
Cytochrome bd oxidase: an emerging anti-tubercular drug target

Abstract
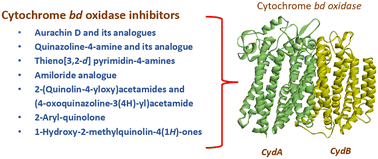
Cytochrome bd (cyt-bd) oxidase, one of the two terminal oxidases in the Mycobacterium tuberculosis (Mtb) oxidative phosphorylation pathway, plays an indispensable role in maintaining the functionality of the metabolic pathway under stressful conditions. However, the absence of this oxidase in eukaryotic cells allows researchers to select it as a potential drug target for the synthesis of anti-tubercular (anti-TB) molecules. Cyt-bd inhibitors have often been combined with cytochrome bcc/aa3 super-complex inhibitors in anti-TB drug regimens to achieve a desired bactericidal response. The functional redundancy between both the terminal oxidases is responsible for this. The cryo-EM structure of cyt-bd oxidase from Mtb (PDB ID: 7NKZ) further accelerated the research to identify its inhibitor. Herein, we have summarized the reported anti-TB cyt-bd inhibitors, insight into the rationale behind targeting cyt-bd oxidase, and an outline of the architecture of Mtb cyt-bd oxidase.
 Please wait while we load your content...
Please wait while we load your content...

