
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Latest developments in coumarin-based anticancer agents: mechanism of action and structure–activity relationship studies
Manankar
Koley
 a,
Jianlin
Han
b,
Vadim A.
Soloshonok
cd,
Subhajit
Mojumder
a,
Ramin
Javahershenas
e and
Ata
Makarem
*f
a,
Jianlin
Han
b,
Vadim A.
Soloshonok
cd,
Subhajit
Mojumder
a,
Ramin
Javahershenas
e and
Ata
Makarem
*f
aCSIR-Central Glass & Ceramic Research Institute, Kolkata, India
bCollege of Chemical Engineering, Nanjing Forestry University, Nanjing, China
cDepartment of Organic Chemistry I, University of the Basque Country, San Sebastián, Spain
dIKERBASQUE, Basque Foundation for Science, Bilbao, Spain
eDepartment of Organic Chemistry, Faculty of Chemistry, Urmia University, Urmia, Iran
fInstitute of Pharmacy, University of Hamburg, Hamburg, Germany. E-mail: ata.makarem@uni-hamburg.de
First published on 20th October 2023
Abstract
Many researchers around the world are working on the development of novel anticancer drugs with different mechanisms of action. In this case, coumarin is a highly promising pharmacophore for the development of novel anticancer drugs. Besides, the hybridization of this moiety with other anticancer pharmacophores has emerged as a potent breakthrough in the treatment of cancer to decrease its side effects and increase its efficiency. This review aims to provide a comprehensive overview of the recent development of coumarin derivatives and their application as novel anticancer drugs. Herein, we highlight and describe the largest number of research works reported in this field from 2015 to August 2023, along with their mechanisms of action and structure–activity relationship studies, making this review different from the other review articles published on this topic to date.
 Manankar Koley | Manankar Koley earned his B.Sc. Degree in Chemistry from Vidyasagar College, University of Calcutta, India in 2020, and his M.Sc. Degree in Organic Chemistry from the University College of Science, Technology, and Agriculture, University of Calcutta, India in 2022. After completing his M.Sc., he worked as a Project Trainee in the Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, India. He is now working as a project student in the Functional Materials & Devices Division, CSIR- Central Glass & Ceramic Research Institute, Kolkata, India. His research interests include catalysis, multistep organic synthesis, medicinal chemistry, and materials sciences. |
 Jianlin Han | Jianlin Han received his Ph.D. in Organic Chemistry in 2007 from Nanjing University. Then he carried out postdoctoral studies for one year at Texas Tech University. In 2008, he moved to the University of Oklahoma to continue postdoctoral research for nearly one year. In 2009 he took the position of Associate Professor at Nanjing University. In 2019, he moved to Nanjing Forestry University and became a Professor there. His research topics include organic fluorine chemistry, radical reactions, and asymmetric synthesis. |
 Vadim Soloshonok | Vadim A. Soloshonok graduated from Kiev State University in 1983 and received his Ph.D. in 1987 from the Ukrainian Academy of Sciences. He is an Ikerbasque research professor at the University of Basque Country, San Sebastian, Spain. He is currently serving as a member of the advisory editorial board of the Journal of Fluorine Chemistry, Editor-in-Chief of Organic Section of the Molecules (MDPI), Past-Chair of the ACS Fluorine Division and author of 360+ research papers. His current research interests are fluorine chemistry, tailor-made amino acids, asymmetric synthesis, and self-disproportionation of enantiomers. |
 Subhajit Mojumder | Subhajit Mojumder has received his M.Sc. Degree in Physics from the Indian Institute of Engineering Science and Technology, Shibpur, India in 2018. Presently he is working as a Junior Research Fellow at Functional Materials and Devices Division, CSIR-Central Glass and Ceramic Research Institute, Kolkata, India. His research interest is focused on nanomaterials and nanotechnology, especially metal oxide gas and humidity sensors, sensor fabrication, chemical synthesis of oxide nanoparticles, and characterization of nanomaterials. |
 Ramin Javahershenas | Ramin Javahershenas was born in Urmia, Iran, in 1971. He received his B.Sc. Degree in Applied Chemistry from Tabriz University, Tabriz, Iran, in 1993, his M.Sc. Degree in Organic Chemistry from Urmia University, Urmia, Iran, under the supervision of Professor Naser Ardabilchi in 1999, and his Ph.D. in Organic Chemistry from Urmia University, Urmia, Iran under the supervision of Professor Jabbar Khalafy, in 2017. His research interests include organic synthesis, heterocyclic synthesis, asymmetric synthesis, natural products synthesis, synthetic methodology, and applications of various catalysts in MCRs. |
 Ata Makarem | Ata Makarem was born in 1987 in Tehran. He studied chemistry in Iran and received his Master's Degree in Organic Chemistry in 2011. He pursued his academic education in Germany, and in 2015 completed his PhD in the field of organic synthesis and coordination chemistry at the University of Heidelberg under the supervision of Professor Bernd Straub. Then, Makarem worked as a Postdoctoral Researcher at the German Cancer Research Center (Heidelberg), where he was later, in 2019, promoted to Principal Scientist and established his independent research in the field of bio-organometallic chemistry. Finally, in 2020, he joined the University of Hamburg (Department of Chemistry) as a Principal Investigator. His current research focus is on bio-organometallic systems for cancer theragnostic. |
1. Introduction
Cancer can occur in any organ or tissue of the body, and the two properties that make cancer cells particularly dangerous are that they can abnormally divide and colonize regions normally reserved for normal cells. According to recent WHO reports,1 in 2020, 10 million deaths were caused by cancer. In 2020, the most common (in terms of new cases of cancer) was breast cancer with 2.26 million new cases and the most common cause of death due to cancer was lung cancer with 1.80 million deaths. Besides, according to the American Cancer Society, an estimated 1.9 million new cancer cases will be diagnosed and 609![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 360 deaths will occur in the United States alone.2 Cancers of female breasts (2.26 million), lung (2.21 million), and colon and rectum (1.93 million) were the top three cancer types in terms of incidence in 2020, while in terms of mortality, lung (1.80 million), colon and rectum (916000), and liver (830000) cancers topped the list. Therefore, it remains a challenge for medicinal chemists to develop new strategies for preparing novel anticancer drugs to fight this fatal disease.
360 deaths will occur in the United States alone.2 Cancers of female breasts (2.26 million), lung (2.21 million), and colon and rectum (1.93 million) were the top three cancer types in terms of incidence in 2020, while in terms of mortality, lung (1.80 million), colon and rectum (916000), and liver (830000) cancers topped the list. Therefore, it remains a challenge for medicinal chemists to develop new strategies for preparing novel anticancer drugs to fight this fatal disease.
Around 80% of the approved anticancer drugs are natural products.3 Coumarin (2H-1-benzopyran-2-one, Fig. 1) and its derivatives possess a wide variety of pharmacological properties, such as anti-inflammatory,4,5 antibacterial,6–9 antiviral,10 antioxidant,11–13 antirhombotic,14 anti-Alzheimer,15,16 and anticancer17,18 activities. Accordingly, many researchers have thoroughly investigated the various mechanisms of action of different classes of coumarin-based anticancer agents, such as alkylating agents, angiogenesis inhibitors, kinase inhibitors, topoisomerase inhibitors, telomerase inhibitors, antimitotic activity, human carbonic anhydrase inhibitors, aromatase inhibitors, monocarboxylate transporter inhibitors, and hormonal antagonists.
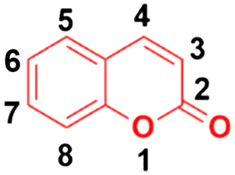 | ||
| Fig. 1 Chemical structure of coumarin. | ||
In the search for novel anticancer drugs, natural products are always a major source. Many coumarin derivatives derived from natural sources show potential in vitro and in vivo anticancer activity.19–22 Moreover, the concept of molecular hybridization, where two or more potential pharmacophores are combined into a single molecular framework, may provide fruitful results in the treatment with the coumarin moiety given that it may lead to the generation of new anticancer drugs with low toxicity, improved specificity, and enhanced effectivity.23
In this review, we aim to provide an overview of the recent developments of coumarin-derived hybrids as potentially important anticancer drugs, focusing on their structure–activity relationship and mechanisms of action and highlighting articles published between 2015 and August 2023. In contrast to other review articles published on this topic, our article presents the greatest number of examples reported in this field during the past 8 years. Although the recently published reviews presented an interesting overview regarding the progress of coumarin-based anticancer agents in the past,24,25 they only discussed a selected number of coumarin hybrids, not describing this topic as extensively as done herein. However, in this context, recently Cardona-Galeano and co-workers provided an interesting bibliometric analysis of coumarin hybrids.26 In another review article, Al-Warhi et al. discussed a group of coumarin anticancer hybrids based on their mechanism of action.27
2. Coumarin hybrids
2.1. Coumarin-artemisinin hybrids
Artemisinin (Fig. 2), having an endoperoxide-bridged sesquiterpene lactone architecture, was discovered in 1972 by Tu Youyou. It is extracted from the plant Artemisia annua, a herb used in traditional Chinese medicine and has been widely used in the treatment of malaria, which is caused by to Plasmodium falciparum. Furthermore, artemisinin also exhibits potent activity against cancer in vivo.28,29 The activity of artemisinin and its derivatives is attributed to the presence of the endoperoxide 1,2,4-trioxane ring, which can cause oxidative stress and damage to cancer cells.30 Hence, the hybridization of coumarin with artemisinin is a useful strategy to design anticancer drugs with enhanced effectiveness. | ||
| Fig. 2 Chemical structure of artemisinin. | ||
The coumarin-artemisinin hybrid 1a (Fig. 3) showed potential activities against four cancer cell lines, i.e., HepG2 (IC50 = 3.05 ± 1.60 μM), Hep3B (IC50 = 3.76 ± 1.76 μM), A2780 (IC50 = 5.82 ± 2.28 μM), and OVCAR-3 (IC50 = 4.60 ± 1.81 μM). It was also reported that hybrid 1a is more potent than hybrids 1b and c, which has a linker between the piperazinyl and carbonyl groups. Fluorescence images revealed that the hybrids localized mainly in the mitochondria and their enhanced potency is due to their strong ability to accumulate in the mitochondria, which enhances the intracellular reactive oxygen species (ROS) level and triggers cell death.31
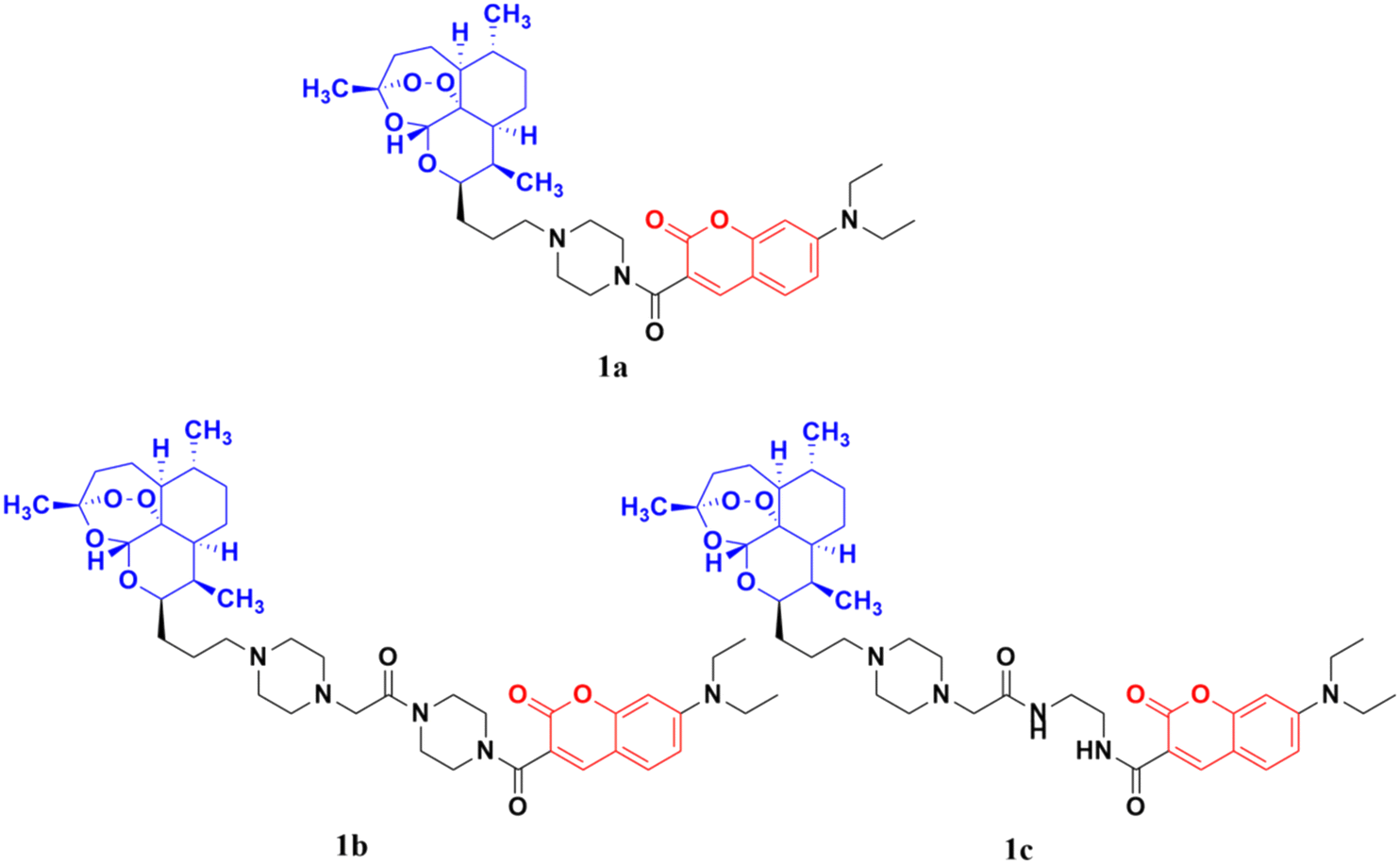 | ||
| Fig. 3 Chemical structures of coumarin-artemisinin hybrid 1a–c. | ||
Four series of twenty coumarin-artemisinin hybrids were developed using click chemistry, which showed efficient activity (IC50 = 0.05–125.40 μM) when assessed (MTT assay) under normoxic or anoxic conditions against three cancer cell lines (HCT-116, MDA-MB-231, and HT-29).32,33 It is worth noting that click chemistry has a wide application in medicinal chemistry.34–37 The structure–activity relationship (SAR) studies showed that the 3-chloro and 4-methyl substituents in the coumarin moiety exhibited greater activity, whereas the 3-ethoxycarbonyl group in the coumarin ring exhibited reduced effectivity. It was also found that all these hybrids exhibited greater activity against the HT-29 cancer cell line under anoxic conditions. The first series of compounds (2a–e) (Fig. 4) exhibited IC50 values in the range of 0.05–91.21 μM, while the hybrids in the second (3a–e), third (4a–e), and fourth (5a–e) series showed IC50 values in the range of 1.22–120.72 μM, 2.46–125.40 μM, and 0.43 > 100 μM, respectively. It was examined that the cytotoxic activities of most of these targeted compounds are 1–10-fold greater under anoxic conditions than that under normoxic conditions.
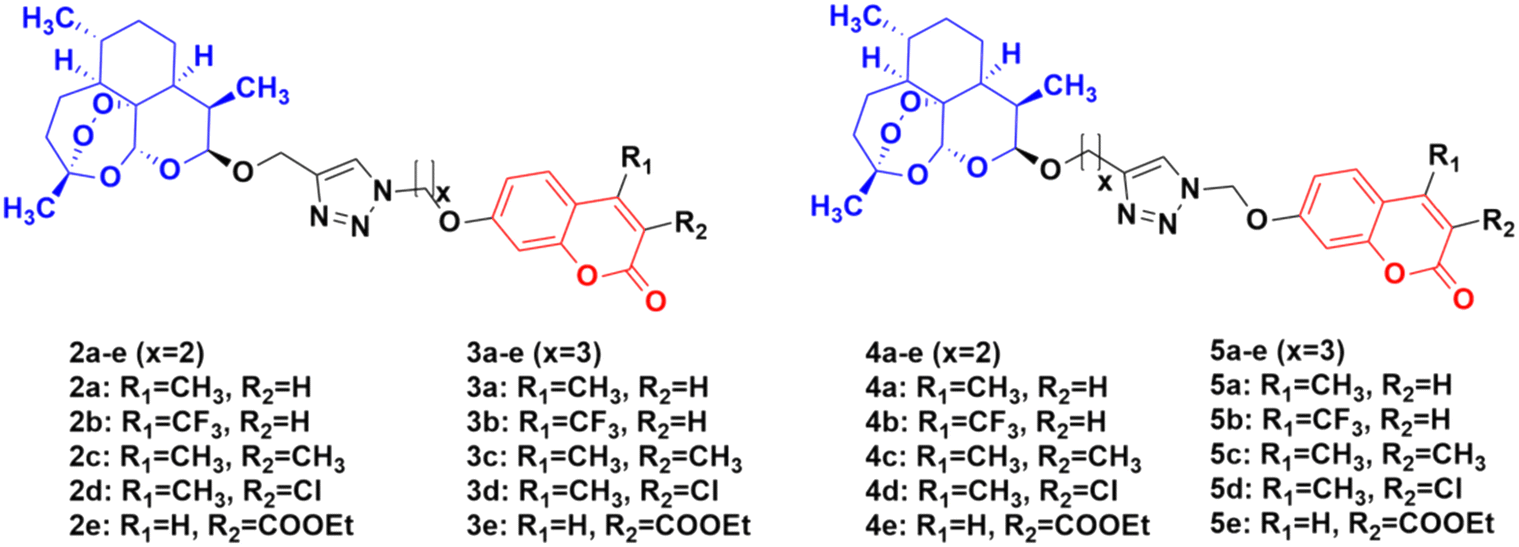 | ||
| Fig. 4 Chemical structures of coumarin-artemisinin hybrids 2a–e, 3a–e, 4a–e and 5a–e. | ||
The potential activity of the hybrids when the linker is moved from the C-7 position to the C-4 position of the coumarin moiety was also examined and compared to doxorubicin (DOX) and DHA.38 A total of thirty novel hybrids were designed and developed and their cytotoxicity against four cancer cell lines (HCT-116, MDA-MB- 231, HT-29, and A-549) was investigated. It was proven that the series of compounds 10 (IC50 = 1.282 > 100 μM) and 11 (IC50 = 0.039–93.53 μM) showed a better cytotoxicity effect than the others, indicating that the 4-oxygen group in the coumarin ring as a part of the linker can enhance the potency (Fig. 5). The compounds in series 8 (8.57 > 100 μM) and 9 (9.33 > 100 μM) also showed significant activity against the HT-29 cancer cell line. In contrast, the cytotoxicity effect of compounds in series 6 and 7 was not very promising.
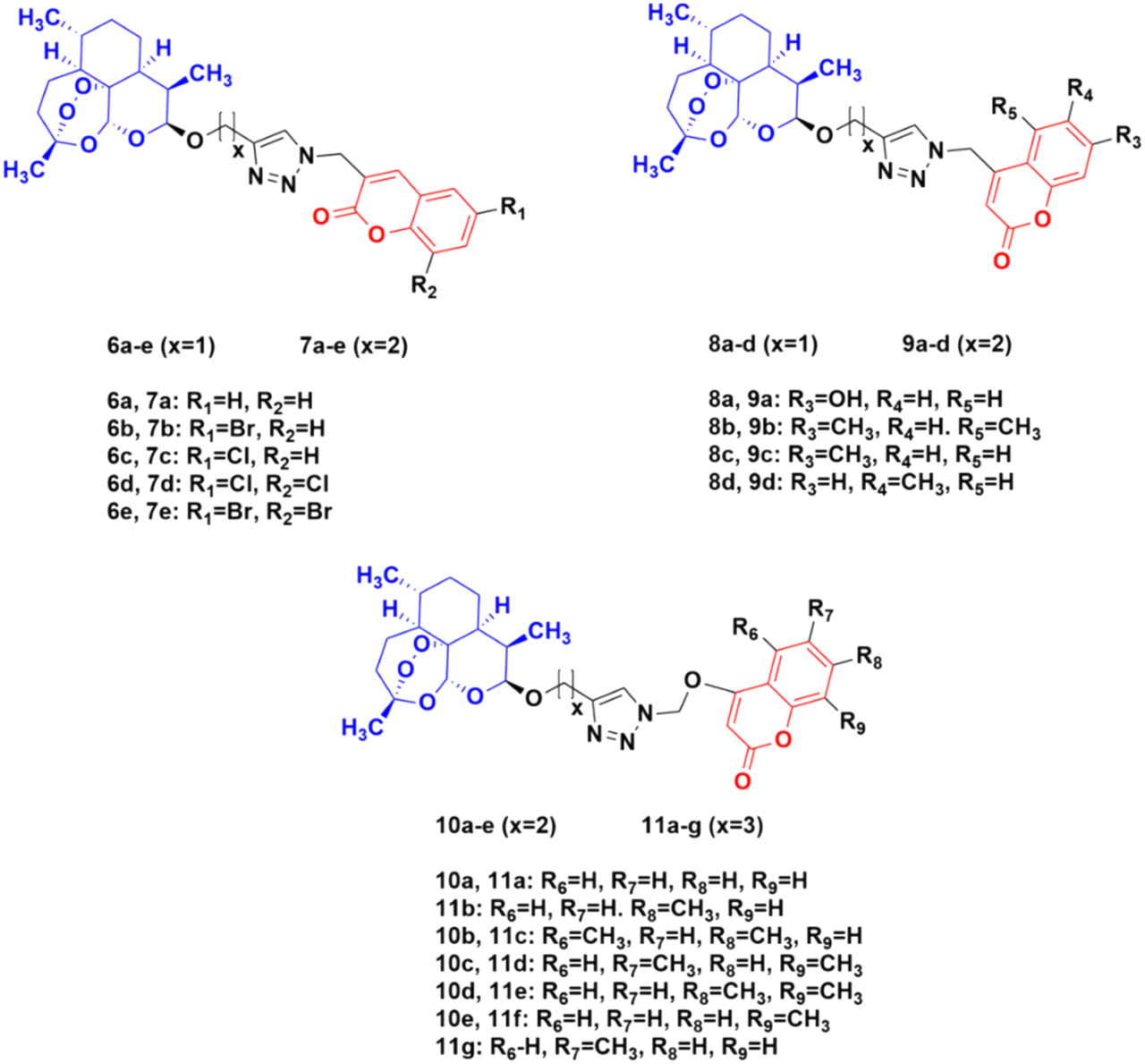 | ||
| Fig. 5 Chemical structures of coumarin-artemisinin hybrid 6a–e, 7a–e, 8a–d, 9a–d, 10a–e and 11a–g. | ||
These hybrids inhibited the proliferation of HT-29 cells, arrested their G0/G1 phase, reduced the migration of tumor cells, and induced both apoptosis and ferroptosis in the HT-29 cancer cell line.
2.2. Coumarin-azole hybrids
Azoles are considered one of the most important heterocycles, consisting of a five-membered ring containing one nitrogen atom and at least one other non-carbon atom (i.e., nitrogen, oxygen, and sulfur). Different types of azoles (Fig. 6) possess significant biological effects39 and they have been fused with the coumarin moiety to develop novel anticancer drugs with increased anticancer properties.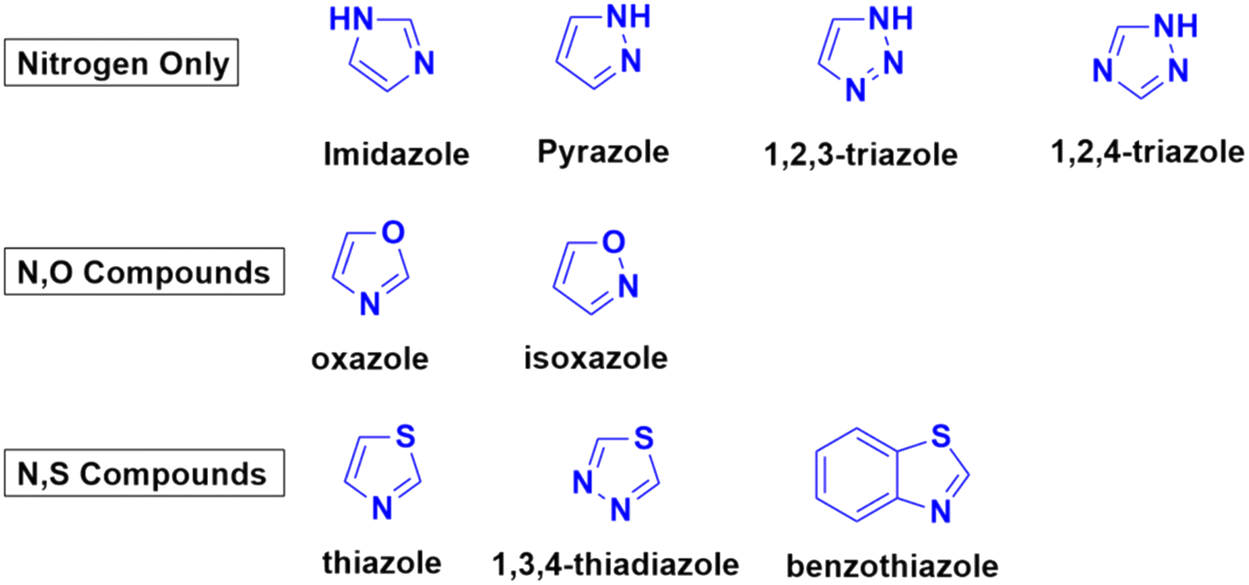 | ||
| Fig. 6 Chemical structures of some azoles possessing biological importance. | ||
Three coumarin–1,2,3-triazole hybrids (12a–c) (Fig. 7) were synthesized and examined for their potent activity against three cancer cell lines (PC3, MGC803, and HepG2).42 Among them, 12c was found to be the most effective with IC50 values of 0.34 ± 0.04 μM, 0.13 ± 0.01 μM, and 1.74 ± 0.54 μM against PC3, MGC803, and HepG2 cancer cell lines, respectively. This hybrid was found to inhibit MGC803 cell growth, induce G2/M phase arrest and apoptosis, and regulate the expression of apoptosis-related proteins.
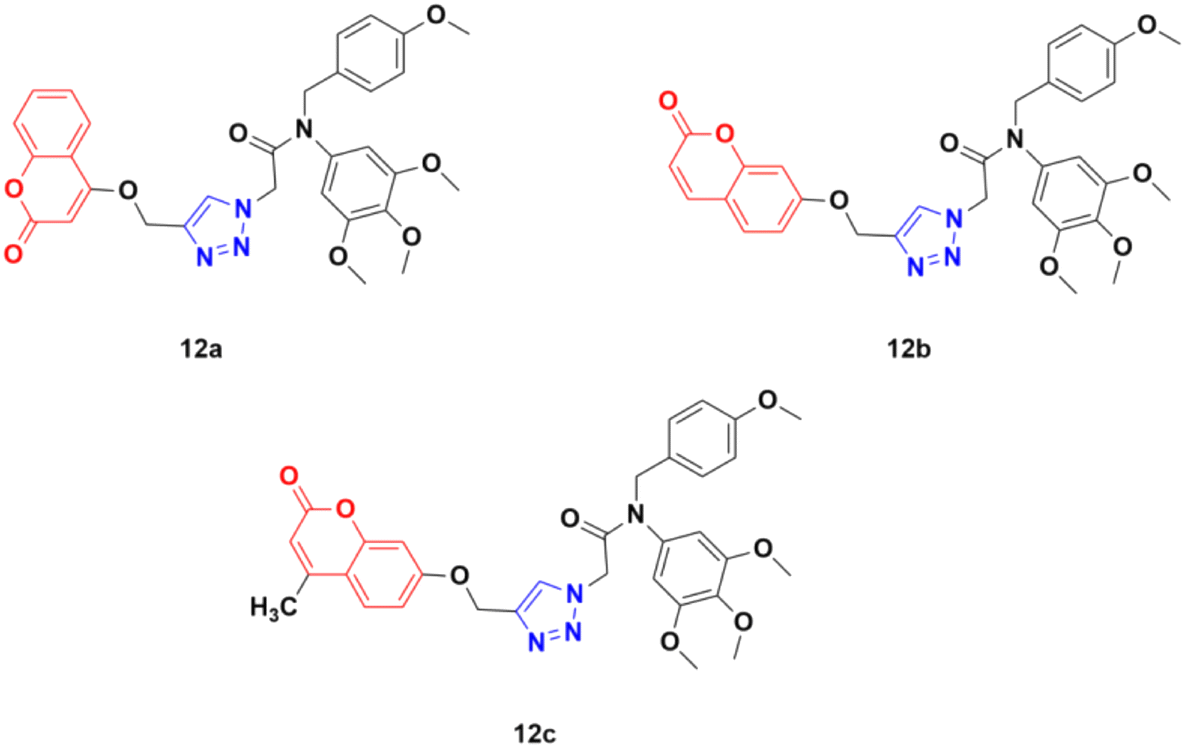 | ||
| Fig. 7 Chemical structures of coumarin–1,2,3-triazole hybrid 12a–c. | ||
Thirty-two novel coumarin–1,2,3-triazole hybrids (Fig. 8) were designed and synthesized via the eco-friendly copper(I) catalyzed Huisgen 1,3-dipolar cycloaddition under microwave irradiation and their potency was evaluated against five cancer cell lines (A549, HepG2, CFPAC-1, HeLa, and SW620).43 Among them, the hybrids containing phenylethyl (13a) and 3,5-difluorophenyl (13b) showed the maximum potent activity against the A549 cell line (IC50 = 24.78 μM and 21.06 μM, respectively). Also, 13c containing 5-iodoindole exhibited significant potency against the HepG2 cancer cell line with an IC50 value of 8.57 μM. Compound 13d was highlighted as a lead with the highest cytotoxicity against the HepG2 cell line and an IC50 value of 0.90 μM. This antiproliferative activity of 13d is due to the suppression of 5-lipoxygenase activity and perturbation of sphingolipid signaling by interfering with intracellular acid ceramidase activity. It induced cell death by early apoptosis.
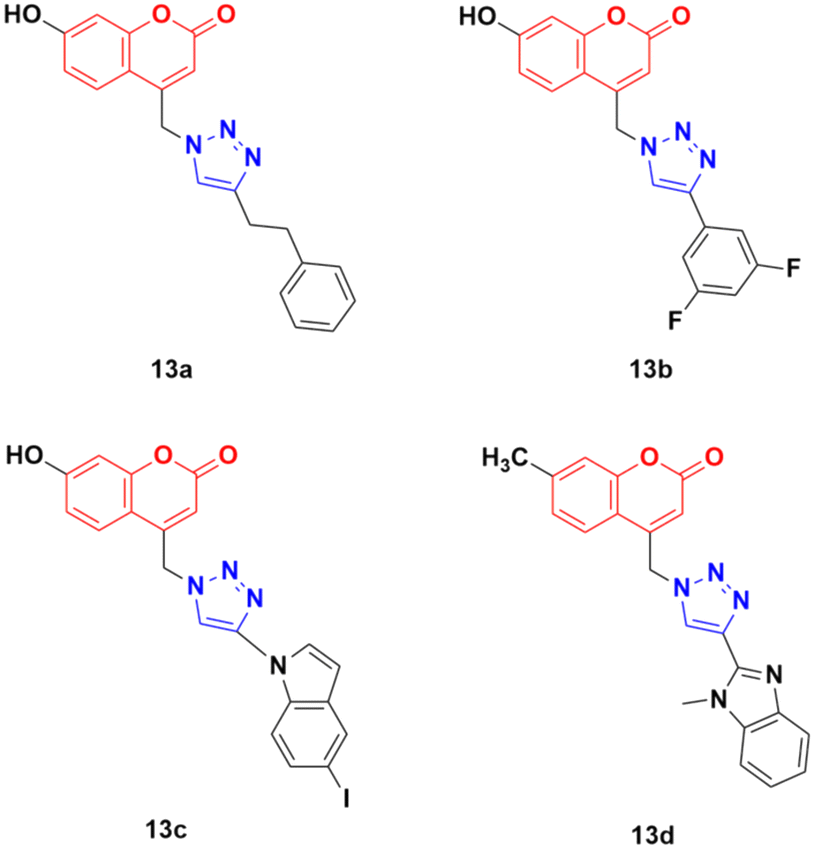 | ||
| Fig. 8 Chemical structures of coumarin–1,2,3-triazole hybrid 13a–d. | ||
A series of other coumarin–1,2,3-triazole hybrids was developed via a similar procedure to that previously mentioned and their antiproliferative activities against three cancer cell lines (HeLa, CaCo-2, and K562) and normal kidney MDCK1 cells were investigated.44 Among them, compounds 14a–d (Fig. 9) showed the most pronounced activity, although they were also found to be cytotoxic against normal MDCK1 cells. 14c possessed high activity against the K562 cell line with an IC50 of value 17.9 ± 5.0 μM, whereas 14d showed significant activity against CaCo-2 cell lines with IC50 value 9.7 ± 1.3 μM.
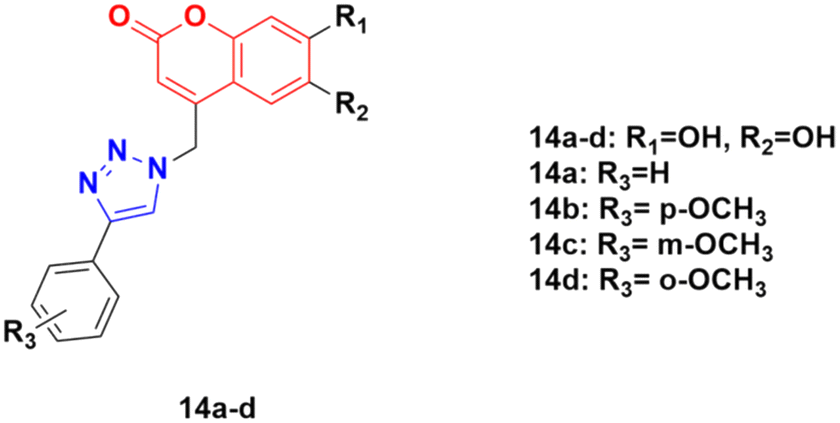 | ||
| Fig. 9 Chemical structures of coumarin–1,2,3-triazole hybrid 14a–d. | ||
Fifteen amide-containing coumarin–1,2,3-triazole hybrids were synthesized and tested in vitro for their anticancer activity against the MDA-MB-231 cancer cell line under both normoxic and hypoxic conditions.45 Among them, compound 15a (Fig. 10) showed the maximum effectivity against the MDA-MB-231 cell line both under hypoxia (IC50 = 0.03 μM) and normoxia (IC50 = 1.34 μM), and it was proven to be more potent than doxorubicin (IC50 = 0.60 μM under hypoxia and IC50 = 1.07 μM under normoxia), cis-platin (IC50 = 4.68 μM under hypoxia and IC50 = 7.87 μM under normoxia), and hydroxycoumarin (IC50 ≥ 100 μM under hypoxia and IC50 ≥ 100 μM under normoxia). Compound 15b also showed moderate activity under hypoxia with an IC50 value of 0.25 μM. Molecular docking analysis revealed that the anticancer activity of 15a is attributed to its potential to inhibit carbonic anhydrase IX.
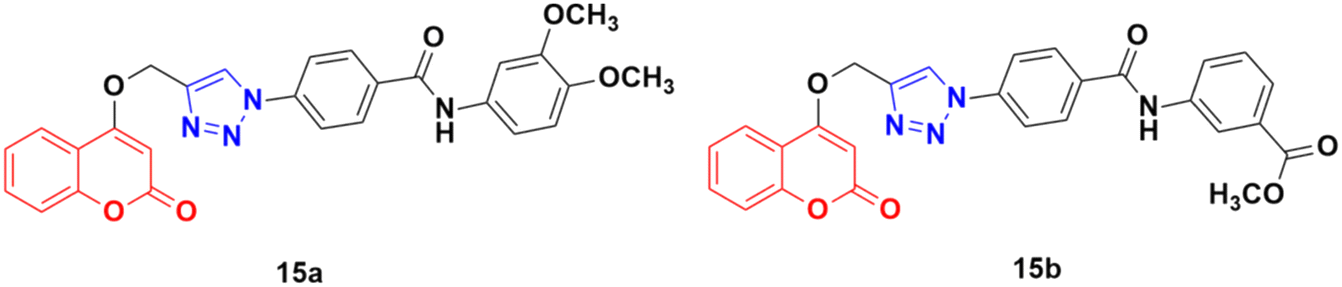 | ||
| Fig. 10 Chemical structures of coumarin–1,2,3-triazole hybrid 15a and b. | ||
A total of fifteen coumarin-tagged β-lactam 1,2,3-triazole hybrids was designed and their anticancer activities evaluated against three cancer cell lines (A549, MCF-7, and MDA-MB-231) together with the HEK-293 normal cell line.46 Among them, 16a and 16b (Fig. 11) showed prominent antiproliferative activity against the MCF-7 cell line with IC50 = 53.55 and 58.62 μM, respectively. Molecular docking studies revealed that these two compounds target estrogen receptor-α.
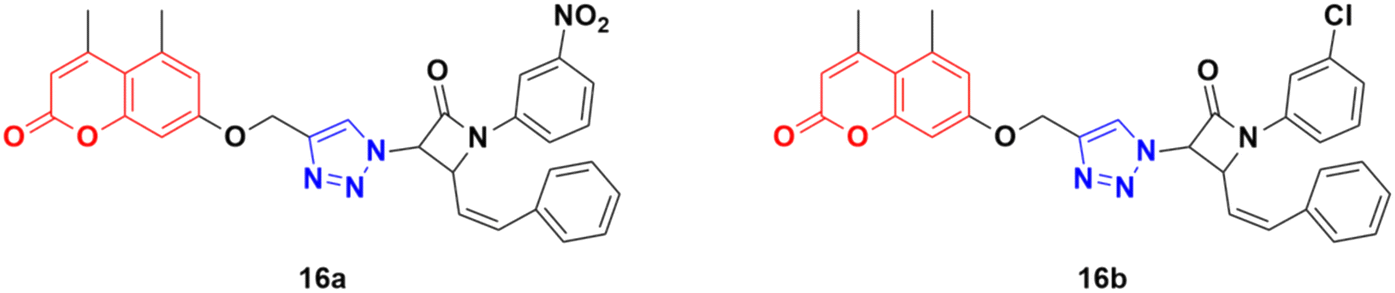 | ||
| Fig. 11 Chemical structures of coumarin–1,2,3-triazole hybrid 16a and b. | ||
Some other coumarin fused 1,2,3-triazole macrocycles were also developed and found to be active against cancer cells.47–50 The most active hybrid 17 (Fig. 12) with IC50 = 49 μM only showed moderate activity against the MCF-7 cancer cell line.
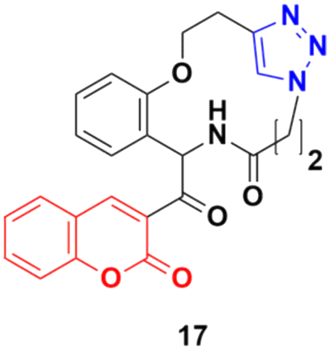 | ||
| Fig. 12 Chemical structure of coumarin–1,2,3-triazole macrocycle hybrid 17. | ||
Four coumarin–triazole hybrids were chosen and examined for their cytotoxic activity on several cancer cell lines and their in vitro toxicity was evaluated on the 3T3 (healthy fibroblasts) cell line.51 They all showed significant cytotoxic activity against the MCF7 breast cancer cell line with an IC50 value lower than that of cisplatin, while 18c (Fig. 13) was the best among them with IC50 = 2.66 μM.
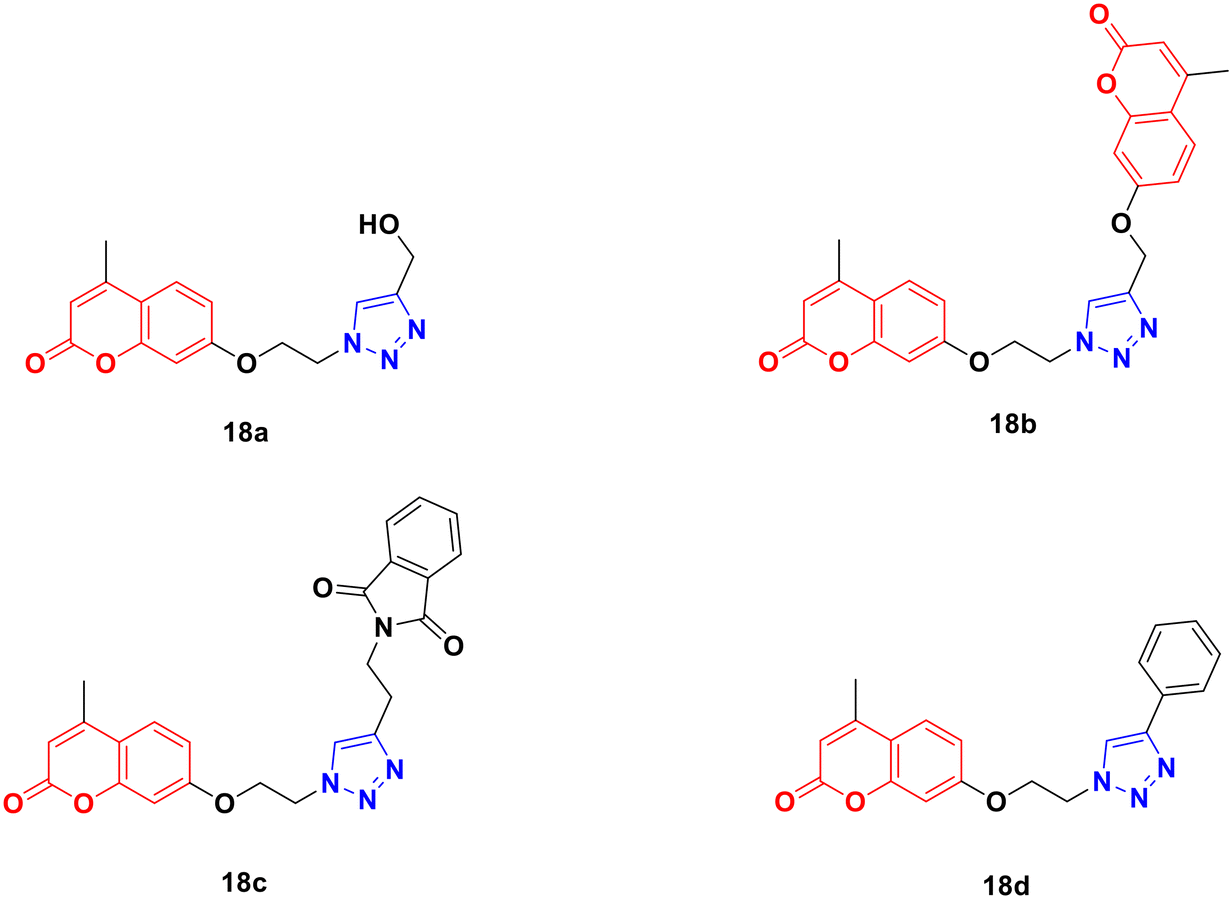 | ||
| Fig. 13 Chemical structures of coumarin–1,2,3-triazole hybrid 18a–d. | ||
A 1,2,3-triazole-tagged glycoside of 4-hydroxy coumarin base (19) (Fig. 14) was synthesized utilizing click chemistry and its cytotoxicity was tested against liver cancer cell lines.52 The IC50 value was found to be 106.81 μg mL−1.
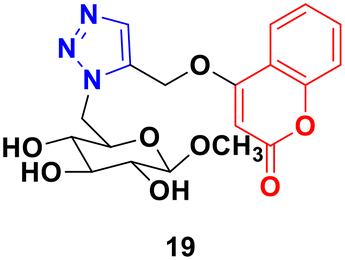 | ||
| Fig. 14 Chemical structure of coumarin–1,2,3-triazole hybrid 19. | ||
1,2,4-Triazole has been evaluated as a novel anticancer,53,54 antifungal,55 and antibacterial56 activities. This nucleus is stable to metabolic degradation and is target-specific with a broad spectrum of pharmacological activities. Furthermore, given that it is polar, it can enhance the solubility of the ligand and improve its activity. Thus, this system is a likely choice for the preparation of novel anticancer agents.
Eighteen coumarin–1,2,4-triazole hybrids were synthesized under microwave irradiation and conventional heating techniques.57 The compounds were investigated for their anticancer activities against four cancer cell lines (BT20 human breast carcinoma, SK-Me1 128 melanoma, DU-145 prostate carcinoma, and A549 lung carcinoma) and HFC normal cell line together with the evaluation of the selectivity index (SI). Among the hybrids, 20a (Fig. 15) showed the highest potency against the BT20 cell line with an IC50 value of 6.4 μg mL−1 and SI = 5.2. 20b was proven to be the most effective against the DU-145 (IC50 = 3.7 μg mL−1 and SI = 9.9) and SK-Me1 128 cell lines (IC50 = 12.3 μg mL−1 and SI = 3.0). 20c was the most effective against the A549 cancer cell line (IC50 = 7.5 μg mL−1 and SI = 4.2). The hybrids showed comparable activity to the reference cis-platin, but in general, they were not superior to the reference against the tested cancer cell lines.
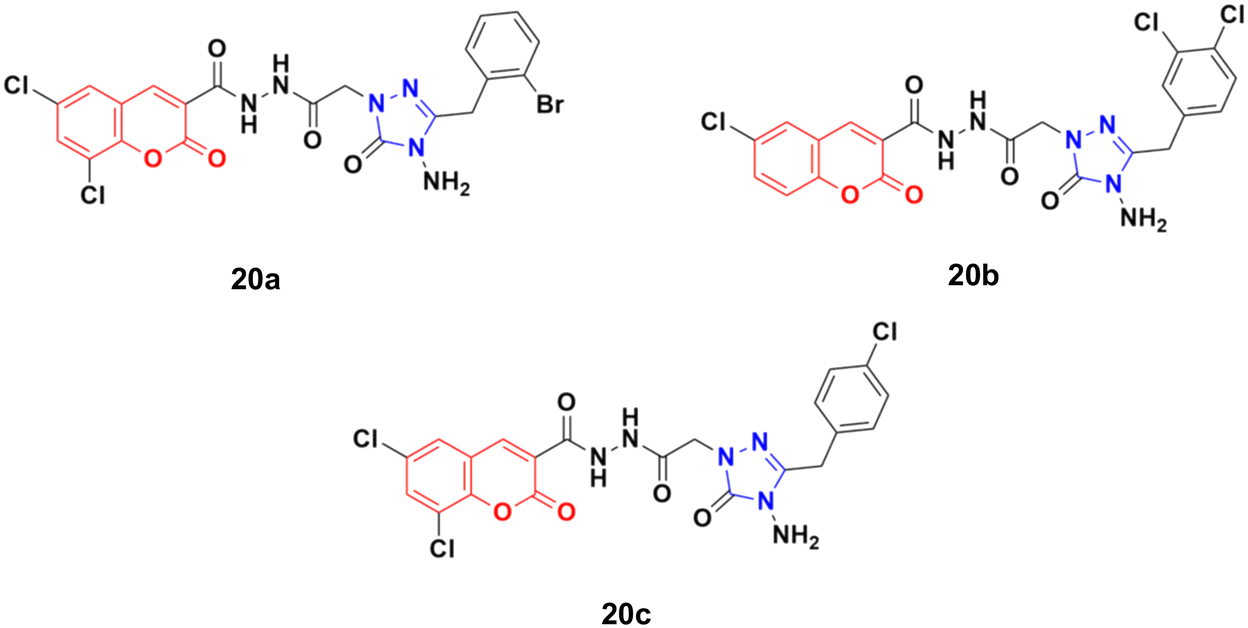 | ||
| Fig. 15 Chemical structures of coumarin–1,2,4-triazole hybrid 20a–c. | ||
 | ||
| Fig. 16 Chemical structure of imidazole. | ||
Four coumarin–benzimidazole hybrids 22a–d (Fig. 17) were screened for their anticancer activity via the evaluation of their GI50 values against fourteen cancer cell lines, including AGS, KATO-III, SNU-1 (stomach cancer), SKOV3, OVCAR-8 (ovarian cancer), BXPC-3, PANC-1 (pancreatic cancer), T24 (bladder cancer), WiDr (colon cancer), HePG2 (liver cancer), SN12C (lung cancer), K562 (leukemia), MCF-7 (breast cancer) and HeLa.59 They were all fairly potent against most of the cell lines excluding MCF-7. Compound 22a possessed maximum activity against thirteen of the fourteen cell lines (GI50 below 0.41 μmol L−1). Further investigation showed that the hybrids have potent activity in inhibiting the PI3K-AKT-mTOR pathway and inducing cancer cell apoptosis.
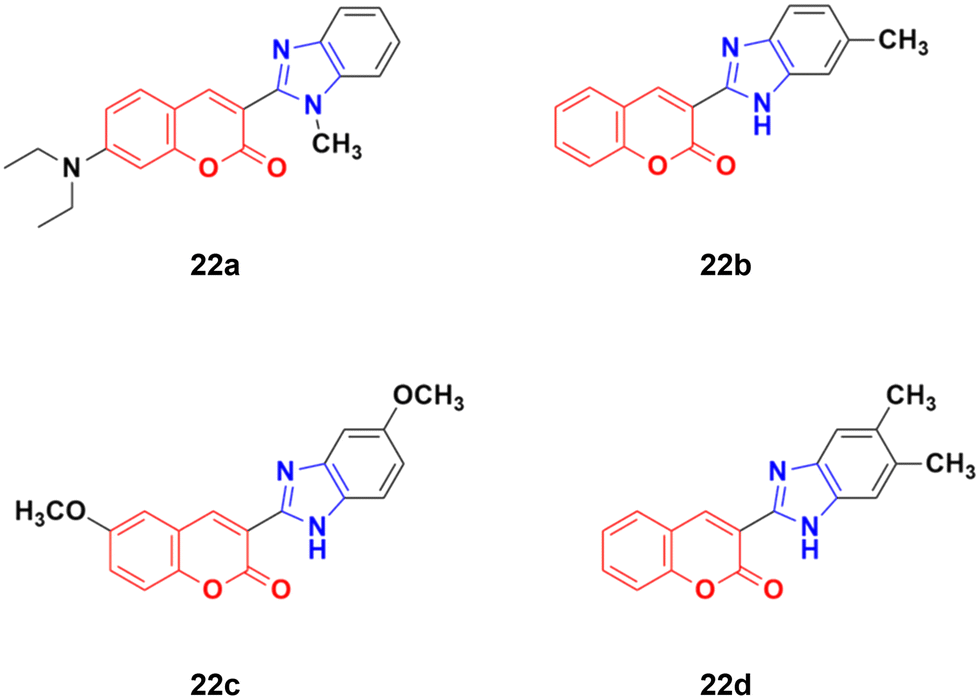 | ||
| Fig. 17 Chemical structures of coumarin–benzimidazole hybrid 22a–d. | ||
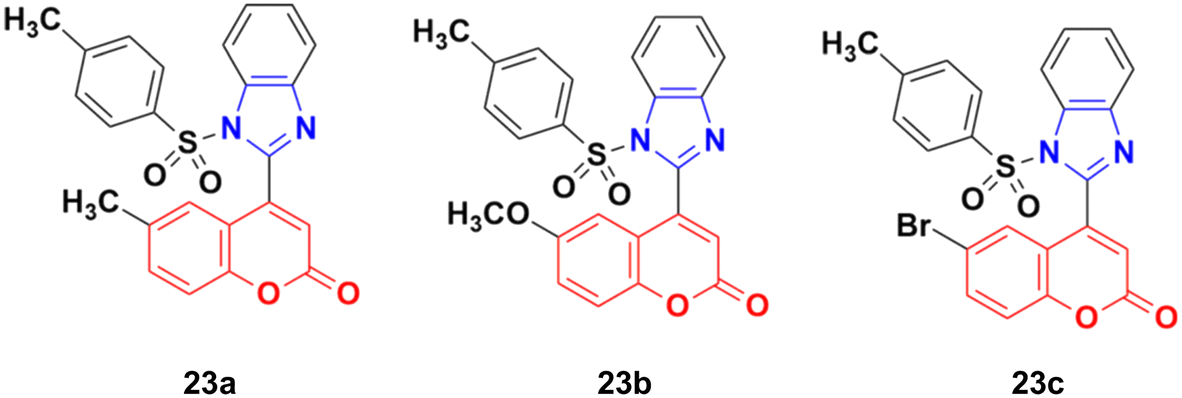
A new series of coumarin–benzimidazole hybrids was designed and their anticancer activity was investigated in HeLa and HT29 cancer cell lines.60 Among them, 23a and 23b (Fig. 18) showed good potency against the HeLa cell line with GI50 values of 36.2 and 35.3, respectively, whereas 23a and 23c possessed marked activity against the HT 29 cell line.
 | ||
| Fig. 18 Chemical structures of coumarin–benzimidazole hybrid 23a–c. | ||
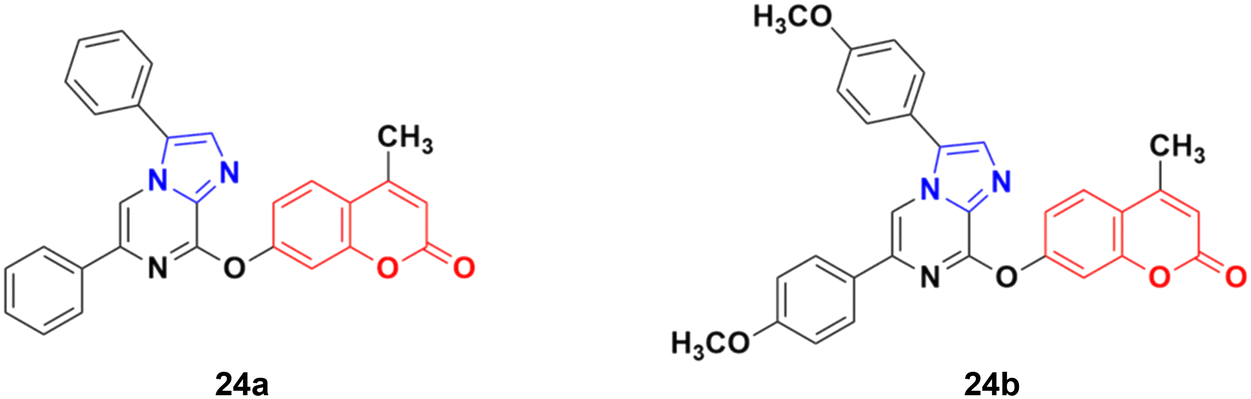
A series of imidazo[1,2-a]pyrazine–coumarin hybrids was synthesized via the Suzuki–Miyaura coupling reaction and screened for their in vitro anticancer activity against sixty human cancer cell lines.61 Among them, compounds 24a and 24b (Fig. 19) showed a broad spectrum of activity against most of the cell lines and found to be more active than 5-fluorouracil.
 | ||
| Fig. 19 Chemical structures of imidazo[1,2-a]pyrazine–coumarin hybrid 24a and b. | ||
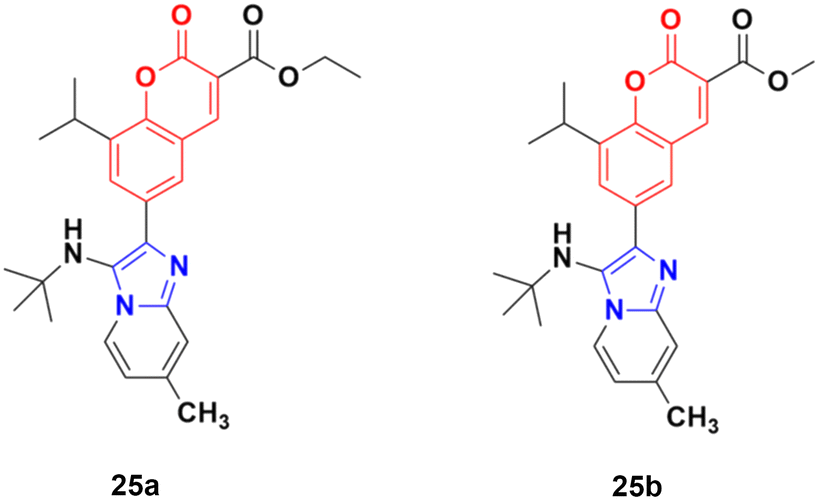
A novel series of coumarin–imidazo[1,2-a]pyridine derivatives was developed using silver(I)-catalyzed Groebke–Blackburn–Bienaymé multicomponent reaction and their antitumor activity was analyzed against three cancer cell lines (MCF-7, MDAMB-231, and Ishikawa).62 Among them, compounds 25a and 25b (Fig. 20) showed the maximum potency. Also, 25b having IC50 = 14.12 ± 3.69 μM against MDA-MB-231 not only induced apoptosis in cells but also induced cell cycle arrest at the G0|G1 phase in the cell. It is worth noting that we recently published some interesting review articles regarding multicomponent reactions.63,64
 | ||
| Fig. 20 Chemical structures of imidazo[1,2-a]pyridine–coumarin hybrid 25a and b. | ||
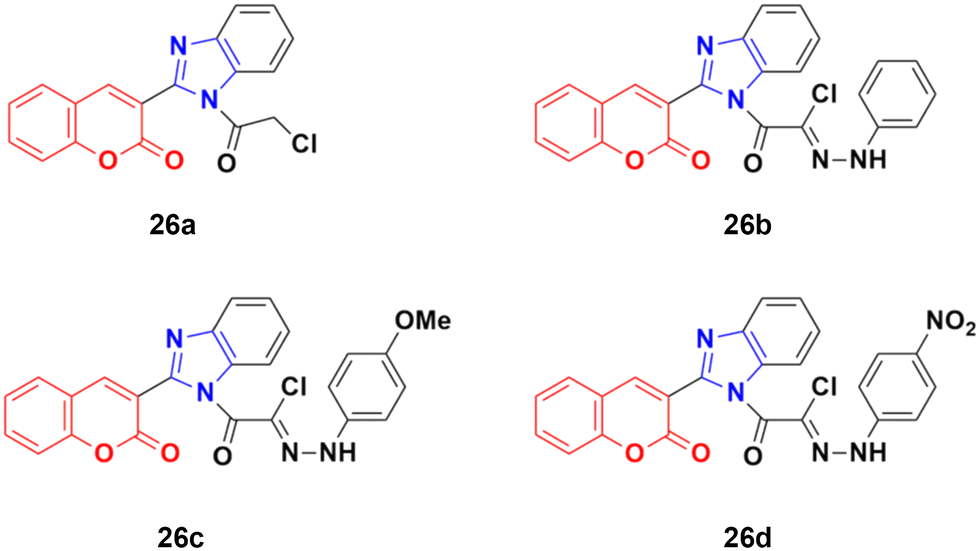
A novel series of coumarin–benzimidazole hybrids was obtained and their growth inhibitory effect (in vitro) was studied against six cancer cell lines (A549, H460, HT29, MKN-45, U87MG, and SMMC-7721) using foretinib as the standard reference.65 The studies indicated that compounds 26a–d (Fig. 21) were the most potent against the six cancer cell lines, while compound 26a was very toxic and 26b was harmful against the tested organism. Among the non-toxic compounds, compound 26d showed the highest potency against the A549 (IC50 = 0.28 ± 0.04 μM) cancer cell line.
 | ||
| Fig. 21 Chemical structures of coumarin–benzimidazole hybrid 26a–d. | ||
Another coumarin-tagged benzimidazole derivative, 2-(2-oxo-2H-chromen-3-yl)-N-phenyl-1H-benzo[d]imidazole-1-carbothioamide (27) (Fig. 22), was synthesized and its activity was investigated in three cancer cell lines (MCF-7, NCI-H460, and SF-268).66 This compound showed almost similar activity to the reference doxorubicin but also toxicity in normal cell lines.
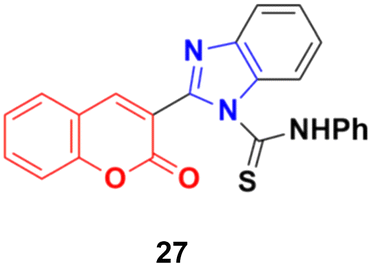 | ||
| Fig. 22 Chemical structure of coumarin–benzimidazole hybrid 27. | ||
Eight platinum(II) complexes with a coumarin–benzimidazole hybrid were synthesized and their biological activity was evaluated against several cancer cell lines (HeLa, Hep-G2, and SK-OV-3/DDP).67 Among them, compound 28 (Fig. 23) showed excellent potency against the SK-OV-3/DDP cell line with IC50 = 1.01 ± 0.27 μM and was better in comparison to the reference cis-platin. Further investigation indicated that this compound induced apoptosis in SK-OV-3/DDP cells via mitochondria dysfunction signaling pathways and was a telomerase inhibitor targeting c-myc promoter elements.
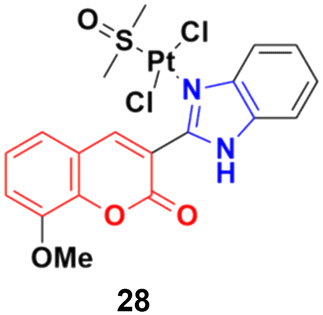 | ||
| Fig. 23 Chemical structure of platinum(II) complex-tagged coumarin–benzimidazole hybrid 28. | ||
Two other platinum(II) complexes, 29a and 29b (Fig. 24), with a coumarin–benzimidazole moiety also possessed excellent anticancer activity against SK-OV-3/DDP cell lines with IC50 values of 10.3 ± 0.3 and 0.5 ± 0.2 μM, respectively.68 Cytotoxic mechanism studies indicated that these two complexes act similarly to the previous platinum-based complex by inhibiting cell cycle progression at the G2/M phase and changing the expression of cell cycle-related proteins.
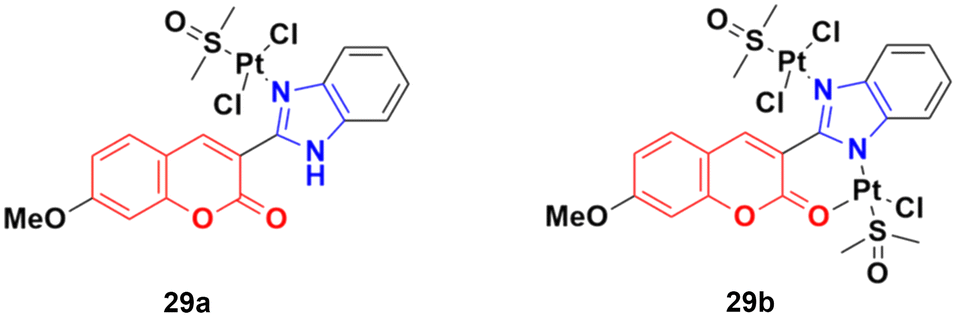 | ||
| Fig. 24 Chemical structures of platinum(II) complex-tagged coumarin–benzimidazole hybrid 29a and b. | ||
Three ruthenium(II) complexes with a coumarin–benzimidazole hybrid were designed and their antitumor efficiency was studied.69 Among them, compound 30 (Fig. 25) showed marked antitumor activity against the NCI-H460 cancer cell line (IC50 = 0.30 ± 0.02 μM) with high selectivity. MTT assay studies revealed that this complex induced apoptosis via telomerase inhibition.
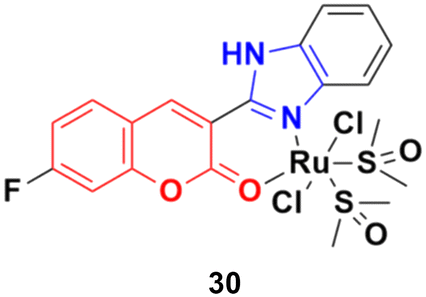 | ||
| Fig. 25 Chemical structure of ruthenium(II) complex-tagged coumarin–benzimidazole hybrid 30. | ||
A new series of novel 3-benzylcoumarin imidazolium salts was prepared together with the evaluation of their anticancer properties against five cancer cell lines (HL-60, SMMC-7721, A-549, MCF-7, and SW-480).70 Among them, compound 31a (Fig. 26) showed the highest efficiency with IC50 values in the range of 2.04–4.51 μM against five human tumor cell lines. Compound 31b was more selective to the SW-480 cell line with an IC50 value 40.0-fold lower than DDP. SAR studies indicated that compound 31a can cause G0/G1 phase cell cycle arrest and apoptosis in the SMMC-7721 cell line.
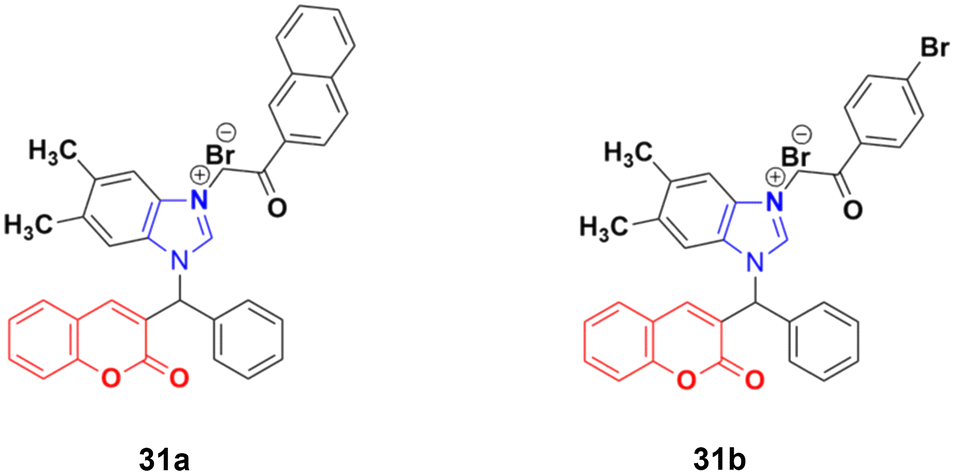 | ||
| Fig. 26 Chemical structures of 3-benzylcoumarin imidazolium salt 31a and b. | ||
A large series of coumarin-substituted benzimidazolium salts (32) (Fig. 27) was developed and their cytotoxic properties studied against PC-3 and A2780 cancer cell lines.71 All the salts showed moderate activity and were less active than docetaxel.
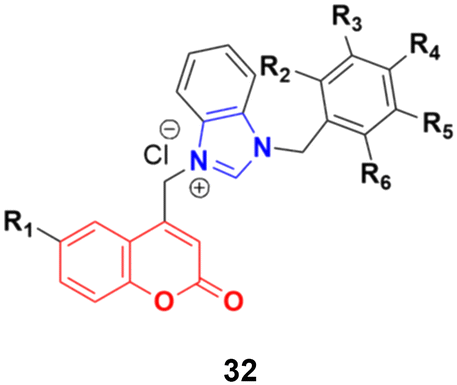 | ||
| Fig. 27 General chemical structure of a coumarin-substituted benzimidazolium salt. | ||
A novel series of sterically encumbered silver(I)-N-heterocyclic carbene complexes with coumarin–benzimidazole hybrids was designed and their cytotoxic activities studied against A549 and H1975 cancer cell lines.72 Complexes 33a–d (Fig. 28) showed promising activity against the above-mentioned cell lines, while complex 33e possessed a promising drug window with the IC50 value of 13.7 ± 2.70 and 14.5 ± 1.20 μM against the H1975 and A549 cancer cell lines, respectively.
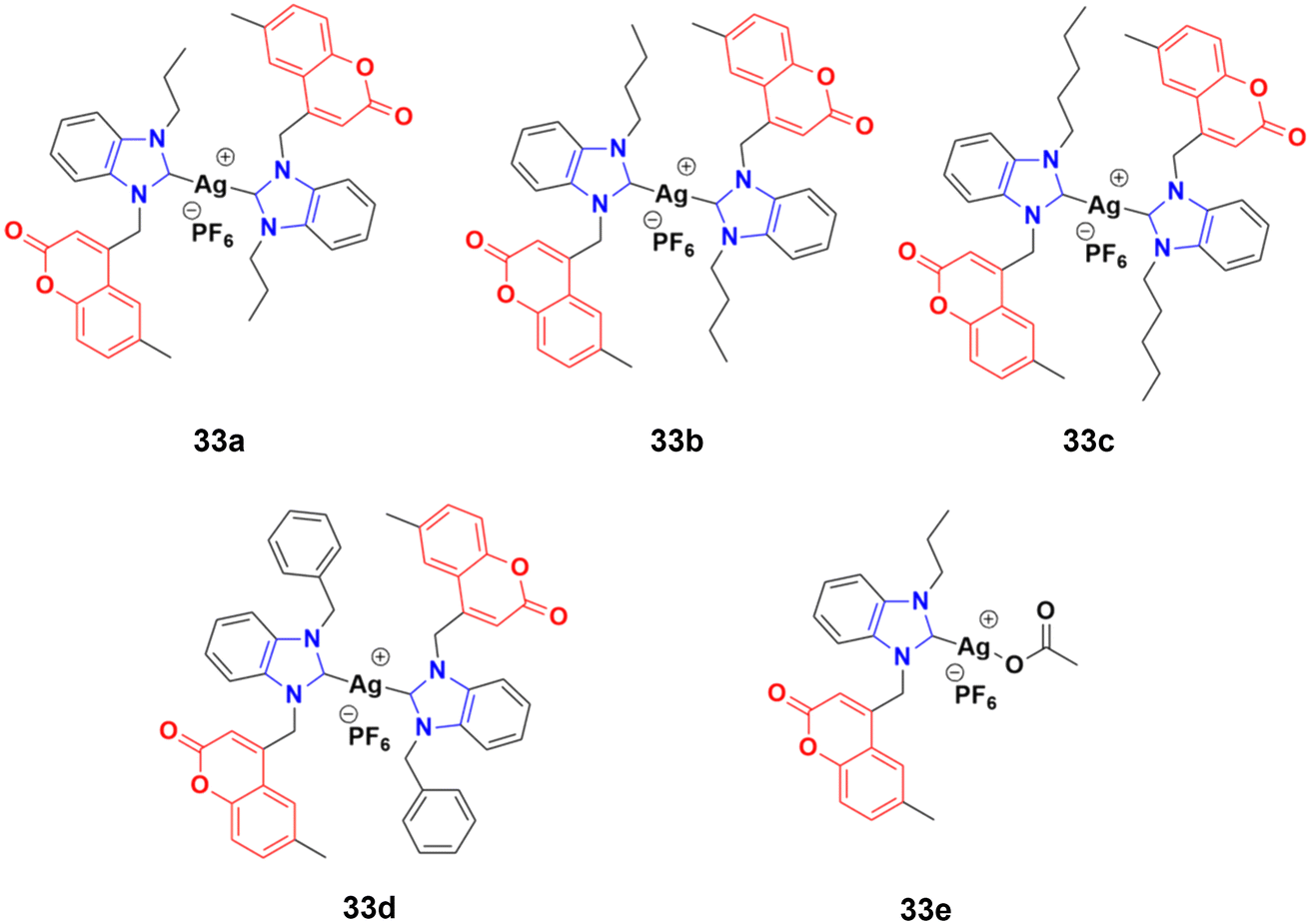 | ||
| Fig. 28 Chemical structures of silver(I)-NHC complexes with coumarin–benzimidazole hybrid 33a–e. | ||
A new series of coumarin–pyrazole hybrids (34a–d) (Fig. 29) was synthesized and their activity screened against two cancer cell lines, i.e., Hep-G2 and MCF-7.73 However, they were not very pharmacologically important given that they were all were less effective than the reference cis-platin and possessed moderate activity against the above-mentioned two cell lines.
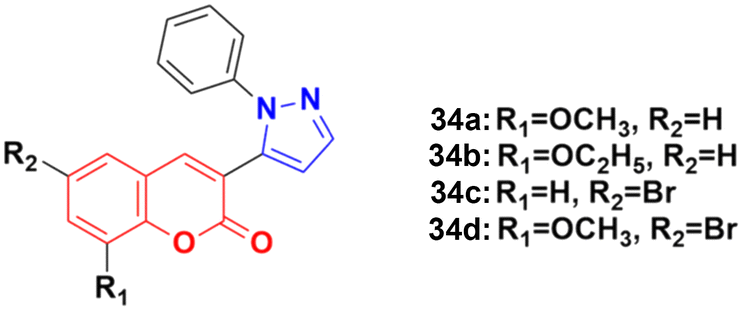 | ||
| Fig. 29 Chemical structures of coumarin–pyrazole hybrid 34a–d. | ||
A series of twenty-two coumarin–pyrazole hybrids was designed and their antiproliferative activities studied in vitro against four cancer cell lines (HepG2, SMMC-7721, U87, and H1299).74 Among them, compound 35 showed excellent anticancer activity against all the cell lines with IC50 values of 2.96 ± 0.25, 2.08 ± 0.32, 3.85 ± 0.41, and 5.36 ± 0.60 μM against the HepG2, SMMC-7721, U87, and H1299 cancer cell lines, respectively. SAR studies revealed that hybrid 35 (Fig. 30) displayed significant anti-metastasis effects by inhibiting cell migration and invasion in the highly metastatic SMMC-7721 cell line and dose-dependent reversed TGF-β1-induced epithelial-mesenchymal transition (EMT). Also, 35 showed low acute toxicity and possible tumor growth inhibitory properties against the SMMC-7721 cell line in vivo.
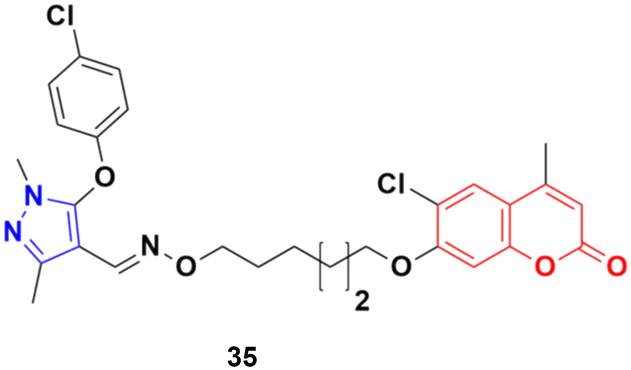 | ||
| Fig. 30 Chemical structure of coumarin–pyrazole hybrid 35. | ||
A new series of fifteen coumarin–pyrazole hybrids was synthesized via a one-pot multicomponent Vilsmeier–Haack reaction with good yield and their anticancer activities were screened against three cancer cell lines, i.e., DU-145, MCF-7, and HeLa.75 All the derivatives exhibited appreciable cytotoxic activity but were not better than the reference doxorubicin. Compounds 36a and 36b (Fig. 31) showed good activity against the HeLa cell line with IC50 values of 5.75 and 6.25 μM, respectively.
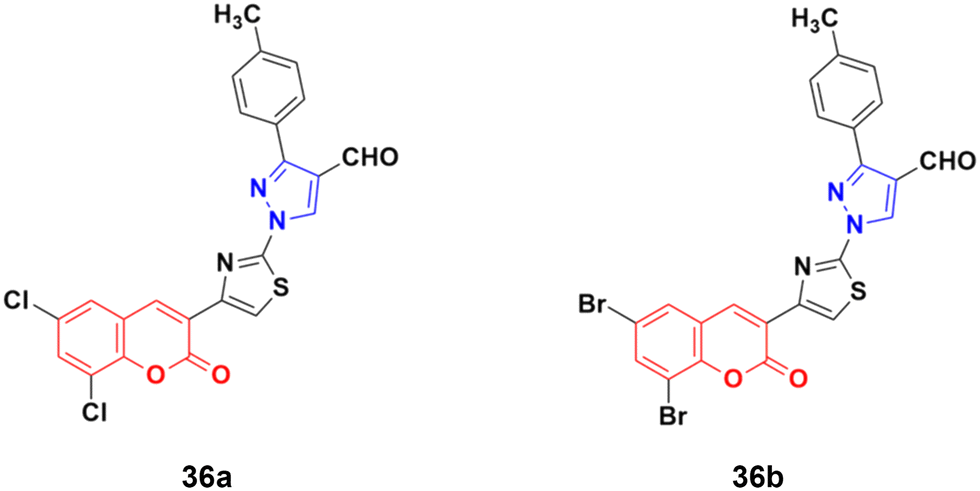 | ||
| Fig. 31 Chemical structures of coumarin–pyrazole hybrid 36a and b. | ||
A similar series of coumarin–pyrazole hybrids was designed and their anticancer properties screened against five cancer cell lines (L1210, CEM, DU- 145, HeLa, and MCF-7).76 Among them, compound 37a (Fig. 32) possessed the maximum potency against the DU-145 cell line with an IC50 value of 7 ± 1 μM, while compound 37b showed the maximum efficiency against the MCF-7 cell line with an IC50 value of 8 ± 2 μM.
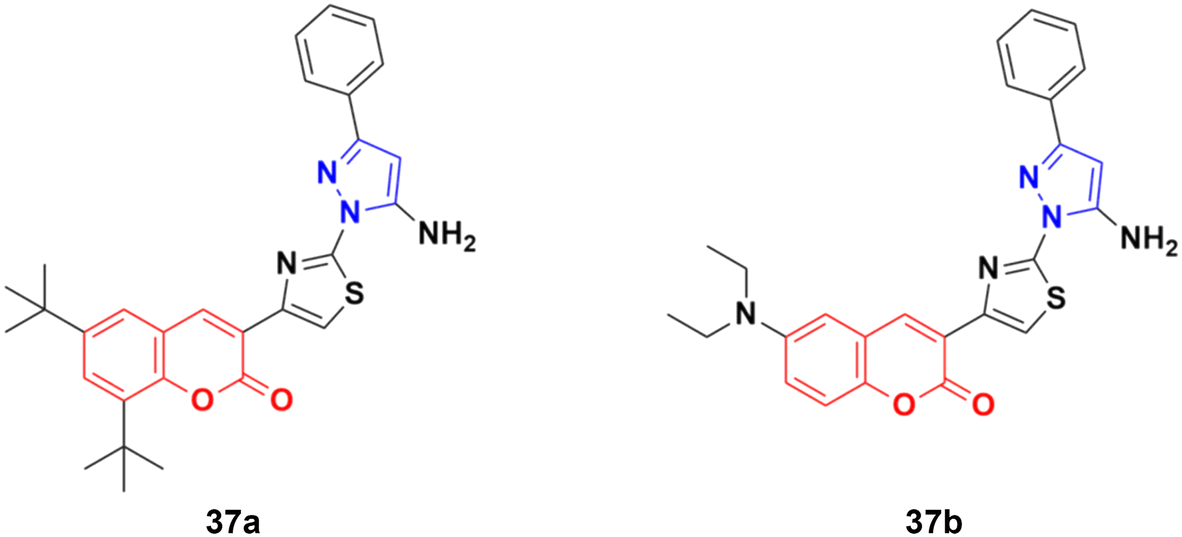 | ||
| Fig. 32 Chemical structures of coumarin–pyrazole hybrid 37a and b. | ||
A novel group of coumarin-tagged 1,3,4-oxadizaole hybrids was prepared and their biological activities studied against the MDA-MB-231 and MCF-7 breast cancer cell lines.77 Compound 38a (Fig. 33) showed excellent cytotoxicity against the MCF-7 cell line with an IC50 value of less than 5 μM, whereas compounds 38b and 38c possessed significant potency against the MDA-MB-231 cell line with an IC50 value of 7.07 μM for both of them. Docking studies revealed that the stronger binding affinity of the designed derivatives is due to the presence of a sulfone unit attached to the substituted benzyl group in their pharmacophores. It is worth noting that oxadiazolyl sulfones are also an emerging tool in bioconjugation methodologies.78,79
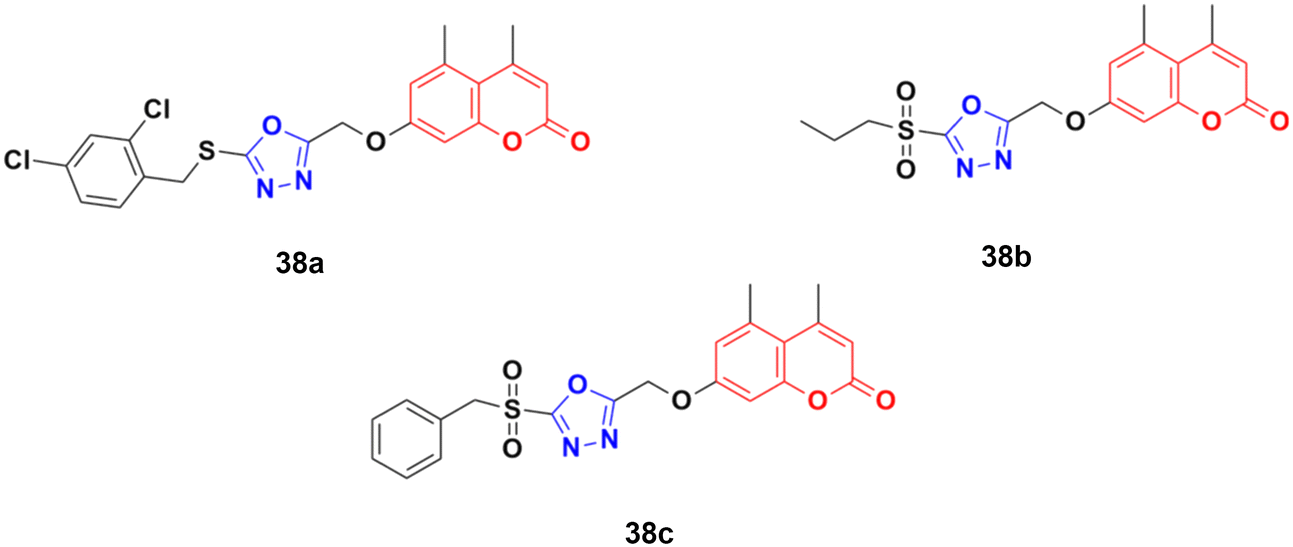 | ||
| Fig. 33 Chemical structures of coumarin–1,3,4-oxadiazole hybrid 38a–c. | ||
A library of twenty coumarin–1,3,4-oxadiazole conjugates was synthesized and analyzed for their inhibitory activity against the four physiologically relevant human carbonic anhydrase (hCA) isoforms CA I, CA II, CA IX, and CA XII.80 Among them, compounds 39a and 39b (Fig. 34) exhibited significant inhibition in lower micromolar potency against hCA XII (Ki of 0.16 μM) and hCA IX (Ki of 2.34 μM), respectively. Hence, these two compounds can serve as promising leads for designing novel anticancer agents by acting through hCA IX and XII inhibition. Besides their pharmaceutical properties, thioethers and thioesters are very useful building blocks in synthetic methodologies, providing unique pathways for building new molecules.81
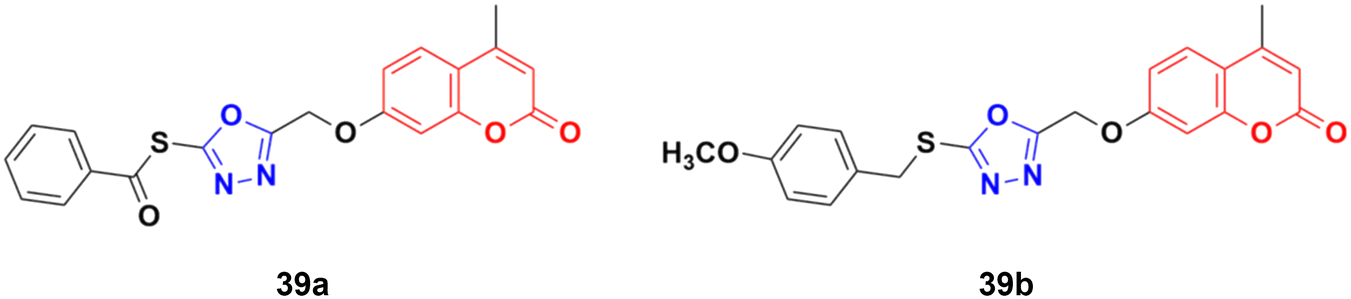 | ||
| Fig. 34 Chemical structures of coumarin–1,3,4-oxadiazole hybrid 39a and b. | ||
A novel series of coumarin–thiazole hybrids was designed and tested by employing human colon (DLD-1) and liver cancer cell lines (HepG2).86 Among the nine compounds, 40a (Fig. 35) was the most effective against DLD-1 with an EC50 value of 5.79 μM, while compound 40b showed the maximum potency against the HepG2 cell line with an EC50 value of 3.70 μM. The designed compounds act by blocking Hsp90 function and were determined to be valuable C-terminal Hsp90 inhibitors.
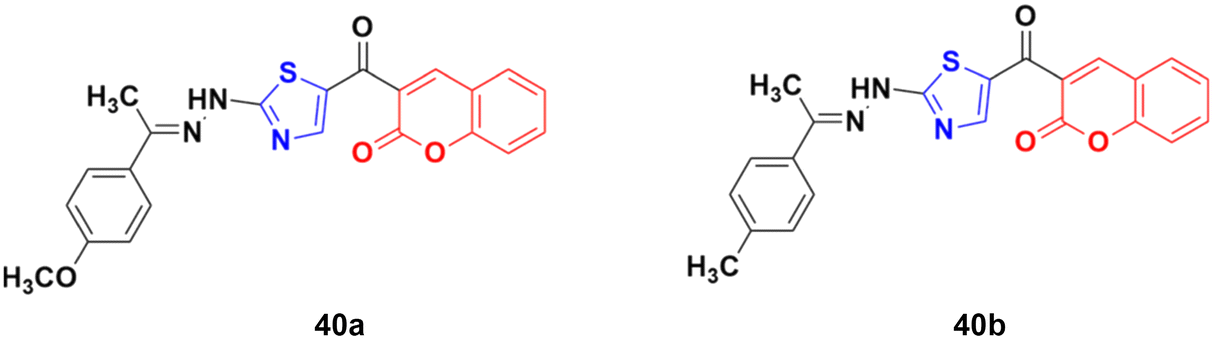 | ||
| Fig. 35 Chemical structures of coumarin–thiazole hybrid 40a and b. | ||
A similar series of coumarin–thiazole conjugates was developed and their cytotoxic activity examined against three human cancer cell lines, i.e., MCF-7, HepG2 and SW480.87 All the compounds showed moderate to low activity against the above-mentioned cell lines but none of them were more potent than the reference etoposide. Compound 41a (Fig. 36) showed significant efficiency (IC50 values of 7.5 ± 0.7, 16.9 ± 0.7, and 13.0 ± 0.6 μg mL−1 against MCF-7, HepG2, and SW480, respectively). Compound 41b possessed the maximum potency against the HepG2 cell line with IC50 = 12.2 ± 2.3 μg mL−1.
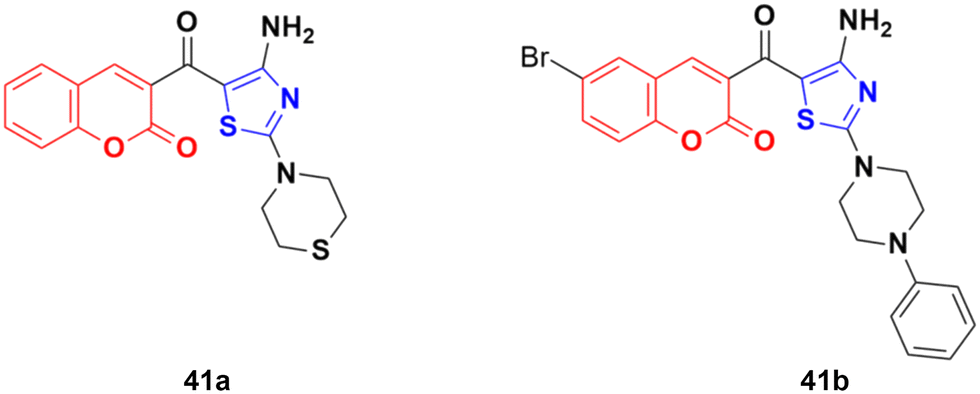 | ||
| Fig. 36 Chemical structures of coumarin–thiazole hybrid 41a and b. | ||
A novel series of thiazolopyrazolyl coumarin derivatives was designed and their cytotoxicity examined against four cancer cell lines (MCF-7, A549, PC3, and HepG2) together with a normal cell line, HFB4.88 Among the synthesized hybrids, compounds 42a–e (Fig. 37) showed significant activity towards the MCF-7 cell line (IC50 = 5.41–10.75 μM) together with a low cytotoxic effect on the normal cell line. Several theoretical and experimental studies revealed the molecular mechanisms that control breast carcinoma metastasis. The mechanistic effectiveness in cell cycle progression, apoptotic induction, and gene regulation was analyzed for compound 42e due to its significant cytotoxicity against MCF-7 and potent VEGFR-2 inhibition. Flow cytometric analysis showed that compound 42e induced cell growth cessation at the G2/M phase and enhanced the percentage of cells in the pre-G1 phase, stimulating the apoptotic death of MCF-7 cells. Furthermore, real-time PCR assay showed that compound 42e upregulated p53 gene expression and elevated the Bax/Bcl-2 ratio. Moreover, the apoptotic induction of MCF-7 breast cancer cells was enhanced effectively through the activation of caspase-7 and 9 by compound 42e. Therefore, 42e can be considered a potent lead for the development of anti-breast cancer candidates.
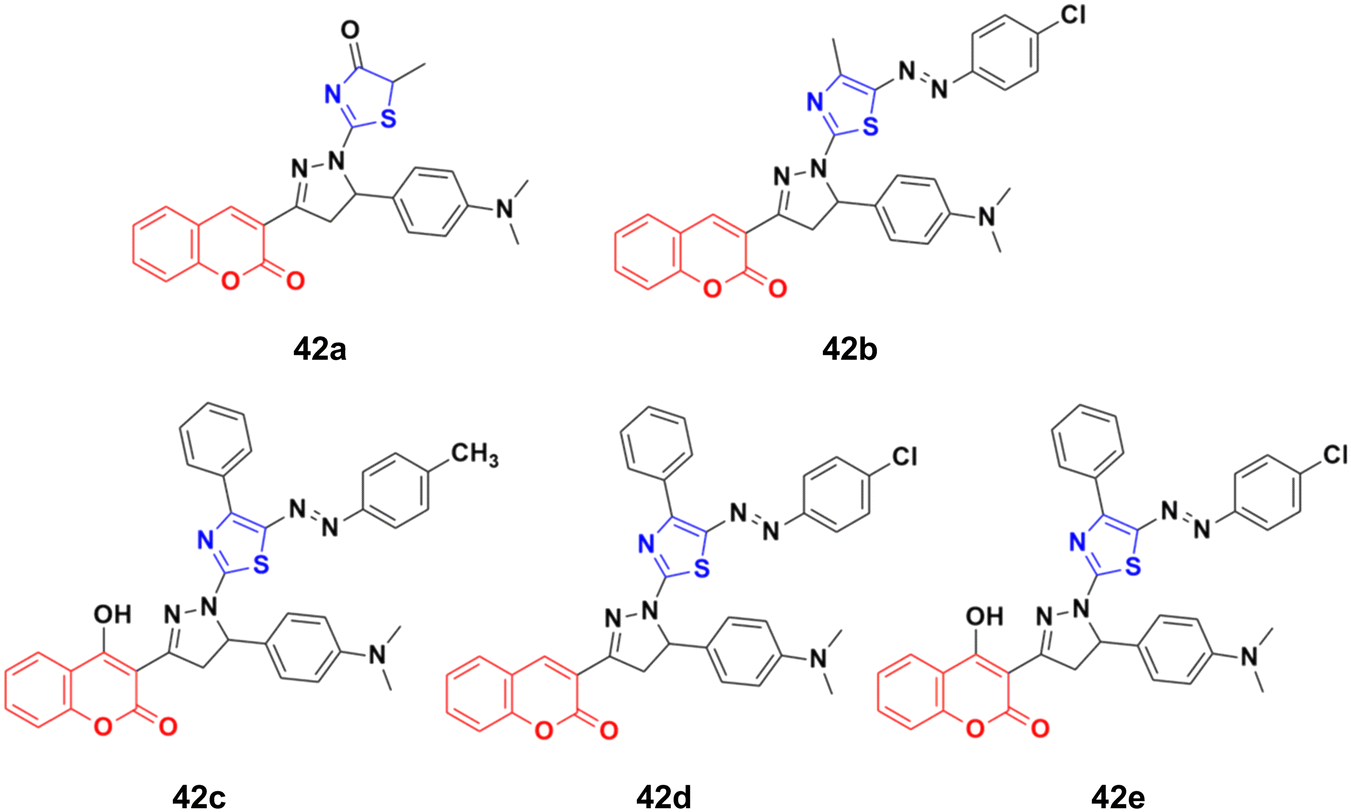 | ||
| Fig. 37 Chemical structures of coumarin–thiazole hybrid 42a–e. | ||
A set of coumarin–thiazole hybrids 43a–d (Fig. 38) showed significant potency against HeLa cells, which was observed to be more potent than the reference doxorubicin.89 Compound 43d possessed maximum antiproliferative activity against the cancer cell lines with an IC50 value of 0.0091 ± 0.0007 cM. The cell cycle investigation showed that compound 43b led to cell cycle cessation at the G0/G1 phase, indicating that the CDK2/E1 complex can be the plausible biological target. The RT-PCR gene expression assay showed that compound 43b increased the levels of the nuclear CDK2 regulators P21 and P27 by 2.30- and 5.7-fold, respectively. The ELISA technique showed also that compound 43b led to the remarkable activation of caspase-9 and -3, inducing cell apoptosis. The molecular docking study for 43a–d rationalized their superior CDK2 inhibitory activity through their hydrogen bonding and hydrophobic interactions with the key amino acids in the CDK2 binding site.
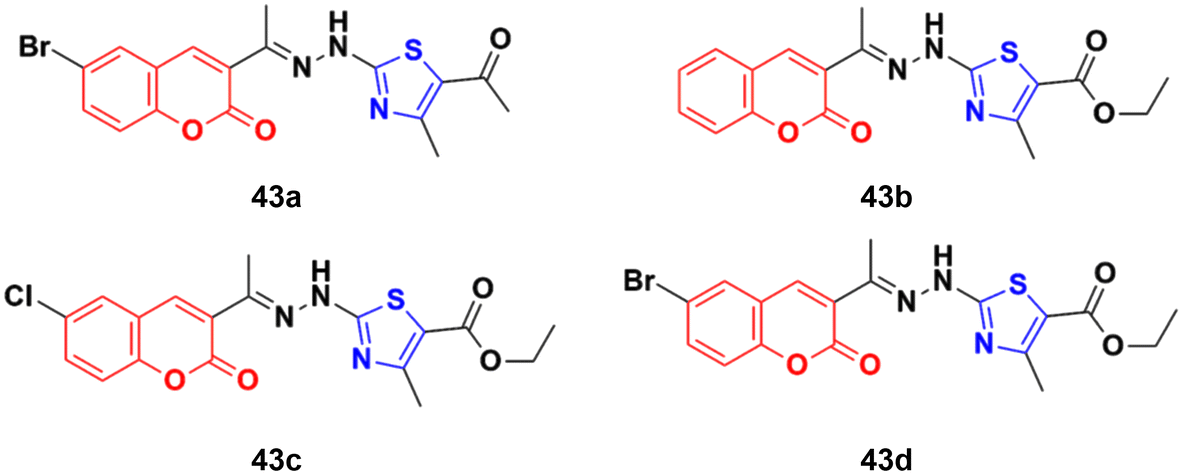 | ||
| Fig. 38 Chemical structures of coumarin–thiazole hybrid 43a–d. | ||
Twelve novel 1-thiazolyl-5-coumarin-3-yl-pyrazole derivatives were developed via a one-pot multicomponent reaction and their anticancer activity was investigated on two cancer cell lines (HepG2 and MCF7).90 Compound 44a (Fig. 39) showed significant cytotoxic activity against the HepG2 cell line with an IC50 value of 3.74 ± 0.02 μM and 44b possessed significant activity against MCF-7 with an IC50 value of 4.03 ± 0.02 μM. 44c was potent against both the HepG2 and MCF-7 cell lines with an IC50 value of 3.06 ± 0.01 μM and 4.42 ± 0.02 μM, respectively.
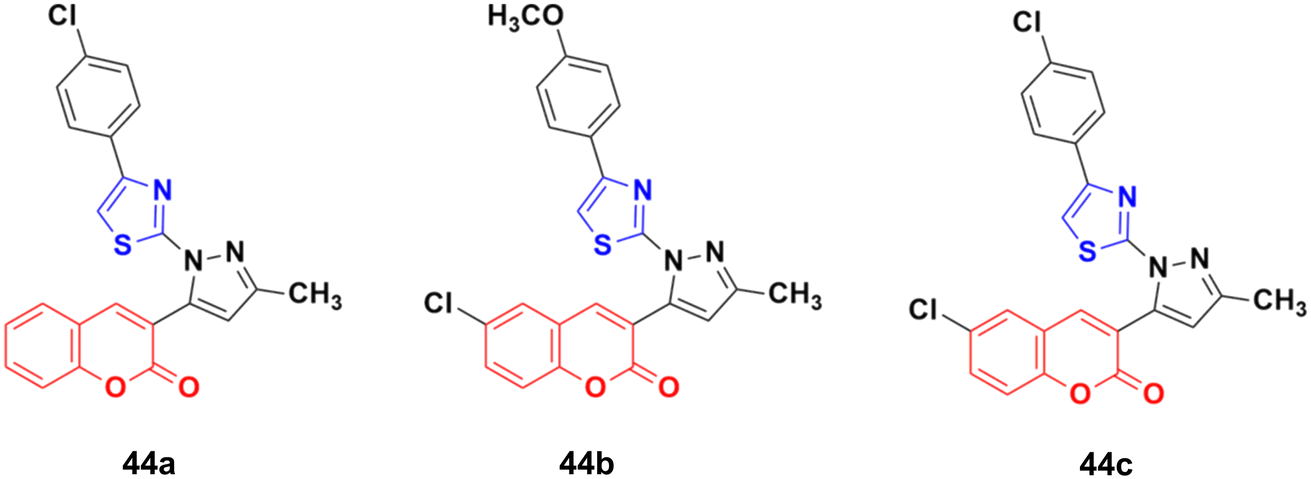 | ||
| Fig. 39 Chemical structures of 1-thiazolyl-5-coumarin-3-yl-pyrazole derivative 44a–c. | ||
Coumarin–benzothiazole hybrids were screened for their antitumor activity at a single dose (10 μm) against a panel of 60 cancer cell lines.91 The most active compound 45 (Fig. 40) was further screened at a five-dose level. It displayed half-maximal growth inhibition (GI50) values of 0.24 and 0.33 μm against the central nervous system (CNS) cancer (SNB-75) and ovarian cancer (OVCAR-4) cell lines, respectively.
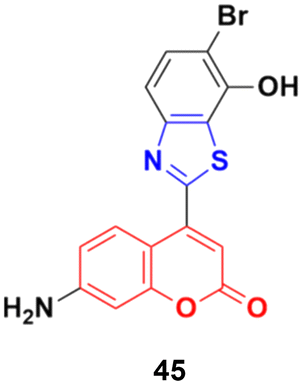 | ||
| Fig. 40 Chemical structure of coumarin–benzothiazole hybrid 45. | ||
Compounds 46a and 46b (Fig. 41) were tested against the most common secondary ALK mutants such as L1196M, G1269A, and G1202R.92 Compound 46a showed potent inhibitory activities against three ALK mutants, L1196M, G1269A, and G1202R, with IC50 of 0.27 μM, 0.30 μM, and 0.59 μM, respectively. Compound 46b displayed an IC50 value of 0.45 μM for L1196M and compatible enzymatic inhibitory activity against G1269A and G1202R with that against WT ALK.
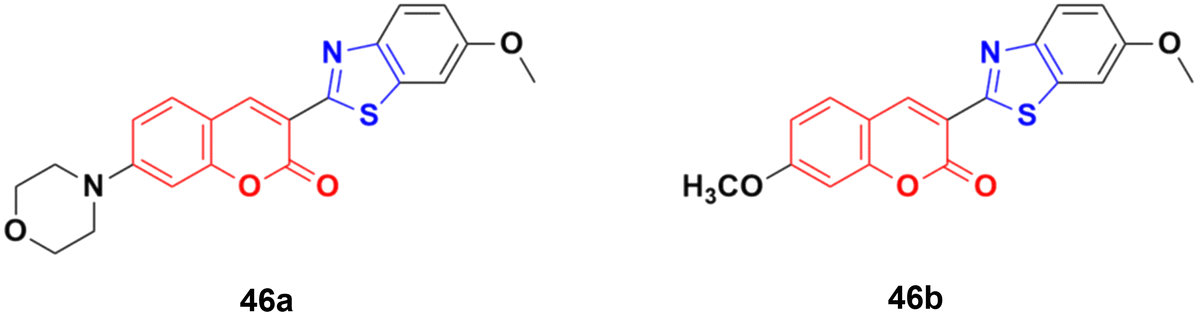 | ||
| Fig. 41 Chemical structures of coumarin–benzothiazole derivative 46a and b. | ||
A series of novel coumarin derivatives having 1,2,4-triazolo[3,4-b][1,3,4]thiadiazole moieties was developed and analyzed in vitro for their anticancer activity against the HCT116 cell line.93 Compound 47 (Fig. 42) possessed significant anticancer activity with an IC50 value of 2.656 μM. Molecular docking studies suggested its possible interaction with tyrosine kinases (CDK2).
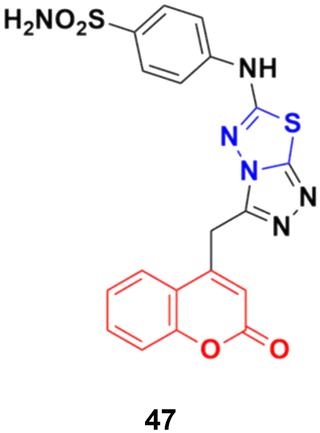 | ||
| Fig. 42 Chemical structure of coumarin–thiadiazole hybrid 47. | ||
A novel series of coumarin–thiadiazole hybrids was developed using nucleophilic substitution reaction and their anticancer activity was tested against HCT-116, MCF-7, and HepG2 but none of them were more potent than the reference cis-platin.94 Compound 48a (Fig. 43) showed maximum potency against the HCT-116 and MCF-7 cell lines with IC50 values of 30.7 and 54.9 μg mL−1, respectively, while 48b was the most potent against HepG2 with an IC50 value of 24.9 μg mL−1.
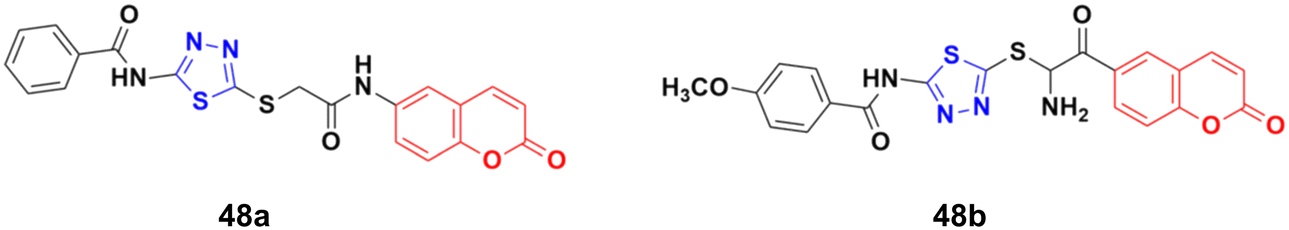 | ||
| Fig. 43 Chemical structures of coumarin–thiadiazole derivative 48a and b. | ||
A series of 2-(3-substituted-4-methyl-2-oxo-2H-chromen-7-yloxy)-2-methylpropanoic acid derivatives was developed by base-catalyzed dehydrohalogenative cyclization followed by Hantzsch synthesis and their in vitro cytotoxicity examined against the MCF-7, MDA-231, and HT29 cancer cell lines.95 Thiazole derivative 49 (Fig. 44) possessed the maximum potency against the MDA-231 and MCF-7 cell lines with IC50 values of 4.84 ± 0.17 and 2.39 ± 0.03 μM, respectively.
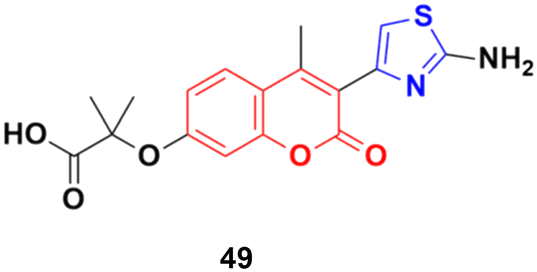 | ||
| Fig. 44 Chemical structure of coumarin–thiazole derivative 49. | ||
A series of coumarin-3-yl-thiazol-3-yl-1,2,4-triazolin-3-ones was synthesized using a microwave-assisted multicomponent protocol and their anticancer activity investigated against four cancer cell lines, including A549, MDA-MBA-231, HeLa and K562.96 Most of the derivatives showed better or comparative cytotoxic effects against all the cancer cell lines compared to the reference doxorubicin. Among them, compound 50a (Fig. 45) was more potent against three of the four cancer cell lines with IC50 = 0.16 μM against MDA-MBA-231, IC50 = 0.17 μM against A549, IC50 = 0.31 μM against K562, and IC50 = 0.25 μM against the HeLa cell line. Compound 50b showed the maximum efficiency against the HeLa cell line with IC50 = 0.21 μM.
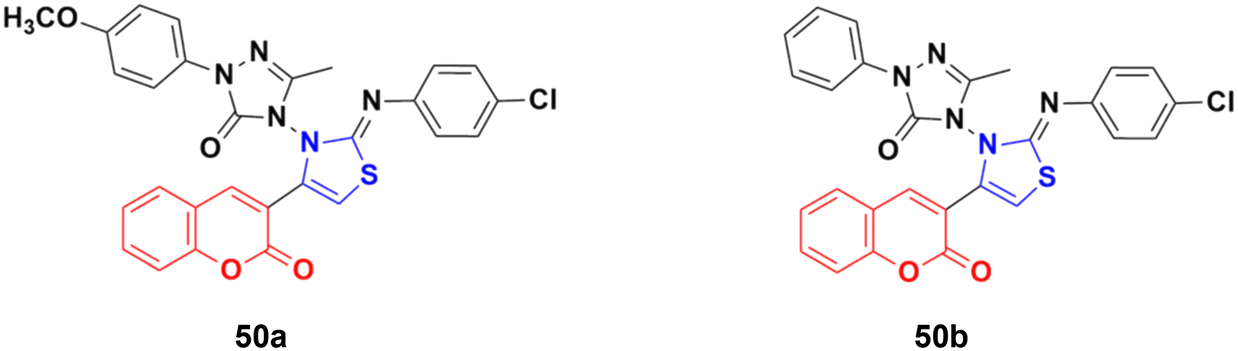 | ||
| Fig. 45 Chemical structures of coumarin–thiazole derivative 50a and b. | ||
The anticancer activity of a series of coumarin–thiazole derivatives was screened in vitro against two cancer cell lines (HeLa and COS-7) together with a normal cell line (W138).97 Three compounds, 51a–c (Fig. 46), showed better cytotoxic ability than doxorubicin. Also, 51c possessed the best potency against the HeLa cell line with IC50 = 1.29 μM, while 51b and 51c showed significant cytotoxicity against COS-7 (IC50 = 1.96 and 1.66 μM, respectively). In silico studies revealed that the compounds meet the optimal needs for good oral absorption with no expected toxicity hazards.
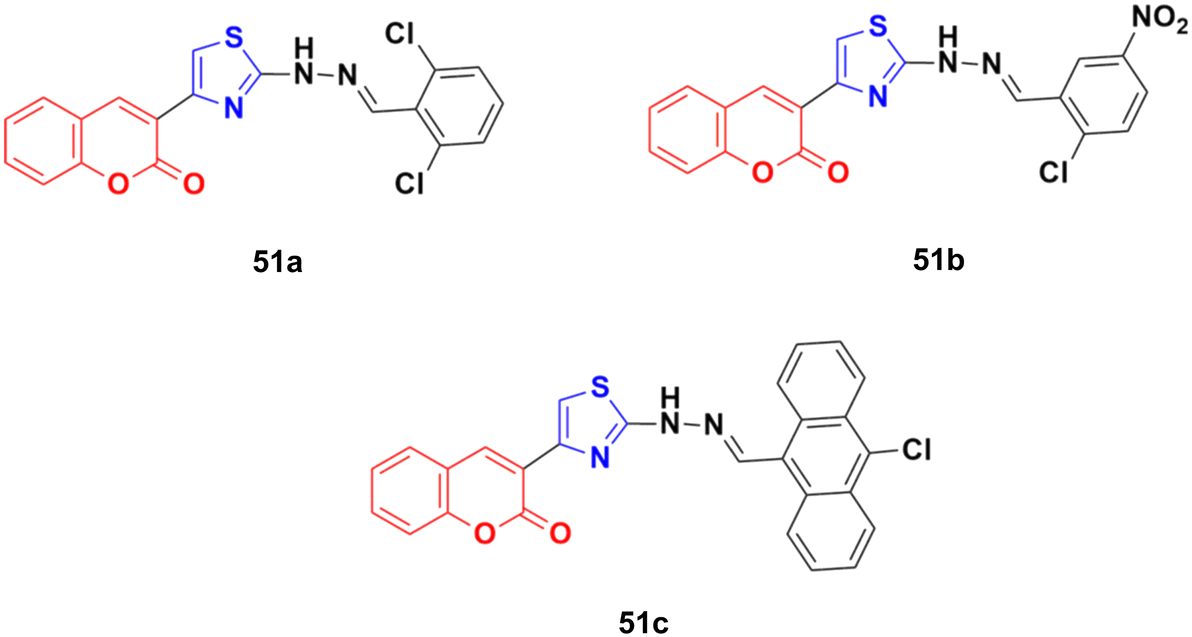 | ||
| Fig. 46 Chemical structures of coumarin–thiazole derivative 51a–c. | ||
The coumarin–aminothiazole hybrids were examined for their cytotoxic activity against the HCT-116 and HT-29 cancer cell lines.98 Among them, compounds 52a–e (Fig. 47) were effective against both cell lines with IC50 values in the range of 0.25 to 0.38 μM. However, compound 52d was the most potent with IC50 = 0.25 ± 0.004 μM against HT-29 and IC50 = 0.26 ± 0.016 μM against HCT-116. Further biological investigation of 52a using Western blotting, caspase activity, glucose uptake, ROS production, and NADPH/NADP levels showed the ability of this lead drug candidate to induce cancer cell death via energy restriction. Moreover, the assessment of the synergistic activity of 52a with cisplatin showed promising outcomes.
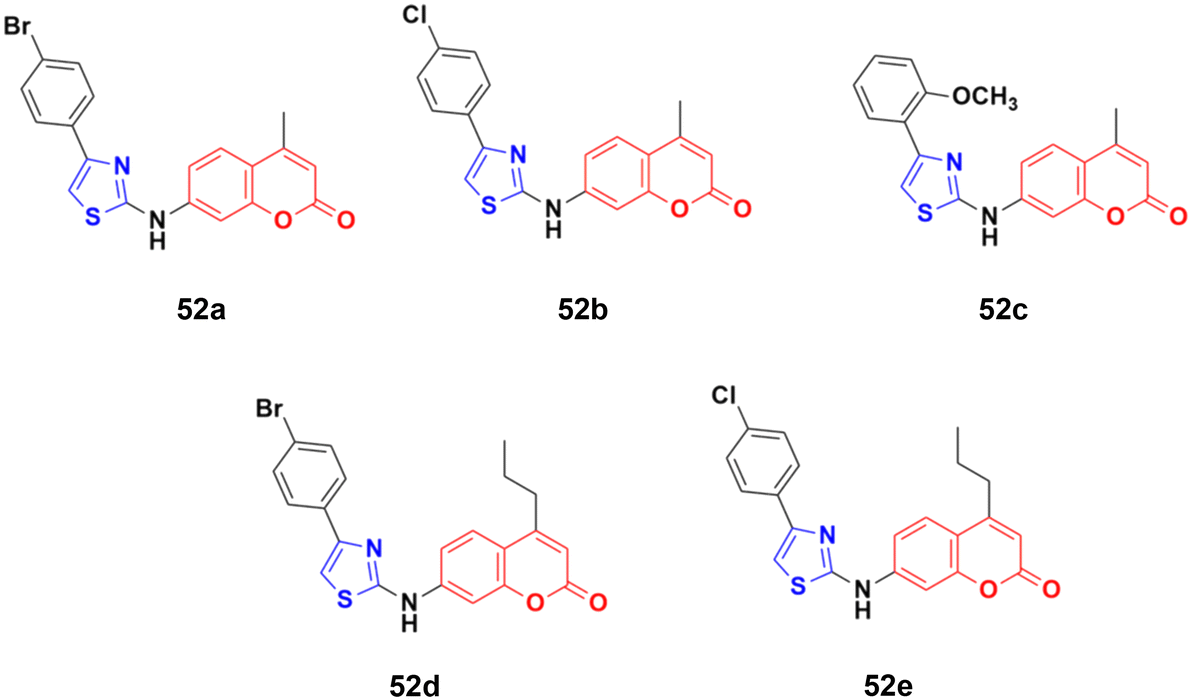 | ||
| Fig. 47 Chemical structures of coumarin–aminothiazole derivative 52a–e. | ||
A series of nine novel acridine–thiazole bridged coumarin derivatives was prepared and evaluated for their in vitro antiproliferative activity on MDA-MB-231, A-549, and HT29 cell lines.99 All the compounds showed a significant cytotoxicity effect but none of them were more potent than the reference cisplatin. Compound 53a (Fig. 48) showed the maximum efficacy against the MDA-MB-231 cell line with IC50 = 8.03 ± 0.81 μM. Compound 53b was significantly active against A-549 and HT-29 with IC50 values 5.18 ± 1.04 μM and 23.09 ± 1.17 μM, respectively.
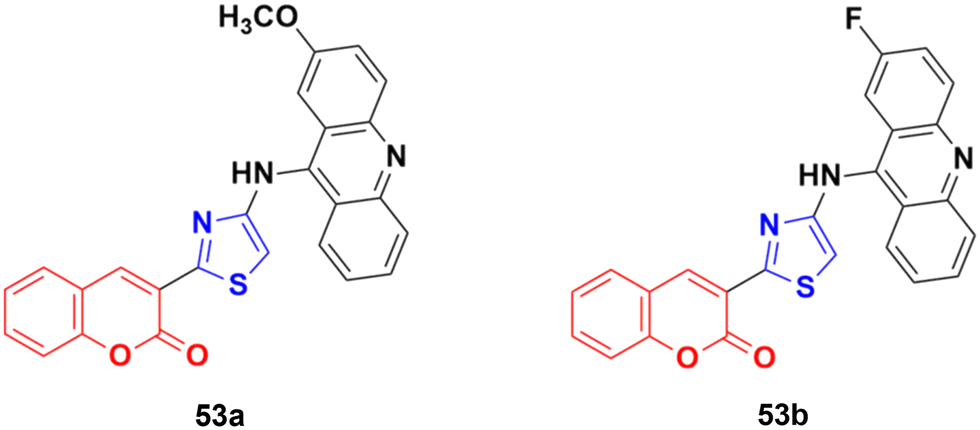 | ||
| Fig. 48 Chemical structures of coumarin–acridine–thiazole derivative 53a and b. | ||
Several coumarin–thiazole derivatives were designed and synthesized and their cytotoxicity assessed on MCF-7 cancer cell lines using sorafenib as the reference drug.100 Among them, 54a and 54b (Fig. 49) demonstrated higher anticancer activities (IC50 = 10.5 ± 0.71 and 11.2 ± 0.80 μM, respectively) than sorafenib (IC50 = 5.10 ± 0.49 μM). These hybrids are thought to inhibit the vascular endothelial growth factor receptor (VEGFR-2) signaling system.
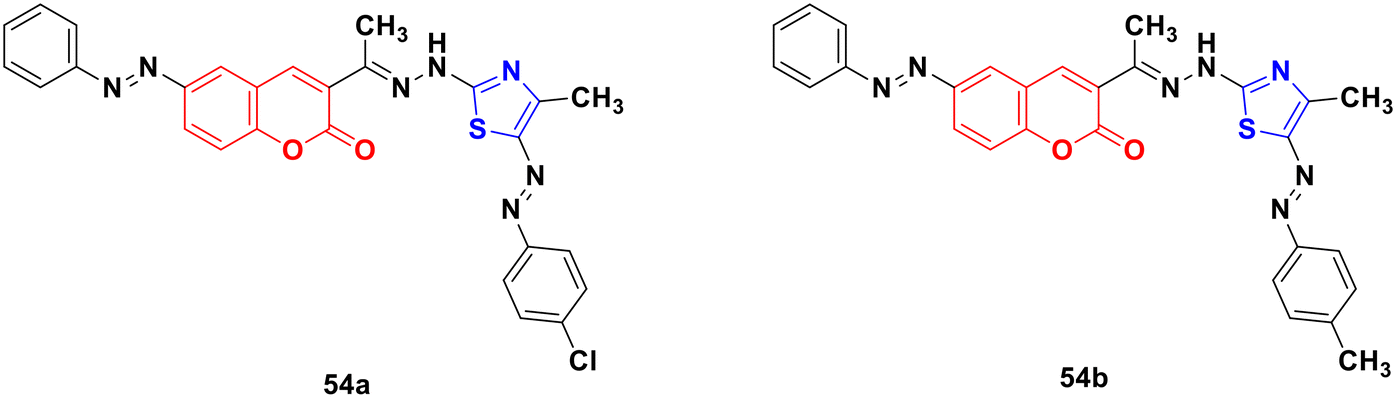 | ||
| Fig. 49 Chemical structures of coumarin–thiazole derivative 54a and b. | ||
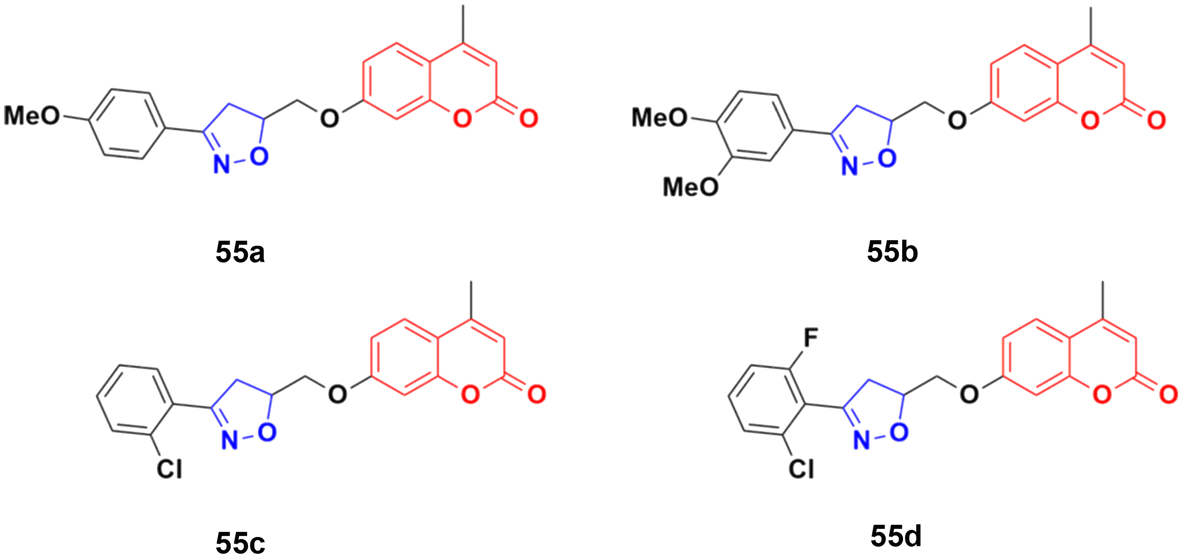 | ||
| Fig. 50 Chemical structures of coumarin-isoxazoline derivative 55a–d. | ||
A series of coumarin–isoxazole derivatives was synthesized from imidoyl chlorides and various substituted 4-(prop-2-yn-1-yloxy)-2H-chromen-2-one and their biological activity evaluated against the HepG2 cell line.102 Among them, 56 (Fig. 51) possessed the best activity (IC50 = 12.85 μM L−1) against the HepG2 cell line. Its toxicity against Vero cells (IC50 = 144.32 μM L−1) was lower than that of cis-platin (IC50 = 28.63 μM L−1). It was observed that the presence of Cl or Br at the 6-position of the coumarin moiety enhances the bioactivity. The docking result showed that the compound can fruitfully interact with the protein.
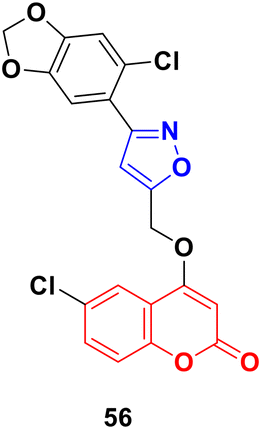 | ||
| Fig. 51 Chemical structure of coumarin–isoxazole derivative 56. | ||
A series of coumarin–thiazolidin-2,4-dione hybrids was tested for their anticancer activity against the MCF-7, HeLa, HT29, A549, and PC3 cancer cell lines, but none of them were more effective than the reference doxorubicin.103 Among them, compound 57 (Fig. 52) was the most potent against the MCF-7, HeLa, and A549 cell lines with IC50 values in the range of 0.95 to 3.20 μM.
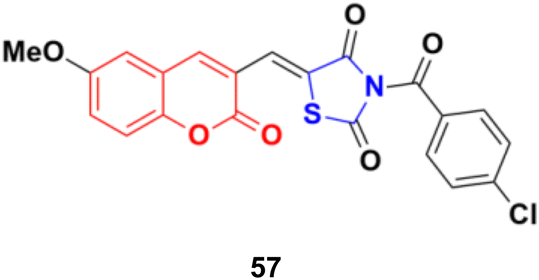 | ||
| Fig. 52 Chemical structure of coumarin–thiazolidin-2,4-dione hybrid 57. | ||
A novel series of coumarin–thiazolidinone derivatives was synthesized using the coupling methodology and their in vitro cytotoxicity screened against the MCF-7 cancer cell line.104 Among them, compounds 58a and 58b (Fig. 53) possessed noteworthy activity with IC50 values of 15.65 ± 0.28 μg mL−1 and 12.15 ± 0.05 μg mL−1, respectively. The structure–activity relationship studies indicated that the presence of an electron-releasing methoxy group increases the cytotoxic activity. Kinase inhibition and suitable binding are responsible for their significant biological property.
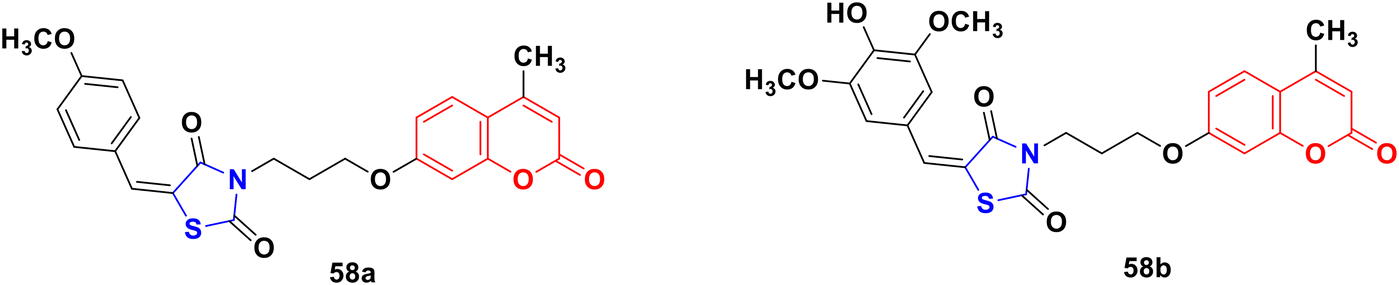 | ||
| Fig. 53 Chemical structures of coumarin–thiazolidinone 58a and b. | ||
2.3. Coumarin–furoxan hybrids
Furoxan (1,2,5-oxadiazole-2-oxide), a nitric oxide donor, is a heterocycle of the isoxazole family and an amine oxide derivative of furazan (Fig. 54). Nitric oxide plays an essential role in cardiovascular regulation, nerve transmission delivery, and immune response, and a high concentration of nitric oxide has potentially significant anticancer effects. Hence, the hybridization of coumarin and furoxan may be a lead for generating new anticancer agents.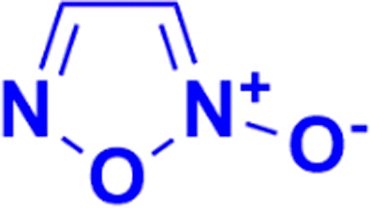 | ||
| Fig. 54 Chemical structure of furoxan. | ||
A series of sixteen furoxan-based coumarin derivatives was synthesized and their antiproliferative activities investigated against several cancer cell lines including A549, HeLa, A2780, A2780/CDDP, and HUVEC.105 They all showed moderate to excellent anticancer activity against the above-mentioned cell lines, but compound 59 (Fig. 55) was observed to be the most potent with IC50 values of 0.024, 0.053, 0.014, 0.062, and 0.034 μM, respectively. This compound was again investigated for its biological activity against some drug-resistant cell lines (MDA-MB-231, MDA-MB-231/Gem, SKOV3, and SKOV3/CDDP) and proved to be very effective against them also. Furthermore, 55 inhibited the growth of A2780 in vivo and displayed lower toxicity on non-tumorigenesis T29. Preliminary pharmacological studies revealed that 59 acts by inducing apoptosis, arresting the cell cycle at the G2/M phase in the A2780 cell line, and disrupting the phosphorylation of MEK1 and ERK1. Compound 59 was further studied to reveal its potential for apoptosis and autophagy induction in lung adenocarcinoma cells.106 The cytotoxicity and apoptosis of A549 and H1299 cells induced by compound 59 were detected by MTT, microscopy, and western blot analysis. Significant growth inhibition and caspase-dependent apoptosis were observed in the compound 59-treated A549 and H1299 cells. Then, it was confirmed that this compound induced autophagy by autophagosome formation, upregulated the expression of autophagy-related protein LC3-II, and autophagic flux. Importantly, abolishing autophagy using inhibitors and ATG5 siRNA enhanced the cytotoxicity of compound 59, indicating the cytoprotective role of autophagy in lung adenocarcinoma. Further mechanistic investigations suggested that the Akt/mTOR and Erk signaling pathways contributed to autophagy induction by compound 59.
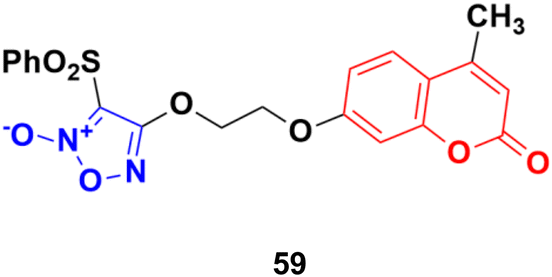 | ||
| Fig. 55 Chemical structure of coumarin–furoxan hybrid 59. | ||
Five phenylsulfonylfuroxan-merging 3-benzyl coumarins were designed and evaluated for their anticancer activities.107 Among them, compound 60 (Fig. 56) showed the most potent antiproliferation activities with IC50 values ranging from 0.5 nM to 143 nM against nine drug-sensitive (HeLa, SKOV3, A549, OVCA429, OVCA433, A2780, MDA-MB-231, MCF-7 and KB) and four drug-resistant cancer cell lines (A2780/CDDP, MDA-MB-231/Gem, MCF-7/ADR, and KB-V). Preliminary pharmacological studies revealed that compound 60 acts by inducing early apoptosis and affecting the cell cycle. Furthermore, it gave 559- and 294-fold selectivity antiproliferation activity in the P-gp overexpressed drug-resistant cancer cell lines MCF-7/ADR and KB-V compared to the drug-sensitive MCF-7 and KB, implying that this compound may have an extra mechanism of anti-MDR-cancer with P-gp overexpression. Here, compound 60 contains fluorine. It is noteworthy to mention that fluorine-containing drugs are tremendously important and are successfully being used in the treatment of many diseases, e.g., multiple myeloma, lymphoma, HIV, chronic heart failure, chronic myeloid leukemia, (ANCA)-associated vasculitis, migraines, von Hippel–Lindau disease, and non-small cell lung cancer.108–115
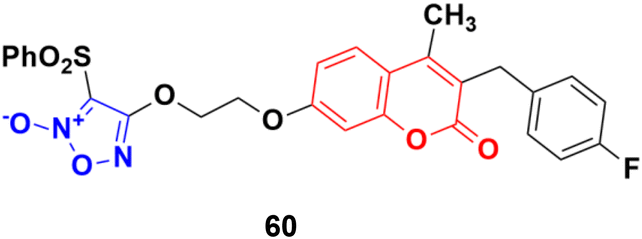 | ||
| Fig. 56 Chemical structure of 3-benzylcoumarin–furoxan hybrid 60. | ||
A novel series of eleven furoxan–coumarin hybrids was developed and their antiproliferative activity studied on five human cancer cell lines including HeLa, SW620, HepG2, HCT116, and MCF7.116 Among them, compounds 61a–c (Fig. 57) showed potent anticancer activity and some evaluated to be more potent than the reference doxorubicin. Compound 61a was the most potent against the HepG2 cell line with IC50 = 3.86 μM, while compound 61b showed the maximum activity against the SW620 and HCT116 cell lines with IC50 values of 1.86 and 3.46 μM, respectively. Compound 61c was observed to be the most effective against the HeLa and MCF7 cell lines with IC50 values of 0.88 and 0.61 μM, respectively.
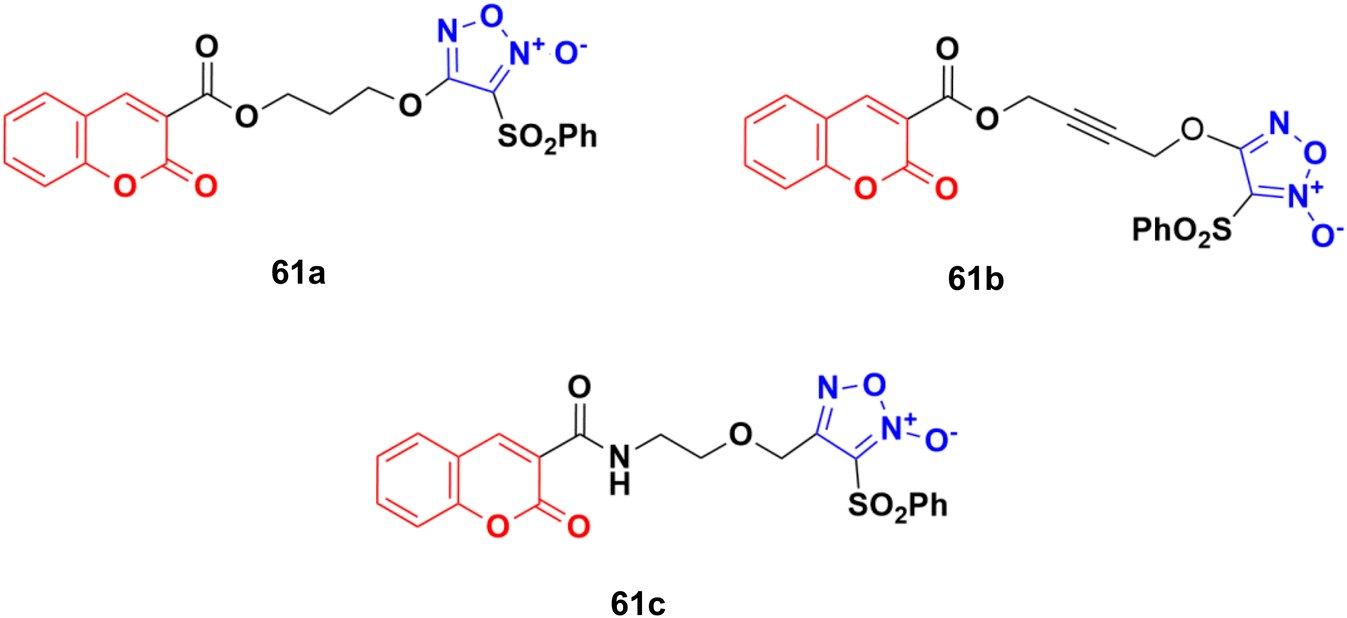 | ||
| Fig. 57 Chemical structures of coumarin–furoxan hybrid 61a–c. | ||
A series of furoxan conjugates of N,N-dialkyl carboxy coumarins was developed as potential anticancer agents and tested for their in vitro antiproliferative activities on various cell lines (MDA-MB-231, 4T1, WiDr, MCF10A, and HDFa cell lines).117 Among them, compound 62 (Fig. 58) showed the highest potency with IC50 values in the range of 0.02 to 38.6 μM. The in vitro mechanistic studies indicated that these compound generated substantial nitric oxide, inhibited colony formation, and caused apoptosis in cancer cells.
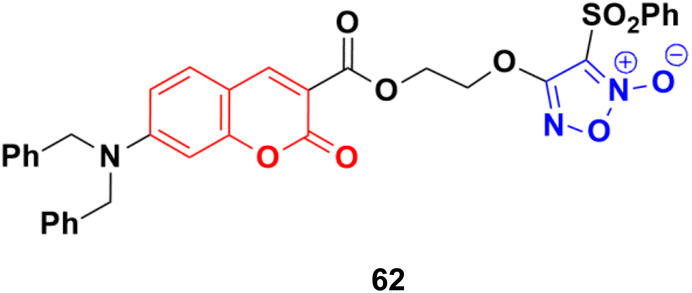 | ||
| Fig. 58 Chemical structure of coumarin–furoxan hybrid 62. | ||
2.4. Coumarin–pyridine/pyrimidine hybrids
Pyridine (Fig. 59), one of the most basic heterocyclic compounds with widespread biological occurrence, forms the nucleus of numerous drugs. Pyridine derivatives are known to possess a variety of biological activities, namely, anti-asthmatic, antibacterial, anticonvulsant, antimalarial, antimuscarinic, antiprotozoal, anticancer, antidiabetic, and anti-inflammatory.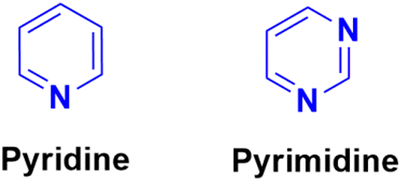 | ||
| Fig. 59 Chemical structures of pyridine and pyrimidine. | ||
Similarly, pyrimidine (Fig. 59), which is structurally related to pyridine, the building unit of DNA and RNA, has also been found to possess marked pharmacological effects.
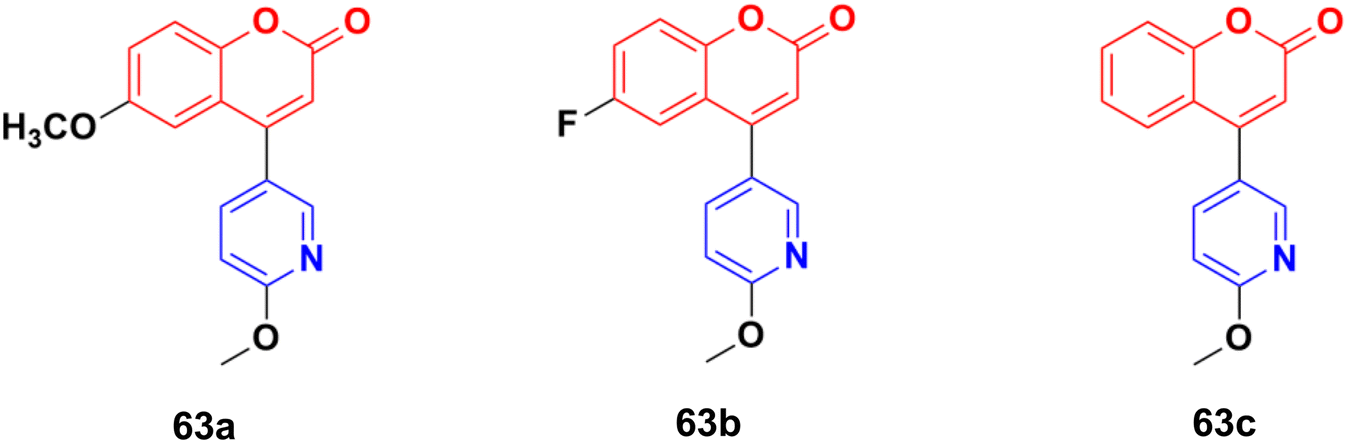
The coumarin–pyridine hybrids 63a–c (Fig. 60) showed weak to moderate activity against the A549 cancer cell line with IC50 values in the range of 34.2 > 80 μM.118 The compounds were found to potently inhibit in vitro microtubule formation via a substoichiometric mode such as CA-4.
 | ||
| Fig. 60 Chemical structures of coumarin–pyridine hybrid 63a–c. | ||
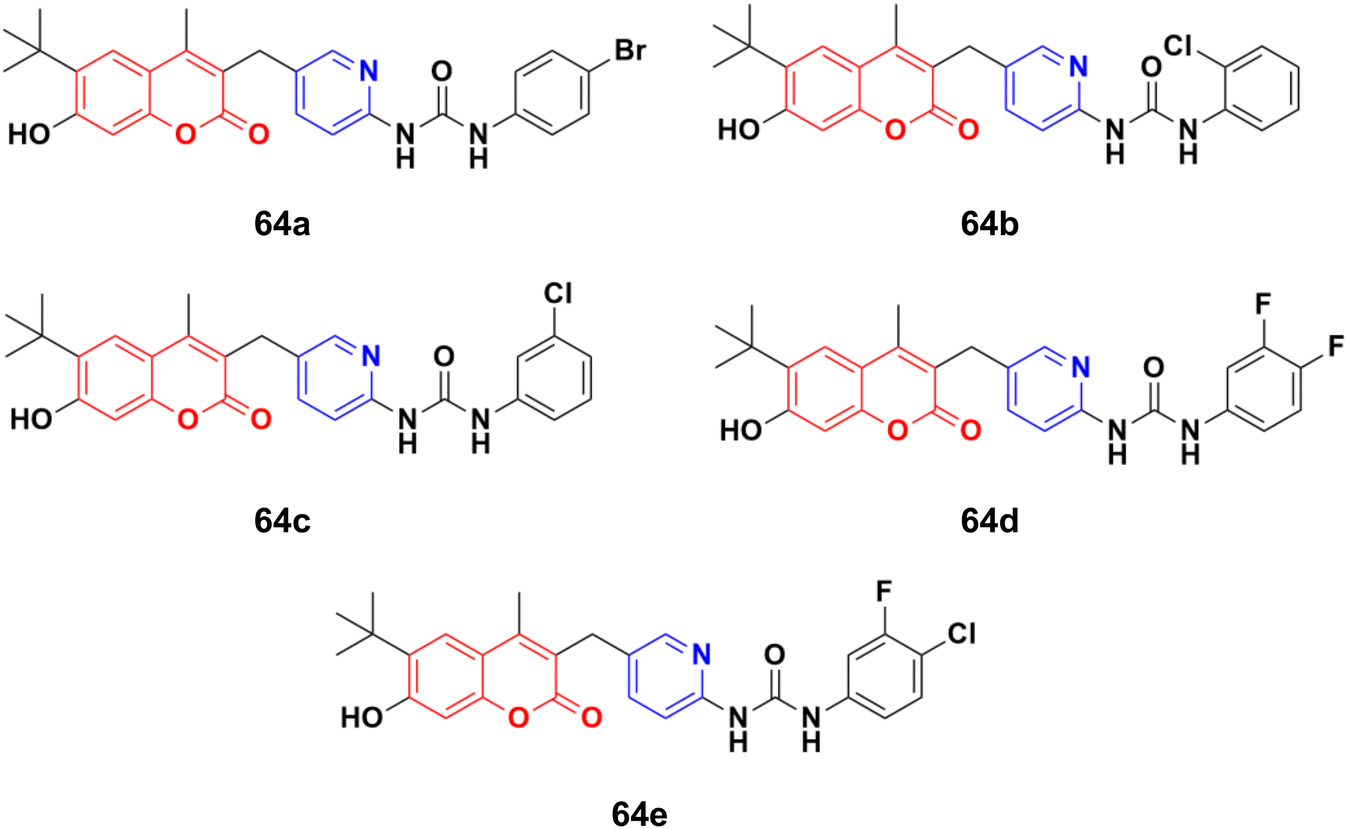
A novel series of thirteen coumarin–pyridine derivatives was designed and their cytotoxic activities examined on four cancer cell lines including K562, HeLa, A549, and MCF7.119 Among them, compounds 64a–e (Fig. 61) showed the maximum potency against the MCF7, A549, HeLa, and K562 (64d and e) cell lines with IC50 values of 2.56 ± 0.17, 4.38 ± 0.09, 2.17 ± 0.45, 1.66 ± 0.09 and 1.66 ± 0.15 μM, respectively. Further investigation revealed that compounds 64a and 64c were much more potent PI3K inhibitors than S14161 and BENC-511 (reference). In addition, 64a was more selective to PI3Kα/β over PI3Kδ/γ, while 64c was a selective PI3Kα/β/δ inhibitor. 64c could also suppress the phosphorylation of Akt and induce K562 cell apoptosis.
 | ||
| Fig. 61 Chemical structures of coumarin–pyridine hybrid 64a–e. | ||
Some other coumarin–pyridine hybrids such as 65a and b (Fig. 62) (IC50: 69.1–377.8 mM against both A549 and MCF-7 cancer cell lines, MTT assay) were also active against the tested cancer cell lines, but most of them were much less potent than the references.120–124
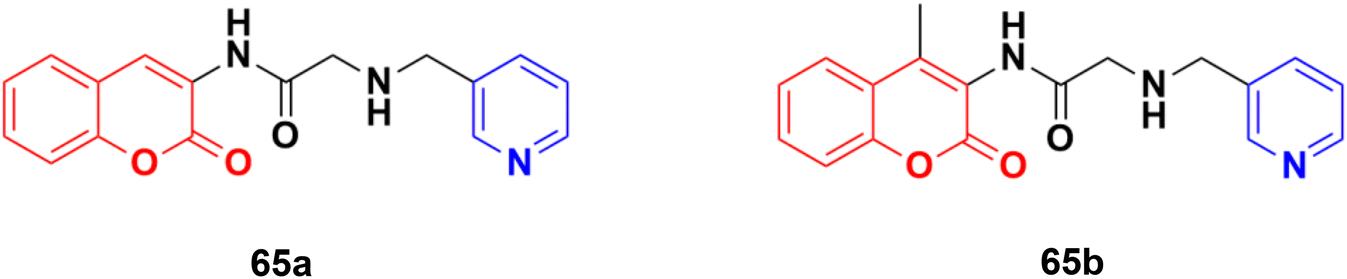 | ||
| Fig. 62 Chemical structures of coumarin–pyridine hybrid 65a and b. | ||
A novel series of fifteen coumarin–pyridine hybrids was synthesized via a one-pot four-component coupling reaction under a neat microwave method and their antiproliferative properties investigated against several cancer cell lines.125 Among them, compounds 66a and 66b (Fig. 63) exhibited promising anticancer activity at low concentrations (10−5 M) against the NCI-60 cell line. These two potent anticancer molecules were screened for their CT-DNA cleavage and fluorescence quenching with BSA transport protein.
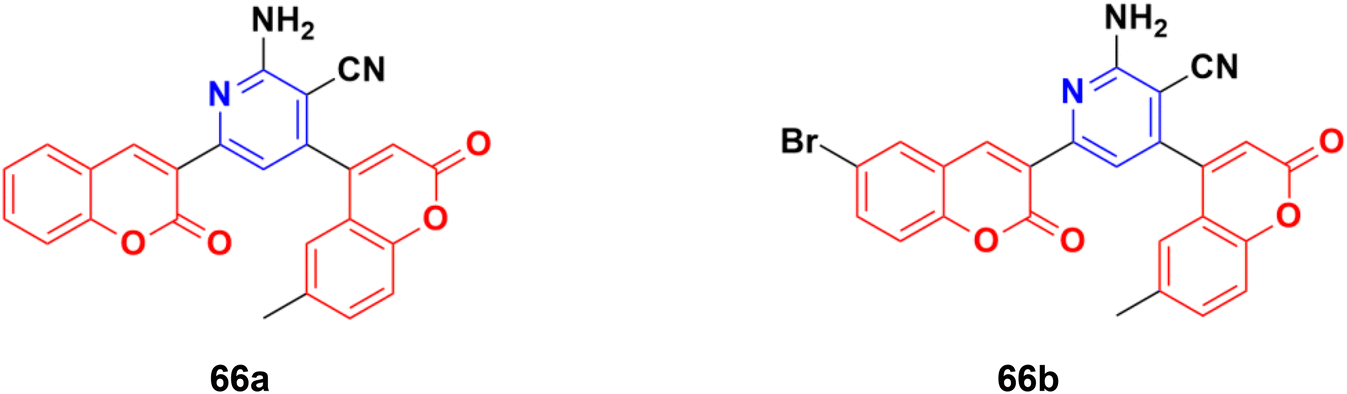 | ||
| Fig. 63 Chemical structures of coumarin–pyridine hybrid 66a and b. | ||
The similar synthesis of coumarin–pyridine conjugates was accomplished via conventional heating and microwave irradiation and the designed compounds were tested for their in vitro cytotoxicity.126 The preliminary screening results showed that most of the compounds had moderate cytotoxic activity against the HCT-116 and MCF-7 cell lines, although compound 67 (Fig. 64) exhibited potent activity against both cell lines with IC50 values of 9.9 ± 0.82 and 14.1 ± 1.14 μM, respectively, which was comparable with the standard drug 5-fluorouracil.
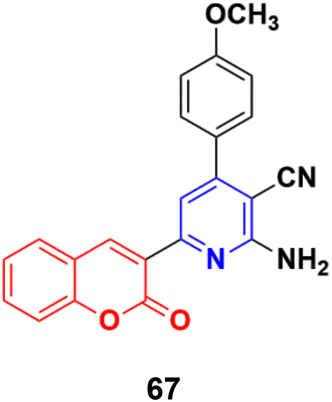 | ||
| Fig. 64 Chemical structures of coumarin–pyridine hybrid 67. | ||
A series of coumarin–pyridine hybrids was synthesized and their anticancer activity evaluated against the MCF-7, HCT-116, HepG-2, and A549 human cancer cell lines.127 Among them, compounds 68a–c (Fig. 65) showed the most potent growth inhibitory activities with IC50 values in the range of 1.1 to 2.4 μM against the MCF-7 cell line. Flow cytometric analysis revealed that these compounds induced cell cycle arrest in the G2/M phase followed by apoptotic cell death. Furthermore, the activity of caspase-3 in MCF-7 cells was tested. The results indicated that compounds 68a–c increased the caspase-3 activity significantly compared to the control group.
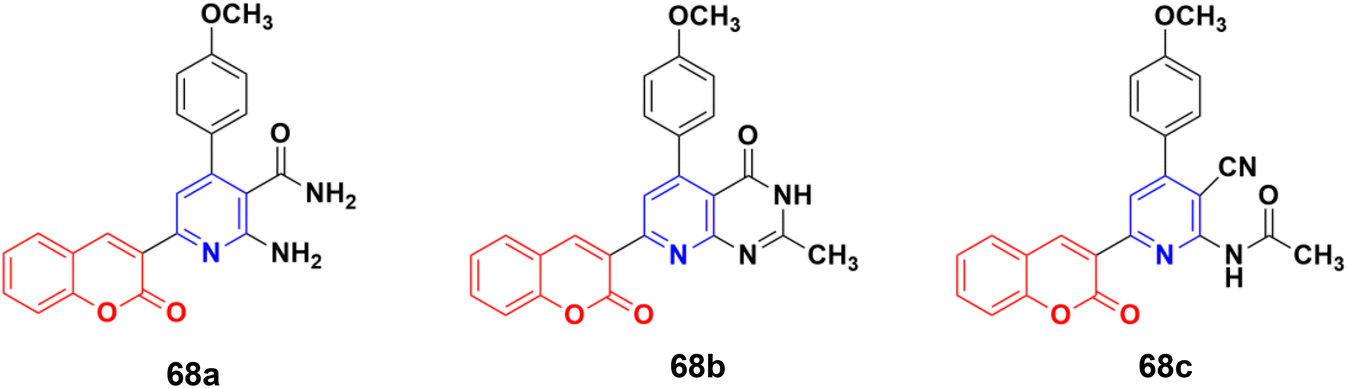 | ||
| Fig. 65 Chemical structures of coumarin–pyridine hybrid 68a–c. | ||
Several thio/furo-fused pyridine moieties having a coumarin scaffold were synthesized using an FeCl3-catalyzed modified Pictet–Spengler reaction as the crucial final step and their biological activities were evaluated against three cancer cell lines including DU145, B16F10, and MCF-7.128 Compound 69a (Fig. 66) possessed significant anticancer activity against the DU145 and B16F10 cell lines with IC50 values of 20.88 and 12.98 μM, respectively, whereas compound 69b was the most potent against the MCF-7 cell line (IC50 = 8.00 μM).
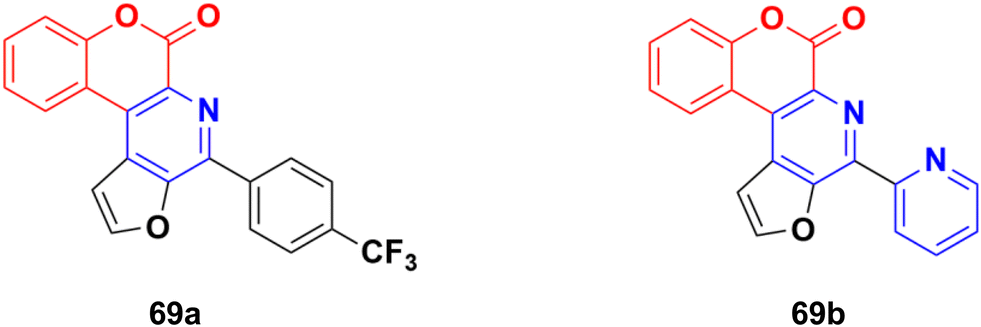 | ||
| Fig. 66 Chemical structures of coumarin–pyridine hybrid 69a and b. | ||
A series of twelve coumarin–pyrimidine conjugates was synthesized under microwave irradiation and their cytotoxic activities evaluated against the A-549 and MDA-MB-231 cancer cell lines.129 Some of them were observed to be more potent than the reference cis-platin. Compound 70a (Fig. 67) showed the maximum potency against A549 with IC50 = 2.15 ± 0.12 μM, while 70b was the most potent against the MCF-7 cell line with IC50 = 2.23 ± 0.19 μM. The DNA cleavage study by the gel electrophoresis method revealed that compounds 70a and b inhibited the growth of the pathogenic organism by cleaving the genome given that no traces of DNA were found.
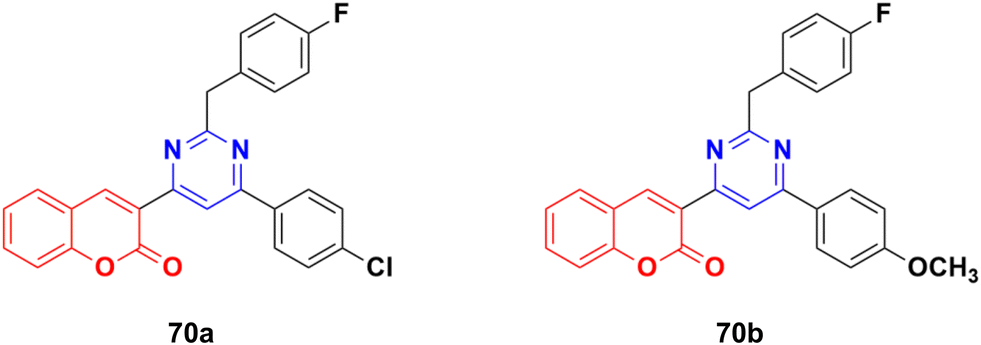 | ||
| Fig. 67 Chemical structures of coumarin–pyrimidine hybrid 70a and b. | ||
Coumarin–pyrimidine hybrid 71 (Fig. 68) showed significant antiproliferative activity against the HePG2 and MCF-7 cancer cell lines with IC50 values of 5.5 ± 0.19 and 6.9 ± 0.38 μg mL−1, respectively.130
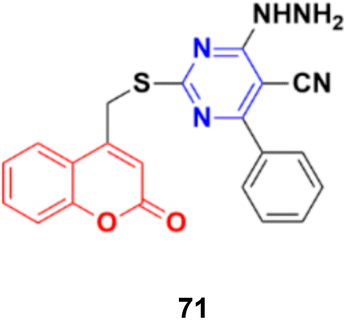 | ||
| Fig. 68 Chemical structure of coumarin–pyrimidine hybrid 71. | ||
A series of 2-phenylpyrimidine coumarin derivatives with potential telomerase-inhibiting activity was designed and synthesized and all the compounds were screened for their antiproliferative activity against the CNE2, KB, and Cal27 cell lines in vitro.131 Among them, compound 72 (Fig. 69) exhibited the best activity (IC50 = 1.92 ± 0.13, 3.72 ± 0.54, and 1.97 ± 0.51 against the CNE2, KB, and Cal27 cell lines, respectively). Flow cytometry revealed that this compound can inhibit CNE2 proliferation. The molecular docking results indicated that compound 72 bonded with telomerase reverse transcriptase (TERT) through multiple interactions, including hydrogen bonding and hydrophobic interactions.
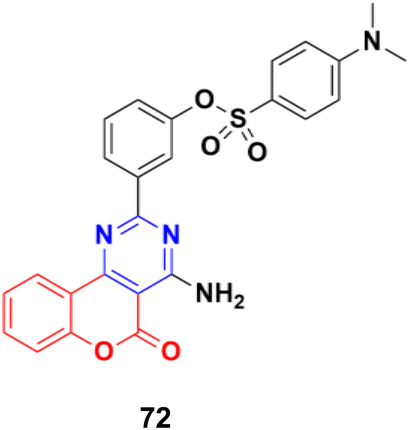 | ||
| Fig. 69 Chemical structure of coumarin–pyrimidine hybrid 72. | ||
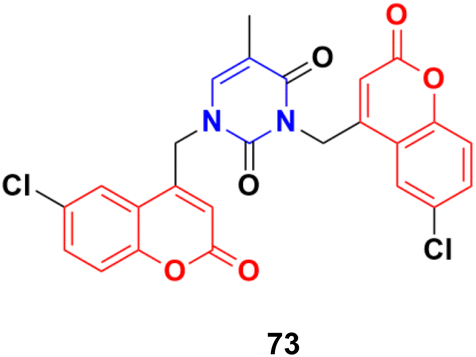
A series of C4–C4′ biscoumarin–pyrimidine hybrids was synthesized via SN2 reaction of substituted 4-bromomethyl coumarin with thymine and screened for in vitro anticancer activity against C6 rat glioma cells.132 Among the screened compounds, compound 73 (Fig. 70) was recognized to be the best antiproliferative candidate, having an IC50 value of 4.85 μM. All the compounds were found to be nontoxic toward healthy human embryonic kidney cells (HEK293). Furthermore, compound 73 displayed strong binding interactions with the drug carrier protein, human serum albumin, and exhibited good solution stability at biological pH conditions.
 | ||
| Fig. 70 Chemical structure of coumarin–pyrimidine hybrid 73. | ||
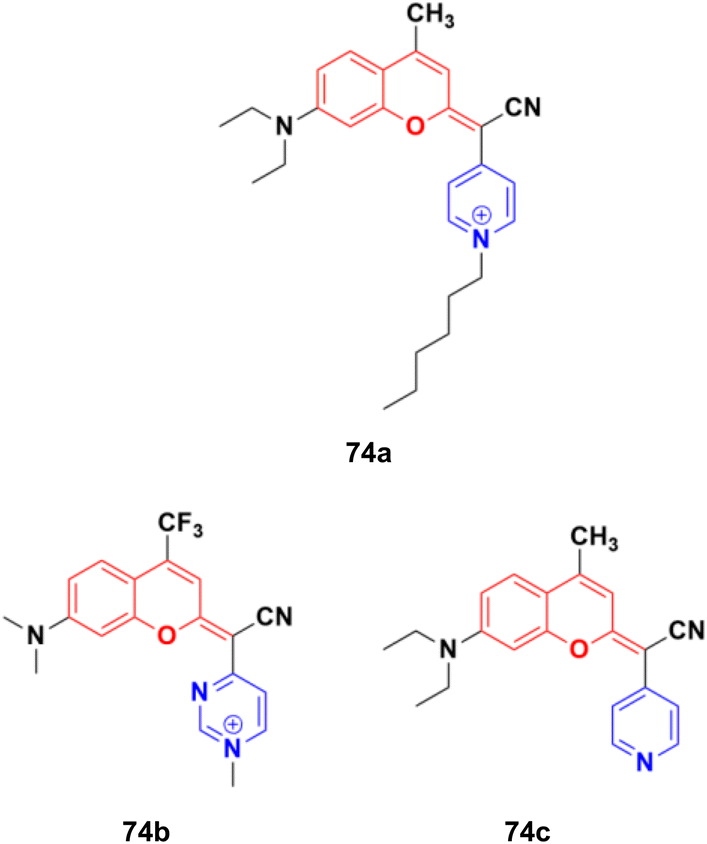
A fascinating family of low-molecular-weight coumarins (COUPYs) was developed, in which the carbonyl group of the lactone function of the classical coumarin scaffold was replaced by cyano(4-pyridine/pyrimidine)methylene moieties, and investigated as potential photodynamic therapy (PTT) anticancer tools.133 Among them, three compounds, 74a–c (Fig. 71), exhibited effective in vitro anticancer activities upon visible-light irradiation under both normoxia and hypoxia (phototherapeutic index of up to 71) and minimal toxicity toward normal cells. In addition, their cytotoxicity was also evaluated in non-tumorigenic ovarian tissue-derived cells (CHO) to determine their differential selectivity for cancer cell lines. Compound 74a showed excellent cytotoxicity against the HeLa and A2780 cell lines with IC50 of values 0.19 ± 0.03, 0.09 ± 0.02 μM, respectively, with a selectivity factor of 15.6. Furthermore, compounds 74b and 74c were also effective against HeLa and A2780 with IC50 values 1.1 ± 0.1 and 1.1 ± 0.3 μM, respectively, but their selectivity factor was low, and even lower than the reference cis-platin. Acting as excellent theranostic agents targeting mitochondria, the mechanism of action of these compounds was investigated in detail in HeLa cells. The generation of cytotoxic ROS and induction of apoptosis and/or autophagy were identified as the cell death modes triggered after irradiation with low doses of visible light.
 | ||
| Fig. 71 Chemical structures of potential PTT COUPY 74a–c. | ||
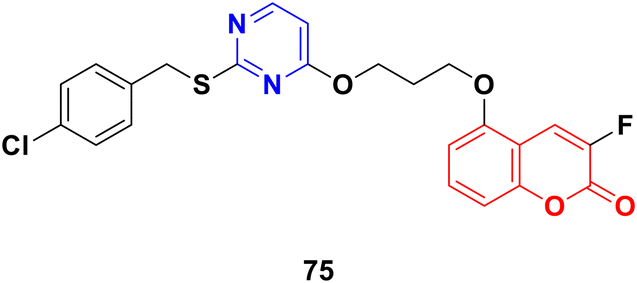
Coumarin-tagged pyrimidine scaffold 75 (Fig. 72) was found to selectively impede the proliferation of HER2-positive BC cells.134 It induced DNA damage and apoptosis in HER-2-positive BC cells more effectively compared to HER-2 negative BC cells. In silico and theoretical calculations revealed that compound 75 could interact with c-Jun N-terminal kinase (JNK), and in vitro studies showed that it increased JNK phosphorylation through ROS generation.
 | ||
| Fig. 72 Chemical structure of coumarin–pyrimidine hybrid 75. | ||
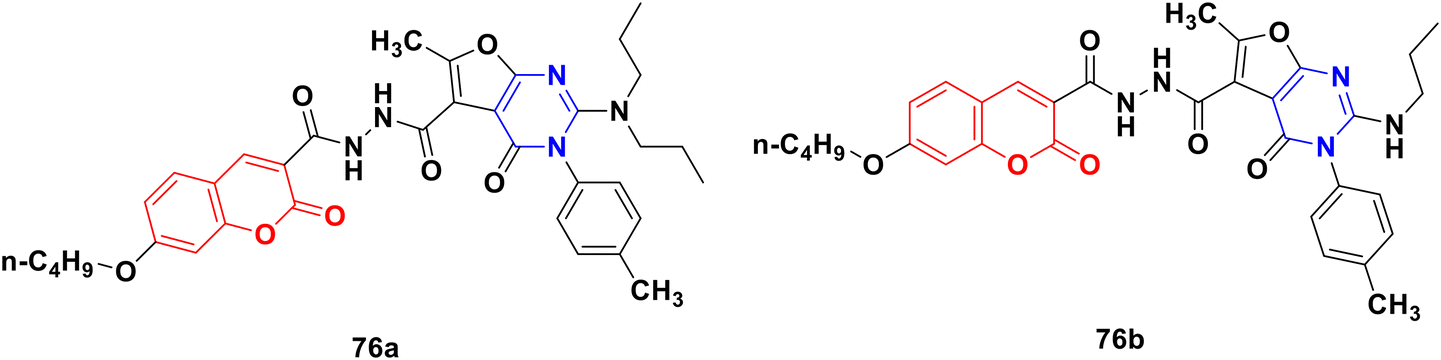
A novel library of coumarin-furo[2,3-d]pyrimidinone hybrid derivatives was synthesized and assessed for their antiproliferative activities against the HepG2 and HeLa cell lines in vitro.135 Compound 76a (Fig. 73) showed maximum potency against HepG2 with an IC50 value of 4.87 μmol L−1. The kinase activity assay revealed that compound 76a may be a multi-target inhibitor. Alternatively, compound 76b was the most potent against the HeLa cell line with an IC50 value of 6.47 μmol L−1. The structure–activity relationship study showed that a more bulky and electro-positive group at the C-2 position the of furo[2,3-d]pyrimidinone ring enhanced the bioactivity.
 | ||
| Fig. 73 Chemical structures of coumarin-furo[2,3-d]pyrimidinone hybrid 76a and b. | ||
2.5. Coumarin–indole/isatin hybrids
Indole (Fig. 74), consisting of a six-membered benzene ring fused to a five-membered pyrrole ring, is an important structural scaffold of various drugs.136,137 More than 200 indole derivatives have already been marketed as drugs (e.g., melatonin, indirubin, and sunitinib) or are in advanced stages of clinical trials. Similarly, isatin (Fig. 74), which is a derivative of indole, has also been observed to have efficient biological activity. This derivative possesses anticancer activities and can induce cell death.138–140 Hence, coumarin–indole/isatin hybrids may be important in the search for novel anticancer candidates.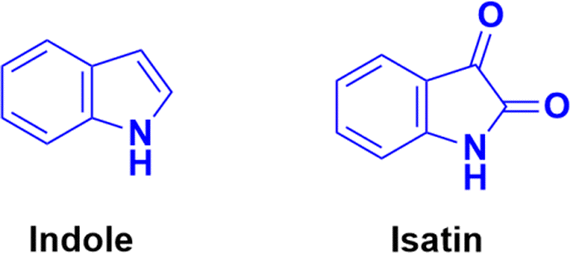 | ||
| Fig. 74 Chemical structures of indole and isatin. | ||
A series of indole-incorporated thiazolyl coumarins was developed and evaluated for their anticancer activities in vitro.141 Among them, compound 77 (NSC: 768621/1) (Fig. 75) showed excellent antiproliferative properties against the full panel of 60 human tumor cell lines. The five dose-level activity results revealed that compound 77 was active against all the cell lines. It showed potent activity against CCRFCEM (GI50: 0.33 μM), NCI-H522 (GI50: 1.03 μM), HCT116 (GI50: 1.60 μM), SF-539 (GI50: 1.58 μM), MALME-3 M (GI50: 1.59 μM), OVCAR-3 (GI50: 1.16 μM), UO-31 (GI50: 0.76 μM), PC-3 (GI50: 0.82 μM) and BT-549 (GI50: 1.13 μM).
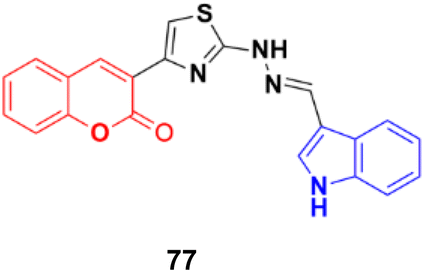 | ||
| Fig. 75 Chemical structure of coumarin–indole hybrid 77. | ||
6-(6-Fluoro-1H-indol-2-yl)-7-hydroxy-4,8-dimethyl-2H-chromen-2-one (78), a coumarin–indole conjugate (Fig. 76), showed the highest level of antimitotic activity with mean GI50/TGI values of 3.28/13.24 mM and certain sensitivity profile towards the non-small cell lung cancer cell line HOP-92 (GI50/TGI/LC50 values 0.95/4.17/29.9 mM).142
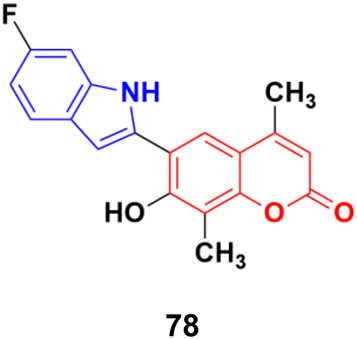 | ||
| Fig. 76 Chemical structure of coumarin–indole hybrid 78. | ||
Another series of coumarin–indole derivatives was synthesized and their cytotoxic activities investigated in vitro against an MCF-7 cancer cell line together with a normal cell line.143 Among the characterized compounds, 79 (Fig. 77) showed the maximum potency against the MCF-7 cell line with IC50 = 7.4 μM. Flow cytometric cell cycle analysis of 79 exhibited the apoptotic mode of cell death due to cell cycle arrest in the G2/M phase.
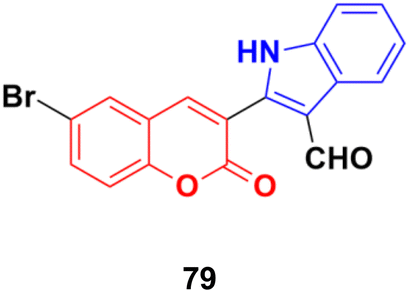 | ||
| Fig. 77 Chemical structure of coumarin–indole hybrid 79. | ||
Further investigation revealed that hybrid 80 (Fig. 78) with a bromine atom in position-7 of the coumarin ring displayed excellent dose-dependent cytotoxic activity with high selectivity for MCF-7 cells in the MTT assay.144 Flow cytometry analysis of 80 showed cell cycle arrest in the S phase and the accumulation of cells in the subG1 phase. The apoptotic mode of cell death induced by 80 was further confirmed by annexin-V staining assay. The wound healing assay revealed a profound impairment in the migration of MCF-7 cells presumably due to the down-regulation of Bcl-2 and Bcl-xL proteins induced by 80, as observed in the immunoblotting analysis. 80 was found to bind favorably to Bcl-2 and Bcl-xL in the docking simulation analysis, suggesting that it is a probable small molecule Bcl-2/Bcl-xL inhibitor and a potential lead for breast cancer chemotherapy with apoptotic and anti-metastatic properties.
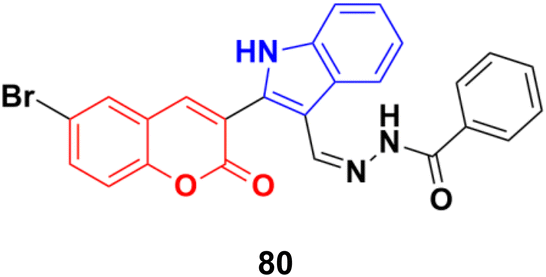 | ||
| Fig. 78 Chemical structure of coumarin–indole hybrid 80. | ||
A new series of indole–triazole–coumarin hybrids was developed via copper(I)-catalyzed [3 + 2] azide–alkyne cycloaddition and showed excellent binding affinity towards CDK2 kinase with cytotoxicity against the human breast cancer cell line MCF-7.145 The IC50 value (17.5 μM) and binding affinity (−11.2 kcal mol−1) obtained for 81 (Fig. 79) against MCF-7 cells are promising and it can act as a lead to generate more potential anticancer moieties.
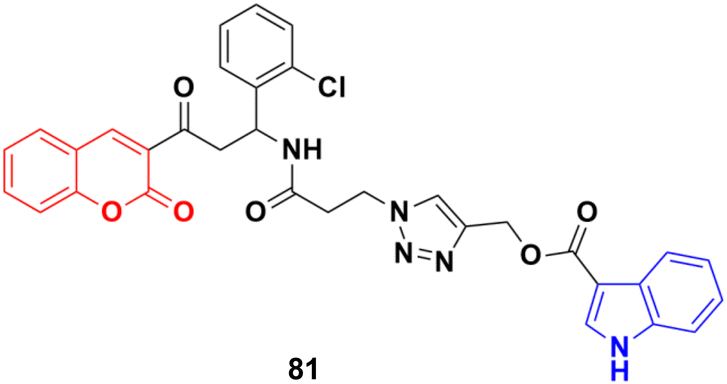 | ||
| Fig. 79 Chemical structure of coumarin–indole hybrid 81. | ||
Another coumarin–indole hybrid, 3-(1-(5-(3-(1H-indol-3-yl)propyl)-1,3,4-thiadiazol-2-ylimino)ethyl)-6-bromo-2H-chromen-2-one (IPTBC) (82) (Fig. 80), exhibited dose-dependent cytotoxicity in breast adenocarcinoma (MCF-7) cells.146 This compound induced cell apoptosis through the active involvement of caspases.
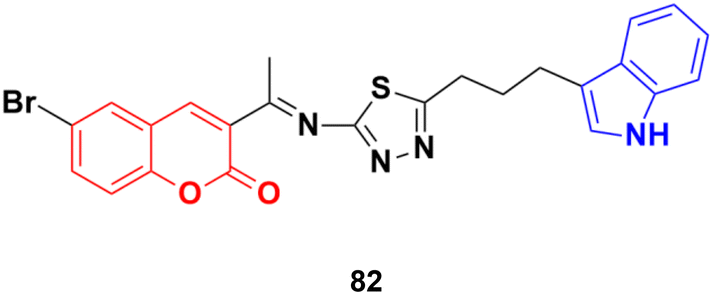 | ||
| Fig. 80 Chemical structure of coumarin–indole hybrid 82. | ||
The coumarin-β-carboline system 83 (Fig. 81) showed antiproliferative activity against the HeLa cell line with a GI50 value of 23.4 μg mL−1.147In silico studies indicated the binding properties of 83 with the kinesin spindle protein (KSP) and tubulin protein. Gel electrophoresis studies revealed that compound 83 completely cleaved the CT-DNA.
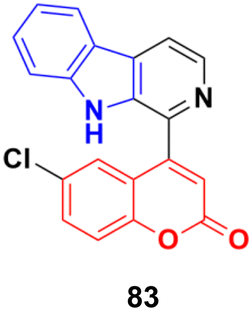 | ||
| Fig. 81 Chemical structure of coumarin-β-carboline system 83. | ||
A novel series of twelve ethylene glycol-tethered coumarin–isatin hybrids were designed and evaluated for their in vitro anticancer activities against HepG2, HeLa, A549, DU145, SKOV3, and MCF-7, and drug-resistant MCF-7/DOX (doxorubicin-resistant MCF7) by SRB assay.148 Among them, compound 84a showed the maximum potency against HepG2, A549, DU145, MCF-7, and MCF-7/DOX with IC50 values of 10.28, 10.92, 20.80, 11.29, and 14.45 μM, respectively. Compounds 84b and 84c possessed the highest efficiency against the HeLa and SKOV3 cell lines with IC50 values of 11.54 and 18.63 μM, respectively (Fig. 82).
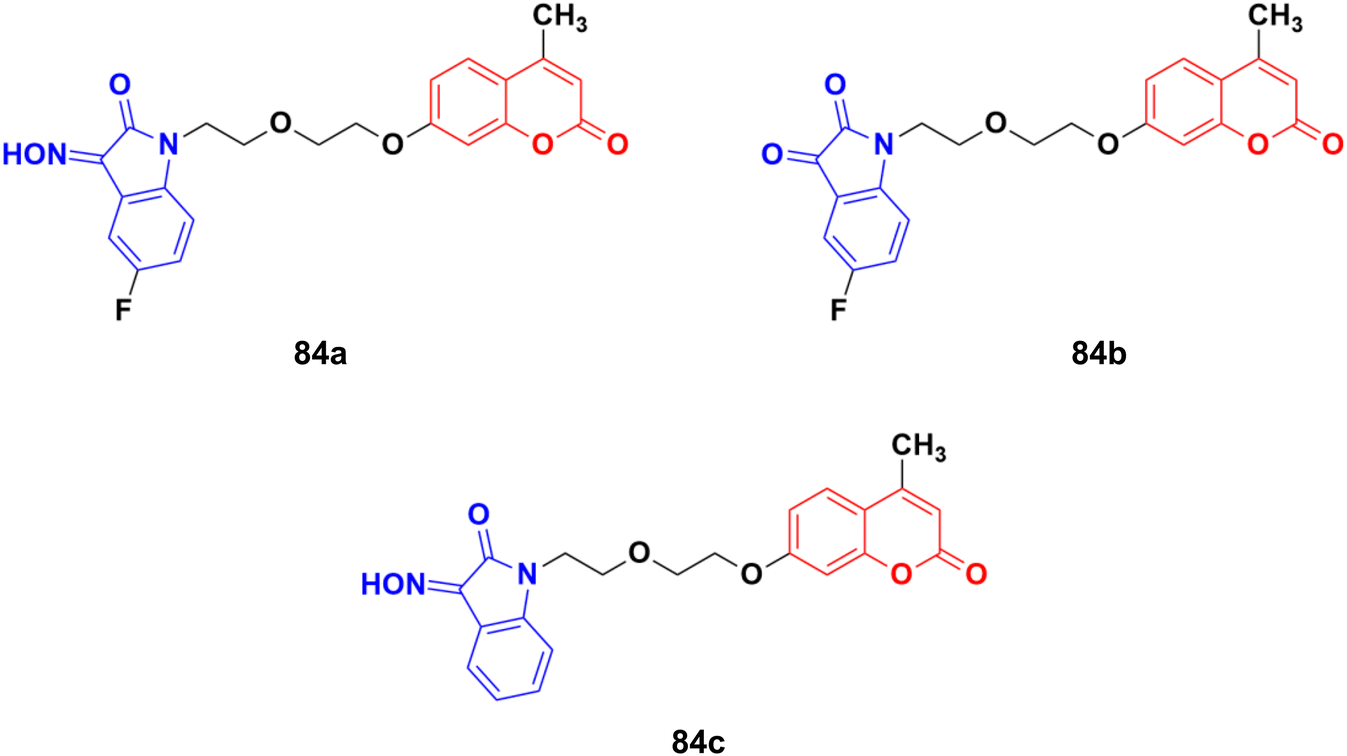 | ||
| Fig. 82 Chemical structures of coumarin–isatin hybrid 84a–c. | ||
Another series of twelve ethylene glycol-tethered coumarin–isatin hybrids was developed and examined for their in vitro cytotoxic activities against HepG2, HeLa, A549, DU145, SKOV3, and MCF-7 as well as drug-resistant MCF-7/DOX (doxorubicin-resistant MCF-7) human cancer cell lines.149 Most of them had very little or no anticancer activities. Compound 85 (Fig. 83) showed moderate activity against the SKOV3, and MCF-7 and MCF-7/DOX cell lines with IC50 values of 23.76, 11.90, and 18.85 μM, respectively.
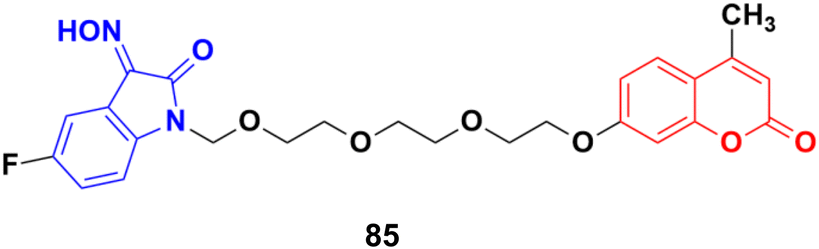 | ||
| Fig. 83 Chemical structure of coumarin–isatin hybrid 85. | ||
A series of twelve diethylene glycol-tethered isatin–1,2,3-triazole–coumarin hybrids was synthesized and investigated for their in vitro anticancer activities against HepG2, HeLa, A549, DU145, SKOV3, MCF-7, and MCF-7/DOX human cancer cell lines.150 Among them, compound 86a (Fig. 84) showed excellent potency against six of the seven cell lines (IC50 values of 19.89, 21.32, 18.67, 31.50, 17.96, and 15.46 μM against HepG2, HeLa, A549, DU145, SKOV3, and MCF-7/DOX cell lines, respectively). Compound 86b was most effective against the MCF-7 cell line with IC50 = 28.74 μM.
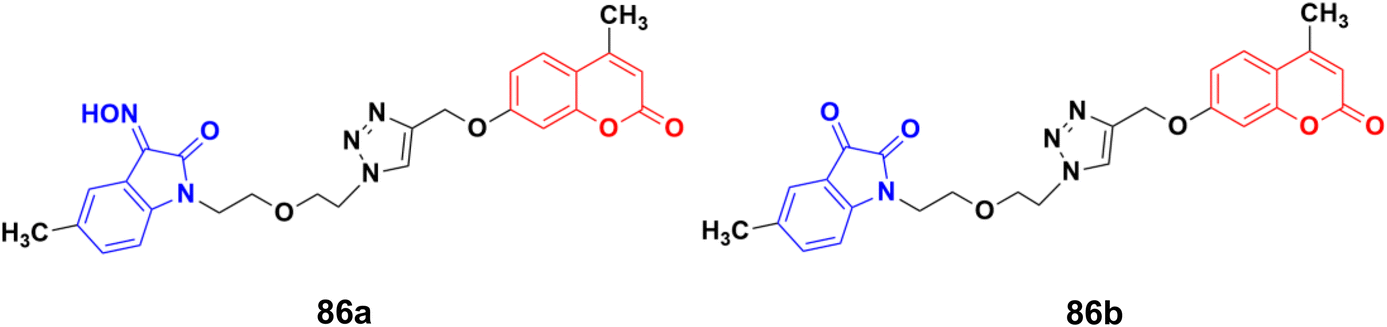 | ||
| Fig. 84 Chemical structures of coumarin–isatin hybrid 86a and b. | ||
A similar study was carried out on tetraethylene glycol-tethered isatin–1,2,3-triazole–coumarin hybrids.151 Among them, compound 87 (Fig. 85) possessed the maximum anticancer activity (IC50 values of 26.11, 25.49, 28.74, 33.42, 35.28, 29.25, and 20.09 against HepG2, HeLa, A549, DU145, SKOV3, MCF-7, and MCF-7/DOX, respectively).
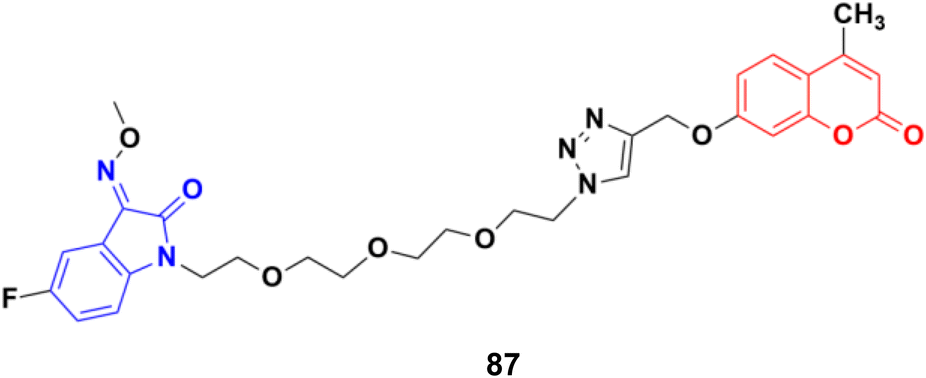 | ||
| Fig. 85 Chemical structure of coumarin–isatin hybrid 87. | ||
A sulphonic acid-functionalized nitrogen sulfur co-doped graphite (SO3H-NSG)-based catalyst was prepared by coating the surface of carbon with a sulphonic acid-bearing ionic liquid, which was used for the synthesis of biologically active coumarin-substituted bis(indolyl)methanes that were finally evaluated for their toxicity and anticancer properties.152 The cytotoxicities of compounds 88a–d (Fig. 86) were investigated against the human breast carcinoma cell line (MCF-7), osteosarcoma cell line (HOS), and normal kidney epithelial cell line (NKE) by the Amber blue reduction assay. Among them, 88a–c showed significant cytotoxicity towards the breast cancer cell line with IC50 of 19.75, 28.04, and 21.19 μM, respectively, whereas 88d was practically nontoxic. Similarly, 88a, b, and d showed significant cytotoxicity towards the osteosarcoma cell line with IC50 of 20.76, 8.75, and 12.23 μM, respectively, whereas 88c was practically nontoxic. These compounds were also evaluated for their possible cytotoxicity against normal human cell lines (NKE). However, they did not significantly affect the growth of normal human kidney cells (IC50 values of >50 μM).
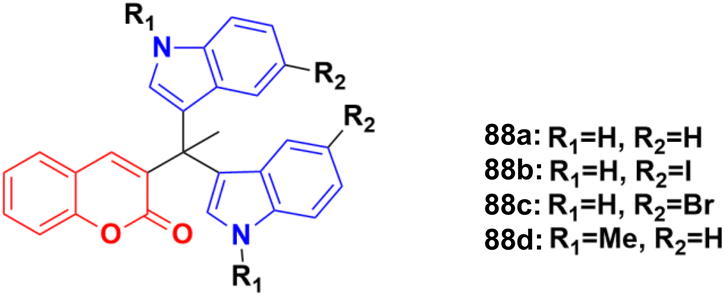 | ||
| Fig. 86 Chemical structures of coumarin–isatin hybrid 88a–d. | ||
A novel family of coumarin–indole derivatives was synthesized and evaluated as tubulin polymerization inhibitors targeting the colchicine binding site.153 Among them, compound MY-413 (89) (Fig. 87) displayed the most potent inhibitory activities against the gastric cancer cell line MGC-803 with an IC50 value of 0.011 μM. Furthermore, the IC50 values of compound 89 on fifteen cancer cell lines were lower than 100 nM and the IC50 values of 9 cancer cell lines were less than 50 nM. Compound 89 effectively inhibited tubulin polymerization (IC50 = 2.46 μM) by binding to the colchicine site. Screening for the inhibitory effects of the compound on 61 kinases revealed that compound MY-413 could inhibit MAPK 39 pathway-related kinases. Because of the inhibitory effects of compound MY-413 on tubulin polymerization and MAPK pathways, compound 89 induced cell apoptosis, arrested the cell cycle in the G2/M phase, induced cell proliferation inhibition and cell migration inhibition in the gastric cancer cell lines MGC-803 and HGC-27. In addition, compound MY-413 could significantly inhibit tumor growth in MGC-803 with tumor growth inhibition (TGI) rates of 70% (15 mg kg−1), 45% and 80% (30 mg kg−1) without obvious toxicity. Consistent with the in vitro results, compound MY-413 also inhibited the MAPK signaling pathway, and induced apoptosis and proliferation inhibition in vivo.
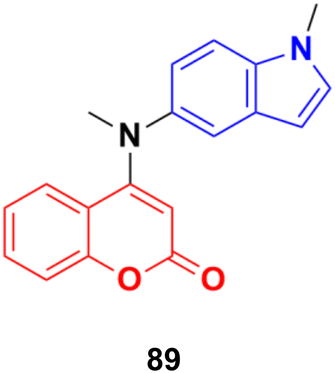 | ||
| Fig. 87 Chemical structure of coumarin–isatin hybrid 89 (MY-413). | ||
2.6. Coumarin–chalcone hybrids
Chalcone (Fig. 88), an α,β-unsaturated ketone, is biologically important and considered a privileged scaffold in medicinal chemistry. The beneficial effect of these substances has been studied in diabetes mellitus. Chalcone derivatives have been linked with anti-inflammatory, analgesic, antipyretic, antimutagenic, antileishmanial, antiproliferative, and antifungal effects.154–160 Therefore, the chalcone skeleton can be considered a useful scaffold, and its hybridization with the coumarin moiety may lead to the discovery of new potent anticancer drugs. | ||
| Fig. 88 Chemical structure of chalcone. | ||
A novel series of coumarin–chalcone hybrids was synthesized and evaluated for anti-proliferative activity against the estrogen receptor-positive MCF-7 and negative MDA-MB-435 breast cancer cell lines.161 Compounds 90a–c (Fig. 89) showed significant potency against the MCF-7 cell line with GI50 values of <10, 18.9, and 32.5 μg mL−1, respectively. Moreover, compound 90a was observed to be superior to N-methyl nitrosourea in vivo in terms of latency (5.5 weeks vs. 4.5 weeks) and reducing the tumor burden (3.1 vs. 4.45) and volume (3.3 mm3vs. 4.8 mm3), suggesting that it can act as a lead for the generation of more potent anticancer candidates.
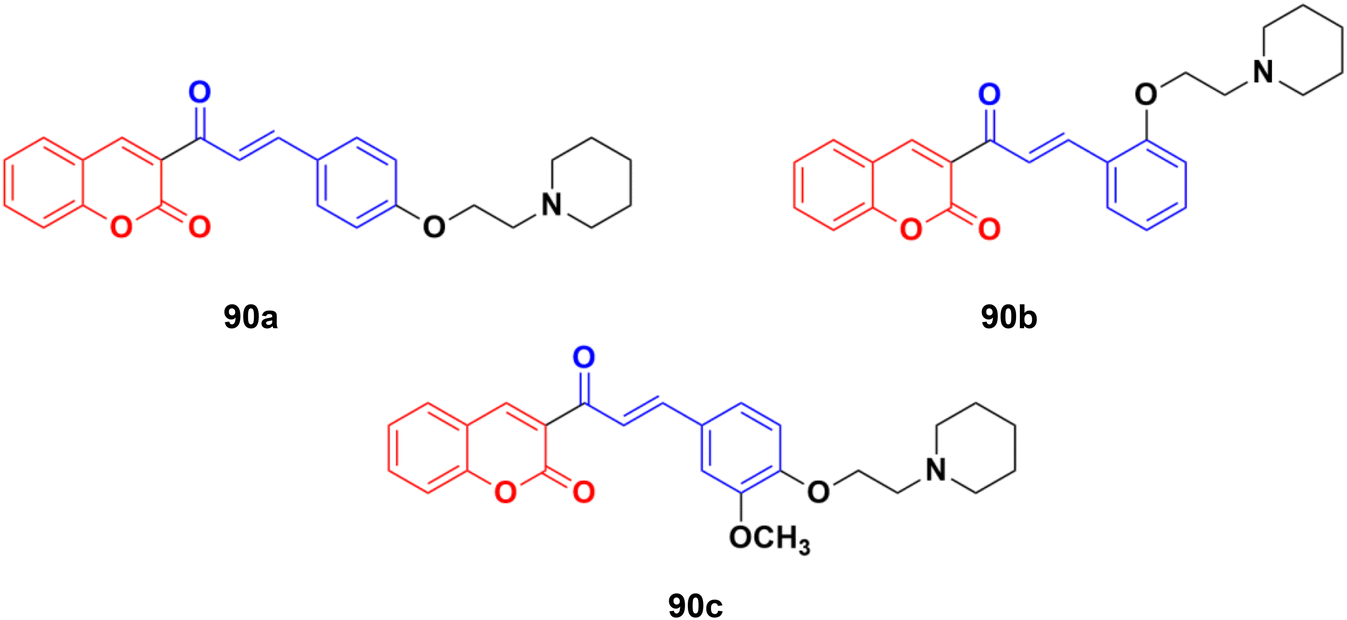 | ||
| Fig. 89 Chemical structures of coumarin–chalcone hybrid 90a–c. | ||
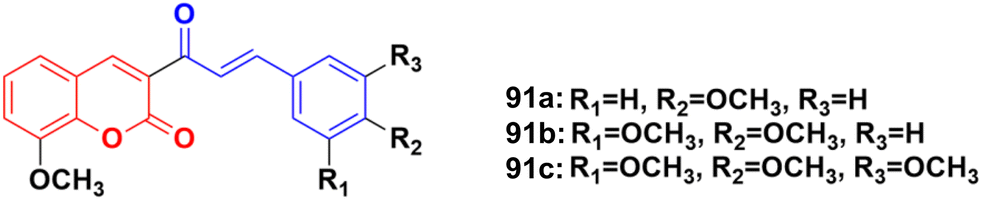
Coumarin–chalcone hybrids 91a–c (Fig. 89) showed anticancer activity against the HepG2 and leukemia K562 cell lines.162 Compound 91a was the most potent against HepG2 with IC50 = 0.65 μM, while 91b showed the maximum efficiency against leukemia K562 with IC50 = 0.93 μM. Furthermore, cell cycle analysis of 91a showed the activation of apoptotic signals as a result of cell cycle arrest in the G2/M phase (Fig. 90).
 | ||
| Fig. 90 Chemical structures of coumarin–chalcone hybrid 91a–c. | ||
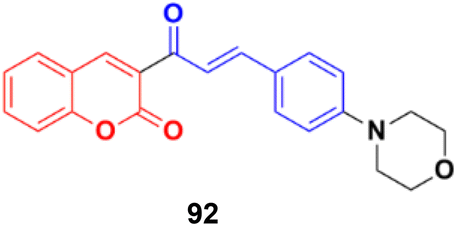
Another coumarin–chalcone hybrid (92) (Fig. 91), which is structurally similar to 90, was synthesized and its anticancer activity was investigated against the T47D breast cancer cell line and cervix cancer cell line HeLa.163 This compound has an IC50 of 0.90 μM for the T47D breast cancer cell line and 2.32 μM for the HeLa cervix cancer cell line.
 | ||
| Fig. 91 Chemical structure of coumarin–chalcone hybrid 92. | ||
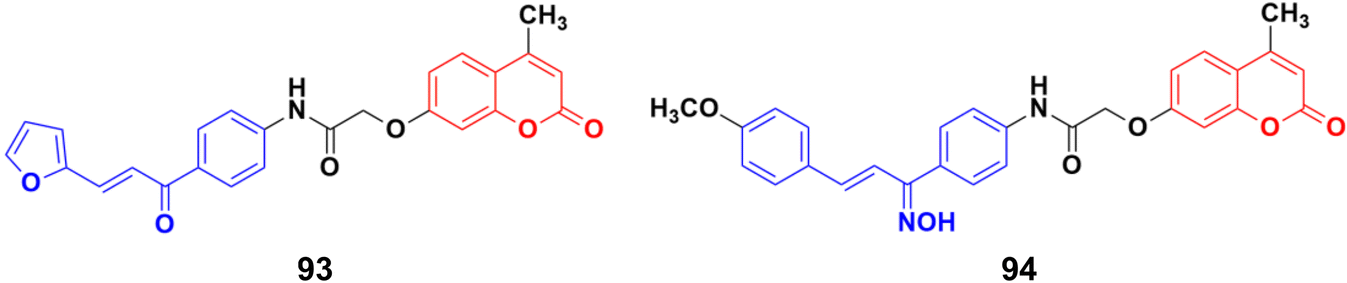
A novel series of coumarin–chalcone conjugates and their NO hybrids was designed and their antiproliferative properties investigated against the MCF-7 cancer cell line.164 The coumarin–chalcone hybrid 93 (Fig. 92) showed the maximum potency with an IC50 value of 9.62 μg mL−1. Among the NO hybrids, compound 94 was the most effective with an IC50 value of 20.9 μg mL−1.
 | ||
| Fig. 92 Chemical structures of coumarin–chalcone hybrid 93 and NO hybrid 94. | ||
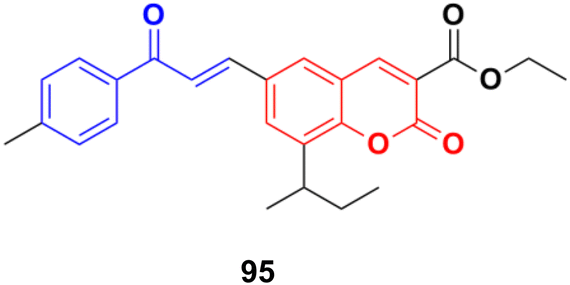
S009-131 (95) (Fig. 93), a coumarin–chalcone hybrid, possessed anti-proliferative and anti-tumor effects by triggering apoptosis.165 S009-131 caused DNA damage by potential binding to the minor groove, which led to the phosphorylation and activation of ATM and DNA-PK, but not ATR. S009-131 induced the DNA-damage-response-triggered activation of p53 through phosphorylation at its key residues.
 | ||
| Fig. 93 Chemical structure of coumarin–chalcone hybrid 95 (S009-131). | ||
A series of novel coumarin–chalcone derivatives containing a urea moiety was developed and screened for their in vitro antiproliferative activities against cancer cell lines (H4IIE and HepG2).166 In addition, the compounds were also tested on a normal cell line (CHO). Among them, compound 96a (Fig. 94) exhibited the maximum potency against H4IIE compared to sorafenib with an IC50 value of 1.62 ± 0.57 μM. Compound 96b also showed better inhibition against HepG2 than sorafenib with an IC50 value of 2.326 ± 0.23 μM. Particularly, 96a induced H4IIE apoptosis and arrested cell cycle in the S phase.
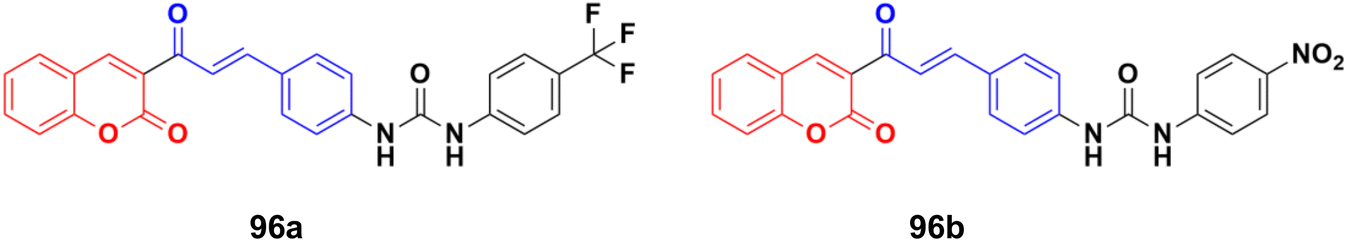 | ||
| Fig. 94 Chemical structures of coumarin–chalcone hybrid 96a and b. | ||
A series of coumarin–chalcone hybrids was synthesized as selenoprotein thioredoxin reductase (TrxRs) inhibitors.167 Most of them exhibited enhanced anticancer activity compared to xanthohumol (Xn). Among them, compound 97 (Fig. 95) (IC50 = 3.6 μM), a fluorescence agent, down-regulated the expression of TrxR and remarkably induced ROS accumulation to activate the mitochondrial apoptosis pathway. Furthermore, it inhibited cancer cell metastasis and abolished the colony formation ability of cancer cells.
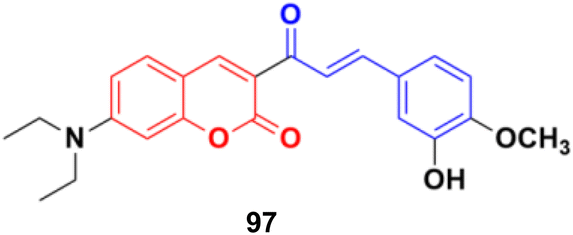 | ||
| Fig. 95 Chemical structure of coumarin–chalcone hybrid 97. | ||
A new series of thirteen coumarin-yl–chalcone derivatives was synthesized and in silico studies were performed to predict the anticancer activity of the compounds against Src, Alb tyrosine kinase, and homology model protein (PDB ID: 4csv).168 Derivatives 98a and b (Fig. 96) showed moderate binding energies. The in vitro cytotoxic activity was analyzed for these two compounds against three human cell lines, including A549, Jurkat, and MCF-7. The results indicated that the hybrids displayed significant anticancer activity but are less cytotoxic than the standard imatinib.
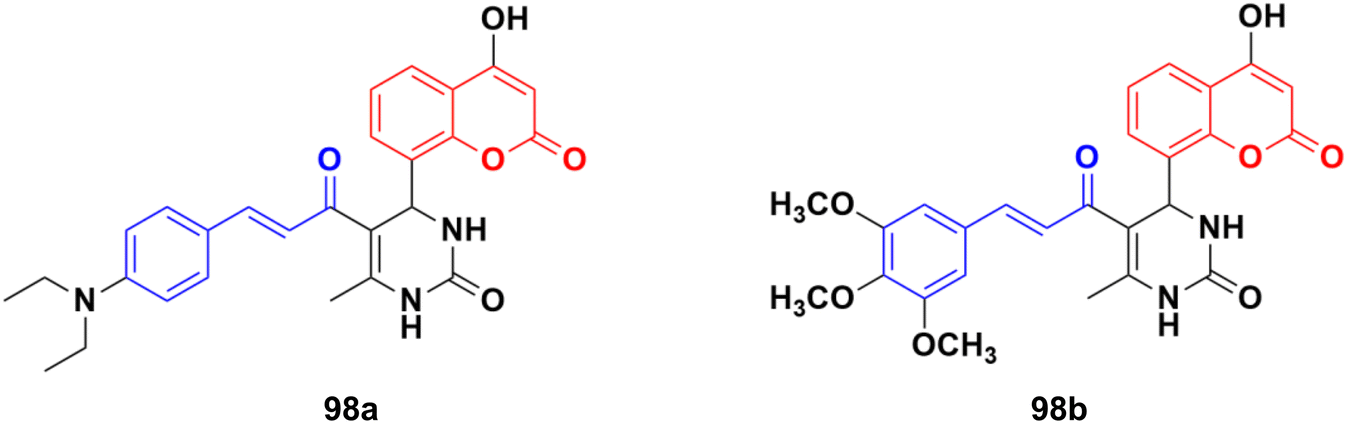 | ||
| Fig. 96 Chemical structures of coumarin–chalcone hybrid 98a and b. | ||
A water-soluble chemo-sensor (99) (Fig. 97) consisting of a chalcone–coumarin framework, which displayed excellent selectivity and sensitivity towards Al3+ ions, showed significant anticancer activity against the MCF-7 cancer cell line with an IC50 value of 15.38 μM.169
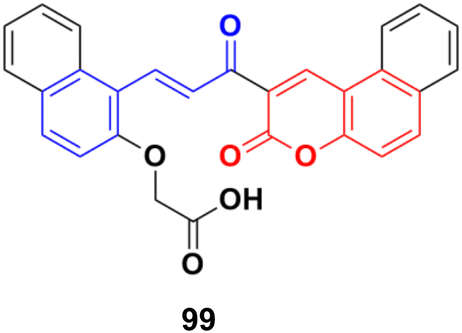 | ||
| Fig. 97 Chemical structure of coumarin–chalcone hybrid 99. | ||
2.7. Coumarin-imine hybrids
Imine (Fig. 98), consisting of a carbon–nitrogen double bond, is regarded as an important pharmacophore and has been used in the synthesis of many drugs.170 It can bind with the various active sites of living organisms through non-covalent interaction, which can be used to design new drugs effectively.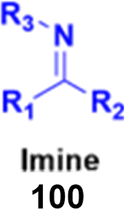 | ||
| Fig. 98 Chemical structure of imine. | ||
A similar series of nine coumarin-imine hybrids (101a–i) (Fig. 99) was synthesized and their antiproliferative profile evaluated against fibroblast cell lines and A549 cancer cell line.171 The percentage of viable cells was determined at different concentrations in the range of 12.5 to 200 μg mL−1. In terms of the WST-1 results, the concentrations of the compounds did not have a prominent effect on cell mortality in the cell line. For the fibroblast cells, the results were significant for only 101a and 101b. However, for the other samples (101c–i), increasing the concentrations of the compounds caused an increase in cell death.
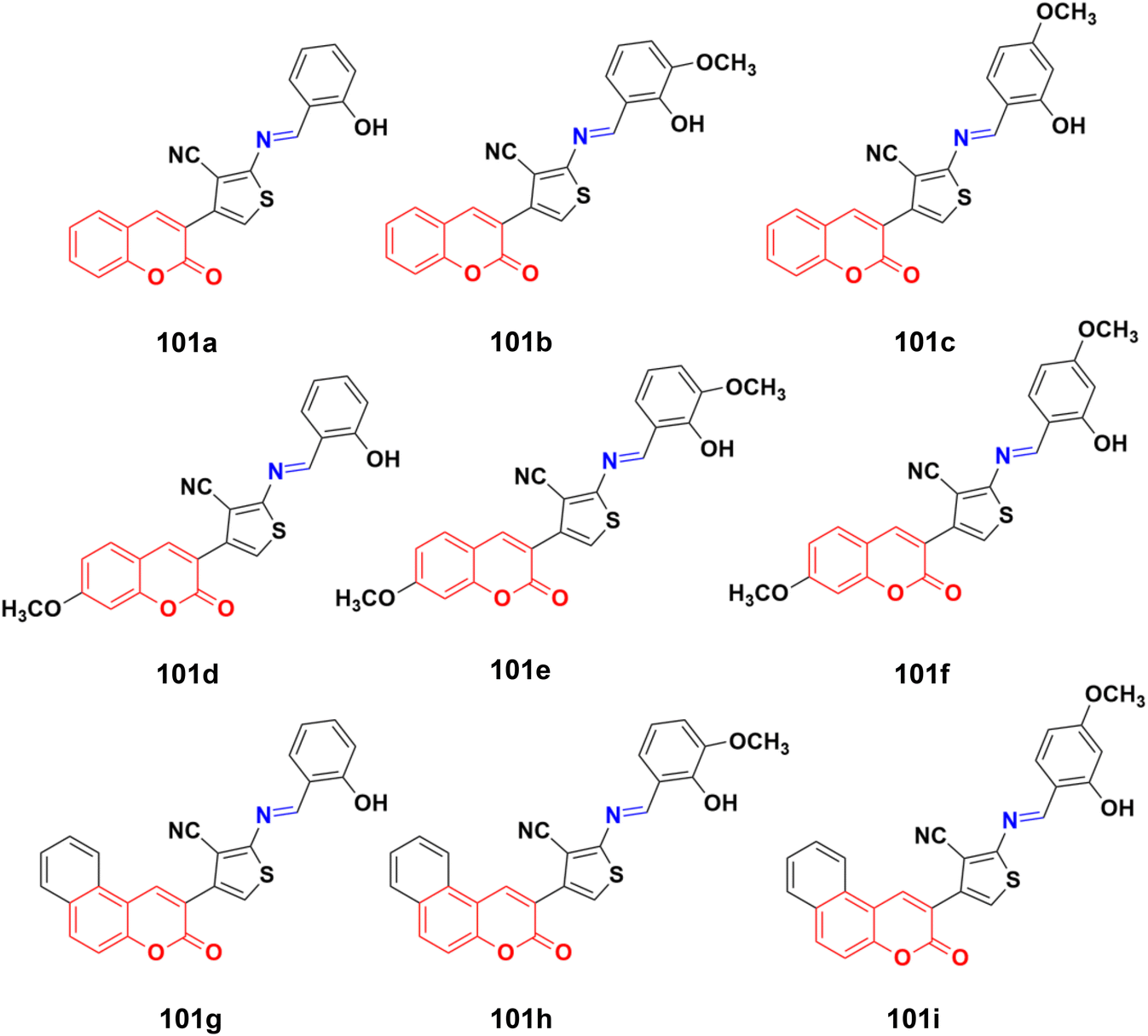 | ||
| Fig. 99 Chemical structures of coumarin-imine hybrids 101a–i. | ||
A series of coumarin–hydrazone hybrids was designed and evaluated for their anticancer activities against four cancer cell lines.172 Among them, compound 102 (Fig. 100) showed the most potency with IC50 = 2.9 ± 0.4, 5.3 ± 1.1, 7.2 ± 0.9, and 9.1 ± 1.2 μM against the HL-60, KE-37, K-562, and MDA-MB-231 cell lines, respectively.
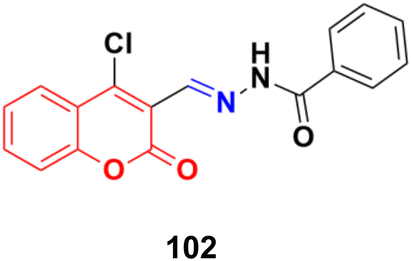 | ||
| Fig. 100 Chemical structure of coumarin-imine hybrid 102. | ||
2.8. Coumarin-sulfonamide/sulfamate/sulfonate hybrids
A novel series of coumarin sulfonamide derivatives was designed to improve the biological activities of COX-2 inhibition and anticancer.173 Among the synthesized compounds, 103 (Fig. 101) possessed the most powerful selective inhibitory and antiproliferative activity (IC50 = 0.09 μM for COX-2, 48.20 μM for COX-1, and 0.36 μM against HeLa cells), which is comparable to the control positive compound celecoxib (0.31 μM, 43.37 μM, and 7.79 μM). Compound 103 effectively induced HeLa cell apoptosis in a dose- and time-dependent manner. Moreover, it could significantly suppress cancer cell adhesion, migration, and invasion. The docking simulation results further indicated that 103 could bind well to the COX-2 active site and guided the reasonable design of a selective COX-2 inhibitor with an anticancer nature shortly.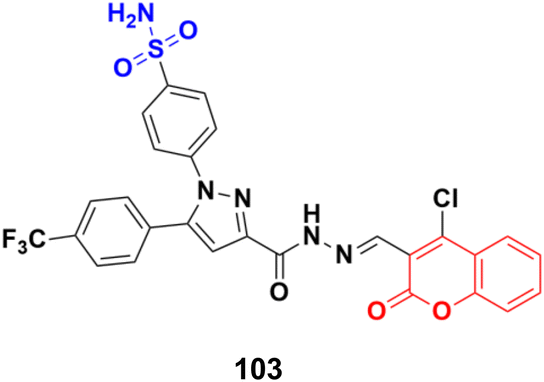 | ||
| Fig. 101 Chemical structure of coumarin-sulfonamide hybrid 103. | ||
Coumarin-sulfonamide hybrid 104 (Fig. 102) was designed to obtain novel dual inhibitors of COX-2 and 5-LOX.174 Compound 104 (IC50 = 0.23 ± 0.16 μM for COX-2, 0.87 ± 0.07 μM for 5-LOX, and 4.48 ± 0.57 μM against A549) showed preliminary superiority compared with the positive controls celecoxib (IC50 = 0.41 ± 0.28 μM for COX-2 and 7.68 ± 0.55 μM against A549) and zileuton (IC50 = 1.35 ± 0.24 μM for 5-LOX). Further investigation confirmed that 104 could induce apoptosis and cell cycle arrest at the G2 phase in a dose-dependent manner in human non-small cell lung cancer A549 cells.
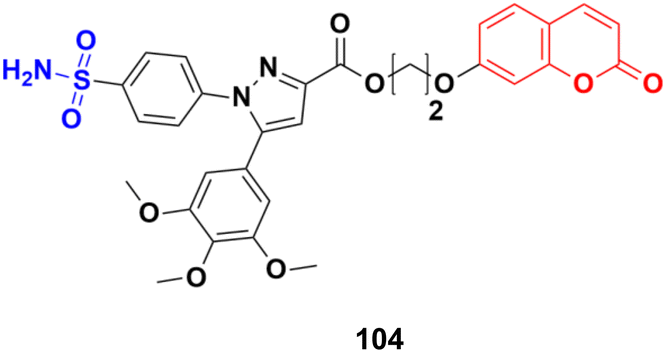 | ||
| Fig. 102 Chemical structure of coumarin-sulfonamide hybrid 104. | ||
Different novel sets of coumarin-6-sulfonamide derivatives were synthesized and their growth inhibitory activity evaluated in vitro towards the proliferation of three cancer cell lines, i.e., HepG2, MCF-7, and Caco-2.175 Compounds 105a and 105b (Fig. 103) emerged as the most active members against HepG2 cells (IC50 = 3.48 ± 0.28 and 5.03 ± 0.39 mM, respectively). These compounds could induce apoptosis in HepG2 cells, as demonstrated by the upregulation of Bax and downregulation of Bcl-2, besides boosting the caspase-3 levels. Besides, compound 105a induced a significant increase in the percentage of cells at pre-G1 by 6.4-fold, with concurrent significant arrest in the G2-M phase by 5.4-fold compared to the control. Also, 105a displayed a significant increase in the percentage of annexin V-FITC-positive apoptotic cells from 1.75% to 13.76%.
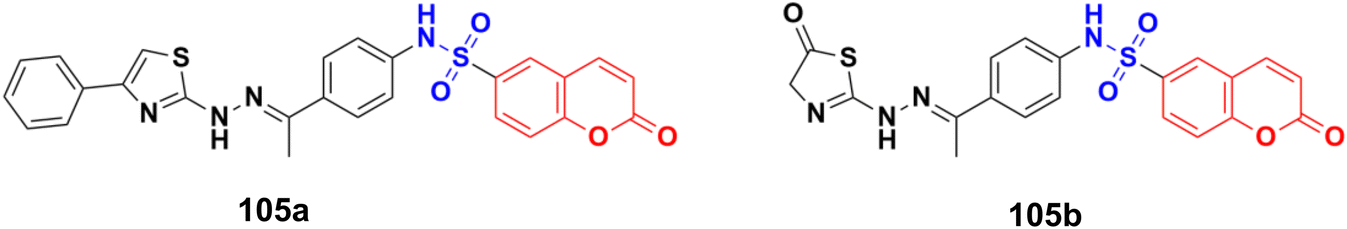 | ||
| Fig. 103 Chemical structures of coumarin-sulfonamide hybrid 105a and b. | ||
Coumarin-sulfonamide hybrid 106 (Fig. 104) showed excellent antiproliferative properties against the MCF-7 cancer cell line with IC50 = 2.53 μM, together with the selective index (SI) of 59.26.176
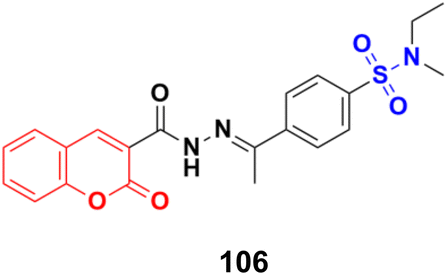 | ||
| Fig. 104 Chemical structure of coumarin-sulfonamide hybrid 106. | ||
Coumarin-sulfonamide hybrid 107 (Fig. 105), synthesized as a new chemotype of BRD4 inhibitors, possessed excellent cytotoxic ability against A549 (IC50 = 4.63 μM), HepG2 (IC50 = 4.75 μM), PANC-1 (IC50 = 7.02 μM), and SGC-7901 (IC50 = 6.39 μM) cell lines.177 Moreover, compound 107 exhibited potent BRD4 binding affinity and cell proliferation inhibitory activity, and especially displayed a favorable PK profile with high oral bioavailability (F = 49.38%) and metabolic stability (T1/2 = 4.2 h), meaningfully making it a promising lead compound for further drug development.
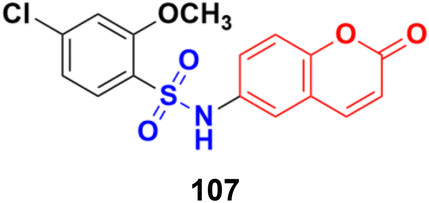 | ||
| Fig. 105 Chemical structure of coumarin-sulfonamide hybrid 107. | ||
A novel series of coumarin-sulfamate hybrids was prepared as potential STS inhibitors.178 The inhibitory effects of the synthesized compounds were tested on STS isolated from the human placenta and against estrogen receptor-(ER)-positive MCF-7 and T47D cells, as well as ER-negative MDA-MB-231 and SkBr3 cancer cell lines. Among the synthesized compounds, 108a and 108b (Fig. 106) showed the highest inhibitory effect in enzymatic STS assays, both with IC50 values of 0.18 μM. Compound 108b exhibited the highest potency against the MCF-7 and T47D cell lines (15.9 μM and 8.7 μM, respectively).
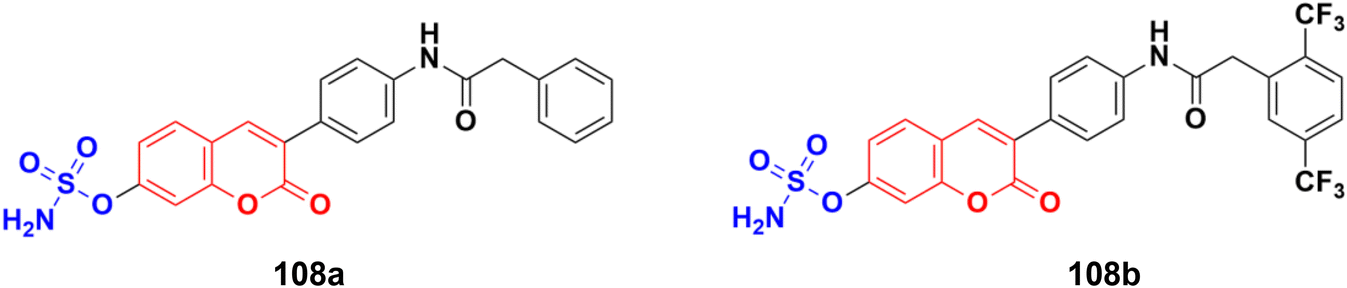 | ||
| Fig. 106 Chemical structures of coumarin-sulfamate hybrid 108a and b. | ||
Potent bicyclic nonsteroidal sulfamate-based active-site-directed inhibitors of the enzyme steroid sulfatase (STS), an emerging target in the treatment of post-menopausal hormone-dependent diseases, including breast cancer, were designed.179 The compounds were examined for STS inhibition in intact MCF-7 breast cancer cells and in placental microsomes. 3-Hexyl-4-methylcoumarin-7-O-sulfamate 109a and 3-benzyl-4-methylcoumarin-7-O-sulfamate 109b (Fig. 107) were particularly effective inhibitors with IC50 values of 0.68 and 1 nM in intact MCF-7 cells and 8 and 32 nM for placental microsomal STS, respectively. They were docked in the STS active site for comparison with estrone 3-O-sulfamate and irosustat, showing their sulfamate group close to the catalytic hydrated formylglycine residue and their pendant group lying between the hydrophobic side-chains of L103, F178, and F488.
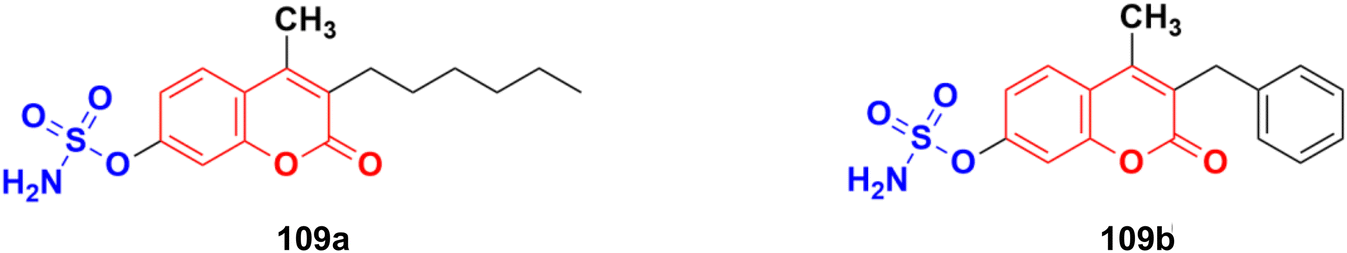 | ||
| Fig. 107 Chemical structures of coumarin-sulfamate hybrid 109a and b. | ||
A series of STAT3 inhibitors was developed and their anti-proliferative activity against four cancer cells investigated.180 Among them, compound 110 (Fig. 108) was the most potent with IC50 = 1.43 ± 0.30, 1.89 ± 0.42, 2.88 ± 0.69, and 3.33 ± 0.23 μM against the MDA-MB-231, HCT-116, HepG2, and MCF-7 cancer cell lines, respectively. STAT3 phosphorylation was inhibited by compound 110 at both Tyr705 and Ser727 residues. Compound 110 inhibited STAT3 phosphorylation, whereas it did not influence the phosphorylation levels of STAT1, 26 JAK2, Src, and Erk1/2, indicating its good selectivity. Moreover, compound 110 down-regulated the expression of the STAT3-target genes Bcl-2 and cyclin D1, increased ROS production, and remarkably reduced the mitochondrial membrane potential to induce the mitochondrial apoptotic pathway. It also suppressed breast cancer 4T1-implanted tumor growth in vivo.
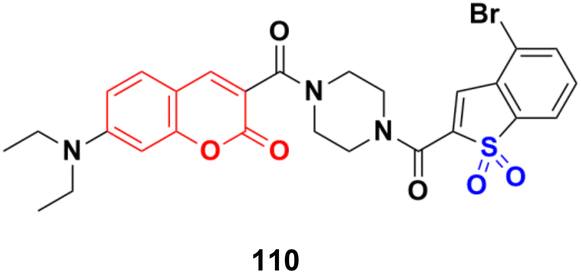 | ||
| Fig. 108 Chemical structure of coumarin–sulfonate hybrid 110. | ||
Fourteen new cycloalkane-fused tricyclic coumarin sulfonate derivatives were developed and evaluated for in vitro anticancer activity against the NCI-57 cancer cell line panel of nine different cancer types.181 Among the compounds, 111a, 111b, and 111c (Fig. 109) showed the highest activities. Compound 111b exerted the highest percentage of growth inhibition (91.91%) against the SNB-75 CNS cancer cell line at 10 μM concentration and was more active than carmustine against this cell line. Compound 111a also showed strong activity against HT29 colon, ACHN renal, and PC-3 prostate cancer cell lines. Furthermore, compound 111c was selective toward the HT29 colon cancer cell line.
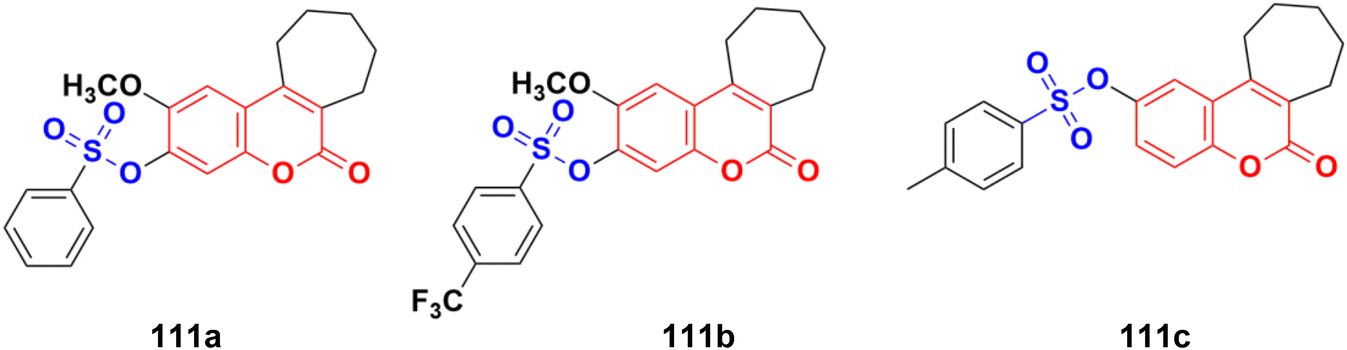 | ||
| Fig. 109 Chemical structure of coumarin–sulfonate hybrid 111a–c. | ||
A series of coumarin-sulfonamide hybrids was synthesized by the condensation reaction of appropriate N-heteroaryl-4-amino benzenesulfonamide with derivatives of 3-acetyl coumarin and their antiproliferative property was screened against the MDA-MB-231, MIA PaCa-2, and H357 cancer cell lines.182 Among them, compound 112 (Fig. 110) demonstrated significant activity against the MDA-MB-231 cell line (IC50 = 7.78 ± 3.78 μM) and H357 cell line (IC50 = 8.68 ± 1.10 μM) after 72 h.
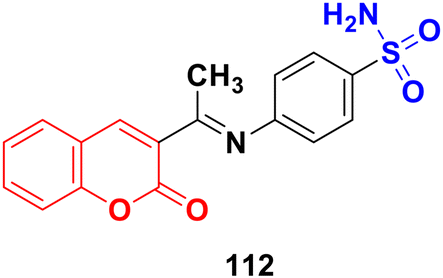 | ||
| Fig. 110 Chemical structure of coumarin-sulfonamide hybrid 112. | ||
2.9. Dihydroxycoumarins
Dihydroxycoumarins are an important class of benzopyrones with different pharmacological properties such as antimicrobial and anticancer.A novel series of coumarin-based nonsteroidal-type fluorescence ligands for drug–target binding imaging was designed and developed.183 Among the synthesized compounds, 113 (Fig. 111) showed potent antiproliferative activity against the MCF-7 cancer cell line with IC50 = 16.1 ± 0.7 μM and against MDA-MB-453 with IC50 = 8.03 ± 0.6 μM. Furthermore, compound 113 could cross the cell membrane, localize, and image drug–target interaction in real time without cell washing.
 | ||
| Fig. 111 Chemical structure of dihydroxycoumarin 113. | ||
A shikonin derivative, PMMB232 (114) (Fig. 112), showed antiproliferation activity with an IC50 value of 3.25 ± 0.35 μM.184 Further, the treatment of HeLa cells with a variety of concentrations of 114 resulted in a dose-dependent event marked by apoptosis. To identify the detailed role and mechanism of PMMB232 in the progression of human cervical cancer, the expression of HIF-1α and E-cadherin in HeLa cells was detected. The results revealed that expression of HIF-1α was downregulated, while E-cadherin protein was upregulated. Meanwhile, glycolysis-related protein PDK1 decreased in the HeLa cells. Conversely, the expression of PDH-E1α was upregulated. The docking simulation results further indicated that PMMB232 can be well bound to HIF-1α.
 | ||
| Fig. 112 Chemical structure of dihydroxycoumarin 114. | ||
Six 4-hydroxycoumarin derivatives were synthesized and their cytotoxic activities were investigated against four cancer cell lines (SMMC-7721, Bel-7402, MHCC97, and Hep3B).185 Compound 115 (Fig. 113) showed the maximum potency with IC50 values = 6 ± 1.4, 8 ± 2.0, 7 ± 1.7, 9 ± 2.0 μM against SMMC-7721, Bel-7402, MHCC97 and Hep3B, respectively.
 | ||
| Fig. 113 Chemical structure of dihydroxycoumarin 115. | ||
A series of nine coumarin derivatives was synthesized and their anticancer activity tested against four human breast cancer cells in vitro using the MTT assay.186 Among them, compound 116 (Fig. 114) showed the maximum potency with IC50 = 25.3 ± 2.3, 15.2 ± 2.4, 25.7 ± 2.2, and 20.2 ± 3.0 μM against the MDA-MB-231, MDA-MB-468, Hst578, and HCC1937 cell lines, respectively.
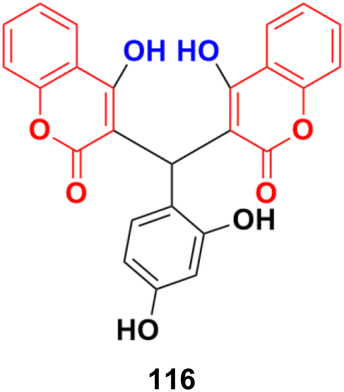 | ||
| Fig. 114 Chemical structure of dihydroxycoumarin 116. | ||
A series of novel 4,7-dihydroxycoumarin-based acryloylcyanohydrazone derivatives was synthesized and evaluated for their antiproliferative activity against four different cancer cell lines (A549, HeLa, SKNSH, and MCF7).187 Compound, 117 (Fig. 115) was the most active with IC50 values of 4.31 ± 0.04, 5.14 ± 0.16, 6.09 ± 0.32, and 3.42 ± 0.52 μM against A549, HeLa, SKNSH, and MCF7, respectively. Further results revealed that compound 117 induced cell cycle arrest at the G2/M phase and inhibited tubulin polymerization. The experimental data from the tubulin polymerization inhibition assay was validated by the molecular docking technique and the results exhibited strong hydrogen bonding interactions with amino acids (ASN-101, TYR-224, ASN-228, and LYS-254) of tubulin.
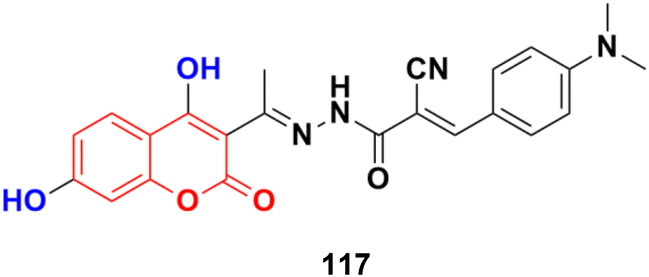 | ||
| Fig. 115 Chemical structure of dihydroxycoumarin 117. | ||
2.10. Anilinocoumarin hybrids
In recent years, it has been observed that when the aniline moiety is tagged with a coumarin scaffold, the anticancer property of the hybrid increases. Thus, designing anilinocoumarin hybrids is an important strategy for the synthesis of new anticancer drugs.A series of novel 4-substituted coumarin derivatives was synthesized and their antiproliferative activity toward a panel of tumor cell lines was investigated.188 Among them, compounds 118a–g (Fig. 116) showed potent antiproliferative ability. 118f was the most potent (IC50 values = 7–47 nM) and retained full activity in multidrug-resistant cancer cells. 118f caused G2/M phase arrest and interacted with the colchicine-binding site in tubulin, reducing the cell migration and disrupting capillary-like tube formation in HUVEC cells. Importantly, compound 118f significantly and dose-dependently reduced tumor growth in four xenograft models including paclitaxel-sensitive and resistant ovarian tumors (A2780s and A2780/T) and adriamycin-sensitive and resistant breast tumors (MCF-7 and MCF-7/ADR, respectively).
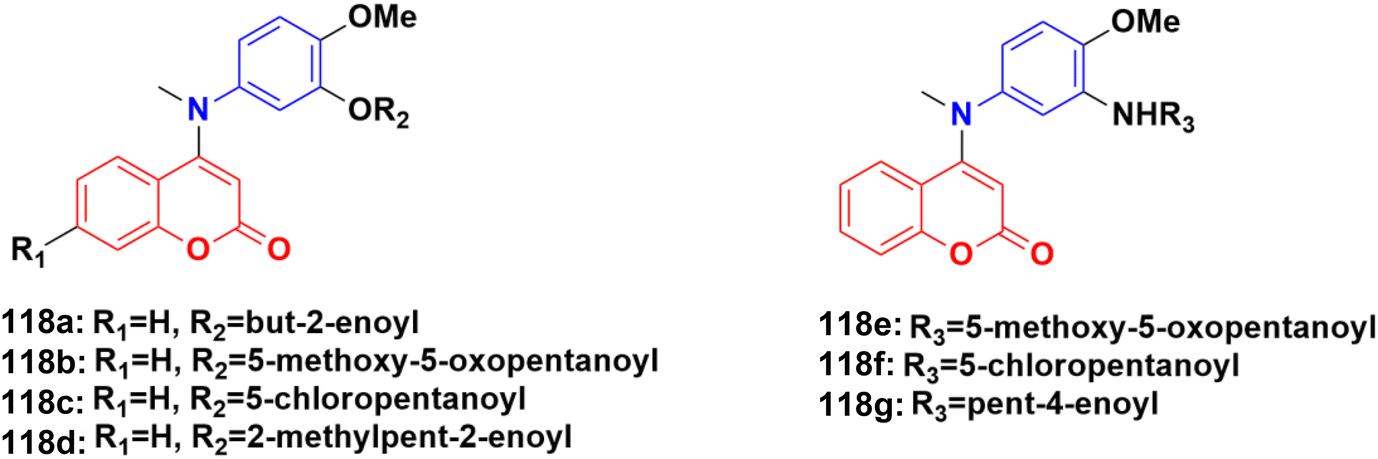 | ||
| Fig. 116 Chemical structures of anilinocoumarin 118a–g. | ||
Eighteen selective ERα modulators (SERMs) were designed and their biological activity investigated against MCF-7 and Ishikawa cell lines.189 The piperidyl-substituted compounds such as 119a and 119b (Fig. 117) demonstrated strong ERα binding affinities and excellent anti-proliferative activities. Compound 119b displayed the most potent ERα binding affinity with an RBA value of 2.83%, while 119a exhibited the best anti-proliferative activity against MCF-7 cells with an IC50 value of 4.52 ± 2.47 μM.
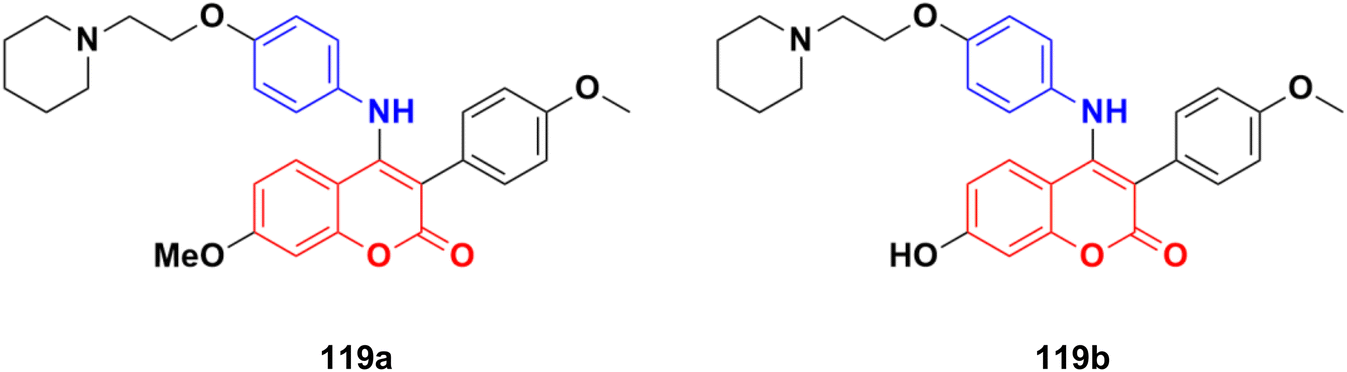 | ||
| Fig. 117 Chemical structures of anilinocoumarin 119a and b. | ||
A novel series of 3-(N-substituted)aminocoumarins was developed rapidly and efficiently and their antiproliferative activity examined against human cancer cell lines.190 Compound 120 (Fig. 118) showed excellent anticancer activity against MT-4, MDA-MB-231, and MCF-7 cancer cell lines with GI50 values of 12.6 ± 0.9, 11.8 ± 1.1 and 10.5 ± 1.2 μM, respectively.
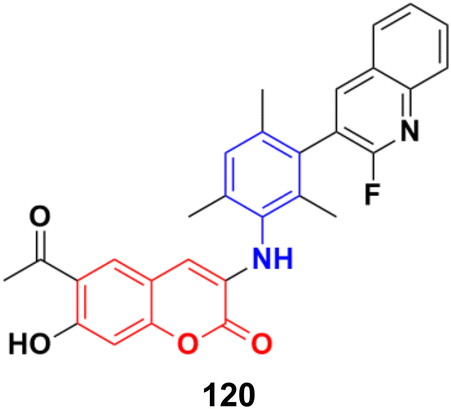 | ||
| Fig. 118 Chemical structure of anilinocoumarin 120. | ||
Various 3-substituted 4-anilino-coumarin derivatives were designed and synthesized and their in vitro cytotoxicity screening performed against MCF-7, HepG2, HCT116, and Panc-1 cancer cell lines by the MTT assay.191 Most of the synthesized compounds exhibited comparable anti-proliferative activity to the positive control 5-fluorouracil against the four tested cancer cell lines. Among the different substituents at the C-3 position the of coumarin scaffold, the 3-trifluoroacetyl group showed the most promising results. Especially, compounds 121a (Fig. 119) (IC50 = 16.57, 5.45, 4.42 and 5.16 μM) and 121b (IC50 = 20.14, 6.71, 4.62 and 5.62 μM) showed excellent anti-proliferative activities on MCF-7, HepG2, HCT116 and Panc-1 cell lines, respectively. In addition, cell cycle analysis and apoptosis activation revealed that 121a induced G2/M phase arrest and apoptosis in MCF-7 cells in a dose-dependent manner. The low toxicity of compounds 121a and 121b was observed against human umbilical vein endothelial cells (HUVECs), suggesting their acceptable safety profiles in normal cells. Furthermore, the results of in silico ADME studies indicated that both 121a and 121b exhibited good pharmacokinetic properties.
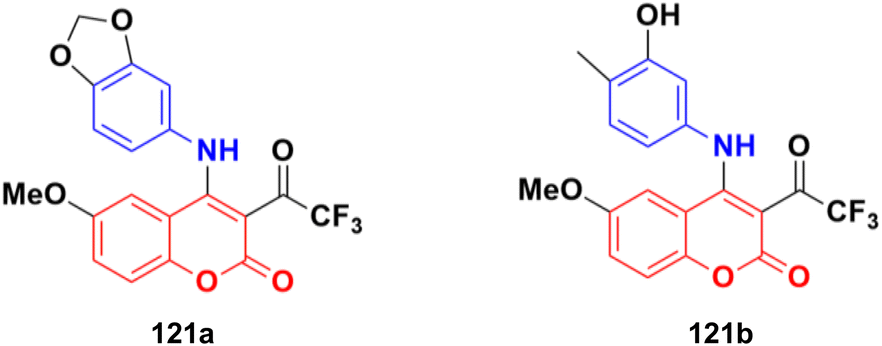 | ||
| Fig. 119 Chemical structures of anilinocoumarin 121a and b. | ||
A series of substituted aminomethyl benzocoumarin derivatives was synthesized and tested for their anticancer activity against the A549, MCF7, and A375 cancer cell lines.192 Among them, the anilinocoumarin compound 122 (Fig. 120) showed excellent growth inhibitory activity against the A549, MCF7, and A375 cancer cell lines with IC50 values of 4.29 μM, 5.17 μM, and 9.02 μM, respectively. Compound 122 was also found to be quite promising at very low concentrations as an anticancer agent against the MCF7 and A549 cell lines.
 | ||
| Fig. 120 Chemical structure of anilinocoumarin 122. | ||
Twenty-five coumarin-based derivatives were developed and investigated for their in vitro anticancer activity against the MCF-7 breast and PC-3 prostate cancer cell lines and further assessed for their in vitro VEGFR-2 kinase inhibitory activity.193 Among them, compound 123 (Fig. 121) (IC50 = 1.24 μM) exhibited exceptional activities superior to the positive control staurosporine (IC50 = 8.81 μM). Further study revealed that compound 123 was capable of inducing preG1 apoptosis, cell growth arrest at the G2/M phase, and activating caspase-9. A molecular docking study suggested that the most active anti-VEGFR-2 derivative 123 demonstrated the ability to interact with the key amino acids in the target VEGFR-2 kinase binding site.
 | ||
| Fig. 121 Chemical structure of anilinocoumarin 123. | ||
A series of substituted coumarins was synthesized directly from coumarins and azides in the presence of Pr(OTf)3 without any additives or ligands and the cytotoxic activity of the compounds was tested against the MGC-803, A549, and NCI-H460 cancer cell lines.194 Compound 124 (Fig. 122) showed maximum potency having IC50 values of 10.19 ± 1.12, 8.75 ± 1.10, and 9.25 ± 1.28 μM against the MGC-803, A549, and NCI-H460 cell lines, respectively.
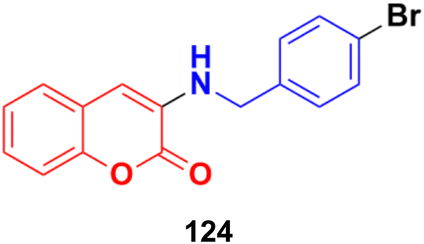 | ||
| Fig. 122 Chemical structure of anilinocoumarin 124. | ||
2.11. Coumarin-metal hybrids
Metal complexes play a vital role in various biochemical phenomena. The presence of metal ions accelerates the drug action, providing better drug delivery, and thus has become important in recent years for better pharmacological effect and enhanced efficiency of a particular metal-based drug. | ||
| Fig. 123 Chemical structure of ferrocene. | ||
A series of tailored novobiocin–ferrocene conjugates was prepared and investigated for in vitro anticancer activity against the MDA-MB-231 breast cancer line.196 They all showed moderate antiproliferative character. Compounds 125a and 125b (Fig. 124) showed maximum potency with IC50 values of 11.7 and 11.8 μM, respectively.
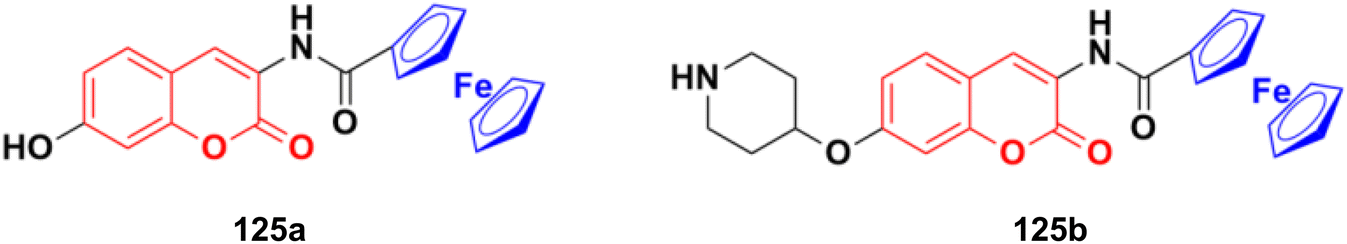 | ||
| Fig. 124 Chemical structures of coumarin–ferrocene hybrid 125a and b. | ||
The coumarin–ferrocene hybrid 126 (Fig. 125) showed good anticancer activity when examined against the HCC38 cancer cell line with IC50 = 1.06 μM.197
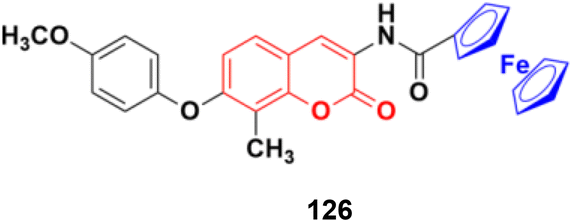 | ||
| Fig. 125 Chemical structure of coumarin–ferrocene hybrid 126. | ||
Seven novel coumarin–ferrocene conjugates were synthesized and their biological activities thoroughly investigated against several human cancer cell lines.198 Most of the hybrids showed moderate and good activity compared to the reference adriamycin. Compound 127a (Fig. 126) showed good potency against the BIU-87 and MCF-7 cancer cell lines with the IC50 values of 1.09 and 12.10 μM, respectively, while 127b was the most effective against SGC-7901 with IC50 = 3.56 μM.
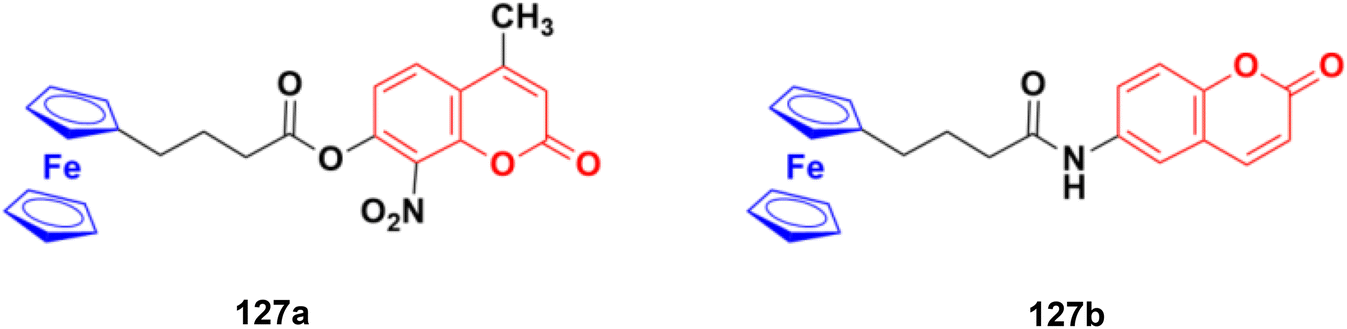 | ||
| Fig. 126 Chemical structures of coumarin–ferrocene hybrid 127a and b. | ||
 | ||
| Fig. 127 Chemical structures of coumarin–organotin hybrid 128a and b. | ||
A series of novel organoplatinum(II) complexes was designed and their cytotoxic effects on various cancer cell lines and drug-resistant cancer cell lines examined.200 Among them, complex 129 showed marked potency against the HeLa and A549/DDP cell lines with IC50 values of 0.15 ± 0.09 and 0.10 ± 0.05 μM, respectively (Fig. 128).
 | ||
| Fig. 128 Chemical structure of organoplatinum(II) complex with coumarin–quinoline moiety 129. | ||
Three coumarin-appended phosphorescent cyclometalated iridium(III) complexes, 130a–c (Fig. 129), were explored as mitochondria-targeted theranostic anticancer agents.201 All three complexes could specifically target mitochondria and show better antiproliferative activities than cisplatin against various cancer cells including cisplatin-resistant cells. They could penetrate human cervical carcinoma (HeLa) cells quickly and efficiently, and carried out theranostic functions by simultaneously inducing and monitoring the morphological changes in the mitochondria. The mechanism studies showed that they exert their anticancer efficacy by initiating a cascade of events related to mitochondrial dysfunction. Genome-wide transcriptional and connectivity map analyses revealed that the cytotoxicity of complex 130c is associated with pathways involved in mitochondrial dysfunction and apoptosis.
 | ||
| Fig. 129 Chemical structures of iridium(III) complex 130a–c. | ||
Three families, namely isoselenocyanate, selenocarbamates, and selenoureas, were designed and tagged with the coumarin moiety, and their antiproliferative properties were analyzed.202 Among them, selenourea 131 (Fig. 130) showed significant cytotoxic activities with GI50 values of 3.0 ± 0.5, 2.3 ± 0.7, 5.2 ± 0.4, 2.9 ± 1.1, and 3.8 ± 1.4 μM against the A549, HBL-100, HeLa, T-47D, WiDr cell lines, respectively. Moreover, dimer 132 also showed prominent cytotoxic activities with GI50 values of 3.2 ± 0.7, 3.5 ± 1.4, 2.9 ± 0.1, 3.6 ± 0.4, and 4.5 ± 1.0 μM against the above-mentioned cancer cell lines, respectively.
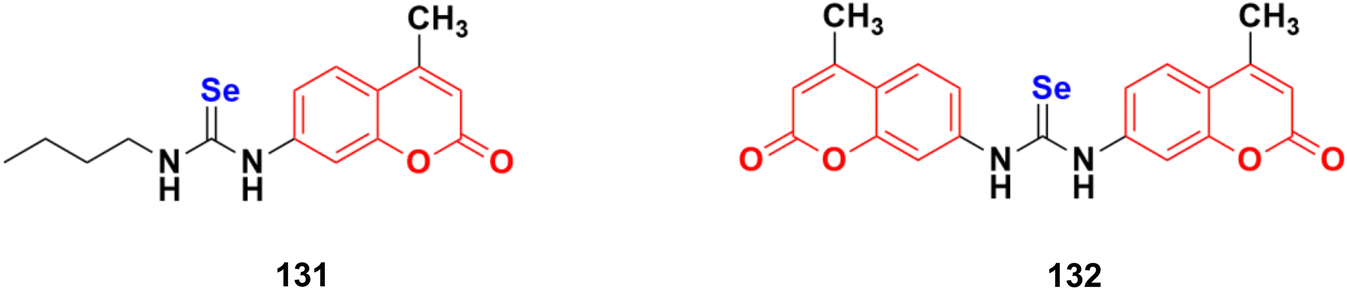 | ||
| Fig. 130 Chemical structures of selenocoumarin 131 and 132. | ||
Three new gold(I)-coumarin-based trackable therapeutic complexes (133a–c) (Fig. 131) were synthesized and their antiproliferative properties on several types of cancer cell lines including colon, breast, and prostate investigated.203 They all displayed moderate anticancer activities against MDAMB-231, PC3, SW480, and HEK293T.
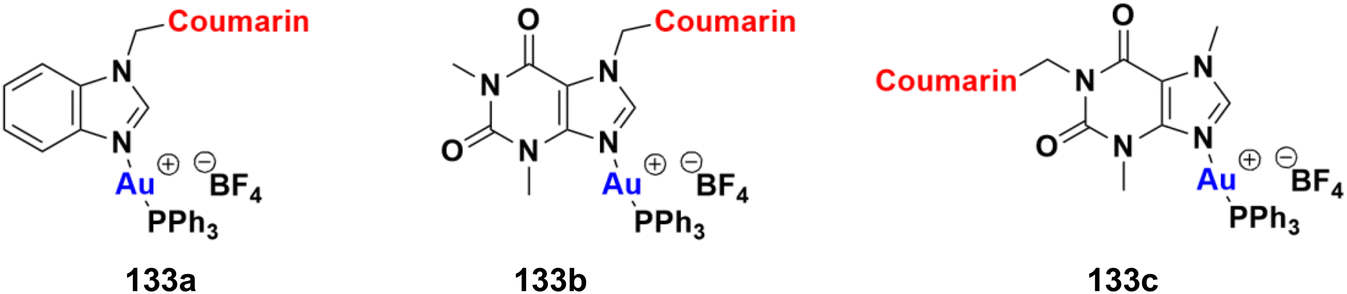 | ||
| Fig. 131 Chemical structures of gold(I)-coumarin 133a–c. | ||
The zinc(II) complex of 3-acetylcoumarin thiosemicarbazone (134) (Fig. 132) showed significant cytotoxicity against human liver carcinoma (HepG-2) and lymphoblastoid multiple myeloma (IM-9) cell lines with the IC50 value of 25 μg mL−1.204 The spectroscopic results suggested that the complex interacted with CT-DNA through the intercalative binding mode.
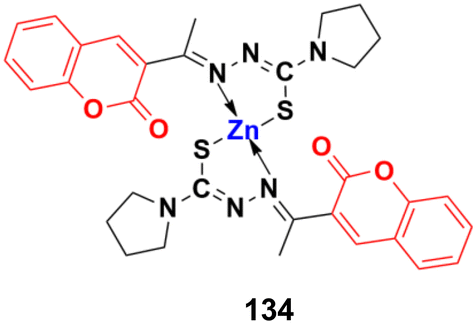 | ||
| Fig. 132 Chemical structure of zinc(II)-coumarin complex 134. | ||
Two new copper(II) (135) and nickel(II) (136) (Fig. 133) complexes with a new coumarin derivative were synthesized and their cytotoxic activities determined by the MTT assay.205 The results showed that the designed drugs have significant cytotoxic activity against the HepG2, HL60, and PC3 cell lines. Cell apoptosis was detected by annexin V/PI flow cytometry and the results showed that the two complexes can induce apoptosis of the three human tumor cells.
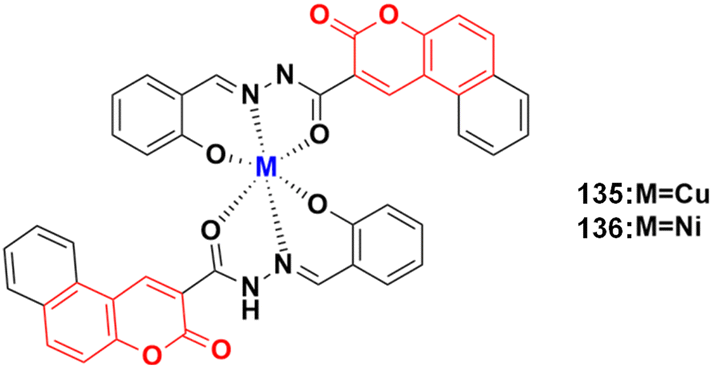 | ||
| Fig. 133 Chemical structure of copper(II)-coumarin complex 135 and nickel(II)-coumarin complex 136. | ||
A Co(II) complex of coumarin [Co(CUAP)(H2O)2Cl] (137) (Fig. 134) exhibited significant antiproliferative properties against the MCF-7 and K-562 cancer cell lines with IC50 values of less than 10 μg mL−1.206
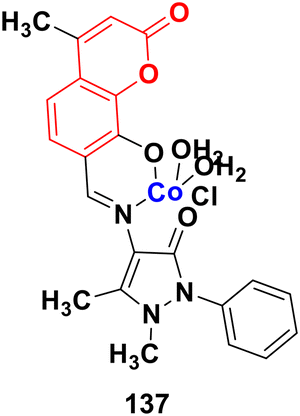 | ||
| Fig. 134 Chemical structure of cobalt(II)-coumarin complex 137. | ||
A Ru(III) complex synthesized from a coumarin derivative (138) (Fig. 135) was observed to be potent against the MCF-7 cancer cell line with an IC50 value of less than 10 μg mL−1.207
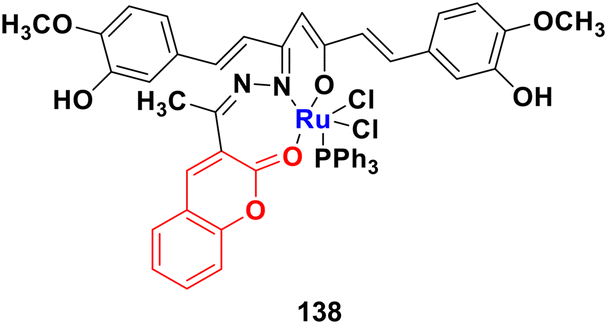 | ||
| Fig. 135 Chemical structure of Ru(III)-coumarin complex 138. | ||
Besides these findings, various metal-based coumarin complexes show significant cytotoxic activities against various cancer cell lines and this strategy is becoming increasingly important, which has a bright prospect.208
2.12. Miscellaneous coumarin hybrids
The coumarin derivative bis(4-hydroxy-2H-chromen-2-one)coumarin 139 (Fig. 136) possessed marked antitumor activity against the MCF-7 cancer cell line by inducing cell cycle arrest in the G2/M phase.209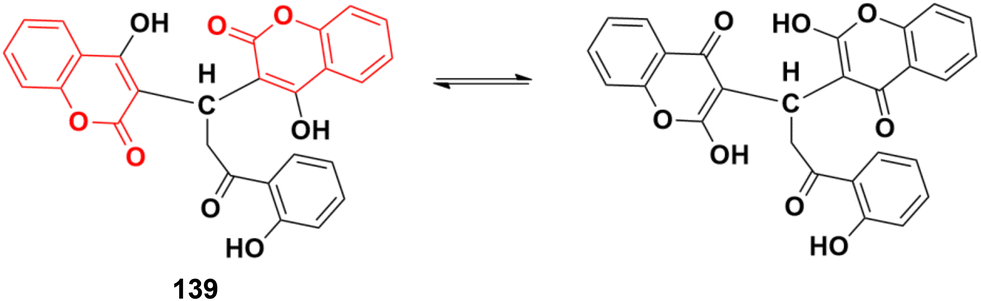 | ||
| Fig. 136 Chemical structure of coumarin dimer 139. | ||
A series of hydrazide–hydrazone and amide-substituted coumarin derivatives was synthesized and evaluated in vitro for their antitumor activity.210 Among them, compound 140a (Fig. 137) showed the maximum potency against the Panc-1 cell line with IC50 = 0.129 ± 0.019 μM and selectivity ratio of >387.60. Compound 140b possessed significant effectivity against the HepG2 cell line with IC50 = 4.892 ± 0.086 μM and selectivity ratio of >10.22. Compound 140c showed marked potency against CCRF cells with IC50 = 3.108 ± 0.439 μM and selectivity ratio of >16.09. Besides, compound 140d was effective against all the cell lines with IC50 values of 5.449 ± 1.380, 9.417 ± 0.548, and 7.448 ± 4.579 against Panc-1, HepG2, and CCRF cells with the corresponding selective ratio of >9.18, >5.31, and >6.71, respectively.
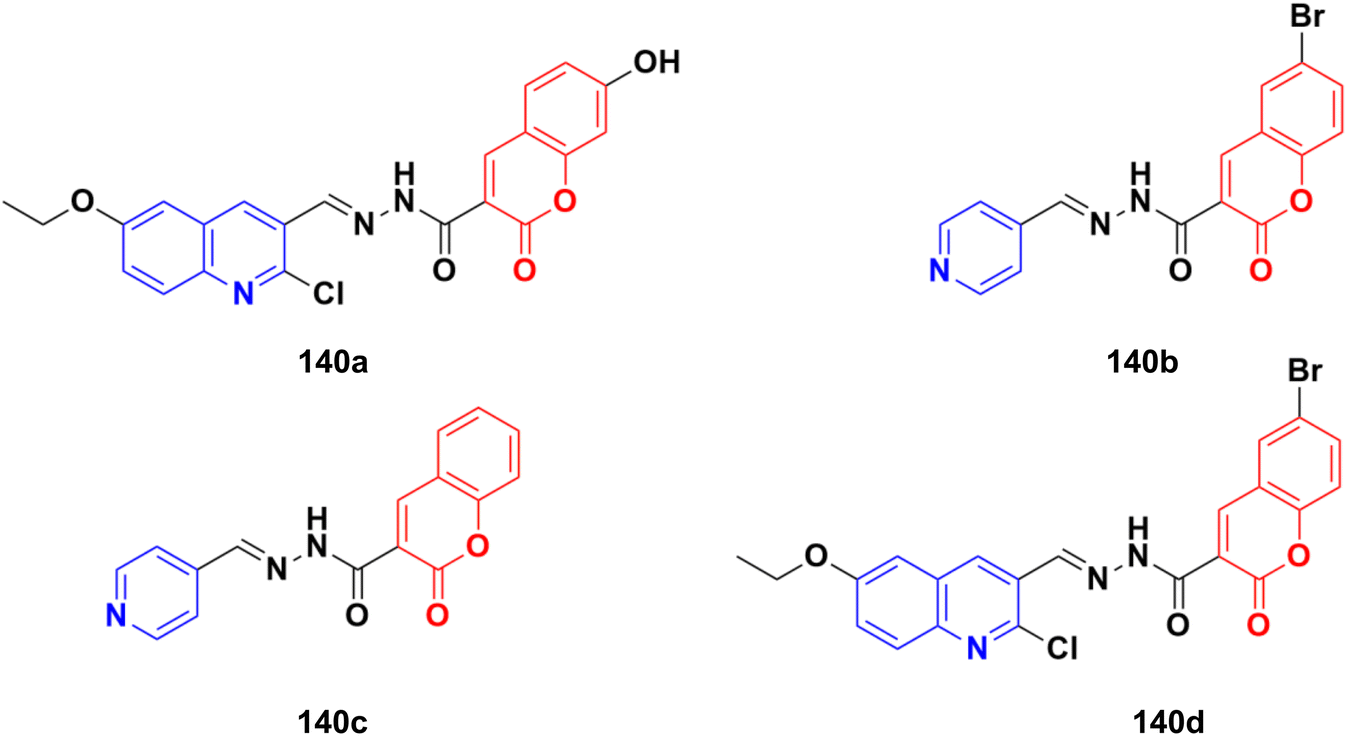 | ||
| Fig. 137 Chemical structures of coumarin derivative 140a–d. | ||
Tacrine-coumarin hybrids 141a and 141b (Fig. 138) showed significant anti-metabolic activity against the 4T1 cell line, with IC50 values of 5.7 μM and 7.0 μM, respectively, while compound 141a also showed promising activity against the MCF-7 cell line, with an IC50 value of 6.0 μM.211
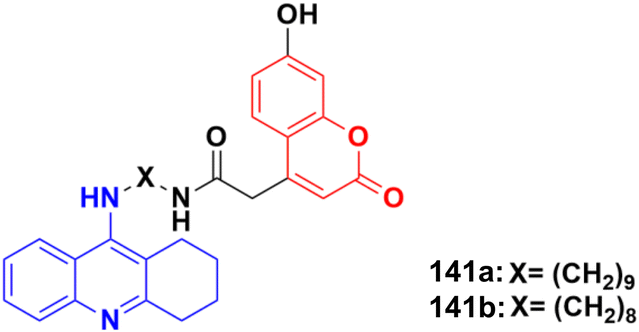 | ||
| Fig. 138 Chemical structures of coumarin-tacrine hybrid 141a and b. | ||
A series of quinoline and thiazole-containing coumarin analogs was developed and their cytotoxic ability examined on mouse leukemic cells.212 The result indicated that 142 (Fig. 139) showed potent activity against EAC and DLA cells in the MTT assay (15.3 μM), trypan blue (15.6 μM), and LDH (14.2 μM) leak assay with 5-fluorouracil as the standard. The experimental data showed that compound 142 induced apoptotic cell death by activating apoptotic factors such as caspase-8 &-3, CAD, cleaved PARP, γ-H2AX, and by degrading genomic DNA of cancer cells, and thereby decreasing the ascitic tumor development in mice. The molecular docking study revealed that compound 142 has a very good interaction with caspase 3 protein by binding with the Arg 207 amino acid through a hydrogen bond.
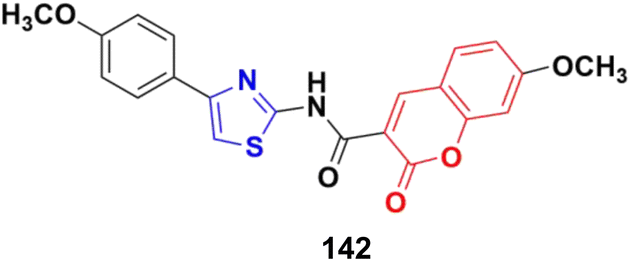 | ||
| Fig. 139 Chemical structure of coumarin–thiazole hybrid 142. | ||
An interesting fact can be nicely shown from the work by Zwergel et al.213 The antiproliferative property of a particular coumarin-based hybrid can be significantly enhanced by slight modification in the coumarin moiety (Fig. 140, Table 1).
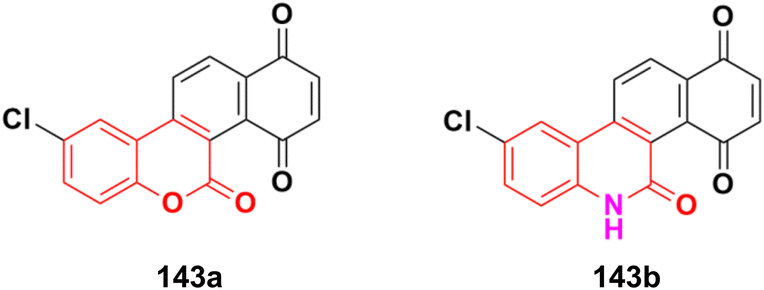 | ||
| Fig. 140 Chemical structures of coumarin derivative 143a and b. | ||
| Compound | Inhibition data (IC50, μM) | Viability (MTT assay, IC50, μM) | ||||||
|---|---|---|---|---|---|---|---|---|
| CDC25A | CDC25C | A549 | MCF7 | PC3 | U373n | Hs683 | SKMEL28 | |
| 143a | 2.57 ± 0.2 | 1.44 ± 0.2 | 25 | 13 | 19 | 12 | 6 | 33 |
| 143b | 4.33 ± 0.4 | 5.72 ± 0.5 | 0.3 | 1.2 | 0.3 | 0.1 | 0.2 | 0.3 |
The styryl coumarin hybrids 3-SC1 (144a), 7-SC2 (144b), and 7-SC3 (144c) (Fig. 141) decreased the cell viability of SW480 in a time- and concentration-dependent manner (IC50-SW480/48 h = 6.92; 1.01 and 5.33 μM, respectively) with high selectivity indices after 48 h of treatment (>400; 67.8 and 7.2, respectively).214 Among them, the most active molecule 7-SC2 induced a greater production of ROS in comparison with the control (p < 0.05) together with a significant increase in the expression of p53 and caspase-3, and a significant reduction in the production of interleukin-6 of SW480 cells. When colon carcinogenesis was induced in Balb/c mice by intraperitoneal injection of azoxymethane, a significant reduction (p < 0.05) in the number of preneoplastic lesions of the mice treated with styryl coumarin hybrid 7-SC2 was observed in the control group. Moreover, no side effects were associated with the administration of the compound. All these in vitro results and the effective reduction of preneoplastic lesions in vivo suggest that styryl coumarin 7-SC2 induces apoptosis in primary tumor cells and implies its potential ability at the early post-initiation phases of colon carcinogenesis.
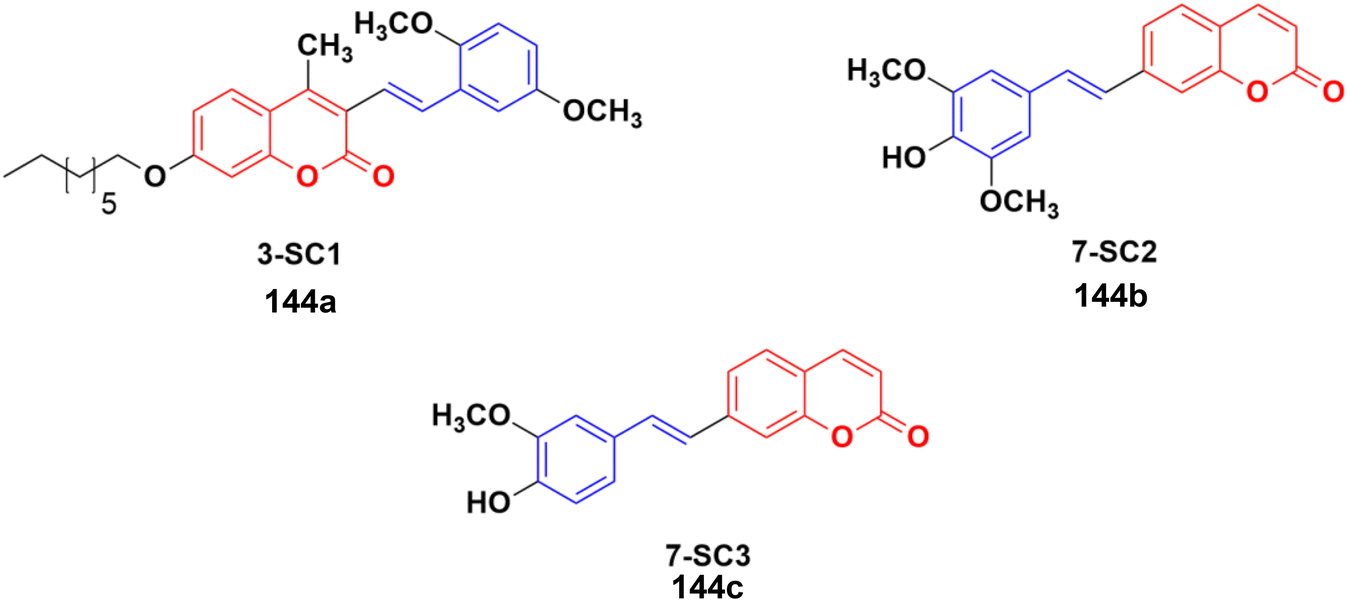 | ||
| Fig. 141 Chemical structures of styryl coumarin 144a–c. | ||
A series of methylene thio-linked coumarin derivatives was prepared by the reaction of substituted 4-(bromomethyl)-2H-chromen-2-one with various heterocyclic mercapto compounds via SN2 reactions in the presence of K2CO3 as a catalyst and their in vitro anticancer activity screened against the MCF-7 cancer cell line.215 Compound 145 (Fig. 142) with a methoxy-substituted benzimidazole ring was found to be the most effective with an IC50 value of 0.18 μM.
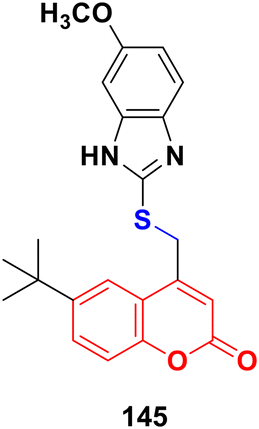 | ||
| Fig. 142 Chemical structure of methylene thio-linked coumarin derivative 145. | ||
Twenty porphyrin–coumarin compounds were synthesized via the condensation reaction of porphyrins with coumarin derivatives and their cytotoxic activity evaluated against A549 and HepG2 cells under light irradiation.216 The structure–activity relationship studies indicated that the coumarin derivatives with shorter alkyl chains to porphyrins exhibited both photodynamic therapy (PDT) and chemotherapy. Moreover, the insertion of metal Zn in the porphyrins also increased the PDT effect of the compounds. Compound 146 (Fig. 143) was found to be the most effective against the HepG2 cell line with an IC50 value of 67.66 ± 0.61 μmol L−1. Alternatively, the Zn-containing compound 147 exhibited the maximum potency against the A549 cell line with an IC50 value of 52.37 ± 1.17 μmol L−1 (both under light irradiation).
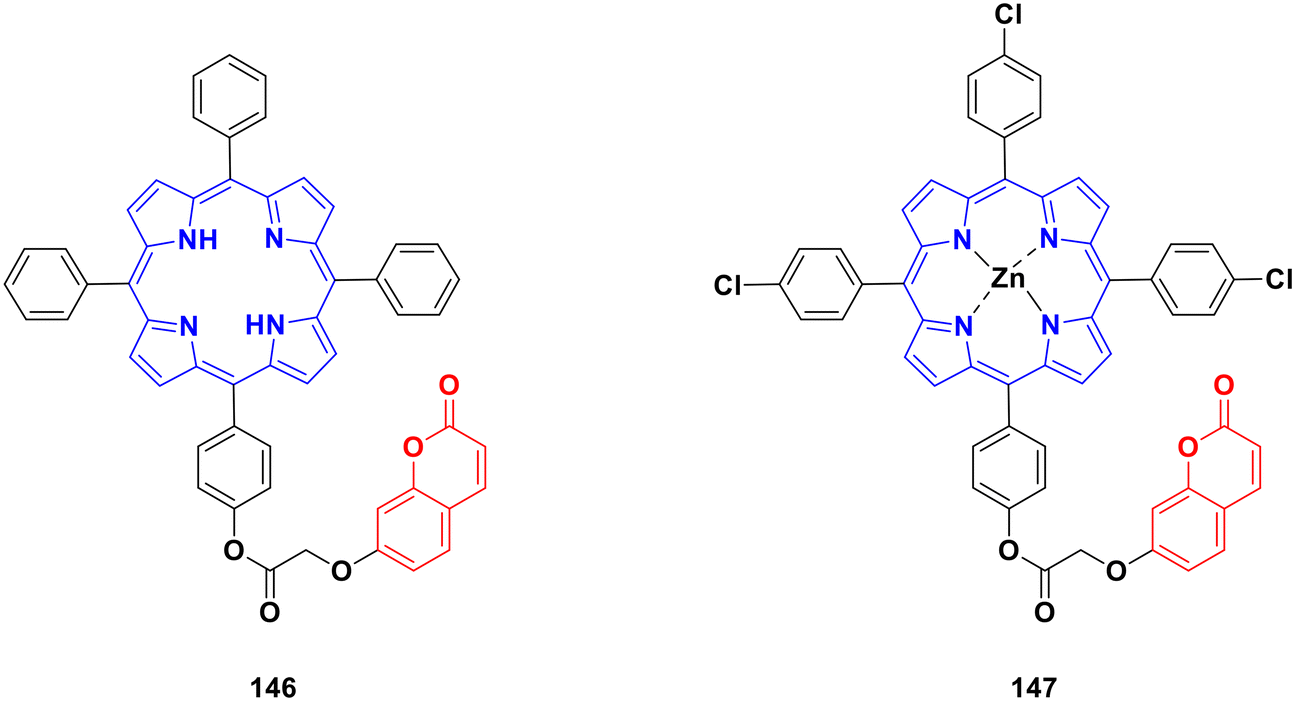 | ||
| Fig. 143 Chemical structures of coumarin–porphyrin complex 146 and zinc-coumarin–porphyrin complex 147. | ||
Besides the above-mentioned coumarin hybrids, coumarin-hydroxamic acid,217 coumarin–ergosterol peroxide,218 coumarin–formonontin,219 coumarin–costunolide,220 coumarin–curcuminoid,221 coumarin–furan,222 coumarin–maltol,223 coumarin–carbazole,224 coumarin–quinazoline,225 coumarin-steroidal,226,227 coumarin-sugar,228–232 coumarin-thiazin-2-thione,233 coumarin–thiophosphate,234 coumarin-triazolothiadiazines,235N-heterocyclocoumarin,236 hydroxymercapto-methylcoumarin,237 triazole-tethered coumarin–isatin,238 and others239–241 also showed certain anticancer activities.
3. Patents
Coumarin hybrids have been successfully used as potential anticancer agents and many international patents have been filed in this regard.In 2015, some triazole-modified coumarin-based compounds (148) were patented, which were proposed to be used in the treatment of various cancers such as breast and prostate cancer.242 The general structure of the compounds is shown in Fig. 144.
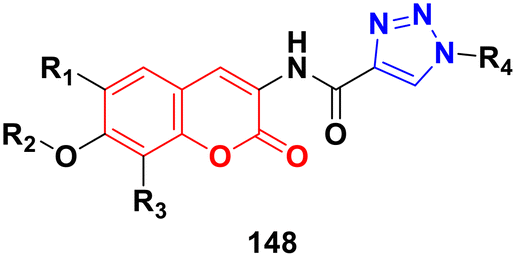 | ||
| Fig. 144 General chemical structure of triazole-modified coumarin-based compound 148. | ||
In 2016, various coumarin derivatives 149a–c with substituents at the 6-position with five or more than five carbon atoms were patented, which were used to treat pancreatic cancer.243 The structures of some of the compounds are shown in Fig. 145.
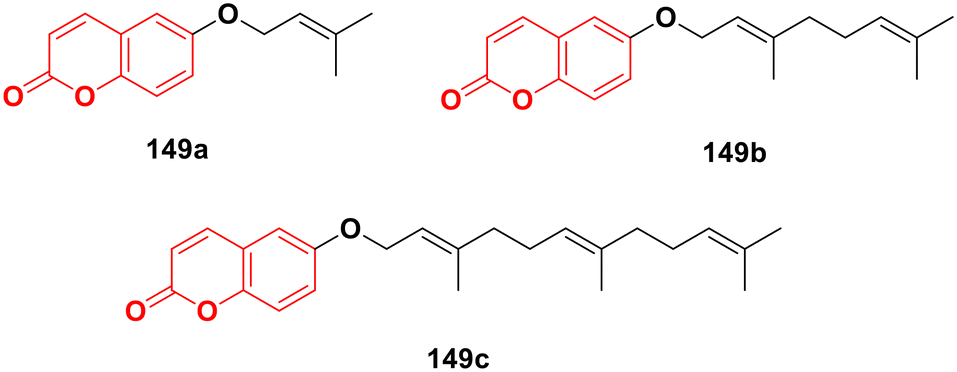 | ||
| Fig. 145 Chemical structures of 6-substituted coumarins 149a–c. | ||
In 2017, the design of different coumarin–gossypol hybrids with antitumor activities was patented,244 which had two different general structures (150a and 150b), as shown in Fig. 146.
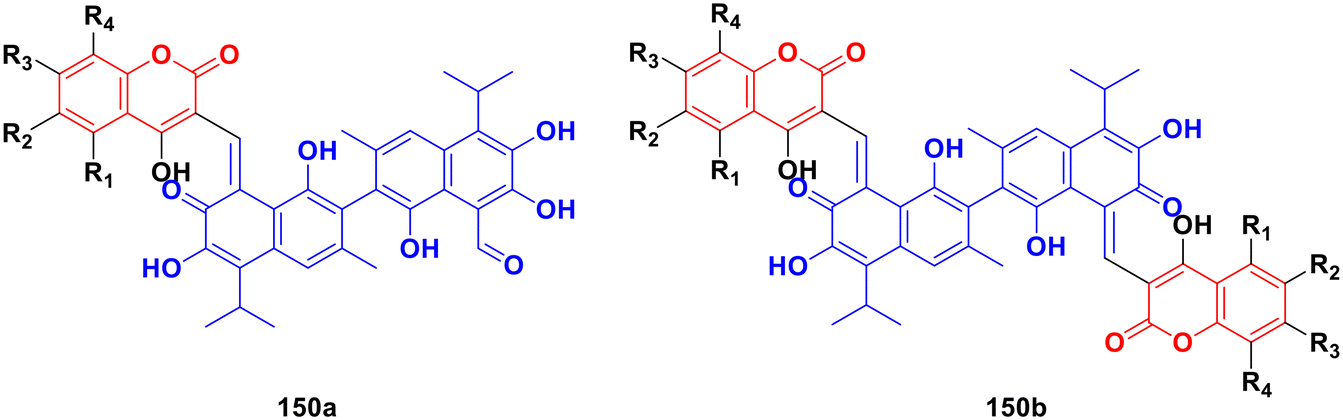 | ||
| Fig. 146 Chemical structures of coumarin–gossypol hybrid 150a and b. | ||
In 2018, the method for the synthesis of a library of selenium-containing coumarin derivatives (151) was patented.245 The general structure of the compounds is presented in Fig. 147. These compounds were proposed to prevent primary cancer.
 | ||
| Fig. 147 Chemical structure of selenium-containing coumarin derivative 151. | ||
In 2019, a patent was filed for the use of terpenic coumarin derivatives as potential anticancer agents with high selectivity and low side effects.246 The general structures of two types of these derivatives are shown in Fig. 148.
 | ||
| Fig. 148 Chemical structure of terpenic coumarin derivative 152a and b. | ||
In 2020, a patent was filed for several novel 4-phenyl-coumarin derivatives.247 The compounds were prepared and used as specific mitochondrial RNA polymerase inhibitors for cancer treatment. The general structure of compound 153 is presented in Fig. 149.
 | ||
| Fig. 149 General chemical structure of 4-phenylcoumarin derivative 153. | ||
In 2022, the preparation of some coumarin derivatives (154) used for their anticancer and antioxidant properties was patented.248 The general structure of the compounds is shown in Fig. 150.
 | ||
| Fig. 150 General chemical structure of coumarin derivative 154. | ||
4. Conclusion
Presently, the high mortality rate of cancer represents a great concern in society. In this case, the coumarin scaffold has become a privileged molecule in the design of anticancer drugs in recent years. Besides, the strategy of its hybridization with other pharmacophores has also become widely accepted in the case of enhanced efficacy. However, the preclinical trials, i.e., the study of in vivo, toxicity, specificity, and interaction of drugs is time-consuming and not cost-effective for researchers of non-profit organizations. Several coumarin derivatives with high anticancer potency were highlighted in this review but the discovery of novel therapeutic drugs is still pending. Moreover, the redox features of the dihydroxy group attached to simple coumarin may not be favorable in vivo and lead to side effects. Further, modification of the pyrone rings is needed to overcome these side effects.The above-mentioned studies also revealed that coumarin azole derivatives bind directly to DNA, which can lead to further improvement in oxidative stress. Coumarin-metal hybrids will also flourish to synthesize new drugs.
We hope, in the future, that the problems of side effects and toxicity regarding the coumarin scaffold will be overcome and coumarin-based anticancer drugs will be available in the market to fight against cancer and help cost-effectively eradicate cancer.
Conflicts of interest
There is no conflict of interest to declare.References
- World Health Organization, Cancer, https://www.who.int/news-room/fact-sheets/detail/cancer Search PubMed.
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Ca-Cancer J. Clin., 2022, 72, 7–33 CrossRef.
- D. C. G. A. Pinto and A. M. S. Silva, Curr. Top. Med. Chem., 2017, 17, 3190–3198 CAS.
- M.-B. Nayeli, H.-R. Maribel, J.-F. Enrique, B.-P. Rafael, A.-F. Margarita, F.-M. Macrina, M.-D. Ivan and G.-C. Manasés, Nat. Prod. Res., 2020, 34, 3244–3248 CrossRef CAS PubMed.
- C. Mu, M. Wu and Z. Li, Chem. Biodiversity, 2019, 16, 1800559 CrossRef.
- Y. Hu, Y. Shen, X. Wu, X. Tu and G.-X. Wang, Eur. J. Med. Chem., 2018, 143, 958–969 CrossRef CAS PubMed.
- K. Bhagat, J. Bhagat, M. K. Gupta, J. V. Singh, H. K. Gulati, A. Singh, K. Kaur, G. Kaur, S. Sharma, A. Rana, H. Singh, S. Sharma and P. M. Singh Bedi, ACS Omega, 2019, 4, 8720–8730 CrossRef CAS.
- A. Singh, J. V. Singh, A. Rana, K. Bhagat, H. K. Gulati, R. Kumar, R. Salwan, K. Bhagat, G. Kaur, N. Singh, R. Kumar, H. Singh, S. Sharma and P. M. S. Bedi, ACS Omega, 2019, 4, 11673–11684 CrossRef CAS PubMed.
- B. M. Chougala, S. Samundeeswari, M. Holiyachi, N. S. Naik, L. A. Shastri, S. Dodamani, S. Jalalpure, S. R. Dixit, S. D. Joshi and V. A. Sunagar, Eur. J. Med. Chem., 2018, 143, 1744–1756 CrossRef CAS PubMed.
- M. Z. Hassan, H. Osman, M. A. Ali and M. J. Ahsan, Eur. J. Med. Chem., 2016, 123, 236–255 CrossRef CAS.
- Y. Al-Majedy, A. Al-Amiery, A. A. Kadhum and A. BakarMohamad, Syst. Rev. Pharm., 2016, 8, 24–30 CrossRef.
- Y. Al-Majedy, D. Al-Duhaidahawi, K. Al-Azawi, A. Al-Amiery, A. Kadhum and A. Mohamad, Molecules, 2016, 21, 135 CrossRef PubMed.
- R. Nagamallu, B. Srinivasan, M. B. Ningappa and A. K. Kariyappa, Bioorg. Med. Chem. Lett., 2016, 26, 690–694 CrossRef CAS PubMed.
- K. Kasperkiewicz, M. B. Ponczek and E. Budzisz, Pharmacol. Rep., 2018, 70, 1057–1064 CrossRef CAS PubMed.
- S.-S. Xie, J.-S. Lan, X. Wang, Z.-M. Wang, N. Jiang, F. Li, J.-J. Wu, J. Wang and L.-Y. Kong, Bioorg. Med. Chem., 2016, 24, 1528–1539 CrossRef CAS PubMed.
- N. Jiang, Q. Huang, J. Liu, N. Liang, Q. Li, Q. Li and S.-S. Xie, Eur. J. Med. Chem., 2018, 146, 287–298 CrossRef CAS PubMed.
- L. Zhang and Z. Xu, Eur. J. Med. Chem., 2019, 181, 111587 CrossRef CAS PubMed.
- X. Song, J. Fan, L. Liu, X. Liu and F. Gao, Arch. Pharm., 2020, 353, e2000025 CrossRef PubMed.
- L. Huang, Z.-L. Feng, Y.-T. Wang and L.-G. Lin, Chin. J. Nat. Med., 2017, 15, 881–888 CAS.
- J. C. J. M. D. S. Menezes and M. F. Diederich, Eur. J. Med. Chem., 2019, 182, 111637 CrossRef CAS PubMed.
- Q. Ma, J.-G. Jiang, X. Yuan, K. Qiu and W. Zhu, Food Chem. Toxicol., 2019, 125, 422–429 CrossRef CAS PubMed.
- T. Kubrak, M. Czop, P. Kołodziej, M. Ziaja-Sołtys, J. Bogucki, A. Makuch-Kocka, D. Aebisher, J. Kocki and A. Bogucka-Kocka, Molecules, 2019, 24, 1824 CrossRef CAS.
- H. Singh, J. V. Singh, K. Bhagat, H. K. Gulati, M. Sanduja, N. Kumar, N. Kinarivala and S. Sharma, Bioorg. Med. Chem., 2019, 27, 3477–3510 CrossRef CAS PubMed.
- A. Rawat and A. Vijaya Bhaskar Reddy, Eur. J. Med. Chem. Rep., 2022, 5, 100038 CAS.
- R. Bhatia, A. Singh, B. Kumar and R. K. Rawal, in Key Heterocyclic Cores for Smart Anticancer Drug–Design Part II, Bentham Science Publishers, 2022, pp. 35–78 Search PubMed.
- W. C. Galeano, H. R. Malule and D. G. Ríos, J. Appl. Pharm. Sci., 2023, 13, 204–212 Search PubMed.
- T. Al-Warhi, A. Sabt, E. B. Elkaeed and W. M. Eldehna, Bioorg. Chem., 2020, 103, 104163 CrossRef CAS PubMed.
- Y. Tu, Angew. Chem., Int. Ed., 2016, 55, 10210–10226 CrossRef CAS PubMed.
- X. Liu, J. Cao, G. Huang, Q. Zhao and J. Shen, Curr. Top. Med. Chem., 2019, 19, 205–222 CrossRef CAS PubMed.
- B. Zhang, Arch. Pharm., 2020, 353(2) DOI:10.1002/ardp.201900240.
- X. Zhang, Q. Ba, Z. Gu, D. Guo, Y. Zhou, Y. Xu, H. Wang, D. Ye and H. Liu, Chem. – Eur. J., 2015, 21, 17415–17421 CrossRef CAS.
- Y. Tian, Z. Liang, H. Xu, Y. Mou and C. Guo, Molecules, 2016, 21, 758 CrossRef.
- H. Yu, Z. Hou, Y. Tian, Y. Mou and C. Guo, Eur. J. Med. Chem., 2018, 151, 434–449 CrossRef CAS.
- D. Bauer, S. M. Sarrett, J. S. Lewis and B. M. Zeglis, Nat. Protoc., 2023, 18, 1659–1668 CrossRef CAS PubMed.
- A. Makarem, M. K. Sarvestani, K. D. Klika and K. Kopka, Synlett, 2019, 30, 1795–1798 CrossRef CAS.
- A. Makarem, K. D. Klika, G. Litau, Y. Remde and K. Kopka, J. Org. Chem., 2019, 84, 7501–7508 CrossRef CAS PubMed.
- K. D. Klika, R. Alsalim, M. Eftekhari and A. Makarem, Dalton Trans., 2022, 51, 12436–12441 RSC.
- H. Yu, Z. Hou, X. Yang, Y. Mou and C. Guo, Molecules, 2019, 24, 1672 CrossRef CAS PubMed.
- D. Rani, V. Garg and R. Dutt, Anti-Cancer Agents Med. Chem., 2021, 21, 1957–1976 CrossRef CAS PubMed.
- B. Zhang, Eur. J. Med. Chem., 2019, 168, 357–372 CrossRef CAS PubMed.
- F. Gao, T. Wang, J. Xiao and G. Huang, Eur. J. Med. Chem., 2019, 173, 274–281 CrossRef CAS PubMed.
- D.-J. Fu, P. Li, B.-W. Wu, X.-X. Cui, C.-B. Zhao and S.-Y. Zhang, Eur. J. Med. Chem., 2019, 165, 309–322 CrossRef CAS.
- T. G. Kraljević, A. Harej, M. Sedić, S. K. Pavelić, V. Stepanić, D. Drenjančević, J. Talapko and S. Raić-Malić, Eur. J. Med. Chem., 2016, 124, 794–808 CrossRef PubMed.
- A. Bistrović, N. Stipaničev, T. Opačak-Bernardi, M. Jukić, S. Martinez, L. Glavaš-Obrovac and S. Raić-Malić, New J. Chem., 2017, 41, 7531–7543 RSC.
- R. An, Z. Hou, J.-T. Li, H.-N. Yu, Y.-H. Mou and C. Guo, Molecules, 2018, 23, 2281 CrossRef PubMed.
- S. Dhawan, P. Awolade, P. Kisten, N. Cele, A. Pillay, S. Saha, M. Kaur, S. B. Jonnalagadda and P. Singh, Chem. Biodiversity, 2020, 17 DOI:10.1002/cbdv.201900462.
- I. Ansary, H. Roy, A. Das, D. Mitra and A. K. Bandyopadhyay, ChemistrySelect, 2019, 4, 3486–3494 CrossRef CAS.
- P. J. Raj and D. Bahulayan, Tetrahedron Lett., 2017, 58, 2122–2126 CrossRef CAS.
- S. Chekir, M. Debbabi, A. Regazzetti, D. Dargère, O. Laprévote, H. Ben Jannet and R. Gharbi, Bioorg. Chem., 2018, 80, 189–194 CrossRef CAS PubMed.
- S. Sinha, A. P. Kumaran, D. Mishra and P. Paira, Bioorg. Med. Chem. Lett., 2016, 26, 5557–5561 CrossRef CAS PubMed.
- L. V. Augsten, G. Göethel, B. Gauer, M. Feiffer Charão, G. von Poser, R. F. S. Canto, M. D. Arbo, V. L. Eifler-Lima and S. C. Garcia, RSC Med. Chem., 2023, 14, 869–879 RSC.
- R. W. Adam and E. H. Zimam, J. Popul. Ther. Clin. Pharmacol., 2023, 30(9), 345–356 Search PubMed.
- P. Kaur and A. Chawla, Int. Res. J. Pharm., 2017, 8, 10–29 CrossRef CAS.
- R. Kaur, A. Ranjan Dwivedi, B. Kumar and V. Kumar, Anti-Cancer Agents Med. Chem., 2016, 16, 465–489 CrossRef CAS PubMed.
- M. K. Kathiravan, A. B. Salake, A. S. Chothe, P. B. Dudhe, R. P. Watode, M. S. Mukta and S. Gadhwe, Bioorg. Med. Chem., 2012, 20, 5678–5698 CrossRef CAS.
- J. Zhang, S. Wang, Y. Ba and Z. Xu, Eur. J. Med. Chem., 2019, 174, 1–8 CrossRef CAS PubMed.
- B. Kahveci, F. Yılmaz, E. Menteşe and S. Ülker, Arch. Pharm., 2017, 350, 1600369 CrossRef PubMed.
- S. S. Alghamdi, R. S. Suliman, K. Almutairi, K. Kahtani and D. Aljatli, Drug Des., Dev. Ther., 2021, 15, 3289–3312 CrossRef PubMed.
- H. Liu, Y. Wang, A. Sharma, R. Mao, N. Jiang, B. Dun and J.-X. She, Anti-Cancer Drugs, 2015, 26, 667–677 CrossRef CAS PubMed.
- M. Holiyachi, S. L. Shastri, B. M. Chougala, L. A. Shastri, S. D. Joshi, S. R. Dixit, H. Nagarajaiah and V. A. Sunagar, ChemistrySelect, 2016, 1, 4638–4644 CrossRef CAS.
- R. Goel, V. Luxami and K. Paul, RSC Adv., 2015, 5, 37887–37895 RSC.
- K. V. Sashidhara, L. R. Singh, D. Choudhary, A. Arun, S. Gupta, S. Adhikary, G. R. Palnati, R. Konwar and R. Trivedi, RSC Adv., 2016, 6, 80037–80048 RSC.
- S. Imeni, A. Makarem and R. Javahershenas, Asian J. Org. Chem., 2023, 12 DOI:10.1002/ajoc.202300303.
- R. Javahershenas and S. Nikzat, RSC Adv., 2023, 13, 16619–16629 RSC.
- R. M. Mohareb, A. E. M. Abdallah and A. A. Mohamed, Chem. Pharm. Bull., 2018, 66, 309–318 CrossRef CAS PubMed.
- R. M. Mohareb and M. S. Gamaan, Bull. Chem. Soc. Ethiop., 2018, 32, 541 CrossRef.
- T. Meng, Q.-P. Qin, Z.-R. Wang, L.-T. Peng, H.-H. Zou, Z.-Y. Gan, M.-X. Tan, K. Wang and F.-P. Liang, J. Inorg. Biochem., 2018, 189, 143–150 CrossRef CAS PubMed.
- Q.-P. Qin, S.-L. Wang, M.-X. Tan, Z.-F. Wang, X.-L. Huang, Q.-M. Wei, B.-B. Shi, B.-Q. Zou and H. Liang, Metallomics, 2018, 10, 1160–1169 CrossRef CAS PubMed.
- Q.-P. Qin, Z.-F. Wang, X.-L. Huang, M.-X. Tan, B.-B. Shi and H. Liang, ACS Med. Chem. Lett., 2019, 10, 936–940 CrossRef CAS.
- X.-Q. Wang, X.-B. Chen, P.-T. Ye, Z.-X. Yang, M.-J. Bai, S.-Y. Duan, Y. Li and X.-D. Yang, Bioorg. Med. Chem. Lett., 2020, 30, 126896 CrossRef CAS PubMed.
- M. O. Karataş, S. Tekin, B. Alici and S. Sandal, J. Chem. Sci., 2019, 131, 69 CrossRef.
- G. Achar, C. R. Shahini, S. A. Patil, J. G. Małecki, S.-H. Pan, A. Lan, X.-R. Chen and S. Budagumpi, J. Inorg. Biochem., 2018, 183, 43–57 CrossRef CAS PubMed.
- K. R. A. Abdellatif, M. A. Abdelgawad, H. A. H. Elshemy, N. M. Kahk and D. M. El Amir, Lett. Drug Des. Discovery, 2017, 14, 773–781 CAS.
- H. Dai, M. Huang, J. Qian, J. Liu, C. Meng, Y. Li, G. Ming, T. Zhang, S. Wang, Y. Shi, Y. Yao, S. Ge, Y. Zhang and Y. Ling, Eur. J. Med. Chem., 2019, 166, 470–479 CrossRef CAS PubMed.
- K. Vaarla, R. K. Kesharwani, K. Santosh, R. R. Vedula, S. Kotamraju and M. K. Toopurani, Bioorg. Med. Chem. Lett., 2015, 25, 5797–5803 CrossRef CAS PubMed.
- K. Vaarla, S. Karnewar, D. Panuganti, S. R. Peddi, R. R. Vedula, V. Manga and S. Kotamraju, ChemistrySelect, 2019, 4, 4324–4330 CrossRef CAS.
- S. Dhawan, N. Kerru, P. Awolade, A. Singh-Pillay, S. T. Saha, M. Kaur, S. B. Jonnalagadda and P. Singh, Bioorg. Med. Chem., 2018, 26, 5612–5623 CrossRef CAS PubMed.
- P. Adumeau, M. Davydova and B. M. Zeglis, Bioconjugate Chem., 2018, 29, 1364–1372 CrossRef CAS PubMed.
- K. D. Klika, C. Da Pieve, K. Kopka, G. Smith and A. Makarem, Org. Biomol. Chem., 2021, 19, 1722–1726 RSC.
- S. G. Narella, M. G. Shaik, A. Mohammed, M. Alvala, A. Angeli and C. T. Supuran, Bioorg. Chem., 2019, 87, 765–772 CrossRef CAS.
- B. Akhlaghinia and A. Makarem, J. Sulfur Chem., 2011, 32, 575–581 CrossRef CAS.
- M. K. Kumawat, Curr. Drug Discovery Technol., 2018, 15, 196–200 CrossRef CAS PubMed.
- Y. Li, J. Geng, Y. Liu, S. Yu and G. Zhao, ChemMedChem, 2013, 8, 27–41 CrossRef CAS.
- P. K. N. Sarangi, J. Sahoo, B. D. Swain, S. K. Paidesetty and G. P. Mohanta, Indian Drugs, 2016, 53, 5–11 Search PubMed.
- A. Aliabadi, Anti-Cancer Agents Med. Chem., 2016, 16, 1301–1314 CrossRef CAS PubMed.
- İ. Koca, M. Gumuş, A. Ozgur, A. Disli and Y. Tutar, Anti-Cancer Agents Med. Chem., 2015, 15, 916–930 CrossRef PubMed.
- A. Ayati, T. Oghabi Bakhshaiesh, S. Moghimi, R. Esmaeili, K. Majidzadeh-A, M. Safavi, L. Firoozpour, S. Emami and A. Foroumadi, Eur. J. Med. Chem., 2018, 155, 483–491 CrossRef CAS.
- T. K. Mohamed, R. Z. Batran, S. A. Elseginy, M. M. Ali and A. E. Mahmoud, Bioorg. Chem., 2019, 85, 253–273 CrossRef CAS PubMed.
- S. S. Abd El-Karim, Y. M. Syam, A. M. El Kerdawy and T. M. Abdelghany, Bioorg. Chem., 2019, 86, 80–96 CrossRef CAS PubMed.
- R. Velpula, R. Deshineni, R. Gali and R. Bavantula, Res. Chem. Intermed., 2016, 42, 1729–1740 CrossRef CAS.
- M. T. Gabr, Heterocycl. Commun., 2018, 24, 243–247 CrossRef CAS.
- S. Mah, J. Jang, D. Song, Y. Shin, M. Latif, Y. Jung and S. Hong, Org. Biomol. Chem., 2019, 17, 186–194 RSC.
- L. H. Al-Wahaibi, H. M. Abu-Melha and D. A. Ibrahim, J. Chem., 2018, 2018, 1–8 CrossRef.
- K. M. Amin, A. M. Taha, R. F. George, N. M. Mohamed and F. F. Elsenduny, Arch. Pharm., 2018, 351, 1700199 CrossRef PubMed.
- K. Venkata Sairam, B. M. Gurupadayya, B. I. Vishwanathan, R. S. Chandan and D. K. Nagesha, RSC Adv., 2016, 6, 98816–98828 RSC.
- S. K. J. Shaikh, M. S. Sannaikar, M. N. Kumbar, P. K. Bayannavar, R. R. Kamble, S. R. Inamdar and S. D. Joshi, ChemistrySelect, 2018, 3, 4448–4462 CrossRef CAS.
- M. T. Gabr, N. S. El-Gohary, E. R. El-Bendary, M. M. El-Kerdawy and N. Ni, EXCLI J., 2017, 16, 1114–1131 Search PubMed.
- F. Hersi, H. A. Omar, R. A. Al-Qawasmeh, Z. Ahmad, A. M. Jaber, D. M. Zaher and T. H. Al-Tel, Sci. Rep., 2020, 10, 2893 CrossRef CAS PubMed.
- S. G. Mane, K. S. Katagi, N. S. Kadam, M. C. Akki and S. D. Joshi, Polycyclic Aromat. Compd., 2022, 42, 371–390 CrossRef CAS.
- T. Z. Abolibda, M. Fathalla, B. Farag, M. E. A. Zaki and S. M. Gomha, Molecules, 2023, 28, 689 CrossRef CAS PubMed.
- G. S. Lingaraju, K. S. Balaji, S. Jayarama, S. M. Anil, K. R. Kiran and M. P. Sadashiva, Bioorg. Med. Chem. Lett., 2018, 28, 3606–3612 CrossRef CAS PubMed.
- L.-M. Jia, S.-L. Huang, W.-G. Pan, Y.-H. Huang and P. Luo, J. Mol. Struct., 2023, 1290, 135892 CrossRef CAS.
- M. B. Palkar, S. S. Jalalpure, R. A. Rane, H. M. Patel, M. S. Shaikh, G. A. Hampannavar, W. S. Alwan, G. S. Bolakatti and R. Karpoormath, Anti-Cancer Agents Med. Chem., 2015, 15, 970–979 CrossRef CAS PubMed.
- R. R. Manda, R. V. Nadh, T. L. Viveka, G. Angajala and V. Aruna, J. Mol. Struct., 2023, 1285, 135453 CrossRef CAS.
- M.-M. Liu, X.-Y. Chen, Y.-Q. Huang, P. Feng, Y.-L. Guo, G. Yang and Y. Chen, J. Med. Chem., 2014, 57, 9343–9356 CrossRef CAS PubMed.
- Q. Wang, Y. Guo, S. Jiang, M. Dong, K. Kuerban, J. Li, M. Feng, Y. Chen and L. Ye, Phytomedicine, 2018, 39, 160–167 CrossRef CAS PubMed.
- Y. Guo, Y. Wang, H. Li, K. Wang, Q. Wan, J. Li, Y. Zhou and Y. Chen, ACS Med. Chem. Lett., 2018, 9, 502–506 CrossRef CAS PubMed.
- J. He, Z. Li, G. Dhawan, W. Zhang, A. E. Sorochinsky, G. Butler, V. A. Soloshonok and J. Han, Chin. Chem. Lett., 2023, 34, 107578 CrossRef CAS.
- Y. Yu, A. Liu, G. Dhawan, H. Mei, W. Zhang, K. Izawa, V. A. Soloshonok and J. Han, Chin. Chem. Lett., 2021, 32, 3342–3354 CrossRef CAS.
- H. Mei, A. M. Remete, Y. Zou, H. Moriwaki, S. Fustero, L. Kiss, V. A. Soloshonok and J. Han, Chin. Chem. Lett., 2020, 31, 2401–2413 CrossRef CAS.
- Q. Wang, J. Han, A. Sorochinsky, A. Landa, G. Butler and V. A. Soloshonok, Pharmaceuticals, 2022, 15, 999 CrossRef CAS PubMed.
- J. Han, L. Kiss, H. Mei, A. M. Remete, M. Ponikvar-Svet, D. M. Sedgwick, R. Roman, S. Fustero, H. Moriwaki and V. A. Soloshonok, Chem. Rev., 2021, 121, 4678–4742 CrossRef CAS PubMed.
- J. Han, A. M. Remete, L. S. Dobson, L. Kiss, K. Izawa, H. Moriwaki, V. A. Soloshonok and D. O'Hagan, J. Fluorine Chem., 2020, 239, 109639 CrossRef CAS.
- H. Mei, J. Han, S. White, D. J. Graham, K. Izawa, T. Sato, S. Fustero, N. A. Meanwell and V. A. Soloshonok, Chem. – Eur. J., 2020, 26, 11349–11390 CrossRef CAS PubMed.
- C. Da Pieve, A. Makarem, S. Turnock, J. Maczynska, G. Smith and G. Kramer-Marek, Molecules, 2020, 25, 1562 CrossRef CAS PubMed.
- Z. Zhang, Z.-W. Bai, Y. Ling, L.-Q. He, P. Huang, H.-X. Gu and R.-F. Hu, Med. Chem. Res., 2018, 27, 1198–1205 CrossRef CAS.
- G. L. Nelson, C. T. Ronayne, L. N. Solano, S. K. Jonnalagadda, S. Jonnalagadda, T. J. Schumacher, Z. S. Gardner, H. Palle, C. Mani, J. Rumbley and V. R. Mereddy, Bioorg. Med. Chem. Lett., 2021, 52, 128411 CrossRef CAS PubMed.
- P. Mutai, G. Breuzard, A. Pagano, D. Allegro, V. Peyrot and K. Chibale, Bioorg. Med. Chem., 2017, 25, 1652–1665 CrossRef CAS PubMed.
- C.-C. Ma and Z.-P. Liu, Anti-Cancer Agents Med. Chem., 2017, 17, 395–403 CrossRef CAS PubMed.
- S. D. Durgapal, R. Soni, S. Umar, B. Suresh and S. S. Soman, Chem. Biol. Drug Des., 2018, 92, 1279–1287 CrossRef CAS PubMed.
- K. Ostrowska, D. Grzeszczuk, D. Maciejewska, I. Młynarczuk-Biały, A. Czajkowska, A. Sztokfisz, Ł. Dobrzycki and H. Kruszewska, Monatsh. Chem., 2016, 147, 1615–1627 CrossRef CAS.
- S. D. Naik, K. M. Hosamani and S. K. Vootla, Chem. Data Collect., 2018, 15–16, 207–222 CrossRef.
- A. A. Fadda, K. S. Mohamed, H. M. Refat and E. E. El-Bialy, Heterocycles, 2015, 91, 134 CrossRef CAS.
- R. M. Mohareb and N. Y. Megally Abdo, Chem. Pharm. Bull., 2015, 63, 678–687 CrossRef CAS PubMed.
- B. M. Chougala, S. S, M. Holiyachi, N. S. Naik, L. A. Shastri, S. Dodamani, S. Jalalpure, S. R. Dixit, S. D. Joshi and V. A. Sunagar, ChemistrySelect, 2017, 2, 5234–5242 CrossRef CAS.
- A. M. El-Naggar, M. M. Hemdan and S. R. Atta-Allah, J. Heterocycl. Chem., 2017, 54, 3519–3526 CrossRef CAS.
- E. A. Fayed, R. Sabour, M. F. Harras and A. B. M. Mehany, Med. Chem. Res., 2019, 28, 1284–1297 CrossRef CAS.
- T. U. Kumar, Y. Bobde, S. Pulya, K. Rangan, B. Ghosh and A. Bhattacharya, ChemistrySelect, 2019, 4, 10726–10730 CrossRef CAS.
- K. M. Hosamani, D. S. Reddy and H. C. Devarajegowda, RSC Adv., 2015, 5, 11261–11271 RSC.
- S. A. Morsy, A. A. Farahat, M. N. A. Nasr and A. S. Tantawy, Saudi Pharm. J., 2017, 25, 873–883 CrossRef PubMed.
- N. Lv, M. Sun, C. Liu and J. Li, Bioorg. Med. Chem. Lett., 2017, 27, 4578–4581 CrossRef CAS PubMed.
- D. S. Reddy, M. Kongot, V. Singh, M. A. Siddiquee, R. Patel, N. K. Singhal, F. Avecilla and A. Kumar, Arch. Pharm., 2021, 354 DOI:10.1002/ardp.202000181.
- E. Ortega-Forte, A. Rovira, A. Gandioso, J. Bonelli, M. Bosch, J. Ruiz and V. Marchán, J. Med. Chem., 2021, 64, 17209–17220 CrossRef CAS PubMed.
- N. Y. Kim, D. Vishwanath, Z. Xi, O. Nagaraja, A. Swamynayaka, K. Kumar Harish, S. Basappa, M. Madegowda, V. Pandey, G. Sethi, P. E. Lobie, K. S. Ahn and B. Basappa, Molecules, 2023, 28, 3450 CrossRef CAS PubMed.
- Y. Jin, S. He, F. Wu, C. Luo, J. Ma and Y. Hu, Eur. J. Pharm. Sci., 2023, 188, 106520 CrossRef CAS PubMed.
- A. Kumari and R. K. Singh, Bioorg. Chem., 2019, 89, 103021 CrossRef CAS PubMed.
- A. Kumari and R. K. Singh, Bioorg. Chem., 2020, 96, 103578 CrossRef CAS PubMed.
- H. Guo, Eur. J. Med. Chem., 2019, 164, 678–688 CrossRef CAS PubMed.
- Z. Xu, S. Zhang, C. Gao, J. Fan, F. Zhao, Z.-S. Lv and L.-S. Feng, Chin. Chem. Lett., 2017, 28, 159–167 CrossRef CAS.
- S. A. Patil, R. Patil and D. D. Miller, Future Med. Chem., 2012, 4, 2085–2115 CrossRef CAS.
- R. Gali, J. Banothu, R. Gondru, R. Bavantula, Y. Velivela and P. A. Crooks, Bioorg. Med. Chem. Lett., 2015, 25, 106–112 CrossRef CAS PubMed.
- O. Galayev, Y. Garazd, M. Garazd and R. Lesyk, Eur. J. Med. Chem., 2015, 105, 171–181 CrossRef CAS.
- P. R. Kamath, D. Sunil, A. A. Ajees, K. S. R. Pai and S. Das, Bioorg. Chem., 2015, 63, 101–109 CrossRef CAS PubMed.
- P. R. Kamath, D. Sunil, A. A. Ajees, K. S. R. Pai and S. Biswas, Eur. J. Med. Chem., 2016, 120, 134–147 CrossRef CAS PubMed.
- R. Pathoor and D. Bahulayan, New J. Chem., 2018, 42, 6810–6816 RSC.
- P. R. Kamath, D. Sunil, M. M. Joseph, A. A. Abdul Salam and S. T.T., Eur. J. Med. Chem., 2017, 136, 442–451 CrossRef CAS PubMed.
- S. Samundeeswari, M. V. Kulkarni, S. D. Joshi, S. R. Dixit, S. Jayakumar and R. M. Ezhilarasi, ChemistrySelect, 2016, 1, 5019–5024 CrossRef.
- Y. Fan, Z. Huang and M. Liu, J. Heterocycl. Chem., 2018, 55, 2722–2726 CrossRef CAS.
- Z. Xu, S.-J. Zhao, J.-L. Deng, Q. Wang, Z.-S. Lv and Y.-L. Fan, J. Heterocycl. Chem., 2019, 56, 400–405 CrossRef CAS.
- Q. Diao, H. Guo and G. Wang, J. Heterocycl. Chem., 2019, 56, 1667–1671 CrossRef CAS.
- Z. Xu, S. Zhao, Z. Lv, F. Gao, Y. Wang, F. Zhang, L. Bai, J. Deng, Q. Wang and Y. Fan, J. Heterocycl. Chem., 2019, 56, 1127–1132 CrossRef CAS.
- V. Singh, A. Dogra, J. Das, P. Manna and N. Gupta, FlatChem, 2021, 29, 100279 CrossRef CAS.
- J. Song, Y.-F. Guan, W.-B. Liu, C.-H. Song, X.-Y. Tian, T. Zhu, X.-J. Fu, Y.-Q. Qi and S.-Y. Zhang, Eur. J. Med. Chem., 2022, 238, 14467 CrossRef PubMed.
- V. R. Yadav, S. Prasad, B. Sung and B. B. Aggarwal, Int. Immunopharmacol., 2011, 11, 295–309 CrossRef CAS PubMed.
- M. Pilatova, L. Varinska, P. Perjesi, M. Sarissky, L. Mirossay, P. Solar, A. Ostro and J. Mojzis, Toxicol. In Vitro, 2010, 24, 1347–1355 CrossRef CAS PubMed.
- Y. Luo, R. Song, Y. Li, S. Zhang, Z.-J. Liu, J. Fu and H.-L. Zhu, Bioorg. Med. Chem. Lett., 2012, 22, 3039–3043 CrossRef CAS PubMed.
- A. Kamal, S. Prabhakar, M. Janaki Ramaiah, P. Venkat Reddy, C. Ratna Reddy, A. Mallareddy, N. Shankaraiah, T. Lakshmi Narayan Reddy, S. N. C. V. L. Pushpavalli and M. Pal-Bhadra, Eur. J. Med. Chem., 2011, 46, 3820–3831 CrossRef CAS PubMed.
- U. M. Kocyigit, Y. Budak, F. Eligüzel, P. Taslimi, D. Kılıç, İ. Gulçin and M. Ceylan, Arch. Pharm., 2017, 350, 1700198 CrossRef PubMed.
- U. M. Kocyigit, Y. Budak, M. B. Gürdere, F. Ertürk, B. Yencilek, P. Taslimi, İ. Gülçin and M. Ceylan, Arch. Physiol. Biochem., 2018, 124, 61–68 CrossRef CAS PubMed.
- S. Burmaoglu, A. O. Yilmaz, M. F. Polat, R. Kaya, İ. Gulcin and O. Algul, Bioorg. Chem., 2019, 85, 191–197 CrossRef CAS PubMed.
- S. N. Mokale, A. Begum, N. S. Sakle, V. R. Shelke and S. A. Bhavale, Biomed. Pharmacother., 2017, 89, 966–972 CrossRef CAS PubMed.
- H. A. H. Elshemy and M. A. Zaki, Bioorg. Med. Chem., 2017, 25, 1066–1075 CrossRef CAS PubMed.
- H. Suwito, H. Hardiyanti, K. Ul Haq, A. Kristanti and M. Khasanah, Mol. Ther., 2018, 2018, M1027 Search PubMed.
- H. A. El-Sherief, G. E.-D. A. Abuo-Rahma, M. E. Shoman, E. A. Beshr and R. M. Abdel-baky, Med. Chem. Res., 2017, 26, 3077–3090 CrossRef CAS.
- R. Ashraf, Hamidullah, M. Hasanain, P. Pandey, M. Maheshwari, L. R. Singh, M. Q. Siddiqui, R. Konwar, K. V. Sashidhara and J. Sarkar, Sci. Rep., 2017, 7, 45287 CrossRef CAS PubMed.
- B. Z. Kurt, N. Ozten Kandas, A. Dag, F. Sonmez and M. Kucukislamoglu, Arabian J. Chem., 2020, 13, 1120–1129 CrossRef CAS.
- Y. Wang, W. Zhang, J. Dong and J. Gao, Bioorg. Chem., 2020, 95, 103530 CrossRef CAS PubMed.
- K. Sathish Kumar, V. Kotra, P. K. Kola, CH. B. Praveena Devi, N. Anusha, B. Hari Babu, S. F. Adil, M. R. Shaik, M. Khan, A. Al-Warthan, O. Alduhaish and M. Mujahid Alam, Saudi J. Biol. Sci., 2021, 28, 386–394 CrossRef CAS PubMed.
- Y.-H. Chan, G.-Y. Yeap, Shanmugapriya and S. Sasidharan, Tetrahedron, 2022, 103, 132568 CrossRef CAS.
- M. A. Motaleb and A. A. Selim, Bioorg. Chem., 2019, 82, 145–155 CrossRef CAS PubMed.
- I. Yahaya, M. Chemchem, B. Aydıner, N. Seferoğlu, F. Erva Tepe, L. Açık, N. Aytuna Çerçi, M. Türk and Z. Seferoğlu, J. Photochem. Photobiol., A, 2019, 368, 296–306 CrossRef CAS.
- V. T. Angelova, N. G. Vassilev, B. Nikolova-Mladenova, J. Vitas, R. Malbaša, G. Momekov, M. Djukic and L. Saso, Med. Chem. Res., 2016, 25, 2082–2092 CrossRef CAS.
- X.-Y. Lu, Z.-C. Wang, S.-Z. Ren, F.-Q. Shen, R.-J. Man and H.-L. Zhu, Bioorg. Med. Chem. Lett., 2016, 26, 3491–3498 CrossRef CAS PubMed.
- F.-Q. Shen, Z.-C. Wang, S.-Y. Wu, S.-Z. Ren, R.-J. Man, B.-Z. Wang and H.-L. Zhu, Bioorg. Med. Chem. Lett., 2017, 27, 3653–3660 CrossRef CAS PubMed.
- A. Sabt, O. M. Abdelhafez, R. S. El-Haggar, H. M. F. Madkour, W. M. Eldehna, E. E.-D. A. M. El-Khrisy, M. A. Abdel-Rahman and L. A. Rashed, J. Enzyme Inhib. Med. Chem., 2018, 33, 1095–1107 CrossRef CAS PubMed.
- K. F. Debbabi, S. A. Al-Harbi, H. M. Al-Saidi, E. H. Aljuhani, S. M. Abd El-Gilil and M. S. Bashandy, J. Enzyme Inhib. Med. Chem., 2016, 31, 7–19 CrossRef CAS PubMed.
- Z. Zhang, L. Gu, B. Wang, W. Huang, Y. Zhang, Z. Ma, S. Zeng and Z. Shen, J. Enzyme Inhib. Med. Chem., 2019, 34, 808–817 CrossRef CAS PubMed.
- M. Daśko, M. Przybyłowska, J. Rachon, M. Masłyk, K. Kubiński, M. Misiak, A. Składanowski and S. Demkowicz, Eur. J. Med. Chem., 2017, 128, 79–87 CrossRef PubMed.
- D. Ganeshapillai, L. W. L. Woo, M. P. Thomas, A. Purohit and B. V. L. Potter, ACS Omega, 2018, 3, 10748–10772 CrossRef CAS PubMed.
- G. Cai, W. Yu, D. Song, W. Zhang, J. Guo, J. Zhu, Y. Ren and L. Kong, Eur. J. Med. Chem., 2019, 174, 236–251 CrossRef CAS PubMed.
- M. I. El-Gamal, D. Baek and C.-H. Oh, Bull. Korean Chem. Soc., 2016, 37, 184–191 CrossRef CAS.
- M. Mahapatra, P. Mohapatra, S. K. Sahoo, A. K. Bishoyi, R. N. Padhy and S. K. Paidesetty, J. Mol. Struct., 2023, 1283, 135190 CrossRef CAS.
- L. Yang, Z. Hu, J. Luo, C. Tang, S. Zhang, W. Ning, C. Dong, J. Huang, X. Liu and H.-B. Zhou, Bioorg. Med. Chem., 2017, 25, 3531–3539 CrossRef CAS PubMed.
- H.-W. Han, C.-S. Zheng, S.-J. Chu, W.-X. Sun, L.-J. Han, R.-W. Yang, J.-L. Qi, G.-H. Lu, X.-M. Wang and Y.-H. Yang, Biomed. Pharmacother., 2018, 97, 656–666 CrossRef CAS PubMed.
- H. Han, C. Li, Z.-F. Zhang and G.-F. An, Main Group Chem., 2019, 18, 193–201 CAS.
- H. Han, Z.-F. Zhang, J.-F. Zhang and B. Zhang, Main Group Chem., 2019, 18, 71–79 CAS.
- P. Govindaiah, N. Dumala, P. Grover and M. Jaya Prakash, Bioorg. Med. Chem. Lett., 2019, 29, 1819–1824 CrossRef CAS PubMed.
- D. Cao, Y. Liu, W. Yan, C. Wang, P. Bai, T. Wang, M. Tang, X. Wang, Z. Yang, B. Ma, L. Ma, L. Lei, F. Wang, B. Xu, Y. Zhou, T. Yang and L. Chen, J. Med. Chem., 2016, 59, 5721–5739 CrossRef CAS PubMed.
- G. Luo, M. Chen, W. Lyu, R. Zhao, Q. Xu, Q. You and H. Xiang, Bioorg. Med. Chem. Lett., 2017, 27, 2668–2673 CrossRef CAS PubMed.
- A. V. Lipeeva, D. O. Zakharov, Y. V. Gatilov, M. A. Pokrovskii, A. G. Pokrovskii and E. E. Shults, ChemistrySelect, 2019, 4, 10197–10201 CrossRef CAS.
- G. Luo, M. Muyaba, W. Lyu, Z. Tang, R. Zhao, Q. Xu, Q. You and H. Xiang, Bioorg. Med. Chem. Lett., 2017, 27, 867–874 CrossRef CAS PubMed.
- R. Soni, S. Umar, N. N. Shah, S. Balkrishnan and S. S. Soman, J. Heterocycl. Chem., 2017, 54, 2501–2510 CrossRef CAS.
- E. Y. Ahmed, N. A. Abdel Latif, M. F. El-Mansy, W. S. Elserwy and O. M. Abdelhafez, Bioorg. Med. Chem., 2020, 28, 115328 CrossRef CAS PubMed.
- J. Li, D. Hu, X. Liang, Y.-C. Wang, H.-S. Wang and Y. Pan, J. Org. Chem., 2017, 82, 9006–9011 CrossRef CAS PubMed.
- R. Wang, H. Chen, W. Yan, M. Zheng, T. Zhang and Y. Zhang, Eur. J. Med. Chem., 2020, 190, 112109 CrossRef CAS PubMed.
- M. Mbaba, J.-A. de la Mare, J. N. Sterrenberg, D. Kajewole, S. Maharaj, A. L. Edkins, M. Isaacs, H. C. Hoppe and S. D. Khanye, JBIC, J. Biol. Inorg. Chem., 2019, 24, 139–149 CrossRef CAS PubMed.
- M. Mbaba, A. N. Mabhula, N. Boel, A. L. Edkins, M. Isaacs, H. C. Hoppe and S. D. Khanye, J. Inorg. Biochem., 2017, 172, 88–93 CrossRef CAS PubMed.
- J.-N. Wei, Z.-D. Jia, Y.-Q. Zhou, P.-H. Chen, B. Li, N. Zhang, X.-Q. Hao, Y. Xu and B. Zhang, J. Organomet. Chem., 2019, 902, 120968 CrossRef CAS.
- H. Wang, L. Hu, W. Du, X. Tian, Q. Zhang, Z. Hu, L. Luo, H. Zhou, J. Wu and Y. Tian, ACS Biomater. Sci. Eng., 2017, 3, 836–842 CrossRef CAS PubMed.
- Q.-P. Qin, Z.-F. Wang, X.-L. Huang, M.-X. Tan, B.-Q. Zou and H. Liang, Eur. J. Med. Chem., 2019, 184, 111751 CrossRef CAS PubMed.
- R.-R. Ye, C.-P. Tan, L.-N. Ji and Z.-W. Mao, Dalton Trans., 2016, 45, 13042–13051 RSC.
- I. Lagunes, P. Begines, A. Silva, A. R. Galán, A. Puerta, M. X. Fernandes, I. Maya, J. G. Fernández-Bolaños, Ó. López and J. M. Padrón, Eur. J. Med. Chem., 2019, 179, 493–501 CrossRef CAS PubMed.
- A. Trommenschlager, F. Chotard, B. Bertrand, S. Amor, P. Richard, A. Bettaïeb, C. Paul, J.-L. Connat, P. Le Gendre and E. Bodio, ChemMedChem, 2018, 13, 2408–2414 CrossRef CAS PubMed.
- K. N. A. Rahman, J. Haribabu, C. Balachandran, N. S. P. Bhuvanesh, R. Karvembu and A. Sreekanth, Polyhedron, 2017, 135, 26–35 CrossRef CAS.
- T. Zhu, Y. Wang, W. Ding, J. Xu, R. Chen, J. Xie, W. Zhu, L. Jia and T. Ma, Chem. Biol. Drug Des., 2015, 85, 385–393 CrossRef CAS PubMed.
- N. Sunitha, C. I. S. Raj and B. S. Kumari, J. Mol. Struct., 2023, 1285, 135443 CrossRef CAS.
- M. Sumi, N. T. Nevaditha and B. Sindhu Kumari, Bioorg. Chem., 2023, 136, 106555 CrossRef CAS PubMed.
- S. Balcıoğlu, M. Olgun Karataş, B. Ateş, B. Alıcı and İ. Özdemir, Bioorg. Med. Chem. Lett., 2020, 30, 126805 CrossRef PubMed.
- L. H. Ramdani, O. Talhi, C. Decombat, M. Vermerie, A. Berry, A. Silva, K. Bachari, M.-P. Vasson, L. Delort and F. Caldefie-Chézet, Anticancer Res., 2019, 39, 6107–6114 CrossRef CAS PubMed.
- T. Nasr, S. Bondock, H. M. Rashed, W. Fayad, M. Youns and T. M. Sakr, Eur. J. Med. Chem., 2018, 151, 723–739 CrossRef CAS PubMed.
- Z. Solárová, M. Kello, S. Hamuľaková, L. Mirossay and P. Solár, Acta Chim. Slov., 2018, 65, 875–881 CrossRef.
- T. Prashanth, B. R. V. Avin, P. Thirusangu, V. L. Ranganatha, B. T. Prabhakar, J. N. N. Sharath Chandra and S. A. Khanum, Biomed. Pharmacother., 2019, 112, 108707 CrossRef CAS PubMed.
- C. Zwergel, B. Czepukojc, E. Evain-Bana, Z. Xu, G. Stazi, M. Mori, A. Patsilinakos, A. Mai, B. Botta, R. Ragno, D. Bagrel, G. Kirsch, P. Meiser, C. Jacob, M. Montenarh and S. Valente, Eur. J. Med. Chem., 2017, 134, 316–333 CrossRef CAS PubMed.
- A. Herrera-R, T. W. Naranjo, M. E. Maldonado, G. Moreno-Q, A. Yepes and W. Cardona-G, Med. Chem. Res., 2020, 29, 377–395 CrossRef CAS.
- P. R. Kadam, Y. D. Bodke, M. B, I. Pushpavathi, N. D. Satyanarayan and B. N. Nippu, J. Mol. Struct., 2023, 1278, 134918 CrossRef CAS.
- Z. Tian, L. Shi, Z. Liu, H. Li, L. Yang and Y. Liu, J. Mol. Struct., 2023, 1288, 135826 CrossRef CAS.
- F. Yang, N. Zhao, J. Song, K. Zhu, C. Jiang, P. Shan and H. Zhang, Molecules, 2019, 24, 2569 CrossRef CAS PubMed.
- M. Bu, H. Li, H. Wang, J. Wang, Y. Lin and Y. Ma, Molecules, 2019, 24, 3307 CrossRef PubMed.
- J.-N. Yao, X.-X. Zhang, Y.-Z. Zhang, J.-H. Li, D.-Y. Zhao, B. Gao, H.-N. Zhou, S.-L. Gao and L.-F. Zhang, Invest. New Drugs, 2019, 37, 1300–1308 CrossRef CAS PubMed.
- Ch. Pavan Kumar, A. Devi, P. Ashok Yadav, R. Rao Vadaparthi, G. Shankaraiah, P. Sowjanya, N. Jain and K. Suresh Babu, J. Asian Nat. Prod. Res., 2016, 18, 1063–1078 CrossRef CAS PubMed.
- H. Singh, M. Kumar, K. Nepali, M. K. Gupta, A. K. Saxena, S. Sharma and P. M. S. Bedi, Eur. J. Med. Chem., 2016, 116, 102–115 CrossRef CAS PubMed.
- B. M. Chougala, S. L. Shastri, M. Holiyachi, L. A. Shastri, S. S. More and K. V. Ramesh, Med. Chem. Res., 2015, 24, 4128–4138 CrossRef CAS.
- S. Koparde, K. M. Hosamani, D. A. Barretto and S. D. Joshi, Chem. Data Collect., 2018, 15–16, 41–53 CrossRef.
- M. Patel, N. Pandey, J. Timaniya, P. Parikh, A. Chauhan, N. Jain and K. Patel, RSC Adv., 2021, 11, 27627–27644 RSC.
- S. Vodnala, A. K. D. Bhavani, R. Kamutam, V. G. M. Naidu, Promila and C. Prabhakar, Bioorg Med Chem Lett, 2016, 26, 3973–3977 CrossRef CAS PubMed.
- M. Bu, T. Cao, H. Li, M. Guo, B. B. Yang, C. Zeng and L. Hu, ChemMedChem, 2017, 12, 466–474 CrossRef CAS PubMed.
- B. Yu, P.-P. Qi, X.-J. Shi, R. Huang, H. Guo, Y.-C. Zheng, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2016, 117, 241–255 CrossRef CAS PubMed.
- H. Zhu, L. Yu, J. Liu, M. Wang, T. Zhang and F. Qiu, Chin. Herb. Med., 2019, 11, 113–115 CrossRef.
- Z.-M. Feng, Z.-Z. Liu, K. Xu, Y.-N. Yang, J.-S. Jiang and P.-C. Zhang, ChemInform, 2016, 47 DOI:10.1002/chin.201633189.
- K. B. Pal, M. Mahanti, X. Huang, S. Persson, A. P. Sundin, F. R. Zetterberg, S. Oredsson, H. Leffler and U. J. Nilsson, Org. Biomol. Chem., 2018, 16, 6295–6305 RSC.
- E. S. Moustafa, N. F. Swilam, O. B. Ghanem, A. N. Hashim, M. A. Nawwar, U. Lindequist and M. W. Linscheid, Pharmazie, 2018, 73, 241–243 CAS.
- L.-L. Lou, R. Guo, Z.-Y. Cheng, P. Zhao, G.-D. Yao, X.-B. Wang, X.-X. Huang and S.-J. Song, Phytochem. Lett., 2018, 24, 15–20 CrossRef CAS.
- A. Sharma, S. Gudala, S. R. Ambati, S. penta, Y. Bomma, V. R. Janapala, A. Jha and A. Kumar, J. Chin. Chem. Soc., 2018, 65, 810–821 CrossRef CAS.
- W. Kozak, M. Daśko, M. Masłyk, B. Gielniewski, J. Rachon and S. Demkowicz, J. Asian Nat. Prod. Res., 2015, 17, 1091–1096 CrossRef CAS PubMed.
- A. Ibrar, S. Zaib, F. Jabeen, J. Iqbal and A. Saeed, Arch. Pharm., 2016, 349, 553–565 CrossRef CAS PubMed.
- K. Wada, M. Goto, K.-H. Lee and H. Yamashita, Chem. Pharm. Bull., 2023, 71, 52–57 CrossRef CAS PubMed.
- E. F. Bruna-Haupt, M. D. Perretti, H. A. Garro, R. Carrillo, F. Machín, I. Lorenzo-Castrillejo, L. Gutiérrez, E. G. Vega-Hissi, M. Mamberto, M. Menacho-Marquez, C. O. Fernández, C. García and C. R. Pungitore, ACS Omega, 2023, 8, 26479–26496 CrossRef CAS PubMed.
- H. Singh, J. V. Singh, M. K. Gupta, A. K. Saxena, S. Sharma, K. Nepali and P. M. S. Bedi, Bioorg. Med. Chem. Lett., 2017, 27, 3974–3979 CrossRef CAS PubMed.
- A. S. Abd-El-Aziz, A. T. Alsaggaf, R. M. Okasha, H. E. A. Ahmed, R. Bissessur, A. A. Abdelghani and T. H. Afifi, ChemistrySelect, 2016, 1, 5025–5033 CrossRef.
- M. Patel and K. Patel, Heterocycl. Commun., 2019, 25, 146–151 CAS.
- S. Prateeptongkum, W. Mahavorasirikul and N. Duangdee, ARKIVOC, 2018, 2018, 73–85 Search PubMed.
- J. Zhao, H. Zhao and B. S. J. Blagg, WO2015/192099Al, 2015.
- D. Carrico-Moniz, US Pat., US9388155BI, 2016 Search PubMed.
- C. Liang, D. Tian, M. Jia, X. Wang, X. Xie and D. Zhang, US Pat., US9758503BI, 2017 Search PubMed.
- P. Arsenjans, J. Vasiljeva, I. Domracheva, I. Shestakova and I. Kalvins, PCT Int. Appl. WO2018/015788Al, 2018.
- F. Tosun and M. Miski, PCT Int. Appl. WO2019/209232A2, 2019.
- R. D. Lucrezia, T. Bergbrede, P. Nussbaumer, U. Koch, B. Klebl, A. Choidas, A. Unger, N. Larsson, M. Falkenberg-Gustafsson and C. M. Gustafsson, US Pat., US2020/0031794Al, 2020 Search PubMed.
- A. R. Temmanaboyina, PCT Int. Appl. WO2022/238741Al, 2022.
| This journal is © The Royal Society of Chemistry 2024 |