
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Re-engineering lysozyme solubility and activity through surfactant complexation†
Jiaming
Mu
 a,
Leran
Mao
b,
Gavin P.
Andrews
a and
Sheiliza
Carmali
*a
a,
Leran
Mao
b,
Gavin P.
Andrews
a and
Sheiliza
Carmali
*a
aSchool of Pharmacy, Queen's University Belfast, BT9 7BL, UK. E-mail: s.carmali@qub.ac.uk
bDepartment of Chemical Engineering, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA
First published on 1st October 2024
Abstract
Hydrophobic ion-pairing is an established solubility engineering technique that uses amphiphilic surfactants to modulate drug lipophilicity and facilitate encapsulation in polymeric and lipid-based drug delivery systems. For proteins, surfactant complexation can also lead to unfolding processes and loss in bioactivity. In this study, we investigated the impact of two surfactants, sodium dodecyl sulphate (SDS) and dioctyl sulfosuccinate (DOSS) on lysozyme's solubility, activity, and structure. SDS and DOSS were combined with lysozyme at increasing charge ratios (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1, 1:1, 1:2 and 1:4) via hydrophobic ion pairing at pH 4.5. Maximum complexation efficiency at the 1:1 charge ratio was confirmed by protein quantitation assays and zeta potential measurements, showing a near neutral surface charge. Lysozyme lipophilicity was successfully increased, withlogD n-octanol/PBS values up to 2.5 with SDS and 1.8 with DOSS. Bioactivity assays assessing lysis of M. lysodeikticus cell walls showed up to a 2-fold increase in lysozyme's catalytic ability upon complexation with SDS at ratios less than stoichiometric, suggesting favourable mechanisms of stabilisation. Secondary structural analysis using Fourier-transform infrared spectroscopy indicated that lysozyme underwent a partial unfolding process upon complexation with low SDS concentrations. Molecular dynamic simulations further confirmed that at these low concentrations, a positive conformation was obtained with the active site residue Glu 35 more solvent-exposed. Combined, this suggested that sub-stoichiometric SDS altered the active site's secondary structure through increased backbone flexibility, leading to higher substrate accessibility. For DOSS, low surfactant concentrations retained lysozyme's native function and structure while still increasing the protein's lipophilic character. Our research findings demonstrate that modulation of protein activity can be related to surfactant chemistry and that controlled ion-pairing can lead to re-engineering of lysozyme solubility, activity, and structure. This has significant implications for advanced protein applications in healthcare, particularly towards the development of formulation strategies for oral biotherapeutics.
:1, 2:1, 1:1, 1:2 and 1:4) via hydrophobic ion pairing at pH 4.5. Maximum complexation efficiency at the 1:1 charge ratio was confirmed by protein quantitation assays and zeta potential measurements, showing a near neutral surface charge. Lysozyme lipophilicity was successfully increased, withlogD n-octanol/PBS values up to 2.5 with SDS and 1.8 with DOSS. Bioactivity assays assessing lysis of M. lysodeikticus cell walls showed up to a 2-fold increase in lysozyme's catalytic ability upon complexation with SDS at ratios less than stoichiometric, suggesting favourable mechanisms of stabilisation. Secondary structural analysis using Fourier-transform infrared spectroscopy indicated that lysozyme underwent a partial unfolding process upon complexation with low SDS concentrations. Molecular dynamic simulations further confirmed that at these low concentrations, a positive conformation was obtained with the active site residue Glu 35 more solvent-exposed. Combined, this suggested that sub-stoichiometric SDS altered the active site's secondary structure through increased backbone flexibility, leading to higher substrate accessibility. For DOSS, low surfactant concentrations retained lysozyme's native function and structure while still increasing the protein's lipophilic character. Our research findings demonstrate that modulation of protein activity can be related to surfactant chemistry and that controlled ion-pairing can lead to re-engineering of lysozyme solubility, activity, and structure. This has significant implications for advanced protein applications in healthcare, particularly towards the development of formulation strategies for oral biotherapeutics.
Introduction
Protein therapeutics have revolutionised the treatment of cancer, infectious diseases, and various metabolic disorders. More than 40 years after the approval of Humulin, the first clinically approved therapeutic protein, protein-based pharmaceuticals now account for two-thirds of the top-selling drugs.1,2 In 2023, leading the sales were Keytruda (pembrolizumab, Merck) used in cancer immunotherapy and the glucagon-like peptide-1 (GLP-1) receptor agonist Ozempic (semaglutide, Novo Nordisk).2 Despite growing success, more than 90% of biotherapeutics are still administered parenterally.3,4 Although effective, frequent injections can be inconvenient and painful, thereby impacting patient compliance. Additionally, parenteral administration often involves higher healthcare costs due to the need for trained medical personnel. Clinical translation of oral biotherapeutics remains a significant challenge due to poor intestinal absorption and enzymatic instability in the gastrointestinal tract.5,6 A notable example is Rybelsus (Novo Nordisk), an oral formulation of semaglutide with a bioavailability of <1%, further highlighting the obstacles in developing oral protein formulations.7,8One approach favoured for successful development of oral biotherapeutics is the use of lipid-based nanocarriers, including liposomes, self-emulsifying drug delivery systems, solid lipid nanoparticles and nanostructured lipid carriers.6,9–11 These lipid-based formulations protect proteins from enzymatic degradation, improve their transmucosal transport and provide controlled release. Ongoing research within this landscape has resulted in the approval of oral peptide drugs such as Neoral (cyclosporine A, Novartis) and Mycapssa (octreotide, Chiasma), with several more currently under clinical evaluation.9,12,13
To facilitate the solubilisation (or encapsulation) of hydrophilic proteins into lipid-based carriers, hydrophobic ion-pairing (HIP) is often employed to enhance protein lipophilicity.14–16 At a molecular level, HIP involves the stoichiometric association between the protein's ionisable groups (e.g., basic amino acids, such as lysine or arginine residues) with oppositely charged surfactants at a suitable pH. The increased lipophilicity stems from the reversible neutralisation of the protein's charge and is dependent on surfactant chemistry and structure. For example, sulphonate- and sulphate-based surfactants have been shown to substantially increase the lipophilicity of insulin, bovine serum albumin and horseradish peroxidase.17 In addition to the surfactant headgroup, the structure and flexibility of the hydrophobic tail are also important factors, with rigid alkyl moieties resulting in lower protein lipophilicity enhancements in contrast to more flexible, linear surfactant analogues. Pre-clinical studies have also shown that surfactant type impacts oral bioavailability, with increased lipophilicity leading to improved intestinal absorption.18
Paradoxically, surfactant complexation can also lead to unfavourable unfolding processes, which disrupt the protein's structure and lead to a loss of bioactivity and reduced therapeutic efficacy.19 Electrostatic and hydrophobic interactions drive surfactant complexation, with the mode and strength of these interactions resulting in altered protein structures and dynamics, and consequently, function.20 Above the surfactant's critical micellar concentration (CMC), hydrophobic interactions dominate, causing proteins to unfold. However, at surfactant concentrations similar to those used in HIP, complexation can yield protein conformations with favourable activities and/or stabilities. We hypothesised that by adjusting the type and concentration of surfactants during the HIP process, we can achieve a spectrum of protein structures, each with its own customised lipophilicity and activity characteristics.
In this study, we investigated the impact of two anionic surfactants, sodium dodecyl sulphate (SDS) and dioctyl sulfosuccinate (DOSS) on the structure and activity of lysozyme. Lysozyme, an antimicrobial enzyme, and an important component of the innate immune system, has been commonly used in formulation studies, including for hydrophobic ion pairing. It has a well characterised three-dimensional structure and an established enzymatic assay.21–23 These factors make lysozyme an ideal model to unravel the effects of surfactant complexation on protein structure and function. Initially, we ion-paired lysozyme with either SDS or DOSS at increasing surfactant concentrations. We then assessed the lipophilic properties of the resulting complexes using a shake-flask method. The catalytic activity of lysozyme and lysozyme-surfactant complexes was measured using a M. lysodeikticus cell wall degradation assay. We then correlated activity data with changes to lysozyme's secondary structure, as determined by Fourier-Transform Infrared Spectroscopy (FT-IR), and thermal resistance, as measured by differential scanning calorimetry (DSC). To gain further insight, we compared wet-lab findings with molecular dynamic simulations. These simulations were performed with lysozyme and lysozyme-surfactant complexes at surfactant concentrations that produced optimal lipophilicity and activity profiles.
Experimental
Materials
Lysozyme from chicken egg white (lyophilized powder, protein ≥90%, ≥20000 units per mg dry weight), Micro BCATM Protein Assay Kit and dimethylsulfoxide (DMSO) were purchased from ThermoFisher Scientific (United Kingdom). Micrococcus lysodeikticus lyophilized cells, sodium dodecyl sulphate (SDS), dioctyl sulfosuccinate (DOSS), sodium acetate, acetic buffer ≥ 99%, potassium phosphate monobasic and dibasic solutions, and phosphate buffered saline (PBS) tablets were obtained from Sigma-Aldrich. All chemicals were used without further purification. Buffers were filtered through 0.2 μm polyethersulfone (PES) membrane before use. Deionized water was used for all the experiments.
Lysozyme-surfactant ion-pairing process
Lysozyme solution (5 mg mL−1, as determined by spectrophotometry at 280 nm, ε1%280 26.424) was prepared with 10 mM acetate buffer pH 4.5 to achieve a net positive charge and maximum complexation efficiency (Fig. S1, ESI†). SDS was dissolved in deionised water (20 mg mL−1) while DOSS was prepared as an aqueous solution with 2% DMSO (15 mg mL−1) to ensure sufficient solubilisation. Surfactant aqueous solutions (1 mL) were then added, dropwise at room temperature, to the lysozyme solution, to achieve the desired surfactant: lysozyme ratios (Table 1) in separate vessels and allowed to mix for 20 min at 550 rpm (Eppendorf 5382 ThermoMixer C v.3.5.0).| Surfactant (mM) | Lysozyme:surfactant |
|
|---|---|---|
| Molar ratio | Charge ratio | |
| 0.8 | 4:9 |
8:1 |
| 1.6 | 2:9 |
4:1 |
| 3.1 | 1:9 |
2:1 |
| 6.3 | 1:18 |
1:1 |
| 12.5 | 1:36 |
1:2 |
| 25.0 | 1:72 |
1:4 |
White precipitates in solution indicated HIP complexation. Complexes were recovered by centrifugation of cloudy solution at 13500 rpm for 10 min at 4 °C (AXYSPIN Refrigerated microcentrifuge). The obtained precipitates were washed with deionised water, followed by lyophilisation (Edwards Modulyo Freeze Dryer) and stored at −20 °C.
Complexation efficiency (CE) was determined by quantification of non-complexed lysozyme in supernatant with MicroBCA assay (Table S1, ESI†) and eqn (1):
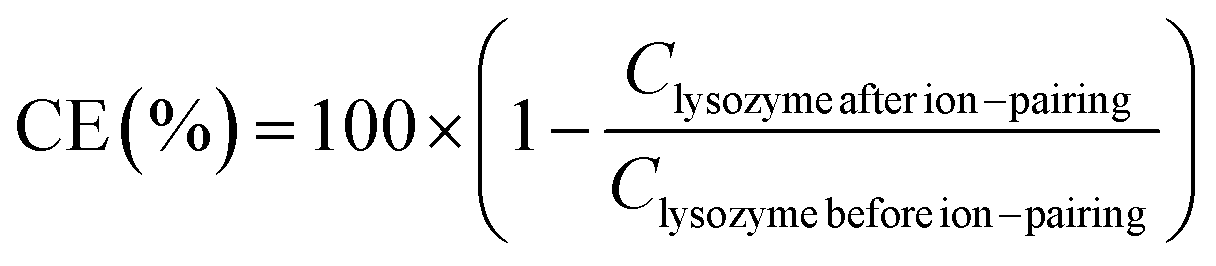 | (1) |
Characterisation of lysozyme-surfactant complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) logD.
Distribution studies using 1-octanol and PBS was adapted from Phan and co-workers.25 1-Octanol was saturated with PBS by mixing of both solvents for 24 hours at 25 °C. After this time, the organic phase was separated by centrifugation under 4000 rpm for 20 min (SIGMA® Laboratory Centrifuge 6–15H). Each lysozyme-surfactant complex (1 mg) was dissolved in 500 μL of PBS saturated 1-octanol. Subsequently, the same volume of the PBS aqueous phase was added to the organic phase, after which the mixture was mixed at 550 rpm for 3 hours at 4 °C. After this time, aqueous and organic phases were separated by centrifugation at 13500 rpm for 10 min. Lysozyme concentration in the aqueous phase was determined by Micro BCA assay and the partition coefficientlogD was determined based on eqn (2):
logD.
Distribution studies using 1-octanol and PBS was adapted from Phan and co-workers.25 1-Octanol was saturated with PBS by mixing of both solvents for 24 hours at 25 °C. After this time, the organic phase was separated by centrifugation under 4000 rpm for 20 min (SIGMA® Laboratory Centrifuge 6–15H). Each lysozyme-surfactant complex (1 mg) was dissolved in 500 μL of PBS saturated 1-octanol. Subsequently, the same volume of the PBS aqueous phase was added to the organic phase, after which the mixture was mixed at 550 rpm for 3 hours at 4 °C. After this time, aqueous and organic phases were separated by centrifugation at 13500 rpm for 10 min. Lysozyme concentration in the aqueous phase was determined by Micro BCA assay and the partition coefficientlogD was determined based on eqn (2):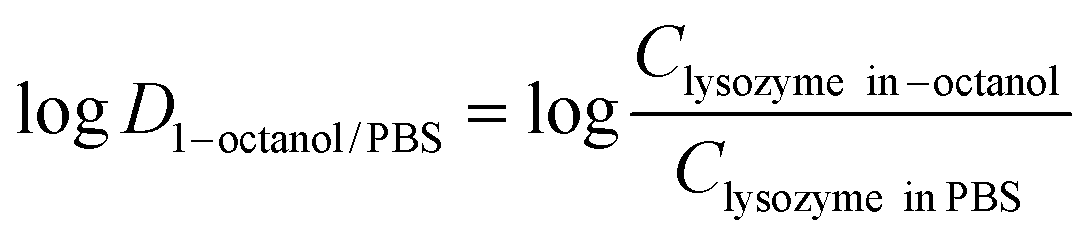 | (2) |
The inverted second-derivative spectra were obtained from the derivative function of peak analysis and fitted with Gaussian band profiles26 with OriginPro 2023b. The fraction of α-helix in infrared second-derivative amide spectra was determined by computing the area of the component peak divided by the sum of areas of all the component peaks of the amide I band around 1650 cm−1.
Molecular dynamics simulations
The starting structure for lysozyme was obtained from the protein data bank (PDB ID 6LYZ). Protein protonation at pH 6.5 was determined using PDB2PQR continuum electrostatics.27 Surfactant structures were built in Avogadro and energy-minimized using the universal force field. Lysozyme was modelled with the CHARMM C36m force field with WYF parameters for cation–pi interactions using CHARMM GUI in a water box fitted to the protein size (∼66–68 Å).28–30 SDS and DOSS topology files were generated using CGenFF parameters.31,32 Protein structures were solvated with TIP3 explicit solvent, and the system was neutralized using 50 mM K+ and PO32− ions to better represent experimental settings.Molecular dynamic simulation was run on GROMACS 2020.1.33,34 The protein structure was energy minimized using the steepest descent approach consisting of 5000 steps followed by NVT equilibration with Nose–Hoover temperature coupling for 125 ps. Simulations for lysozyme with and without the addition of surfactant were run for 35 ns with an NPT ensemble using Nose–Hoover temperature coupling and Parrinello–Rahman isotropic pressure coupling at 293.15 K. Electrostatics were modelled using the Particle Mesh Ewald method in an automatically generated grid. The production run was analysed for root-mean-squared deviation (RMSD) and radial probability distribution (G(r)) using VMD.35 The averaged PDB structure in each 5 ns simulation sequence was exported and visualized in Biovia Discovery Studio (Dassault Systems) for secondary structure analysis and solvent-accessible surface area (SASA) analysis (ESI†).
Results and discussion
Preparation and characterisation of lysozyme-surfactant complexes
Lysozyme is a small globular protein consisting of 129 amino acids cross-linked with four disulphide bridges.22 Due to its high isoelectric point (pI 11.35), lysozyme's acidic groups (7 aspartic acid and 2 glutamic acid residues) remain non-ionised and its basic groups (11 arginine, 1 histidine and 6 lysine residues) become protonated at low pH. As a result, these 18 positively charged residues can non-covalently interact with negatively charged surfactants (Fig. 1A and B). In this study, lysozyme was ion paired with anionic sodium dodecyl sulphate (SDS) and dioctyl sulfosuccinate (DOSS) at pH 4.5. These two surfactants were selected due to their similar, stabilising kosmotropic headgroups (SO4− and SO3−, respectively) and distinct hydrophobic, tail groups (linear vs. branched).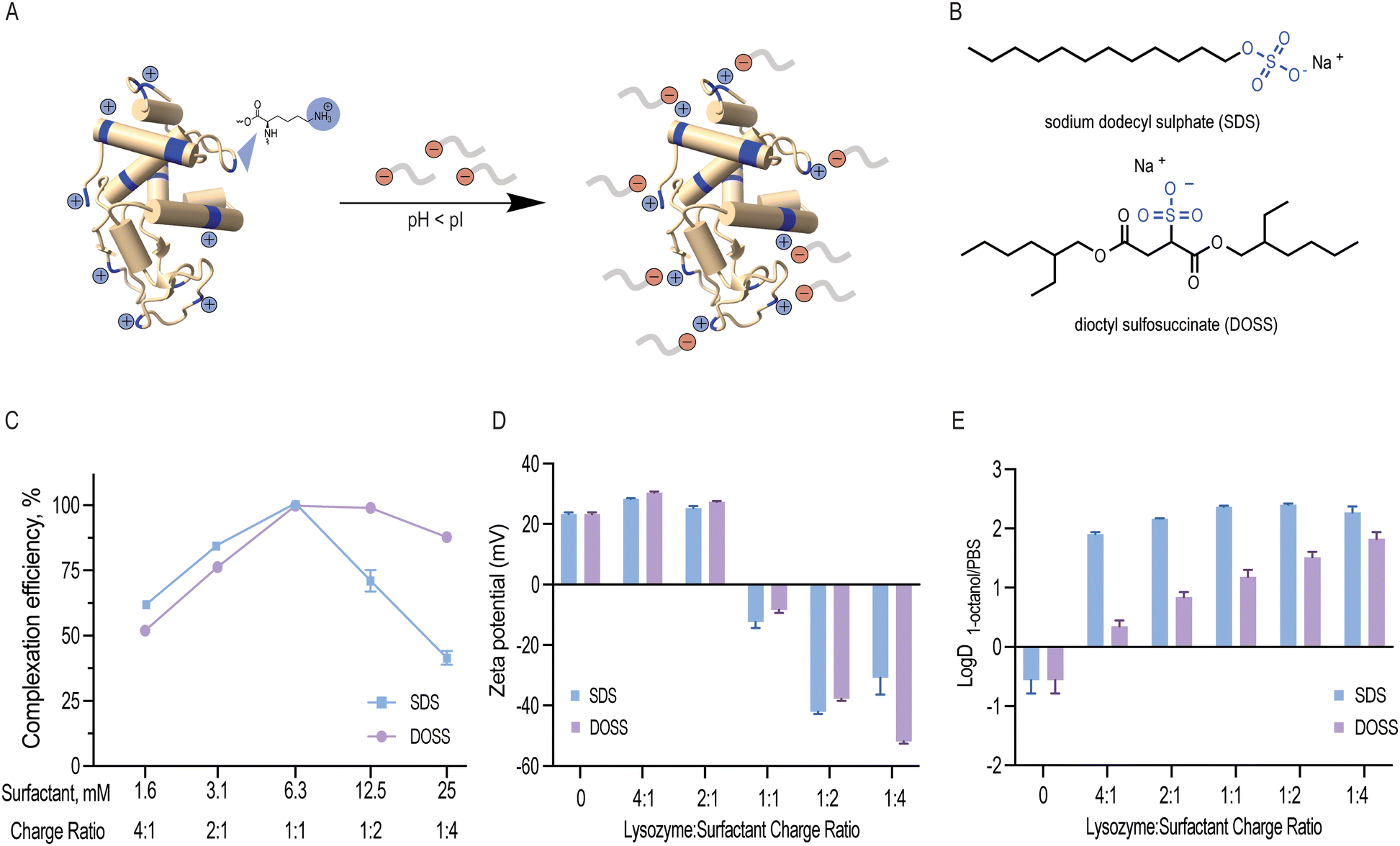 | ||
| Fig. 1 (A) At low pH conditions, lysozyme is positively charged and can associate with anionic surfactants primarily through non-covalent electrostatic interactions, forming lysozyme-surfactant complexes; (B) chemical structures of surfactants sodium dodecyl sulphate (SDS) and dioctyl sulfosuccinate (DOSS) used in this study; (C) impact of surfactant concentration on lysozyme-surfactant complexation efficiency; (D) apparent surface charge variation as a function of lysozyme: surfactant charge ratio; (E) impact of lysozyme: surfactant charge ratio on lysozyme hydrophobicity as determined by the partition coefficient (logD) of lysozyme following surfactant addition. | ||
:1 (Fig. 1C). At this ratio, we expected 18 surfactant molecules to bind to 1 lysozyme molecule, which corresponds to a surfactant concentration of 6.3 mM. When surfactant concentration exceeds this binding saturation point, micelles form, and proteins can be re-solubilised.37 We experimentally determined the CMC values for SDS and DOSS under the conditions used in this study and found them to be 7.2 and 4.8 mM, respectively (Fig. S3 and S4, ESI†). This further confirmed that the observed decrease in both complexation efficiency and solution turbidity above the surfactant concentration of 6.3 mM led to protein re-solubilisation. Dynamic light scattering experiments also showed that complexes with surfactant concentration, namely at the 1:4 charge ratio, were found to have similar particle sizes to native lysozyme (Table S1, ESI†). These results are also in agreement with previous studies that have shown that a stoichiometric or slightly higher binding ratio is optimal for hydrophobic ion pairing.16
Both SDS and DOSS have negatively charged head groups that can interact ionically with the basic residues of lysozyme, as shown in Fig. 1B. However, SDS and DOSS have distinct chemical and structural properties. SDS has a linear structure, while DOSS is a branched and more lipophilic surfactant, withlogP 3.86 and 4.36, respectively (calculated by ALOGPS 2.1).36 We hypothesized that these chemical and structural differences would affect how the surfactants interact with the surface of lysozyme, with DOSS involving more hydrophobic interactions. To further understand this, we used zeta potential as a proxy for surface charge. We noted a decreasing trend with increasing surfactant concentration for both SDS and DOSS, as shown in Fig. 1D This trend suggests that the primary mode of interaction for both surfactants is ionic. At the stoichiometric binding point, we observed an apparent charge neutralisation effect due to near complete complexation at all positively charged residues of lysozyme. Beyond this point, an overall negative surface charge was observed, attributed to the presence of excess anionic surfactants.
:1 charge ratio, as shown in Fig. 2B and C, exhibited a more rigid and rough surface texture. Moreover, upon surfactant dissociation, we noted that this rigidity was maintained, indicating that surfactant complexation had irreversibly altered lysozyme's morphology (Fig. 2D and E).
 | ||
| Fig. 2 (A) SEM image of Native lysozyme; (B) L-SDS complex at the charge ratio of 1:1; (C) L-DOSS complex at the charge ratio of 1:1; (D) dissociated lysozyme from the L-SDS complex; (E) dissociated lysozyme from the L-DOSS complex. Sample was freeze-dried before testing at the magnitude of 250×. Images were processed with Fiji ImageJ 1.54 h. | ||
Impact of surfactant complexation on lysozyme bioactivity
An important aspect of hydrophobic ion pairing with proteins is to ensure functional integrity. During complexation, lysozyme precipitation may result in enzyme deactivation due to irreversible aggregation. Moreover, ionic surfactants, such as SDS and DOSS, are usually associated with protein denaturation due to their charged head groups, but in some cases, they can heighten activity, where partially unfolded proteins retain their overall native shape, and consequently function.40To investigate how surfactant complexation impacted lysozyme function, we measured lysozyme's activity via bacterial cell wall lysis, following surfactant dissociation (Fig. 3A). For both SDS and DOSS, lysozyme showed a catalytic enhancement at low surfactant concentrations. For lysozyme-SDS complexes, a near two-fold enhancement was observed when a maximum of 9 surfactant molecules were bound to 1 molecule of lysozyme (0.8–3.1 mM SDS concentration). For DOSS complexes, this increase in activity was less accentuated. At higher concentrations, complexes formed with SDS and DOSS led to a loss in activity. This can be attributed to protein unfolding due to micelle formation and the loss of lysozyme's positive charge in the active site, leading to alterations in substrate recognition.
 | ||
| Fig. 3 (A) Impact of SDS and DOSS concentration on lysozyme lytic activity; (B) comparative study between native lysozyme (L), lysozyme pre-complexed with 1.6 mM SDS or DOSS (L-SDS and L-DOSS 4:1, respectively) and control samples with lysozyme in the presence of 1.6 mM SDS or DOSS (C-SDS and C-DOSS, respectively). Shown are three individual experiments ± SEM. *p < 0.05 by ordinary one-way ANOVA (Šídák's multiple comparisons test). | ||
To better understand whether the observed heightened activity was a result of surfactant presence or ion-pairing, we conducted control studies measuring the activity of non-complexed lysozyme in the presence of SDS and DOSS at a concentration of 1.6 mM, or 4:1 ratio (Fig. 3B). We selected this surfactant concentration as these complexes displayed similar activity profiles. Results showed that the activity of pre-formed SDS complexes (L-SDS) was significantly different from that of native lysozyme (L) and lysozyme in the presence of SDS (C-SDS). This indicated that ion-pairing with 1.6 mM SDS, and the subsequent increase in activity, was due to surfactant complexation which may have induced positive conformational changes. In contrast, for the DOSS complex (L-DOSS), no statistically significant difference was observed between native lysozyme (L) and lysozyme in the presence of 1.6 mM DOSS (C-DOSS). This suggested DOSS complexation did not impact lysozyme's catalytic activity, retaining its original native function. Previous reports have shown that the increase in lysozyme's bioactivity can be related to increased hydrophobic interactions between lysozyme and the cell substrate.41 Our findings are consistent with these reports, with SDS complexes showing increased lipophilicity and activity properties.
To gain further insight into the source of lysozyme's catalytic enhancement upon surfactant addition, we used molecular dynamics (MD) to simulate our experimental system with 9 molecules of either SDS or DOSS interacting with lysozyme at pH 6.5. Analysis of the MD trajectory showed a higher degree of backbone flexibility of the active site residues throughout the simulation time, particularly for SDS molecules. Conformational flexibility has been shown to correlate strongly with bioactivity, which for lysozyme may also relate to increased substrate access.42
Increasing protein hydrophobicity can also lead to partial unfolding, with lysozyme's active site residues Glu 35 and Asp 52 slightly more solvent-exposed, contributing to an apparent catalytic enhancement.43 Solvent accessibility calculations showed that lysozyme with 9 molecules of surfactant led to an increase in exposure of Glu 35 but not for Asp 52, suggesting the enhanced activity effect primarily stemmed from conformational changes in Glu 35. A closer analysis revealed that at the 2:1 lysozyme:surfactant charge ratio complex, Glu 35 was predominately located in a β-turn secondary structure, while complexes with charge ratios where activity was lost, an α-helix structure was observed. The β-turn structure has been shown to increase protein stability and dynamics and increased solvent exposure.44 Interestingly, no difference in secondary structure was observed for the catalytic residue Asp 52. Combined, these findings suggest that sub-stoichiometric concentrations of SDS likely altered the secondary structure of the lysozyme active site by modulating the active site's backbone flexibility, leading to higher substrate accessibility.
Impact of surfactant interactions on lysozyme structure
After determining how the catalytic activity of lysozyme varies with surfactant type and concentration, we now sought to explore how the structure of lysozyme changes upon hydrophobic ion-pairing. We first used FTIR spectroscopy to investigate changes to lysozyme's secondary structure upon surfactant association. We focused on analysis of the amide I band (1600–1700 cm−1), which is due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of peptide bonds and is influenced by the secondary structure.45
O stretching vibrations of peptide bonds and is influenced by the secondary structure.45
As shown in Fig. 4A, both SDS and DOSS association led to distinct modifications in lysozyme's secondary structure. For SDS, lysozyme complexes initially underwent a partial unfolding process, as observed by a decrease in α-helical content. This was followed by an increase in helical structure at higher surfactant concentrations.
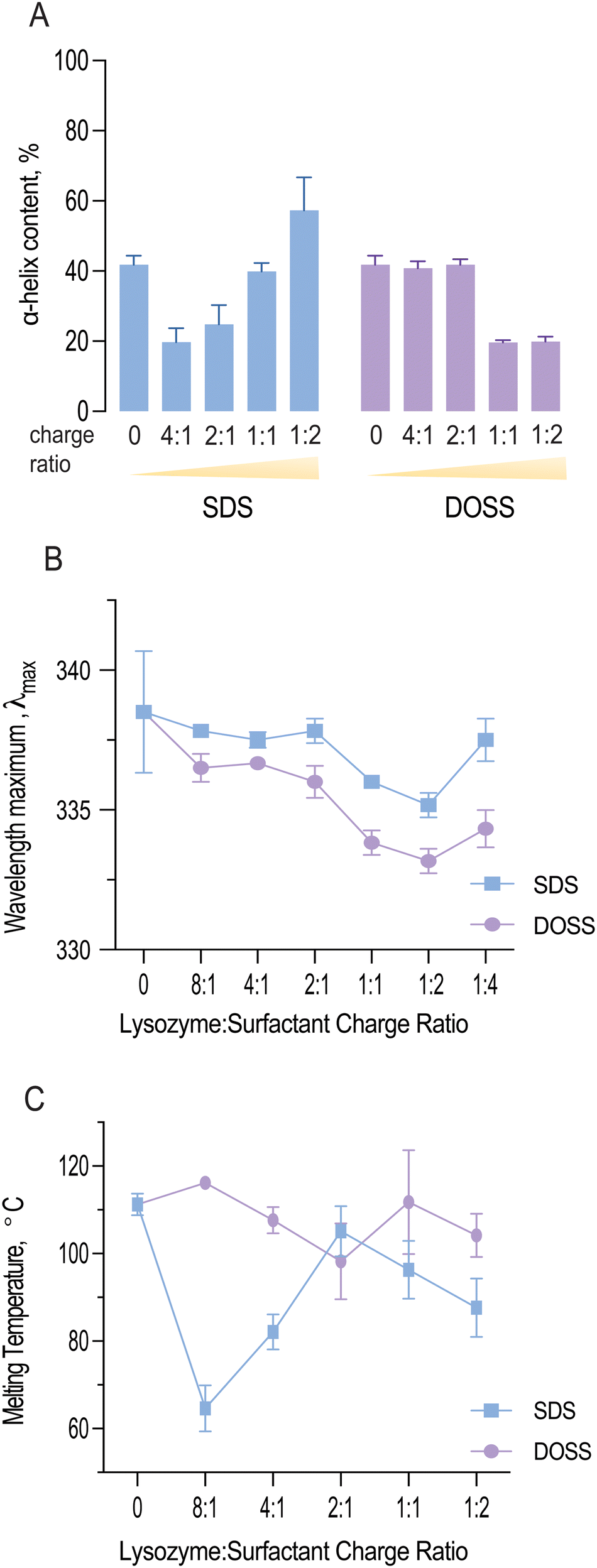 | ||
| Fig. 4 (A) Impact of surfactant concentration on lysozyme secondary structure as determined by the content of α helix at amide I band, [Lysozyme] = 0.35 mM; (B) changes in wavelength maximum (lmax) of lysozyme complexes with increasing concentrations of SDS and DOSS; (C) the effect of surfactant complexation on lysozyme's thermal resistance. | ||
Quantitative analysis of the deconvoluted amide I band revealed that the native lysozyme contained approximately 41.8% α-helix content, which increased to 57.3% in the presence of excess SDS. This observation aligns with previous studies, where SDS binding has been found to induce a molten globule state, characterised by high α-helical content but lack of tertiary structure.46
In contrast, lysozyme-DOSS complexes at low surfactant concentrations retained their α-helical content (41.8%), possibly due to predominant electrostatic interactions between DOSS's negatively charged headgroup and lysozyme's cationic residues. However, in the presence of excess DOSS, hydrophobic interactions can also occur, which was observed by a significant loss in α-helical content (19.9%).
Analysis of the variation of intrinsic fluorescence properties of lysozyme in the presence of surfactant also provided us with some further insight into the observed conformational changes. Tryptophan fluorescence is dependent on the polarity of its local environment, with changes in wavelength maximum and fluorescence intensity roughly correlated to solvent exposure. Lysozyme contains 6 tryptophan residues, with Trp 62 and 108 responsible for most of the protein's emission.47
Fig. 4B shows the wavelength maximum (λmax) of lysozyme at a fixed lysozyme concentration (5 mg mL−1) with increasing surfactant concentrations. For both SDS and DOSS, a shift in λmax was observed, further confirming the occurrence of protein conformational changes. For lysozyme-SDS complexes, the λmax first underwent an increasing blue shift until reaching the 1:2 charge ratio or surfactant concentration up to 12.5 mM. These findings indicate that the tryptophan residues in lysozyme may have experienced a more hydrophobic microenvironment, in agreement with our complexation and lipophilicity results shown in Fig. 1C and E. Above the SDS concentration of 12.5 mM, a red shift in the wavelength maximum back to 337 nm was noted for lysozyme, corresponding to the re-solubilisation of lysozyme and SDS micellar re-folding. For lysozyme-DOSS complexes a similar trend was initially observed, albeit without the complete red shift in the presence of excess surfactant, suggesting that DOSS leads to a distinct unfolding pathway, without the formation of a molten globule state.
Impact of surfactant complexation on lysozyme thermal stability
Previous studies have established a connection between protein stability, thermal resistance, and factors such as protein electrostatics, hydropathy and core packing.48 Hyperthermophilic proteins are characterised by enhanced hydrophobic interactions and salt bridge formations which are important in their ability to withstand elevated temperatures.49 Since surfactant complexation increased lysozyme's hydrophobicity, we now aimed to understand the impact on lysozyme's thermal stability. We characterised lysozyme and resulting complexes’ thermal properties using differential scanning calorimetry, and analysed thermal resistance as defined by the melting temperature (Tm).
Fig. 4C illustrates how the melting temperature of lysozyme fluctuates with the concentration of SDS and DOSS. Typically, a higher Tm value indicates a more stable protein structure.50 In the case of lysozyme-SDS complexes, a significant drop in the melting temperature was noted initially. However, this was followed by a rise at the 2:1 charge ratio (or 3.1 mM), bringing it close to the original melting temperature of native lysozyme (Tm = 111.23 ± 2.5 °C). Subsequently, we observed a slow decline in thermal resistance. Previous studies have demonstrated a connection between protein helicity and thermal stability.51 A detailed examination of the variations in thermal resistance and helical content in lysozyme-SDS complexes indeed confirms this correlation. The initial decrease in helicity coincides with the same concentration range as the reduction in lysozyme's melting temperature. Upon reaching an SDS concentration of 3.1 mM, we observed an increase in thermal resistance, which corresponds with the rise in helical content due to SDS-induced helical folding.
Furthermore, we noted that the initial decline in thermal stability was linked to an increase in lysozyme's catalytic activity. SDS has been shown to stabilize the β- strand secondary structure at low concentrations.52 Our MD studies revealed that the active site residue, Glu 35, was in a β-turn secondary structure at concentrations where lysozyme remained functional and analysis of lysozyme-surfactant interactions showed that in contrast to DOSS, SDS displayed less contacts with residues located in β-turns, which are important for stability. Therefore, we hypothesise that at sub-stoichiometric ratios, SDS enhances lysozyme's catalytic activity while reducing its thermal stability, exemplifying a typical ‘stability-activity trade-off’.
Analysis of thermal resistance of lysozyme-DOSS complexes showed a subtle stabilisation effect at the 8:1 charge ratio. This was subsequently followed by a steady decrease, reaching its minimum at the 2:1 charge ratio. After this point, we observed an increase in the melting temperature, which remained close to the original Tm of native lysozyme. As mentioned previously, changes in protein solubility can often suggest a variation in the protein's melting temperature. For lysozyme complexes with DOSS, lipophilic and Tm changes were less pronounced in comparison to lysozyme-SDS complexes, further highlighting the dependency of both parameters.
Conclusions
In this study, we formed ion pairs between lysozyme and two surfactants, SDS and DOSS, at various charge ratios. This resulted in a variety of lysozyme-surfactant complexes, each with unique characteristics in terms of lipophilicity, activity, and structure. Complexation with either SDS or DOSS increased lysozyme's hydrophobicity, however only the controlled addition of SDS in sub-stoichiometric amounts, below the 1:1 ratio, led to complexes with increased activity. This increased activity was attributed to partial unfolding and greater exposure of the active site, thereby enhancing substrate accessibility. Our study underscores that surfactant chemistry can influence protein activity and that controlled ion-pairing can modify lysozyme solubility while enhancing bioactivity. These insights are currently being applied in the development of lipid-based formulation strategies for oral biotherapeutics, potentially leading to more effective, and patient-friendly treatments.
Author contributions
Conceptualisation: S. C., and G. P. A.; data curation: J. M. and L. M.; data analysis and interpretation: S. C., J. M. and L. M.; manuscript writing/editing and visualisation: S. C., J. M. and L. M.Data availability
Data for this article has been deposited in the Queen's University Belfast repository - Queen's University Research Portal at https://pure.qub.ac.uk/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Queen's University/China Scholarship Council PhD Scholarships is acknowledged.References
- FDA Drug Bull., 1982, 12, 18–19 Search PubMed.
- P. Verdin, Nat. Rev. Drug Discovery, 2024, 23, 240 CrossRef CAS.
- A. Patel, K. Cholkar and A. K. Mitra, Ther. Delivery, 2014, 5, 337–365 CrossRef CAS.
- R. J. Kulchar, R. Singh, S. Ding, E. Alexander, K. W. Leong and H. Daniell, Biomaterials, 2023, 302, 122312 CrossRef CAS.
- H. Peng, J. Wang, J. Chen, Y. Peng, X. Wang, Y. Chen, D. L. Kaplan and Q. Wang, Expert Opin. Drug Delivery, 2023, 20, 1349–1369 CrossRef CAS PubMed.
- G. Noh, T. Keum, V. Raj, J. Kim, C. Thapa, K. Shakhakarmi, M. J. Kang, Y. T. Goo, Y. W. Choi and S. Lee, Int. J. Biol. Macromol., 2023, 225, 911–922 CrossRef CAS PubMed.
- S. Hughes and J. J. Neumiller, Clin. Diabetes Publ. Am. Diabetes Assoc., 2020, 38, 109–111 CrossRef.
- R. V. Overgaard, A. Navarria, S. H. Ingwersen, T. A. Bækdal and R. J. Kildemoes, Clin. Pharmacokinet., 2021, 60, 1335–1348 CrossRef CAS.
- S. Haddadzadegan, F. Dorkoosh and A. Bernkop-Schnürch, Adv. Drug Delivery Rev., 2022, 182, 114097 CrossRef CAS PubMed.
- E. Muntoni, E. Marini, N. Ahmadi, P. Milla, C. Ghè, A. Bargoni, M. T. Capucchio, E. Biasibetti and L. Battaglia, Acta Diabetol., 2019, 56, 1283–1292 CrossRef CAS.
- J. Mudassir, A. Raza, M. A. Khan, H. Hameed, G. A. Shazly, A. Irfan, S. J. Rana, K. Abbas, M. S. Arshad, S. Muhammad and Y. A. Bin Jardan, Pharmaceutics, 2023, 15, 1973 CrossRef CAS.
- N. Parquet, O. Reigneau, H. Humbert, M. Guignard, P. Ribaud, G. Socié, A. Devergie, H. Espérou and E. Gluckman, Bone Marrow Transplant., 2000, 25, 965–968 CrossRef CAS PubMed.
- A. Labadzhyan, L. B. Nachtigall, M. Fleseriu, M. B. Gordon, M. Molitch, L. Kennedy, S. L. Samson, Y. Greenman, N. Biermasz, M. Bolanowski, A. Haviv, W. Ludlam, G. Patou and C. J. Strasburger, Pituitary, 2021, 24, 943–953 CrossRef CAS PubMed.
- J. D. Meyer and M. C. Manning, Pharm. Res., 1998, 15, 188–193 CrossRef CAS.
- K. D. Ristroph and R. K. Prud’homme, Nanoscale Adv., 2019, 1, 4207–4237 RSC.
- J. Griesser, G. Hetényi, M. Moser, F. Demarne, V. Jannin and A. Bernkop-Schnürch, Int. J. Pharm., 2017, 520, 267–274 CrossRef CAS.
- V. Claus, M. Sandmeier, N. Hock, H. Spleis, S. Lindner, M. Kalb and A. Bernkop-Schnürch, Int. J. Pharm., 2023, 647, 123507 CrossRef CAS.
- S. Bonengel, M. Jelkmann, M. Abdulkarim, M. Gumbleton, V. Reinstadler, H. Oberacher, F. Prüfert and A. Bernkop-Schnürch, J. Controlled Release, 2018, 273, 21–29 CrossRef CAS.
- Y. Sun, P. L. O. Filho, J. C. Bozelli, J. Carvalho, S. Schreier and C. L. P. Oliveira, Soft Matter, 2015, 11, 7769–7777 RSC.
- M. D. Lad, V. M. Ledger, B. Briggs, R. J. Green and R. A. Frazier, Langmuir, 2003, 19, 5098–5103 CrossRef CAS.
- A. N. Smolelis and S. E. Hartsell, J. Bacteriol., 1949, 58, 731–736 CrossRef CAS PubMed.
- L. N. Johnson, Sci. Prog., 1966, 54, 367–385 CAS.
- A. A. A. Hassan, T. Sovány, K. Pamlényi, M. Deák, V. Hornok, E. Csapó, G. Regdon, I. Csóka and K. Kristó, Pharmaceutics, 2024, 16, 589 CrossRef CAS.
- K. C. Aune and C. Tanford, Biochemistry, 1969, 8, 4579–4585 CrossRef CAS PubMed.
- T. N. Q. Phan, R. Ismail, B. Le-Vinh, S. Zaichik, F. Laffleur and A. Bernkop-Schnürch, ACS Biomater. Sci. Eng., 2020, 6, 5032–5039 CrossRef CAS.
- A. Dong, P. Huang and W. S. Caughey, Biochemistry, 1990, 29, 3303–3308 CrossRef CAS.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. Liles, M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G.-W. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Protein Sci. Publ. Protein Soc., 2018, 27, 112–128 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks, A. D. MacKerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. I. Brooks, A. D. MacKerell, J. B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS PubMed.
- W. Yu, X. He, K. Vanommeslaeghe and A. D. MacKerell, J. Comput. Chem., 2012, 33, 2451–2468 CrossRef CAS PubMed.
- H. Bekker, H. Brendsen, E. Dijkstra, S. Achterop, R. Vondrumen, D. Vanderspoel, A. Sijbers, H. Keegstra and M. Renardus, Phys. Comput., 1993, 92, 252–256 Search PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14(33–38), 27–28 Search PubMed.
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Y. Tanchuk and V. V. Prokopenko, J. Comput. Aided Mol. Des., 2005, 19, 453–463 CrossRef CAS.
- S. H. Choi and T. G. Park, Int. J. Pharm., 2000, 203, 193–202 CrossRef CAS.
- H. D. Lu, P. Rummaneethorn, K. D. Ristroph and R. K. Prud’homme, Mol. Pharm., 2018, 15, 216–225 CrossRef CAS.
- X. Pan, H. Wang, C. Li, J. Z. H. Zhang and C. Ji, J. Chem. Inf. Model., 2021, 61, 3159–3165 CrossRef CAS PubMed.
- Y. Sun, P. L. O. Filho, J. C. Bozelli, J. Carvalho, S. Schreier and C. L. P. Oliveira, Soft Matter, 2015, 11, 7769–7777 RSC.
- R. A. Ivanov, O. A. Soboleva, S. A. Smirnov and P. A. Levashov, Russ. J. Bioorganic Chem., 2015, 41, 260–265 CrossRef CAS PubMed.
- K. Garajová, A. Balogová, E. Dušeková, D. Sedláková, E. Sedlák and R. Varhač, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 281–288 CrossRef.
- J. Held and S. van Smaalen, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1136–1146 CrossRef CAS PubMed.
- K. Fujiwara, S. Ebisawa, Y. Watanabe, H. Fujiwara and M. Ikeguchi, BMC Struct. Biol., 2015, 15, 21 CrossRef.
- C. Jones, B. Mulloy and A. H. Thomas, Analysis of Polypeptide and Protein Structures Using Fourier Transform Infrared Spectroscopy, Humana Press, New Jersey, 1993, vol. 22 Search PubMed.
- K. Kuwajima, Biomolecules, 2020, 10, 407 CrossRef CAS PubMed.
- L. Förster, J. Biol. Chem., 1975, 250, 3738–3745 CrossRef.
- F. Desantis, M. Miotto, L. Di Rienzo, E. Milanetti and G. Ruocco, Sci. Rep., 2022, 12, 12087 CrossRef CAS.
- H. Dong, A. Mukaiyama, T. Tadokoro, Y. Koga, K. Takano and S. Kanaya, J. Mol. Biol., 2008, 378, 264–272 CrossRef CAS.
- J. W. Bye and R. J. Falconer, Protein Sci. Publ. Protein Soc., 2013, 22, 1563–1570 CrossRef CAS.
- A. P. Yakimov, A. S. Afanaseva, M. A. Khodorkovskiy and M. G. Petukhov, Acta Nat., 2016, 8, 70–81 CrossRef CAS.
- L. Zhong and W. C. Johnson, Jr, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4462–4465 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00720d |
| This journal is © The Royal Society of Chemistry 2024 |