl)ethyl | 70 |
| 5 | 12 | (S)-(−)-1-(1-Naphthyl)ethyl | 55 |
| 6 | 13 | (R)-(+)-1-(1-Naphthyl)ethyl | 58 |
| 7 | 14 | 4-Fluorobenzyl | 61 |
| 8 | 15 | 4-Methoxybenzyl | 50 |
| 9 | 16 | 2-(1H-Indol-3-yl)ethyl | 55 |
| 10 | 17 | 3-(1H-Imidazol-1-yl)propyl | 54 |
| 11 | 18 | 5-Bromo-2-hydroxybenzyl | 53 |
| 12 | 19 | Quinolin-2-ylmethyl | 51 |
| 13 | 20 | Quinolin-4-ylmethyl | 55 |
| 14 | 21 | Pyridin-4-ylmethyl | 46 |
2.1.2.1. Synthesis route of the critical intermediates for regioisomer preparation (mesylated alcohol 23 and the primary aminoalcohol 25). Within the framework of our synthetic plan, the next step includes the building of regioisomers of aminoalcohols 8–21. For this purpose and starting from the considerable application of derivatives bearing mesyl, a good leaving group,28 the alcoholic function of compound 5 was quantitatively transformed into the mesyl derivative by the reaction with methanesulfonyl chloride in the presence of dry pyridine. Since sodium borohydride is well known as a highly reactive and chemoselective reducing agent under specific conditions,29 the NaBH4-mediated ketone reduction of compound 22 was accomplished effectively and quantitatively in a mixture of DCM and MeOH (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). As the stereoselectivity of the reduction of the prochiral ketone can be influenced by the reaction temperature, product 23 was obtained as a single product under ice cooling. Next, the azidation of compound 23 with sodium azide was fully and preferably performed in dry DMF at 90 °C. Then, the favoured primary 1,3-aminoalcohol 25 was efficiently prepared by hydrogenation of 1,3-azido alcohol 24 catalysed by Pd/C and H2 at 10 bar30 (Scheme 4).
:1). As the stereoselectivity of the reduction of the prochiral ketone can be influenced by the reaction temperature, product 23 was obtained as a single product under ice cooling. Next, the azidation of compound 23 with sodium azide was fully and preferably performed in dry DMF at 90 °C. Then, the favoured primary 1,3-aminoalcohol 25 was efficiently prepared by hydrogenation of 1,3-azido alcohol 24 catalysed by Pd/C and H2 at 10 bar30 (Scheme 4).
 | ||
| Scheme 4 Preparation of primary 1,3-aminoalcohol 25. (i) MsCl (6 eq.), dry pyridine, 25 °C, 12 h, 76%; (ii) NaBH4 (2 eq.), DCM, MeOH (1:1), 0 °C, 3 h, 76%; (iii) NaN3 (2 eq.), dry DMF, reflux, 12 h, 90%; (iv) Pd/C (10%), H2 (10 atm), MeOH, 25 °C, 12 h, 75%. | ||
2.1.2.2. Preparation of 1,3-aminoalcohols via either MsO/primary amine exchange or Schiff base formation. As mesylates are well-known to be excellent leaving groups in nucleophilic substitution reactions, the exchange of the MsO function to form primary amino derivatives was successfully accomplished in the presence of triethylamine, yielding N-substituted 1,3-aminoalcohols 26–29 with acceptable yields.31 Another series of 1,3-aminoalcohols 30–37 was prepared in a similar manner by reductive alkylation of primary aminoalcohol 25 (Scheme 5 and Table 2).
 | ||
| Scheme 5 (i) R1NH2 (4 eq.), Et3N, MeCN (1:1), reflux, 72 h, 41–50%; (ii) (1) aldehydes (1.2 eq.), dry EtOH, 25 °C, 3 h; (2) NaBH4 (2 eq.), dry EtOH, 25 °C, 3 h, 43–66%; (iii) acetylenes (1.5 eq.), Cu(OAc)2·H2O (2 mol%), sodium ascorbate (10 mol%), t-BuOH, H2O (2:1), 25 °C, 12 h, 90%. | ||
| Entry | Compound | R1/R2 | Yield (%) |
|---|---|---|---|
| 1 | 26 | (S)-(−)-1-(2-Naphthyl)ethyl | 50 |
| 2 | 27 | (R)-(−)-1-(2-Naphthyl)ethyl | 50 |
| 3 | 28 | (S)-(−)-1-(1-Naphthyl)ethyl | 46 |
| 4 | 29 | (R)-(+)-1-(1-Naphthyl)ethyl | 41 |
| 5 | 30 | Benzyl | 66 |
| 6 | 31 | 1-Naphthylmethyl | 50 |
| 7 | 32 | 4-Fluorobenzyl | 55 |
| 8 | 33 | 4-Methoxybenzyl | 50 |
| 9 | 34 | 5-Bromo-2-hydroxybenzyl | 53 |
| 10 | 35 | Quinoline-2-ylmethyl | 43 |
| 11 | 36 | Quinoline-4-ylmethyl | 55 |
| 12 | 37 | Pyridine-4-ylmethyl | 50 |
:H2O (2:1)] to afford 1,4-disubstituted 1,2,3-triazoles 38 and 39 in a regioselective manner (Scheme 4 and Table 3).
| Entry | Compound | R3 | Yield (%) |
|---|---|---|---|
| 1 | 38 | Phenyl | 90 |
| 2 | 39 | Benzyl | 90 |
2.2. Determination of relative configuration of synthesised 1,3-aminoalcohols
The relative configuration of the new stereocenter of the synthesised 1,3-aminoalcohols at position 16 was determined by means of NOESY experiments.For aminoalcohols 8–21, three clear NOE signals were observed between H-15 and H-16, H-15 and H-7, and H-15 and H-19. Therefore, the structure of N-substituted 1,3-aminoalcohols was determined as shown in Fig. 2. Besides the NOESY experiments, the relative configuration of primary aminoalcohol 7 was transformed by reductive alkylation with benzaldehyde yielding a compound that was identical to compound 8 obtained by reductive amination of 5.
 | ||
| Fig. 2 The relative configuration of compound 10 (1,3-aminoalcohols 8–21). | ||
In addition, the relative configuration of 26–39 was defined by NOE spectral analysis of compound 23. Based on the observed signals between H-15 and H-16 and between H-15 and H-10, the structure of 23 was determined, as shown in Fig. 3.
 | ||
| Fig. 3 The relative configuration of compound 23. | ||
3. Antiproliferative activity
The in vitro antiproliferative activities of the synthesised 1,3-aminoalcohols 8–21 and 26–37 and hydroxy triazoles 38 and 39 against a panel of different human cancer cell lines, including cervical (SiHA and HeLa), breast (MCF7 and MDA-MB-231) and ovary (A2780) cancers as well as non-cancerous fibroblast cells were assayed by the MTT method.34 Cisplatin, which is one of the most effective anticancer agents, was used as the reference drug and the results are summarised in Fig. 4 (Table 4). | ||
| Fig. 4 Antiproliferative properties of the selected allo-gibberic acid-based aminoalcohols against cancer cells and NIH/3T3 fibroblasts. *Data from ref. 35. | ||
| Compound | Calculated IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| HeLa | SiHa | MCF-7 | MDA-MB-231 | A2780 | NIH/3T3 | |
| 12 | 20.70 | 22.34 | 3.43 | 10.38 | 6.17 | 6.02 |
| 13 | 18.55 | 22.73 | 7.07 | 19.29 | 4.57 | 14.47 |
| 16 | 4.01 | 4.12 | 3.14 | 4.71 | 4.57 | 12.95 |
| 18 | 5.06 | 21.87 | 3.85 | 6.48 | 5.08 | 19.71 |
| 26 | 5.10 | 4.36 | 3.66 | 4.47 | 4.57 | 4.62 |
| 32 | 18.78 | 8.40 | 4.40 | 7.71 | 15.03 | 15.17 |
| 33 | 12.91 | 17.67 | 4.08 | 6.50 | 13.83 | 14.74 |
| 34 | 4.71 | 20.55 | 2.42 | 4.99 | 5.39 | 13.02 |
| Cisplatin | 12.14 | 4.29 | 8.34 | 25.56 | 5.27 | 5.50 |
4. Structure–activity relationship (SAR)
The in vitro antiproliferative activities of the prepared N-substituted 1,3-aminoalcohols against a group of cancer cell lines, involving cervical (SiHA and HeLa), breast (MCF-7 and MDA-MB 231) and ovary (A2780) cancers were evaluated by the MTT method, and the obtained results are summarised as the following:Both N-benzyl-substituted aminoalcohol 8 and its corresponding regioisomer 30 showed modest suppression against all tested cancer cell lines. As the aromatic ring had the tendency to form hydrophobic and/or π interactions with the potential target, we further explored the activity by introducing either electron-withdrawing groups (F, Br) or electron-donating groups (OH, OCH3). The results indicated that compounds 14 and 15 with fluoro and methoxy substituents at the para position, respectively, displayed a cell line-dependent influence, while their related regioisomers 32 and 33 elicited substantial enhancement in inhibitory activity with high selectivity towards MCF-7 and MDA-MB-231 as well as low cytotoxicity against NIH fibroblast cells. However, the introduction of both o-OH and m-Br (para to each other) in compounds 18 and 34 led to a further rise in activity but with mixed effects on cancer selectivity. Based on their calculated IC50 values, the latter molecules (18, 32–34) are more potent than or comparable with the reference agent cisplatin (Fig. 5).
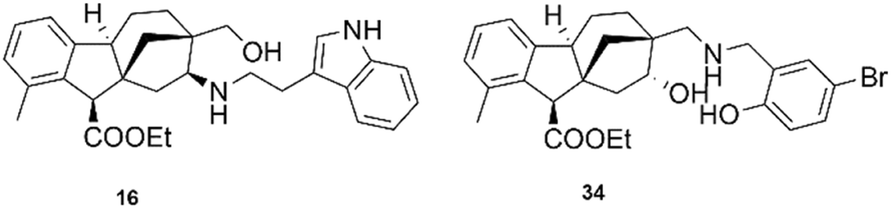 | ||
| Fig. 5 The structure of the most promising allo-gibberic acid derivatives. | ||
For N-1-naphthylmethyl-substituted aminoalcohol 9, the inhibition values were substantial only at higher concentration (30 μM), whereas its related regioisomer 31 showed more pronounced growth in activity with marked selectivity towards SiHa, MCF-7 and MDA-MDB 231. For both regioisomers, the introduction of a methyl group at the α position (compounds 12, 13, 28 and 29) resulted in highly active compounds. In particular, the R-diastereoisomers 13 and 29 were endowed with marked selectivity indicated by limited cytotoxicity against NIH/3T3. On the other hand, the inhibition activity of R- and S-α-methyl-2-naphthylmethyl derivatives 10 and 11 has decreased, whilst their regioisomers 26 and 27 were marked with both potent inhibition of cell growth and high cytotoxicity on non-cancerous cells.
Among all prepared aminoalcohols with heterocyclic substituents (quinoline, pyridine and 1,2,3-trizole), compound 35 with a quinolin-2-ylmethyl substituent showed potent inhibition activity against breast cancers (MCF-7 and MDA-MB-231) with much less cytotoxicity against fibroblasts, while compound 16 with the N-2-(1H-indol-3-yl)ethyl substituent exhibited lower than 5 μM IC50 values against all utilized cancerous cell lines but substantially higher value (12.95 μM) against fibroblasts, indicating a cancer-selective antiproliferative property of the molecule. Based on these findings, 16 may be regarded as a lead compound utilizable for the design and synthesis of further anticancerous gibberellic acid analogues.
5. Experimental
Commercially available compounds were used as obtained from suppliers (Molar Chemicals Ltd., Halásztelek, Hungary; Merck Ltd., Budapest, Hungary, and VWR International Ltd., Debrecen, Hungary), while solvents were dried according to standard procedures. Optical rotations were measured in MeOH at 20 °C using a PerkinElmer 341 polarimeter (PerkinElmer Inc., Shelton, CT, USA). Chromatographic separations and monitoring of reactions were carried out on a Merck Kieselgel 60 instrument (Merck Ltd., Budapest, Hungary). HR-MS flow injection analysis was performed using a Thermo Scientific Q Exactive Plus hybrid quadrupole-Orbitrap (Thermo Fisher Scientific, Waltham, MA, USA) mass spectrometer coupled to a Waters Acquity I-Class UPLC™ system (Waters, Manchester, UK). Melting points were determined on a Kofler apparatus (Nagema, Dresden, Germany) and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker Avance DRX 500 spectrometer (Bruker Biospin, Karlsruhe, Baden Württemberg, Germany) [500 MHz (1H) and 125 MHz (13C), δ = 0 (TMS)]. Chemical shifts are expressed in ppm (δ) relative to TMS as the internal reference. J values are given in Hz.Gibberellic acid 1 is commercially available from Merck with ee% = 90% ([α]20D = +78.0, c 2, MeOH). Allo-gibberic acid 2 was prepared from gibberellic acid 1 according to a literature method.12 Compounds 3 and 4 were prepared as mentioned in our previous work.22 All spectroscopic data were similar to those described therein.
Preparation of hydroxymethyl ketone derivative 5
Method A. Epoxide 4 (2.00 g, 6.09 mmol) was suspended in 50 mL HCl (3 N) solution. The mixture was stirred for 72 hours at room temperature and then the solvent was evaporated. The residue was diluted with water (30 mL) and extracted with EtOAc (3 × 50 mL). After that, the organic phase was washed with brine solution (3 × 50 mL), dried over Na2SO4, filtered and then evaporated. The crude product was purified by column chromatography using n-hexane
:EtOAc (1:1) as eluent.
Method B. To the solution of epoxide 4 (2.00 g, 6.09 mmol) in toluene (50 mL), boron trifluoride diethyl etherate (0.50 mL) was added and the mixture was stirred at room temperature for 12 hours. After that, the organic phase was washed with water (50 mL) and brine solution (3 × 50 mL), dried over Na2SO4, filtered and then evaporated. The crude product was purified by column chromatography using n-hexane
:EtOAc (1:1) as eluent.
Yield: (method A: 80%, method B: 95%); white crystals; m.p.: 157–160 °C; [α]20D = +4.4 (c 0.14, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.20 (t, J = 7.5 Hz, 1H), 7.06 (d, J = 7.5 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 4.27 (m, 2H), 4.17 (s, 1H), 3.70 (dd, J = 6.6, 11.2 Hz, 1H), 3.49 (dd, J = 5.0, 11.3 Hz, 1H), 3.05 (t, J = 8.0 Hz, 1H), 2.71 (d, J = 18.0 Hz, 1H), 2.51 (dd, J = 3.2, 18.0 Hz, 1H), 2.19 (s, 3H), 2.17–2.10 (m, 1H), 2.08–2.03 (m, 1H), 1.93 (dd, J = 3.2, 12.1 Hz, 1H), 1.90–1.65 (m, 3H), 1.58 (s, 1H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 222.0, 171.6, 145.8, 137.5, 136.1, 129.0, 128.2, 120.4, 65.3, 60.8, 56.0, 53.7, 51.9, 51.6, 49.4, 33.2, 29.0, 22.7, 19.4, 14.4; HR-MS (ESI): m/z calcd for C20H25O4 [M + H]+: 329.1675; found: 329.1744.
Preparation of primary 1,3-aminoalcohol 7
:EtOAc (1:3) as eluent.
Ethyl (4bS,7R,9aS,10R,E)-8-(hydroxyimino)-7-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 6. Yield: 93%; colourless oily compound; [α]20D = +66.8 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.48 (s, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.03 (d, J = 7.5 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 4.30–4.18 (m, 2H), 4.10 (s, 1H), 3.65–3.58 (m, 2H), 3.01–2.98 (m, 1H), 2.87–2.79 (m, 2H), 2.17 (s, 3H), 2.12–2.03 (m, 1H), 1.95–1.73 (m, 3H), 1.65–1.61 (m, 1H), 1.30 (t, J = 7.2 Hz, 3H), 1.25 (d, J = 12.3 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 171.7, 169.6, 146.0, 138.0, 135.0, 128.4, 127.9, 120.1, 67.9, 60.8, 55.8, 51.9, 51.6, 48.5, 40.1, 35.9, 31.9, 21.8, 19.4, 14.4; HR-MS (ESI): m/z calcd for C20H26NO4 [M + H]+: 344.1784; found: 344.1857.
:Et2O (1:1).
Ethyl (4bS,7R,8S,9aS,10R)-8-amino-7-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 7. Yield: 72%; white crystals; m.p.: 129–132 °C; [α]20D = +14.2 (c 0.14, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 8.03 (s, 2H), 7.17–7.13 (m, 1H), 6.99 (dd, J = 4.0, 7.2 Hz, 2H), 4.18–4.13 (m, 2H), 4.10 (s, 1H), 3.49–3.43 (m, 1H), 3.39 (d, J = 11.1 Hz, 1H), 3.28 (d, J = 11.0 Hz, 1H), 3.14–3.08 (m, 1H), 2.5–2.45 (m, 1H), 2.15–2.07 (m, 1H), 2.06 (s, 3H),1.88–1.82 (m, 1H), 1.69–1.60 (m, 1H), 1.44–1.30 (m, 3H), 1.22 (t, J = 7.0 Hz, 3H), 0.98 (d, J = 12.1 Hz, 1H); 13C NMR (125 MHz, DMSO-d6): δ = 172.0, 147.4, 138.3, 134.5, 128.5, 128.2, 121.0, 65.5, 60.5, 56.6, 53.4, 52.0, 52.2, 46.5, 42.7, 34.5, 23.6, 22.3, 19.3, 14.7; HR-MS (ESI): m/z calcd for C20H28NO3 [M + H]+: 330.2063; found: 330.2064.
Preparation of primary 1,3-aminoalcohol 25
:EtOAc (1:1) as eluent and then it was crystallised in Et2O to afford the product as a white solid (1.4 g, 76%).
Ethyl (4bS,7S,9aS,10R)-1-methyl-7-(((methylsulfonyl)oxy)methyl)-8-oxo-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 22. Yield: 76%; white crystals; m.p.: 183–185 °C; [α]20D = +1.7 (c 0.14, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.21 (t, J = 7.6 Hz, 1H), 7.07 (d, J = 7.6 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 4.31–4.26 (m, 3H), 4.18 (s, 1H), 4.05 (d, J = 9.9 Hz, 1H), 3.08–3.03 (m, 1H), 2.97 (s, 3H), 2.72 (d, J = 18.0 Hz, 1H), 2.53 (dd, J = 3.4, 17.9 Hz, 1H), 2.19 (s, 3H), 2.17–2.12 (m, 1H), 2.07 (dd, J = 3.4, 12.7 Hz, 1H), 1.92–1.83 (m, 1H), 1.71–1.65 (m, 3H), 1.32 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ = 217.3, 171.4, 145.3, 137.4, 135.2, 129.1, 128.3, 120.4, 70.3, 61.0, 55.9, 51.7, 51.5, 51.1, 49.1, 37.0, 33.1, 28.7, 22.4, 19.4, 14.4; HR-MS (ESI): m/z calcd for C21H26NaO6S [M + Na]+: 429.1348; found: 429.1336.
:1) (200 mL) and NaBH4 (0.37 g, 9.84 mmol) was added to the solution in small portions under ice cooling. Then, the mixture was stirred at 0 °C for 3 hours. After completion of the reaction (as monitored by TLC), the mixture was evaporated to dryness. The residue was diluted with water (50 mL) and then extracted with DCM (3 × 50 mL). The organic phase was washed with brine solution (3 × 50 mL), dried over Na2SO4, filtered and then evaporated. The crude product was purified by column chromatography on silica gel using n-hexane:EtOAc (1:1) as eluent.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-(((methylsulfonyl)oxy)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 23. Yield: 76%; colourless oily compound; [α]20D = −8.4 (c 0.15, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 7.13 (t, J = 7.5 Hz, 1H), 6.97 (d, J = 7.5 Hz, 2H), 4.93 (d, J = 3.8 Hz, 1H), 4.21–4.11 (m, 2H), 4.07–3.97 (m, 3H), 3.93–3.87 (m, 1H), 3.12 (s, 3H), 2.95 (t, J = 8.5 Hz, 1H), 2.48–2.44 (m, 1H), 2.20–2.11 (m, 1H), 2.05 (s, 3H), 2.04–1.98 (m, 1H), 1.64–1.58 (m, 1H), 1.45–1.37 (m, 2H), 1.29–1.24 (m, 1H), 1.22 (t, J = 7.1 Hz, 3H), 0.86 (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, DMSO-d6): δ = 172.0, 147.4, 138.5, 134.5, 128.5, 128.0, 120.8, 75.2, 71.6, 60.4, 56.5, 53.8, 52.2, 46.4, 45.9, 36.8, 34.0, 22.33, 22.31, 19.4, 14.7; HR-MS (ESI): m/z calcd for C21H29O6S [M + H]+: 408.1607; found: 408.1612.
:EtOAc (2:1) as eluent.
Ethyl (4bS,7S,8R,9aS,10R)-7-(azidomethyl)-8-hydroxy-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 24. Yield: 90%; colourless oily compound; [α]20D = −18.7 (c 0.15, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.16 (t, J = 7.5 Hz, 1H), 7.00 (d, J = 7.4 Hz, 1H), 6.97 (d, J = 7.4 Hz, 1H), 4.23 (q, J = 7.1 Hz, 2H), 4.06 (m, 1H), 4.00 (s, 1H), 3.37 (d, J = 11.7 Hz, 1H), 3.28 (d, J = 11.7 Hz, 1H), 3.03 (t, J = 8.2 Hz, 1H), 2.67 (dd, J = 10.5, 14.0 Hz, 1H), 2.25–2.15 (m, 2H), 2.14 (s, 3H), 1.92 (d, J = 3.6 Hz, 1H), 1.70–1.61 (m, 2H), 1.55–1.51 (m, 1H), 1.41–1.35 (m, 1H), 1.29 (t, J = 7.1 Hz, 3H), 0.85 (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 146.7, 137.9, 134.7, 128.5, 127.8, 120.3, 75.7, 61.0, 60.5, 56.7, 53.9, 52.6, 46.1, 45.6, 35.1, 24.4, 21.8, 19.4, 14.4; HR-MS (ESI): m/z calcd for C20H26N3O3 [M + H]+: 355.1896; found: 355.1904.
:MeOH (4:1) as eluent.
Ethyl (4bS,7S,8R,9aS,10R)-7-(aminomethyl)-8-hydroxy-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 25. Yield: 75%; colourless oily compound; [α]20D = −15.3 (c 0.15, MeOH); 1H NMR (500 MHz, CD3OD): δ = 7.14 (t, J = 7.5 Hz, 1H), 6.98 (d, J = 7.5 Hz, 2H), 4.25 (q, J = 6.9 Hz, 2H), 4.02 (s, 1H), 3.93 (dd, J = 4.8, 10.5 Hz, 1H), 3.01 (t, J = 8.6 Hz, 1H), 2.70–2.58 (m, 3H), 2.28–2.21 (m,1H), 2.13 (s, 4H), 1.72–1.65 (m, 1H), 1.63–1.50 (m, 2H), 1.49–1.41 (m, 1H), 1.33 (t, J = 7.1 Hz, 3H), 0.76 ppm (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ = 173.0, 147.1, 137.8, 134.2, 127.9, 127.5, 120.1, 75.0, 60.4, 56.8, 54.2, 52.2, 50.0, 46.5, 46.2, 34.2, 22.9, 22.2, 18.2, 13.3; HR-MS (ESI): m/z calcd for C20H28NO3 [M + H]+: 330.2063; found: 330.2058.
Preparation of 1,3-aminoalcohol regioisomers
:EtOAc (1:1 then 1:3) as eluent.
Ethyl (4bS,7R,8S,9aS,10R)-8-(benzylamino)-7-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 8. Prepared with benzylamine, yield: 65%; colorless oily compound; [α]20D = −1.2 (c 0.14, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.36–7.31 (m, 4H), 7.29–7.26 (m, 1H), 7.15 (t, J = 7.1 Hz, 1H), 6.99 (d, J = 7.2 Hz, 1H), 6.95 (d, J = 7.3 Hz, 1H), 4.25–4.19 (m, 2H), 4.01 (s, 1H), 3.89 (d, J = 12.8 Hz, 1H), 3.78 (d, J = 12.8 Hz, 1H), 3.55 (s, 2H), 3.05 (dd, J = 6.0, 10.5 Hz, 1H), 2.96 (t, J = 8.3 Hz, 1H), 2.75–2.69 (m, 1H), 2.15 (s, 3H), 2.13–2.10 (m, 1H), 1.98–1.89 (m, 1H), 1.74–1.64 (m, 2H), 1.59–1.54 (m, 1H), 1.41 (dd, J = 1.9, 11.7 Hz, 1H), 1.30–1.24 (m, 4H), 0.74 (d, J = 11.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 146.7, 138.8, 138.1, 134.7, 128.51, 128.50, 128.2, 127.85, 127.83, 127.22, 127.21, 120.3, 73.8, 64.5, 60.4, 57.1, 54.9, 53.5, 52.8, 45.5, 44.9, 34.9, 24.9, 21.9, 19.4, 14.4; HR-MS (ESI): m/z calcd for C27H34NO3 [M + H]+: 420.2533; found: 420.2528.
Ethyl (4bS,7R,8S,9aS,10R)-7-(hydroxymethyl)-1-methyl-8-((naphthalen-1-ylmethyl)amino)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 9. Prepared with 1-naphthylmethylamine, yield: 62%; white powder; m.p.: 78–81 °C; [α]20D = −46.2 (c 0.14, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.12 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 7.9 Hz, 1H), 7.57–7.41 (m, 4H), 7.14 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.6 Hz, 2H), 4.33 (d, J = 12.7 Hz, 1H), 4.25–4.07 (m, 3H), 3.63 (s, 1H), 3.53–3.43 (m, 3H), 3.30 (dd, J = 5.7, 10.3 Hz, 1H), 2.56 (dd, J = 10.4, 13.6 Hz, 1H), 2.24 (3H, s), 2.10–2.01 (1H, m), 2.00–1.93 (1H, m), 1.89–1.81 (1H, m), 1.63 (1H, m), 1.55–1.48 (1H, m), 1.31 (dd, J = 2.1, 11.5 Hz, 1H), 1.24 (t, J = 7.1 Hz, 3H), 1.05 (d, J = 11.3 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.6, 147.4, 138.7, 136.5, 134.4, 133.8, 131.8, 128.7, 128.1, 127.8, 127.6, 126.4, 126.3, 125.8, 125.3, 123.6, 120.4, 73.3, 65.1, 60.4, 58.0, 54.1, 52.0, 50.7, 45.5, 41.2, 40.3, 25.9, 21.0, 18.7, 14.4; HR-MS (ESI): m/z calcd for C31H36NO3 [M + H]+: 470.2689; found: 470.2688.
Ethyl (4bS,7R,8S,9aS,10R)-7-(hydroxymethyl)-1-methyl-8-(((S)-1-(naphthalen-2-yl)ethyl)amino)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 10. Prepared with (S)-(−)-1-(2-naphthyl)ethylamine, yield: 60%; white wax; m.p.: 57–61 °C; [α]20D = −43.6 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.85 (dd, J = 8.8, 14.0 Hz, 3H), 7.73 (s, 1H), 7.53–7.43 (m, 3H), 7.15 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 8.5 Hz, 2H), 4.07–3.98 (m, 4H), 3.43 (d, J = 10.1 Hz, 1H), 3.30 (d, J = 10.2 Hz, 1H), 3.00 (t, J = 8.2 Hz, 1H), 2.74–2.62 (m, 2H), 2.20–2.13 (m, 1H), 2.12 (s, 3H), 1.97–1.90 (m, 1H), 1.79–1.59 (m, 3H), 1.44 (d, J = 7.0 Hz, 3H), 1.32–1.27 (m, 1H), 1.04 (t, J = 7.2 Hz, 3H), 0.52 (d, J = 11.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 171.9, 146.7, 141.9, 138.1, 134.7, 133.4, 133.0, 128.6, 128.5, 127.75, 127.74, 127.73, 126.0, 125.9, 125.6, 124.4, 120.2, 73.3, 62.0, 60.3, 57.1, 56.8, 55.1, 53.4, 45.2, 44.6, 34.6, 25.1, 24.8, 21.9, 19.4, 14.2; HR-MS (ESI): m/z calcd for C32H38NO3 [M + H]+: 484.2773; found: 484.2856.
Ethyl (4bS,7R,8S,9aS,10R)-7-(hydroxymethyl)-1-methyl-8-(((R)-1-(naphthalen-2-yl)ethyl)amino)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 11. Prepared with (R)-(−)-1-(2-naphthyl)ethylamine, yield: 70%; oily compound; [α]20D = −63.1 (c 0.14, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.82 (d, J = 8.2 Hz, 3H), 7.73 (s, 1H), 7.50–7.44 (m, 3H), 7.13 (t, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 1H), 6.92 (d, J = 7.5 Hz, 1H), 4.24–4.15 (m, 2H), 4.01 (q, J = 6.5 Hz, 1H), 3.89 (s, 1H), 3.63 (dd, J = 10.1, 25.4 Hz, 2H), 3.18 (dd, J = 6.1, 10.2 Hz, 1H), 2.86 (t, J = 8.3 Hz, 1H), 2.53 (dd, J = 10.4, 13.6 Hz, 1H), 2.10 (s, 3H), 2.09–2.03 (m, 1H), 1.98–1.86 (m, 1H), 1.81–1.72 (m, 1H), 1.68–1.52 (m, 1H), 1.47 (d, J = 6.6 Hz, 3H), 1.42–1.31 (m, 2H), 1.25 (t, J = 7.2 Hz, 3H), 0.71 (d, J = 11.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 146.7, 143.4, 138.1, 134.6, 133.4, 132.9, 128.5, 128.4, 127.78, 127.77, 127.70, 126.1, 125.7, 125.0, 124.7, 120.3, 74.1, 63.2, 60.4, 57.3, 57.0, 55.0, 53.4, 45.6, 45.4, 34.7, 24.8, 23.0, 22.0, 19.4, 14.4; HR-MS (ESI): m/z calcd for C32H38NO3 [M + H]+: 484.2846; found: 484.2837.
Ethyl (4bS,7R,8S,9aS,10R)-7-(hydroxymethyl)-1-methyl-8-(((S)-1-(naphthalen-1-yl)ethyl)amino)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 12. Prepared with (S)-(−)-1-(1-naphthyl)ethylamine, yield: 55%; colorless oily compound; [α]20D = +27.5 (c 0.12, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.22 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 7.9 Hz, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.67 (d, J = 7.0 Hz, 1H), 7.56–7.45 (m, 3H), 7.15 (t, J = 7.6 Hz, 1H), 6.98 (t, J = 7.0 Hz, 2H), 4.76 (q, J = 6.5 Hz, 1H), 4.05–3.98 (m, 2H), 3.95 (s, 1H), 3.45 (d, J = 10.2 Hz, 1H), 3.35 (d, J = 10.4 Hz, 1H), 3.03–2.97 (m, 1H), 2.78 (dd, J = 6.0, 10.1 Hz, 1H), 2.60 (dd, J = 10.1, 13.2 Hz, 1H), 2.22–2.14 (m, 1H), 2.11 (s, 3H), 1.99–1.92 (m, 1H), 1.74–1.62 (m, 3H), 1.53 (d, J = 6.5 Hz, 3H), 1.33–1.29 (m, 1H), 0.98 (t, J = 7.2 Hz, 3H), 0.56 (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 171.9, 146.7, 140.3, 138.2, 134.7, 134.0, 131.5, 129.1, 128.5, 127.7, 127.4, 125.9, 125.8, 125.4, 123.4, 122.6, 120.2, 72.7, 61.8, 60.3, 57.0, 55.1, 53.3, 51.9, 45.5, 45.2, 34.6, 24.8, 24.6, 22.0, 19.4, 14.1; HR-MS (ESI): m/z calcd for C32H38NO3 [M + H]+: 484.2773; found: 484.2853.
Ethyl (4bS,7R,8S,9aS,10R)-7-(hydroxymethyl)-1-methyl-8-(((R)-1-(naphthalen-1-yl)ethyl)amino)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 13. Prepared with (R)-(+)-1-(1-naphthyl)ethylamine, yield: 58%; white powder; m.p.: 50–53 °C; [α]20D = −7.6 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.16 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 8.2 Hz, 1H), 7.61 (d, J = 7.2 Hz, 1H), 7.56–7.45 (m, 3H), 7.13 (t, J = 7.5 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 6.93 (d, J = 7.5 Hz, 1H), 4.72 (q, J = 6.4 Hz, 1H), 4.28–4.16 (m, 2H), 3.95 (s, 1H), 3.65 (d, J = 9.9 Hz, 1H), 3.59 (d, J = 9.9 Hz, 1H), 3.29–3.29 (m, 1H), 2.89–2.83 (m, 1H), 2.70–2.63 (m, 1H), 2.13 (s, 3H), 2.10–1.99 (m, 1H), 1.95–1.86 (m, 1H), 1.81–1.72 (m, 1H), 1.69–1.59 (m, 1H), 1.55 (d, J = 6.5 Hz, 3H), 1.50–1.39 (m, 2H), 1.27 (t, J = 7.1 Hz, 3H), 0.75 (d, J = 11.6 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 146.7, 141.7, 138.1, 134.6, 133.9, 130.6, 129.1, 128.5, 127.8, 127.6, 126.1, 125.7, 125.5, 123.1, 122.6, 120.3, 74.0, 62.8, 60.4, 57.1, 55.0, 53.4, 51.6, 45.5, 45.1, 34.8, 24.8, 22.3, 21.9, 19.4, 14.4; HR-MS (ESI): m/z calcd for C32H38NO3 [M + H]+: 484.2773; found: 484.2858.
Ethyl (4bS,7R,8S,9aS,10R)-8-((4-fluorobenzyl)amino)-7-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 14. Prepared with 4-fluorobenzylamine, yield: 61%; oily compound; [α]20D = +1.5 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.32–7.26 (m, 2H), 7.16 (t, J = 7.5 Hz, 1H), 7.05–6.94 (m, 4H), 4.25–4.18 (m, 2H), 4.01 (s, 1H), 3.86 (d, J = 12.8 Hz, 1H), 3.73 (d, J = 13.0 Hz, 1H), 3.54 (dd, J = 9.9, 15.0 Hz, 2H), 3.06–2.94 (m, 2H), 2.71 (dd, J = 10.5, 13.4 Hz, 1H), 2.15 (s, 3H), 2.13–2.09 (m, 1H), 1.97–1.89 (m, 1H), 1.74–1.64 (m, 2H), 1.59–1.53 (m, 1H), 1.41 (d, J = 11.4 Hz, 1H), 1.28 (t, J = 7.1 Hz, 3H), 0.75 (d, J = 11.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 163.0, 146.7, 138.1, 135.5, 134.7, 129.7, 129.7, 128.5, 127.8, 120.3, 115.4, 115.2, 73.6, 64.4, 60.5, 57.1, 54.9, 53.5, 52.0, 45.5, 44.9, 34.9, 24.8, 21.9, 19.4, 14.4; 19F NMR (470 MHz, CDCl3): δ = −115.4 (Cq-F); HR-MS (ESI): m/z calcd for C27H33FNO3 [M + H]+: 438.2366; found: 438.2445.
Ethyl (4bS,7R,8S,9aS,10R)-7-(hydroxymethyl)-8-((4-methoxybenzyl)amino)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 15. Prepared with 4-methoxybenzylamine, yield: 50%; oily compound; [α]20D = −7.2 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.25–7.22 (m, 2H), 7.15 (t, J = 7.5 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 6.95 (d, J = 7.4 Hz, 1H), 6.89–6.85 (m, 2H), 4.28–4.17 (m, 2H), 4.01 (s, 1H), 3.85–3.78 (m, 4H), 3.71 (d, J = 12.5 Hz, 1H), 3.55 (s, 2H), 3.06–2.93 (m, 2H), 2.73 (dd, J = 10.4, 13.4 Hz, 1H), 2.15 (s, 3H), 2.13–2.08 (m, 1H), 1.97–1.88 (m, 1H), 1.74–1.63 (m, 2H), 1.58–1.52 (m, 1H), 1.41 (dd, J = 1.8, 11.7 Hz, 1H), 1.29 (t, J = 7.1 Hz, 3H), 0.73 (d, J = 12.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 158.8, 146.7, 138.2, 134.7, 131.9, 129.4, 129.3, 128.5, 127.7, 120.2, 113.94, 113.91, 73.9, 64.5, 60.4, 57.1, 55.2, 54.9, 53.6, 52.2, 45.4, 45.0, 34.9, 24.9, 21.9, 19.4, 14.4; HR-MS (ESI): m/z calcd for C28H36NO4 [M + H]+: 450.2638; found: 450.2631.
Ethyl (4bS,7R,8S,9aS,10R)-8-((2-(1H-indol-3-yl)ethyl)amino)-7-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 16. Prepared with tryptamine, yield: 55%; white powder; m.p.: 61–65 °C; [α]20D = −1.3 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.05 (s, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.19 (t, J = 7.24 Hz, 1H), 7.15–7.10 (m, 2H), 7.05 (d, J = 2.0 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 6.92 (d, J = 7.5 Hz, 1H), 4.25–4.19 (m, 2H), 3.96 (s, 1H), 3.54 (s, 2H), 3.10–2.80 (m, 6H), 2.68 (dd, J = 10.5, 13.4 Hz, 1H), 2.13 (s, 3H), 2.02–1.92 (m, 1H), 1.87–1.77 (m, 1H), 1.70–1.56 (m, 2H), 1.45–1.35 (m, 2H), 1.28 (t, J = 7.2 Hz, 3H), 0.72 (d, J = 11.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 146.7, 138.1, 136.4, 134.7, 128.5, 127.7, 127.3, 122.1, 121.9, 120.2, 119.3, 118.7, 113.8, 111.2, 73.9, 65.3, 60.4, 57.1, 54.8, 53.5, 48.8, 45.3, 44.7, 34.8, 26.1, 24.9, 21.8, 19.4, 14.5; HR-MS (ESI): m/z calcd for C30H37N2O3 [M + H]+: 473.2726; found: 473.2804.
Ethyl (4bS,7R,8S,9aS,10R)-8-((3-(1H-imidazol-1-yl)propyl)amino)-7-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 17. Prepared with 1-(3-aminopropyl)imidazole, yield: 54%; colorless oily compound; [α]20D = +30.2 (c 0.11, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.51 (s, 1H), 7.16 (t, J = 7.5 Hz, 1H), 7.07 (s, 1H), 7.02 (d, J = 7.4 Hz, 1H), 6.96 (d, J = 7.7 Hz, 1H), 6.93 (s, 1H), 4.22 (q, J = 7.3 Hz, 2H), 4.04 (t, J = 6.7 Hz, 2H), 3.95 (s, 1H), 3.58–3.53 (m, 2H), 2.98–2.91 (m, 2H), 2.76–2.62 (m, 2H), 2.55–2.49 (m, 1H), 2.14 (s, 3H), 2.12–2.09 (m, 1H), 2.01–1.84 (m, 3H), 1.73–1.63 (m, 2H), 1.51–1.40 (m, 2H), 1.29 (t, J = 7.1 Hz, 3H), 0.76 (d, J = 12.0 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 146.6, 138.1, 137.2, 134.7, 129.6, 128.5, 127.8, 120.3, 118.8, 73.4, 65.0, 60.5, 57.1, 54.9, 53.4, 45.5, 45.2, 44.9, 44.6, 34.9, 31.5, 24.8, 21.9, 19.4, 14.5; HR-MS (ESI): m/z calcd for C26H36N3O3 [M + H]+: 438.2678; found: 438.2767.
:MeOH 19:1 then n-hexane:EtOAc 1:2 as eluent.
Ethyl (4bS,7R,8S,9aS,10R)-8-((5-bromo-2-hydroxybenzyl)amino)-7-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 18. Prepared with 5-bromosalicylaldehyde (5-bromo-2-hydroxybenzaldehyde), yield: 53%; white powder; m.p.: 82–85 °C; [α]20D = +4.2 (c 0.14, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.25–7.23 (m, 1H), 7.18–7.12 (m, 2H), 7.00 (d, J = 7.5 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.71 (d, J = 8.7 Hz, 1H), 4.23 (q, J = 7.1 Hz, 2H), 4.01 (t, J = 6.9 Hz, 2H), 3.88 (d, J = 13.9 Hz, 1H), 3.52 (d, J = 10.3 Hz, 1H), 3.47 (d, J = 10.3 Hz, 1H), 3.10 (dd, J = 5.3, 10.3 Hz, 1H), 3.00 (t, J = 8.0 Hz, 1H), 2.65–2.58 (m, 1H), 2.18–2.15 (m, 1H), 2.14 (s, 3H), 1.96–1.87 (m, 1H), 1.74–1.65 (m, 2H), 1.56–1.44 (m, 2H), 1.29 (t, J = 7.2 Hz, 3H), 0.93 (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 157.3, 146.5, 138.0, 134.7, 131.5, 130.7, 128.6, 127.8, 124.9, 120.3, 118.1, 110.8, 70.7, 62.1, 60.5, 56.8, 54.3, 53.1, 51.4, 46.5, 44.0, 36.1, 24.7, 21.7, 19.4, 14.4; HR-MS (ESI): m/z calcd for C27H33BrNO4 [M + H]+: 516.1494; found: 516.1558.
Ethyl (4bS,7R,8S,9aS,10R)-7-(hydroxymethyl)-1-methyl-8-((quinolin-2-ylmethyl)amino)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 19. Prepared with 2-quinoline carboxaldehyde, yield: 51%; oily compound; [α]20D = +17.2 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.16 (t, J = 8.5 Hz, 2H), 7.82 (d, J = 8.1 Hz, 1H), 7.75–7.70 (m, 1H), 7.56–7.52 (m, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.16 (t, J = 7.6 Hz,1H), 7.01–6.97 (m, 2H), 4.30 (d, J = 16.4 Hz, 1H), 4.20–4.09 (m, 3H), 3.98 (s, 1H), 3.60 (dd, J = 11.2, 13.3 Hz, 2H), 3.16–3.04 (m, 2H), 2.59–2.54 (m. 1H), 2.31–2.24 (m, 2H), 2.13 (s, 3H), 1.78–1.64 (m, 3H), 1.43–1.39 (m, 1H), 1.21 (t, J = 7.1 Hz, 3H), 0.75 ppm (d, J = 11.6 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.3, 160.2, 147.3, 147.1, 138.3, 137.0, 134.6, 129.9, 128.38, 128.30, 127.7, 127.6, 127.3, 126.3, 120.7, 120.3, 73.4, 62.6, 60.4, 57.0, 54.4, 53.7, 53.5, 46.4, 45.5, 35.7, 25.2, 21.9, 19.3, 14.4; HR-MS (ESI): m/z calcd for C30H35N2O3 [M + H]+: 471.2642; found: 471.2647.
Ethyl (4bS,7R,8S,9aS,10R)-7-(hydroxymethyl)-1-methyl-8-((quinolin-4-ylmethyl)amino)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 20. Prepared with 4-quinoline carboxaldehyde, yield: 55%; oily compound; [α]20D = +4.1 (c 0.15, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.89 (d, J = 4.3 Hz, 1H), 8.15 (d, J = 8.2 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.72 (t, J = 7.0 Hz, 1H), 7.59 (t, J = 7.7 Hz, 1H), 7.47 (d, J = 4.3 Hz, 1H), 7.17 (t, J = 7.5 Hz, 1H), 7.00 (d, J = 7.4 Hz, 1H), 6.97 (d, J = 7.4 Hz, 1H), 4.37 (d, J = 14.2 Hz, 1H), 4.28–4.21 (m, 3H), 4.04 (s, 1H), 3.60–3.52 (m, 2H), 3.18 (dd, J = 5.6, 10.2 Hz, 1H), 3.05–2.98 (m, 1H), 2.78 (dd, J = 10.5, 13.2 Hz, 1H), 2.20–2.16 (m, 1H), 2.15 (s, 3H), 2.04–1.98 (m, 1H), 1.74–1.64 (m, 3H), 1.49–1.43 (m, 1H), 1.29 (t, J = 7.1 Hz, 3H), 0.81 (d, J = 12.0 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 150.3, 148.2, 146.6, 145.2, 138.1, 134.7, 130.2, 129.2, 128.6, 127.8, 126.9, 126.7, 123.1, 120.3, 120.1, 73.0, 64.6, 60.5, 57.0, 54.8, 53.5, 49.2, 46.0, 44.8, 35.0, 24.8, 21.8, 19.4, 14.5; HR-MS (ESI): m/z calcd for C30H35N2O3 [M + H]+: 471.2642; found: 471.2633.
Ethyl (4bS,7R,8S,9aS,10R)-7-(hydroxymethyl)-1-methyl-8-((pyridin-4-ylmethyl)amino)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 21. Prepared with 4-pyridine carboxaldehyde, yield: 46%; oily compound; [α]20D = −13.9 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.56 (d, J = 5.8 Hz, 2H), 7.29–7.26 (m, 2H), 7.16 (t, J = 7.5 Hz, 1H), 7.00 (d, J = 7.5 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 4.27–4.19 (m, 2H), 4.01 (s, 1H), 3.90 (d, J = 14.7 Hz, 1H), 3.78 (d, J = 14.3 Hz, 1H), 3.59–3.52 (m, 2H), 3.06–2.95 (m, 2H), 2.68 (dd, J = 10.3, 13.4 Hz, 1H), 2.20–2.16 (m, 1H), 2.15 (s, 3H), 2.01–1.95 (m, 1H), 1.74–1.65 (m, 2H), 1.61–1.55 (m, 1H), 1.45–1.41 (m, 1H), 1.28 (t, J = 7.2 Hz, 3H), 0.78 (d, J = 11.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 149.92, 149.93, 149.0, 146.6, 138.1, 134.7, 128.5, 127.8, 122.91, 122.90, 120.3, 73.1, 64.2, 60.5, 57.0, 54.8, 53.4, 51.6, 45.8, 44.8, 35.0, 24.7, 21.9, 19.4, 14.4; HR-MS (ESI): m/z calcd for C26H33N2O3 [M + H]+: 421.2485; found: 421.2464.
Ethyl (4bS,7S,8R,9aS,10R)-7-((benzylamino)methyl)-8-hydroxy-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 30. Prepared with benzaldehyde, yield: 66%; oily colorless compound; [α]20D = −2.7 (c 0.14, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.35–7.23 (m, 5H), 7.14 (t, J = 7.5 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 7.5 Hz, 1H), 4.21 (m, 2H), 4.03–3.98 (m, 2H), 3.81–3.72 (m, 2H), 3.04–2.95 (m, 1H), 2.85 (d, J = 11.1 Hz, 1H), 2.66 (dd, J = 10.6, 13.8 Hz, 1H), 2.38 (d, J = 11.1 Hz, 1H), 2.26–2.16 (m, 2H), 2.13 (s, 3H), 1.68–1.62 (m, 1H), 1.60–1.46 (m, 2H), 1.41–1.34 (m, 1H), 1.28 (t, J = 7.2 Hz, 3H), 0.65 ppm (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.4, 147.2, 138.7, 138.1, 134.6, 128.53, 128.52, 128.4, 128.14, 128.13, 127.8, 127.1, 120.4, 78.7, 60.5, 60.4, 57.1, 54.8, 54.7, 52.6, 45.2, 44.8, 35.0, 23.9, 22.2, 19.4, 14.4; HR-MS (ESI): m/z calcd for C27H34NO3 [M + H]+: 420.2533; found: 420.2525.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-(((naphthalen-1-ylmethyl)amino)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 31. Prepared with 1-naphthaldehyde, yield: 50%; colorless oily compound; [α]20D = −18.4 (c 0.12, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.10 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.80–7.76 (m, 1H), 7.55–7.40 (m, 4H), 7.14 (t, J = 7.5 Hz, 1H), 6.98 (d, J = 7.4 Hz, 1H), 6.93 (d, J = 7.4 Hz, 1H), 4.25–4.14 (m, 4H), 4.04–3.99 (m, 2H), 3.02–2.95 (m, 2H), 2.66 (dd, J = 10.6, 13.8 Hz, 1H), 2.51 (d, J = 11.6 Hz, 1H), 2.23–2.14 (m, 2H), 2.13 (s, 3H), 1.65–1.49 (m, 3H), 1.39–1.31 (m, 1H), 1.29 (t, J = 7.1 Hz, 3H), 0.68 (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.4, 147.2, 138.1, 135.3, 134.6, 133.9, 131.8, 128.7, 128.4, 128.1, 127.8, 126.34, 126.31, 125.7, 125.3, 123.5, 120.4, 78.8, 61.2, 60.4, 57.1, 54.8, 52.8, 52.6, 45.3, 44.8, 35.1, 23.9, 22.2, 19.4, 14.5; HR-MS (ESI): m/z calcd for C31H36NO3 [M + H]+: 470.2689; found: 470.2681.
Ethyl (4bS,7S,8R,9aS,10R)-7-(((4-fluorobenzyl)amino)methyl)-8-hydroxy-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 32. Prepared with 4-fluorobenzaldehyde, yield: 55%; oily compound; [α]20D = −22.5 (c 0.15, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.25–7.21 (m, 2H), 7.14 (t, J = 7.5 Hz, 1H), 7.03–6.93 (m, 4H), 4.24–4.17 (m, 2H), 4.02 (s, 1H), 3.96 (dd, J = 5.8, 8.7 Hz, 1H), 3.76 (d, J = 13.3 Hz, 1H), 3.68 (d, J = 13.1 Hz, 1H), 3.03–2.98 (m, 1H), 2.81 (d, J = 11.3 Hz, 1H), 2.69–2.63 (m, 1H), 2.35 (d, J = 11.3 Hz, 1H), 2.25–2.15 (m, 2H), 2.13 (s, 3H), 1.67–1.47 (m, 3H), 1.41–1.35 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H), 0.64 ppm (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.4, 147.1, 138.1, 135.67, 135.65, 134.6, 129.6, 129.5, 128.4, 127.8, 120.4, 115.3, 115.2, 78.7, 60.4, 60.3, 57.1, 54.8, 53.9, 52.6, 45.3, 44.8, 35.0, 23.9, 22.2, 19.4, 14.4; 19F NMR (470 MHz, CDCl3): δ = −115.6 (Cq-F); HR-MS (ESI): m/z calcd for C27H33FNO3 [M + H]+: 438.2438; found: 438.2429.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-7-(((4-methoxybenzyl)amino)methyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 33. Prepared with 4-methoxybenzaldehyde, yield: 50%; oily compound; [α]20D = −7.1 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.22–7.18 (m, 2H), 7.14 (t, J = 7.4 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 7.5 Hz, 1H), 6.86 (d, J = 8.7 Hz, 2H), 4.25–4.17 (m, 2H), 4.02 (s, 1H), 3.97 (dd, J = 5.6, 10.7 Hz, 1H), 3.80 (s, 3H), 3.72 (d, J = 13.0 Hz, 1H), 3.67 (d, J = 13.0 Hz, 1H), 3.00 (t, J = 8.5 Hz, 1H), 2.82 (d, J = 11.2 Hz, 1H), 2.69–2.62 (m, 1H), 2.36 (d, J = 11.2 Hz, 1H), 2.24–2.17 (m, 2H), 2.13 (s, 3H), 1.68–1.46 (m, 3H), 1.38–1.33 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H), 0.64 ppm (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.4, 158.8, 147.2, 138.1, 134.63, 131.97, 129.33, 129.32, 128.4, 127.8, 120.4, 113.91, 113.90, 78.7, 60.4, 60.3, 57.1, 55.2, 54.8, 54.1, 52.6, 45.2, 44.8, 35.0, 24.0, 22.2, 19.4, 14.4; HR-MS (ESI): m/z calcd for C28H36NO4 [M + H]+: 450.2638; found: 450.2631.
Ethyl (4bS,7S,8R,9aS,10R)-7-(((5-bromo-2-hydroxybenzyl)amino)methyl)-8-hydroxy-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 34. Prepared with 5-bromosalicylaldehyde (5-bromo-2-hydroxybenzaldehyde), yield: 53%; colourless oily compound; [α]20D = −5.2 (c 0.15, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.23 (dd, J = 2.3, 8.7 Hz, 1H), 7.15 (t, J = 7.3 Hz, 1H), 7.07 (d, J = 2.2 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.69 (d, J = 8.5 Hz, 1H), 4.22 (q, J = 7.1 Hz, 2H), 4.05–4.00 (m, 1H), 3.99 (s, 1H), 3.95–3.88 (m, 2H), 3.02 (t, J = 8.3 Hz, 1H), 2.73–2.62 (m, 2H), 2.55 (d, J = 11.1 Hz, 1H), 2.25–2.15 (m, 2H), 2.13 (s, 3H), 1.67–1.53 (m, 3H), 1.50–1.44 (m, 1H), 1.29 (t, J = 7.1 Hz, 3H), 0.77 ppm (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 157.3, 146.8, 137.9, 134.6, 131.4, 130.9, 128.5, 127.9, 124.2, 120.4, 118.1, 110.7, 76.8, 60.5, 59.1, 56.8, 54.1, 52.9, 52.5, 47.0, 45.5, 35.8, 24.2, 22.1, 19.3, 14.4; HR-MS (ESI): m/z calcd for C27H33BrNO4 [M + H]+: 516.1494; found: 516.1561.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-(((quinolin-2-ylmethyl)amino)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 35. Prepared with 2-quinoline carboxaldehyde, yield: 43%; oily compound; [α]20D = −15.4 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.13 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.81 (d, J = 7.9 Hz, 1H), 7.74–7.69 (m, 1H), 7.53 (t, J = 7.1 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.14 (t, J = 7.5 Hz, 1H), 6.99–6.94 (m, 2H), 4.22–4.16 (m, 2H), 4.12–4.01 (m, 4H), 3.06–3.00 (m, 1H), 2.94 (d, J = 11.1 Hz, 1H), 2.68 (dd, J = 10.6, 13.8 Hz, 1H), 2.46 (d, J = 11.1 Hz, 1H), 2.36–2.28 (m, 1H), 2.25–2.18 (m, 1H), 2.13 (s, 3H), 1.71–1.65 (m, 1H), 1.62–1.48 (m, 3H), 1.27 (t, J = 7.2 Hz, 3H), 0.68 ppm (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.4, 158.5, 147.6, 147.2, 138.1, 136.7, 134.6, 129.6, 128.9, 128.4, 127.8, 127.6, 127.3, 126.2, 120.5, 120.3, 78.4, 60.7, 60.4, 57.1, 56.1, 54.8, 52.6, 45.4, 45.0, 35.1, 23.9, 22.2, 19.4, 14.4; HR-MS (ESI): m/z calcd for C30H35N2O3 [M + H]+: 471.2642; found: 471.2636.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-(((quinolin-4-ylmethyl)amino)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 36. Prepared with 4-quinoline carboxaldehyde, yield: 55%; oily compound; [α]20D = −2.78 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.87 (d, J = 4.3 Hz, 1H), 8.13 (d, J = 8.2 Hz, 1H), 8.06 (d, J = 8.2 Hz, 1H), 7.75–7.70 (m, 1H), 7.61–7.57 (m, 1H), 7.37 (d, J = 4.4 Hz, 1H), 7.15 (t, J = 7.5 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 1H), 4.25–4.18 (m, 4H), 4.05–4.00 (m, 2H), 3.05–2.98 (m, 1H), 2.95–2.87 (m, 1H), 2.67 (dd, J = 10.6, 13.8 Hz, 1H), 2.51 (d, J = 11.1 Hz, 1H), 2.25–2.17 (m, 2H), 2.14 (s, 3H), 1.67–1.52 (m, 3H), 1.44–1.39 (m, 1H), 1.28 (t, J = 7.2 Hz, 3H), 0.70 ppm (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 150.2, 148.3, 147.1, 145.0, 138.0, 134.6, 130.3, 129.2, 128.5, 127.8, 127.0, 126.7, 123.1, 120.4, 120.0, 78.5, 61.0, 60.4, 57.0, 54.7, 52.6, 51.2, 45.5, 45.0, 35.1, 24.0, 22.2, 19.4, 14.5; HR-MS (ESI): m/z calcd for C30H35N2O3 [M + H]+: 471.2642; found: 471.2631.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-(((pyridin-4-ylmethyl)amino)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 37. Prepared with 4-pyridine carboxaldehyde, yield: 50%; oily colorless compound; [α]20D = −17.5 (c 0.12, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.56 (d, J = 5.5 Hz, 2H), 7.22 (d, J = 4.8 Hz, 2H), 7.15 (t, J = 7.5 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 1H), 4.20 (q, J = 7.0 Hz, 2H), 4.04–3.98 (m, 2H), 3.85 (d, J = 14.3 Hz, 1H), 3.71 (d, J = 14.1 Hz, 1H), 3.05–2.99 (m, 1H), 2.80 (d, J = 11.1 Hz, 1H), 2.67 (dd, J = 10.5, 13.7 Hz, 1H), 2.36 (d, J = 11.2 Hz, 1H), 2.27–2.16 (m, 2H), 2.13 (s, 3H), 1.69–1.49 (m, 3H), 1.45–1.37 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H), 0.66 ppm (d, J = 12.1 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.3, 150.0, 149.9, 148.9, 147.1, 138.0, 134.6, 128.5, 127.8, 123.0, 122.9, 120.4, 78.5, 60.44, 60.43, 57.0, 54.7, 53.4, 52.6, 45.4, 45.0, 35.1, 23.9, 22.2, 19.4, 14.4; HR-MS (ESI): m/z calcd for C26H33N2O3 [M + H]+: 421.2485; found: 421.2482.
:MeCN (1:1) (2 mL), then a primary amine (0.96 mmol, 4 eq.) was added to the solution. The mixture was stirred under reflux for 72 hours. After the completion of the reaction (as monitored by TLC), the mixture was evaporated to dryness, the residue was diluted with H2O (5 mL) and then extracted with DCM (3 × 10 mL). The organic phase was washed with brine solution (3 × 25 mL), dried over Na2SO4, filtered and then evaporated. The crude product was purified by column chromatography on silica gel using n-hexane:EtOAc (1:2) as eluent.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-((((S)-1-(naphthalen-2-yl)ethyl)amino)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 26. Prepared with (S)-(−)-1-(2-naphthyl)ethylamine, yield: 50%; oily compound; [α]20D = −24.8 (c 0.14, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.84–7.79 (m, 3H), 7.64 (s, 1H), 7.50–7.42 (m, 2H), 7.39–7.36 (m, 1H), 7.13 (t, J = 7.6 Hz, 1H), 6.97 (d, J = 7.4 Hz, 1H), 6.92 (d, J = 7.4 Hz, 1H), 4.25–4.16 (m, 2H), 4.09–4.03 (m, 1H), 4.02 (s, 1H), 3.84 (q, J = 6.6 Hz, 1H), 3.03–2.97 (m, 1H), 2.72–2.63 (m, 2H), 2.34 (d, J = 11.4 Hz, 1H), 2.25–2.13 (m, 2H), 2.12 (s, 3H), 1.69–1.62 (m, 1H), 1.51–1.40 (m, 5H), 1.33–1.29 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H), 0.63 (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.4, 147.2, 142.6, 138.1, 134.6, 133.4, 132.7, 128.5, 128.4, 127.8, 127.7, 127.6, 126.1, 125.6, 125.0, 124.2, 120.4, 78.7, 60.3, 59.1, 58.9, 57.1, 54.9, 52.6, 45.2, 44.9, 34.8, 23.9, 23.7, 22.2, 19.4, 14.4; HR-MS (ESI): m/z calcd for C32H38NO3 [M + H]+: 484.2846; found: 484.2838.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-((((R)-1-(naphthalen-2-yl)ethyl)amino)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 27. Prepared with (R)-(−)-1-(2-naphthyl)ethylamine, yield: 50%; oily compound; [α]20D = +1.3 (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.86–7.80 (m, 3H), 7.70 (s, 1H), 7.51–7.45 (m, 3H), 7.13 (t, J = 7.5 Hz, 1H), 6.96 (m, 2H), 4.12–4.05 (m, 2H), 3.97 (s, 1H), 3.85–3.77 (m, 2H), 3.03–2.97 (m, 1H), 2.75 (d, J = 11.1 Hz, 1H), 2.57 (dd, J = 10.6, 13.8 Hz, 1H), 2.31–2.24 (m, 1H), 2.22–2.17 (m, 2H), 2.10 (s, 3H), 1.65–1.51 (m, 2H), 1.47–1.39 (m, 5H), 1.16 (t, J = 7.2 Hz, 3H), 0.48 (d, J = 12.1 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.3, 147.2, 142.4, 138.2, 134.6, 133.4, 132.9, 128.5, 128.4, 127.78, 127.75, 127.70, 126.0, 125.64, 125.61, 124.5, 120.4, 78.6, 60.3, 59.2, 58.9, 57.1, 54.8, 52.6, 45.1, 44.7, 34.9, 24.8, 24.0, 22.2, 19.3, 14.3; HR-MS (ESI): m/z calcd for C32H38NO3 [M + H]+: 484.2846; found: 484.2834.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-((((S)-1-(naphthalen-1-yl)ethyl)amino)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 28. Prepared with (S)-(−)-1-(1-naphthyl)ethylamine, yield: 46%; oily compound; [α]20D = +8.3 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.10 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.76 (dd, J = 2.6, 6.7 Hz, 1H), 7.53–7.46 (m, 4H), 7.14 (t, J = 7.5 Hz, 1H), 6.97 (d, J = 7.4 Hz, 1H), 6.93 (d, J = 7.5 Hz, 1H), 4.55 (q, J = 6.6 Hz, 1H), 4.23–4.16 (m, 2H), 4.07 (dd, J = 6.5, 10.7 Hz, 1H), 4.02 (s, 1H), 3.05–2.98 (m, 1H), 2.80 (d, J = 11.3 Hz, 1H), 2.71–2.65 (m, 1H), 2.42 (d, J = 11.4 Hz, 1H), 2.28–2.14 (m, 2H), 2.13 (s, 3H), 1.69–1.63 (m, 1H), 1.57–1.52 (m, 1H), 1.51 (d, J = 6.5 Hz, 3H), 1.48–1.44 (m, 1H), 1.38–1.32 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H), 0.65 (d, J = 12.1 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.4, 147.2, 140.7, 138.1, 134.6, 133.9, 131.1, 129.0, 128.4, 127.8, 127.4, 126.0, 125.58, 125.56, 122.6, 122.2, 120.4, 78.7, 60.4, 59.1, 57.1, 54.8, 54.0, 52.6, 45.2, 44.9, 34.9, 24.0, 23.0, 22.2, 19.4, 14.4; HR-MS (ESI): m/z calcd for C32H38NO3 [M + H]+: 484.2846; found: 484.2837.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-((((R)-1-(naphthalen-1-yl)ethyl)amino)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 29. Prepared with (R)-(+)-1-(1-naphthyl)ethylamine, yield: 41%; oily compound; [α]20D = −19.3 (c 0.14, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.14 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.66 (d, J = 7.0 Hz, 1H), 7.53–7.45 (m, 3H), 7.14 (t, J = 7.5 Hz, 1H), 6.99–6.94 (m, 2H), 4.55 (q, J = 6.5 Hz, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.99 (s, 1H), 3.95 (dd, J = 5.6, 10.6 Hz, 1H), 3.01 (t, J = 8.7 Hz, 1H), 2.77 (d, J = 11.0 Hz, 1H), 2.63 (dd, J = 10.6, 13.8 Hz, 1H), 2.33–2.25 (m, 2H), 2.23–2.16 (m, 1H), 2.11 (s, 3H), 1.67–1.63 (m, 1H), 1.59–1.54 (m, 1H), 1.49–1.43 (m, 5H), 1.20 (t, J = 7.1 Hz, 3H), 0.54 ppm (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.3, 147.2, 140.4, 138.1, 134.6, 134.0, 131.4, 129.0, 128.4, 127.8, 127.3, 125.87, 125.80, 125.4, 122.6, 120.4, 78.6, 60.4, 59.2, 57.1, 54.8, 54.3, 52.6, 45.2, 44.8, 35.0, 24.0, 23.8, 22.2, 19.3, 14.3; HR-MS (ESI): m/z calcd for C32H38NO3 [M + H]+: 484.2846; found: 484.2838.
:1). The mixture was stirred at room temperature for 12 hours, then t-BuOH was evaporated. The residue was dissolved in water (10 mL) and extracted with EtOAc (3 × 10 mL). The organic phase was washed with brine (3 × 10 mL), dried over Na2SO4, and then evaporated at low pressure. The crude product was purified by column chromatography on silica gel using n-hexane:EtOAc (1:1) as eluent.
Ethyl (4bS,7S,8R,9aS,10R)-8-hydroxy-1-methyl-7-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 38. Prepared with phenylacetylene, yield: 90%; white crystals; m.p.: 172–174 °C; [α]20D = −18.5 (c 0.15, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.80 (d, J = 7.3 Hz, 2H), 7.71 (s, 1H), 7.45–7.40 (m, 2H), 7.34 (t, J = 7.4 Hz, 1H), 7.16 (t, J = 7.5 Hz, 1H), 7.00 (d, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 1H), 4.33–4.18 (m, 5H), 4.02 (s, 1H), 3.25 (d, J = 2.9 Hz, 1H), 3.06 (t, J = 8.2 Hz, 1H), 2.73 (dd, J = 10.5, 13.9 Hz, 1H), 2.25–2.17 (m, 1H), 2.14 (s, 3H), 2.07–1.99 (m, 1H), 1.81–1.75 (m, 1H), 1.68–1.61 (m, 2H), 1.33–1.28 (m, 1H), 1.27 (t, J = 7.1 Hz, 3H), 1.01 (d, J = 11.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 147.4, 146.6, 137.9, 134.7, 130.2, 128.91, 128.90, 128.6, 128.3, 127.9, 125.72, 125.71, 121.6, 120.3, 75.9, 60.6, 58.3, 56.5, 53.6, 52.3, 45.9, 45.1, 36.4, 23.7, 21.5, 19.4, 14.5; HR-MS (ESI): m/z calcd for C28H32N3O3 [M + H]+: 458.2438; found: 458.2429.
Ethyl (4bS,7S,8R,9aS,10R)-7-((4-benzyl-1H-1,2,3-triazol-1-yl)methyl)-8-hydroxy-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulene-10-carboxylate 39. Prepared with 3-phenyl-1-propyne, yield: 90%; white crystals; m.p.: 139–140 °C; [α]20D = −38.6 (c 0.13, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.32–7.20 (m, 5H), 7.17–7.11 (m, 2H), 6.99 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 7.5 Hz, 1H), 4.33–4.28 (m, 1H), 4.24–4.06 (m, 6H), 4.01 (s, 1H), 3.38 (d, J = 3.1 Hz, 1H), 3.04 (t, J = 8.2 Hz, 1H), 2.71 (dd, J = 10.6, 13.8 Hz, 1H), 2.21–2.16 (m, 1H), 2.14 (s, 3H), 2.01–1.93 (m, 1H), 1.79–1.73 (m, 1H), 1.63–1.53 (m, 2H), 1.28 (t, J = 7.2 Hz, 3H), 1.23–1.16 (m, 1H), 0.95 (d, J = 11.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 172.2, 147.3, 146.7, 138.8, 138.0, 134.7, 128.69, 128.68, 128.67, 128.66, 128.65, 127.8, 126.5, 123.6, 120.3, 76.1, 60.5, 58.4, 56.5, 53.6, 52.3, 45.8, 45.1, 36.4, 32.1, 23.6, 21.4, 19.4, 14.5; HR-MS (ESI): m/z calcd for C29H34N3O3 [M + H]+: 472.2594; found: 472.2584.
Conclusions
A new library of allo-gibberic acid-based aminoalcohol regioisomers was synthesised stereoselectively starting from commercially available gibberellic acid. The successful formation of hydroxymethyl ketone derivative 5 by acid-mediated rearrangement of previously prepared epoxide provided the desired 1,3-aminoalcohols through Schiff base formation. To obtain the desired regioisomers, the primary alcohol functionality of 5 was subjected to mesylation, then replaced with either primary amine or azide function. The formed azide derivative was subjected to either CuAAC reaction to obtain 1,2,3-triazoles or to Pd-catalysed hydrogenolysis to obtain primary aminoalcohol, which was further transformed into 1,3-aminoalcohols by reductive alkylation. The antiproliferative effects were assayed by the MTT method with several N-substituted derivatives showing remarkable inhibition of cell growth on human cancer cell lines (HeLa, SiHa, A2780, MCF-7 and MDA-MB-231). A significant difference was observed in the antiproliferative activity between the regioisomers. Some compounds exerted outstanding activities against the malignant cells with limited action on fibroblasts, indicating considerable cancer selectivity. These agents seem to be superior to the clinically utilised cisplatin. Consequently, some selected compounds could be regarded as potential hit compounds and may be subjected to further investigations.Author contributions
Z. S. (Zsolt Szakonyi) and I. Z. conceived and designed the experiments. Z. A. K. and Z. S. (Zsuzsanna Schelz) performed the experiments, analysed the data and wrote the experimental part. Z. S. (Zsolt Szakonyi), T. M. L. and I. Z. discussed the results and contributed to the writing of the paper. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.Acknowledgements
We are grateful for financial support from the Hungarian Research Foundation (NKFI K138871). Project no. TKP2021-EGA-32 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme and also by the University of Szeged Open Access Fund (grant No. 6656).References
- E. J. Barreiro, C. A. M. Fraga and L. M. Lima, in Plant Bioactives and Drug Discovery, ed. V. Cechinel-Filho, Wiley, 1st edn, 2012, pp. 81–126 Search PubMed.
- J. Khazir, B. A. Mir, S. A. Mir and D. Cowan, J. Asian Nat. Prod. Res., 2013, 15, 764–788 CrossRef CAS.
- T. S. Ibrahim, P. Khongorzul, M. Muyaba and R. N. Alolga, Front. Pharmacol., 2023, 14, 1227574 CrossRef CAS PubMed.
- P. S. Riehl, Y. C. DePorre, A. M. Armaly, E. J. Groso and C. S. Schindler, Tetrahedron, 2015, 71, 6629–6650 CrossRef CAS.
- E. Kataev, R. N. Khaybullin, R. R. Sharipova and I. Yu. Strobykina, Rev. J. Chem., 2011, 1, 93–160 CrossRef.
- H.-D. Sun, S.-X. Huang and Q.-B. Han, Nat. Prod. Rep., 2006, 23, 673 RSC.
- E. Fujita, Y. Nagao, K. Kaneko, S. Nakazawa and H. Kuroda, Chem. Pharm. Bull., 1976, 24, 2118–2127 CrossRef CAS PubMed.
- Y. Yang, H. Sun, Y. Zhou, S. Ji and M. Li, Nat. Prod. Res., 2009, 23, 1007–1012 CrossRef CAS PubMed.
- B. A. Owona and H. J. Schluesener, Drugs RD, 2015, 15, 233–244 CrossRef CAS PubMed.
- J. Huang, L. Wu, S. Tashiro, S. Onodera and T. Ikejima, J. Pharmacol. Sci., 2008, 107, 370–379 CrossRef CAS PubMed.
- J. Qin, J. Tang, L. Jiao, J. Ji, W.-D. Chen, G.-K. Feng, Y.-H. Gao, X.-F. Zhu and R. Deng, Life Sci., 2013, 93, 655–663 CrossRef CAS PubMed.
- R. W. Huigens Iii, K. C. Morrison, R. W. Hicklin, T. A. Flood Jr, M. F. Richter and P. J. Hergenrother, Nat. Chem., 2013, 5, 195–202 CrossRef PubMed.
- J. R. Hanson, J. Chem. Res., 2018, 42, 285–290 CrossRef CAS.
- J. Chen, Z. Sun, Y. Zhang, X. Zeng, C. Qing, J. Liu, L. Li and H. Zhang, Bioorg. Med. Chem. Lett., 2009, 19, 5496–5499 CrossRef CAS PubMed.
- M.-J. Wu, D.-M. Wu, J.-B. Chen, J.-F. Zhao, L. Gong, Y.-X. Gong, Y. Li, X.-D. Yang and H. Zhang, Bioorg. Med. Chem. Lett., 2018, 28, 2543–2549 CrossRef CAS PubMed.
- S. Zhu, F. Luo and P.-H. Sun, Med. Chem. Res., 2020, 29, 1341–1354 CrossRef CAS.
- B. Goel, E. Chatterjee, B. Dey, N. Tripathi, N. Bhardwaj, A. Khattri, S. K. Guru and S. K. Jain, ACS Omega, 2021, 6, 8253–8260 CrossRef CAS PubMed.
- S. M. Lait, D. A. Rankic and B. A. Keay, Chem. Rev., 2007, 107, 767–796 CrossRef CAS PubMed.
- D. Ozsvár, V. Nagy, I. Zupkó and Z. Szakonyi, Int. J. Mol. Sci., 2021, 22, 11232 CrossRef PubMed.
- M. Raji, T. M. Le, T. Huynh, A. Szekeres, V. Nagy, I. Zupkó and Z. Szakonyi, Chem. Biodiversity, 2022, 19, e202200077 CrossRef CAS PubMed.
- F. Z. Bamou, T. M. Le, B. A. Tayeb, S. A. S. Tahaei, R. Minorics, I. Zupkó and Z. Szakonyi, ChemistryOpen, 2022, 11, e202200169 CrossRef CAS PubMed.
- Z. A. Khdar, T. M. Le, Z. Schelz, I. Zupkó and Z. Szakonyi, Int. J. Mol. Sci., 2022, 23, 10366 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- K. Schreiber, G. Schneider and G. Sembdner, Tetrahedron, 1968, 24, 73–78 CrossRef CAS.
- Y. Fujimoto, Y. Kanzawa, Y. Ikuina, K. Kakinuma and N. Ikekawa, J. Chem. Soc., Chem. Commun., 1989, 1107 RSC.
- W. E. Rosen and M. J. Green, J. Org. Chem., 1963, 28, 2797–2804 CrossRef CAS.
- S. Patil, S. D. Jadhav, M. B. Deshmukh and U. P. Patil, Int. J. Org. Chem., 2012, 02, 166–171 CrossRef.
- S. Sezer, Y. Gümrükçü, İ. Bakırcı, M. Yağız Ünver and C. Tanyeli, Tetrahedron: Asymmetry, 2012, 23, 662–669 CrossRef CAS.
- D. E. Ward and C. K. Rhee, Can. J. Chem., 1989, 67, 1206–1211 CrossRef CAS.
- A. Causero, C. Troll and B. Rieger, Ind. Eng. Chem. Res., 2020, 59, 15464–15477 CrossRef CAS.
- R. Pöhler, J. H. Krahn, J. van den Boom, G. Dobrynin, F. Kaschani, H. Eggenweiler, F. T. Zenke, M. Kaiser and H. Meyer, Angew. Chem., Int. Ed., 2018, 57, 1576–1580 CrossRef PubMed.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- D. Bai, Z. Schelz, D. Erdős, A. K. Kis, V. Nagy, I. Zupkó, G. T. Balogh and Z. Szakonyi, Int. J. Mol. Sci., 2023, 24, 1121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00665d |
| This journal is © The Royal Society of Chemistry 2024 |