
Highly efficient, catalyst-free, one-pot sequential four-component synthesis of novel spiroindolinone-pyrazole scaffolds as anti-Alzheimer agents: in silico study and biological screening†
Hormoz
Pourtaher
a,
Yasaman
Mohammadi
bc,
Alireza
Hasaninejad
*a and
Aida
Iraji
 *bc
*bc
aDepartment of Chemistry, Faculty of Sciences, Persian Gulf University, Bushehr, Iran. E-mail: alirezahasaninejad@gmail.com; a_hasaninejad@yahoo.com
bResearch Center for Traditional Medicine and History of Medicine, Department of Persian Medicine, School of Medicine, Shiraz University of Medical Sciences, Shiraz 7134845794, Iran. E-mail: aida.iraji@gmail.com; iraji@sums.ac.ir
cStem Cells Technology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
First published on 4th December 2023
Abstract
Alzheimer's disease is a neurodegenerative disorder that impacts memory, thinking, and behavior, and currently, there is no effective cure available for its treatment. This study explored a one-pot strategy for synthesizing spiroindolinone-pyrazole derivatives through a sequential four-component condensation reaction. These derivatives were further investigated for their potential as anti-Alzheimer's disease agents. The developed synthetic procedure provides remarkable advantages, including a clean reaction profile, abundant starting materials, operational simplicity, and easy purification without traditional methods with good to excellent yields (84–96%). Next, the biological potencies of the newly synthesized spiroindolinone-pyrazole derivatives against AChE and BChE as Alzheimer's disease-related targets were determined. Also, the kinetic study and cytotoxicity of the most potent derivative were investigated. Furthermore, molecular docking and molecular dynamics evaluations were performed employing in silico tools to investigate the interaction, orientation, and conformation of the potent analog over the active site of the enzyme.
1. Introduction
Alzheimer's disease (AD) is the most common cause of dementia, especially in the elder population, which causes memory and cognitive impairments with behavioral abnormalities. AD is estimated to affect 115.4 million people worldwide by 2050, which requires urgent attention.1,2The pathogenesis of AD is still inconclusive and undefined, and different proposed mechanisms, including amyloid beta (Aβ), hyperphosphorylation of T-protein, protein deposits, oxidative stress, and metal dyshomeostasis, were defined. The consistent loss of cholinergic neurons and a considerable decline in choline acetyltransferase activity are the most prominent phenotypes of AD progression.3,4 As a result, cholinergic system dysfunction happens with synaptic damage accompanied by neuronal loss in the brain, particularly in the hippocampus and cerebral cortex.5
Acetylcholine (ACh) is known as one of the main neurotransmitters in the central nervous system (CNS), synthesized from acetyl coenzyme A and choline by the enzyme choline acetyltransferase. It regulates many processes, including memory, learning, attention, and behavior.6 In detail, ACh exerts excitatory effects at the neuromuscular junction by binding to either nicotinic or muscarinic ACh receptors, thereby initiating its action. The termination of ACh's activity within the synaptic cleft is facilitated by two primary cholinesterases (ChEs), namely acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). These enzymes hydrolyze ACh into choline and acetate, halting synaptic transmission.7,8
The loss of cholinergic neurons and the reduction of ACh in patients with AD make AChE and BChE key therapeutic targets to modify the condition of the patients. It was reported that as the disease progresses, AChE activity decreases and BChE activity increases, making it an ideal target for the late stage of the disease.7 Approved ChE inhibitors are tacrine (withdrawal), donepezil, rivastigmine, galantamine, and huperzine A (approved by CFDA). However, these drugs are associated with severe side effects, including nausea, vomiting, diarrhea, muscle weakness, and weight loss. As a result, excessive efforts have been made to design novel Che inhibitors.9
Multicomponent reactions (MCRs) are versatile protocols in which multiple reactions are combined in one synthetic operation and are widely used to synthesize a variety of complex heterocycles and natural products.10 These reactions are characterized by convergence, ease of execution, efficiency, reducing waste, high yields, and atom economy.11,12 With the rising interest in generating heterocyclic compound libraries, exploring fresh multicomponent and synthetic reactions for academic and industrial research is an engaging research endeavor.
Pyrazolines are one of the important classes of nitrogen heterocyclic compounds, and their analogs are of great importance in medicinal chemistry.13–15 They are one of the groups of studied compounds among the azole family. Indeed, various synthesis methods and synthetic analogs have been reported over the years. The presence of the pyrazole core in different structures leads to various applications in different fields such as technology, agriculture, and medicine. These kinds of compounds possess a broad spectrum of biological activity, such as anticancer,16 antimicrobial,17 anti-inflammatory,18 anticonvulsant19 antitumor,20 and antiviral21 activities.
Isatin, also known as 1H-indole-2,3-dione, was initially synthesized by Erdmann and Laurent through the oxidation of indigo using nitric acid and/or chromic acid.22,23 Isatin and its derivatives occur naturally in certain plants like the Isatis genus, Calanthe discolor LINDL,24 and Couroupita guianensis Aubl.25 It has also been studied in the parotid gland of Bufo frogs.26 In humans, isatin is a metabolic byproduct of adrenaline.27–29 Isatin and its derivatives are fascinating and versatile building blocks widely used to synthesize important and bioactive heterocyclic compounds.30–35
The spirooxindole framework36,37 is a significant structural arrangement found in various bioactive natural compounds, including horsfiline, coerulescine, spirotryprostatin A, welwitindolinone A, elacomine, and alstonisine. Spirotryprostatin A, a natural alkaloid obtained from the fermentation broth of Aspergillus fumigatus, has been recognized as a new inhibitor of microtubule assembly. Additionally, pteropodine and isopteropodine have demonstrated the ability to regulate the function of muscarinic serotonin receptors38–40 (Fig. 1).
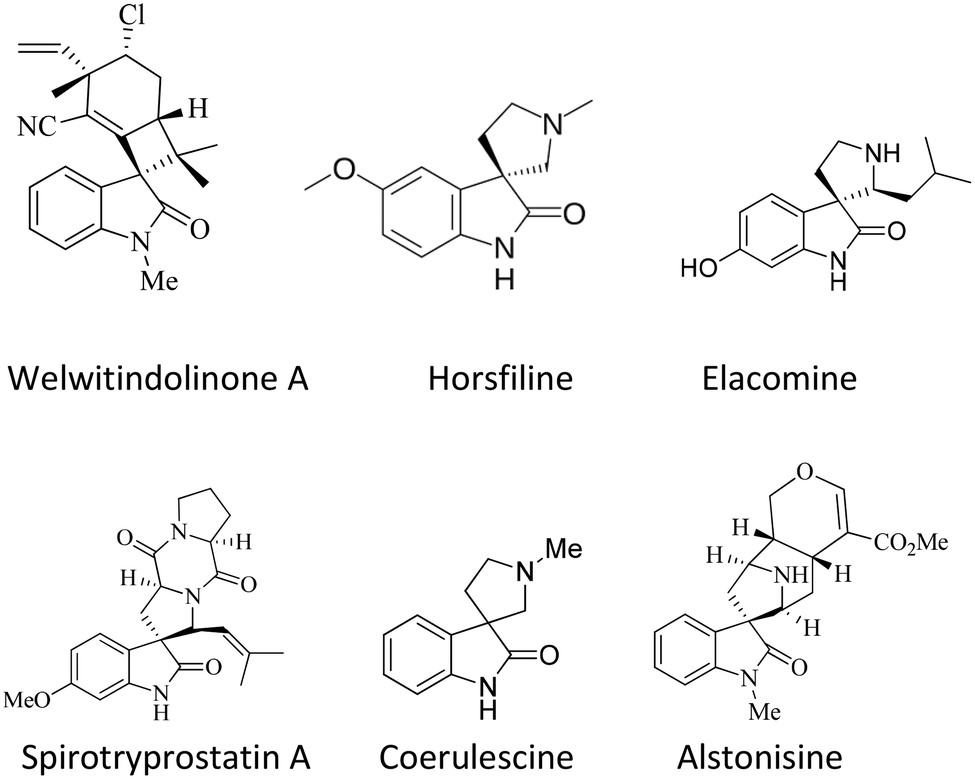 | ||
| Fig. 1 Representative structure of spiroxindole derivatives. | ||
The significance of the indole core in medicinal and organic chemistry has led to a heightened focus on synthesizing oxo-indole derivatives. Various methods for crafting spiroxindole derivatives have been documented.41–44
Most of these synthetic routes can be categorized based on the following key reactions: (1) Mannich/Pictet–Spengler;45,46 (2) 1,3-dipolar cycloaddition;47,48 (3) Morita–Baylis–Hillman,49,50 intermolecular alkylation;51,52 (4) electrocyclization;53 (5) sigmatropic rearrangement reactions.54,55 For instance, Hongyu Guo and coworkers reported the synthesis of spirooxindole-pyrazolines and pyrazolones via P(NMe2)3-mediated substrate-controlled annulations of azoalkenes with α-dicarbonyl compounds.56 Assem Barakat and coworkers reported a new series of functionalized spirooxindoles linked with the 3-acylindole scaffold in methanol solvent.57 Alizadeh et al. have also reported the synthesis of these compounds using isatin derivatives, BMTN, ammonia, 1,3-dicarbonyl, and p-TSA as catalysts in water.19
In continuation of our research on multicomponent reaction and synthesis of spirooxindole derivatives and also considering the possible biological effects of pyrazoline and spiroxindole parts in an organic compound, synthesizing a series of new spiropyrazoline derivatives can be useful. In line with our ongoing interest in synthesizing biologically active heterocyclic compounds as anti-AD agents, we present a concise and efficient method for synthesizing novel spiro pyrazoline derivatives. This one-pot, sequential four-component synthesis involves the reaction of 1,1-bis(methylthio)-2-nitroethylene (BMTNE), arylamines, hydrazine hydrate, and isatine derivatives, all under catalyst-free conditions in EtOH at reflux. Next, all the derivatives were screened for their anti-AChE and BuChE activities. A kinetic study was also performed on the most potent compound against the respective enzyme. Molecular docking and molecular dynamics simulations were carried out on the active analogs to gain insights into the binding interactions. Furthermore, the neurotoxicity of these compounds was evaluated against the SH-SY5Y neuroblastoma cell line.
2. Experimental section
2.1 General
All chemicals were purchased from Merck or Fuluka chemical companies. 1H-NMR 300 MHz and 13C-NMR (75 MHz) spectra were run on a Bruker Avance 300 MHz instrument in DMSO-d6. A Unicom Galaxy Series FT-IR 4600 spectrophotometer was used to obtain infra-red spectra. Melting points were recorded on a Buchi B-545 apparatus in open capillary tubes. Reaction progress was screened by TLC using silica gel polygram SIL G/UV254 plates.2.2 General procedure for the synthesis of compounds 5a–q
Initially, to prepare N-aryl-1-(methylthio)-2-nitroethenamine 3a, a mixture of BMTNE 1 (1 mmol) and arylamine 2 (1 mmol) was stirred in ethanol (5 mL) at reflux for 12 h. After that, isatin derivatives 4 (1 mmol) and NH2NH2 (80% aq.) (1.2 mmol) were added and were heated at reflux. After completion of the reaction (monitored by TLC analysis), the crude solid formed, was filtered and washed with ethanol (5 ml) to give the pure product 5.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, Rf = 0.272); IR (KBr): 3359, 3286, 3055, 2973, 1714, 1641, 1619, 1598, 1477, 1355, 1226, 1199, 1108, 1077, 871, 630; 1H NMR (300 MHz, DMSO-d6) δ: 10.60 (1H, s, NH), 9.90 (1H, s, NH), 7.64–7.52 (2H, m, Ar), 7.42–7.28 (3H, m, Ar), 7.22 (1H, td, J = 7.7, 1.2 Hz, Ar), 6.80 (1H, d, J = 7.8 Hz, Ar), 6.68 (1H, t, J = 7.6 Hz, Ar), 5.95 (2H, s, NH), 2.32 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.7, 159.0, 146.1, 143.7, 136.6, 133.5, 132.0, 131.1, 127.4, 127.1, 126.7, 126.4, 121.8, 117.6, 110.4, 71.6, 18.2; anal. calcd for C17H15N5O3: C, 60.53; H, 4.48; N, 20.76. Found: C, 60.88; H, 4.64; N, 20.93.
:4, Rf = 0.269); IR (KBr): 3326, 3026, 3162, 1722, 1644, 1573, 1475, 1353, 1299, 1255, 1203, 1153, 1106, 877, 815, 626. 1H NMR (300 MHz, DMSO-d6) δ: 10.69 (1H, s, NH), 10.36 (1H, s, NH), 8.24 (1H, d, J = 7.5 Hz, Ar), 7.72 (2H, d, J = 8.2 Hz, Ar), 7.35–7.28 (3H, m, Ar), 6.97 (1H, t, J = 7.6 Hz, Ar), 6.88 (1H, d, J = 7.7 Hz, Ar), 5.89 (2H, s, NH), 2.35 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.7, 157.4, 145.7, 144.0, 136.4, 134.4, 132.4, 129.9, 126.6, 122.2, 121.5, 117.4, 110.7, 72.8, 21.0.
:4, Rf = 0.265); IR (KBr): 3289, 3261, 3170, 3077, 2952, 1716, 1635, 1590, 1514, 1494, 1427, 1259, 1106, 1018, 941, 854, 800, 713; 1H NMR (300 MHz, DMSO-d6) δ: 13.42 (1H, s, OH), 11.27 (1H, s, NH), 10.86 (1H, s, NH), 8.22 (1H, d, J = 7.6 Hz, Ar), 7.45–735 (3H, m, Ar), 7.10 (1H, t, J = 7.6 Hz, Ar), 6.93 (2H, dd, J = 15.7, 7.7 Hz, Ar), 6.98–6.75 (3H, m, Ar), 6.17 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 163.0, 159.3, 151.7, 145.5, 142.9, 135.9, 134.3, 132.1, 129.6, 122.9, 122.5, 119.9, 117.1, 111.7, 74.9; anal. calcd for C16H13N5O4: C, 56.64; H, 3.86; N, 20.64. Found: C, 56.23; H, 3.65; N, 20.85.
:4, Rf = 0.265); IR (KBr): 3293, 3218, 3075, 2971, 1716, 1645, 1508, 1477, 1334, 1218, 1108, 1068, 900, 815, 732; 1H NMR (300 MHz, DMSO-d6) δ: 10.65 (1H, s, NH), 10.32 (1H, s, NH), 8.19 (1H, dd, J = 7.7, 1.3 Hz, Ar), 7.78–7.67 (2H, m, Ar), 7.31 (1H, td, J = 7.7, 1.3 Hz, Ar), 7.10–7.05 (2H, m, Ar), 6.97 (1H, td, J = 7.6, 1.0 Hz, Ar), 6.86 (1H, d, J = 7.7 Hz, Ar), 5.86 (2H, s, NH), 3.82 (3H, s, OMe); 13C NMR (76 MHz, DMSO-d6) δ: 165.7, 157.5, 156.8, 145.3, 143.9, 132.2, 131.8, 126.7, 123.3, 122.2, 117.5, 114.6, 110.6, 72.7, 55.8; anal. calcd for C17H15N5O4: C, 57.79; H, 4.28; N, 19.82. Found: C, 57.85; H, 4.10; N, 19.88. Found: C, 57.53; H, 4.10; N, 19.66.
:4, Rf = 0.268); IR (KBr): 3322, 3265, 3162, 3027, 1725, 1644, 1299, 1260, 1205, 1106, 813, 688, 625; 1H NMR (300 MHz, DMSO-d6) δ: 10.68 (1H, s, NH), 10.32 (1H, s, NH), 8.32 (1H, dd, J = 7.7, 1.2 Hz, Ar), 7.83 (1H, d, J = 2.3 Hz, Ar), 7.43–7.27 (2H, m, Ar), 7.23 (1H, d, J = 8.2 Hz, Ar), 6.96 (1H, td, J = 7.6, 1.0 Hz, Ar), 6.88 (1H, d, J = 7.8 Hz, Ar), 5.88 (2H, s, NH), 2.31 (3H, s, Me), 2.27 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.7, 157.6, 145.6, 144.0, 137.2, 136.6, 133.2, 132.4, 130.3, 126.5, 122.7, 122.0, 118.9, 117.4, 110.7, 72.8, 19.9, 19.4; anal. calcd for C18H17N5O3: C, 61.53; H, 4.88; N, 19.93. Found: C, 61.04; H, 4.56; N, 20.21.
:4, Rf = 0.272); IR (KBr): 3361, 3288, 3170, 3075, 2977, 1722, 1640, 1600, 1523, 1482, 1336, 1228, 1130, 1073, 918, 835, 634; 1H NMR (300 MHz, DMSO-d6) δ: 10.70 (1H, s, NH), 10.40 (1H, s, NH), 8.24 (1H, d, J = 7.6 Hz, Ar), 7.81 (1H, s, Ar), 7.50 (2H, t, J = 7.8 Hz, Ar), 7.32 (1H, td, J = 7.7, 1.3 Hz, Ar), 7.24 (1H, t, J = 7.4 Hz, Ar), 6.95 (1H, t, J = 7.6 Hz, Ar), 6.88 (1H, d, J = 7.8 Hz, Ar), 5.90 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 165.6, 157.5, 146.1, 144.1, 138.9, 132.6, 129.5, 126.7, 125.1, 122.2, 121.6, 117.3, 110.8, 72.8; anal. calcd for C16H12ClN5O3: C, 53.72; H, 3.38; N, 19.58. Found: C, 53.49; H, 3.45; N, 19.25.
:4, Rf = 0.269); IR (KBr): 3307, 3261, 3120, 3062, 2970, 1735, 1644, 1604, 1508, 1477,1388, 1245, 1216, 1184, 1066, 905, 845, 777, 628; 1H NMR (300 MHz, DMSO-d6) δ: 10.71 (1H, s, NH), 10.48 (1H, s, NH), 8.17 (1H, dd, J = 7.6, 1.2 Hz, Ar), 7.90–7.82 (2H, m, Ar), 7.60–7.54 (2H, m, Ar), 7.34 (1H, td, J = 7.7, 1.3 Hz, Ar), 7.01 (1H, td, J = 7.7, 1.1 Hz, Ar), 6.88 (1H, d, J = 7.7 Hz, Ar), 5.88 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 165.5, 157.2, 146.5, 144.2, 137.9, 132.7, 129.4, 128.7, 126.7, 123.0, 122.4, 117.2, 110.8, 62.5.
:4, Rf = 0.255); IR (KBr): 3313, 3253, 3080, 2992, 1722, 1633, 1513, 1426, 1344, 1261, 1072, 850, 808; 1H NMR (300 MHz, DMSO-d6) δ: 10.47 (1H, s, NH), 10.27 (1H, s, NH), 7.53 (2H, d, J = 8.1 Hz, Ar), 7.25–7.19 (2H, m, Ar), 6.98 (1H, d, J = 2.4 Hz, Ar), 6.81–6.66 (2H, m, Ar), 5.68 (2H, s, NH), 3.88 (3H, s, OMe), 3.74 (3H, s, OMe); 13C NMR (76 MHz, DMSO-d6) δ: 177.7, 161.3, 155.6, 153.3, 150.6, 136.0, 135.0, 131.9, 124.7, 118.6, 116.7, 112.8, 110.7, 109.6, 60.6, 56.0, 55.9; anal. calcd for C18H17N5O5: C, 56.39; H, 4.47; N, 18.27. Found: C, 56.69; H, 4.41; N, 18.63.
:4, Rf = 0.262); IR (KBr): 3345, 3288, 3166, 3096, 2992, 1722, 1644, 1608, 1504, 1357, 1292, 1236, 1201, 1139, 1020, 730, 568; 1H NMR (300 MHz, DMSO-d6) δ: 10.46 (1H, s, NH), 10.28 (1H, s, NH), 7.88 (1H, d, J = 2.6 Hz, Ar), 7.61 (1H, s, Ar), 7.48 (1H, dd, J = 8.1, 2.3 Hz, Ar), 7.20 (1H, d, J = 8.1 Hz, Ar), 6.91 (1H, dd, J = 8.5, 2.7 Hz, Ar), 6.77 (1H, d, J = 8.4 Hz, Ar), 5.87 (2H, s, NH), 3.59 (3H, s, OMe), 2.28 (3H, s, Me), 2.25 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.8, 157.7, 155.0, 145.8, 137.6, 137.4, 136.5, 133.4, 130.4, 122.9, 119.3, 118.0, 117.8, 112.9, 111.0, 72.7, 55.9, 19.8, 19.3; anal. calcd for C19H19N5O4: C, 59.84; H, 5.02; N, 18.36. Found: C, 59.21; H, 4.97; N, 18.66.
:4, Rf = 0.272); IR (KBr): 3396, 3360, 3268, 1741, 1646, 1506, 1479, 1335, 1191, 1106, 1066, 898, 644; 1H NMR (300 MHz, DMSO-d6) δ: 11.35 (1H, s, NH), 10.10 (1H, s, NH), 8.92 (1H, d, J = 2.5 Hz, Ar), 8.22 (1H, dd, J = 8.7, 2.5 Hz, Ar), 7.73 (1H, d, J = 3.0 Hz, Ar), 7.14 (1H, d, J = 9.1 Hz, Ar), 7.03 (1H, d, J = 8.6 Hz, Ar), 6.87 (1H, dd, J = 9.0, 3.1 Hz, Ar), 6.04 (2H, s, NH), 3.88 (3H, s, OMe), 3.62 (3H, s, OMe); 13C NMR (75 MHz, DMSO-d6) δ: 165.8, 159.2, 153.6, 149.0, 145.3, 143.5, 142.4, 128.4, 127.0, 121.9, 117.2, 113.1, 111.4, 110.8, 110.6, 72.3, 56.1, 55.5; anal. calcd for C18H16N6O7: C, 50.47; H, 3.76; N, 19.62. Found: C, 50.68; H, 3.97; N, 19.21.
:4, Rf = 0.270); IR (KBr): 3343, 3282, 3143, 2923, 1716, 1644, 1504, 1349, 1191, 1016, 887, 757; 1H NMR (300 MHz, DMSO) δ: 11.40 (1H, s, NH), 10.82 (1H, s, NH), 9.00 (1H, d, J = 2.4 Hz, Ar), 8.29–8.21 (1H, m, Ar), 7.91–7.80 (2H, m, Ar), 7.60–7.51 (2H, m, Ar), 7.04 (1H, d, J = 8.7 Hz, Ar), 5.92 (2H, s, NH); 13C NMR (75 MHz, DMSO) δ: 165.7, 158.9, 149.4, 143.9, 142.5, 137.3, 129.7, 129.3, 128.7, 123.4, 121.8, 116.8, 110.9, 72.9; anal. calcd for C16H11ClN6O5: C, 47.71; H, 2.75; N, 20.87. Found: 48.09; H, 2.62; N, 20.51.
:4, Rf = 0.270); IR (KBr): 3315, 3289, 3162, 3052, 2960, 1712, 1641, 1550, 1348, 1297, 1249, 1114, 1031, 890, 817; 1H NMR (300 MHz, DMSO-d6) δ: 11.09 (1H, s, NH), 10.21 (1H, s, NH), 7.84 (1H, d, J = 7.6 Hz, Ar), 7.54–7.46 (2H, m, Ar), 7.44–7.31 (3H, m, Ar), 6.83 (1H, t, J = 7.8 Hz, Ar), 5.98 (2H, s, NH), 2.31 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 166.0, 160.1, 143.4, 140.5, 140.5, 136.4, 133.7, 131.2, 130.6, 127.5, 126.9, 126.6, 121.8, 119.0, 111.5, 111.0, 71.6, 18.1; anal. calcd for C18H14F3N5O3: C, 53.34; H, 3.48; N, 17.28. Found: C, 52.89; H, 3.30; N, 17.13.
:4, Rf = 0.262); IR (KBr): 3396, 3366, 3265, 1731, 1646, 1508, 1475, 1332, 1191, 1108, 1066, 890, 634; 1H NMR (300 MHz, DMSO-d6) δ: 11.23 (1H, s, NH), 10.77 (1H, s, NH), 8.53 (1H, d, J = 7.6 Hz, Ar), 8.24 (1H, t, J = 2.0 Hz, Ar), 7.64 (1H, d, J = 8.1 Hz, Ar), 7.58–7.46 (2H, m, Ar), 7.34 (1H, dt, J = 7.2, 1.9 Hz, Ar), 7.16 (1H, t, J = 7.8 Hz, Ar), 5.92 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 165.8, 158.5, 152.3, 144.2, 141.3, 140.0, 138.1, 133.8, 131.3, 129.8, 127.1, 125.9, 125.0, 121.4, 120.1, 118.6, 72.7; anal. calcd for C17H11ClF3N5O3: C, 47.96; H, 2.60; N, 16.45; O, 11.27. Found: C, 48.55; H, 2.78; N, 16.79.
:4, Rf = 0.265); 1H NMR (300 MHz, DMSO-d6) δ: 11.05 (1H, s, NH), 10.75 (1H, s, NH), 8.20–8.06 (2H, m, Ar), 8.02 (1H, d, J = 8.1 Hz, Ar), 7.79 (1H, d, J = 7.3 Hz, Ar), 7.72–7.56 (3H, m, Ar), 7.40 (1H, d, J = 8.1 Hz, Ar), 7.31 (1H, d, J = 7.6 Hz, Ar), 6.56 (1H, t, J = 7.8 Hz, Ar), 6.10 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 165.9, 160.6, 143.7, 140.5, 134.3, 133.9, 130.6, 128.7, 127.7, 127.0, 126.9, 126.0, 123.9, 123.5, 121.2, 118.7, 111.4, 110.9, 71.8; anal. calcd for C21H14 F3N5O3: C, 57.15; H, 3.20; F, 12.91; N, 15.87. Found: C, 57.44; H, 3.63; N, 15.88.
:4, Rf = 0.282); IR (KBr): 3380, 3320, 3215, 3093, 2967, 1735, 1714, 1650, 1585, 1480, 1360, 1240, 1216, 1070, 934, 734, 588; 1H NMR (300 MHz, DMSO-d6) δ: 10.78 (1H, s, NH), 10.51 (1H, s, NH), 8.22 (1H, d, J = 2.2 Hz, Ar), 7.64 (2H, d, J = 8.2 Hz, Ar), 7.39–7.25 (3H, m, Ar), 6.85 (1H, d, J = 8.3 Hz, Ar), 5.90 (2H, s, NH), 2.35 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.4, 158.4, 144.6, 142.4, 137.7, 135.9, 135.0, 131.5, 129.9, 126.3, 126.0, 122.3, 118.5, 114.2, 112.0, 72.5, 21.1; anal. calcd for C17H14ClN5O3: C, 54.92; H, 3.80; N, 18.84. Found: 54.49; H, 3.82; N, 19.11.
:4, Rf = 0.282); IR (KBr): 3378, 3338, 3261, 1741, 1716, 1650, 1481, 1334, 1194, 1097, 1068, 838, 694; 1H NMR (300 MHz, DMSO-d6) δ: 10.57 (1H, s, NH), 9.88 (1H, s, NH), 7.60 (1H, d, J = 7.6 Hz, Ar), 7.43–7.16 (4H, m, Ar), 6.80 (1H, d, J = 7.7 Hz, Ar), 6.68 (1H, t, J = 7.6 Hz, Ar), 5.95 (2H, s, NH), 2.32 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.7, 159.0, 146.1, 143.7, 136.7, 133.5, 132.0, 131.1, 127.4, 127.1, 126.7, 126.5, 121.8, 117.7, 110.4, 71.6, 18.2; anal. calcd for: C16H13N5O4.
:4, Rf = 0.272); IR (KBr): 3359, 3286, 3055, 2973, 1714, 1641, 1619, 1598, 1477, 1355, 1226, 1199, 1108, 1077, 871, 630; 1H NMR (300 MHz, DMSO-d6) δ: 10.60 (1H, s, NH), 9.90 (1H, s, NH), 7.64–7.52 (2H, m, Ar), 7.42–7.28 (3H, m, Ar), 7.22 (1H, td, J = 7.7, 1.2 Hz, Ar), 6.80 (1H, d, J = 7.8 Hz, Ar), 6.68 (1H, t, J = 7.6 Hz, Ar), 5.95 (2H, s, NH), 2.32 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.7, 159.0, 146.1, 143.7, 136.6, 133.5, 132.0, 131.1, 127.4, 127.1, 126.7, 126.4, 121.8, 117.6, 110.4, 71.6, 18.2; anal. calcd for C17H15N5O3: C, 60.53; H, 4.48; N, 20.76. Found: C, 60.88; H, 4.64; N, 20.93.
:4, Rf = 0.269); IR (KBr): 3326, 3026, 3162, 1722, 1644, 1573, 1475, 1353, 1299, 1255, 1203, 1153, 1106, 877, 815, 626. 1H NMR (300 MHz, DMSO-d6) δ: 10.69 (1H, s, NH), 10.36 (1H, s, NH), 8.24 (1H, d, J = 7.5 Hz, Ar), 7.72 (2H, d, J = 8.2 Hz, Ar), 7.35–7.28 (3H, m, Ar), 6.97 (1H, t, J = 7.6 Hz, Ar), 6.88 (1H, d, J = 7.7 Hz, Ar), 5.89 (2H, s, NH), 2.35 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.7, 157.4, 145.7, 144.0, 136.4, 134.4, 132.4, 129.9, 126.6, 122.2, 121.5, 117.4, 110.7, 72.8, 21.0.
:4, Rf = 0.265); IR (KBr): 3289, 3261, 3170, 3077, 2952, 1716, 1635, 1590, 1514, 1494, 1427, 1259, 1106, 1018, 941, 854, 800, 713; 1H NMR (300 MHz, DMSO-d6) δ: 13.42 (1H, s, OH), 11.27 (1H, s, NH), 10.86 (1H, s, NH), 8.22 (1H, d, J = 7.6 Hz, Ar), 7.45–735 (3H, m, Ar), 7.10 (1H, t, J = 7.6 Hz, Ar), 6.93 (2H, dd, J = 15.7, 7.7 Hz, Ar), 6.98–6.75 (3H, m, Ar), 6.17 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 163.0, 159.3, 151.7, 145.5, 142.9, 135.9, 134.3, 132.1, 129.6, 122.9, 122.5, 119.9, 117.1, 111.7, 74.9; anal. calcd for C16H13N5O4: C, 56.64; H, 3.86; N, 20.64. Found: C, 56.23; H, 3.65; N, 20.85.
:4, Rf = 0.265); IR (KBr): 3293, 3218, 3075, 2971, 1716, 1645, 1508, 1477, 1334, 1218, 1108, 1068, 900, 815, 732; 1H NMR (300 MHz, DMSO-d6) δ: 10.65 (1H, s, NH), 10.32 (1H, s, NH), 8.19 (1H, dd, J = 7.7, 1.3 Hz, Ar), 7.78–7.67 (2H, m, Ar), 7.31 (1H, td, J = 7.7, 1.3 Hz, Ar), 7.10–7.05 (2H, m, Ar), 6.97 (1H, td, J = 7.6, 1.0 Hz, Ar), 6.86 (1H, d, J = 7.7 Hz, Ar), 5.86 (2H, s, NH), 3.82 (3H, s, OMe); 13C NMR (76 MHz, DMSO-d6) δ: 165.7, 157.5, 156.8, 145.3, 143.9, 132.2, 131.8, 126.7, 123.3, 122.2, 117.5, 114.6, 110.6, 72.7, 55.8; anal. calcd for C17H15N5O4: C, 57.79; H, 4.28; N, 19.82. Found: C, 57.85; H, 4.10; N, 19.88. Found: C, 57.53; H, 4.10; N, 19.66.
:4, Rf = 0.268); IR (KBr): 3322, 3265, 3162, 3027, 1725, 1644, 1299, 1260, 1205, 1106, 813, 688, 625; 1H NMR (300 MHz, DMSO-d6) δ: 10.68 (1H, s, NH), 10.32 (1H, s, NH), 8.32 (1H, dd, J = 7.7, 1.2 Hz, Ar), 7.83 (1H, d, J = 2.3 Hz, Ar), 7.43–7.27 (2H, m, Ar), 7.23 (1H, d, J = 8.2 Hz, Ar), 6.96 (1H, td, J = 7.6, 1.0 Hz, Ar), 6.88 (1H, d, J = 7.8 Hz, Ar), 5.88 (2H, s, NH), 2.31 (3H, s, Me), 2.27 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.7, 157.6, 145.6, 144.0, 137.2, 136.6, 133.2, 132.4, 130.3, 126.5, 122.7, 122.0, 118.9, 117.4, 110.7, 72.8, 19.9, 19.4; anal. calcd for C18H17N5O3: C, 61.53; H, 4.88; N, 19.93. Found: C, 61.04; H, 4.56; N, 20.21.
:4, Rf = 0.272); IR (KBr): 3361, 3288, 3170, 3075, 2977, 1722, 1640, 1600, 1523, 1482, 1336, 1228, 1130, 1073, 918, 835, 634; 1H NMR (300 MHz, DMSO-d6) δ: 10.70 (1H, s, NH), 10.40 (1H, s, NH), 8.24 (1H, d, J = 7.6 Hz, Ar), 7.81 (1H, s, Ar), 7.50 (2H, t, J = 7.8 Hz, Ar), 7.32 (1H, td, J = 7.7, 1.3 Hz, Ar), 7.24 (1H, t, J = 7.4 Hz, Ar), 6.95 (1H, t, J = 7.6 Hz, Ar), 6.88 (1H, d, J = 7.8 Hz, Ar), 5.90 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 165.6, 157.5, 146.1, 144.1, 138.9, 132.6, 129.5, 126.7, 125.1, 122.2, 121.6, 117.3, 110.8, 72.8; anal. calcd for C16H12ClN5O3: C, 53.72; H, 3.38; N, 19.58. Found: C, 53.49; H, 3.45; N, 19.25.
:4, Rf = 0.269); IR (KBr): 3307, 3261, 3120, 3062, 2970, 1735, 1644, 1604, 1508, 1477,1388, 1245, 1216, 1184, 1066, 905, 845, 777, 628; 1H NMR (300 MHz, DMSO-d6) δ: 10.71 (1H, s, NH), 10.48 (1H, s, NH), 8.17 (1H, dd, J = 7.6, 1.2 Hz, Ar), 7.90–7.82 (2H, m, Ar), 7.60–7.54 (2H, m, Ar), 7.34 (1H, td, J = 7.7, 1.3 Hz, Ar), 7.01 (1H, td, J = 7.7, 1.1 Hz, Ar), 6.88 (1H, d, J = 7.7 Hz, Ar), 5.88 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 165.5, 157.2, 146.5, 144.2, 137.9, 132.7, 129.4, 128.7, 126.7, 123.0, 122.4, 117.2, 110.8, 62.5.
:4, Rf = 0.255); IR (KBr): 3313, 3253, 3080, 2992, 1722, 1633, 1513, 1426, 1344, 1261, 1072, 850, 808; 1H NMR (300 MHz, DMSO-d6) δ: 10.47 (1H, s, NH), 10.27 (1H, s, NH), 7.53 (2H, d, J = 8.1 Hz, Ar), 7.25–7.19 (2H, m, Ar), 6.98 (1H, d, J = 2.4 Hz, Ar), 6.81–6.66 (2H, m, Ar), 5.68 (2H, s, NH), 3.88 (3H, s, OMe), 3.74 (3H, s, OMe); 13C NMR (76 MHz, DMSO-d6) δ: 177.7, 161.3, 155.6, 153.3, 150.6, 136.0, 135.0, 131.9, 124.7, 118.6, 116.7, 112.8, 110.7, 109.6, 60.6, 56.0, 55.9; anal. calcd for C18H17N5O5: C, 56.39; H, 4.47; N, 18.27. Found: C, 56.69; H, 4.41; N, 18.63.
:4, Rf = 0.262); IR (KBr): 3345, 3288, 3166, 3096, 2992, 1722, 1644, 1608, 1504, 1357, 1292, 1236, 1201, 1139, 1020, 730, 568; 1H NMR (300 MHz, DMSO-d6) δ: 10.46 (1H, s, NH), 10.28 (1H, s, NH), 7.88 (1H, d, J = 2.6 Hz, Ar), 7.61 (1H, s, Ar), 7.48 (1H, dd, J = 8.1, 2.3 Hz, Ar), 7.20 (1H, d, J = 8.1 Hz, Ar), 6.91 (1H, dd, J = 8.5, 2.7 Hz, Ar), 6.77 (1H, d, J = 8.4 Hz, Ar), 5.87 (2H, s, NH), 3.59 (3H, s, OMe), 2.28 (3H, s, Me), 2.25 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.8, 157.7, 155.0, 145.8, 137.6, 137.4, 136.5, 133.4, 130.4, 122.9, 119.3, 118.0, 117.8, 112.9, 111.0, 72.7, 55.9, 19.8, 19.3; anal. calcd for C19H19N5O4: C, 59.84; H, 5.02; N, 18.36. Found: C, 59.21; H, 4.97; N, 18.66.
:4, Rf = 0.272); IR (KBr): 3396, 3360, 3268, 1741, 1646, 1506, 1479, 1335, 1191, 1106, 1066, 898, 644; 1H NMR (300 MHz, DMSO-d6) δ: 11.35 (1H, s, NH), 10.10 (1H, s, NH), 8.92 (1H, d, J = 2.5 Hz, Ar), 8.22 (1H, dd, J = 8.7, 2.5 Hz, Ar), 7.73 (1H, d, J = 3.0 Hz, Ar), 7.14 (1H, d, J = 9.1 Hz, Ar), 7.03 (1H, d, J = 8.6 Hz, Ar), 6.87 (1H, dd, J = 9.0, 3.1 Hz, Ar), 6.04 (2H, s, NH), 3.88 (3H, s, OMe), 3.62 (3H, s, OMe); 13C NMR (75 MHz, DMSO-d6) δ: 165.8, 159.2, 153.6, 149.0, 145.3, 143.5, 142.4, 128.4, 127.0, 121.9, 117.2, 113.1, 111.4, 110.8, 110.6, 72.3, 56.1, 55.5; anal. calcd for C18H16N6O7: C, 50.47; H, 3.76; N, 19.62. Found: C, 50.68; H, 3.97; N, 19.21.
:4, Rf = 0.270); IR (KBr): 3343, 3282, 3143, 2923, 1716, 1644, 1504, 1349, 1191, 1016, 887, 757; 1H NMR (300 MHz, DMSO) δ: 11.40 (1H, s, NH), 10.82 (1H, s, NH), 9.00 (1H, d, J = 2.4 Hz, Ar), 8.29–8.21 (1H, m, Ar), 7.91–7.80 (2H, m, Ar), 7.60–7.51 (2H, m, Ar), 7.04 (1H, d, J = 8.7 Hz, Ar), 5.92 (2H, s, NH); 13C NMR (75 MHz, DMSO) δ: 165.7, 158.9, 149.4, 143.9, 142.5, 137.3, 129.7, 129.3, 128.7, 123.4, 121.8, 116.8, 110.9, 72.9; anal. calcd for C16H11ClN6O5: C, 47.71; H, 2.75; N, 20.87. Found: 48.09; H, 2.62; N, 20.51.
:4, Rf = 0.270); IR (KBr): 3315, 3289, 3162, 3052, 2960, 1712, 1641, 1550, 1348, 1297, 1249, 1114, 1031, 890, 817; 1H NMR (300 MHz, DMSO-d6) δ: 11.09 (1H, s, NH), 10.21 (1H, s, NH), 7.84 (1H, d, J = 7.6 Hz, Ar), 7.54–7.46 (2H, m, Ar), 7.44–7.31 (3H, m, Ar), 6.83 (1H, t, J = 7.8 Hz, Ar), 5.98 (2H, s, NH), 2.31 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 166.0, 160.1, 143.4, 140.5, 140.5, 136.4, 133.7, 131.2, 130.6, 127.5, 126.9, 126.6, 121.8, 119.0, 111.5, 111.0, 71.6, 18.1; anal. calcd for C18H14F3N5O3: C, 53.34; H, 3.48; N, 17.28. Found: C, 52.89; H, 3.30; N, 17.13.
:4, Rf = 0.262); IR (KBr): 3396, 3366, 3265, 1731, 1646, 1508, 1475, 1332, 1191, 1108, 1066, 890, 634; 1H NMR (300 MHz, DMSO-d6) δ: 11.23 (1H, s, NH), 10.77 (1H, s, NH), 8.53 (1H, d, J = 7.6 Hz, Ar), 8.24 (1H, t, J = 2.0 Hz, Ar), 7.64 (1H, d, J = 8.1 Hz, Ar), 7.58–7.46 (2H, m, Ar), 7.34 (1H, dt, J = 7.2, 1.9 Hz, Ar), 7.16 (1H, t, J = 7.8 Hz, Ar), 5.92 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 165.8, 158.5, 152.3, 144.2, 141.3, 140.0, 138.1, 133.8, 131.3, 129.8, 127.1, 125.9, 125.0, 121.4, 120.1, 118.6, 72.7; anal. calcd for C17H11ClF3N5O3: C, 47.96; H, 2.60; N, 16.45; O, 11.27. Found: C, 48.55; H, 2.78; N, 16.79.
:4, Rf = 0.265); 1H NMR (300 MHz, DMSO-d6) δ: 11.05 (1H, s, NH), 10.75 (1H, s, NH), 8.20–8.06 (2H, m, Ar), 8.02 (1H, d, J = 8.1 Hz, Ar), 7.79 (1H, d, J = 7.3 Hz, Ar), 7.72–7.56 (3H, m, Ar), 7.40 (1H, d, J = 8.1 Hz, Ar), 7.31 (1H, d, J = 7.6 Hz, Ar), 6.56 (1H, t, J = 7.8 Hz, Ar), 6.10 (2H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ: 165.9, 160.6, 143.7, 140.5, 134.3, 133.9, 130.6, 128.7, 127.7, 127.0, 126.9, 126.0, 123.9, 123.5, 121.2, 118.7, 111.4, 110.9, 71.8; anal. calcd for C21H14 F3N5O3: C, 57.15; H, 3.20; F, 12.91; N, 15.87. Found: C, 57.44; H, 3.63; N, 15.88.
:4, Rf = 0.282); IR (KBr): 3380, 3320, 3215, 3093, 2967, 1735, 1714, 1650, 1585, 1480, 1360, 1240, 1216, 1070, 934, 734, 588; 1H NMR (300 MHz, DMSO-d6) δ: 10.78 (1H, s, NH), 10.51 (1H, s, NH), 8.22 (1H, d, J = 2.2 Hz, Ar), 7.64 (2H, d, J = 8.2 Hz, Ar), 7.39–7.25 (3H, m, Ar), 6.85 (1H, d, J = 8.3 Hz, Ar), 5.90 (2H, s, NH), 2.35 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.4, 158.4, 144.6, 142.4, 137.7, 135.9, 135.0, 131.5, 129.9, 126.3, 126.0, 122.3, 118.5, 114.2, 112.0, 72.5, 21.1; anal. calcd for C17H14ClN5O3: C, 54.92; H, 3.80; N, 18.84. Found: 54.49; H, 3.82; N, 19.11.
:4, Rf = 0.282); IR (KBr): 3378, 3338, 3261, 1741, 1716, 1650, 1481, 1334, 1194, 1097, 1068, 838, 694; 1H NMR (300 MHz, DMSO-d6) δ: 10.57 (1H, s, NH), 9.88 (1H, s, NH), 7.60 (1H, d, J = 7.6 Hz, Ar), 7.43–7.16 (4H, m, Ar), 6.80 (1H, d, J = 7.7 Hz, Ar), 6.68 (1H, t, J = 7.6 Hz, Ar), 5.95 (2H, s, NH), 2.32 (3H, s, Me); 13C NMR (75 MHz, DMSO-d6) δ: 165.7, 159.0, 146.1, 143.7, 136.7, 133.5, 132.0, 131.1, 127.4, 127.1, 126.7, 126.5, 121.8, 117.7, 110.4, 71.6, 18.2; anal. calcd for: C16H13N5O4.
2.3 Screening of ChE inhibitory activity
The assay was performed according to previously reported procedures.58,592.4 Enzyme kinetic studies
The mode of inhibition of the most active compound 5f, identified with the lowest IC50, was investigated against AChE activity with different concentrations of acetylthiocholine (0.1–1 mM) as the substrate in the absence and presence of 5f at different concentrations. A Lineweaver–Burk plot was generated to identify the type of inhibition, and the Michaelis–Menten constant (Km) value was determined from the plot between the reciprocal of the substrate concentration (1/[S]) and reciprocal of the enzyme rate (1/V) over the various inhibitor concentrations. The experimental inhibitor constant (Ki) value was constructed by secondary plots of the 5f concentration versus Km.602.5 Molecular docking study
The SMILE format of 5f was converted to a three-dimensional structure within the Maestro software package. The X-ray structures of AChE (PDB code: 4EY7). and BChE (PDB code: 4BDS) were prepared with the Protein Preparation Wizard interface of Maestro via removing the ligand and water molecules, adding hydrogen atoms, and optimizing their position according to PROPKA prediction at pH 7.0. The molecular docking was performed using IFD mode with the ligand as flexible, the force field was set as OPLS-2005, and all other parameters were set to default. The binding site was used to generate the grid for IFD calculation. The maximum 5 poses with receptor and ligand van der Waals radii of 0.7 and 0.5, respectively were considered. Residues within 15 Å of the crystallographic ligands at the active site were refined, followed by side-chain optimization. Structures with prime energy of more than 30 kcal mol−1 are eliminated. A re-docking experiment to validate the used docking protocol was done, and the RMSD value of 0.79 indicates that the docking experiment is reliable.2.6 Molecular dynamics simulation
The molecular dynamics (MD) simulation of this study was performed by using the Desmond v5.3 module (https://www.schrodinger.com/products/desmond) implemented in the Maestro interface (from Schrödinger 2018-4 suite). The appropriate pose for the MD simulation procedure of the compounds was obtained by the IFD method. To build the system for MD simulation, the protein–ligand complexes were solvated with SPC explicit water molecules and placed in the center of an orthorhombic box of appropriate size in the periodic boundary condition. Sufficient counter-ions and a 0.15 M solution of NaCl were also utilized to neutralize the system and to simulate the real cellular ionic concentrations, respectively. The MD protocol involved minimization, pre-production, and production MD simulation steps. In the minimization procedure, the system was allowed to relax for 2500 steps by the steepest descent approach. Then the system's temperature was raised from 0 to 300 K with a small force constant on the enzyme to restrict any drastic changes. MD simulations were performed via the NPT (constant number of atoms, constant pressure i.e. 1.01325 bar, and constant temperature i.e. 300 K) ensemble. The Nose–Hoover chain method was used as the default thermostat with a 1.0 ps interval and Martyna–Tobias–Klein as the default barostat with a 2.0 ps interval by applying the isotropic coupling style. Long-range electrostatic forces were calculated using the particle-mesh-based Ewald approach with the cut-off radius for coulombic forces set to 9.0 Å. Finally, the system was subjected to produce MD simulations for 100 ns for the protein–ligand complex. During the simulation, every 1000 ps of the actual frame was stored. The systems' dynamic behavior and structural changes were analyzed by calculating the root mean square deviation (RMSD) and RMSF. Subsequently, the energy-minimized structure calculated from the equilibrated trajectory system was evaluated to investigate each ligand–protein complex interaction.2.7 Toxicity assay on SH-SY5Y
SH-SY5Y cells were maintained in Dulbecco's modified Eagle medium with Ham's F12 medium (DMEM/F12) containing 15% fetal bovine serum, 100 units per ml penicillin and 100 μg ml−1 streptomycin. Cells were seeded into flasks containing the supplemented medium and maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Cell viability, virtually the mitochondrial activity of living cells, was measured by quantitative colorimetric assay with MTT, as described previously. The MTT reagent, at a final concentration of 0.5 mg ml−1, was added to each well at the end of the incubation period, and the plate was placed in a humidified incubator for an additional two h periods. Metabolically active cells convert the yellow MTT tetrazolium compound to a purple formazan product. Then, the insoluble formazan was dissolved with dimethylsulfoxide; colorimetric determination of MTT reduction was measured at 540 nm. Control cell treated media were taken as 100% viability.33. Results and discussion
3.1 Design
Structural features and key binding sites of AChE and BChE were clarified by X-ray crystallographic studies with known inhibitors. It was demonstrated that both enzymes consist of the peripheral anionic site (PAS), and catalytic active site (CAS) which contain anionic subsite (AS), oxyanion hole, and acyl binding pocket. The PAS is located at the gorge's entrance, and the CAS at the bottom of the gorge. It was confirmed that interactions with the CAS and PAS induce significant changes in the activity of the enzymes.61 Donepezil (Fig. 2) as a ChE inhibitor mimics the binding mode of the ACh neurotransmitter so that its inden-1-one moiety occupies the PAS pocket and benzyl piperidine occupies the CAS pocket. Regarding the structural future of donepezil, different ChE inhibitors were developed. For years, the indole and indolinone scaffolds have proven invaluable to medicinal chemistry researchers in developing various ChE inhibitors. These scaffolds have been particularly effective in targeting the PAS pocket, facilitating the design of potent inhibitors.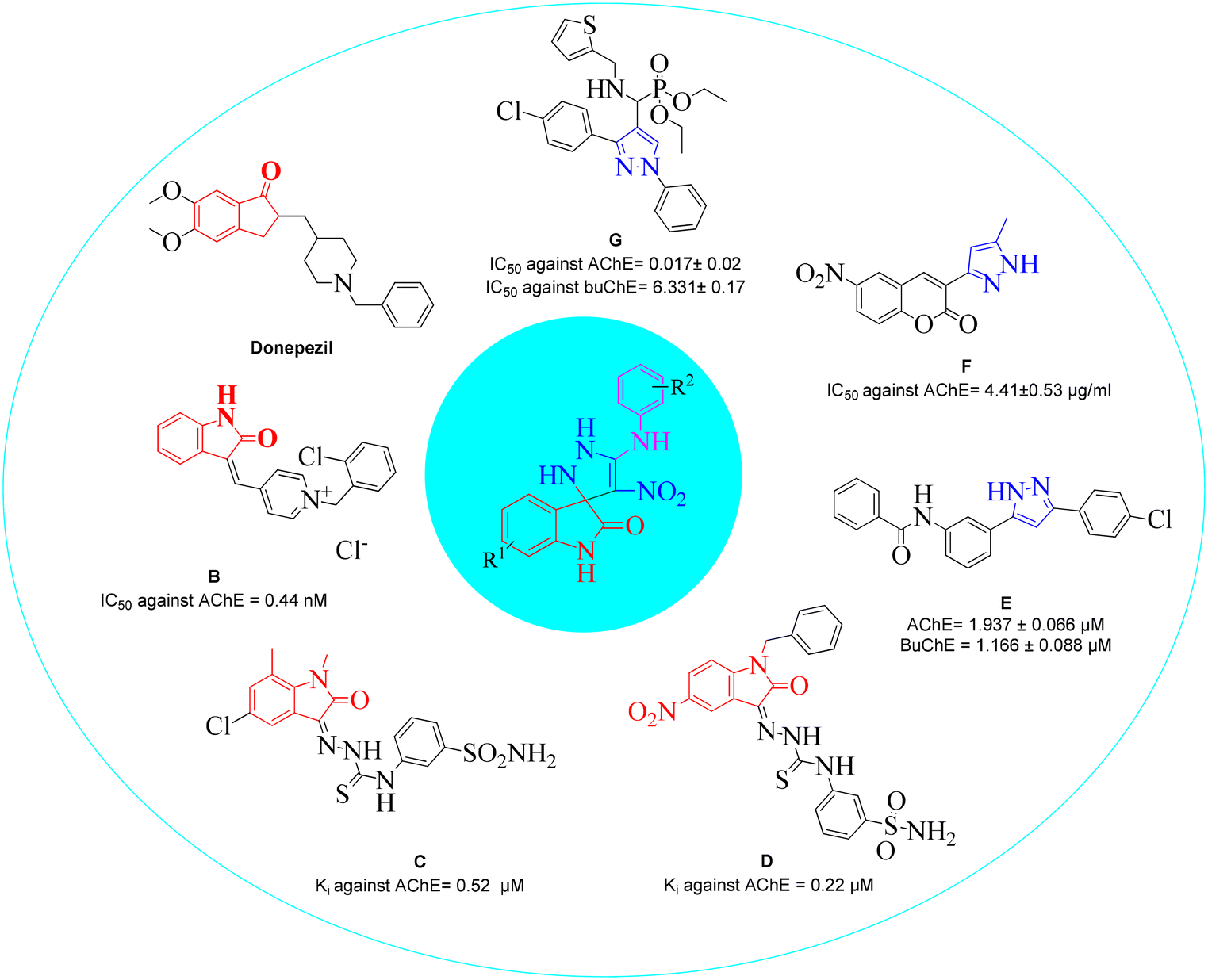 | ||
| Fig. 2 The design strategy of spiroindolinone-pyrazole derivatives. | ||
Indolinone-bearing pyridine derivatives were designed (Fig. 2, compound B) as dual-binding inhibitors of AChE, and the most potent derivative was 32-fold more potent than donepezil as a reference drug.62 A library of indolinone-based hydrazinecarbothioamide derivatives (Fig. 2, compound C) was synthesized and tested for their anti-ChE activities in the past year. The most active analog exhibited Ki = 0.52 ± 0.11 μM with a selectivity of SI = 37.69 for AChE over BuChE.63 In another study, a series of benzyl-indolinones (Fig. 2, compound D) were designed using molecular modeling. In vitro evaluations exhibited significant inhibition of derivatives, in which the most potent analog exhibited more than 2-fold potency compared with donepezil.64 The structure–activity relationship (SAR) also demonstrated that substitution at the different positions of the indolidinone ring significantly affects the potency.
It has been reported in the literature that various compounds carrying the pyrazole skeleton have ChE inhibitory effects.65 Gutti et al. presented a linear pyrazole derivative as a ChE inhibitor (compound E). The most potent compound demonstrated significant inhibition of ChE activity and effectively reduced metal-induced aggregation of Aβ1–42 at a concentration of 20 μM. Furthermore, the compound exhibited a high cell viability of nearly 90%.66 A set of hybrid derivatives bearing a pyrazole and coumarin scaffold and the most potent entry, compound F, showed high potential AChE inhibitory activities with IC50 values of 4.41 ± 0.53 μg ml−1.67 Another study in 2020 reported that compound G was the most potent mixed-type inhibitor acting as a dual site inhibitor by occupying the CAS and PAS, which was further supported by enzyme kinetic study. The compounds did not show any cytotoxicity against HEK-293 cells.68 In a review, the limited SAR of pyrazole was developed, and it was exhibited that the smaller groups attached to the pyrazole ring promote ChE inhibition compared to bulky ones. Also, electron-withdrawing atoms increase activity.69
In light of this information, we aimed to develop and synthesize an efficient and convenient one-pot reaction of novel spiroindolinone-pyrazole derivatives in the present study. To investigate the structure–activity relationships (SARs) of ChE inhibition, a substituted phenyl ring (small ring) was coupled with the pyrazole group. Additionally, the designed scaffold featured a nitro moiety known for its strong electron-withdrawing properties, which could enhance interactions with the enzyme's binding site. This incorporation provided an added value to the scaffold, potentially contributing to increased potency in ChE inhibition. All the synthesized compounds were evaluated for their ChE activities, and the kinetic study of the most potent derivative was executed. In addition, docking studies and molecular dynamic simulations of the most potent compound were performed. The present work aimed to identify agents that may lead to further development as potential anti-Alzheimer drugs.
3.2 Chemistry
During our laboratory efforts on developing multicomponent reactions for synthesizing biologically important heterocyclic compounds, new spiro pyrazoline derivatives were synthesized in the present work. Initially, BMTNE 1 (1 mmol) and arylamine 2 (1 mmol) were reacted in EtOH (5 mL) at reflux to form N-aryl-1-(methylthio)-2-nitroethenamine 3, after 12 h, NH2NH2 (80% aq.) (1.2 mmol) and isatin derivatives 4 (1 mmol) were added to obtain the desired product 5. To optimize the reaction conditions for the synthesis of 4′-nitro-5′-(o-tolylamino)-1′,2′-dihydrospiro[indoline-3,3′-pyrazol]-2-one 5a, initially intermediate 2-methyl-N-(1-(methylthio)-2-nitrovinyl)aniline 3a was prepared according to the reported procedure;70 then, NH2NH2 (80% aq.) and isatin 4a were used as substrates in the model reaction to determine the optimal reaction conditions. Different solvents and reaction temperatures were carefully evaluated. The results are shown in Table 1. The product yield was low when the reaction was carried out in water (entry 1). Ethanol at reflux condition provided higher yields than other organic solvents (compare entry 5 with entries 2–4). It should be mentioned that the reaction was carried out in ethanol at different temperatures (room temperature, 40 °C, 60 °C Table 1 entries 6–8). These studies showed that refluxing ethanol provided the highest yield. Based on the optimal reaction conditions, we studied two-step one-pot sequential synthesis of 5a from BMTNE 1, 2-methylaniline 2a, NH2NH2 (80% aq.), and isatin 4a, and we were able to get the same result for the final product (93%). At first, we added BMTNE 1 (1 mmol) and 2-methylaniline 2a (1 mmol) in refluxing ethanol for 12 h then, the other components were added to form the final product 5a.|
|
|||
|---|---|---|---|
| Entry | Solvent | T (°C) | Yieldb (%) |
| a Reaction conditions: 2-methyl-N-(1-(methylthio)-2-nitrovinyl)aniline (1 mmol), NH2NH2 (80% aq.) (1.2 mmol), isatin (1 mmol), solvent 5 mL. b Isolated yield. | |||
| 1 | H2O | 80 | 25 |
| 2 | CH3CN | Reflux | — |
| 3 | THF | Reflux | 30 |
| 4 | Toluene | 80 | 45 |
| 5 | EtOH | Reflux | 93 |
| 6 | EtOH | r.t | 30 |
| 7 | EtOH | 40 | 55 |
| 8 | EtOH | 60 | 80 |
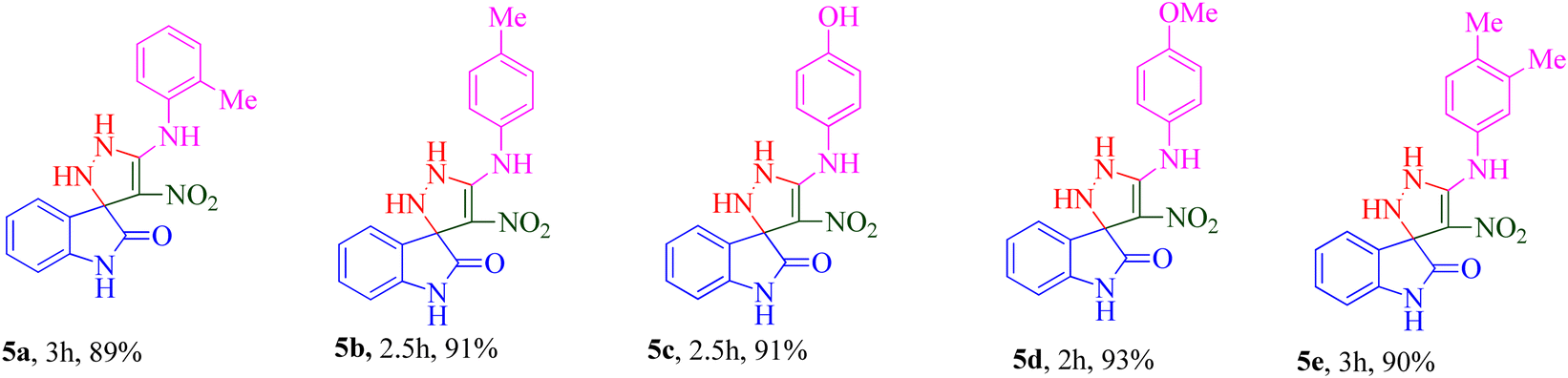
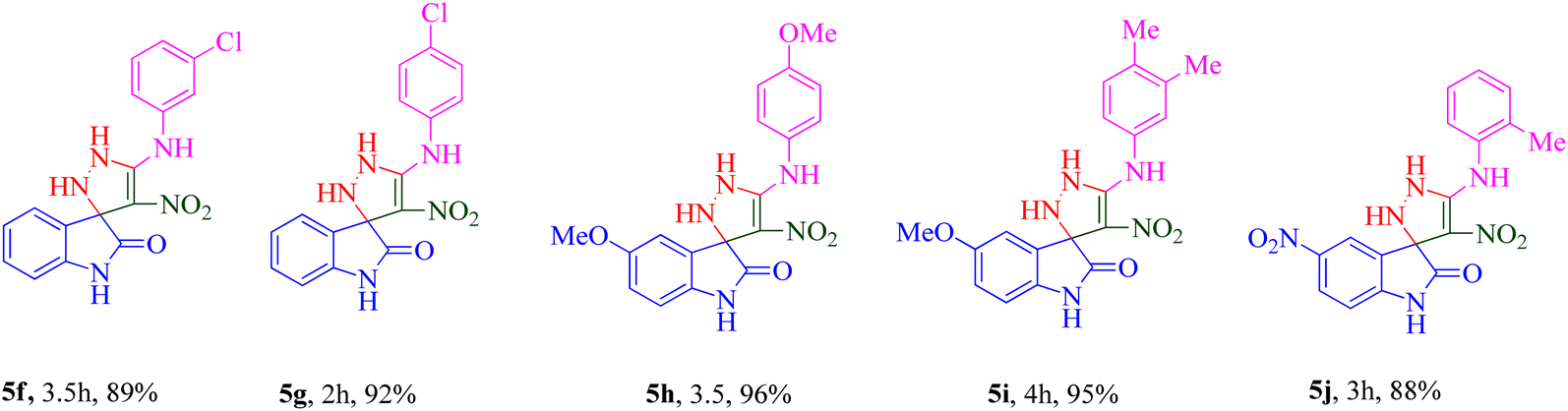
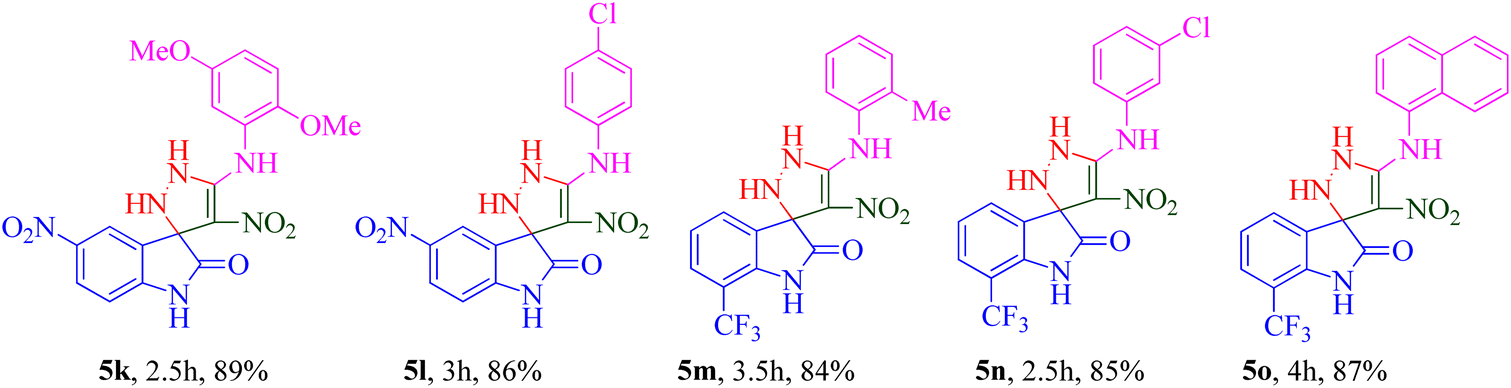
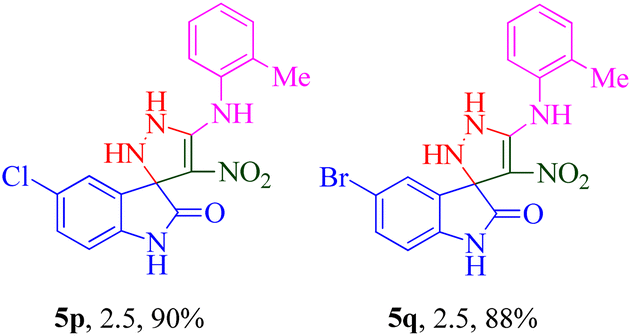
The scope and efficiency of the reaction were discovered using a range of structurally diverse aryl amine and isatin derivatives to form the corresponding products 5a–q.
With the optimal reaction conditions, the substrate scope of N-aryl-1-(methylthio)-2-nitroethenamine 3 and isatin derivatives 4 was investigated. As shown in Table 1, the results showed that various substituent groups of N-aryl-1-(methylthio)-2-nitroethenamine 3 usually had a slight effect on the yields, and we could not ascertain the effect of any specific substituent. All the N-aryl-1-(methylthio)-2-nitroethenamine substrates can be used in the reaction, producing compound 5 in good to excellent yields. The effect of substituted groups (R1) of isatins was also evaluated. The results revealed that different isatin substrates 4 could be transformed into the desired product 5 (Table 1). The substrates with electron-donating groups (R1 = OMe) or electron-withdrawing groups (R1 = Cl, Br, NO2, CF3) and isatin were well-tolerated during this transformation. The substrates with electron-donating groups usually provided the target compounds in higher yields than isatin or substrates with electron-withdrawing groups (Table 2).
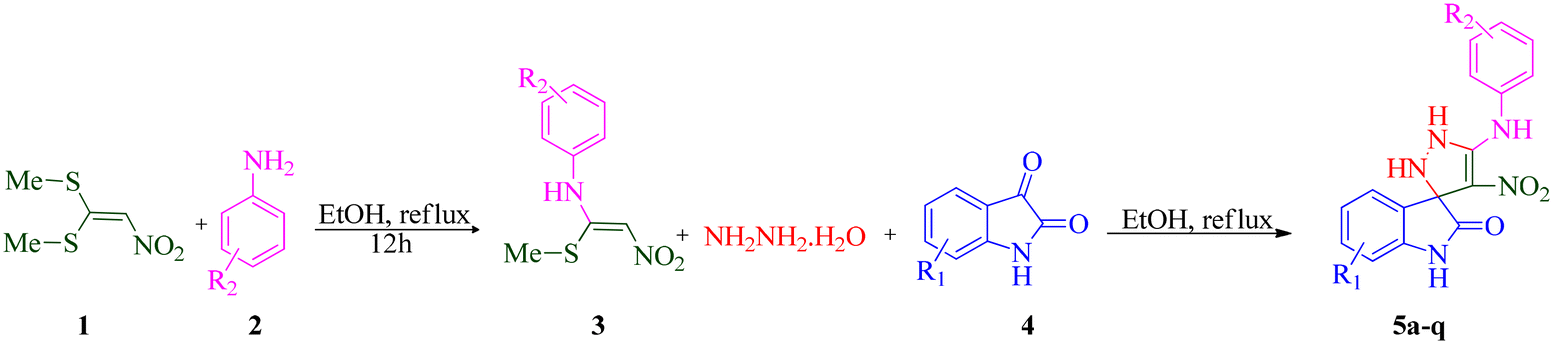
According to a literature survey,71 a plausible mechanism for forming product 5 is shown in Scheme 1. The first synthetic pathway to produce 5 is initiated with intermediate 6 formed from the nucleophilic substitution of the NH2 group of arylamine and the NH2 group of the hydrazine molecule with two methylsulfanyl groups of BMTNE 1. Then, the one-component 1,1-enediamines 6 and the isatin derivatives 4 undergo an aza-ene reaction to give intermediate 7, resulting in the formation of product 5.
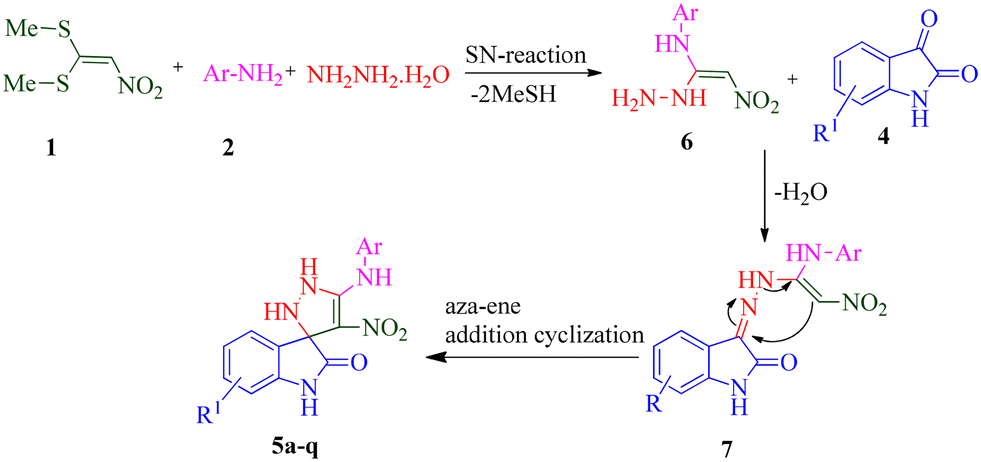 | ||
| Scheme 1 Proposed mechanism for the formation of compound 5 | ||
3.3 AChE inhibitory activity of 5a–q derivatives
All the synthesized compounds were evaluated for their inhibitory activity against AChE. The results are summarized in Table 3.|
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | AChE mean ± S.E | BChE | ||
| % inhibition at 50 μM | IC50 (μM) | % inhibition at 50 μM | IC50 (μM) | |||
| a Data presented here are mean ± S.E. b Positive control. | ||||||
| 5a | H | 2-CH3 | 35.38 ± 4.61 | — | 60.07 ± 4.62 | 19.95 ± 2.23 |
| 5b | H | 4-CH3 | 30.48 ± 5.82 | — | 56.49 ± 2.53 | 25.08 ± 1.59 |
| 5c | H | 4-OH | 40.96 ± 4.22 | — | 12.91 ± 1.79 | — |
| 5d | H | 4-OCH3 | 47.77 ± 3.29 | — | 53.35 ± 8.21 | 34.67 ± 3.68 |
| 5e | H | 3,4-diCH3 | 29.44 ± 3.89 | — | 31.08 ± 1.08 | — |
| 5f | H | 3-Cl | 75.47 ± 3.68 | 6.02 ± 2.59 | 13.94 ± 3.68 | — |
| 5g | H | 4-Cl | 13.95 ± 2.18 | — | 14.90 ± 5.73 | — |
| 5h | 5-OCH3 | 4-OCH3 | 29.74 ± 1.19 | — | Not active | |
| 5i | 5-OCH3 | 3,4-diCH3 | Not active | — | 52.29 ± 5.76 | 47.30 ± 5.75 |
| 5j | 5-NO2 | 2-CH3 | 71.57 ± 8.94 | 15.48 ± 2.69 | 38.94 ± 7.36 | — |
| 5k | 5-NO2 | 2,5-diOCH3 | 55.15 ± 6.84 | 42.55 ± 4.11 | 25.13 ± 1.58 | — |
| 5l | 5-NO2 | 4-Cl | 65.89 ± 4.73 | 37.15 ± 3.52 | 21.84 ± 2.10 | — |
| 5m | 7-CF3 | 2-CH3 | 63.13 ± 4.32 | 16.98 ± 2.43 | 37.53 ± 7.53 | |
| 5n | 7-CF3 | 3-Cl | 20.63 ± 1.57 | — | 18.8 ± 4.58 | |
| 5o | 7-CF3 | Naphtyl | 39.07 ± 4.73 | — | Not active | — |
| 5p | 5-Cl | 2-CH3 | 21.92 ± 1.71 | — | 73.76 ± 1.95 | 9.13 ± 1.82 |
| 5q | 5-Br | 2-CH3 | 51.76 ± 2.21 | 41.68 ± 6.93 | 34.00 ± 6.37 | |
| Donepezilb | 0.079 ± 0.05 | 10.6 ± 2.1 | ||||
Among 5a–e bearing H at the R1 position and an electron-donating group at R2, it was understood that 5d (R2 = 4-OCH3) recorded 47.77% inhibition at 50 μM and bioestric replacement of methoxy with hydroxyl resulted in 5c with a slight reduction in the activity (inhibition at 50 μM = 40.96%). Ortho (5a) and para methyl (5b) substitution, as well as 3,4-diCH3 (5e) substitution, did not significantly improve the activity compared with 5d. Contrarily, among analogs featuring halogen substitutions at the R2 position, the compound bearing a 3-Cl substitution at R2 (5f) demonstrated the highest potency, with an IC50 value of 6.02 ± 2.59 μM. However, when the substitution was shifted from the meta to the para position (5g), the potency considerably decreased to 13.95 ± 2.18% inhibition at 50 μM, indicating a significant deterioration in activity.
As can be seen in 5h and 5i, the introduction of 5-OCH3 as an electron-donating group at the R1 position, regardless of the type of substitution at R2, brought a reduction in the inhibitory activities compared to unsubstituted derivatives (5a–g; R1 = H).
A notable enhancement in potency was observed in compounds 5j, 5k, and 5l upon replacing the OCH3 group at the R1 position with the strong electron-withdrawing group NO2, possessing both resonance and inductive effects. Comparatively, these substitutions exhibited higher potencies than others at the same position. Additionally, the trend observed for R2 substitutions was as follows: 2-CH3 (5j, IC50 = 15.48 ± 2.69 μM) > 4-Cl (5l, IC50 = 37.15 ± 3.52 μM) > 2,5-diOCH3 (5k, IC50 = 42.55 ± 4.11 μM). This indicates that the presence of a methyl group at the 2-position (R2) resulted in the highest potency, followed by a chlorine substitution at the 4-position, while the presence of two methoxy groups at the 2 and 5 positions displayed slightly lower potency.
The trifluoromethyl (–CF3) group, one of the most powerful electron-withdrawing groups, was substituted at the 7 position of the indolinone ring (5m–o). Among –CF3 containing derivatives, 2-methyl substitution recorded better potency (5m; IC50 = 16.98 ± 2.43 μM) in comparison with chlorine (5n) as electron-withdrawing and naphthyl (5o) as bulk moieties.
It was observed that the presence of a 2-methyl group at the R2 position exhibited better potency when combined with electron-donating groups such as NO2 and CF3 at the R1 position. Building upon this observation, the 2-methyl moiety was retained, and chlorine (5p) and bromine (5q) halogen groups were introduced at the R1 position. Interestingly, the bulkier bromine moiety was more favorable than the chlorine group. This suggests that the size and steric properties of the substituents play a crucial role in the overall activity and interaction of the compound with the target site.
3.4 BChE inhibitory activity of 5a–q derivatives
Next, all derivatives 5a–q were screened against BChE, and results are presented in Table 3. Assessments of 5a–g (R1 = H) showed that 5a bearing a 2-CH3 moiety was the most potent derivative in R1 = H derivatives. Replacement of the position from ortho to para lightly reduced the activity in 5b with IC50 = 25.08 ± 1.59 μM. Also, multi-methyl substitutions (5e) deteriorated the potency to 31.08 ± 1.08% inhibition at 50 μM. Also, replacing 4-CH3 with 4-OH (5c) and 4-OCH3 (5d) did not improve the activity vs.5a. On the other hand, halogen substitutions significantly reduced the activity. In this group, it was understood that the electron-donating group at the R2 position is more favorable.The results obtained from the investigation of compound 5h (R2: 4-OCH3) and compound 5i (R2: 3,4-diCH3), both possessing a 5-OCH3 substituent at R1, revealed that the introduction of the 3,4-diCH3 substitution resulted in a more potent BChE inhibitor with an IC50 value of 47.30 ± 5.75 μM. These findings suggest that increased lipophilicity, conferred by the 3,4-diCH3 substitution, improves BChE inhibition.
Relative to the activity of 5j–l with a 5-nitro substituent at the R1 position, similar to R1 = H derivatives, the introduction of 2-CH3 substitution increased the activity in comparison with 2,5-diOCH3 (5k) and 4-Cl (5l).
Among 7-CF3 containing derivatives (5m–o), similar results were recorded in which the 2-CH3 moiety was identified as a potent analog followed by 5n (R2: 3-Cl; 18.8% inhibition at 50 μM). Also, 5o was completely inactive.
Moreover, 5-Cl (5p) and 5-Br (5q) groups as halogen-substituted derivatives did function positively in the anti-BChE assay, and the best biological results were seen in 5p (R1: 5-Cl, R2: 2-CH3) with an IC50 value of 9.13 ± 1.82 μM.
3.5 Summary of SARs
In the case of the electron-withdrawing group at R1, introducing a 2-methyl substitution was found to be more favorable in most cases. However, when R1 was not substituted (R1 = H), the 3-Cl moiety was identified as the most potent analog among the tested compounds. These results suggest that the specific substitution pattern and electronic properties of the group attached to R1 play a significant role in determining the inhibitory potency of the compounds. The anti-BChE result reflects the good impact of the 2-methyl substitution to get the optimum BChE activity. 5f showed a remarkable improvement in AChE inhibition (6.02 ± 2.59 μM) with no BChE inhibitory activity. 5p exhibited selective and significant BChE inhibitory potency (IC50 = 9.13 ± 1.82 μM). Also, the presence of a nitro moiety increases the selectivity against AChE.3.6 Enzyme kinetic studies of 5f against AChE
According to Fig. 3a, the Lineweaver–Burk plot showed that the Km gradually increased and Vmax changed with increasing inhibitor concentration, indicating a mix-type inhibition. The results showed that 5f might bind to the free enzyme or the enzyme that is already bound to the substrate. Furthermore, the slope plot versus different inhibitor concentrations gave an estimate of the inhibition constant, Ki of 5.59 μM (Fig. 3b).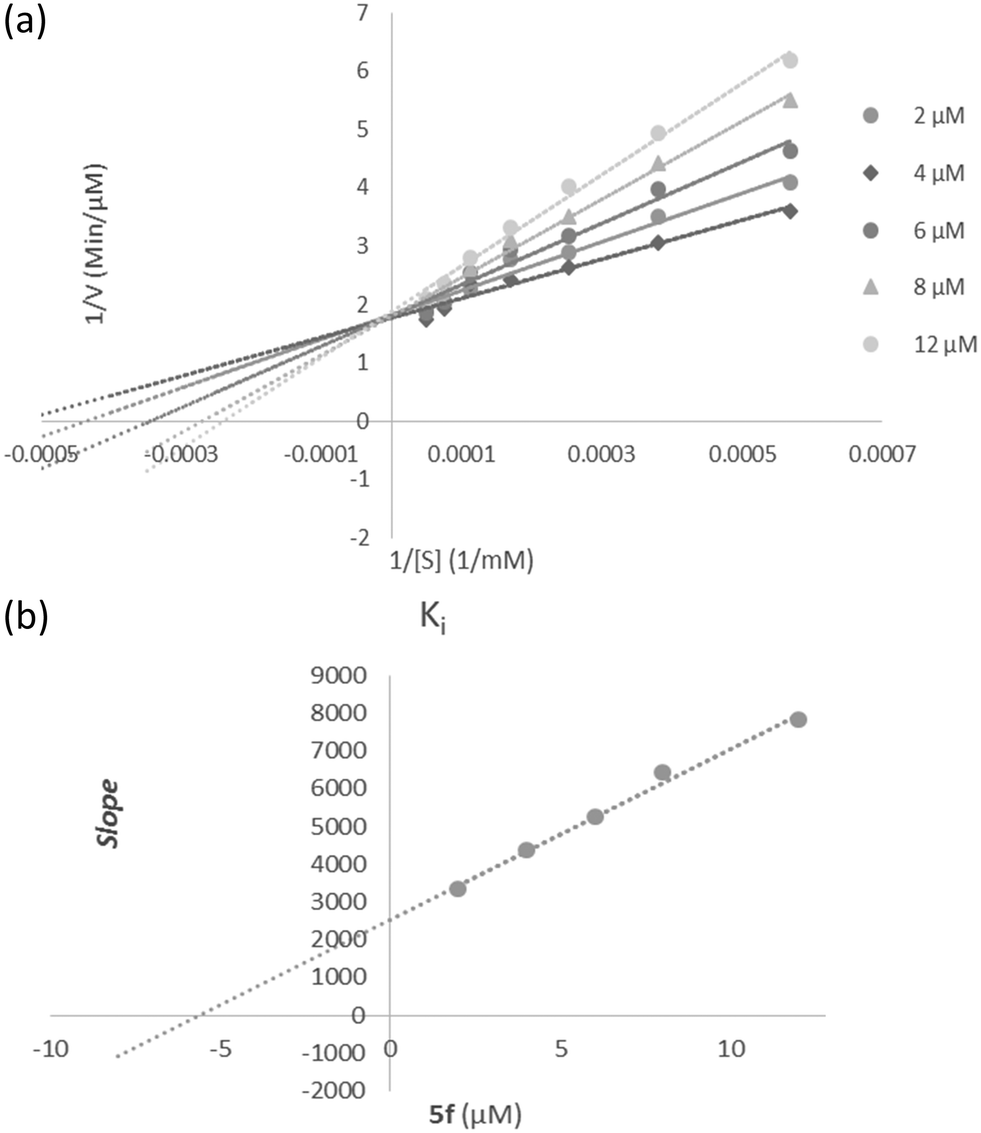 | ||
| Fig. 3 Kinetics of AChE inhibition by 5f. (a) The Lineweaver–Burk plot in the absence and presence of different concentrations of sample 5f; (b) the secondary plot between slope and various concentrations of 5f. | ||
3.7 Molecular docking
AChE consists of two main pockets, a PAS near the entrance of the gorge and a CAS at the bottom of the gorge, approximately 20 Å deep. The PAS consists of amino acids of tryptophan 286, tyrosine 337, and phenylalanine 338, which guide molecules to the CAS pocket. CAS comprises the catalytic triad with glutamic acid 334, serine 203, and histidine 447 as the main residues and the anionic subsite of tryptophan 72. Also, there are two residues at the bottleneck, tyrosine 121 and phenylalanine 330, of the enzyme's binding site that guide molecules toward the active site.First, the validity and reliability of molecular docking were assessed by redocking the AChE and BChE natural ligands inside the binding site of this relevant enzyme. RMSD less than 2 Å was recorded as the ideal value.
In the pursuit of identifying the most potent AChE inhibitor, compound 5f has emerged as the primary candidate, warranting further investigation. It is worth noting that 5f exists as an enantiomeric mixture, necessitating a comprehensive analysis of both its R and S isomers within the AChE active site. Molecular docking assessments were conducted on both the S and R enantiomers of compound 5f to elucidate their respective modes of action. This research aimed to uncover these enantiomers' binding orientations and poses within the AChE binding pocket. The results of the molecular docking calculations yielded significant insights. The S enantiomer exhibited a binding energy of −9.87 kcal mol−1, while the R enantiomer displayed a binding energy of −7.63 kcal mol−1. These findings were derived from a detailed examination of the binding interactions and their associated energetics within the AChE binding site.
The interactions of S and R isomers of 5f and in the AChE binding pocket are presented in Fig. 4 and 5, respectively.
 | ||
| Fig. 4 2D interaction diagram of the S isomer of 5f in the AChE binding site. | ||
 | ||
| Fig. 5 2D interaction diagram of the R isomer of 5f in the BChE binding site. | ||
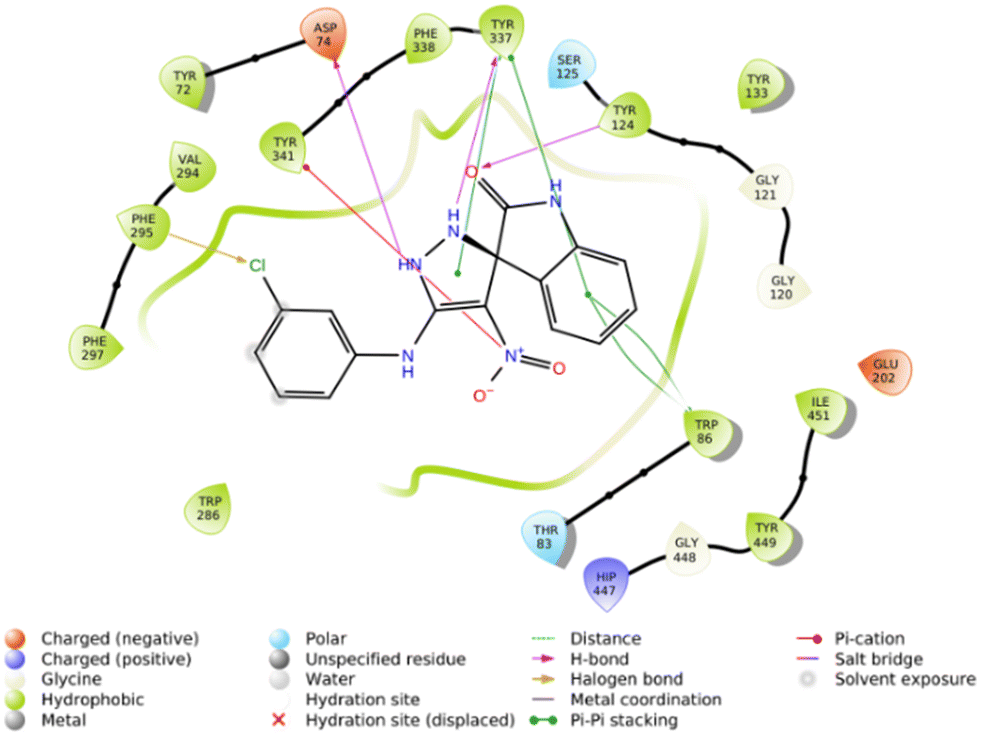
The molecular docking study showed that the S enantiomer of 5f was appropriately oriented in the active site of AChE (Fig. 4). The indolineone moiety participated in H-bound interaction with Tyr124 (2.94 Å) and pi–pi stacking interaction with Trp86 (3.78 Å). Pyrazole recorded two H-bound interactions with Asp74 (2.60 Å) and Tyr337 (2.28 Å) and one pi–pi stacking interaction with Tyr337 (4.20 Å). The nitro moiety of 5f also participated in pi–cation interaction with Tyr341 (6.10 Å), and the 3-Cl substituted group displayed a halogen bond with Phe295 (3.51 Å).
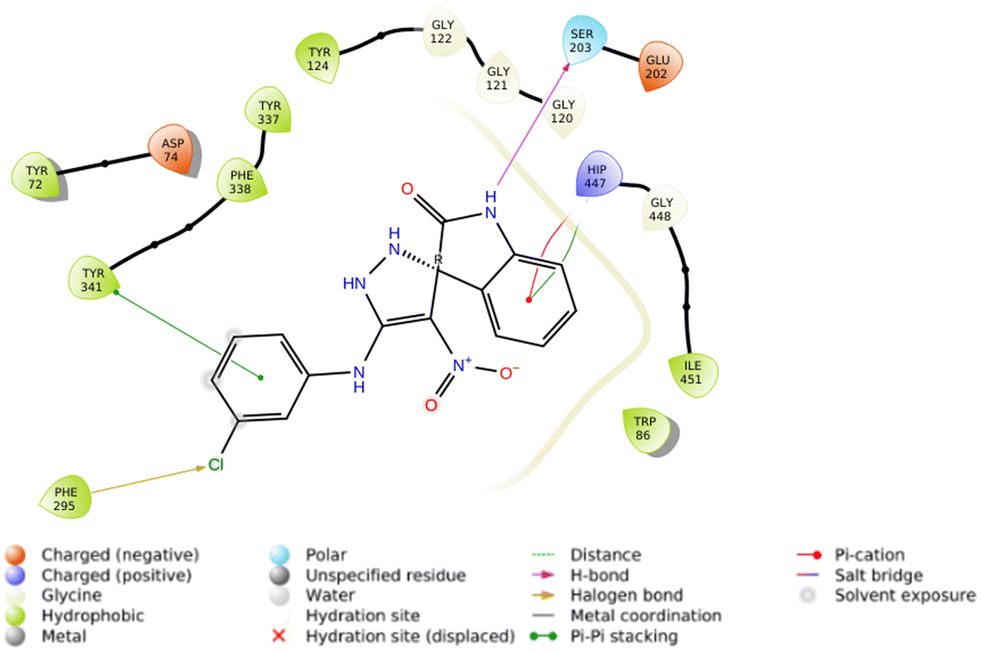
On the other hand, the R isomer of 5f against the AChE binding site (Fig. 5) displayed notable interactions with key residues, including Phe295 (3.46 Å, halogen interaction), Ser203 (2.97 Å, hydrogen bond), Tyr341 (3.76 Å, pi–pi stacking), and His447 (4.10 Å, pi–cation) and His447 (4.36 Å, pi–pi stacking).
Furthermore, the molecular docking study results involving all derivatives against both AChE and BChE, along with details regarding the types of interactions and corresponding distances, were comprehensively presented in the ESI within Tables S1 and S2.†
3.8 Molecular dynamics simulation
Molecular dynamics simulations were conducted for both the R and S enantiomers of 5f to evaluate protein flexibility.To study the stability of the system's dynamic behavior, the best IFD pose of R and S enantiomers of compound 5f and apo enzyme was implemented to predict the motion of complex systems at an atomistic level. Root mean square deviation (RMSD) values indicate the conformational stability and perturbations of the system, and RMSD value changes around 1–3 Å are optimum values for small globular protein and the complex has reached equilibrium.21
The RMSD plot for AChE (in blue) displayed a notable initial increase followed by fluctuations around 1.5 Å. Meanwhile, the AChE-R isomer of complex 5f (in green) exhibited a slightly lower RMSD value of 1.2 Å. It is worth noting that the R isomer displayed comparatively lower stability. In contrast, the AChE-S isomer of complex 5f (in orange) displayed remarkable stability throughout the simulation, with an RMSD value of 0.7 Å maintained over the entire 100 ns duration.
These observations suggest that the simulation time employed was sufficient to attain an equilibrium structure during the simulations. Additionally, it appears that the S isomer exhibited greater stability compared to the R isomer (Fig. 6).
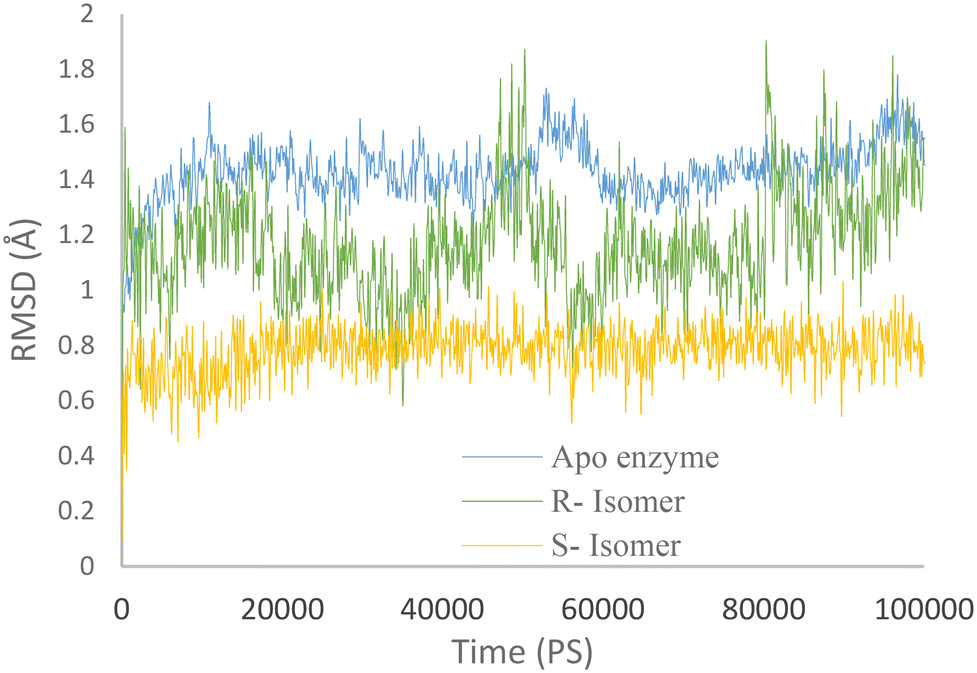 | ||
| Fig. 6 RMSD values of the AChE-S isomer of complex 5f (orange), the AChE-R isomer of complex 5f (green), and AChE (blue) throughout the simulation time. | ||
RMSF values illustrate the protein backbone flexibility throughout the simulation time. Notably, the RMSF values indicate that the S isomer of compound 5f plays a substantial role in stabilizing crucial regions, including the PAS pocket (indicated by the blue dashed line), the catalytic triad (highlighted by the green dashed line), and the anionic subsite (represented by the red dashed line), as illustrated in Fig. 7.
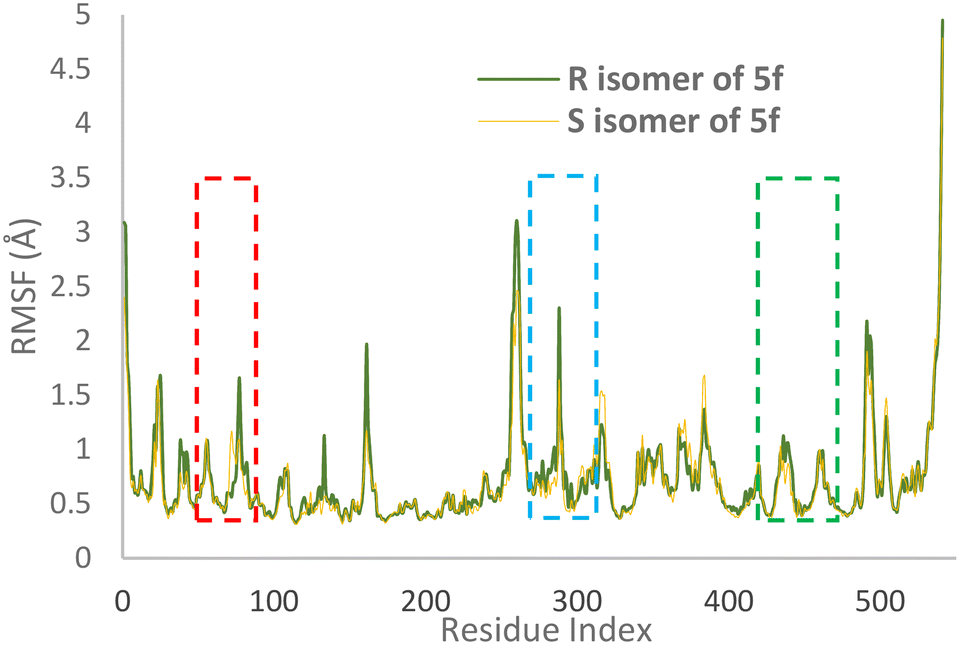 | ||
| Fig. 7 RMSF values of the AChE-S isomer of complex 5f (orange) and the AChE-R isomer of complex 5f (green) in angstroms throughout the simulation. | ||
Furthermore, the 2D interaction diagram of the S isomer of compound 5f in complex with the enzyme is depicted in Fig. 8. These interactions happened for at least 30% of the duration of the MD simulation time. Fig. 8 shows that the carbonyl oxygen of the indolinone moiety participated in two H-bound interactions with Tyr72 (32% of MD simulation time) and Asn87 (63% of MD simulation time) of the anionic subsite. Also, NH of indolinone interacted with Ser125 through hydrogen bonds for about 82% of the MD simulation time. The pyrazole ring demonstrated two H-bound interactions with Tyr337 (95% of the simulation time) and Tyr 341 (74% of the MD simulation time) of the PAS. Also, the 3-chlorophenyl group stabilized through hydrophobic π–π stacking non-bonding interaction by the Tyr341 residue for about 34% of MD simulation time. All these results aligned with the kinetic study confirming mix-type inhibition of the 5f derivative.
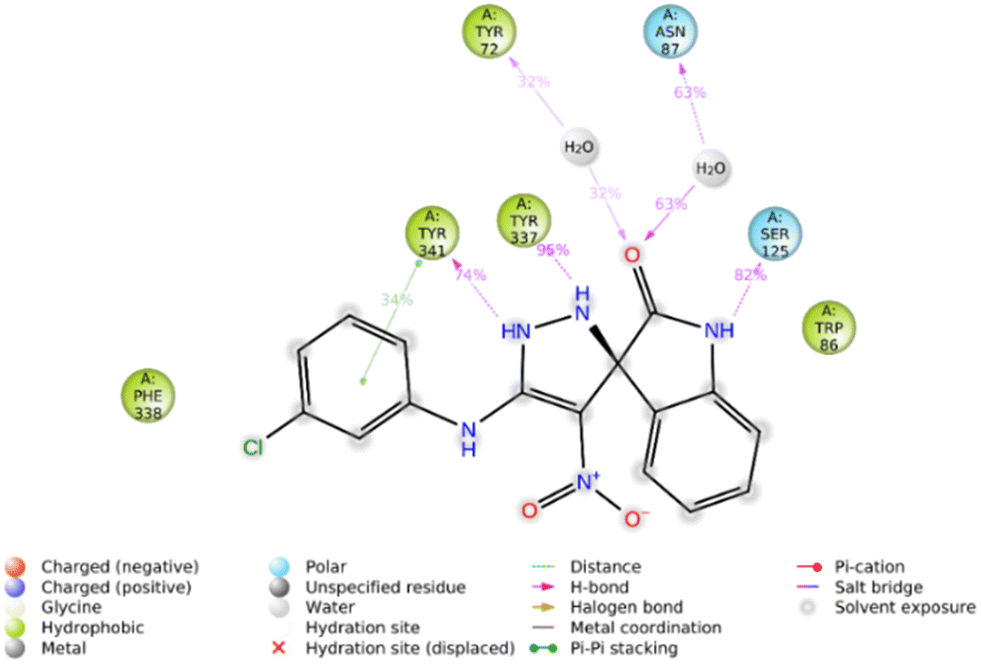 | ||
| Fig. 8 2D interaction diagram of the S isomer of compound 5f within the AChE binding site occurring over 30% of MD simulation time. | ||
Furthermore, the 2D interaction diagram of the R isomer of compound 5f in complex with the enzyme is depicted in Fig. 9. Briefly, indolinone demonstrated pi–cation interaction for around 81% of MD run plus pi–pi stacking interaction (48% of MD simulation time) with His447. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of indolinone also recorded H-bound interaction with Ser203 mediated with water (35% of MD duration time). The nitro moiety also participates in another H-bound interaction with Tyr133 (35% of MD duration time). Another pi–pi stacking interaction was also seen between imidazole and Tyr337 (31% of MD duration time).
O moiety of indolinone also recorded H-bound interaction with Ser203 mediated with water (35% of MD duration time). The nitro moiety also participates in another H-bound interaction with Tyr133 (35% of MD duration time). Another pi–pi stacking interaction was also seen between imidazole and Tyr337 (31% of MD duration time).
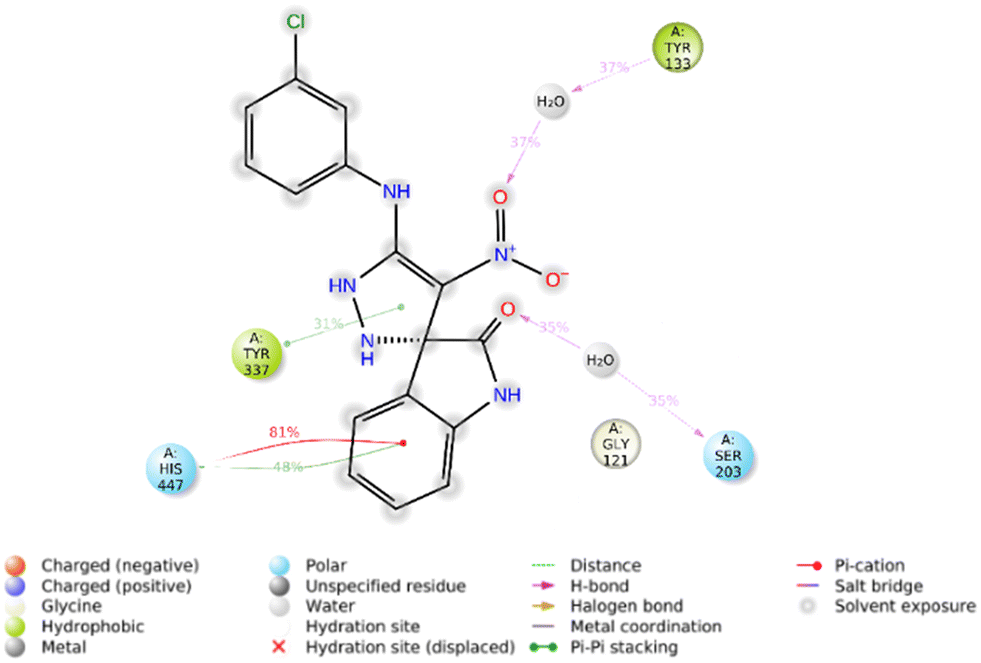 | ||
| Fig. 9 2D interaction diagram of the R isomer of compound 5f within the AChE binding site occurring over 30% of MD simulation time. | ||
3.9 Effect of 5f on SH-SY5Y cell viability
The SH-SY5Y neuroblastoma cell line is widely used as an in vitro neuronal model for evaluating neurodegenerative disorders. As a result, the toxicity of 5f as the most potent derivative was evaluated against the SH-SY5Y cell line, which was examined. As shown in Fig. 10, the compound exhibited 64.81 ± 0.11% viability at 50 μM with no significant toxicity at the lower dose.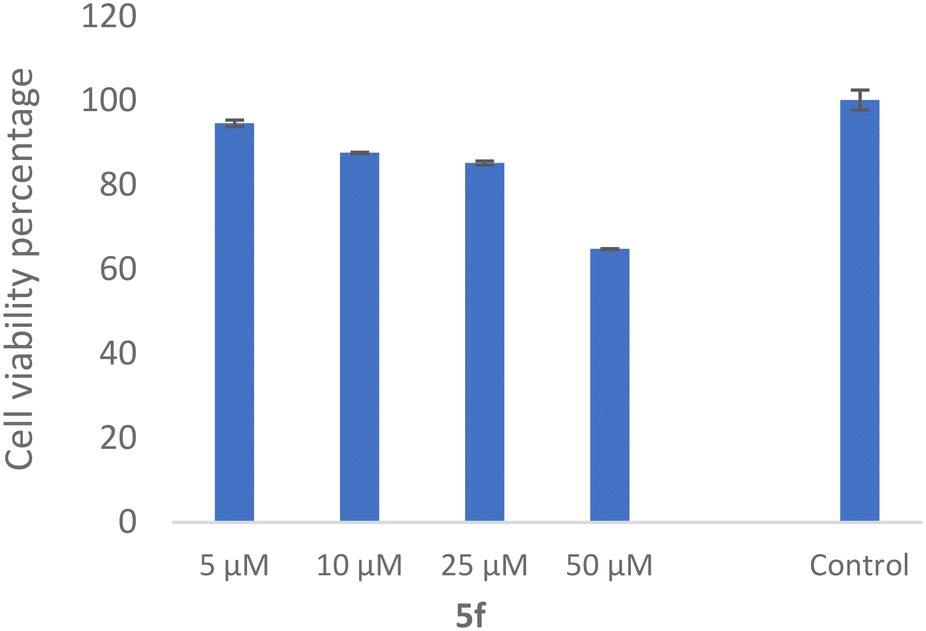 | ||
| Fig. 10 Cell viability of 5f on the SHSY-5Y cell line. | ||
3.10 ADMET properties and in silico toxicity
ADMET encompasses the assessment of absorption, distribution, metabolism, excretion, and toxicity of pharmaceutical compounds within a biological system. These properties are crucial for drug developers in evaluating the safety and effectiveness of drugs during both the preclinical and clinical development stages. Table 4 demonstrates drug-likeness prediction for 5a–q, and Table 5 presents the ADMET predictions for all the compounds, calculated using the pkCSM and SwissADME online servers. The estimated human intestinal absorption (HIA) for the selected compounds indicates a high likelihood of efficient absorption within the gastrointestinal tract. Furthermore, all the compounds are projected to possess Caco-2 permeability levels suitable for oral administration. Regarding metabolism, it is noteworthy that none of the compounds are predicted to inhibit CYP2D6, which is a favorable characteristic. However, they are expected to act as inhibitors of CYP3A4. The derivatives exhibit acceptable toxicity profiles, making them suitable candidates for further drug design and refinement.| Compound | Molecular weight | LogP |
Rotatable bonds | Acceptors | Donors | Surface area |
|---|---|---|---|---|---|---|
| 5a | 337.339 | 1.80812 | 3 | 6 | 4 | 142.596 |
| 5b | 337.339 | 1.80812 | 3 | 6 | 4 | 142.596 |
| 5c | 339.311 | 1.2053 | 3 | 7 | 5 | 141.025 |
| 5d | 353.338 | 1.5083 | 4 | 7 | 4 | 147.710 |
| 5e | 351.366 | 2.11654 | 3 | 6 | 4 | 148.961 |
| 5f | 357.757 | 2.1531 | 3 | 6 | 4 | 146.534 |
| 5g | 357.757 | 2.1531 | 3 | 6 | 4 | 146.534 |
| 5h | 383.364 | 1.5169 | 5 | 8 | 4 | 159.188 |
| 5i | 381.392 | 2.12514 | 4 | 7 | 4 | 160.439 |
| 5j | 382.336 | 1.71632 | 4 | 8 | 4 | 157.249 |
| 5k | 428.361 | 1.4251 | 6 | 10 | 4 | 173.841 |
| 5l | 402.754 | 2.0613 | 4 | 8 | 4 | 161.187 |
| 5m | 405.336 | 2.82692 | 3 | 6 | 4 | 161.458 |
| 5n | 425.754 | 3.1719 | 3 | 6 | 4 | 165.396 |
| 5o | 441.369 | 3.6717 | 3 | 6 | 4 | 177.774 |
| 5p | 371.784 | 2.46152 | 3 | 6 | 4 | 152.899 |
| 5q | 416.235 | 2.57062 | 3 | 6 | 4 | 156.464 |
| Compd. | Absorptiona | Distributiona | Metabolisma | Excretiona | Toxicitya | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HIA% | Caco2 permeability | VDss (log L kg−1) | BBB permeability | CYP3A4 inhibition | CYP2D6 inhibition | CYP2C9 inhibition | CYP2C19 inhibition | Total clearance | Oral rat acute toxicity (LD50) | Oral rat chronic toxicity (LOAEL) | hERG1 inhibitor | |
| a HIA (human intestinal absorption): >80% is high and <30% is poor; VDss (steady-state volume of distribution): log L kg−1: >0.45 is high and <−0.15 is low. The software could not predict parameters for these compounds (−). | ||||||||||||
| 5a | 81.109 | 0.137 | 0.048 | −0.492 | No | No | No | No | 0.509 | 2.127 | 1.877 | No |
| 5b | 81.109 | 0.137 | 0.048 | −0.492 | No | No | No | No | 0.509 | 2.127 | 1.877 | No |
| 5c | 77.373 | −0.253 | 0.238 | −0.663 | No | No | No | No | 0.371 | 2.308 | 2.411 | No |
| 5d | 81.98 | −0.117 | −0.065 | −0.681 | No | No | No | No | 0.437 | 2.076 | 1.971 | No |
| 5e | 81.578 | 0.2 | 0.074 | −0.504 | No | No | No | No | 0.509 | 2.154 | 1.759 | No |
| 5f | 81.817 | 0.024 | −0.01 | −0.646 | No | No | No | No | −0.375 | 2.112 | 1.924 | No |
| 5g | 81.817 | 0.024 | −0.01 | −0.646 | No | No | No | No | −0.375 | 2.112 | 1.924 | No |
| 5h | 78.877 | −0.057 | −0.235 | −0.677 | No | No | No | No | 0.427 | 2.003 | 1.483 | No |
| 5i | 78.475 | 0.112 | −0.082 | −0.5 | No | No | No | No | 0.458 | 2.082 | 1.241 | No |
| 5j | 78.490 | −0.05 | −0.316 | −0.594 | No | No | No | No | 0.453 | 2.704 | 2.187 | No |
| 5k | 75.561 | −0.175 | −0.495 | −0.981 | No | No | No | No | 0.461 | 2.795 | 2.058 | No |
| 5l | 79.198 | −0.07 | −0.362 | −0.747 | No | No | No | No | −0.417 | 2.737 | 2.233 | No |
| 5m | 83.776 | 0.164 | −0.105 | −0.681 | Yes | No | Yes | Yes | 0.124 | 2.036 | 1.403 | No |
| 5n | 84.484 | 0.052 | −0.161 | −0.835 | Yes | No | Yes | Yes | −0.256 | 2.023 | 1.45 | No |
| 5o | 90.348 | −0.081 | −0.328 | −0.676 | Yes | No | Yes | Yes | 0.142 | 2.294 | 1.329 | No |
| 5p | 82.467 | 0.11 | 0.038 | −0.542 | No | No | No | Yes | −0.477 | 2.066 | 1.715 | No |
| 5q | 82.203 | 0.115 | 0.048 | −0.563 | Yes | No | Yes | Yes | −0.499 | 2.074 | 1.698 | No |
4. Conclusions
In conclusion, we have successfully developed a one-pot sequential four-component reaction for synthesizing spiro pyrazoline derivatives under catalyst-free conditions. This reaction provides a novel, rapid, and efficient route for the preparation of a variety of spiropyrazoline derivatives in good to excellent yields from readily available building blocks of N-aryl-1-(methylthio)-2-nitroethenamine, hydrazine hydrate and isatin derivatives. Next, all the derivatives were tested as possible AChE and BChE inhibitors. Results exhibited the promising potency and selectivity of 5f bearing H at R1 and 3-Cl at R2 with an IC50 value of 6.02 ± 2.59 μM with no significant activity against BChE. On the other hand 5p (R1: 5-Cl, R2: 2-CH3) recorded IC50 = 9.13 ± 1.82 μM against BChE. SAR suggested that the specific substitution pattern and electronic properties of the group attached to R1 and R2 played a significant role in determining the inhibitory potency of the compounds. The kinetic study of 5f demonstrated mix-type inhibition with a Ki of 5.59 μM.Furthermore, based on the MD studies, the S enantiomer of compound 5f depicted noticeable interactions with Tyr72 (H-bound) and Asn87 (H-bound) of the anionic subsite plus Tyr337 (H-bound) and Tyr 341 (H-bound & pi–pi stacking) of the PAS confirming its high potency. Also, 5f was tested against the SHSY-5Y cell line, exhibiting no significant toxicity at 25 μM. It can be understood that this set of compounds can serve as structural outlines to design and expand potential anti-AD agents.
Consents
There are no consents to declare.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the financial support of the Vice-Chancellor for Research of Shiraz University of Medical Sciences (grant number: IR.SUMS.REC.1401.728).References
- A. E. Israilovich and I. S. Oybekovna, Clinical and Neurological Approach to Dementia of the Alzheimer's Type, Central Asian Journal of Medical and Natural Science, 2023, 4(1), 7–11 Search PubMed.
- D. F. Weaver, Druggable targets for the immunopathy of Alzheimer's disease, RSC Med. Chem., 2023, 14(9), 1645–1661 RSC.
- P. Spatz, T. Zimmermann, S. Steinmüller, J. Hofmann, T. Maurice and M. Decker, Novel benzimidazole-based pseudo-irreversible butyrylcholinesterase inhibitors with neuroprotective activity in an Alzheimer's disease mouse model, RSC Med. Chem., 2022, 13(8), 944–954 RSC.
- N. Oliyaei, M. Moosavi-Nasab, N. Tanideh and A. Iraji, Multiple roles of fucoxanthin and astaxanthin against Alzheimer's disease: Their pharmacological potential and therapeutic insights, Brain Res. Bull., 2023, 193, 11–21 CrossRef CAS PubMed.
- Z. R. Chen, J. B. Huang, S. L. Yang and F. F. Hong, Role of Cholinergic Signaling in Alzheimer's Disease, Molecules, 2022, 27(6), 1–23 Search PubMed.
- J. Jasiecki, M. Targońska and B. Wasąg, The Role of Butyrylcholinesterase and Iron in the Regulation of Cholinergic Network and Cognitive Dysfunction in Alzheimer's Disease Pathogenesis, Int. J. Mol. Sci., 2021, 22(4), 2033 CrossRef CAS PubMed.
- A. Nordberg, C. Ballard, R. Bullock, T. Darreh-Shori and M. Somogyi, A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer's disease, Prim. Care Companion CNS Disord., 2013, 15(2), 1–45 Search PubMed.
- H. Pourtaher, A. Hasaninejad, S. Zare, N. Tanideh and A. Iraji, The anti-Alzheimer potential of novel spiroindolin-1,2-diazepine derivatives as targeted cholinesterase inhibitors with modified substituents, Sci. Rep., 2023, 13(1), 11952 CrossRef CAS PubMed.
- H. Yao, G. Uras, P. Zhang, S. Xu, Y. Yin, J. Liu, S. Qin, X. Li, S. Allen and R. Bai, Discovery of Novel Tacrine–Pyrimidone Hybrids as Potent Dual AChE/GSK-3 Inhibitors for the Treatment of Alzheimer's Disease, J. Med. Chem., 2021, 64(11), 7483–7506 CrossRef CAS PubMed.
- C. S. Graebin, F. V. Ribeiro, K. R. Rogério and A. E. Kümmerle, Multicomponent reactions for the synthesis of bioactive compounds: A review, Curr. Org. Synth., 2019, 16(6), 855–899 CrossRef CAS PubMed.
- Q. Guan, L.-L. Zhou and Y.-B. Dong, Construction of Covalent Organic Frameworks via Multicomponent Reactions, J. Am. Chem. Soc., 2023, 145(3), 1475–1496 CrossRef CAS PubMed.
- K. Afratis, J. M. Bateman, B. F. Rahemtulla, O. Hughes, B. C. Milgram, T. A. Mulhern and E. P. A. Talbot, Regioselective Synthesis of Fully Substituted Fused Pyrroles through an Oxidant-Free Multicomponent Reaction, Org. Lett., 2023, 25(3), 461–465 CrossRef CAS PubMed.
- N. Hosseini Nasab, F. Azimian, R. S. Shim, Y. S. Eom, F. H. Shah and S. J. Kim, Synthesis, anticancer evaluation, and molecular docking studies of thiazolyl-pyrazoline derivatives, Bioorg. Med. Chem. Lett., 2023, 80, 129105 CrossRef CAS PubMed.
- K. Haider, M. Shafeeque, S. Yahya and M. S. Yar, A comprehensive review on pyrazoline based heterocyclic hybrids as potent anticancer agents, Eur. J. Med. Chem. Rep., 2022, 5, 100042 CAS.
- M. J. Ahsan, A. Ali, A. Ali, A. Thiriveedhi, M. A. Bakht, M. Yusuf, A. Salahuddin, O. Afzal and A. S. A. Altamimi, Pyrazoline Containing Compounds as Therapeutic Targets for Neurodegenerative Disorders, ACS Omega, 2022, 7(43), 38207–38245 CrossRef CAS PubMed.
- D. Matiadis and M. Sagnou, Pyrazoline hybrids as promising anticancer agents: An up-to-date overview, Int. J. Mol. Sci., 2020, 21(15), 5507 CrossRef CAS PubMed.
- M. Asad, M. N. Arshad, M. Oves, M. Khalid, S. A. Khan, A. M. Asiri, M. Rehan and H. Dzudzevic-Cancar, N-Trifluoroacetylated pyrazolines: Synthesis, characterization and antimicrobial studies, Bioorg. Chem., 2020, 99, 103842 CrossRef CAS PubMed.
- M. Mantzanidou, E. Pontiki and D. Hadjipavlou-Litina, Pyrazoles and pyrazolines as anti-inflammatory agents, Molecules, 2021, 26(11), 3439 CrossRef CAS PubMed.
- N. Zohreh and A. Alizadeh, Uncatalyzed One-Pot Synthesis of Highly Substituted Pyridazines and Pyrazoline-Spirooxindoles via Domino SN/Condensation/Aza-ene Addition Cyclization Reaction Sequence, ACS Comb. Sci., 2013, 15, 278–286 CrossRef CAS PubMed.
- E. A. Abdelsalam, A. A. Abd El-Hafeez, W. M. Eldehna, M. A. El Hassab, H. M. M. Marzouk, M. M. Elaasser, N. A. Abou Taleb, K. M. Amin, H. A. Abdel-Aziz, P. Ghosh and S. F. Hammad, Discovery of novel thiazolyl-pyrazolines as dual EGFR and VEGFR-2 inhibitors endowed with in vitro antitumor activity towards non-small lung cancer, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 2265–2282 CrossRef CAS PubMed.
- S. Upadhyay, A. C. Tripathi, S. Paliwal and S. K. Saraf, 2-pyrazoline derivatives in neuropharmacology: Synthesis, ADME prediction, molecular docking and in vivo biological evaluation, EXCLI J., 2017, 16, 628–649 Search PubMed.
- O. L. Erdmann, Untersuchungen über den Indigo, Arch. Pharm., 1840, 72, 253–285 CrossRef.
- A. Laurent, Indigo Research, Ann. Chim. Phys., 1840, 3, 393–434 Search PubMed.
- M. Yoshikawa, T. Murakami, A. Kishi, T. Sakurama, H. Matsuda, M. Nomura, H. Matsuda and M. Kubo, Novel indole S, O-bisdesmoside, calanthoside, the precursor glycoside of tryptanthrin, indirubin, and isatin, with increasing skin blood flow promoting effects, from two Calanthe species (Orchidaceae), Chem. Pharm. Bull., 1998, 46(5), 886–888 CrossRef CAS PubMed.
- J. Bergman, J.-O. Lindstroem and U. Tilstam, Tetrahedron, 1985, 41, 2879–2881 CrossRef CAS.
- L. Wei, Q. Wang and X. Liu, Application of thin-layer chromatography in quality control of Chinese medicinal preparations. II. Qualitative analysis of some Chinese medicinal preparations of Chansu, Yaowu Fenxi Zazhi, 1982, 2(5), 288–291 CAS.
- M. d'Ischia, A. Palumbo and G. Prota, Adrenalin oxidation revisited. New products beyond the adrenochrome stage, Tetrahedron, 1988, 44(20), 6441–6446 CrossRef.
- A. Palumbo, M. d'Ischia, G. Misuraca and G. Prota, A new look at the rearrangement of adrenochrome under biomimetic conditions, Biochim. Biophys. Acta, Gen. Subj., 1989, 990(3), 297–302 CrossRef CAS.
- J. Halket, P. Watkins, A. Przyborowska, B. Goodwin, A. Clow, V. Glover and M. Sandler, Isatin (indole-2, 3-dione) in urine and tissues: Detection and determination by gas chromatography—mass spectrometry, J. Chromatogr. B: Biomed. Sci. Appl., 1991, 562(1–2), 279–287 CrossRef CAS.
- A. Ashraf, Z. Shafiq, M. S. Khan Jadoon, M. N. Tahir, J. Pelletier, J. Sevigny, M. Yaqub and J. Iqbal, Synthesis, Characterization, and In Silico Studies of Novel Spirooxindole Derivatives as Ecto-5′-Nucleotidase Inhibitors, ACS Med. Chem. Lett., 2020, 11(12), 2397–2405 CrossRef CAS.
- W. Liu, S. Chen, F. Zhang, S. He, S. Wang and C. Sheng, Design, synthesis and biological evaluation of novel antitumor spirodihydrothiopyran-oxindole derivatives, Bioorg. Med. Chem. Lett., 2019, 29(13), 1636–1642 CrossRef CAS PubMed.
- K. Nikoofar and S. M. Dizgarani, A concise synthesis of isatin-based aromatic compounds in water as the reaction medium resulting in an approach to green chemistry, Monatsh. Chem., 2015, 146, 1161–1204 CrossRef CAS.
- M. Bortolami, F. Leonelli, M. Feroci and F. Vetica, Step economy in the stereoselective synthesis of functionalized oxindoles via organocatalytic domino/one-pot reactions, Curr. Org. Chem., 2021, 25(11), 1321–1344 CAS.
- S. Baddepuri, B. S. Allaka, R. K. Gamidi, M. Faizan, R. Pawar and S. Basavoju, An ultrasound assisted green protocol for the synthesis of quinoxaline based bisspirooxindoles: Crystal structure analysis, enone umpolung, DFT calculations, anti-cancer activity, and molecular docking studies, Synth. Commun., 2023, 53(11), 835–854 CrossRef CAS.
- N. Nivetha, R. M. Martiz, S. M. Patil, R. Ramu, S. Sreenivasa and S. Velmathi, Benzodioxole grafted spirooxindole pyrrolidinyl derivatives: Synthesis, characterization, molecular docking and anti-diabetic activity, RSC Adv., 2022, 12(37), 24192–24207 RSC.
- C. Marti and E. M. Carreira, Construction of Spiro [pyrrolidine-3, 3′-oxindoles]− recent applications to the synthesis of oxindole alkaloids, Eur. J. Org. Chem., 2003, 2003(12), 2209–2219 CrossRef.
- A. B. Dounay, K. Hatanaka, J. J. Kodanko, M. Oestreich, L. E. Overman, L. A. Pfeifer and M. M. Weiss, Catalytic asymmetric synthesis of quaternary carbons bearing two aryl substituents. Enantioselective synthesis of 3-alkyl-3-aryl oxindoles by catalytic asymmetric intramolecular heck reactions, J. Am. Chem. Soc., 2003, 125(20), 6261–6271 CrossRef CAS PubMed.
- M. Y. Chang, C. L. Pai and Y. H. Kung, Synthesis of (.+−.)-Coerulescine (Ia) and a Formal Synthesis of (.+−.)-Horsfiline (Ib), ChemInform, 2006, 37(12), 8463–8465 Search PubMed.
- P. S. Baran and J. M. Richter, Enantioselective total syntheses of welwitindolinone A and fischerindoles I and G, J. Am. Chem. Soc., 2005, 127(44), 15394–15396 CrossRef CAS PubMed.
- C.-B. Cui, H. Kakeya, G. Okada, R. Onose and H. Osada, Novel Mammalian Cell Cycle Inhibitors, Tryprostatins A, B and Other Diketopiperazines Produced by Aspergillus Fumigatus I. Taxonomy, Fermentation, Isolation and Biological Properties, J. Antibiot., 1996, 49(6), 527–533 CrossRef CAS PubMed.
- A. M. S. El-Sharief, Y. A. Ammar, A. Belal, M. A. S. El-Sharief, Y. A. Mohamed, A. B. Mehany, G. A. E. Ali and A. Ragab, Design, synthesis, molecular docking and biological activity evaluation of some novel indole derivatives as potent anticancer active agents and apoptosis inducers, Bioorg. Chem., 2019, 85, 399–412 CrossRef CAS PubMed.
- G. S. Reddy and M. Pal, Indole derivatives as anti-tubercular agents: An overview on their synthesis and biological activities, Curr. Med. Chem., 2021, 28(22), 4531–4568 CAS.
- B. Suliphuldevara Mathada, N. Gunavanthrao Yernale and J. N. Basha, The Multi-Pharmacological Targeted Role of Indole and its Derivatives: A review, ChemistrySelect, 2023, 8(1), e202204181 CrossRef CAS.
- Z. A. Homoud, M. Taha, F. Rahim, N. Iqbal, M. Nawaz, R. K. Farooq, A. Wadood, M. Alomari, I. Islam and S. Algheribe, Synthesis of indole derivatives as Alzheimer inhibitors and their molecular docking study, J. Biomol. Struct. Dyn., 2022, 1–14 CrossRef PubMed.
- A. Sengupta, S. Maity, A. Mondal, P. Ghosh, S. Rudra and C. Mukhopadhyay, Pseudo five component reaction towards densely functionalized spiro [indole-3, 2′-pyrrole] by picric acid, an efficient syn-diastereoselective catalyst: insight into the diastereoselection on C (sp 3)–C (sp 3) axial conformation, Org. Biomol. Chem., 2019, 17(5), 1254–1265 RSC.
- A. Riesco-Domínguez, N. van der Zwaluw, D. Blanco-Ania and F. P. Rutjes, An Enantio-and Diastereoselective Mannich/Pictet–Spengler Sequence To Form Spiro [piperidine-pyridoindoles] and Application to Library Synthesis, Eur. J. Org. Chem., 2017, 2017(3), 662–670 CrossRef.
- A. S. Girgis, J. Stawinski, N. S. Ismail and H. Farag, Synthesis and QSAR study of novel cytotoxic spiro [3H-indole-3, 2′(1′ H)-pyrrolo [3, 4-c] pyrrole]-2, 3′, 5′(1H, 2′ aH, 4′ H)-triones, Eur. J. Med. Chem., 2012, 47, 312–322 CrossRef CAS PubMed.
- A. S. Girgis, A. F. Mabied, J. Stawinski, L. Hegazy, R. F. George, H. Farag, E. M. Shalaby and I. A. Farag, Synthesis and DFT studies of an antitumor active spiro-oxindole, New J. Chem., 2015, 39(10), 8017–8027 RSC.
- P. Shanmugam, B. Viswambharan, K. Selvakumar and S. Madhavan, A facile and efficient synthesis of highly functionalised 3, 3′-dispiropyrrolidine-and 3, 3′-dispiropyrrolizidine bisoxindoles via [3+ 2] cycloaddition, Tetrahedron Lett., 2008, 49(16), 2611–2615 CrossRef CAS.
- X.-C. Zhang, S.-H. Cao, Y. Wei and M. Shi, Phosphine-and nitrogen-containing Lewis base catalyzed highly regioselective and geometric selective cyclization of isatin derived electron-deficient alkenes with ethyl 2, 3-butadienoate, Org. Lett., 2011, 13(5), 1142–1145 CrossRef CAS PubMed.
- A. Huang, J. J. Kodanko and L. E. Overman, Asymmetric synthesis of pyrrolidinoindolines. Application for the practical total synthesis of (−)-phenserine, J. Am. Chem. Soc., 2004, 126(43), 14043–14053 CrossRef CAS PubMed.
- T. D. Bagul, G. Lakshmaiah, T. Kawabata and K. Fuji, Total synthesis of spirotryprostatin B via asymmetric nitroolefination, Org. Lett., 2002, 4(2), 249–251 CrossRef CAS PubMed.
- B. Viswambharan, K. Selvakumar, S. Madhavan and P. Shanmugam, Pyridine Core Activation via 1, 5-Electrocyclization of Vinyl Pyridinium Ylides Generated from Bromo Isomerized Morita− Baylis− Hillman Adduct of Isatin and Pyridine: Synthesis of 3-Spirodihydroindolizine Oxindoles, Org. Lett., 2010, 12(9), 2108–2111 CrossRef CAS PubMed.
- T. Kawasaki, A. Ogawa, R. Terashima, T. Saheki, N. Ban, H. Sekiguchi, K.-E. Sakaguchi and M. Sakamoto, Synthesis of diversely functionalized hexahydropyrrolo [2, 3-b] indoles using domino reactions, olefination, isomerization and Claisen rearrangement followed by reductive cyclization, J. Org. Chem., 2005, 70(8), 2957–2966 CrossRef CAS PubMed.
- Z. Mao and S. W. Baldwin, New spirocyclic oxindole synthesis based on a hetero Claisen rearrangement, Org. Lett., 2004, 6(14), 2425–2428 CrossRef CAS PubMed.
- Y. Du, Y. Liu, H. Guo, R. Liu and R. Zhou, Chemo-and Diastereoselective Synthesis of Spirooxindole-pyrazolines and Pyrazolones via P (NMe2) 3-Mediated Substrate-Controlled Annulations of Azoalkenes with α-Dicarbonyl Compounds, Org. Lett., 2023, 4776–4781 CrossRef CAS PubMed.
- M. S. Altowyan, S. M. Soliman, M. Haukka, N. H. Al-Shaalan, A. A. Alkharboush and A. Barakat, Synthesis, Characterization, and Cytotoxicity of New Spirooxindoles Engrafted Furan Structural Motif as a Potential Anticancer Agent, ACS Omega, 2022, 7(40), 35743–35754 CrossRef CAS PubMed.
- M. Saeedi, A. Maleki, A. Iraji, R. Hariri, T. Akbarzadeh, N. Edraki, O. Firuzi and S. S. Mirfazli, Synthesis and bio-evaluation of new multifunctional methylindolinone-1,2,3-triazole hybrids as anti-Alzheimer's agents, J. Mol. Struct., 2021, 1229, 129828 CrossRef CAS.
- M. Saeedi, D. Mohtadi-Haghighi, S. S. Mirfazli, M. Mahdavi, R. Hariri, H. Lotfian, N. Edraki, A. Iraji, O. Firuzi and T. Akbarzadeh, Design and synthesis of selective acetylcholinesterase inhibitors: arylisoxazole-phenylpiperazine derivatives, Chem. Biodiversity, 2019, 16(2), e1800433 CrossRef PubMed.
- H. Pourtaher, A. Hasaninejad and A. Iraji, Design, synthesis, in silico and biological evaluations of novel polysubstituted pyrroles as selective acetylcholinesterase inhibitors against Alzheimer's disease, Sci. Rep., 2022, 12(1), 15236 CrossRef CAS PubMed.
- A. N. Çokuğraş, Butyrylcholinesterase: structure and physiological importance, Turk Biyokim. Derg., 2003, 28(2), 54–61 Search PubMed.
- H. Akrami, B. F. Mirjalili, M. Khoobi, H. Nadri, A. Moradi, A. Sakhteman, S. Emami, A. Foroumadi and A. Shafiee, Indolinone-based acetylcholinesterase inhibitors: Synthesis, biological activity and molecular modeling, Eur. J. Med. Chem., 2014, 84, 375–381 CrossRef CAS PubMed.
- K. Demir-Yazıcı, Ç. B. Apaydın, Ö. Soylu-Eter, N. Özsoy and N. Karalı, Synthesis, molecular modeling and cholinesterase inhibitory effects of 2-indolinone-based hydrazinecarbothioamides, Future Med. Chem., 2021, 13(24), 2133–2151 CrossRef PubMed.
- Ç. B. Apaydın, Ö. Soylu-Eter, P. Eraslan-Elma, N. Özsoy and N. Karalı, Synthesis and molecular modeling studies of 1-benzyl-2-indolinones as selective AChE inhibitors, Future Med. Chem., 2022, 14(23), 1705–1723 CrossRef PubMed.
- R. F. Costa, L. C. Turones, K. V. N. Cavalcante, I. A. Rosa Júnior, C. H. Xavier, L. P. Rosseto, H. B. Napolitano, P. Castro, M. L. F. Neto, G. M. Galvão, R. Menegatti, G. R. Pedrino, E. A. Costa, J. L. R. Martins and J. O. Fajemiroye, Heterocyclic Compounds: Pharmacology of Pyrazole Analogs From Rational Structural Considerations, Front. Pharmacol., 2021, 12, 666725 CrossRef CAS PubMed.
- G. Gutti, D. Kumar, P. Paliwal, A. Ganeshpurkar, K. Lahre, A. Kumar, S. Krishnamurthy and S. K. Singh, Development of pyrazole and spiropyrazoline analogs as multifunctional agents for treatment of Alzheimer's disease, Bioorg. Chem., 2019, 90, 103080 CrossRef CAS PubMed.
- A. Benazzouz-Touami, A. Chouh, S. Halit, S. Terrachet-Bouaziz, M. Makhloufi-Chebli, K. Ighil-Ahriz and A. M. S. Silva, New Coumarin-Pyrazole hybrids: Synthesis, Docking studies and Biological evaluation as potential cholinesterase inhibitors, J. Mol. Struct., 2022, 1249, 131591 CrossRef CAS.
- S. Shaikh, P. Dhavan, G. Pavale, M. M. V. Ramana and B. L. Jadhav, Design, synthesis and evaluation of pyrazole bearing α-aminophosphonate derivatives as potential acetylcholinesterase inhibitors against Alzheimer's disease, Bioorg. Chem., 2020, 96, 103589 CrossRef CAS PubMed.
- R. J. Obaid, N. Naeem, E. U. Mughal, M. M. Al-Rooqi, A. Sadiq, R. S. Jassas, Z. Moussa and S. A. Ahmed, Inhibitory potential of nitrogen, oxygen and sulfur containing heterocyclic scaffolds against acetylcholinesterase and butyrylcholinesterase, RSC Adv., 2022, 12(31), 19764–19855 RSC.
- C. Venkatesh, B. Singh, P. Mahata, H. Ila and H. Junjappa, Heteroannulation of nitroketene N, S-arylaminoacetals with POCl3: A novel highly regioselective synthesis of unsymmetrical 2, 3-substituted quinoxalines, Org. Lett., 2005, 7(11), 2169–2172 CrossRef CAS PubMed.
- N. Zohreh and A. Alizadeh, Uncatalyzed one-pot synthesis of highly substituted pyridazines and pyrazoline-spirooxindoles via domino SN/condensation/aza-ene addition cyclization reaction sequence, ACS Comb. Sci., 2013, 15(6), 278–286 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00255a |
| This journal is © The Royal Society of Chemistry 2024 |