DOI:
10.1039/D3MD00479A
(Research Article)
RSC Med. Chem., 2024,
15, 1283-1294
Design, synthesis, and cytotoxicity of ibuprofen-appended benzoxazole analogues against human breast adenocarcinoma†
Received
9th September 2023
, Accepted 29th December 2023
First published on 2nd January 2024
Abstract
A library of novel ibuprofen-appended benzoxazole analogues (7a–l) was synthesized via a series of nitration, reduction, and condensation–cyclization reactions and screened for their in vitro anticancer activity against human breast cancer MCF-7 and MDA-MB-231 cell lines using doxorubicin as a standard reference. Compounds 7h and 7j displayed outstanding activity against the MCF-7 cell line with an IC50 value of 8.92 ± 0.91 μM and 9.14 ± 8.22 μM, respectively, compared to the doxorubicin IC50 value of 9.29 ± 1.02 μM. Compound 7h also exhibited outstanding activity against the MDA-MB-231 cell line with an IC50 value of 7.54 ± 0.95 μM compared to the doxorubicin IC50 value of 7.68 ± 5.36 μM. Compounds 7h, 7i, 7j, and 7g showed identical morphological changes to those showed by doxorubicin. The molecular docking study against ERα unveiled their best docking scores and binding interactions in agreement to experimental results. Pharmacokinetics prediction envisaged their drug-like properties suitable for therapeutic applications.
Introduction
For several decades, cancer has been a very serious disorder that has caused a significant number of deaths by affecting all age groups worldwide.1,2 It is caused by mutation and abnormal growth of cells and was accountable for 10 million deaths in 2020 globally; this number is expected to reach 13 million by 2030.3–5 There are around 277 distinct forms of cancers identified in an overall context.6 In men, lung, stomach, prostate, colorectal, and liver cancers are prevalent; while in women, breast, colorectal, lung, cervical, and stomach cancers are prevalent.7 Cancer can be prevented by avoiding exposure to carcinogenic agents; augmenting the host defense mechanism; and with chemopreventive agents that inhibit, reverse, or block tumor formation.8 However, it is still a life-threatening disorder and there is tremendous demand for the development of new anticancer drugs.9
Doxorubicin, which is an anthracycline derivative, is considered to be a potent drug and is used in the treatment of several cancers, such as breast cancer, lung cancer, ovarian cancer, thyroid cancer, non-Hodgkin's and Hodgkin's lymphoma, multiple myeloma, sarcoma, and cervical cancer.10–13 The mechanism exhibited by doxorubicin in cancer cells involves the disruption of topoisomerase-II-mediated DNA repair and intercalation into DNA14 and blocking of extracellular signal-regulated protein kinases 1/2 (ERK1/2)15 and fibroblast growth factor receptor 2 (FGFR2).16 The major limitation of doxorubicin is cardiotoxicity.17 It requires additional medication to prevent the cardiotoxicity of doxorubicin.18 Trastuzumab, lapatinib, neratinib, and tucatinib are among the pharmaceutical agents now approved for the therapeutic management of breast cancer. The occurrence of congestive heart failure (CHF) as a result of trastuzumab administration was noted during the initial investigations of a metastatic illness.19 Patients undergoing treatment with lapatinib frequently report diarrhea and dermatologic side effects.20 Neratinib, as a dual inhibitor of HER and EGFR kinases, may have side effects that are comparable to those of lapatinib. The most frequently reported adverse effect is diarrhea, and certain clinicians advise initiating anti-diarrheal prophylaxis with the initial dose.21 Nausea, diarrhea, and fatigue were the three most frequently associated adverse effects with tucatinib.22 Hence, it is urgent to develop drugs that demonstrate competent efficacy and reduced toxicity.
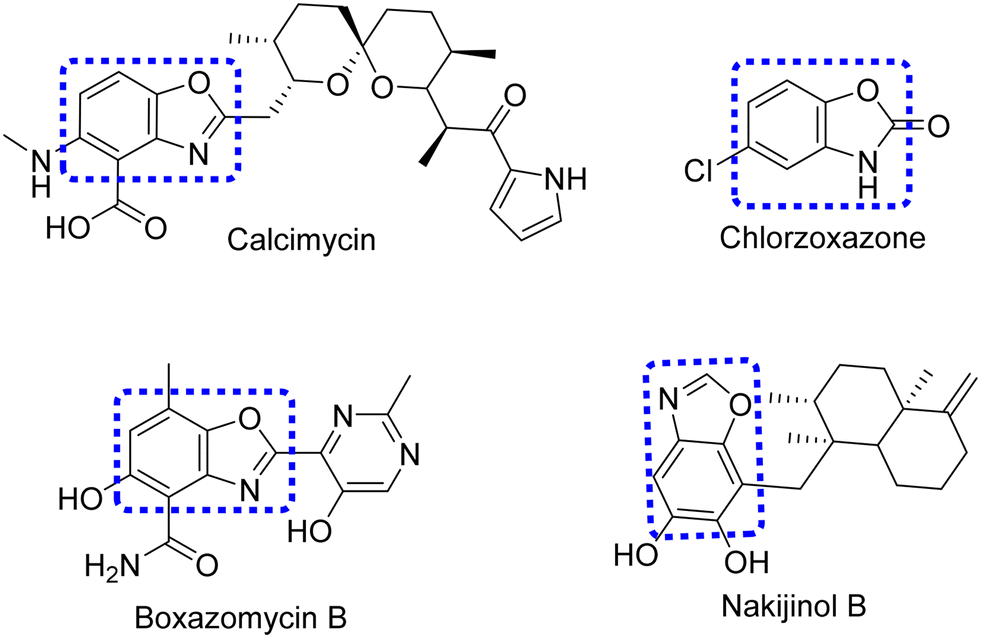
Heterocyclic molecules containing oxygen and nitrogen atoms have gained prominence due to their varied pharmacological activities.23–25 Benzoxazole is one of the most important heterocyclic compounds that exhibit remarkable pharmacological activities26 such as anticancer,27 anthelmintic,28 cyclooxygenase inhibitory,29 antifungal,30 antitubercular,31 5HT3 receptor antagonists,32 antiinflammatory,33 analgesic34 and cyclin-dependent kinase inhibitory,35 5-lipoxygenase inhibitory,36 melatonin receptor agonist,37 antibacterial,38 antiHIV,39 anticonvulsant,40 antiviral,41 antiallergic,42 cyclooxygenase 2 (COX-2) inhibitory,43 antihyperglycemic,44 dopamine D4 agonists,43 herbicidal,45 amyloidogenesis inhibitors,46 rho kinase inhibitors,47 and diarrhea-dependent irritable bowel syndrome property.48 Other than biology, benzoxazoles have various applications such as fluorescent,49 brighteners,50 whitening agents,51 ballistic materials,52 and sensors.53 Several marketed drugs are available having benzoxazole as the core active moiety, like calcimycin54 and boxazomycin B,55 used as antibiotics, and chlorzoxazone,56 a muscle relaxant, which are presented in Fig. 1. Nakijinol B57 is a natural product found in Dactylospongia elegans has shown anticancer properties.
 |
| | Fig. 1 Marketed drugs containing the benzoxazole structural block. | |
Ibuprofen is a non-steroidal antiinflammatory drug, which was introduced in the year 1969.58 It is a propionic acid derivative generally used for the treatment of rheumatoid and osteoarthritis, musculo-skeletal pain, and for mild to moderate pain.59,60 It is widely used as an antiinflammatory, analgesic, and antipyretic drug supplied in dosages of 200 to 800 mg three times a day.61 Several reports are available as researchers synthesized ibuprofen hybrids to enhance its biological activities such as antioxidant,62 antiinflammatory,63 and antimicrobial.64 Previously, our research group synthesized ibuprofen––1,2,3-triazole hybrids and investigated their enhanced antiinflammatory and antibacterial activities65 (Fig. 2).
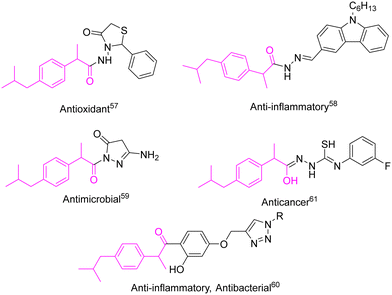 |
| | Fig. 2 Representative examples of ibuprofen hybrids. | |
The process of joining two or more bioactive fragments into a single molecule using proper linkers is known as molecular hybridization. In comparison to their respective components, these new hybrids are always endowed with increased activity or novel biological features.66,67 With an extension to our research work,65 the broad spectrum of pharmacological activities of benzoxazoles and enhanced biological activities of ibuprofen hybrids inspired us to design and synthesize novel ibuprofen-tethered benzoxazole scaffolds (Fig. 3). Further, the novel compounds were screened for their cytotoxicity against human breast cancer cell lines MCF-7 and MDA-MB-231, and molecular docking studies were performed to understand their binding interactions.
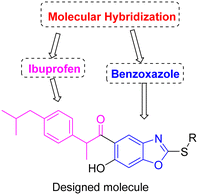 |
| | Fig. 3 Designed molecule of ibuprofen-tethered benzoxazole. | |
Results and discussion
Chemistry
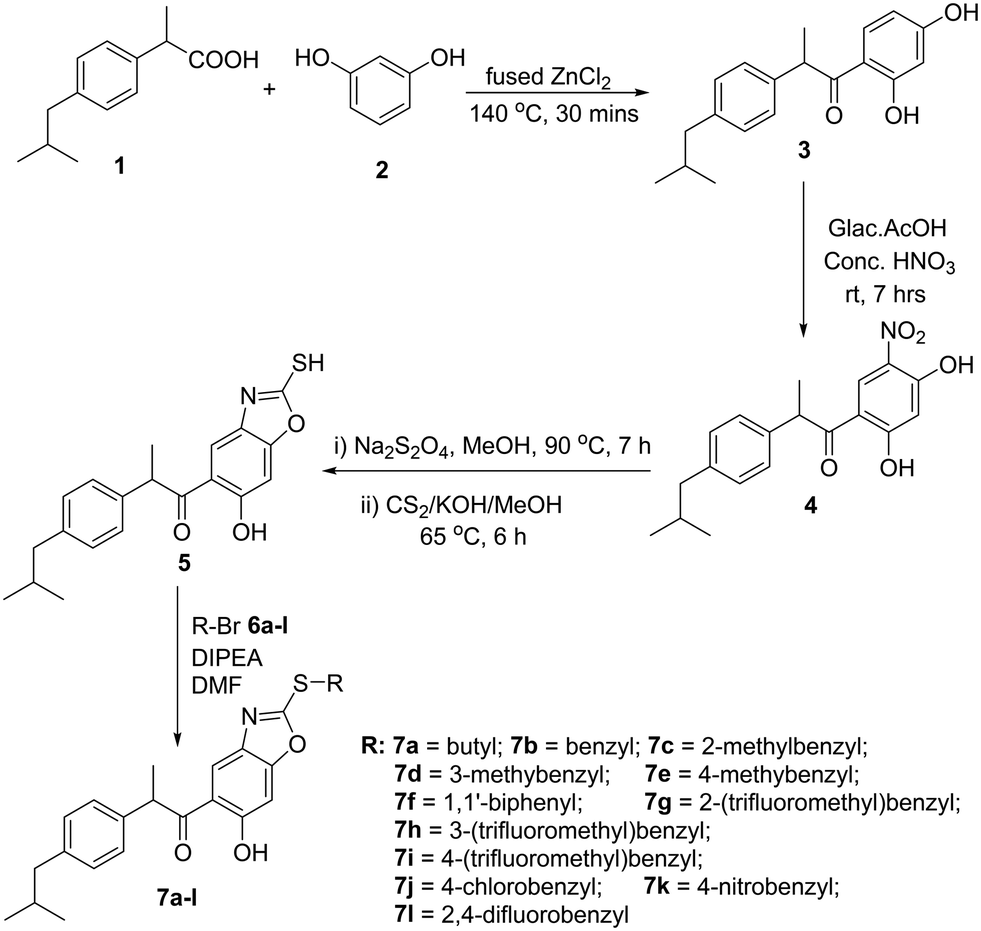
In the present work, the synthesis of 1-(2-(thiosubstituted)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one derivatives 7a–l is presented in Scheme 1. 1-(2,4-Dihydroxyphenyl)-2-(4-isobutylphenyl)propan-1-one 3 is a reported compound synthesized in good yield by the reaction between ibuprofen 1 and resorcinol 2 in the presence of anhydrous ZnCl2. Upon nitration of compound 3, using glacial acetic acid + fuming HNO3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the nitration mixture, 1-(2,4-dihydroxy-5-nitrophenyl)-2-(4-isobutylphenyl)propan-1-one 4 was obtained; the nitration occurs at the 5th position of resorcinol nuclei as this position is meta to carbonyl, and the ortho & para positions to the subsequent –OH groups. The incorporation of the –NO2 group was evidenced by the –N
:1) as the nitration mixture, 1-(2,4-dihydroxy-5-nitrophenyl)-2-(4-isobutylphenyl)propan-1-one 4 was obtained; the nitration occurs at the 5th position of resorcinol nuclei as this position is meta to carbonyl, and the ortho & para positions to the subsequent –OH groups. The incorporation of the –NO2 group was evidenced by the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency signals that appeared near 1572 cm−1 and 1366 cm−1 in the IR spectrum and mass [M + H]+ = 344.35. Compound 4 was refluxed with Na2S2O4 in methanol for 7 h, followed by the addition of CS2 and KOH to obtain 1-(6-hydroxy-2-mercaptobenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one 5. It was evidenced by the appearance of two singlets at δ 7.50 ppm and δ 6.92 ppm in the 1H NMR spectrum corresponding to the two protons of the benzoxazole ring. The –SH and –OH protons appeared as singlets downfield at δ 10.35 ppm and δ 12.90 ppm in the proton 1H NMR spectrum, respectively. The stretching frequency absorption peak of the S–H bond was observed at 2607 cm−1 in the IR spectrum. Compound 5 was allowed to react with various benzyl bromides 6a–l individually in the presence of diisopropylethyl amine in DMF at room temperature to obtain 1-(2-(thiosubstituted)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one derivatives 7a–l (Fig. 4). It was evident that the protons of the –CH2– group planked between the S atom and phenyl group appeared as multiplet at δ 4.51 ppm in the 1H NMR spectrum. The –SH proton signal was absent as the attacking benzyl bromides attached to the S atom by the removal of HBr. In addition, the –SH stretching peak in the IR spectrum at about 2607 cm−1 disappeared. And the –OH stretching frequency absorption peak was present at 3437 cm−1.
O stretching frequency signals that appeared near 1572 cm−1 and 1366 cm−1 in the IR spectrum and mass [M + H]+ = 344.35. Compound 4 was refluxed with Na2S2O4 in methanol for 7 h, followed by the addition of CS2 and KOH to obtain 1-(6-hydroxy-2-mercaptobenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one 5. It was evidenced by the appearance of two singlets at δ 7.50 ppm and δ 6.92 ppm in the 1H NMR spectrum corresponding to the two protons of the benzoxazole ring. The –SH and –OH protons appeared as singlets downfield at δ 10.35 ppm and δ 12.90 ppm in the proton 1H NMR spectrum, respectively. The stretching frequency absorption peak of the S–H bond was observed at 2607 cm−1 in the IR spectrum. Compound 5 was allowed to react with various benzyl bromides 6a–l individually in the presence of diisopropylethyl amine in DMF at room temperature to obtain 1-(2-(thiosubstituted)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one derivatives 7a–l (Fig. 4). It was evident that the protons of the –CH2– group planked between the S atom and phenyl group appeared as multiplet at δ 4.51 ppm in the 1H NMR spectrum. The –SH proton signal was absent as the attacking benzyl bromides attached to the S atom by the removal of HBr. In addition, the –SH stretching peak in the IR spectrum at about 2607 cm−1 disappeared. And the –OH stretching frequency absorption peak was present at 3437 cm−1.
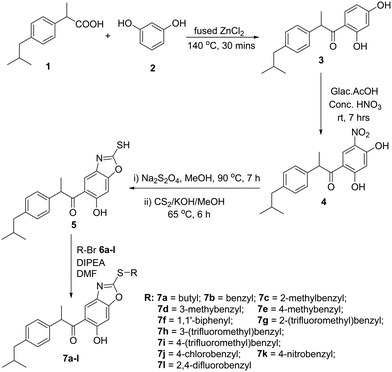 |
| | Scheme 1 Synthesis of 1-(2-(thiosubstituted)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one derivatives (7a–l). | |
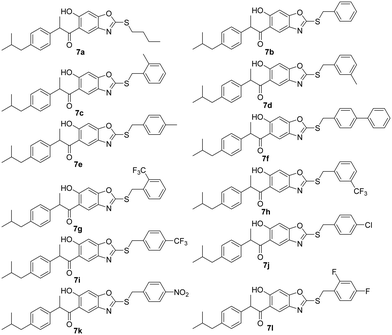 |
| | Fig. 4 Analogues of target compounds (7a–l). | |
Cytotoxicity
The novel ibuprofen-linked benzoxazole analogues 7a–l were screened for their in vitro anticancer activity against the human breast cancer MCF-7 and MDA-MB-231 cell lines. The MTT assay protocol was performed using various micromolar concentrations, viz., 2, 4, 8, 16, 32, 64, and 128 μM, for which the marketed drug doxorubicin was used as the standard. The IC50 values of compounds are tabulated in Table 1. Compound 7h containing (3-trifluoromethyl)benzyl group and compound 7j containing the 4-chlorobenzyl group exhibited outstanding activity against the MCF-7 cell line with an IC50 value of 8.92 ± 0.91 μM and 9.14 ± 8.22 μM, respectively, compared to the doxorubicin IC50 value of 9.29 ± 1.02 μM. Compound 7g containing the (2-trifluoromethyl)benzyl group also showed promising activity against the MCF-7 cell line with an IC50 value of 12.19 ± 1.33 μM, which is closer in value to the doxorubicin IC50 value, whereas other molecules indicated good to moderate activity compared to doxorubicin against the MCF-7 cell line. Compound 7h also implied outstanding activity against the MDA-MB-231 cell line with an IC50 value of 7.54 ± 0.95 μM compared to the doxorubicin IC50 value of 7.68 ± 5.36 μM. Further, compounds 7g and 7i [(2-trifluoromethyl)benzylgroup] showed promising activity against the MDA-MB-231 cell line with an IC50 value of 12.19 ± 1.17 μM and 8.78 ± 0.58 μM, respectively, closer to the doxorubicin IC50 value. All the other compounds indicated good to moderate activity against the MDA-MB-231 cell lines. It may be attributed to the electron withdrawing functions –CF3 and –Cl present on the molecules, which make the molecule electron-deficient; as a result, they likely to bind with the electron-rich site of the biological targets. Also, there is a possibility of halogen bonding and hydrogen acceptor interaction of F and Cl atoms in the cavity of the target molecules. Apart from this, they have a free –OH group, which can act as a hydrogen bond donor as well as acceptor. Also, the planar benzoxazole and phenyl nucleuses can form π–π stacked and T-shaped π interactions with the target sites. The % cell viability of compounds 7a–l against MCF-7 and MDA-MB-231 cell are presented in Fig. 5 and 6. The normal cell line HEK-293 was treated to know their toxicity and it was revealed that there were no adverse effects.
Table 1 IC50 value of compounds 7a–l against the MCF-7 and MDA-MB-231 cell line
| Entry |
IC50 (μM±SD) |
| MCF-7 |
MDA-MB-231 |
HEK-293 |
|
7a
|
62.54 ± 5.33 |
69.53 ± 5.25 |
90.01 ± 6.89 |
|
7b
|
109.27 ± 9.84 |
82.33 ± 7.14 |
89.72 ± 6.12 |
|
7c
|
62.05 ± 7.24 |
55.20 ± 5.69 |
92.44 ± 7.41 |
|
7d
|
83.39 ± 7.92 |
53.25 ± 5.27 |
81.22 ± 5.68 |
|
7e
|
68.24 ± 7.05 |
61.32 ± 5.33 |
87.26 ± 5.22 |
|
7f
|
57.18 ± 6.35 |
55.32 ± 4.98 |
89.57 ± 5.36 |
|
7g
|
12.19 ± 1.33 |
12.19 ± 1.17
|
91.07 ± 6.51 |
|
7h
|
8.92 ± 0.91
|
7.54 ± 0.95
|
90.45 ± 6.92 |
|
7i
|
39.12 ± 3.34 |
8.78 ± 0.58
|
89.41 ± 5.58 |
|
7j
|
9.14 ± 8.22
|
19.98 ± 1.24 |
88.25 ± 6.21 |
|
7k
|
48.73 ± 4.16 |
54.47 ± 4.22 |
85.63 ± 6.15 |
|
7l
|
72.88 ± 62.28 |
59.25 ± 5.67 |
87.54 ± 6.01 |
|
Doxorubicin
|
9.29 ± 1.02 |
7.68 ± 5.36 |
89.27 ± 6.23 |
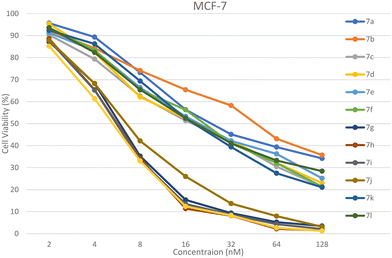 |
| | Fig. 5 % Cell viability of the MCF-7 cell line upon the treatment of 7a–l. The data are derived from three replicates and presented as mean ± SEM. All data were statistically evaluated by one-way ANOVA at a significance level of p < 0.05. | |
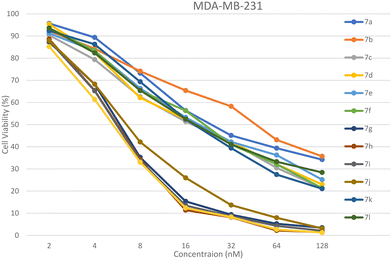 |
| | Fig. 6 % Cell viability of the MDA-MB-231 cell line upon treatment of 7a–l. The data are derived from three replicates and presented as mean ± SEM. All data were statistically evaluated by one-way ANOVA at a significance level of p < 0.05. | |
Morphology
Cell density and maintaining the original form are traits of improved growth and metabolic activity, respectively. The loss of their original size and shape, as well as the deformation of their shape, are signs of metabolic inactivity. Cell death in suspension type cells was described by loss of shine and a lack of spreading capacity, but it was characterized in adherent cells by the rounding of the cells and floating on the growth media. Severe cell damage was visible in smaller fragments of cellular remnants floating on the growth media. A microscope examination of the cells revealed that the treatment with several possible chemicals had caused the morphology of the cells to change from their characteristic elongated form into a spherical shape. In support of the results obtained from cytotoxic studies by the MTT assay, the morphology of the best active compounds was recorded at their IC50 value. In support of the results obtained from the cytotoxic studies by the MTT assay, compounds 7g, 7h, and 7j showed morphological deformation of MCF-7 cells as well as showed morphological changes against the MDA-MB-231 cells compared with the control cells. At their respective IC50 concentrations, the compounds screened and exhibited identical morphological changes to those seen for the effects of doxorubicin on MCF-7 and MDA-MB-231 cells, presented in Fig. 7 and 8, respectively.
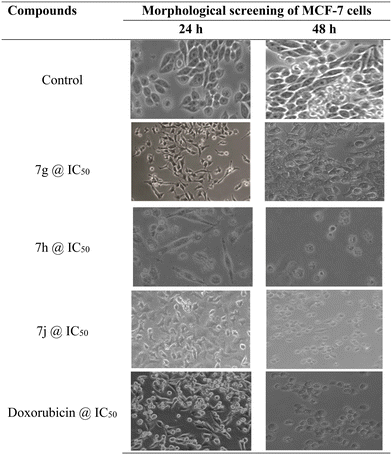 |
| | Fig. 7 Morphological screening of compounds 7g, 7h, and 7j against MCF-7 cell lines. | |
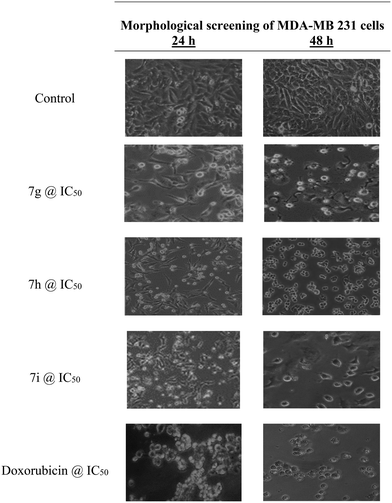 |
| | Fig. 8 Morphological screening of compounds 7g, 7h, and 7i against MDA-MB-231 cell lines. | |
Molecular docking against ERα
ERα plays a role in cell proliferation and has been found in the endometrial, breast cancer, and ovarian stromal cells as well as in the hypothalamus.68 The ligand (tamoxifen)-bound ERα complex prevent the estrogenic effects responsible for cancer cell proliferation. Hence, we have chosen the crystal structure of human estrogen receptor alpha ligand-binding domain in the complex with 4-hydroxytamoxifen (PDB ID: 3ERT) for docking study.69,70 All newly synthesized molecules were docked into the active site pocket of Estrogen Receptor Alpha (ERα). Further, the result was validated by redocking the co-crystallized ligand 4-hydroxy tamoxifen, which presented a RMSD value of 1.027 Å. The docking scores of all the ligand molecules are comparable to 4-hydroxytamoxifen against ERα. The binding affinity scores of compounds 7a–l were ranging from −7.9 to −9.9 kcal mol−1 whereas 4-hydroxytamoxifen scored −9.1 kcal mol−1 (Table 2).
Table 2 Docking scores and binding interactions of compounds 7a–l against ERα (PDB ID: 3ERT)
| Compound |
Binding energy (kcal mol−1) |
Interacting amino acids |
| H-bond |
Hydrophobic |
|
7a
|
−7.9 |
Thr347 |
Met343, Leu346, Leu349, Ala350, Trp383, Leu384, Leu525 |
|
7b
|
−9.0 |
Thr347 |
Met343, Leu346, Ala350, Trp383, Leu384, Met388, Met421, Ile424, Met522, Leu525, Tyr526, Lys529 |
|
7c
|
−9.3 |
Thr347 |
Met343, Leu346, Leu349, Ala350, Asp351, Leu354, Trp383, Leu387, Met388, Leu391, Phe404, Met421, Ile424, Leu428, Gly521, Leu525, Leu536 |
|
7d
|
−9.6 |
Thr347 |
Met343, Leu346, Ala350, Trp383, Leu384, Leu387, Leu391, Phe404, Met522, Leu525, Tyr526, Lys529 |
|
7e
|
−9.0 |
Thr347 |
Met343, Leu346, Thr347, Ala350, Asp351, Trp383, Leu384, Leu387, Leu391, Met522, Leu525, Tyr526, Lys529 |
|
7f
|
−9.5 |
Thr347 |
Met343, Leu346, Ala350, Trp383, Leu387, Met388, Leu391, Phe404, Leu428, Met522, Leu525 |
|
7g
|
−9.6 |
Thr347 |
Leu346, Leu349, Ala350, Met388, Leu391, Phe404, Met421, Ile424, Leu428, Gly521, Met522, Leu525, Lys529 |
|
7h
|
−9.9 |
Arg394 |
Met343, Leu346, Leu349, Ala350, Glu353, Trp383, Leu384, Leu387, Met388, Leu381, Phe404, Met522, Leu525, Tyr526 |
|
7i
|
−9.7 |
Thr347, Arg394 |
Met343, Leu346, Leu349, Ala350, Asp351, Glu353, Trp383, Leu384, Leu387, Met388, Leu391, Phe404, Met522, Leu525, Tyr526, Lys529 |
|
7j
|
−9.5 |
Thr347, His524 |
Met343, Leu346, Ala350, Trp383, Met388, Glu419, Gly420, Met421, Ile424, Gly521, Met522, His524, Leu525, Tyr526, Lys529 |
|
7k
|
−8.9 |
Thr347 |
Met343, Leu346, Thr347, Ala350, Asp351, Trp383, Leu384, Leu387, Leu391, Met522, Leu525, Tyr526, Lys529 |
|
7l
|
−9.3 |
Thr347, His524 |
Met343, Leu346, Ala350, Trp383, Leu384, Met388, Gly420, Met421, Ile424, Gly521, Met522, Leu525, Tyr526, Lys529 |
|
4-Hydroxy tamoxifen
|
−9.1 |
Glu353 |
Met343, Leu346, Leu387, Leu391, Met421, Leu525 |
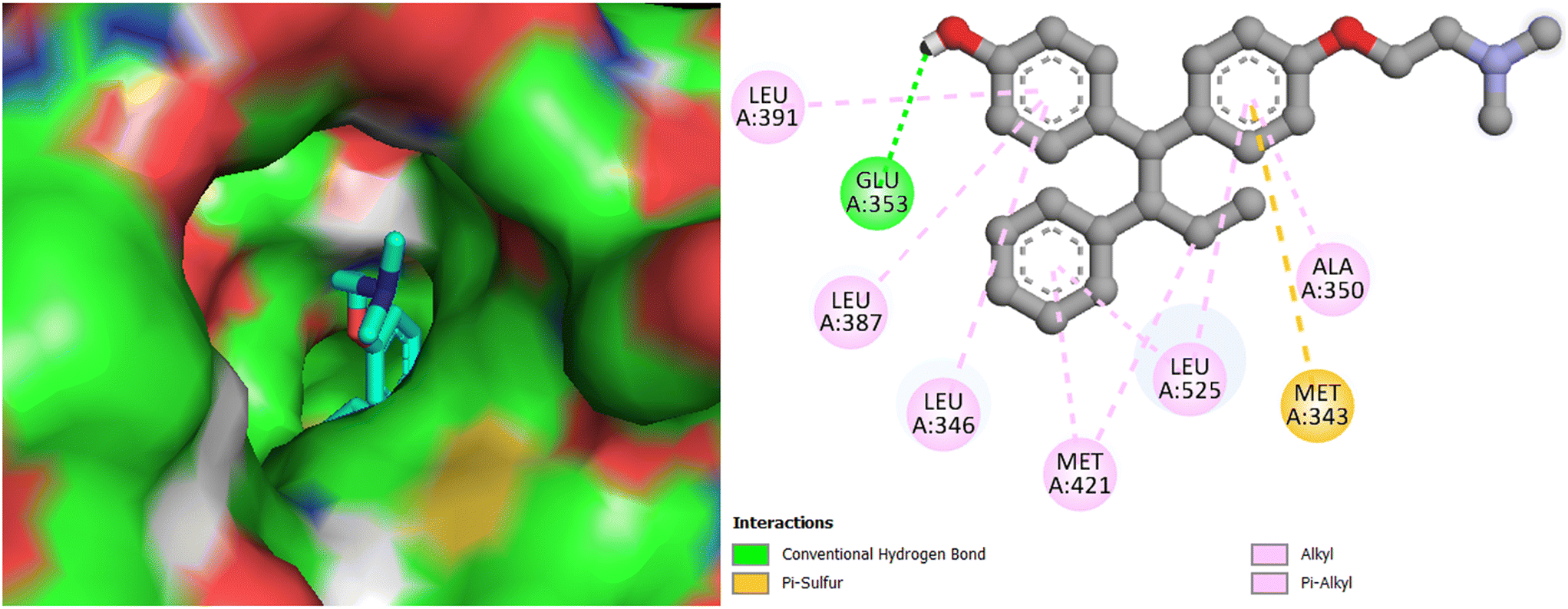
Interestingly, the best active compound 7h scored the highest binding energy value of −9.9 kcal mol−1; it demonstrated two key interactions with Arg394 of Erα, with the bond distance of 2.50 and 2.84 Å. Further, hydrophobic interactions are indicated by 7g with Met343, Leu346, Ala350, Trp383, Leu384, Met388, Gly420, Met421, Ile424, Gly521, Met522, Leu525, Tyr526, and Lys529 of Erα (Fig. 9), among which two π–sulphur bond interactions with Met343, two halogen bond interactions with Glu353 and Phe404, and a π–π stacked interactions with Trp383 are extant. The co-crystallized ligand 4-hydroxytamoxifen scored a binding energy value of −9.1 kcal mol−1. It indicated an H-bond interaction with Glu353, and hydrophobic interactions with Met343, Leu346, Leu387, Leu391, Met421, and Leu525 of ERα (Fig. 10). The other promising molecules 7g, 7i, and 7j also scored the best binding energy values of −9.7, −9.7, and −9.5 kcal mol−1, respectively, higher than that of the 4-hydroxytamoxifen score; moreover, the binding interactions of compounds are same as seen for 4-hydroxytamoxifen, which proves that they could best fit into the cavity of ERα.
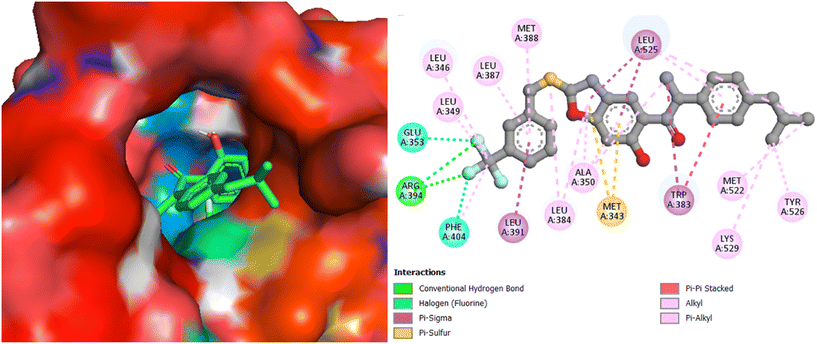 |
| | Fig. 9 Docking pose and binding interactions of compound 7h in the cavity of Erα. | |
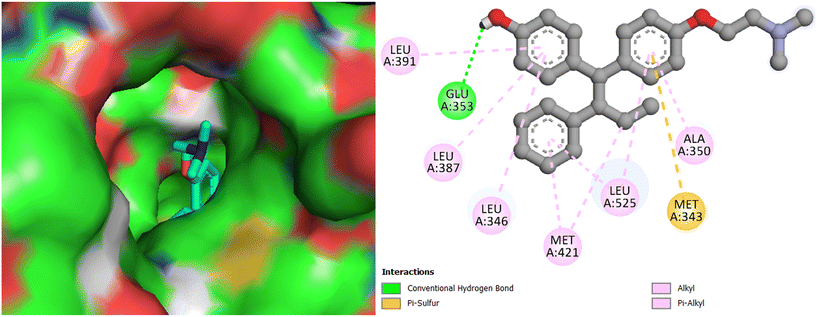 |
| | Fig. 10 Docking pose and binding interactions of 4-hydroxytamoxifen in the cavity of Erα. | |
Prediction of pharmacokinetics properties
The pharmacokinetic properties of the compounds were predicted using SwissADME web server71 and are presented in Table 3. The octanol/water partition coefficient (logPw/o) values were estimated in the range of 2.90–5.01 and as per the essential condition of Lipinski's rule of five.72 The molecular weights of all the compounds were in the range of 411.56–521.67 g mol−1. As the molecular weights are ≤500 g mol−1, they can be easily transported, absorbed, and diffused after administering into the body,73 though some of the compounds molecular weight is more than 500 g mol−1; still, they have showed activity against the target cell line. The predicted rotatable bonds were in the range of 8–9, H-bond acceptors were ≤10, and the H-bond donors were ≤5. The topological polar surface (TSPA) of the analogues were in the range of 88.63–134.45; these lower TSPA values indicate the acceptable range of results. The bioavailability scores of all the compounds are within the prescribed range; they can be easily absorbed and metabolized in the body. The synthetic accessibility score of all the analogues was found to be <10, which confirms that they can be synthesized easily.74 The pharmacokinetic evaluation reveal that all molecules have favorable drug-likeness properties and could be considered as therapeutic agents.
Table 3 Predicted pharmacokinetic properties of compounds 7a–l
| Compound |
Molecular weight (range ≤ 500) |
Rotatable bonds (range 1–10) |
H-bond acceptors (range ≤ 10) |
H-bond donors (range ≤ 5) |
TPSA |
LogPo/w (range ≤ 5) |
QPlogS (solubility) |
Bioavailability score (range 0.4–0.6) |
Synthetic accessibility |
Lipinski violations |
|
7a
|
411.56 |
9 |
4 |
1 |
88.63 |
4.69 |
−6.91 |
0.55 |
4.22 |
0 |
|
7b
|
445.57 |
8 |
4 |
1 |
88.63 |
4.26 |
−7.44 |
0.55 |
4.33 |
1 |
|
7c
|
459.60 |
8 |
4 |
1 |
88.63 |
4.57 |
−7.74 |
0.55 |
4.51 |
1 |
|
7d
|
459.60 |
8 |
4 |
1 |
88.63 |
4.35 |
−7.74 |
0.55 |
4.48 |
1 |
|
7e
|
459.60 |
8 |
4 |
1 |
88.63 |
4.68 |
−7.74 |
0.55 |
4.45 |
1 |
|
7f
|
521.67 |
9 |
4 |
1 |
88.63 |
5.01 |
−8.91 |
0.17 |
4.74 |
2 |
|
7g
|
513.57 |
9 |
7 |
1 |
88.63 |
2.90 |
−8.29 |
0.17 |
4.48 |
2 |
|
7h
|
513.57 |
9 |
7 |
1 |
88.63 |
4.64 |
−8.29 |
0.17 |
4.48 |
2 |
|
7i
|
513.57 |
9 |
7 |
1 |
88.63 |
4.69 |
−8.29 |
0.17 |
4.41 |
2 |
|
7j
|
480.02 |
8 |
4 |
1 |
88.63 |
4.43 |
−8.03 |
0.55 |
4.32 |
1 |
|
7k
|
490.57 |
9 |
6 |
1 |
134.45 |
3.76 |
−7.5 |
0.55 |
4.44 |
0 |
|
7l
|
481.55 |
8 |
6 |
1 |
88.63 |
4.42 |
−7.76 |
0.55 |
4.36 |
1 |
Conclusion
A library of 1-(2-(substitutedthio)-4-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutyl phenyl)propan-1-one analogues 7a–l was synthesized by a series of nitration, reduction, and condensation cyclization reactions. The structure of novel compounds was characterized by spectral analytical data of 1H NMR, 13C NMR, IR, and Mass. All the novel analogues were screened for their in vitro anticancer activity against human breast cancers MCF-7 and MDA-MB-231 cell lines using doxorubicin as the standard reference. Compounds 7h and 7j displayed outstanding activity against the MCF-7 cell line with an IC50 value of 8.92 ± 0.91 μM and 9.14 ± 8.22 μM, respectively, compared to the doxorubicin IC50 value of 9.29 ± 1.02 μM. Compound 7g showed promising activity against the MCF-7 cell line with an IC50 value of 12.19 ± 1.33 μM. Compound 7h also implied outstanding activity against the MDA-MB-231 cell line with an IC50 value of 7.54 ± 0.95 μM compared to the doxorubicin IC50 value of 7.68 ± 5.36 μM. Further, compounds 7g and 7i showed promising activity against the MDA-MB-231 cell line with an IC50 value of 12.19 ± 1.17 μM and 8.78 ± 0.58 μM, respectively, whereas other molecules indicated good to moderate activity against both the cell lines. The morphological screening of compounds 7h, 7i, 7j, and 7g showed identical morphological changes to those seen for the effects of doxorubicin. The molecular docking study against ERα unveiled their best docking scores and binding interactions in agreement to the experimental results. The pharmacokinetic prediction revealed that all molecules have favorable drug-likeness properties. Hence, the novel ibuprofen-appended benzoxazoles could be the lead molecules in drug discovery and can be investigated for the development of chemotherapeutics.
Experimental section
Materials and methods
All synthetic reagents, catalysts, and solvents were bought from commercial vendors Merck, Spectrochem, TCI, SDFine, and Avra chemicals and utilized without any additional purification. The advancement of the response was checked by TLC technique utilizing permeated silica gel Merck 60F254 plates. The melting point of the final compounds were recorded utilizing a Stuart SMP3 liquefying point mechanical assembly and uncorrected. For purifying mixtures, recrystallization technique and section chromatography were utilized with 60–120 mesh silica gel. The design of mixtures laid out utilizing the IR spectra of compounds were recorded on a Shimadzu FTIR 8400 S spectrometer utilizing KBr beads, 1H and 13C NMR run on a Varian 400 spectrometer (400 and 100 MHz, individually) by involving CDCl3 and DMSO as a dissolvable, and TMS as the internal standard. The mass spectra recorded on a Shimadzu GC–MS QP 1000 spectrometer. Elemental analysis was recorded on an Elementar apparatus.
Synthesis of 1-(2,4-dihydroxyphenyl)-2-(4-isobutylphenyl)propan-1-one (3).
To the fused ZnCl2 (0.001 mmol), ibuprofen (0.001 mmol) was added by constant stirring with a glass rod, and then stirring was continued for 20 minutes at 130 °C. To this, resorcinol (0.001 mmol) was added slowly, and the reaction mixture was stirred at 140 °C for another 20 minutes. After completion, the reaction mixture was poured on crushed ice to obtain 1-(2,4-dihydroxyphenyl)-2-(4-isobutylphenyl)propan-1-one (3); the solid was treated with dil. HCl and allowed to soak overnight. The free flow solid was filtered and dried under vacuum. M.P. = 130 °C.
Synthesis of 1-(2,4-dihydroxy-5-nitrophenyl)-2-(4-isobutylphenyl)propan-1-one (4).
2 g 1-(2,4-dihydroxyphenyl)-2-(4-isobutylphenyl)propan-1-one (3) was dissolved in 20 mL glacial acetic acid; after the solution was clear, 2 mL mixture of glacial acetic acid and conc. HNO3 (1:1) was added dropwise by maintaining the temperature below 30 °C. The reaction mixture was stirred for 7 hours to obtain 1-(2,4-dihydroxy-5-nitrophenyl)-2-(4-isobutylphenyl)propan-1-one (4). Monitoring the reaction by TLC, the mixture was poured in ice cold water, and the solid separated was washed with water 3 times and dried under vacuum. Yellow solid. Yield: 65%. M.P.: 135–138 °C. IR (KBr, cm−1): 3223, 2918, 1634, 1572, 1366. 1H NMR (400 MHz, CDCl3), δ, ppm: 13.05 (s, 1H), 10.97 (s, 1H), 8.76 (s, 1H), 7.21 (d, J = 8.0 Hz, 2H), 7.11 (d, J = 7.6 Hz, 2H), 6.54 (s, 1H), 4.64 (q, 1H), 2.41 (d, J = 7.2 Hz, 2H), 1.86–1.75 (m, 1H), 1.54 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.52, 163.88, 162.30, 156.50, 140.94, 138.37, 129.99, 127.31, 124.06, 119.95, 115.95, 99.40, 45.08, 30.24, 22.49, 19.00. ESI-MS m/z [M + H]+: 344.35.
Synthesis of 1-(6-hydroxy-2-mercaptobenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (5).
1 g 1-(2,4-dihydroxy-5-nitrophenyl)-2-(4-isobutylphenyl)propan-1-one (4) was refluxed in methanol (25 mL) till the solution was clear. Now, sodium dithionite (2.0 eq.) was added and dissolved in water dropwise. The reaction mixture was refluxed for 2 hours, monitored by TLC, another lot of solid sodium dithionite was added, and the reaction was continued. After completion, 0.89 g potassium hydroxide (1.0 eq.) was added and dissolved in water, followed by the dropwise addition of carbon disulphide (3.0 eq); then, the reaction mixture was refluxed at 65 °C for 6 hours to get 1-(6-hydroxy-2-mercaptobenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (5). After the completion of the reaction, the pH of the solution was adjusted, extracted into ethyl acetate, and dried under vacuum. Brown solid. Yield: 75%. M.P.: 137–139 °C. IR (KBr, cm−1): 3439, 2607, 1635. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.90 (s, 1H), 10.33 (s, 1H), 7.50 (s, 1H), 7.16–7.08 (m, 4H), 6.92 (s, 1H), 4.57 (q, 1H), 2.41 (d, J = 7.2 Hz, 2H), 1.84–1.78 (m, 1H), 1.53 (d, J = 6.8 Hz, 3H), 0.86 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 205.50, 181.00, 162.64, 153.27, 141.11, 138.06, 130.13, 127.02, 122.73, 115.40, 110.03, 100.51, 47.85, 44.96, 30.09, 22.37, 19.37. ESI-MS: m/z [M + H]+: 356.13.
General procedure for the synthesis of 1-(2-(thiosubstituted)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutyl phenyl)propan-1-one derivatives (7a–l).
To 0.05 g 1-(6-hydroxy-2-mercaptobenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (5) dissolved in DMF, substituted benzyl bromides (6a–l) (1.0 eq.) was added individually in the presence of diisopropylethylamine, the reaction mixture was stirred for 1 hour at room temperature, and 1-(2-(thiosubstituted)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl) propan-1-one derivatives (7a–l) were obtained in good yields.
Spectra
1-(2-(Butylthio)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (7a).
White solid. Yield 79%. M.P.: 142–145 °C. IR (KBr, cm−1): 3439, 1637. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.86 (s, 1H), 8.02 (s, 1H), 7.21 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.0 Hz, 2H), 6.95 (s, 1H), 4.74 (q, 1H), 3.28–3.24 (m, 2H), 2.40 (d, J = 7.2 Hz, 2H), 1.82–1.76 (m, 3H), 1.54 (d, J = 6.8 Hz, 3H), 1.52–1.46 (m, 2H), 0.96 (t, 3H), 0.86 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.56, 165.90, 162.11, 156.42, 140.84, 138.33, 134.80, 129.95, 127.34, 119.53, 115.89, 99.21, 47.27, 45.11, 32.20, 31.25, 30.23, 22.50, 21.91, 19.58, 13.66. ESI-MS m/z [M + H]+: 412.37. Elemental analysis calculated for C24H29NO3S: C, 70.04; H, 7.10; N, 3.40%. Found: C, 69.97; H, 7.04; N, 3.35%.
1-(2-(Benzylthio)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (7b).
White solid. Yield: 76%. M.P.: 145–147 °C. IR (KBr, cm−1): 3437, 1637, 1512. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.87 (s, 1H), 8.05 (s, 1H), 7.42 (d, J = 6.8 Hz, 2H), 7.35–7.28 (m, 3H), 7.22 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 8.0 Hz, 2H), 6.96 (s, 1H), 4.74 (q, 1H), 4.53–4.46 (m, 2H), 3.28–3.24 (m, 1H), 2.41 (d, J = 7.2 Hz, 2H), 1.84–1.78 (m, 3H), 1.55 (d, J = 6.8 Hz, 3H), 0.86 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.55, 165.12, 162.20, 156.46, 140.88, 138.32, 135.51, 134.68, 129.98, 129.15, 128.95, 128.18, 127.34, 119.76, 116.00, 99.30, 47.33, 45.12, 36.79, 30.24, 22.52, 19.58. ESI-MS m/z [M + H]+: 446.17. Elemental analysis calculated for C27H27NO3S: C, 72.78; H, 6.11; N, 3.14%. Found: C, 72.74; H, 6.07; N, 3.09%.
1-(6-Hydroxy-2-((2-methylbenzyl)thio)benzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (7c).
White solid. Yield: 78%. M.P.: 146–148 °C. IR (KBr, cm−1): 3437, 1637, 1512. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.87 (s, 1H), 8.06 (s, 1H), 7.38 (d, J = 7.2 Hz, 1H), 7.24 (d, J = 6.0 Hz, 2H), 7.20–7.13 (m, 3H), 7.08 (d, J = 8.0 Hz, 2H), 6.96 (s, 1H), 4.74 (q, 1H), 4.56–4.49 (m, 2H), 2.42 (s, 3H), 2.41 (d, J = 7.2 Hz, 2H), 1.84–1.76 (m, 1H), 1.55 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.54, 165.24, 162.20, 156.41, 140.86, 138.33, 137.25, 134.69, 132.90, 130.87, 130.29, 129.98, 128.63, 127.33, 126.54, 119.71, 115.99, 99.30, 47.33, 45.11, 35.13, 30.23, 22.51, 19.57, 19.31. ESI-MS m/z [M + H]+: 460.44. Elemental analysis calculated for C28H29NO3S: C, 73.17; H, 6.36; N, 3.05%. Found: C, 73.12; H, 6.31; N, 3.00%.
1-(6-Hydroxy-2-((3-methylbenzyl)thio)benzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (7d).
White solid. Yield: 80%. M.P.: 146–148 °C. IR (KBr, cm−1): 3437, 1637, 1512. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.87 (s, 1H), 8.05 (s, 1H), 7.25–7.22 (m, 4H), 7.08 (d, J = 8.0 Hz, 4H), 6.96 (s, 1H), 4.73 (q, 1H), 4.47 (m, 2H), 2.40 (d, J = 7.2 Hz, 2H), 2.33 (s, 3H), 1.86–1.76 (m, 1H), 1.55 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.53, 165.22, 162.19, 156.45, 140.87, 138.71, 138.32, 135.27, 134.70, 129.98, 129.86, 128.97, 128.84, 127.33, 126.20, 119.73, 115.98, 99.29, 47.33, 45.11, 43.45, 36.80, 30.24, 22.52, 19.58. ESI-MS m/z [M + H]+: 460.38. Elemental analysis calculated for C28H29NO3S: C, 73.17; H, 6.36; N, 3.05%. Found: C, 73.13; H, 6.30; N, 3.01%.
1-(6-Hydroxy-2-((4-methylbenzyl)thio)benzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (7e).
White solid. Yield: 80%. M.P.: 147–149 °C. IR (KBr, cm−1): 3435, 1635, 1511. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.87 (s, 1H), 8.04 (s, 1H), 7.30 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 7.6 Hz, 1H), 7.13 (d, J = 7.6 Hz, 3H), 7.08 (d, J = 8.4 Hz, 2H), 6.95 (s, 1H), 4.72 (q, 1H), 4.47 (m, 2H), 2.40 (d, J = 7.2 Hz, 2H), 2.32 (s, 3H), 1.86–1.76 (m, 1H), 1.55 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.53, 165.24, 162.18, 156.44, 140.86, 138.33, 134.71, 132.37, 129.97, 129.62, 129.40, 129.06, 127.33, 119.72, 115.97, 99.27, 47.31, 45.11, 36.61, 30.23, 22.52, 21.27, 19.57. ESI-MS m/z [M + H]+: 460.38. Elemental analysis calculated for C28H29NO3S: C, 73.17; H, 6.36; N, 3.05%. Found: C, 73.10; H, 6.32; N, 3.02%.
1-(2-(([1,1′-Biphenyl]-4-ylmethyl)thio)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (7f).
White solid. Yield: 79%. M.P.: 151–153 °C. IR (KBr, cm−1): 3438, 1636, 1517. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.87 (s, 1H), 8.06 (s, 1H), 7.64 (s, 1H), 7.57–7.50 (m, 3H), 7.45–7.35 (m, 5H), 7.21 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 8.0 Hz, 2H), 6.96 (s, 1H), 4.75–4.70 (q, 1H), 4.56 (m, 2H), 2.40 (d, J = 7.2 Hz, 2H), 1.84–1.77 (m, 1H), 1.55 (d, J = 6.8 Hz, 3H), 0.86 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.53, 165.03, 162.21, 156.47, 141.97, 140.87, 140.65, 138.29, 136.00, 134.68, 129.39, 128.94, 127.98, 127.68, 127.26, 127.01, 119.76, 116.00, 99.32, 47.30, 45.11, 36.85, 30.23, 22.52, 19.57. ESI-MS m/z [M + H]+: 523.48 [M + H]+. Elemental analysis calculated for C33H31NO3S: C, 75.98; H, 5.99; N, 2.69%. Found: C, 75.94; H, 5.95; N, 2.63%.
1-(6-Hydroxy-2-((2-(trifluoromethyl)benzyl)thio)benzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (7g).
White solid. Yield: 82%. M.P.: 150–152 °C. IR (KBr, cm−1): 3432, 1631, 1513. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.87 (s, 1H), 8.07 (s, 1H), 7.70 (dd, J = 8.0, J = 12.4 Hz, 2H), 7.52–7.46 (m, 2H), 7.42 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 5.6 Hz, 1H), 7.09 (d, J = 8.0 Hz, 2H), 6.96 (s, 1H), 4.75 (q, 1H), 4.70–4.66 (m, 2H), 2.41 (d, J = 7.2 Hz, 2H), 1.87–1.77 (m, 1H), 1.56 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.57, 164.94, 162.28, 156.58, 140.92, 138.33, 134.58, 132.50, 131.91, 131.48, 130.01, 128.38, 127.78, 127.36, 126.54, 126.48, 119.84, 116.07, 99.39, 47.34, 45.13, 33.18, 30.25, 22.52, 19.59. ESI-MS m/z [M + H]+: 514.47. Elemental analysis calculated for C28H26F3NO3S: C, 65.48; H, 5.10; N, 2.73%. Found: C, 65.44; H, 5.05; N, 2.68%.
1-(6-Hydroxy-2-((3-(trifluoromethyl)benzyl)thio)benzo[d]oxazol-5-yl)-2-(4-isobutyl phenyl)propan-1-one (7h).
White solid. Yield: 81%. M.P.: 150–152 °C. IR (KBr, cm−1): 3433, 1635, 1514. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.86 (s, 1H), 8.05 (s, 1H), 7.64–7.43 (m, 4H), 7.22 (d, J = 8.0 Hz, 2H), 7.10–7.07 (m, 2H), 6.96 (s, 1H), 4.73 (q, 1H), 4.55–4.48 (m, 2H), 2.41 (d, J = 7.2 Hz, 2H), 1.86–1.76 (m, 1H), 1.55 (d, J = 7.2 Hz, 3H), 0.87 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.57, 164.94, 162.28, 156.58, 140.92, 138.33, 134.58, 132.50, 132.29, 131.91, 131.48, 130.01, 128.38, 127.78, 127.36, 126.48, 119.84, 116.07, 99.39, 47.34, 45.13, 33.18, 30.25, 22.52, 19.59. ESI-MS m/z [M + H]+: 514.29. Elemental analysis calculated for C28H26F3NO3S: C, 65.48; H, 5.10; N, 2.73%. Found: C, 65.43; H, 5.05; N, 2.67%.
1-(6-Hydroxy-2-((4-(trifluoromethyl)benzyl)thio)benzo[d]oxazol-5-yl)-2-(4-isobutyl phenyl)propan-1-one (7i).
White solid. Yield: 81%. M.P.: 151–153 °C. IR (KBr, cm−1): 3434, 1635, 1517. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.86 (s, 1H), 8.04 (s, 1H), 7.59–7.54 (m, 4H), 7.22 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 8.0 Hz, 2H), 6.95 (s, 1H), 4.73 (q, 1H), 4.55–4.47 (m, 2H), 2.41 (d, J = 7.2 Hz, 2H), 1.86–1.76 (m, 1H), 1.55 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.53, 164.37, 162.29, 156.50, 140.93, 140.04, 138.31, 134.52, 130.00, 129.48, 127.33, 125.89, 125.85, 122.70, 119.89, 116.09, 99.38, 47.36, 45.11, 36.00, 30.24, 22.50, 19.57. ESI-MS m/z [M + H]+: 514.29. Elemental analysis calculated for C28H26F3NO3S: C, 65.48; H, 5.10; N, 2.73%. Found: C, 65.45; H, 5.04; N, 2.68%.
1-(2-((4-Chlorobenzyl)thio)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl) propan-1-one (7j).
White solid. Yield: 79%. M.P.: 145–147 °C. IR (KBr, cm−1): 3437, 1632, 1518. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.86 (s, 1H), 8.04 (s, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 8.0 Hz, 2H), 6.95 (s, 1H), 4.73 (q, 1H), 4.48–4.40 (m, 2H), 2.41 (d, J = 7.2 Hz, 2H), 1.84–1.78 (m, 1H), 1.55 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.54, 164.65, 162.24, 156.46, 140.90, 138.31, 134.57, 134.29, 134.07, 130.48, 129.99, 129.09, 127.33, 119.83, 116.04, 99.34, 47.34, 45.11, 35.99, 30.24, 22.52, 19.57. ESI-MS m/z [M + H]+: 480.41. Elemental analysis calculated for C27H26ClNO3S: C, 67.56; H, 5.46; N, 2.92%. Found: C, 67.51; H, 5.40; N, 2.88%.
1-(6-Hydroxy-2-((4-nitrobenzyl)thio)benzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (7k).
Yellow solid. Yield: 77%. M.P.: 145–147 °C. IR (KBr, cm−1): 3439, 1636, 1573, 1517, 1365. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.86 (s, 1H), 8.18 (d, J = 8.8 Hz, 2H), 8.04 (s, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 8.0 Hz, 2H), 6.95 (s, 1H), 4.73 (q, 1H), 4.58–4.50 (m, 2H), 2.41 (d, J = 7.2 Hz, 2H), 1.86–1.76 (m, 1H), 1.55 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.52, 163.88, 162.30, 156.50, 147.63, 145.24, 143.66, 140.94, 138.26, 134.38, 130.02, 127.31, 124.06, 119.95, 116.11, 99.40, 47.33, 45.08, 35.64, 30.24, 22.49, 19.56. ESI-MS m/z [M + H]+: 491.34. Elemental analysis calculated for C27H26N2O5S: C, 66.11; H, 5.34; N, 5.71%. Found: C, 66.05; H, 5.29; N, 5.68%.
1-(2-((2,4-Difluorobenzyl)thio)-6-hydroxybenzo[d]oxazol-5-yl)-2-(4-isobutylphenyl)propan-1-one (7l).
Yellow solid. Yield: 79%. M.P.: 145–148 °C. IR (KBr, cm−1): 3435, 1637, 1572. 1H NMR (400 MHz, CDCl3), δ, ppm: 12.86 (s, 1H), 8.06 (s, 1H), 7.28–7.23 (m, 3H), 7.09 (d, J = 8.0 Hz, 2H), 7.04–6.96 (m, 3H), 4.75 (q, 1H), 4.52–4.40 (m, 2H), 2.41 (d, J = 7.2 Hz, 2H), 1.85–1.78 (m, 1H), 1.56 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 206.58, 164.41, 162.28, 156.57, 140.93, 138.30, 134.56, 130.01, 127.36, 119.93, 117.79, 117.51, 116.94, 116.70, 116.61, 116.37, 116.28, 116.09, 99.38, 47.34, 45.13, 30.25, 29.63, 22.53, 19.56. ESI-MS m/z [M + H]+: 482.43. Elemental analysis calculated for C27H25N2F2O3S: C, 67.34; H, 5.23; N, 2.91%. Found: C, 67.29; H, 5.18; N, 2.88%.
Cytotoxicity
Cell culture.
Human breast adenocarcinoma cell lines MCF-7 (ER Positive) and MDA-MB-231 cells (Triple negative) were procured from the National Centre for Cell Sciences (NCCS), Pune India, and were sub-cultured in-house at PGP Life Sciences, Hyderabad. The original source of the cell line is an American-type culture collection (ATCC). Initially, MCF-7 and MDA-MB 231 cells were grown in 25 cm2 bottle canted neck flasks (Corning) with Dulbecco modified essential medium F12 (DMEM-F12) medium supplemented with 20% fetal bovine serum (FBS), 3 mM L-glutamine, along with the addition of antibiotic solution consisting of penicillin 500 U mL−1, streptomycin 500 μg mL−1, and amphotericin-B 1.25 μg mL−1 to protect the cell lines from bacterial infections, and the cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C. The cells were passaged enzymatically with 0.25% trypsin-1 mM EDTA and subcultured on 75 cm2 plastic flasks at a density of 2.3 × 104 cells per cm2. The culture medium was replaced every 2 days. Cell confluence (80%) was confirmed by microscopic observance. Experiments were performed 24 hours post-seeding to prevent cell differentiation.
MTT assay protocol
MCF-7 and MDA-MB 231 cells (100 μL per well) were plated and cultured in clear bottom 96-well tissue culture plates (the number of cells were 105 cells per well). The test samples (7a–l) were added with concentrations ranging from 2 to 128 μM (2, 4, 8, 16, 32, 64, and 128 μM) in triplicate after 24 hours of seeding, and the cells were incubated for 24 hours of treatment time. A 20 μL culture medium was used for all the test samples. The medium was removed and cells were washed with PBS twice. 15 μL MTT reagent was added per well, which was made up in PBS medium to a final concentration of 0.5 mg mL−1. The volume of the reagent should be adjusted depending on the volume of the cell culture. Cells were incubated for 3 hours at 37 °C until intracellular purple formazan crystals were visible under a microscope. MTT reagent was removed and 100 μL DMSO was added to each well and mixed gently on an orbital shaker for one hour at room temperature. The volume of DMSO should be adjusted depending on the volume of the cell culture. The absorbance at OD570nm for each well was measured on an absorbance plate reader. Similar procedures were followed in duplicate with regard to the human embryonic kidney cells (HEK-293 cells).
Morphological screening
Human adenocarcinoma cells lines MCF-7 (ER Positive) MDA-MB 231 (Triple negative) were grown in 25 cm2 bottle canted neck flasks (Corning) with Dulbecco modified essential medium F12 (DMEM-F12) supplemented with 20% fetal bovine serum (FBS), 3 mM L-glutamine, along with the addition of antibiotic solution consisting of penicillin 500 U mL−1, streptomycin 500 μg mL−1, and amphotericin-B 1.25 μg mL−1 to protect the cell lines from bacterial infections, and the cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C. The cells were treated enzymatically with 0.25% trypsin-1 mM EDTA and subcultured on 75 cm2 plastic flasks at a density of 2.3 × 104 cells per cm2. The culture medium was replaced every 2 days. Cell confluence (80%) was confirmed by microscopic observance. Experiments were performed 24 hours post-seeding to prevent cell differentiation. Both MCF-7 and MDA-MB 231 cells were treated 24 hours post-seeding with the selected compounds. MCF-7 cells were treated with compounds 7g, 7h, and 7j at their IC50, and MDA-MB 231 cells with compounds 7g, 7h, and 7i at their IC50, were treated along with doxorubicin at their corresponding IC50. Cells were observed for 24 and 48 hours after the treatment of the test samples. Images were taken by an Axiovert 200M phase contrast microscope at 10× magnification. Axiovision Rel.4.2 software was used to acquire the images.
Molecular docking method
Autodock Vina integrated PyRx tool was employed for docking simulations.75–77 The crystal structure of human estrogen receptor alpha ligand-binding domain in the complex with 4-hydroxytamoxifen (PDB ID: 3ERT) was retrieved from Protein Data Bank (https://www.rcsb.org). Initially, water molecules and heteroatoms of the protein were removed, and polar hydrogens were added. The ligands were sketched using ChemDraw Professional 16.0 in MDL file format. The energies of all the ligands were minimized after loading into PyRx and converted to the PDBQT file format.78 The 3D grid box was configured with dimensions of center_x = 23.41, center_y = 35.88, center_z = −6.82, size_x = 18.00, size_y = 22.99, and size_z = 17.64, and docking simulations were performed after assigning the exhaustiveness value of 8. The docking result were visualized using Pymol and Biovia discovery studio visualizer.79
Conflicts of interest
All authors declare that they have no conflict of interest.
Acknowledgements
All authors thank Head, Department of Chemistry, University College of Science, Osmania University for providing lab facilities. We thank AccuAnalyticals, Hyderabad for providing spectral analysis data and PGP Life Sciences, Hyderabad for testing biological activities.
References
- M. Al-Ghorbani, A. Bushra Begum, Z. Zabiulla, S. V. Mamatha and S. A. Khanum, Res. J. Pharm. Technol., 2015, 8, 611–628 CrossRef.
- T. K. Sawyer, Cancer, 2007, 383–405 Search PubMed.
- D. Veeranna, L. Ramdas, G. Ravi, S. Bujji, V. Thumma and J. Ramchander, ChemistrySelect, 2022, 7, e202201758 CrossRef CAS.
- R. R. Ruddarraju, A. C. Murugulla, R. Kotla, M. C. B. Tirumalasetty, R. Wudayagiri, S. Donthabakthuni and R. Maroju, MedChemComm, 2017, 8, 176–183 RSC.
- C. Sun, C. Chen, S. Xu, J. Wang, Y. Zhu, D. Kong, H. Tao, M. Jin, P. Zheng and W. Zhu, Bioorg. Med. Chem., 2016, 24, 3862–3869 CrossRef CAS PubMed.
- S. H. Hassanpour and M. Dehghani, J. Cancer Res. Pract., 2017, 4, 127–129 CrossRef.
-
W. H. Organization, Cancer, https://www.who.int/cancer/en/, (accessed 9 October 2018) Search PubMed.
- C. Y. Lee, E. H. Chew and M. L. Go, Eur. J. Med. Chem., 2010, 45, 2957–2971 CrossRef CAS PubMed.
- D. Vásquez, J. A. Rodríguez, C. Theoduloz, P. B. Calderon and J. A. Valderrama, Eur. J. Med. Chem., 2010, 45, 5234–5242 CrossRef PubMed.
- F. Arcamone, G. Cassinelli, G. Fantini, A. Grein, P. Orezzi, C. Pol and C. Spalla, Biotechnol. Bioeng., 1969, 11, 1101–1110 CrossRef CAS PubMed.
- H. Cortés-Funes and C. Coronado, Cardiovasc. Toxicol., 2007, 7, 56–60 CrossRef PubMed.
- D. X. Robledo-Cadena, J. C. Gallardo-Pérez, V. Dávila-Borja, S. C. Pacheco-Velázquez, J. A. Belmont-Díaz, S. J. Ralph, B. A. Blanco-Carpintero, R. Moreno-Sánchez and S. Rodríguez-Enríquez, Pharmaceuticals, 2020, 13, 1–25 CrossRef PubMed.
- D. A. Rushing, S. R. Raber, K. A. Rodvold, S. C. Piscitelli, G. S. Plank and D. A. Tewksbury, Cancer, 1994, 74, 834–841 CrossRef CAS PubMed.
- S. Y. van der Zanden, X. Qiao and J. Neefjes, FEBS J., 2021, 288, 6095–6111 CrossRef CAS PubMed.
- A. Shukla, J. M. Hillegass, M. B. MacPherson, S. L. Beuschel, P. M. Vacek, H. I. Pass, M. Carbone, J. R. Testa and B. T. Mossman, Mol. Cancer, 2010, 9, 1–13 Search PubMed.
- B. Sergei, D. Pavel, G. Aigul, B. Firyuza, N. Ilmira, M. Ilshat, A. Aida, K. Refat, A. Natalia, S. Elena and G. Vera, Int. J. Mol. Sci., 2020, 21, 352 CrossRef CAS PubMed.
- R. D. Olson and P. S. Mushlin, FASEB J., 1990, 4, 3076–3086 CrossRef CAS PubMed.
- N. Bansal, M. J. Adams, S. Ganatra, S. D. Colan, S. Aggarwal, R. Steiner, S. Amdani, E. R. Lipshultz and S. E. Lipshultz, Cardio-Oncology, 2019, 5, 18 CrossRef PubMed.
- D. F. Hayes and M. H. Picard, J. Clin. Oncol., 2006, 24, 4056–4058 CrossRef CAS PubMed.
- C. Frankel and F. M. Palmieri, Clin. J. Oncol. Nurs., 2010, 14, 223–233 CrossRef PubMed.
- R. N. Hamid, C. S. Ahn and W. W. Huang, J. Dermatol. Treat., 2019, 30, 487–488 CrossRef CAS PubMed.
- V. F. Borges, C. Ferrario, N. Aucoin, C. Falkson, Q. Khan, I. Krop, S. Welch, A. Conlin, J. Chaves, P. L. Bedard, M. Chamberlain, T. Gray, A. Vo and E. Hamilton, JAMA Oncol., 2018, 4, 1214 CrossRef PubMed.
- M. Nagamani, T. Vishnu, P. Jalapathi and M. Srinivas, J. Iran. Chem. Soc., 2022, 19, 1049–1060 CrossRef CAS.
- S. Aitha, V. Thumma, R. Matta, S. Ambala, K. Jyothi, S. Manda and J. Pochampally, Results Chem., 2023, 5, 100987 CrossRef CAS.
- V. Mallikanti, V. Thumma, R. Matta, K. R. Valluru, L. N. S. Konidena, L. S. Boddu and J. Pochampally, Chem. Data Collect., 2023, 45, 101034 CrossRef CAS.
- T. Ghoshal and T. M. Patel, Future J. Pharm. Sci., 2020, 6, 94 CrossRef.
- D. Osmaniye, A. Hıdır, B. N. Sağlık, S. Levent, Y. Özkay and Z. A. Kaplancıklı, Chem. Biodiversity, 2022, 19, 112979 CrossRef PubMed.
- R. V. Satyendra, K. A. Vishnumurthy, H. M. Vagdevi, K. P. Rajesh, H. Manjunatha and A. Shruthi, Eur. J. Med. Chem., 2011, 46, 3078–3084 CrossRef CAS PubMed.
- S. Garrepalli, M. Sarangapani, P. Garrepally and S. Chilukala, Pharm. Lett., 2011, 3, 427–432 CAS.
- K. Zomorodian, S. Khabnadideh, A. Sakhteman, B. B. F. Mirjalili, M. Ranjbar and L. Zamani, Farmacia, 2020, 68, 155–163 CrossRef CAS.
- V. Šlachtová and L. Brulíková, ChemistrySelect, 2018, 3, 4653–4662 CrossRef.
- Z. Yang, D. J. Fairfax, J. H. Maeng, L. Masih, A. Usyatinsky, C. Hassler, S. Isaacson, K. Fitzpatrick, R. J. Deorazio, J. Chen, J. P. Harding, M. Isherwood, S. Dobritsa, K. L. Christensen, J. D. Wierschke, B. I. Bliss, L. H. Peterson, C. M. Beer, C. Cioffi, M. Lynch, W. M. Rennells, J. J. Richards, T. Rust, Y. L. Khmelnitsky, M. L. Cohen and D. D. Manning, Bioorg. Med. Chem. Lett., 2010, 20, 6538–6541 CrossRef CAS PubMed.
- G. Angajala and R. Subashini, Struct. Chem., 2020, 31, 263–273 CrossRef CAS.
- O. Afzal, A. S. A. Altamimi, M. M. Shahroz, H. K. Sharma, Y. Riadi and M. Q. Hassan, Molecules, 2021, 26, 2389 CrossRef CAS PubMed.
- S. Singh, G. Veeraswamy, D. Bhattarai, J. Il Goo, K. Lee and Y. Choi, Asian J. Org. Chem., 2015, 4, 1338–1361 CrossRef CAS.
- H. Song, S. R. Oh, H. K. Lee, G. Han, J. H. Kim, H. W. Chang, K. E. Doh, H. K. Rhee and H. Y. P. Choo, Bioorg. Med. Chem., 2010, 18, 7580–7585 CrossRef CAS PubMed.
- S. Arulmurugan, H. P. Kavitha and J. P. Vennila, Mini-Rev. Org. Chem., 2020, 18, 769–785 CrossRef.
- W. Zhang, J. Liu, J. M. Macho, X. Jiang, D. Xie, F. Jiang, W. Liu and L. Fu, Eur. J. Med. Chem., 2017, 126, 7–14 CrossRef CAS PubMed.
- Y. A. Al-Soud, H. Al-Sa'doni, H. A. S. Amajaour and N. A. Al-Masoudi, Z. fur Naturforsch. – B J. Chem. Sci., 2007, 62, 523–528 CrossRef CAS.
- M. X. Song, Y. Huang, S. Wang, Z. T. Wang and X. Q. Deng, Arch. Pharm., 2019, 352, 1800313 CrossRef PubMed.
- V. S. K. Cheerala, P. Ghanta and S. C. Neelakantan, RSC Adv., 2021, 11, 39328–39342 RSC.
- R. K. Jadhav, A. B. Nikumbh and B. K. Karale, Orient. J. Chem., 2015, 31, 967–972 CrossRef CAS.
- D. Sampson, X. Y. Zhu, S. V. K. Eyunni, J. R. Etukala, E. Ofori, B. Bricker, N. S. Lamango, V. Setola, B. L. Roth and S. Y. Ablordeppey, Bioorg. Med. Chem., 2014, 22, 3105–3114 CrossRef CAS PubMed.
- K. Arakawa, M. Inamasu, M. Matsumoto, K. Okumura, K. Yasuda, H. Akatsuka, S. Kawanami, A. Watanabe, K. Homma, Y. Saiga, M. Ozeki and I. Iijima, Chem. Pharm. Bull., 1997, 45, 1984–1993 CrossRef CAS PubMed.
- D. P. Sangi, Y. G. Meira, N. M. Moreira, T. A. Lopes, M. P. Leite, M. E. Pereira-Flores and E. S. Alvarenga, Pest Manage. Sci., 2019, 75, 262–269 CrossRef CAS PubMed.
- S. M. Johnson, S. Connelly, I. A. Wilson and J. W. Kelly, J. Med. Chem., 2008, 51, 260–270 CrossRef CAS PubMed.
- V. Abbhi and P. Piplani, Curr. Med. Chem., 2018, 27, 2222–2256 CrossRef PubMed.
- Y. J. Oh, D. Kim, S. Oh, E. J. Jang, H. Y. Won, H. Jeong, M. G. Jeong, H. Y. P. Choo and E. S. Hwang, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- A. Reiser, L. J. Leyshon, D. Saunders, M. V. Mijovic, A. Bright and J. Bogie, J. Am. Chem. Soc., 1972, 94, 2414–2421 CrossRef CAS.
- S. Il Um, Dyes Pigm., 2007, 75, 185–188 CrossRef.
- H. Ding, R. Wang, X. Wang and W. Ji, J. Sep. Sci., 2018, 41, 3294–3301 CrossRef CAS PubMed.
- G. A. Holmes, K. Rice and C. R. Snyder, J. Mater. Sci., 2006, 41, 4105–4116 CrossRef CAS.
- M. Taki, J. L. Wolford and T. V. O'Halloran, J. Am. Chem. Soc., 2004, 126, 712–713 CrossRef CAS PubMed.
- H. K. Sarma, B. K. Sharma and S. C. Tiwari, Curr. Sci., 2003, 85, 1401–1403 CAS.
- T. Kusumi, T. Ooi, M. R. Wülchli and H. Kakisawa, J. Am. Chem. Soc., 1988, 110, 2954–2958 CrossRef CAS.
- J. T. Stewart and C. A. Janicki, Anal. Profiles Drug Subst. Excipients, 1987, 16, 119–144 CAS.
- S. P. B. Ovenden, J. L. Nielson, C. H. Liptrot, R. H. Willis, D. M. Tapiolas, A. D. Wright and C. A. Motti, J. Nat. Prod., 2011, 74, 65–68 CrossRef CAS PubMed.
- S. S. Adams, K. F. McCullough and J. S. Nicholson, Arch. Int. Pharmacodyn. Ther., 1969, 178, 115–129 CAS.
- K. S. Albert and C. M. Gernaat, Am. J. Med., 1984, 77, 40–46 CrossRef CAS PubMed.
- M. G. Radford, K. E. Holley, J. P. Grande, T. S. Larson, R. D. Wagoner, J. V. Donadio and J. T. McCarthy, JAMA, 1996, 276, 466–469 CrossRef CAS PubMed.
- R. Bushra and N. Aslam, Oman Med. J., 2010, 25, 155–161 CrossRef PubMed.
- I. M. Vasincu, M. Apotrosoaei, A. T. Panzariu, F. Buron, S. Routier and L. Profire, Molecules, 2014, 19, 15005–15025 CrossRef PubMed.
- C. Gundogdu-Hizliates, H. Alyuruk, M. Gocmenturk, Y. Ergun and L. Cavas, Bioorg. Chem., 2014, 52, 8–15 CrossRef CAS PubMed.
- I. Q. Abdulla, Nat. Sci., 2014, 06, 47–53 CAS.
- K. K. Angajala, S. Vianala, R. Macha, M. Raghavender, M. K. Thupurani and P. J. Pathi, Springerplus, 2016, 5, 423 CrossRef PubMed.
- S. Gali, D. Raghu, V. Mallikanti, V. Thumma and N. Vaddiraju, Mol. Diversity, 2023 DOI:10.1007/s11030-023-10661-3.
- S. Aitha, V. Thumma, S. Ambala, R. Matta, S. Panga and J. Pochampally, ChemistrySelect, 2023, 8, e202300405 CrossRef CAS.
- E. R. Levin, Mol. Endocrinol., 2005, 19, 1951–1959 CrossRef CAS PubMed.
- V. Mallikanti, V. Thumma, K. C. Veeranki, S. Gali and J. Pochampally, ChemistrySelect, 2022, 7, e202204020 CrossRef CAS.
- V. K. Chaitanya, P. Jalapathi, M. R. Chandar, T. Vishnu, M. Veerabhadraiah and M. Raghavender, J. Iran. Chem. Soc., 2023, 20, 995–1006 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- Y. C. Martin, J. Med. Chem., 2005, 48, 3164–3170 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- P. Ertl and A. Schuffenhauer, Aust. J. Chem., 2009, 1, 1–11 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 31(2), 455–461 CrossRef PubMed.
-
S. Dallakyan and A. J. Olson, in Methods in Molecular Biology, Springer, 2015, vol. 1263, pp. 243–250 Search PubMed.
- G. Yaku, D. Ramulu, V. Thumma, A. Paluri and R. Dharavath, ChemistrySelect, 2023, 8, e202300255 CrossRef CAS.
- A. K. Sabhavath, S. Madderla, R. Dharavath, V. Thumma, G. Thara, S. Gundu and A. Dongamanti, ChemistrySelect, 2022, 7, e202203847 CrossRef CAS.
- D. Ashok, G. Thara, B. K. Kumar, G. Srinivas, D. Ravinder, T. Vishnu, M. Sarasija and B. Sushmitha, RSC Adv., 2023, 13(1), 25–40 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Veerabhadraiah
Mallikanti
b,
Raghavender
Matta
c,
Ravinder
Dharavath
d and
Pochampally
Jalapathi
a,
Veerabhadraiah
Mallikanti
b,
Raghavender
Matta
c,
Ravinder
Dharavath
d and
Pochampally
Jalapathi