
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMiRNA-20a-loaded graphene oxide–polyethylenimine enters bone marrow mesenchymal stem cells via clathrin-dependent endocytosis for efficient osteogenic differentiation
Yujie
Ji
 abc,
Qiaoling
Qing
ab,
Zhaoying
Zhang
ab,
Han
Qin
*ab and
Xuerong
Xiang
*ab
abc,
Qiaoling
Qing
ab,
Zhaoying
Zhang
ab,
Han
Qin
*ab and
Xuerong
Xiang
*ab
aStomatological Hospital of Chongqing Medical University, 426 North Songshi Road, Chongqing 401147, China. E-mail: qinhan@hospital.cqmu.edu.cn
bChongqing Key Laboratory of Oral Diseases, Stomatological Hospital of Chongqing Medical University, 426 North Songshi Road, Chongqing 401147, China
cDepartment of Periodontics and Oral Mucosal Disease, Wuxi Stomatological Hospital, Wuxi, Jiangsu, 214000, China
First published on 17th July 2024
Abstract
Precise control of osteogenic differentiation of stem cells through osteogenesis-promoting microRNAs (miRNAs), such as miR-20a, is an exceptionally promising strategy to enhance bone regeneration. However, due to the difficulty of miRNAs in penetrating the negatively charged cell membrane and their susceptibility to degradation by Rnase in vivo, vectors are needed to protect miRNAs and effectively deliver them into cells. Graphene oxide (GO) has gained prominence as a vector for drug and gene delivery due to its outstanding physicochemical properties. In this study, the polyethyleneimine (PEI)-functionalized GO complex (GO–PEI) efficiently delivered miR-20a into rat bone marrow mesenchymal stem cells (BMSCs), resulting in a sustained high level of miR-20a in the cells. The GO–PEI/miR-20a complex could significantly promote osteogenic differentiation of BMSCs under the combined action of GO–PEI and miR-20a. In addition, the cellular uptake mechanism of the GO–PEI/miR-20a complex was investigated, confirming that it entered BMSCs via clathrin-mediated, energy-dependent endocytosis. In summary, the GO–PEI/miR-20a complex represents an effective platform for enhancing osteogenesis, and the elucidation of its cellular internalization mechanism contributes to a deeper understanding of the interactions between GO–PEI and cells.
Introduction
Bone exhibits the intrinsic capacity for regeneration and undergoes continuous self-remodeling throughout life. However, there are complex clinical conditions in which bone regeneration is required in large quantity, such as for skeletal reconstruction of large bone defects, or cases in which the regenerative process is compromised, including avascular necrosis, atrophic non-unions and osteoporosis.1 Recently, numerous research studies have harnessed RNA interference (RNAi) technology as a therapeutic strategy for bone regeneration.2–4 This approach can effectively and specifically regulate the intracellular expression of certain proteins by introducing exogenous short interfering RNAs (siRNAs) or microRNAs (miRNAs), thereby accelerating osteogenesis. MiRNA is a highly conserved type of endogenous non-protein-encoding RNA of about 19–25 nucleotides in length, which plays roles in regulating cell proliferation, differentiation, apoptosis, and development by binding to the 3′ untranslated region of target mRNAs, whereby they can degrade or induce translational silencing.5 It has been reported that miR-20a, a significant regulator of bone metabolism,6 promotes osteogenic differentiation in human bone marrow mesenchymal stem cells (BMSCs),7 adipose stem cells8 and dental pulp stem cells,9 which suggests the potential of utilizing miR-20a in RNAi therapy for bone regeneration based on stem cells. Due to the susceptibility of miRNAs to degradation by RNase, vectors are essential for protecting miRNAs and ensuring their efficient delivery into cells.10Currently, a variety of nanomaterials are attracting increasing attention, such as silica and silicon-based nanoparticles, metal and metal oxide nanoparticles, carbon nanoparticles, dendrimers, polymers, etc.11 They hold the potential to overcome the challenges and barriers in miRNA delivery due to their unique physicochemical properties.12–15 Graphene oxide (GO) is a highly oxidized form of graphene produced by chemical oxidation of large graphite powders, with a thin atomic structure similar to graphene.16 GO is characterised by good biocompatibility, strong mechanical properties, colloidal stability, abundant oxygen-containing functional groups and a large specific surface area.17 These attributes allow GOs to deliver drugs,18 proteins19 and nucleic acids20via π–π stacking, electrostatic or hydrophobic interactions. But the presence of negative charges on both GO and nucleic acids could potentially hinder the efficient loading of nucleic acids. Covalent modification of GO is a common method to enhance the delivery efficiency and improve its stability. Polyethyleneimine (PEI) is a polycationic derivative that can efficiently condense nucleic acids to transfer into the cells and is regarded as a golden standard polymer for endosomal escape of genes.21 It has been reported that PEI functionalized GO (GO–PEI) was successfully employed as a DNA vector, and it showed higher gene delivery efficiency than bare PEI.22 However, GO–PEI was mainly used as a drug and a gene carrier for cancer cells previously; little research has been reported on its use as a miRNA vector for bone regeneration enhancement.23
The present work investigated the potential of using positively charged GO–PEI as a carrier for miR-20a to facilitate the osteogenic differentiation of BMSCs. The cellular delivery efficiency and osteogenic differentiation-promoting ability of GO–PEI/miR-20a were assessed. Moreover, despite the notable progress in the biomedical applications of GO, there remains a lack of in-depth understanding of the cellular uptake mechanism of GO.16,24 Therefore, this study further examined the cellular uptake mechanism of the GO–PEI/miR-20a complex to provide a scientific basis for the design and optimization of GO–PEI-based delivery systems.
Materials and methods
Preparation and characterization of the GO–PEI/miR-20a complex
PEI functionalized GO (GO–PEI) with a diameter ranging from 190 to 320 nm was purchased from Nanjing/Jiangsu XFNANO Materials Tech Co., Ltd (Nanjing, China). The sense strand sequence of miR-20a was 5′-UAAAGUGCUUAUAGUGCAGGUAG (Tsingke Biotechnology Co., Ltd, Beijing, China). After ultrasonic treatment of GO–PEI for 30 minutes, GO–PEI/miR-20a complexes of different N/P ratios (0, 5, 10, 20, 40 and 80) were obtained by homogeneously mixing the appropriate volume of GO–PEI solution (0.5 mg mL−1) and miR-20a solution (10 μM) on ice for 20 min.The morphology of GO–PEI was examined using transmission electron microscopy (TEM; JEOL JEM-F200, Japan). The surface charge and size of the samples in deionized water was measured using a nanoparticle size potentiometer (Malvern Zetasizer Nano ZS90, UK). The UV-Vis absorption spectra from 200 to 800 nm for GO–PEI, miR-20a, and GO–PEI/miR-20a samples were recorded using a UV-Vis spectrophotometer (Shimadzu UV-3600, Japan).
Cell culture
BMSCs were harvested from the bone marrow of two-week-old Sprague-Dawley (SD) rats that were purchased from GemPharmatech Co., Ltd. Cells were cultured in α-minimum essential medium (α-MEM; Hyclone, Logan, UT, United States) supplemented with 10% fetal bovine serum (FBS; Gibco, United States), 100 U mL−1 of penicillin, and 100 μg mL−1 of streptomycin. The BMSCs were cultured in incubation at 37 °C, and the medium was replaced every 3 days. The BMSCs at passages 2–6 were used for subsequent studies.Cell viability measurements
The cellular toxicity of GO–PEI was examined using a CCK-8 assay kit (AbMole, United States). Briefly, BMSCs were cultured in 24-well plates at a density of 5 × 104 per well. 6 h after being seeded, GO–PEI was added to the cells at final concentrations of 5, 10, 20, 30, 40, 50 and 60 μg mL−1. Cells without GO–PEI treatment were set as the control group. After 24 and 48 h, the medium was removed, and the mixture of CCK-8 solution and fresh medium with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 was added to each well. Then, the cells were incubated at 37 °C for 2 h. The absorbance was tested by using a SpectraMAX iD3® Multi-mode microplate reader (Sunnyvale, CA, USA) at 450 nm. The cytotoxicity of cells treated with free miR-20a, GO–PEI (5 μg mL−1), Lipo8000 Transfection Reagent (Lipo; Beyotime, Beijing, China), GO–PEI/miR-20a and Lipo8000/miR-20a (Lipo/miR-20a) for 2, 4, and 6 days was further examined as described above.
:10 was added to each well. Then, the cells were incubated at 37 °C for 2 h. The absorbance was tested by using a SpectraMAX iD3® Multi-mode microplate reader (Sunnyvale, CA, USA) at 450 nm. The cytotoxicity of cells treated with free miR-20a, GO–PEI (5 μg mL−1), Lipo8000 Transfection Reagent (Lipo; Beyotime, Beijing, China), GO–PEI/miR-20a and Lipo8000/miR-20a (Lipo/miR-20a) for 2, 4, and 6 days was further examined as described above.
Confocal microscopy studies
Cy3-labeled miR-20a (miR-20a-cy3; 10 μM) solution was mixed with GO–PEI at different N/P ratios and Lipo for appropriate time periods, respectively. Then, BMSCs were treated with GO–PEI/miR-20a-cy3 (red fluorescence) and Lipo/miR-20a-cy3 complexes at a final miRNA concentration of 50 nM for 6 hours. Next, the cells were extensively washed with cold PBS and fixed with 4% formaldehyde for 30 min. After rinsing twice with PBS, the cells were stained with antifade mounting medium with DAPI (Beyotime, Beijing, China) for cell nucleus (blue fluorescence). Finally, the slides were mounted and observed using a confocal laser scanning microscope (CLSM; Leica, Germany). Cell counts and nomalized relative fluorescence intensity were obtained using ImageJ.Real-time PCR analysis
To determine the relative levels of miR-20a, BMSCs were treated with GO–PEI/miR-20a and Lipo/miR-20a complexes at the same final concentration of miR-20a (50 nM) for 6, 12, 24, 48, 72 and 96 hours. Then, total RNA was extracted from BMSCs using an RNA Isolation Kit (Beyotime, Beijing, China). The relative mRNA expression of miR-20a was determined using an miRcute Plus miRNA First-Strand cDNA Kit and miRcute Plus miRNA qPCR Kit (TIANGEN, China). The reaction and detection were conducted using a CFX96 real-time PCR detection system (Bio-Rad Laboratories, Inc., Hercules, CA, United States). The cycle threshold (Ct) values were collected and normalized to the level of U6, a housekeeping microRNA. The primer sequences of miR-20a and U6 are listed in Table 1.| Gene | Primer sequence (5′–3′) |
|---|---|
| miR-20a | F:GGCTAAAGTGCTTATAGTGCAGGTAG |
| U6 | F:GCTTCGGCAGCACATATACTAAAAT |
| Runx-2 | F:GGCCACTTACCACAGAGCTATTA |
| R:GTGTCTGCCTGGGATCTGTAATC | |
| ALP | F:CCAAAGGCTTCTTCTTGCTAGTG |
| R:TGATCAGCAGTAACCACAGTCAA | |
| OCN | F:GAGGGCAGTAAGGTGGTGAATAG |
| R:GGGTCGAGTCCTGGAGAGTAG | |
| OPN | F:CGCATTACAGCAAACACTCAGAT |
| R:CCGTTTCTTCAGAGGACACAGAA | |
| GAPDH | F:GAAGGTCGGTGTGAACGGAT |
| R:CCCATTTGATGTTAGCGGGAT |
To measure the relative expression levels of osteogenic differentiation markers (Runx-2, ALP, OCN and OPN), BMSCs were treated with free miR-20a, GO–PEI, GO–PEI/miR-20a, Lipo, and Lipo/miR-20a in osteogenic medium for 3, 7, and 14 days. The osteogenic medium contained basal growth medium with 50 μg mL−1 ascorbic acid (Solarbio, Beijing, China), 10 mM β-glycerophosphate (Sigma-Aldrich, Germany), and 10 nM dexamethasone (Solarbio, Beijing, China). The medium was changed every three days, and the complexes and miRNA were added accordingly. The relative levels of genes were quantified using ABScript III RT Master Mix for qPCR and 2× Universal SYBR Green Fast qPCR Mix (ABclonal, Wuhan, China). The relative quantification of target genes was normalized to that of GAPDH, and the 2−ΔΔCt method was used to calculate the gene expression. The primer sequences used were synthesized by Tsingke Biotechnology and are listed in Table 1.
Alkaline phosphatase (ALP) staining and activity
BMSCs were cultured to confluence in 6-well plates and treated with free miR-20a, GO–PEI, Lipo, GO–PEI/miR-20a and Lipo/miR-20a in osteogenic medium for 7 or 14 days. Next, the cells were rinsed with PBS twice, fixed in 4% paraformaldehyde for 30 minutes, and subsequently incubated with BCIP/NBT ALP working solution (Beyotime, Beijing, China) for 5 minutes. The cells were washed thoroughly with dH2O and then images were acquired. As for ALP activity, the cells were lysed with 0.1% Triton X-100, and the protein concentration was measured using a BCA protein assay kit (Beyotime, Beijing, China). ALP activity was determined at 405 nm using a commercially available kit (Beyotime, Beijing, China) according to the manufacturer's instructions.Alizarin Red S (ARS) staining
BMSCs were cultured for 14 and 21 days and then incubated in an Alizarin Red S Staining Kit for Osteogenesis (Beyotime, Beijing, China) for 20 minutes at room temperature. After incubation, the cells were washed thoroughly with dH2O and air-dried to visualize calcium deposits. For quantitative analysis, the staining was eluted using 10% cetylpyridinium chloride (Solarbio, Beijing, China) at room temperature for 30 minutes. The eluted dye solution was collected and the absorbance was read at 562 nm.Cellular uptake mechanism analysis of the GO–PEI/miR-20a complex
BMSCs were seeded in 6-well culture plates at a density of 105 cells per mL, and then cultured at 37 °C in 5% CO2 humidified incubator. After 24 h, different endocytosis inhibitors were added separately to the culture medium as follows: (1) to inhibit the clathrin-dependent endocytosis pathway, 15 μg mL−1 chlorpromazine (CPZ; AbMole, United States) was used for 0.5 h;25 (2) to inhibit the macropinocytosis pathway, 75 μg mL−1 amiloride (Solarbio, Beijing, China) was used for 1 h;26 and (3) to inhibit caveolae-mediated endocytosis, 10 mM methyl-β-cyclodextrin (MβCD; Solarbio, Beijing, China) was used for 0.5 h.27 Subsequently, the culture medium was removed and the cells were thoroughly washed twice with PBS. Next, fresh medium with GO–PEI/miR-20a-cy3 (N/P = 20) was added and maintained for 6 h at 37 °C. To hinder the energy-dependent endocytosis pathway, another group of cells without inhibitors was treated with GO–PEI/miR-20a-cy3 for 6 hours at 4 °C.28 The final concentration of miR-20a in all groups is 50 nM. The cells without inhibitor pretreatment and cultured at 37 °C were set as the control. Next, the cells were extensively washed with cold PBS and fixed with 4% formaldehyde for 30 min. After rinsing twice with PBS, the cells were stained with Actin-Tracker Green-488 (Beyotime, Beijing, China) for cell skeleton (green fluorescence), and finally, DAPI for cell nucleus (blue fluorescence). The slides were observed under an upright fluorescence microscope (ZEISS, Germany). Cell counts and nomalized relative fluorescence intensity were obtained using ImageJ.Statistical analysis
All experiments were repeated at least three times to confirm the reliability of the study. Data were provided as mean ± standard deviation (SD). Statistical comparisons between different groups or two groups were evaluated by one-way ANOVA test or t-test comparison. In all of the statistical evaluations, *p< 0.05, **p< 0.01 and ***p< 0.001 were considered statistically significant.Results
Characterization of GO–PEI and the GO–PEI/miR-20a complex
GO has been extensively researched in the field of nanomedicine owing to its exceptional physicochemical, electrical, and optical properties. A physiologically stable polymer-functionalized nano-GO conjugate (GO–PEI) was obtained by covalently conjugating PEI with GO through amide bonds. Other studies have reported in detail the synthesis of GO–PEI.29 GO–PEI and miR-20a interact electrostatically to form GO–PEI/miR-20a nanoparticles. The GO–PEI complex is stable in both PBS and cell culture medium without significant aggregation. TEM images demonstrated the overlapping surfaces of GO–PEI (Fig. 1a). The nanoparticle size potentiometer analysis demonstrated that both GO–PEI and GO–PEI/miR-20a were positively charged. The zeta potential of GO–PEI was 11.7 ± 4.58 mV, while that of GO–PEI/miR-20a was slightly lower at 8.11 ± 5.44 mV (Fig. 1b). Dynamic light scattering (DLS) analysis showed that the average diameters of GO–PEI and GO–PEI/miR-20a were 165.1 ± 17.99 nm and 423.0 ± 62.52 nm, respectively (Fig. 1c). The UV-vis spectrum exhibited UV absorption peaks at about 260 nm for miRNA and GO–PEI/miR-20a (Fig. 1d). The results suggested that positively charged GO–PEI complexes could bind and encapsulate negatively charged miRNAs.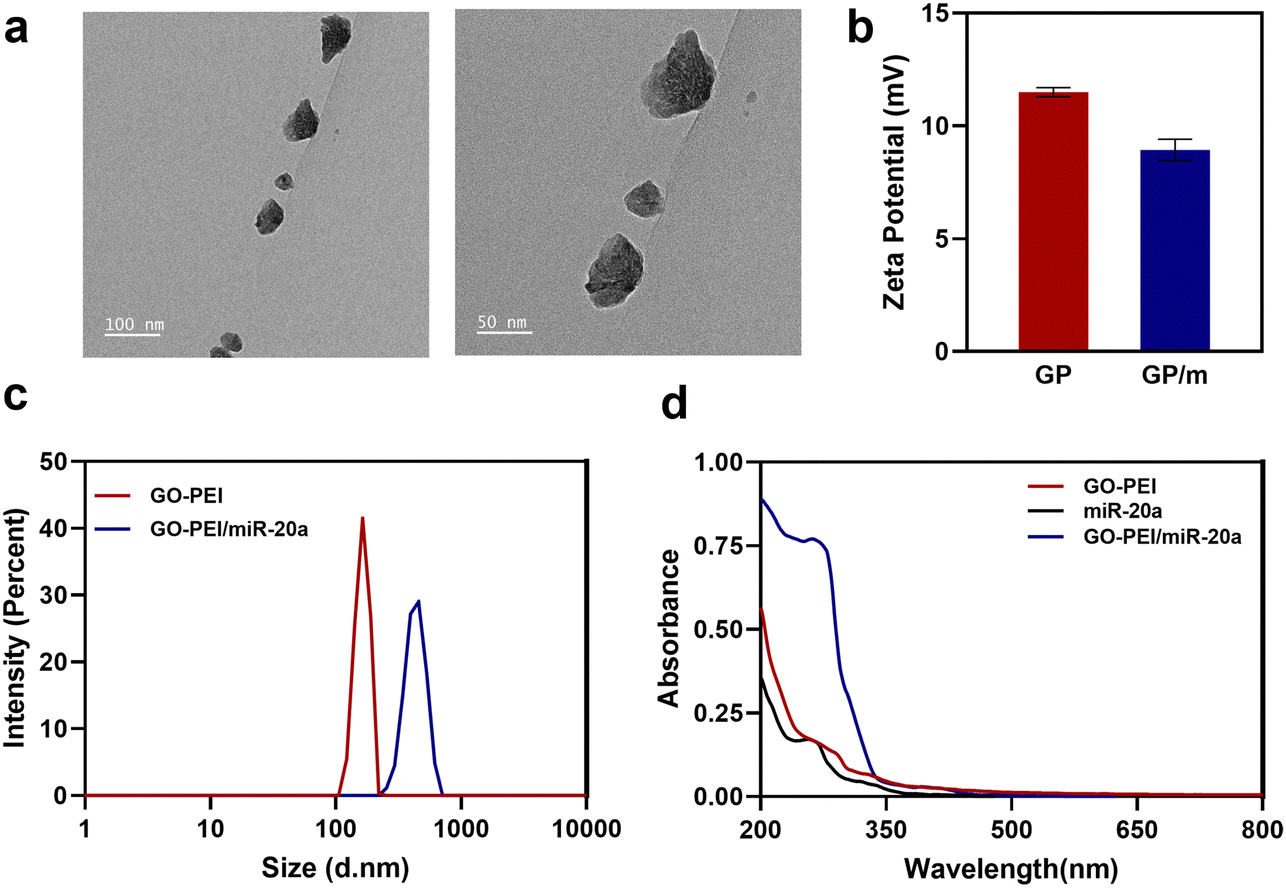 | ||
| Fig. 1 Characterization of GO–PEI and GO–PEI/miR-20a complexes. (a) TEM images of GO–PEI. Scale bars: 100 nm and 50 nm. (b) Measurement of the zeta potential of GO–PEI (GP) and GO–PEI/miR-20a (GP/m) complexes. (c) Size distributions of GO–PEI and GO–PEI/miR-20a measured by DLS analysis. (d) UV-vis absorption spectra from 200 to 800 nm for GO–PEI (red line), free miR-20a (black line), and the GO–PEI/miR-20a complex (blue line) in phosphate buffer. | ||
Biocompatibility and delivery efficiency of the GO–PEI/miR-20a complex
Low cytotoxicity is a prerequisite for potential nucleic acid delivery vectors. The cell proliferation ability of BMSCs treated with different concentrations of GO–PEI was examined using Cell Count Kit-8 (CCK-8). The results showed that the viability of BMSCs was not significantly affected by GO–PEI at a concentration of up to 40 μg mL−1 after 24 and 48 hours (Fig. 2a). Interestingly, 10 μg mL−1 GO–PEI showed a proliferative effect on the cells after 48 hours. In the following study, the maximum working concentration of GO–PEI used was less than 40 μg mL−1. The cytotoxicity of GO–PEI and commonly used lipid transfection agents was further compared. After 4 and 6 days, the absorbance of cells treated with lipo and lipo/miR-20a was significantly lower compared to those treated with GO–PEI and the GO–PEI/miR-20a complex (Fig. 2b), indicating that GO–PEI showed lower long-term cytotoxicity.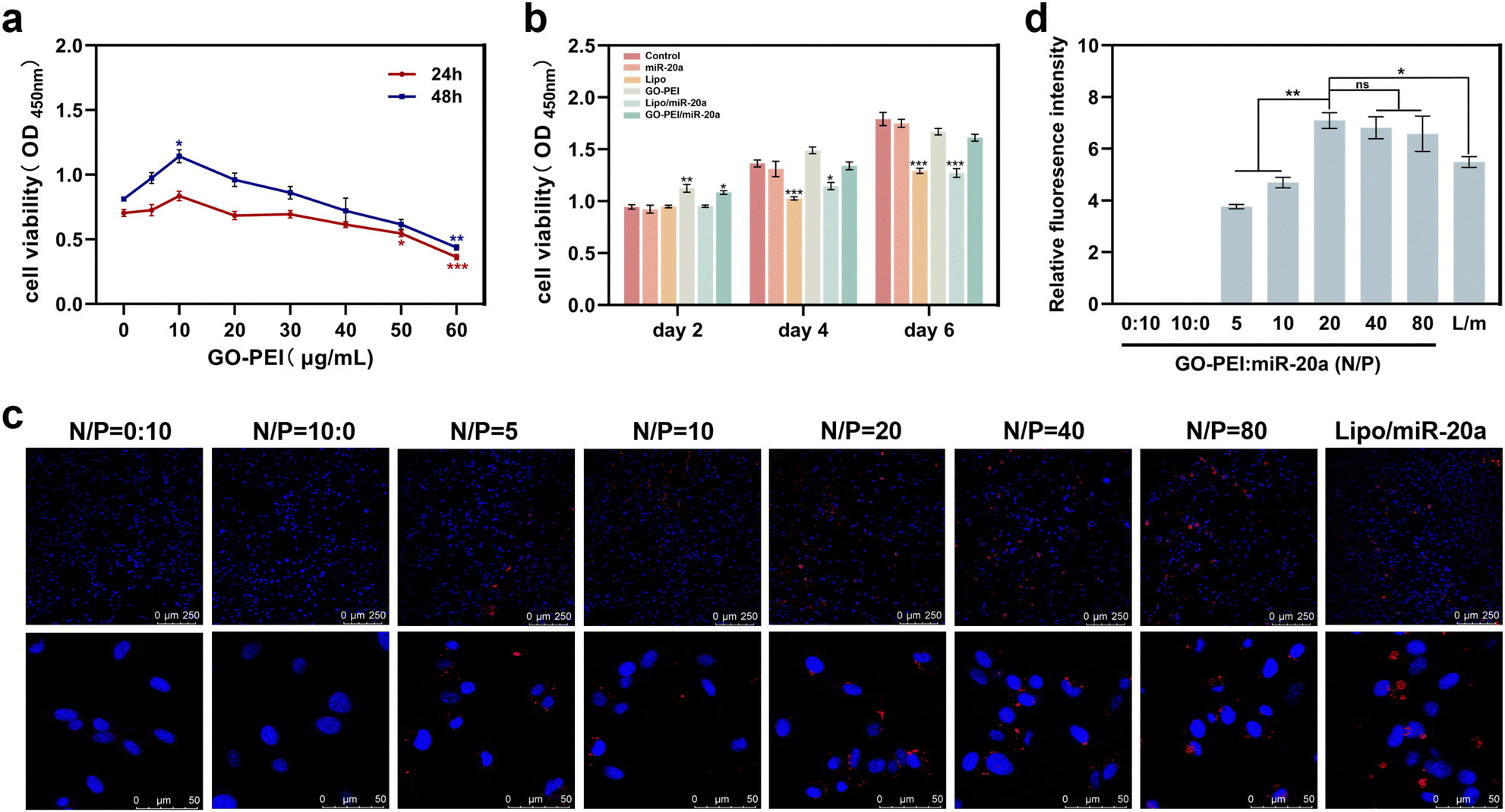 | ||
| Fig. 2 Biocompatibility and delivery effectiveness of the GO–PEI/miR-20a complex. (a) The viability of BMSCs was measured by the CCK-8 assay after treatment with GO–PEI at various concentrations (0, 5, 10, 20, 30, 40, 50 and 60 μg mL−1) for 24 and 48 h. (b) The viability of BMSCs was measured from different groups after 2, 4, and 6 days of culture. (c) Confocal microscopy images of BMSCs after treatment with GO–PEI/miR-20a-cy3 at different N/P ratios and lipo/miR-20a-cy3. MiR-20a was labeled with cy3 (red) and the nuclei were stained with DAPI (blue). (d) Relative fluorescence intensity of BMSCs after treatment with GO–PEI/miR-20a-cy3 at different N/P ratios and lipo/miR-20a-cy3 (L/m). | ||
Confocal microscopy studies were carried out to examine the delivery of miR-20a. The images indicated that free miR-20a-cy3 was unable to enter the cells, whereas dispersed red fluorescence signals were clearly observed in the cells treated with GO–PEI/miR-20a-cy3 and lipo/miR-20a-cy3 (Fig. 2c). In samples with N/P ratios below 20, the relative fluorescence intensity of miR-20a-cy3 in cells was weak. The relative fluorescence intensities in samples with N/P ratios of 20, 40 and 80 were not significantly different and were higher than those of groups treated with lipo/miR-20a-cy3 (Fig. 2d). To ensure high delivery efficiency and low cytotoxicity, the GO–PEI/miR-20a complex with an N/P ratio of 20 was used. The levels of miR-20a in cells at different time points were further evaluated by qPCR. Compared to the control group, the relative levels of miR-20a in BMSCs treated with GO–PEI/miR-20a and lipo/miR-20a increased continuously over time, peaking at 24 hours at approximately 140-fold and 70-fold, respectively (Fig. 3). The level of miR-20a gradually decreased after 48 h and then increased again after the reintroduction of GO–PEI/miR-20a and lipo/miR-20a. At each time point, the levels of miR-20a in the GO–PEI/miR-20a group were 2–3 times higher than those in the Lipo/miR-20a group. All these results indicated that GO–PEI could deliver miR-20a efficiently.
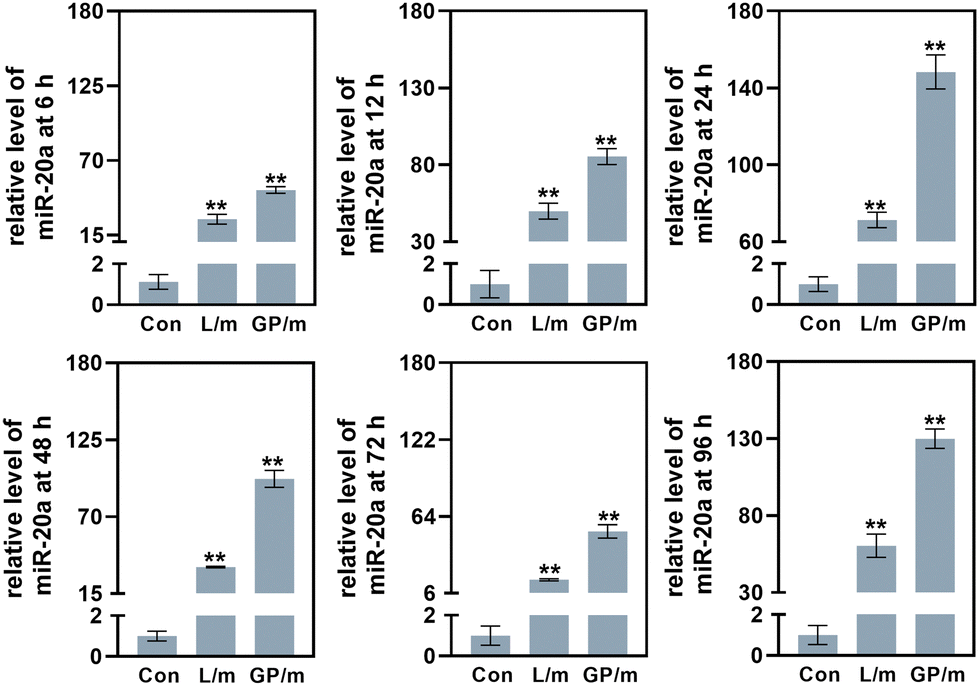 | ||
| Fig. 3 The levels of miR-20a in cells treated with GO–PEI/miR-20a (GP/m) and lipo/miR-20a (L/m) at 6, 12, 24, 48, 72 and 96 h. | ||
GO–PEI/miR-20a complex facilitated osteogenic differentiation of BMSCs
To evaluate the effects of GO–PEI/miR-20a and GO–PEI on the osteogenic differentiation of BMSCs, key markers of early differentiation (Runx2 and ALP) and mineralization (OCN and OPN) stages were measured by qRT-PCR. Compared with the control group, the levels of the four markers did not significantly change in cells treated with free miR-20a and Lipo groups at each tested time points (3, 7, and 14 days). While the levels of these markers obviously elevated to varying degrees in the GO–PEI, lipo/miR-20a, and GO–PEI/miR-20a groups (Fig. 4). Among them, the cells treated with GO–PEI/miR-20a consistently displayed the highest levels of osteogenic differentiation markers at all time points.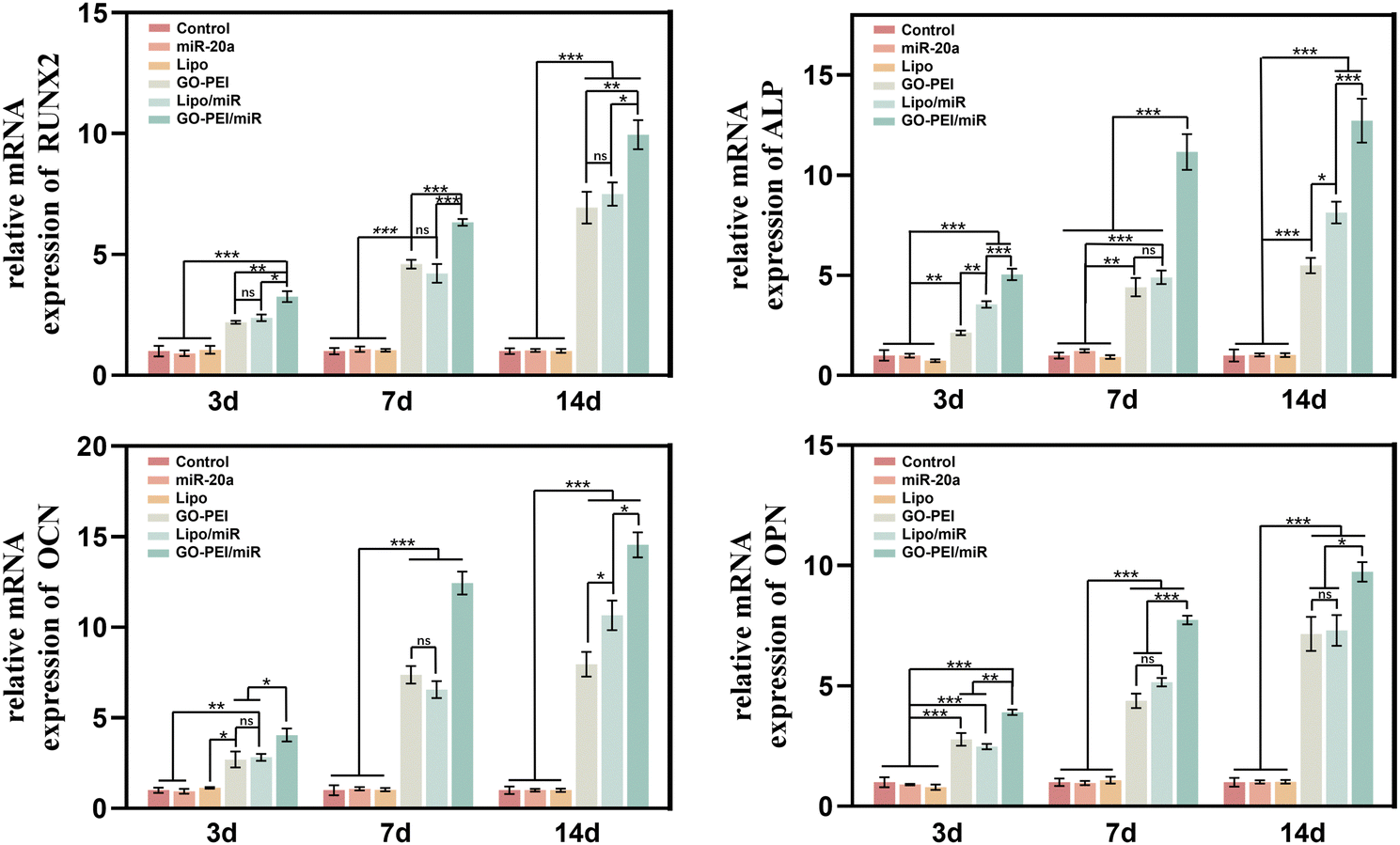 | ||
| Fig. 4 Relative expression levels of osteogenesis-related genes, Runx-2 (a), ALP (b), OCN (c), and OPN (d), at days 3, 7 and 14. | ||
ALP is an osteogenesis marker that is often highly expressed in osteoblasts and is associated with the early onset of mineralization in newly formed bone.30 ALP staining results showed a similar trend with the gene expression assays. Cells treated with GO–PEI, Lipo/miR-20a and GO–PEI/miR-20a appeared significantly darker in blue-purple color than the control, free miRNA and Lipo groups, indicating higher ALP activity in these groups (Fig. 5a). The GO–PEI/miR-20a group exhibited the highest ALP activity at both 7 and 14 days. The results of the quantitative study of ALP were consistent with the staining results (Fig. 5b). Furthermore, alizarin red S staining revealed that small mineralized nodules were present in the control, free miRNA, and Lipo groups, whereas calcium deposition was significantly greater in cells treated with GO–PEI, GO–PEI/miR-20a, and Lipo/miR-20a for 14 and 21 days (Fig. 6a). The highest level of calcium deposition was observed in cells treated with GO–PEI/miR-20a. Notably, a large number of mineralized nodules were also detected in the GO–PEI group, which was almost double of the control group after 21 days (Fig. 6b). These results suggested that GO–PEI could promote osteogenic differentiation of BMSCs, and the GO–PEI/miR-20a complex had a more excellent sustaining effect on osteogenic induction.
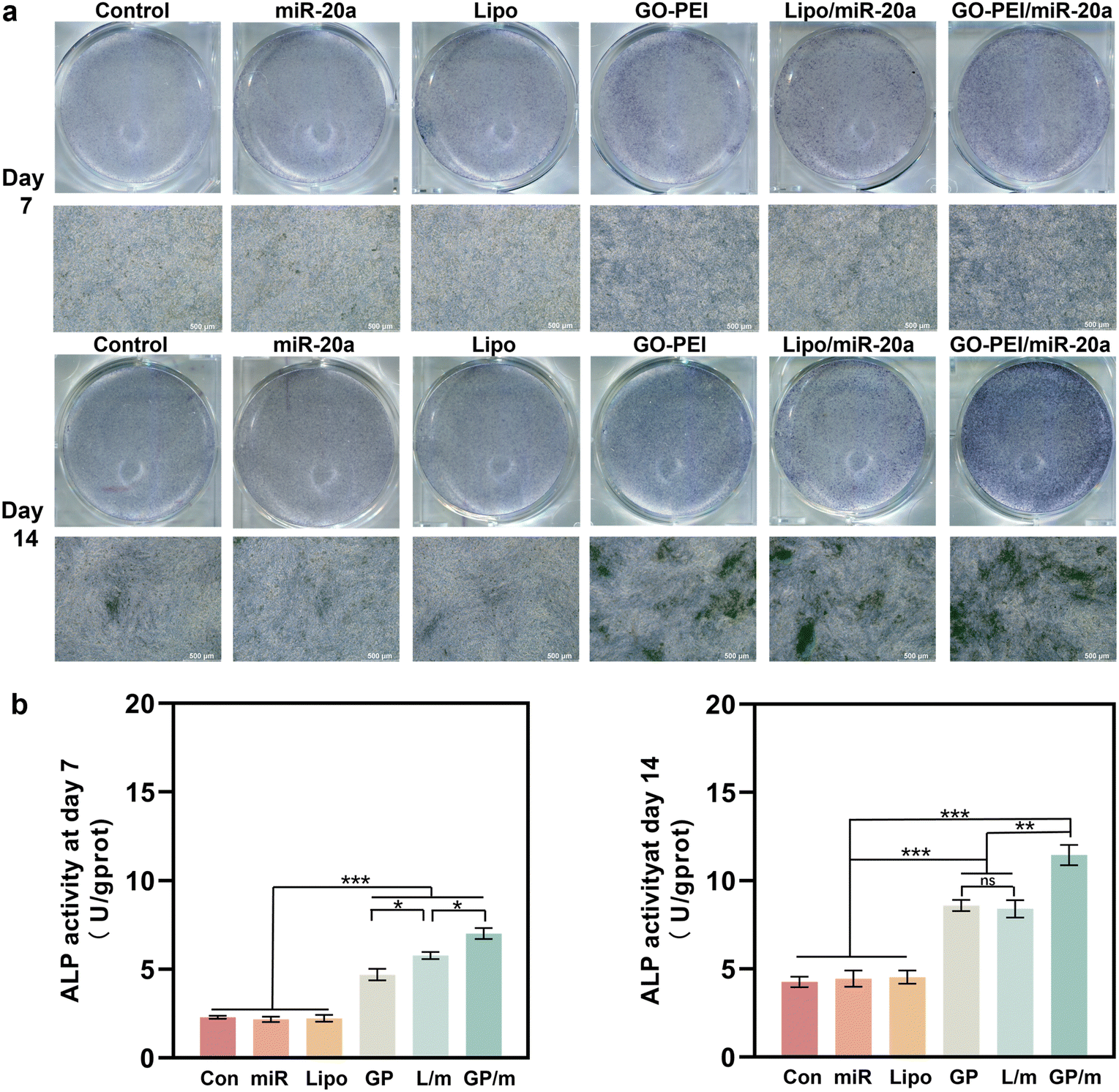 | ||
| Fig. 5 Staining image (a) and quantitative study (b) of ALP activities of BMSCs after 7 and 14 days. | ||
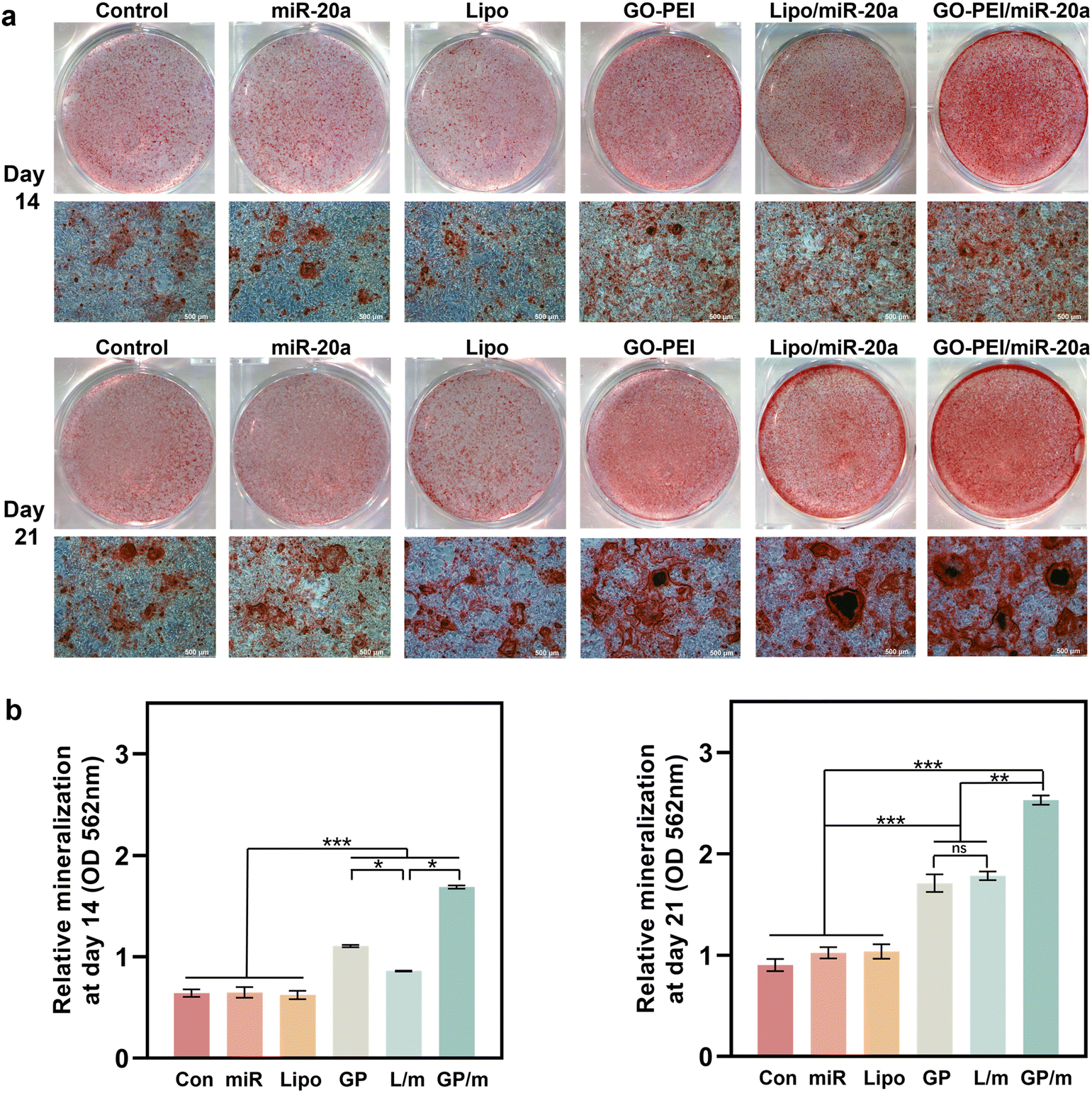 | ||
| Fig. 6 Staining image (a) and quantitative study (b) of ARS staining of BMSCs after 14 and 21 days. | ||
Cellular uptake mechanism of the GO–PEI/miR-20a complex
To elucidate the potential cellular uptake mechanism of the GO–PEI/miR-20a complex, the cells were pretreated with several specific endocytosis inhibitors.31,32 The energy dependency of cellular uptake was also investigated by incubating the cells under low temperature conditions (4 °C).28,33 In the control group (without inhibitor pretreatment), GO–PEI/miR-20a-cy3 entered the cells after mere 30 minutes (Fig. 7a) and accumulated in the cells after 1 hour (Fig. 7b). Fluorescent particles (indicated by arrows) were frequently observed on the cell membrane (Fig. 7c) and in the cytoplasm near the nucleus (Fig. 7d). Cellular viability assays demonstrated minimal cytotoxicity of the three inhibitors including CPZ (clathrin inhibitor), amiloride (actin inhibitor), and MβCD (caveolae inhibitor) at their working concentrations (Fig. 7e). In cells pretreated with CPZ, the red fluorescence was significantly reduced (Fig. 7g) and the relative fluorescence intensity decreased to approximately 12% of the control group (Fig. 7f). The relative fluorescence intensities of the cells pretreated with amiloride and MβCD showed no significant reduction. Furthermore, the cells cultured under low temperature conditions internalized only a very small amount of GO–PEI/miR-20a-cy3. The results suggested that the GO–PEI/miR-20a complex rapidly entered BMSCs mainly via clathrin-mediated endocytosis, which was an energy dependent process.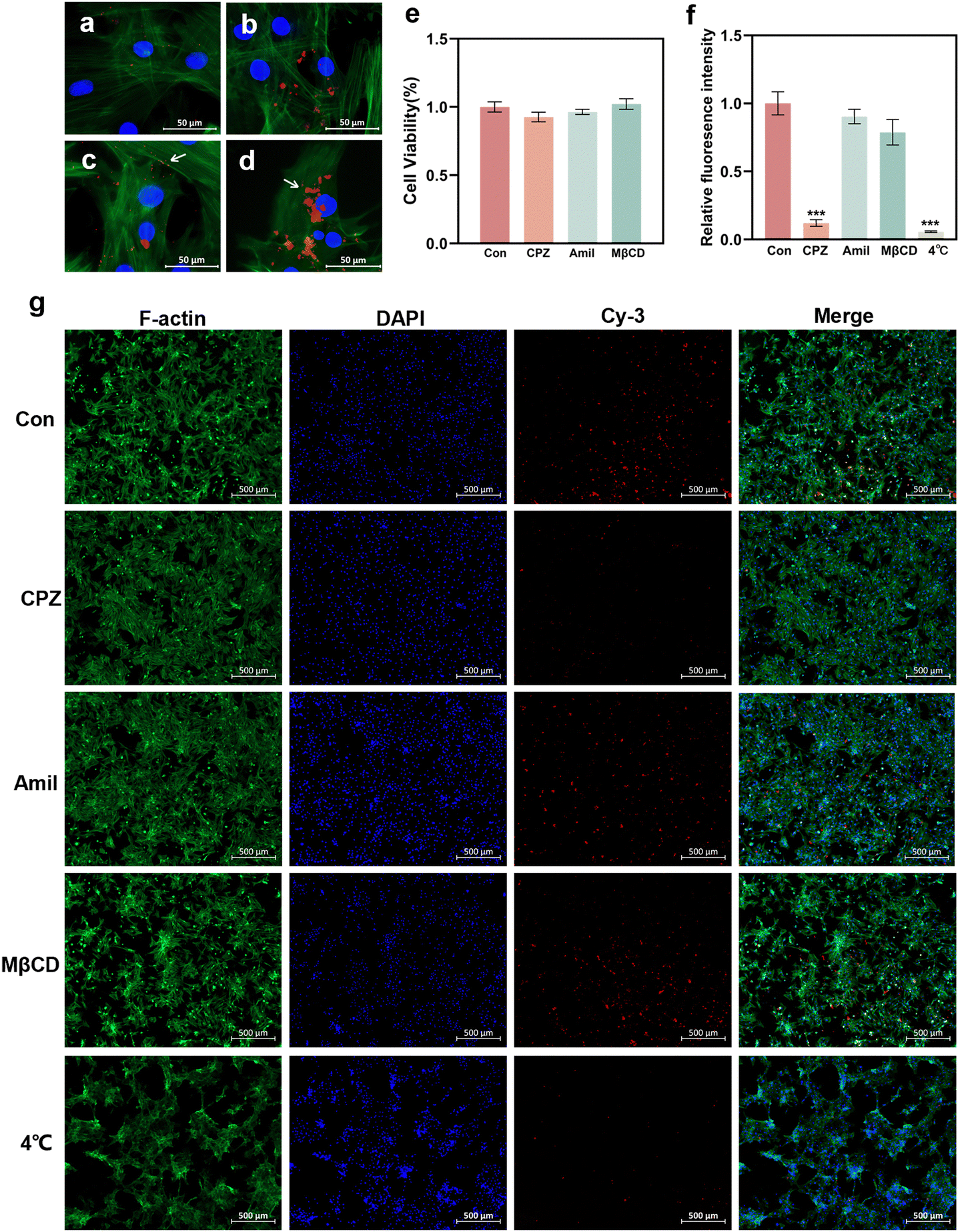 | ||
| Fig. 7 Cellular uptake mechanism of the GO–PEI/miR-20a complex. Fluorescence microscopy images of cells without inhibitor pretreatment: (a) 30 min after incubation with GO–PEI/miR-20a-cy3; (b) 1 h after incubation with GO–PEI/miR-20a-cy3; (c) fluorescent particles (labeled with arrows) located on the cell membrane; (d) fluorescent particles (labeled with arrows) located in the cytoplasm near the nucleus. (e) Cytotoxicity of endocytosis inhibitors. (f) Relative fluorescence intensities of cells with specific endocytosis inhibitor pretreatment or under low temperature conditions. (g) Fluorescence microscopy images. Red: GO–PEI/miR-20a-cy3; blue: nucleus; and green: cytoplasmic F-actin filaments. | ||
Discussion
Increasing evidence indicates that miRNAs are critical regulators of stem cell differentiation and the use of miRNA mimics is a potential strategy to improve bone formation.34 MiR-20a is a member of the miR-17–92 gene cluster, which plays a crucial role in bone development and osteogenic differentiation.35 MiR-20a promotes osteogenic differentiation by directly down-regulating several inhibitors of osteoblast differentiation.6 Researches have shown that miR-20a could enhance bone formation in rat calvarial bone defects36 and promote rat cranial bone augmentation.37 In addition, miR-20a can negatively regulate osteoclast proliferation and differentiation through different pathways.38,39 Despite the important role of miR-20a in bone homeostasis, the challenges associated with miRNA delivery, such as its difficulty in crossing the cell membrane and rapid degradation, have limited its application in RNAi therapy.In recent years, numerous studies have concentrated on the development of carbon nanomaterials as delivery platforms.15 GO is a representative two-dimensional carbon nanomaterial, characterized by its ease of synthesis, controlled particle size, and large specific surface area.40 The current study indicated that prolonged exposure to water induces the gradual degradation of GO flakes, converting them into humic acid-like structures.41 Graphene did not cause appreciable toxicity to treated mice when appropriately functionalized and administered in controlled dosages.42 GO could be gradually cleared primarily through kidneys and feces excretion pathways.43,44 Good biocompatibility and clearance have laid the foundation for GO as a novel miRNA vector. However, both GO and nucleic acids carry negative charges, leading to electrostatic repulsion between them. Cationic PEI can preserve the biological activity of the miRNA by inducing endosomal release.45 The escape from the endo-/lysosomal compartment is attributed to the interplay of three effects, namely of the proton sponge effect, umbrella effect, and disruptive interaction with the endosomal membrane.46 The high cytotoxicity of PEI due to its high positive charge density and non-degradability is a major obstacle that limits its application in RNAi therapy. The cytotoxicity of PEI depends on its molecular weight, structure and concentration.21
In this study, PEI-functionalized GO was employed, it was beneficial for combining the attractive characteristics of GO and PEI to achieve an efficient nanocarrier. PEI endowed the complex with a positive charge, thereby facilitating its binding to miRNA and enhancing both its cellular uptake and delivery efficiency. Meanwhile, GO–PEI at a low mass ratio was significantly less cytotoxic than PEI, which might be due to the greater biocompatibility of GO than PEI.47 The CCK-8 assay showed that the cytotoxicity of GO–PEI was concentration-dependent. The viability of BMSCs was not significantly affected by GO–PEI at a concentration of up to 40 μg mL−1. In addition, GO–PEI demonstrated a tendency to promote cell proliferation at low concentration. This could be attributed to the abundant oxygen-containing functional groups present in GO, which enhance the polarity and hydrophilicity of the surface, thereby facilitating the adhesion of proteins and cells.48,49 Then, it was shown that GO–PEI efficiently delivered miR-20a into the cells, which resulted in a sustained high level of miR-20a. These results indicate that GO–PEI exhibited significant advantages as a gene delivery vector.
Following the demonstration of consistent and efficient delivery of miR-20a by GO–PEI, the effect of GO–PEI/miR-20a on promoting osteogenic differentiation of BMSCs was further examined. Runx2, a key transcription factor, is crucial for the commitment of mesenchymal stem cells to the osteoblast lineage and regulates the expression of osteoblast-specific genes.50 ALP is an early marker of osteogenic differentiation, OCN and OPN are secreted by osteoblasts and activated in the later stages of bone differentiation, particularly in the mineralization process.51,52 The qRT-PCR assay showed the most significant increase in the expression of osteogenesis-related genes in cells treated with the GO–PEI/miR-20a complex at tested time points (3, 7 and 14 days). ALP staining and activity assays as well as ARS staining and quantification demonstrated a consistent trend with gene upregulation. Given the above, it could be inferred that after entering the cells, the GO–PEI/miR-20a complex escaped the endo-/lysosomal pathway and then effectively increased the level of miR-20a. Subsequently, miR-20a activated the BMP/Runx2 signaling pathway by co-repressing several target genes, including PPARγ, Bambi and Crim1, thereby enhancing the osteogenic differentiation of BMSCs.6,9,53 However, it is not yet clear at which stage the nucleic acid dissociates from the complex and where the free miRNA resides.46 Meanwhile, it is noteworthy that GO–PEI alone also significantly promoted the osteogenic differentiation of BMSCs. This could be attributed to its physical properties, such as porous wrinkled surface, large specific surface area and high Young's modulus. These characteristics could induce changes in cell adhesion, tension and shape, facilitating spontaneous osteoblastic differentiation of cells through the activation of the integrin/FAK axis.54–56 Furthermore, GO can also function as a preconcentration platform for osteoinductive agents, hastening the commitment of BMSCs to the osteogenic lineage.49,57,58 These findings suggested that the GO–PEI/miR-20a complex was highly effective in promoting osteogenic differentiation of BMSCs, with both components, GO–PEI and miR-20a, actively contributing to the process. Recent research studies have applied GO in scaffolds for bone tissue engineering, aiming to improve their mechanical properties, hydrophilicity, immunomodulatory ability, antibacterial ability, and osteoinductive activity.59,60 Further research could combine GO–PEI/miR-20a with bone regeneration scaffolds, such as hydrogels, with a view to enhancing the properties of the composite and achieving more precise and controllable release.61
With the advance of nanotechnology, elucidating the role of endocytosis in nanoparticle internalization is becoming increasingly vital. This would be key to understand the post-internalization destiny and toxicological characteristics of nanoparticles. Endocytosis is generally categorized into two types: clathrin-dependent endocytosis and clathrin-independent endocytosis. Clathrin-independent endocytosis primarily includes caveolin-dependent endocytosis, clathrin and caveolae-independent endocytosis, macropinocytosis and phagocytosis.62 Nanoparticles must bind to the surface of the cell membrane in all these types of transport and are then enveloped by the membrane to form a transport vesicle. Eventually, the transport vesicle detaches from the cell membrane and is directed to the appropriate cell organelle such as endosome or lysosome. This study revealed that the GO–PEI/miR-20a complex was internalized efficiently by BMSCs primarily through clathrin-mediated endocytosis, which is an energy-dependent process. Previous studies have reported that GO is actively internalized via phagocytosis,26 macropinocytosis,63 clathrin-dependent endocytosis,64 caveolin-dependent endocytosis25 or endocytosis mediated by other factors. The size of the nanoparticle is often considered to be one of the main factors influencing cellular uptake. Mammalian cells internalize particles with sizes ranging from 100 nm to about 300 nm via clathrin-mediated endocytosis, particles with sizes ranging from 50 to 80 nm via caveolin-mediated endocytosis and larger particles (0.5–2 μm) via micropinocytosis.65 The average size of GO–PEI in this study was 165.1 ± 17.99 nm, which meets the requirement for clathrin-mediated endocytosis. The average size of GO–PEI/miR-20a was 423.0 ± 62.52 nm, and the increase in the complex size due to the addition of miR-20a did not appear to affect the uptake mechanism of the complex. However, it could not be completely excluded that GO–PEI/miR-20a was internalized through caveolin-mediated endocytosis or macropinocytosis. Whether the difference in the relative fluorescence intensity between cells pretreated with and without amiloride and MβCD was statistically significant or not is difficult to elucidate at this stage. Because multiple entry mechanisms often operate simultaneously during the process of endocytosis, and the inhibitory effect of inhibitors may depend on the type of cell line.65,66 Besides size, the endocytosis process is significantly influenced by the surface charge of the nanoparticles. Neutral and anionic nanoparticles are internalized considerably less efficiently than cationic nanoparticles because of the high affinity of cations with the negatively charged proteoglycans expressed in the surface of most cells.67,68 Therefore, the positive charge on the surface of GO–PEI facilitated its internalization by BMSCs in this study. Moreover, it should be mentioned that the impact of the size of graphene derivatives on their cellular uptake pathways highly depends on their surface charges: the cellular uptake efficacy of positively charged graphene sheets is size-independent and occurs via phagocytosis and clathrin-mediated endocytosis pathways; for graphene sheets with a negative surface charge, the cellular uptake efficiency is significantly affected by their size.28 In addition to the intrinsic properties, the cellular uptake mechanism and efficiency of GO may also depend on cell types.33,63,69 The uptake rate of GO by mouse BMSCs was considerably higher than that of mouse macrophage cells (Raw264.7) and human epithelial cells (A549), which might be accounted for the abundant intracellular vesicles in BMSCs.70 Moreover, it is worth noting that biotransformation phenomena play an important role in biological interactions between cells and nanomaterials. Bioactive compounds such as enzymes may cause defects on the GO surface or reduce its chemical groups.71 Meanwhile, biomolecules could adsorb onto the surface of GO and affect its endocytosis. For instance, Alnasser et al. found that the internalization of graphene is mediated by the interaction of the specific cellular scavenger receptor B1 and the key protein recognition motif apolipoprotein A-I presented on the graphene surface.72 Taken together, the uptake mechanism of GO–PEI is influenced by multiple factors, further studies need to be undertaken to gather comprehensive biological data as well as to gain a better understanding of its behavior in biological systems.
In summary, as illustrated in Fig. 8, the GO–PEI/miR-20a complex was internalized primarily through clathrin-dependent endocytosis and efficiently promoted the osteogenic differentiation of BMSCs owing to the contribution of both components.
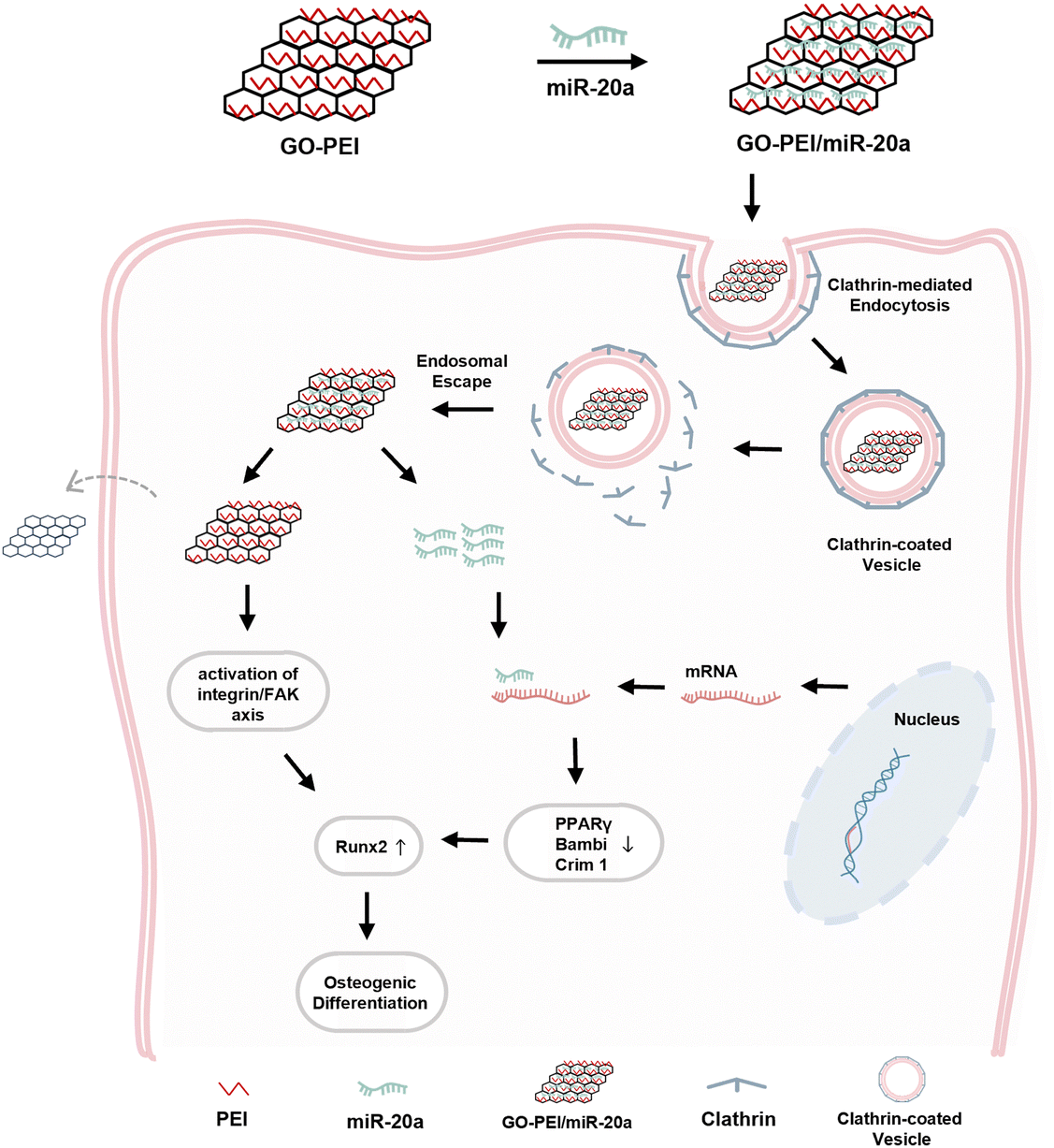 | ||
| Fig. 8 Schematic diagram of GO–PEI/miR-20a entering BMSCs via clathrin-mediated endocytosis and enhancing osteogenic differentiation. | ||
Conclusions
Effective gene delivery can open new avenues for research studies by extending nucleic acid or gene products to the otherwise inaccessible intracellular targets. In this work, the GO–PEI/miR-20a complex served as an effective platform for enhancing osteogenic differentiation, with both GO–PEI and miR-20a playing critical roles. The GO–PEI/miR-20a complex was internalized mainly through clathrin-mediated, energy-dependent endocytosis. The strategy of stem cell-based osteogenesis using the GO–PEI/miR-20a complex represents a promising approach for bone regeneration and deserves further investigation in vivo. The elucidation of its endocytosis mechanism is crucial to gain deeper insights into the cytotoxicity of GO–PEI and the interactions between GO–PEI and cells, which will facilitate its implementation in the field of biomedicine.Author contributions
Yujie Ji: conceptualization, methodology, data curation, formal analysis, software, writing – original draft, and validation. Qiaoling Qing: methodology, validation, and software. Zhaoying Zhang: formal analysis and validation. Han Qin: conceptualization, funding acquisition, project administration, and writing – review & editing. Xuerong Xiang: conceptualization, funding acquisition, and project administration.Data availability
All relevant data are within the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Natural Science Foundation of China (82001063) and the Postdoctoral Science Foundation of Chongqing (cstc2020jcyj-bshX0108).References
- R. Dimitriou, E. Jones, D. McGonagle and P. V. Giannoudis, BMC Med., 2011, 9, 66 CrossRef PubMed.
- N. Z. Laird, T. M. Acri, K. Tingle and A. K. Salem, Adv. Drug Delivery Rev., 2021, 174, 613–627 CrossRef CAS PubMed.
- Q. Leng, L. Chen and Y. Lv, Theranostics, 2020, 10, 3190–3205 CrossRef CAS PubMed.
- S. Ghafouri-Fard, A. Abak, S. Tavakkoli Avval, S. Rahmani, H. Shoorei, M. Taheri and M. Samadian, Biomed. Pharmacother., 2021, 142, 111942 CrossRef CAS PubMed.
- J. Wang, S. Liu, J. Li, S. Zhao and Z. Yi, Stem Cell Res. Ther., 2019, 10, 197 CrossRef PubMed.
- J. F. Zhang, W. M. Fu, M. L. He, W. D. Xie, Q. Lv, G. Wan, G. Li, H. Wang, G. Lu, X. Hu, S. Jiang, J. N. Li, M. C. Lin, Y. O. Zhang and H. F. Kung, RNA Biol., 2011, 8, 829–838 CrossRef CAS PubMed.
- C. T. Huynh, M. K. Nguyen, M. Naris, G. Y. Tonga, V. M. Rotello and E. Alsberg, Nanomedicine, 2016, 11, 1535–1550 CrossRef CAS PubMed.
- T. Luo, X. Yang, Y. Sun, X. Huang, L. Zou and J. Liu, Cells Tissues Organs, 2019, 208, 148–157 Search PubMed.
- X. Cen, X. Pan, B. Zhang, W. Huang, F. Pei, T. Luo, X. Huang, J. Liu and Z. Zhao, Stem Cell Res. Ther., 2021, 12, 421 Search PubMed.
- P. Wang, F. Perche, D. Logeart-Avramoglou and C. Pichon, Int. J. Pharm., 2019, 569, 118594 Search PubMed.
- C. A. Hong and Y. S. Nam, Theranostics, 2014, 4, 1211–1232 CrossRef PubMed.
- M. S. Draz, B. A. Fang, P. Zhang, Z. Hu, S. Gu, K. C. Weng, J. W. Gray and F. F. Chen, Theranostics, 2014, 4, 872–892 CrossRef CAS PubMed.
- S. Hosseinpour, Y. Cao, J. Liu, C. Xu and L. J. Walsh, J. Mater. Chem. B, 2021, 9, 2275–2284 RSC.
- S. Hosseinpour, M. N. Gomez-Cerezo, Y. Cao, C. Lei, H. Dai, L. J. Walsh, S. Ivanovski and C. Xu, Pharmaceutics, 2022, 14, 2302 CrossRef CAS PubMed.
- N. Panwar, A. M. Soehartono, K. K. Chan, S. Zeng, G. Xu, J. Qu, P. Coquet, K. T. Yong and X. Chen, Chem. Rev., 2019, 119, 9559–9656 CrossRef CAS PubMed.
- M. Azizi-Lalabadi and S. M. Jafari, Adv. Colloid Interface Sci., 2021, 292, 102416 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 Search PubMed.
- X. Liu, L. Li, B. Gaihre, S. Park, Y. Li, A. Terzic, B. D. Elder and L. Lu, ACS Nano, 2022, 16, 2741–2755 CrossRef CAS PubMed.
- M. Zou, J. Sun and Z. Xiang, Adv. Healthcare Mater., 2021, 10, e2001502 CrossRef PubMed.
- D. P. Singh, C. E. Herrera, B. Singh, S. Singh, R. K. Singh and R. Kumar, Mater. Sci. Eng., C, 2018, 86, 173–197 CrossRef CAS PubMed.
- M. A. Islam, T. E. Park, B. Singh, S. Maharjan, J. Firdous, M. H. Cho, S. K. Kang, C. H. Yun, Y. J. Choi and C. S. Cho, J. Controlled Release, 2014, 193, 74–89 CrossRef CAS PubMed.
- L. Feng, S. Zhang and Z. Liu, Nanoscale, 2011, 3, 1252–1257 Search PubMed.
- N. Karki, H. Tiwari, C. Tewari, A. Rana, N. Pandey, S. Basak and N. G. Sahoo, J. Mater. Chem. B, 2020, 8, 8116–8148 RSC.
- C. Chung, Y. K. Kim, D. Shin, S. R. Ryoo, B. H. Hong and D. H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- J. Huang, C. Zong, H. Shen, M. Liu, B. Chen, B. Ren and Z. Zhang, Small, 2012, 8, 2577–2584 CrossRef CAS PubMed.
- N. Luo, D. Ni, H. Yue, W. Wei and G. Ma, ACS Appl. Mater. Interfaces, 2015, 7, 5239–5247 CrossRef CAS PubMed.
- U. S. Huth, R. Schubert and R. Peschka-Süss, J. Controlled Release, 2006, 110, 490–504 Search PubMed.
- Z. Tu, K. Achazi, A. Schulz, R. Mülhaupt, S. Thierbach, E. Rühl, M. Adeli and R. Haag, Adv. Funct. Mater., 2017, 27, 1701837 CrossRef.
- B. Wang, X. Su, J. Liang, L. Yang, Q. Hu, X. Shan, J. Wan and Z. Hu, Mater. Sci. Eng., C, 2018, 90, 514–522 Search PubMed.
- C. Wei, Z. Liu, F. Jiang, B. Zeng, M. Huang and D. Yu, Cell Proliferation, 2017, 50, e12367 Search PubMed.
- L. H. Wang, K. G. Rothberg and R. G. Anderson, J. Cell Biol., 1993, 123, 1107–1117 Search PubMed.
- M. A. West, M. S. Bretscher and C. Watts, J. Cell Biol., 1989, 109, 2731–2739 Search PubMed.
- H. Yue, W. Wei, Z. Yue, B. Wang, N. Luo, Y. Gao, D. Ma, G. Ma and Z. Su, Biomaterials, 2012, 33, 4013–4021 Search PubMed.
- J. B. Lian, G. S. Stein, A. J. van Wijnen, J. L. Stein, M. Q. Hassan, T. Gaur and Y. Zhang, Nat. Rev. Endocrinol., 2012, 8, 212–227 Search PubMed.
- D. Penzkofer, A. Bonauer, A. Fischer, A. Tups, R. P. Brandes, A. M. Zeiher and S. Dimmeler, PLoS One, 2014, 9, e101153 CrossRef PubMed.
- M. K. Nguyen, O. Jeon, P. N. Dang, C. T. Huynh, D. Varghai, H. Riazi, A. McMillan, S. Herberg and E. Alsberg, Acta Biomater., 2018, 75, 105–114 Search PubMed.
- R. Shido, Y. Sumita, M. Hara, M. Iwatake, S. Narahara, M. Umebayashi, K. I. Miura, Y. Kodama and I. Asahina, Regen. Biomater., 2021, 8, rbaa060 Search PubMed.
- H. Wang and Y. Shen, Mol. Med. Rep., 2019, 20, 4271–4276 CAS.
- X. H. Kong, S. F. Shi, H. J. Hu and J. X. Wang, J. Biol. Regul. Homeostatic Agents, 2021, 35, 921–931 CAS.
- M. Hoseini-Ghahfarokhi, S. Mirkiani, N. Mozaffari, M. A. Abdolahi Sadatlu, A. Ghasemi, S. Abbaspour, M. Akbarian, F. Farjadian and M. Karimi, Int. J. Nanomed., 2020, 15, 9469–9496 Search PubMed.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed.
- K. Yang, J. Wan, S. Zhang, Y. Zhang, S. T. Lee and Z. Liu, ACS Nano, 2011, 5, 516–522 Search PubMed.
- D. A. Jasim, C. Ménard-Moyon, D. Bégin, A. Bianco and K. Kostarelos, Chem. Sci., 2015, 6, 3952–3964 Search PubMed.
- Y. Xiao, Y. X. Pang, Y. Yan, P. Qian, H. Zhao, S. Manickam, T. Wu and C. H. Pang, Adv. Sci., 2023, 10, e2205292 CrossRef PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 Search PubMed.
- J. Casper, S. H. Schenk, E. Parhizkar, P. Detampel, A. Dehshahri and J. Huwyler, J. Controlled Release, 2023, 362, 667–691 Search PubMed.
- B. Chen, M. Liu, L. Zhang, J. Huang, J. Yao and Z. Zhang, J. Mater. Chem., 2011, 21, 7736–7741 Search PubMed.
- S. H. M. Wong, S. S. Lim, T. J. Tiong, P. L. Show, H. F. M. Zaid and H. S. Loh, Int. J. Mol. Sci., 2020, 21, 5202 Search PubMed.
- Y. Luo, H. Shen, Y. Fang, Y. Cao, J. Huang, M. Zhang, J. Dai, X. Shi and Z. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 6331–6339 Search PubMed.
- F. Long, Nat. Rev. Mol. Cell Biol., 2011, 13, 27–38 Search PubMed.
- Y. Li, L. Yang, Y. Hou, Z. Zhang, M. Chen, M. Wang, J. Liu, J. Wang, Z. Zhao, C. Xie and X. Lu, Bioact. Mater., 2022, 18, 213–227 CAS.
- Z. Yan, T. Sun, W. Tan, Z. Wang, J. Yan, J. Miao, X. Wu, P. Feng and Y. Deng, Small, 2023, 19, e2301426 Search PubMed.
- W. Liu, J. Huang, F. Chen, D. Xie, L. Wang, C. Ye, Q. Zhu, X. Li, X. Li and L. Yang, Stem Cell Res. Ther., 2021, 12, 348 Search PubMed.
- J. Qiu, H. Geng, D. Wang, S. Qian, H. Zhu, Y. Qiao, W. Qian and X. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 12253–12263 Search PubMed.
- H. Xie, T. Cao, A. Franco-Obregón and V. Rosa, Int. J. Mol. Sci., 2019, 20, 574 Search PubMed.
- Q. Li and Z. Wang, Int. J. Nanomed., 2020, 15, 4659–4676 Search PubMed.
- W. C. Lee, C. H. Lim, H. Shi, L. A. Tang, Y. Wang, C. T. Lim and K. P. Loh, ACS Nano, 2011, 5, 7334–7341 Search PubMed.
- M. Wu, L. Zou, L. Jiang, Z. Zhao and J. Liu, J. Tissue Eng. Regen. Med., 2021, 15, 915–935 CrossRef CAS PubMed.
- J. Su, Z. Du, L. Xiao, F. Wei, Y. Yang, M. Li, Y. Qiu, J. Liu, J. Chen and Y. Xiao, Mater. Sci. Eng., C, 2020, 113, 110983 CrossRef CAS PubMed.
- Z. Peng, T. Zhao, Y. Zhou, S. Li, J. Li and R. M. Leblanc, Adv. Healthcare Mater., 2020, 9, e1901495 CrossRef PubMed.
- T. Pan, W. Song, H. Xin, H. Yu, H. Wang, D. Ma, X. Cao and Y. Wang, Bioact. Mater., 2022, 10, 1–14 Search PubMed.
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37–44 CrossRef CAS PubMed.
- J. Linares, M. C. Matesanz, M. Vila, M. J. Feito, G. Gonçalves, M. Vallet-Regí, P. A. Marques and M. T. Portolés, ACS Appl. Mater. Interfaces, 2014, 6, 13697–13706 Search PubMed.
- A. Shafiee, S. Iravani and R. S. Varma, MedComm, 2022, 3, e118 Search PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 Search PubMed.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, Mol. Ther., 2010, 18, 561–569 Search PubMed.
- T. Mizuhara, K. Saha, D. F. Moyano, C. S. Kim, B. Yan, Y. K. Kim and V. M. Rotello, Angew. Chem., Int. Ed., 2015, 54, 6567–6570 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- S. Song, H. Fu, B. He, D. Wang, M. Qin, D. Yang, D. Liu, G. Song, Y. Shi, H. Zhang, X. Wang, W. Dai and Q. Zhang, Int. J. Nanomed., 2018, 13, 4391–4404 CrossRef CAS PubMed.
- Y. Xin and B. Wan, Anal. Chim. Acta, 2019, 1079, 103–110 CrossRef CAS PubMed.
- G. P. Kotchey, B. L. Allen, H. Vedala, N. Yanamala, A. A. Kapralov, Y. Y. Tyurina, J. Klein-Seetharaman, V. E. Kagan and A. Star, ACS Nano, 2011, 5, 2098–2108 CrossRef CAS PubMed.
- F. Alnasser, V. Castagnola, L. Boselli, M. Esquivel-Gaon, E. Efeoglu, J. McIntyre, H. J. Byrne and K. A. Dawson, Nano Lett., 2019, 19, 1260–1268 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |