
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical eco-friendly synthesis of photothermal and emissive copper nanoclusters in water: towards sustainable nanomaterials†
Angelo
Ferlazzo
*a,
Stefano
Bonforte
a,
Federica
Florio
a,
Salvatore
Petralia
 *b,
Lorenzo
Sorace
c,
Beatrice
Muzzi
d,
Andrea
Caneschi
*e and
Antonino
Gulino
*a
*b,
Lorenzo
Sorace
c,
Beatrice
Muzzi
d,
Andrea
Caneschi
*e and
Antonino
Gulino
*a
aDepartment of Chemical Sciences and INSTM Research Unit, University of Catania, Viale Andrea Doria 6, 95125 Catania, Italy. E-mail: antonino.gulino@unict.it
bDepartment of Drug and Health Science, University of Catania, Via Santa Sofia 64, 95125 Catania, Italy and Institute of Biomolecular Chemistry, CNR, Via Paolo Gaifami 18, 95126, Catania, Italy. E-mail: salvatore.petralia@unict.it
cDepartment of Chemistry “U. Schiff” and INSTM Research Unit, University of Florence, 50019 Sesto Fiorentino (FI), Italy
dCNR-ICCOM – Istituto di Chimica dei Composti Organometallici, 50019 Sesto Fiorentino (FI), Italy
eDepartment of Industrial Engineering (DIEF) and INSTM Research Unit, University of Florence, 50139 Florence, Italy
First published on 7th August 2024
Abstract
The pursuit of sustainable and environmentally benign approaches to nanomaterial synthesis has gained significant momentum in recent years. Copper is rather cheap and more abundant than gold or silver and small copper nanoclusters (∼1 nm) show unique characteristics which makes them extremely attractive for technological applications. Among these endeavours, the development of eco-friendly methods for the synthesis of copper nanoclusters (Cu NCs) in aqueous environments has emerged as a promising avenue. This shift towards green syntheses not only addresses the growing concerns regarding the environmental impact of conventional synthetic routes but also offers a platform for producing Cu NCs with tailored properties for diverse applications. In the present study, we report on a green photochemical approach to synthesising water-dispersible copper nanoclusters capped with monoethanolamine to confine their size. The obtained material was characterized by a large palette of techniques, including UV-vis spectroscopy, fluorescence spectroscopy, electron paramagnetic resonance, X-ray photoelectron spectroscopy, Z-potential analysis, cyclic voltammetry, field-dependent magnetization, scanning transmission electron microscopy and dynamic light scattering. The results unequivocally demonstrate that the Cu NCs are temporally and thermally stable for months, therefore promising for technological uses.
Introduction
In the last few decades, research into nanomaterials has made countless threads. For example, many studies have focused on the fabrication of carbon and semiconducting quantum dots, ultra-small noble metal particles, and nanosized metal oxides for catalysis, molecular recognition etc.1 To improve the chemical stability and uniformity at the atomic level, enhance their electrical, magnetic, and optical properties and to decrease their possible toxicity, it is essential to investigate new synthetic strategies.2In addition, when the size of these nanomaterials approaches the Fermi wavelength of an electron, unique size-dependent properties have been observed. For instance, gold, silver or copper nanoparticles composed of tens or a few hundred atoms, referred to as nanoclusters (NCs), show optical and catalytic properties as well as other features, much different from those observed for bulk metals.3–5
Noble metal NCs are currently synthesized by exploiting different protective/capping ligands (e.g. polymers, organic molecules, etc.) that also help to confine their size growth.6,7 These ligands interact with the cluster surface atoms in such a way to embed the given cluster to saturate the surface atom coordination sites, thus enhancing the physicochemical stability of NCs.
In this field many studies have been devoted to the fabrication of gold and silver NCs while copper NCs have attracted much lower attention because of their tendency to oxidation; only recently research showed renewed interest toward these systems, finalized to improve their stability and properties.8,9 As a few examples, it has been shown that the excellent fluorescence properties of some Cu NCs can be optimized by using different experimental conditions during their synthesis (solvent, pH, temperature, capping ligands etc.).8 Similarly, Ag NCs show fluorescence intensity inversely proportional to their size,10–12 and also the emission of Au NC aggregates has been ascribed to ligand-to-metal charge transfer or ligand-to-metal–metal charge transfer from the sulphur of the thiolate ligands to the Au atoms and consequent radiative relaxation via a metal-centred triplet state.13
In this general context, some approaches for the synthesis of stable copper NCs have been reported such as electrochemical, milling, etching, reducing (mainly with ascorbic acid), template-assisted (proteins, enzymes, DNA and polymers as templates) and ligand-capped methods.14 Among these, the synthesis of Cu NCs using reducing agents is certainly the most widely used, whereas green syntheses (absence of organic solvents) have been little investigated.
Among the various synthetic approaches, the photochemical method offers the advantage of being reagent-free while allowing easy process control, particularly as regards the exposure time and the intensity of the exciting light. Moreover, the metal precursor's optical properties can be used to tune the size, morphology, and amount of the nanostructures.
There are a few examples in the literature of photochemical reactions to produce Cu NCs using ethanol as a solvent.15–17 To our knowledge, only one example of the synthesis of Cu NCs by photoreduction using copper chloride as a precursor in water has been reported by Gui et al., who precipitated Cu NCs with propanol from a buffer solution and then redissolved them in water.18 The utilization of water as a solvent presents an attractive alternative to traditional chemical routes for the synthesis of Cu NCs. This approach not only mitigates the use of hazardous reagents and organic solvents but also facilitates the integration of Cu NCs into aqueous-based systems, expanding their applicability in biomedical, environmental, and catalytic applications.
Therefore, in our present study, we report on the green photochemical synthesis of Cu NCs, consisting of the UV light (254 nm) irradiation of a copper(II) acetylacetonate (CuII(acac)2) precursor in the presence of a photosensitizer (acetone) and monoethanolamine (MEA) as a capping agent in a deaerated aqueous solution, and we delve into the precise control over size, morphology, and surface properties. By focusing on the eco-friendly synthesis of Cu NCs in water, this paper aims to underscore the importance of sustainable approaches in nanomaterial synthesis while highlighting the potential of Cu NCs as environmentally friendly alternatives for diverse technological applications. The obtained Cu NCs in water were found to be stable for months, and this behaviour is due to the monoethanolamine capping layer which, on the one hand, limits the growth of the nanoclusters by confining them to a few nanometres, and on the other hand, almost seals the obtained nanoclusters, giving them high stability. The Cu NCs exhibited an excellent photothermal conversion efficiency upon blue-light exposure (405 nm).19–22
Experimental
Synthesis of copper nanoclusters
High-grade copper(II) acetylacetonate (hereafter Cu(acac)2) 99.9%, monoethanolamine (MEA), water and acetone for spectrophotometric use were purchased from Sigma Aldrich. We prepared a stock 1.649 × 10−4 M water solution of Cu(acac)2 by adding 4.317 mg of copper(II) acetylacetonate (MM = 261.76 g mol−1; 1.6492 × 10−5 mol) in 100 mL of distilled water while stirring for 2 hours (solubility in water of Cu(acac)2 = 0.2 g L−1). The MX5 Microbalance Metter Toledo© (resolution 0.8–0.9 μg) was used for weighing chemicals. It is necessary to stabilize the as-obtained Cu NCs with an appropriate capping agent and, after some experiments, we have chosen to add to the starting copper solution 60 μL of monoethanolamine (δ = 1.02 g mL−1, MM = 61.084 g mol−1, 1.002 × 10−3 mol) diluted in 6 mL of water, and 7 mL of pure acetone (photosensitizer) before irradiation.23 Therefore, the resulting Cu(II) and MEA concentrations (113 mL total volume) are 1.459 × 10−4 and 8.87 × 10−3 M, respectively. This last solution was sealed in a quartz reaction tube and irradiated in a reactor at 254 nm with four Helios Quartz© lamps (UVC power intensity 4 W cm−1 per each), under a constant argon flux (100 sccm min−1) for 180 minutes and left under argon flux for additional 50 minutes in the dark to remove gaseous reaction products. The reactor is equipped with a fan to prevent overheating. The obtained yellow solution was concentrated to 6 mL using a rotavapor. The concentrate was centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 minutes, filtered through a 0.2 μm filter and lyophilised (T = −20 °C) thus obtaining an oil. Dynamic light scattering measurements evidenced that the dispersion of Cu NCs in water possesses a mean hydrodynamic diameter (volume %) of around 60.9 ± 6.7 nm at 25 °C, with a Z-potential value of −10.2 ± 2.5 mV. The hydrodynamic radius value obtained by dynamic light scattering (DLS) analysis depends on the water capping layer that covers the Cu NCs (already capped with monoethanolamine), thus increasing the overall volume. In fact, the DLS theory states that the electric dipole layer of both the capping layer and the solvent adhering to the surface of the particles influences their movement in the medium. Thus, the hydrodynamic diameter gives information about the inorganic core along with any coating materials and solvent layers attached to the particle. For example, it has been reported that the diameter of some Fe3O4 particles was found to be 25 nm from TEM, whereas their average hydrodynamic size was 164 nm from DLS. Therefore, the size of Cu NCs remains the same as that obtained with TEM measurements (vide infra).24
000 rpm for 30 minutes, filtered through a 0.2 μm filter and lyophilised (T = −20 °C) thus obtaining an oil. Dynamic light scattering measurements evidenced that the dispersion of Cu NCs in water possesses a mean hydrodynamic diameter (volume %) of around 60.9 ± 6.7 nm at 25 °C, with a Z-potential value of −10.2 ± 2.5 mV. The hydrodynamic radius value obtained by dynamic light scattering (DLS) analysis depends on the water capping layer that covers the Cu NCs (already capped with monoethanolamine), thus increasing the overall volume. In fact, the DLS theory states that the electric dipole layer of both the capping layer and the solvent adhering to the surface of the particles influences their movement in the medium. Thus, the hydrodynamic diameter gives information about the inorganic core along with any coating materials and solvent layers attached to the particle. For example, it has been reported that the diameter of some Fe3O4 particles was found to be 25 nm from TEM, whereas their average hydrodynamic size was 164 nm from DLS. Therefore, the size of Cu NCs remains the same as that obtained with TEM measurements (vide infra).24
Characterization methods
:5 nm. The photoluminescence quantum yield (ϕPL) of the dispersion of Cu NCs in water (n = 1.33) was obtained using a solution of quinine sulphate (n = 1.36) as a standard (ϕPL = 0.55).
Results and discussion
Copper nanoclusters can be produced by photochemical approaches, involving the reduction of Cu ions in the presence of stabilizing agents under UV light. This procedure often leads to the formation of very small-sized and well-dispersed nanoclusters with unique properties. For this purpose, initially, we performed some preliminary tests to shed light on the behaviour of the photochemical reduction of the Cu(II) ion from Cu(acac)2 in water using the UV-vis technique. The UV-vis spectrum of a 5.4966 × 10−5 M Cu(acac)2 water solution (obtained by dilution of the initial stock solution) shows two bands at 238.5 nm (ε = 9200 M−1 cm−1) and 292.6 nm (ε = 15860 M−1 cm−1) due to a ligand-to-metal charge transfer (LMCT) and to a π–π* (HOMO → LUMO) transition of the ligand, respectively (Fig. 1).15 No other absorption bands were observed under these conditions since the d–d transition of the Cu(II) ion has an ε value ∼10 M−1 cm−1. The UV-vis spectrum of the Cu(acac)2(MEA)2 complex, where the NH2– group of the MEA capping agent saturates the two free axial positions of the Cu ion, shows a unique band envelope with a main absorption that is shifted from 292.6 to 283.4 nm, due to the reinforced ligand field.31 Then, irradiation at 254 nm of the above solution (Fig. 1) caused a profound spectral change with a spectrum now dominated by a new band at 265.2 nm and two shoulders at ∼315 and 380 nm. Previously reported data on similar Cu NCs indicated the appearance of bands at almost identical positions; the 265.2 nm absorbance has been attributed to an intraligand nN → π* transition (at 260–275 nm for Cu NCs stabilized with glutathione),32 the one at 315 nm to self-assembled Cu NCs (at the identical 315 nm position for Cu NCs stabilized with 4,6-dimethyl-2-mercaptopyrimidine)33 and that at 380 nm to an enhanced interaction between Cu atoms and the electron-rich nitrogen atoms of MEA (reported at the identical 380 nm position for Cu NCs stabilized with citrate anions).34 After 24 h, the UV-vis spectrum is dominated by the 315 nm band, which undergoes a blue shift to 303.9 nm, while the shoulder at 380 nm remains. The presence of this shoulder at 380 nm and the absence of any plasmonic peak suggest the formation of small nanoclusters.
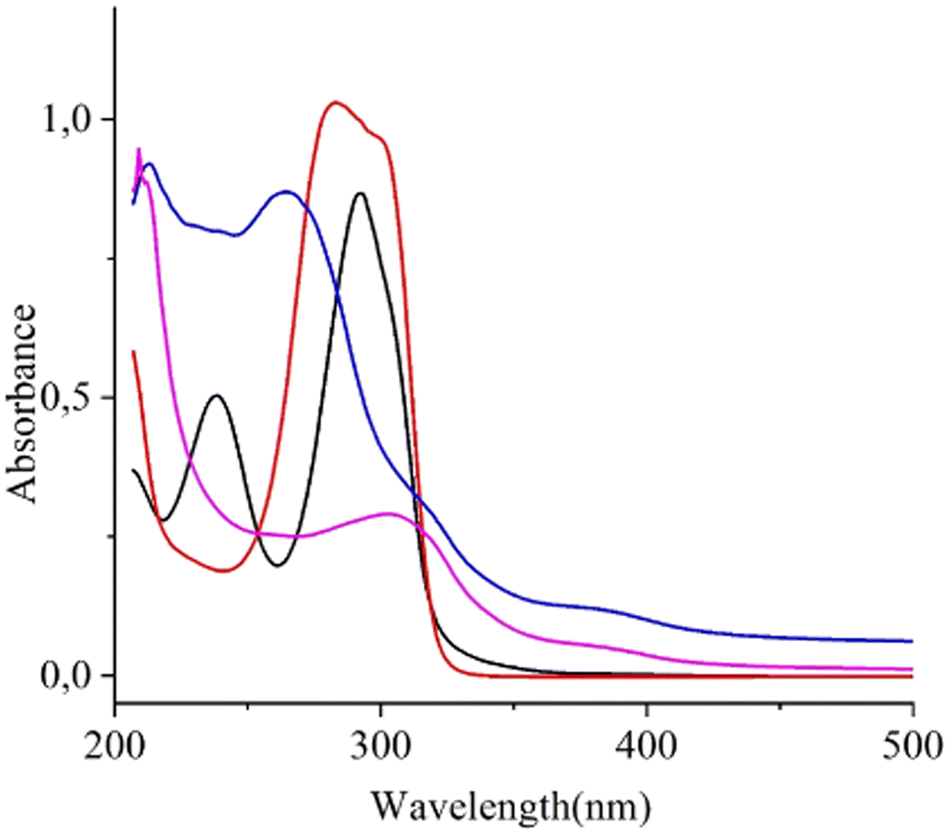 | ||
| Fig. 1 UV-vis spectra of: 5.5 × 10−5 M Cu(acac)2 water solution (black line); 5.5 × 10−5 M Cu(acac)2 and 2.96 × 10−3 M monoethanolamine capping agent water solution (red line); 5.5 × 10−5 M Cu(acac)2 and 2.96 × 10−3 M monoethanolamine capping agent water solution after 180 min irradiation at 254 nm followed by 50 min dwell of the resulting solution in the dark during continuous argon flux (see experimental details) (blue line); final solution after 24 h from the end of the irradiation and after a volume reduction to 6 mL, measured using a 0.1 cm quartz cuvette (pink line). | ||
Fig. 2 shows the emission spectra of the 5.5 × 10−5 M Cu(acac)2 water solution where the intensity was multiplied by 100 since this starting solution shows almost no emission. In contrast, the final Cu NCs solution, after 24 h from the end of irradiation, and after a volume reduction from 100 to 6 mL (vide supra), under the same excitation wavelength, shows an intense emission band at 489.9 nm with a full width at half-maximum (fwhm) of 100 nm and a high energy shoulder at ∼460 nm. The increase in the emission after 24 h has been previously reported in this type of system and is a consequence of Cu NCs self-assembly, due to the coordination of Cu NCs with the capping agent that induces LMCT and/or LMMCT charge transfer.35,36 The results of the emission experiments reported here are almost identical to those reported for the above-mentioned Cu NCs assemblies that exhibit a strong emission peak centred at 490 nm with an fwhm of 86 nm.35 No major differences were observed in the emission spectrum as the excitation wavelength changed in the range of 330–410 nm. The photoluminescence quantum yield (ϕPl) of Cu NCs in water was calculated to be 0.12.
 | ||
| Fig. 2 Emission spectra, λexc = 390 nm, of 5.4966 × 10−5 M Cu(acac)2 water solution (black line where the intensity was multiplied by 100) and final Cu NCs solution after 24 h from the end of the irradiation and after a volume reduction to 6 mL (red line). | ||
Furthermore, the stability of the emission properties of these Cu NCs was confirmed 7, 20, 26 and 32 days after the end of the reaction (Fig. S1, ESI†).
To resume, both UV-vis and emission measurements confirm the formation of small Cu nanoclusters.8 Therefore, it emerged that the emission increases upon the increase of the Cu–Cu inter- and intra-NCs interactions,37 and upon the restriction of the periodic motion (vibration and rotation) of the capping molecules that hampers the nonradiative relaxation of excited states.38–41
Concerning the possible reaction pathway (Scheme 1), irradiation of the photosensitizer (acetone) at 254 nm induces a n → π* (S0 to T1) transition whose de-excitation produces both CH3CO˙ and ˙CH3 reactive species. Interaction of the CH3CO˙ radical with CuII(acac)2 in water results in the formation of CuI(acac), acetylacetone (Hacac) and acetic acid. CuI(acac) further reacts with another CH3CO˙ radical and another H2O molecule to give Cu0, Hacac and, again, acetic acid. The Cu0 metal atoms in the presence of ethanolamine agent form small Cu0 clusters capped by ethanolamine molecules that hamper the cluster size increase, thus avoiding some aggregation. The two ˙CH3 species produce ethane. In tune with our supposed mechanism, we experimentally observed that the final solution had been acidic because of the formation of acetic acid.19,42
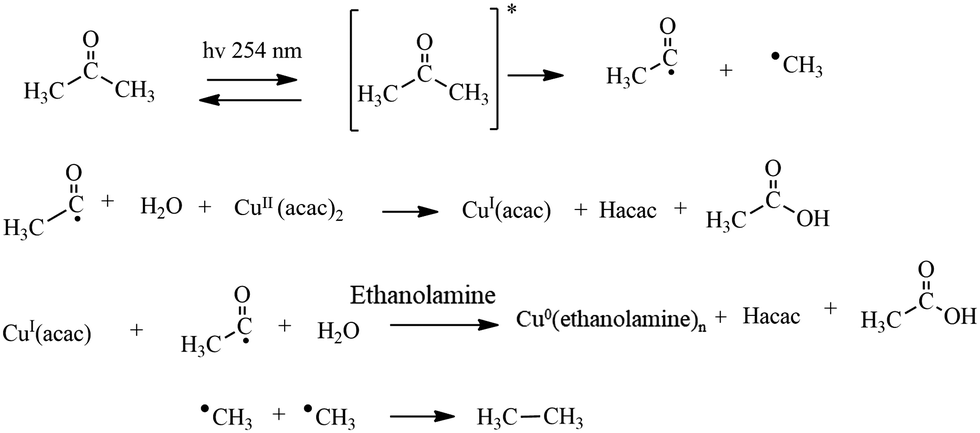 | ||
| Scheme 1 Synthesized copper nanoclusters (Cu NCs) using a photochemical pathway. | ||
To confirm the formation of small Cu NCs, morphological and chemical composition analysis, was performed. STEM-HAADF images (Fig. 3) displayed small spherical polydisperse nanocrystals, with a narrow range size from 2.5 to 5 nm, dispersed in aggregates of ethanolamine. STEM-EDXS elemental mapping distribution, instead, confirmed the composition of the Cu NCs, and highlighted the retaining of Cu(II) ions complexed with ethanolamine, even after the reduction step (Fig. 3). Interestingly, only O of ethanolamine was observed, suggesting that no oxidation of the Cu NCs occurred.
 | ||
| Fig. 3 (top) STEM-HAADF image of Cu NCs capped with ethanolamine and STEM-EDXS elements distribution maps; (bottom) TEM images with different magnifications also show the particle size distribution centred at 3.5 nm. | ||
Fig. 4 shows CV measurements for the starting Cu(acac)2 and Cu NCs species. The surface area of Cu/SPCE is larger than that of Cu2+/SPCE due to the presence of copper nanoparticles, and this fact results in a larger current range of the related CV curve.43 The anodic oxidation peak (Cu0 → Cu2+) is at −0.11 V. This value is consistent with that previously reported (−0.09 V) for a closed system measured with a similar electrode42 and confirms the presence of Cu0. In the cathodic phase, there is evidence of the Cu2+ to Cu+ reduction, consistent with the peak at −0.33 V, followed by the Cu+ → Cu0 reduction at −0.46 V. Both these values are in close agreement with those reported (−0.37 and −0.52 V) for a similar system.44 The CV data indicate an anodic oxidation peak (Ipa) of 32.06 μA and a cathodic reduction peak (Ipc) of 34.95 μA. These two values are consistent with a completely reversible redox reaction; the small current difference of 2.89 μA between Ipa and Ipc can be attributed to the presence of a small amount of Cu2+, less than 9% to the overall copper content in the measured representative Cu NC sample, also in agreement with magnetic measurements (vide infra). Five months after the synthesis, we repeated the CV measurements for the Cu NCs and obtained almost identical results thus demonstrating the long-term stability of the present Cu NCs (Fig. S2, ESI†). In addition, eight months after the synthesis, we recorded again the UV-vis spectrum of the final Cu NCs solution (Fig. S3, ESI†). The band at 303.9 nm remains unaltered, and this observation further confirms the excellent stability of these Cu NCs.
 | ||
| Fig. 4 CV of 5.4966 × 10−5 M Cu(acac)2 water solution (black line and digits) and final Cu NCs solution after 24 h from the end of the irradiation and after a volume reduction to 6 mL (red line and digits). | ||
The successful synthesis of copper nanoclusters was further confirmed by studying their electronic structure using X-ray photoelectron spectroscopy (XPS), which provides information on the oxidation states and the chemical environment of the studied elements, and allows estimation of the surface elemental composition, once the relevant atomic sensitivity factors have been considered.45,46 The two peaks at 932.4 and 952.4 eV observed in the Cu 2p binding energy region of the high-resolution XPS of the representative Cu NCs (Fig. 5A) are readily attributed to the Cu 2p3/2 and 2p1/2 spin–orbit pair and are strongly indicative of the presence of Cu0 states.47,48 Indeed, the presence of relevant Cu2+ species would result in an XPS spectrum which, apart from the two Cu 2p main spin–orbit couple, should show two additional Cu 2p peaks at ∼2 eV higher binding energy and intense satellite features centred at ∼942 and ∼962 eV. These additional peaks and satellites seem to be absent in the present spectrum or hidden under the noisy background, since the very low content of remaining Cu2+ species as detected by CV measurements (less than 9% of the overall copper content). In addition, by considering the reduction potentials of the different copper species in water solution (Cu2+ + e → Cu+ E0 = 0.15 V; Cu+ + e → Cu0 E0 = 0.52 V; Cu2+ + 2e → Cu0 E0 = 0.34 V), no stable Cu+ species is expected in aqueous solution since any oxidant strong enough to convert Cu to Cu+ would also convert Cu+ to Cu2+ and Cu+ ions are unstable toward disproportionation reaction: 2Cu+ → Cu2+ + Cu E0 = 0.37 V, K = 106. Therefore, in an aqueous solution, only low equilibrium concentrations of Cu(I) can exist and XPS confirms that our synthetic procedure was effective in obtaining Cu0 species in water solution. Fig. 5B shows the high-resolution XPS of the representative Cu nanoclusters in the N 1s binding energy region. The unique and rather symmetrical peak at 400.2 eV is due to the nitrogen of the monoethanolamine capping layer that stabilises the Cu nanoclusters.49,50Fig. 5C shows the XPS of the representative Cu nanoclusters in the C 1s binding energy region. A careful deconvolution of the experimental spectral profile required three Gaussians at 285.7, 286.9 and 288.7 eV due to the Cu–NH2–C, –CH2–OH and some –COO– functionalities (due to some acetic acid residues, see Scheme 1), respectively.25,26Fig. 5D shows the high-resolution XPS of the representative Cu nanoclusters in the O 1s binding energy region. The component at 532.0 eV is due to the oxygen of the monoethanolamine capping layer that stabilises the Cu nanoclusters while the component at 533.1 eV is due to the oxygen of the Si/SiO2 substrate.46
 | ||
| Fig. 5 Al-Kα excited XPS of the Cu nanoclusters in the Cu 2p (A) and N 1s (B) binding energy regions. Al-Kα excited XPS of the Cu nanoclusters in the C 1s (C) binding energy region: the blue, cyan and magenta Gaussians refer to the 285.7, 286.9 and 288.7 eV components, respectively. Al-Kα excited XPS of the Cu nanoclusters in the O 1s (D) binding energy region: the blue and cyan Gaussians refer to the 532.0 and 533.1 eV components, respectively. For both C and D figures, the wine line refers to the background and the red line superimposed to the black experimental profile refers to the sum of the two Gaussian components. | ||
Magnetic and EPR characterization
EPR spectroscopy of both the pure oil and the water suspension of the synthesized material was measured at 40 K to evaluate the magnetic features of the sample, where Cu NCs are supposed to be diamagnetic. Both samples show a weak spectrum (Fig. 6), remarkably similar and characteristic of copper(II) in elongated coordination geometry, with g‖ > g⊥ > ge and A‖ > A⊥ (where Aii are the principal components of the hyperfine coupling tensor with the I = 3/2 nuclear spin of 63Cu and 65Cu). The similarity of the spectra in the two samples, with the clearly resolved parallel hyperfine structure, testifies that the paramagnetic centres are not clustered together and can be safely attributed to a paramagnetic impurity fraction of an otherwise diamagnetic sample. The best simulation of the spectra (reported in Fig. 6) provides spin Hamiltonian parameters (gx = 2.044 ± 0.001, gy = 2.056 ± 0.001, gz =2.234 ± 0.001; Ax = Ay = 50 ± 10 MHz, Az = 563 ± 1 MHz) which are consistent with previously reported ones for copper(II) ethanolamine complexes.51,52 | ||
| Fig. 6 X-band (ν ≅ 9.40 GHz) EPR spectra measured at 40 K for the oil (red trace) and water suspension (green trace) together with best simulations (black and blue lines) obtained using parameters reported in the text. | ||
To obtain a quantitative estimate of the amount of paramagnetic fraction, we measured the field-dependent magnetization at 5 K (Fig. 7). When rescaled to the atomic weight of copper (see the Experimental section), the measured values highlight that the magnetic contribution is indeed extremely weak, with a high field value much lower than that expected for a pure S = 1/2 system. This is thus consistent with what is expected for metallic copper nanoclusters with a small impurity of a paramagnetic species Cu(II). The observed behaviour closely follows the expected one for a simple Brillouin system with a very low abundance of paramagnetic centres, which the fit (performed by fixing the g value to the average of the EPR determined ones, gave = 2.111) estimated to be 7.25 ± 0.05%. In summary, the EPR and magnetic characterization unequivocally indicate the presence of a large majority of diamagnetic nanocluster with a small (<10%) amount of copper(II) complexes, in notable agreement with the results of the electrochemical characterization (vide supra).
 | ||
| Fig. 7 Field-dependent magnetization of the oil sample measured at 5 K and best fit using Brillouin function with parameters reported in the text. | ||
Photothermal properties of Cu NCs
Although carbon and metal-based photothermal nanostructures have been investigated,19,53–55 very few examples of photothermal Cu0 nanomaterials are reported in the literature.20–22 To investigate the photothermal properties of Cu NCs, an aqueous dispersion (100 mL, Abs405nm = 0.36) was continuously exposed to a 405 nm laser (210 mW). The temperature changes were monitored using a thermal camera (Fig. 8A). When the temperature of the system reached the maximum value (temperature change = Tmax − Tenvironment = 9 °C), the laser was turned off, and the temperature change was monitored during the cooling process to confirm the heat transfer of the system. Three representative photothermal cycles are reported to confirm the excellent stability of the Cu NCs upon UV (405 nm) light exposition. The power-dependent photothermal behaviour was confirmed by experiments performed with different laser powers of 70, 100, 150 and 210 mW. The temperature difference values recorded were about 3.2 °C, 4.1 °C, 6.5 °C and 9.12 °C, respectively (Fig. 8B). In contrast, when a water dispersion of precursor Cu(acac)2 was exposed to a laser power of 210 mW, only a negligible increase in temperature was observed (black line Fig. 8B), thus confirming the photothermal properties of Cu NCs. A photothermal conversion efficiency (η) value of about 38%, with a photothermal time constant (τs) values of about 110 s were calculated using an already reported procedure (Fig. S4, ESI†).56 These data confirm the excellent conversion efficiency of Cu NCs compared to similar Cu-nanostructures reported in the literature. In fact, Korgel and coworkers reported for Cu nanocrystals, doped with selenide, a η value of about 27%.57 Zhao et al. synthesized copper-based nanostructures which exhibited a photothermal conversion efficiency of 10.6% under laser irradiation.58 | ||
| Fig. 8 Photothermal experiments for Cu NCs dispersion. (A) Photothermal heating–cooling cycles of the Cu NC nanostructure water dispersion, volume 100 μL, Abs405nm = 0.36 in a glass tube (insets representative thermograph during photothermal experiments in a quartz tube) and (B) photothermal effect of the Cu NCs dispersion at different laser power. | ||
Conclusions
In line with the pursuit of sustainable and environmentally benign approaches to nanomaterial synthesis, we proposed a green photochemical approach to synthesize Cu NCs capped with monoethanolamine in water. These Cu NCs have been characterized by morphological, optical, electric, electronic, and magnetic measurements. Both XPS results and the observation of the anodic oxidation peak (Cu0 → Cu2+ at −0.11 V) confirmed the Cu0 oxidation state in the prepared material. EPR and magnetic characterization, in agreement with electrochemical data, evidenced the persistence of some residual paramagnetic Cu2+ ions. Since STEM-EDXS elemental mapping distribution highlighted that only oxygen of ethanolamine was observed, thus suggesting that no oxidation of the Cu NCs occurred, the presence of Cu2+ ions is due to the incomplete photochemical reduction of the starting Cu(acac)2 precursor, possibly due to the formation of a small fraction of copper(II) ethanolamine complexes before the reduction was complete. On the other hand, the obtained ethanolamine-capped Cu NCs are stable for months. We also observed a strong blue emission (at 460–490 nm) due to LMCT and LMMCT charge transfers in self-assembled Cu NCs.The reported results unequivocally demonstrate that Cu NCs are temporally and thermally stable for months and open the way to produce Cu NCs with adapted properties for potential technological uses.
Author contributions
The manuscript was written with the contribution of all authors.Data availability
The data supporting this article have been included in the main text and as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Catania for the financial support of the PIA.CE.RI. MAF-MOF project 2020–2022 and the B.R.I.T. laboratory of the University of Catania for the availability of the XPS facility. This work has also been partially funded by the European Union (NextGeneration EU) through the MUR-PNRR project SAMOTHRACE (ECS00000022). L. S. acknowledges the support of MUR through Dipartimenti di Eccellenza 2023-2027 (DICUS 2.0, CUP B97G22000740001) to the Department of Chemistry “Ugo Schiff” of the University of Florence.References
- S. Wei, A. Li, J.-C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W.-C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- X. Wang, J. Zhuang, Q. Peng and Y. Li, Nature, 2005, 437, 121–124 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- L. Zhang and E. Wang, Nano Today, 2014, 9, 132–157 CrossRef CAS.
- M. G. Taylor and G. Mpourmpakis, Nat. Commun., 2017, 8, 15988 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- Y. An, Y. Ren, M. Bick, A. Dudek, E. H.-W. Waworuntu, J. Tang, J. Chen and B. Chang, Biosens. Bioelectron., 2020, 154, 112078 CrossRef CAS PubMed.
- X. Liu and D. Astruc, Coord. Chem. Rev., 2018, 359, 112–126 CrossRef CAS.
- L. A. Peyser, A. E. Vinson, A. P. Bartko and R. M. Dickson, Science, 2001, 291, 103–106 CrossRef CAS PubMed.
- C. Vázquez-Vázquez, M. Banobre-Lopez, A. Mitra, M. A. Lopez-Quintela and J. Rivas, Langmuir, 2009, 25, 8208–8216 CrossRef PubMed.
- Y. Chen, T. Yang, H. Pan, Y. Yuan, L. Chen, M. Liu, K. Zhang, S. Zhang, P. Wu and J. Xu, J. Am. Chem. Soc., 2014, 136, 1686–1689 CrossRef CAS PubMed.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- A. Baghdasaryan and T. Bürgi, Nanoscale, 2021, 13, 6283–6340 RSC.
- S. Giuffrida, G. G. Condorelli, L. L. Costanzo, I. L. Fragalà, G. Ventimiglia and G. Vecchio, Chem. Mater., 2004, 16, 1260–1266 CrossRef CAS.
- G. G. Condorelli, L. L. Costanzo, I. L. Fragalà, S. Giuffrida and G. Ventimiglia, J. Mater. Chem., 2003, 13, 2409–2411 RSC.
- X. Zhu, B. Wang, F. Shi and J. Nie, Langmuir, 2012, 28, 14461–14469 CrossRef CAS PubMed.
- R. Gui, J. Sun, X. Cao, Y. Wang and H. Jin, RSC Adv., 2014, 4, 29083–29088 RSC.
- G. M. Consoli, G. Forte, L. Maugeri, V. Consoli, V. Sorrenti, L. Vanella, G. Buscarino, S. Agnello, M. Camarda, G. Granata, L. Ferreri and S. Petralia, ACS Appl. Nano Mater., 2022, 6, 358–369 CrossRef.
- P. Huang, L. Kong, F. Zhang, L. Chen, Y. Zhang, X. Shi, T. Lawson, S. Chou, Y. Liu and W. Wu, Small, 2024, 2312211 CrossRef CAS PubMed.
- J. Wang, Y. Gu, Y. Fan and M. Yang, ACS Appl. Nano Mater., 2023, 6, 13561–13569 CrossRef.
- J. Zhang, P. Li, T. Wang, J. Li, K. Yun, X. Zhang and X. Yang, Pharmacol. Res., 2023, 187, 106632 CrossRef CAS PubMed.
- A. Zhao, Z. Li and H. Wang, Polymer, 2010, 51, 2099–2105 CrossRef CAS.
- C. I. Olariu, H. H. P. Yiu, L. Bouffier, T. Nedjadi, E. Costello, S. R. Williams, C. M. Halloran and M. J. Rosseinsky, J. Mater. Chem., 2011, 21, 12650–12659 RSC.
- A. Gulino, Anal. Bioanal. Chem., 2013, 405, 1479–1495 CrossRef CAS PubMed.
- D. Briggs and J. T. Grant, Surface analysis by Auger and X-ray photoelectron spectroscopy, Chichester, UK, 2003 Search PubMed.
- G. Greczynski and L. Hultman, Angew. Chem., 2020, 59, 5002–5006 CrossRef CAS PubMed.
- S. Sharma, S. Das, K. Kaushik, A. Yadav, A. Patra and C. K. Nandi, J. Phys. Chem. Lett., 2023, 14, 8979–8987 CrossRef CAS PubMed.
- A. Ferlazzo, C. Espro, D. Iannazzo and G. Neri, IEEE Trans. Instrum. Meas., 2023, 72, 9508308 CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- Z. Wang, B. Chen, A. S. Susha, W. Wang, C. J. Reckmeier, R. Chen, H. Zhong and A. Rogach, Adv. Sci., 2016, 3, 1600182 CrossRef PubMed.
- A. Baghdasaryan, R. Grillo, S. Roy Bhattacharya, M. Sharma, E. Reginato, H. Theraulaz, I. Dolamic, M. Dadras, S. Rudaz, E. Varesio and T. Burgi, ACS Appl. Nano Mater., 2018, 1, 4258–4267 CrossRef CAS.
- Q. Sun, Z. Ning, E. Yang, F. Yin, G. Wu, Y. Zhang and Y. Shen, Angew. Chem., 2023, 135, e202312 Search PubMed.
- A. Dutta, U. Goswami and A. Chattopadhyay, ACS Appl. Mater. Interfaces, 2018, 10, 19459–19472 CrossRef CAS PubMed.
- Z. Wu, J. Liu, Y. Gao, H. Liu, T. Li, H. Zou, Z. Wang, K. Zhang, Y. Wang, H. Zhang and B. Yang, J. Am. Chem. Soc., 2015, 137, 12906–12913 CrossRef CAS PubMed.
- Y. Chen, T. Yang, H. Pan, Y. Yuan, L. Chen, M. Liu, K. Zhang, S. Zhang, P. Wu and J. Xu, J. Am. Chem. Soc., 2014, 136, 1686–1689 CrossRef CAS PubMed.
- Q. Benito, X. F. Le Goff, S. Maron, A. Fargues, A. Garcia, C. Martineau, F. Taulelle, S. Kahlal, T. Gacoin, J. P. Boilot and S. Perruchas, J. Am. Chem. Soc., 2014, 136, 11311–11320 CrossRef CAS PubMed.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- P. Duan, N. Yanai, Y. Kurashige and N. Kimizuka, Angew. Chem., 2015, 54, 7544–7549 CrossRef CAS PubMed.
- Y. Yuan, C. J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew. Chem., 2015, 54, 1780–1786 CrossRef CAS PubMed.
- S. Giuffrida, L. L. Costanzo, G. Ventimiglia and C. Bongiorno, J. Nanopart. Res., 2008, 10, 1183–1192 CrossRef CAS.
- A. Ferlazzo, C. Espro, D. Iannazzo, A. Bonavita and G. Neri, Mater. Today Commun., 2023, 35, 106036 CrossRef CAS.
- K. Ngamchuea, S. Wannapaiboon, P. Nongkhunsan, P. Hirunsit and I. Fongkaew, J. Electrochem. Soc., 2022, 169, 020567 CrossRef CAS.
- S. Li, H. Zhang, Z. Liu, J. Xu, G. Fan, W. Li, Q. Li, X. Hu and G. Jing, Appl. Sci., 2022, 12, 1245 CrossRef CAS.
- A. Gulino, G. G. Condorelli, P. Mineo and I. Fragalà, Nanotechnology, 2005, 16, 2170–2175 CrossRef CAS PubMed.
- M. P. Seah, I. S. Gilmore and G. Beamson, Surf. Interface Anal., 1998, 26, 642–649 CrossRef CAS.
- Y. Zhang, J. Zhang, Z. Li, Z. Qin, S. Sharma and G. Li, Commun. Chem., 2023, 6, 24 CrossRef CAS PubMed.
- H. Keisar, G. de Ruiter, A. H. Velders, P. Milko, A. Gulino, G. Evmenenko, L. J. W. Shimon, Y. Diskin-Posner, M. Lahav and M. E. van der Boom, J. Am. Chem. Soc., 2018, 140, 8162–8171 CrossRef CAS PubMed.
- A. Gulino, S. Bazzano, P. Mineo, E. Scamporrino, D. Vitalini and I. Fragalà, Chem. Mater., 2005, 17, 521–526 CrossRef CAS.
- R. Tauler, E. Casassas and B. M. Rode, Inorg. Chim. Acta, 1986, 114, 203–209 CrossRef CAS.
- S. K. Hoffmann, J. Goslar, I. Polus and B. Mazela, Appl. Magn. Reson., 2003, 24, 321–331 CrossRef CAS.
- G. M. L. Consoli, L. Maugeri, G. Forte, G. Buscarino, A. Gulino, L. Lanzanò, P. Bonacci, N. Musso and S. Petralia, J. Mater. Chem. B, 2024, 12, 952–961 RSC.
- G. M. L. Consoli, M. L. Giuffrida, S. Zimbone, L. Ferreri, L. Maugeri, M. Palmieri, C. Satriano, G. Forte and S. Petralia, ACS Appl. Mater. Interfaces, 2023, 15, 5732–5743 CrossRef CAS PubMed.
- G. M. L. Consoli, M. L. Giuffrida, C. Satriano, T. Musumeci, G. Forte and S. Petralia, Chem. Commun., 2022, 58, 3126–3129 RSC.
- G. M. L. Consoli, L. Maugeri, N. Musso, A. Gulino, L. D’Urso, P. Bonacci, G. Buscarino, G. Forte and S. Petralia, Adv. Healthcare Mater., 2024, 2303692 CrossRef CAS PubMed.
- C. M. Hessel, V. P. Pattani, M. Rasch, M. G. Panthani, B. Koo, J. W. Tunnell and B. A. Korgel, Nano Lett., 2011, 11(6), 2560–2566 CrossRef CAS PubMed.
- H. Xiang, F. Xue, T. Yi, H. P. Tham, J.-G. Liu and Y. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 16344–16351 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. Luminescence emission stability of the final Cu NCs; Fig. S2. CV of Cu NCs recorded 5 months after the synthesis. Fig. S3. UV-vis spectrum of the final Cu NC solution eight months after the synthesis. Fig. S4. Photothermal conversion efficiency. See DOI: https://doi.org/10.1039/d4ma00401a |
| This journal is © The Royal Society of Chemistry 2024 |