
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel layered Cu-based perovskite metal–organic framework with 1,2-diaminoethane cations: synthesis, crystal structure, and thermal and magnetic properties†
Asmae
Ben Abdelhadi
 ab,
Rachid
Ouarsal
a,
Morgane
Poupon
c,
Michal
Dusek
c,
Juan Pedro Andrés
González
d,
Luis
Lezama
e,
Brahim
El Bali
f,
Mohammed
Lachkar
*a and
Abderrazzak
Douhal
*b
ab,
Rachid
Ouarsal
a,
Morgane
Poupon
c,
Michal
Dusek
c,
Juan Pedro Andrés
González
d,
Luis
Lezama
e,
Brahim
El Bali
f,
Mohammed
Lachkar
*a and
Abderrazzak
Douhal
*b
aEngineering Laboratory of Organometallic, Molecular Materials, and Environment (LIMOME), Faculty of Sciences, Sidi Mohamed Ben Abdellah University, Fez 30000, Morocco. E-mail: mohammed.lachkar@usmba.ac.ma
bDepartamento de Química Física, Facultad de Ciencias Ambientales y Bioquímica, y INAMOL, Campus Tecnológico de Toledo, Universidad de Castilla-La Mancha (UCLM), Avenida Carlos III, S.N., Toledo 45071, Spain. E-mail: abderrazzak.douhal@uclm.es
cInstitute of Physics of the Czech Academy of Sciences, Na Slovance 2, 8, Praha 182 21, Czech Republic
dDepartamento de Física Aplicada, Instituto Regional de Investigación Científica Aplicada (IRICA), Universidad de Castilla - La Mancha, 13071, Ciudad Real, Spain
eDepartamento de Química Orgánica e Inorgánica, Facultad de Ciencia y Tecnología, Universidad del País Vasco, UPV/EHU, Bo Sarriena s/n, Leioa 48940, Spain
fIndependent Scientist, Marrakech, Morocco
First published on 17th May 2024
Abstract
Lead-free organic–inorganic metal perovskites have recently attracted special attention. Here, we report on the synthesis of single crystals of a new layered perovskite-type formate framework templated by diprotonated ethylene diammonium (NH3(CH2)2NH3)[Cu(HCO2)4]∞ (1) using a mild solution chemistry approach. The as-prepared sample was characterized by single crystal X-ray diffraction (SCXRD), Fourier transform infrared spectrophotometry, thermogravimetric analyses, and differential scanning calorimetry. We also studied its magnetic properties. SCXRD data show that 1 crystallizes as a monoclinic structure in the P21/c space group, and its structure consists of [Cu(HCO2)4]2− anionic squared layers and an (NH3(CH2)2NH3)2+ cation interlayer. In the layers, divalent copper cations Cu2+ have an octahedral geometry and are bridged by the equatorial formate HCO2− in an infinite anti–anti chain. The (NH3(CH2)2NH3)2+ cation interlayer forms N–H⋯Oformate extensive H-bonds to anionic layers. IR absorption bands are characteristic of the ethylene diammonium cation and formate group and are consistent with X-ray diffraction data. TGA results reveal that the studied compound starts to decompose at 373 K. The magnetic study reveals a two-dimensional Heisenberg antiferromagnetic behaviour with moderate interactions between Cu(II) ions through O–C–O bridges. A weak ferromagnetism has also been detected arising from spin canting in an ordered antiferromagnetic material.
1. Introduction
Organic–inorganic materials with perovskite structures are mainly classified into two families: AMX3 (3D-structured perovskites) and A2MX4 (2D-structured or layered perovskites) in which A = organic cations, M = transition metals, and X = anionic ligands.1–4 These hybrid perovskites consist of a metal-anionic inorganic layer (composed of an MX6 octahedron or tetrahedral MX4) containing large enough cavities to accommodate positively charged organic-cations, typically protonated amines, on the A-site. The interlayer cohesion between the adjacent layers of the organic cation and inorganic metallic layer in such materials occurs generally through H-bonding, π–π stacking and van der Waals interactions.5–9 In the case of layered perovskites, different short bridging ligands can be employed in the X-site, such as halides (Cl−, Br− and I−),10,11 formates (HCOO−),12 and azides (N3−),13 hypophosphites (H2POO−),14 thiocyanates (SCN−),15 and dicyanamides (N(CN)2−).16 Hybrid perovskites containing halide atoms in their X-site are very attractive materials due to their outstanding optical properties, making them of great interest in the field of photoelectric devices.17–19 Meanwhile, hybrid perovskites with formate anions in the X-site have been extensively studied due to their magnetic, ferroelectric, multiferroic and luminescent properties.20–22 The formate ion (carboxylate anion, HCOO−) is a short bridging ligand, good for the construction of coordination polymers and can also provide significant magnetic coupling (ferromagnetic, F, or antiferromagnetic, AF) between spin carriers, and resulting magnetic properties depend on different bridging modes (such as syn–syn, syn–anti, and anti–anti) of the formato-ion and metal ion.23 In addition to that, the carboxylate anion is a good acceptor of H-bonds, thus allowing their formation, which is beneficial for dielectric properties.24,25 These chemical and electronic abilities facilitate the formation of interesting layered-metal-formate frameworks with different structures, porosity, magnetism, and even dielectric properties, making them attractive materials in different fields of research in chemistry and physics.26–29Thus, within the past few years, several 3D-structured perovskite-type architecture formate frameworks [AmineHn][M(HCOO)3]n templated by monoprotonated amines have been reported.30–32 For example, it has been reported that multiferroic behavior occurs in this kind of perovskite due to a ferroelectric order–disorder transition involving the monoprotonated amine cation and magnetic ordering between octahedral metal atoms (M-sites).33 In contrast, there are only a few reports on 2D-structured or layered perovskite formate frameworks.28,34,35 To the best of our knowledge, the 2D-perovskite-like metal formate frameworks templated by diamines are limited.36 Typically, the metal formate frameworks incorporating diamines crystallize in different structures. For example, the [NH3(CH2)4NH3][M2(HCOO)6] formate compound, containing 1,4-diaminobutane (NH3(CH2)4NH3)2+ as a counter cation, crystallizes in the centrosymmetric trigonal structure.37 Meanwhile, the structure of a magnesium-based formate framework templated by 1,3-diaminopropane consists of elongated cavities containing a polymeric assembly made of [NH3(CH2)3NH3]2+ and H2O moieties.38 Moreover, a series of metal-formate frameworks templated by 1,2-diaminoethane (DAE) and water with the general formula [NH3(CH2)2NH3]M2(HCOO)6·H2O (where M = Mg, Mn, Co, Ni and Zn) have been synthesized, and four of these compounds crystallize in polar R3 and one in the chiral P6322 space group (Ni-analogue) at room temperature.39
In this regard, this study aims to investigate the synthesis, crystal structure, single crystal X-ray diffraction (SCXRD), infrared spectroscopy (IR), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC) of a novel 2D-layered perovskite-type architecture formate framework templated by an ethylenediamine cation, (NH3(CH2)2NH3)[Cu(HCO2)4] (1). The magnetic properties of 1 were investigated for polycrystalline samples under different applied magnetic fields. The crystal structure consists of a layered perovskite-type structure composed of organic cation interlayers (NH3(CH2)2NH3)2+ and anionic layers [Cu(HCO2)4]2−. Meanwhile, magnetic measurements revealed that 1 exhibits 2D-Heisenberg antiferromagnetic (AF) behavior with moderate interactions between the Cu(II) ions through the O–C–O bridges. 1 also exhibits weak ferromagnetism arising from spin canting in an ordered antiferromagnetic material.
2. Experimental part
2.1. Chemicals
Copper (II) chloride tetrahydrate (CuCl2.4H2O, 99.9%) and formic acid (HCOOH, 98%) were purchased from Fluka (Buchs, Switzerland), anhydrous methanol (99.7%) was purchased from Panreac (Barcelona, Spain) and 1,2-diaminoethane (C2H8N2, 99.5%) was bought from Sigma Aldrich (France). All chemicals and solvents used for the synthesis were of reagent grade quality and were used as received.2.2. Synthesis
Blue rectangular crystals of 1 were prepared using a mild solution chemistry approach by a facile slow diffusion process (Fig. 1). In a typical experiment, the crystals of 1 were obtained from a sealed and undisturbed mixture of two solutions in a glass tube (9 mm inner diameter). The first solution (S1) containing 10 mmol of ethylenediamine and 80 mmol of HCOOH dissolved in 10 mL of anhydrous methanol was placed at the bottom of a glass tube. The second solution (S2), composed of 2 mmol (0.340 g) of CuCl2·4H2O dissolved in 10 mL of anhydrous methanol, was added gently to S1. Blue single-crystals of (NH3(CH2)2NH3)[Cu(HCO2)4] were harvested after one week, washed with methanol and dried at room temperature (Fig. 1).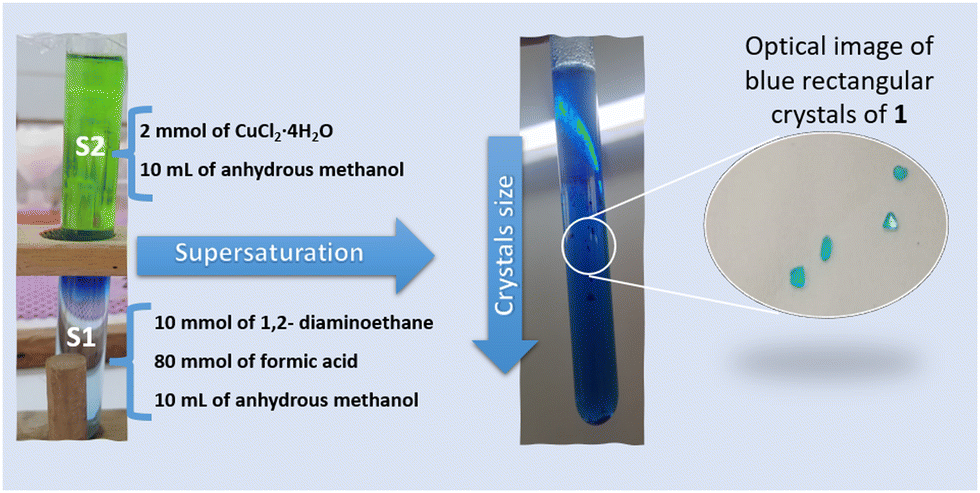 | ||
| Fig. 1 Schematic representation of the synthesis of (NH3(CH2)2NH3)[Cu(HCO2)4] single crystals using a facile slow diffusion process. The copper solution (S2) is gently added on top of a solution containing a mixture of organic linkers (S1). Blue crystals with different sizes are formed at the interface and at the bottom of the tube. | ||
2.3. Crystallographic structure determination, thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), infrared and magnetic measurements
The crystal structure of 1 was determined at room temperature (RT) using a four-circle Oxford Diffraction Gemini CCD diffractometer at 120 K, on a blue rectangular single crystal block of 0.69 × 0.48 × 0.45 mm3. Data collection was performed using Mo Kα radiation (λ = 0.71073 Å) from a classical sealed tube monochromated by graphite and collimated by a fibre-optics Enhance collimator. Technical details and analysis of data are given in the ESI.†Table 1 gives crystal data, data collection and refinement, while Table 2 exhibits the values of the lengths of the H-bonds. In addition to that, Tables S1 and S2 (ESI†) provide, respectively, refined atomic positions and displacement parameters. (CCDC 2332899†).| Chemical formula | C6H14CuN2O8 |
| Molecular mass (g mol−1) | 305.7 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 300 |
| a, b, c (Å) | 9.721 (2), 6.681 (2), 9.161 (2) |
| β (°) | 108.90 (3) |
| V (Å3) | 562.9 (3) |
| Z | 2 |
| Radiation type | Mo Kα |
| μ (mm−1) | 1.97 |
| Crystal size (mm) | 0.69 × 0.48 × 0.45 |
| Data collection | |
| Diffractometer | Xcalibur, AtlasS2, Gemini ultra |
| T min, Tmax | 0.802, 1 |
| No. of measured, independent and observed [I > 3σ(I)] reflections | 9106, 2765, 2177 |
| R int | 0.032 |
(sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ/λ)max (Å−1) θ/λ)max (Å−1) |
0.692 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.024, 0.064, 1.32 |
| No. (reflections, parameters, restraints) | 2765, 155, 2 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.27, −0.27 |
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| Symmetry codes: (v) −x + 1, y + 1/2, −z + 3/2; (vi) x, y + 1, z. | ||||
| N1—H1N1⋯O4v | 0.868 (19) | 1.937 (19) | 2.782 (2) | 164 (2) |
| N1—H2N1⋯O4vi | 0.89 (2) | 1.97 (2) | 2.791 (2) | 154 (2) |
| N1—H3N1⋯O2 | 0.91 (2) | 1.89 (2) | 2.786 (2) | 167 (2) |
TGA and DSC analyses for the investigated compound were performed in the temperature range of 295–1150 K using a LINSEIS TGA PT1600 thermal analyzer. The sample weight was 22 mg, and the heating speed rate was 283 K min−1 using an open platinum crucible under nitrogen flow.
The room-temperature IR (Infrared) spectra in the mid-IR (4000–400 cm−1) range were measured with a Vertex 70 (Bruker) Fourier transform infrared spectrometer (FTIR) using the attenuated total reflectance (ATR) method. The spectral resolution was 4 cm−1.
Magnetic measurements were carried out on an EverCool MPMS SQUID magnetometer (Quantum Design, San Diego, CA, USA) using a well-crushed polycrystalline sample. Zero-field cooled (ZFC) and field-cooled (FC) magnetic susceptibility data were obtained under different applied magnetic fields between 40 Oe and 10 kOe in the temperature range of 5–300 K. Hysteresis loops in ZFC conditions were obtained at different temperatures of 5 and 50 K at magnetic fields ranging from −50 to 50 kOe.
3. Results and discussion
3.1. Single crystal structure description
The blue single crystals of 1 were grown at room temperature by a facile slow diffusion solution process. An SCXRD experiment was used to determine the crystal structure of 1. The analysis reveals a monoclinic type of system and a P21/c space group, with Z = 2, a = 9.721 (2) Å, b = 6.681 (2) Å, c = 9.161 (2) Å, β = 108.90° (3) and volume = 562.9 (3) Å3. The molecular structure of 1 consists of one ethylene diammonium cation and one metal (Cu2+) cation surrounded by six formate (HCOO−) anions (Fig. 2A). The structure is layered perovskite-type, composed of 2D-squared layers of formato-bridged [Cu(HCOO)4]2− and an interlayer of [NH3(CH2)2NH3]2+ organic cations, which is sandwiched between inorganic layers (Fig. 2B). Cu2+ cations are octahedrally coordinated by four equatorial formate ligands that connect neighbouring coordination spheres in the anti–anti mode configuration to form squared layers, and each Cu2+ ion is further coordinated by two apical terminal HCOO− groups, which are in single anti-mode, pointing up and down, forming the H-bonds with [NH3(CH2)2NH3]2+ cations (Fig. 2B). Each octahedron around Cu(II) is linked to four neighbouring Cu(II)-ions through four bridging formate groups forming a 2D (4,4) network in the (bc) plane (Fig. 2C). The network of (NH3(CH2)2NH3)[Cu(HCO2)4] is similar to reported metal-formate networks in 2D metal-formate compounds.28,34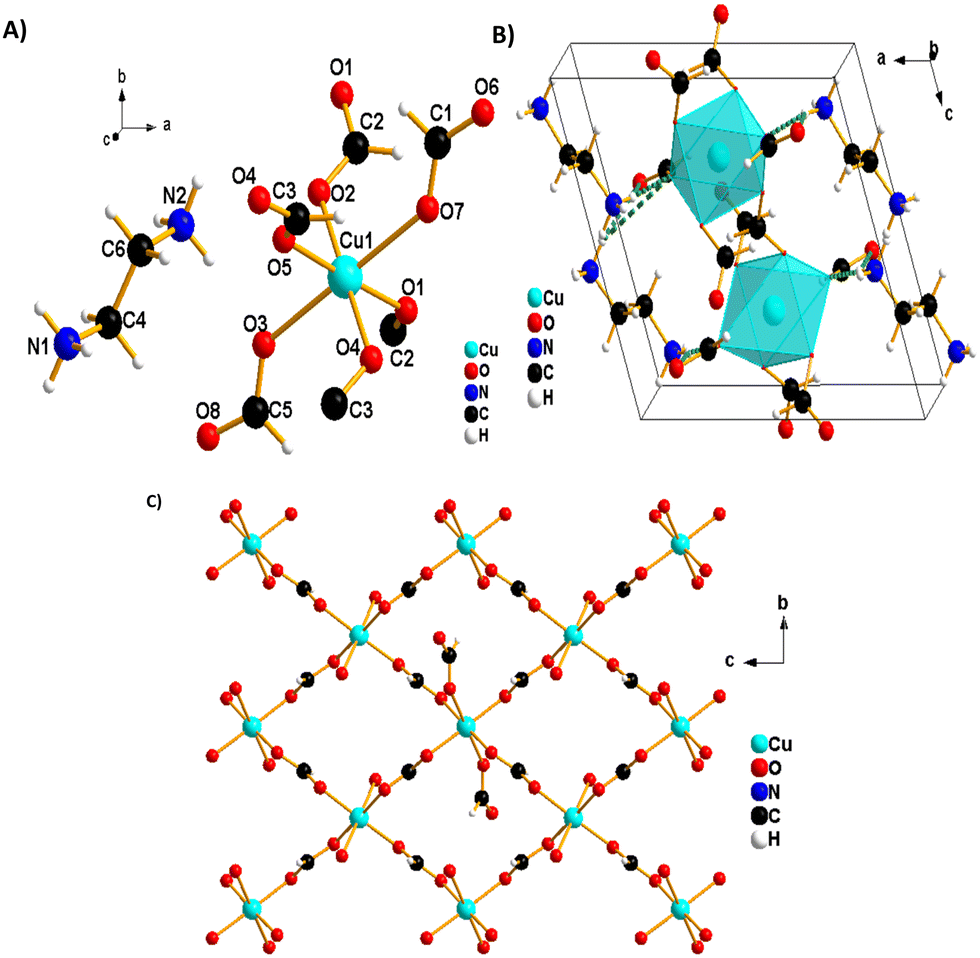 | ||
| Fig. 2 (A) Molecular structure of 1. (B) Packing plots of 1 viewed along the b direction; the H-bonds between donor and acceptor atoms are shown as green dashed lines. (C) View of a 2D infinite layer of the Cu (4,4) network in the (bc) plane. | ||
The observed coordination environment around one independent Cu2+ ion is described as a Jahn–Teller distorted octahedron with four short Cu_O equatorial bond lengths between 1.9831 (13)–1.9880 (11) Å and elongated apical ones of 2.3787 (12) Å (Table S3, ESI†). These distances are comparable to those previously reported for the CuO6 octahedron in other copper-based formate frameworks.40,41 The grid edges of the layer network or Cu⋯Cu distances via the bridging formate (HCOO−) are 5.6692 (2) Å. Meanwhile, the layer separation is approximately 9.7209 (4) Å; this separation is larger due to long axial Cu–O bonds (the apical unidentate formate is involved in the H-bonds) in the CuO6 octahedron and to the large size of diammonium localised between the layers. The [NH3(CH2)2NH3]2+ cations are in front of or above the layer, possessing two NH3 groups as good H-bonding donors, interacting via six conventional N–H⋯O hydrogen bonds with anionic metal-formate [Cu(HCOO)4]2− layers (Fig. 3 and Table 2). All six hydrogen bonds formed play an important role to connect adjacent metal–formate layers together. This pattern of H-bonding is typically observed for the layered perovskites [CH3C(NH2)2]2[M(HCOO)4] (M = Co2+ and Ni2+) templated by acetamidinium cations.28
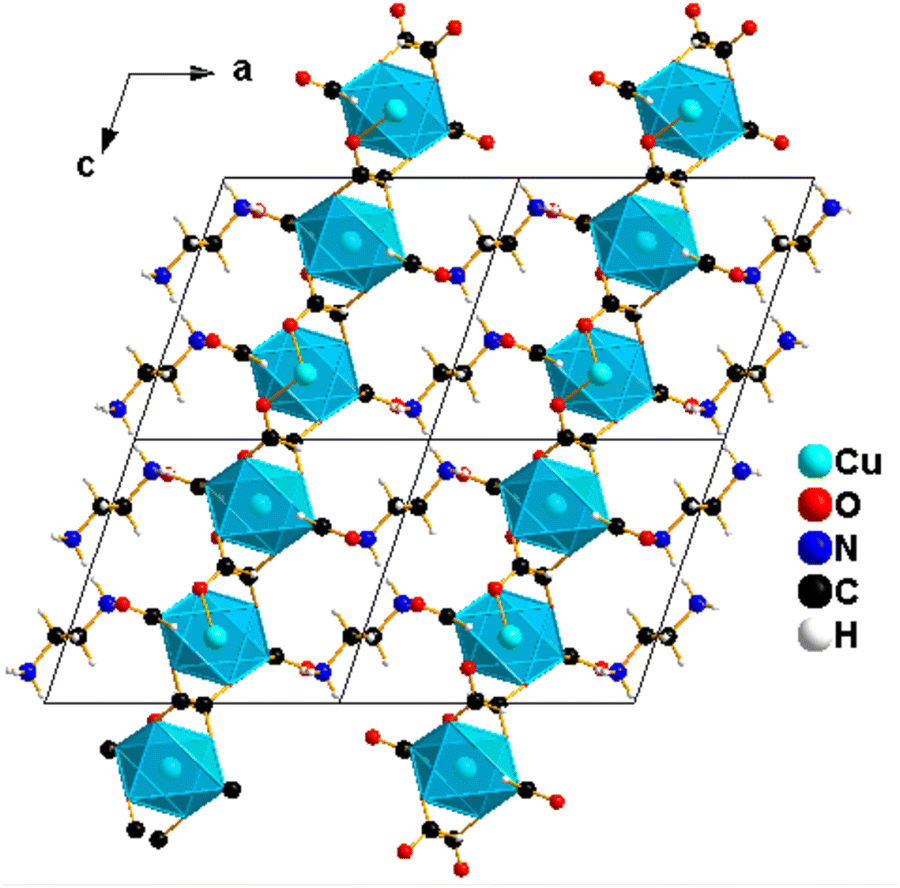 | ||
| Fig. 3 Illustration of the crystal structure of 1 showing the Cu(II)-formate network layer viewed in the b-direction. CuO6 polyhedra are shown in light blue and non-linking formate groups extend from the axial positions to interact with [NH3(CH2)2NH3]2+ cations between the layers. | ||
Two non-linking formats coordinate in the axial positions of metal octahedra, and terminal oxygen atoms extend in between the layers. They extend from the axial positions to interact with the [NH3(CH2)2NH3]2+ cations between the layers, and they are responsible for the charge balance of Cu-based perovskite-like formate frameworks as a counter ion. H-bonding interactions are observed between the non-linking formate oxygen and diammonium group [NH3(CH2)2NH3]2+ as summarized in Table 2.
These H-bonding interactions should lead to a larger stabilization than what was observed for 3D perovskite formats due to the presence of additional groups on the cations allowing H-bonding interactions as well as the presence of uncoordinated formate oxygen atoms.
3.2. Room-temperature vibrational studies
Fourier transform infrared (FT-IR) spectroscopy is a powerful and widely used technique to look at functional groups present in the crystal and to investigate their vibrational behavior. Fig. 4 shows the FT-IR spectrum of ground crystals of 1 recorded at RT, while Table S4 (ESI†) exhibits the values of IR absorption band frequencies and proposed assignment. The IR spectrum shows broad vibration bands in the 3300 to 3000 cm−1 region that reflect the stretching modes of NH3. Meanwhile, the vibration bands between 3000–2800 cm−1 correspond to the symmetric stretching vs (CH2) and antisymmetric stretching vas (CH2) vibrations.39 In addition to these bands, neighbouring weak ones appear in the range 2880–2800 cm−1 that can be attributed to the stretching vibrations of C–H group v1 (HCOO) of the formate coordinating in an anti–anti configuration and to some combinations and overtones of other v (HCOO) modes. Compared to IR-vibration bands in syn–syn mode for the metal-formate frameworks templated by 1,4-diaminobutane (NH3(CH2)4NH3)2+ as a cation,37 these vibration bands are found to blue shift by 40 to 50 cm−1 as a result of the C–O short distance in the case of the anti–anti bridging mode.38 Compared to the other metal-formate frameworks containing 1,2-diaminoethane as a linker, all of the observed vibration bands are similar to reported ones.39 In the medium wavenumber region, the bands observed at 1640 and 1629 cm−1 are attributed to the antisymmetric deformation vibration δas (NH3) of NH3, indicating the involvement of this group in the formation of the H-bond system to stabilize the crystal structure (Fig. 4), which is in agreement with the analysis of the structure obtained by the SCXRD experiment. The two weak bands at 1456 and 1445 cm−1 correspond to the deformation vibration of δ (CH2). The strongest characteristic formate vibrations bands are described in detail in the ESI.† These characteristic formate vibration bands coincide well with those previously reported for other organic metal-formate frameworks.27,28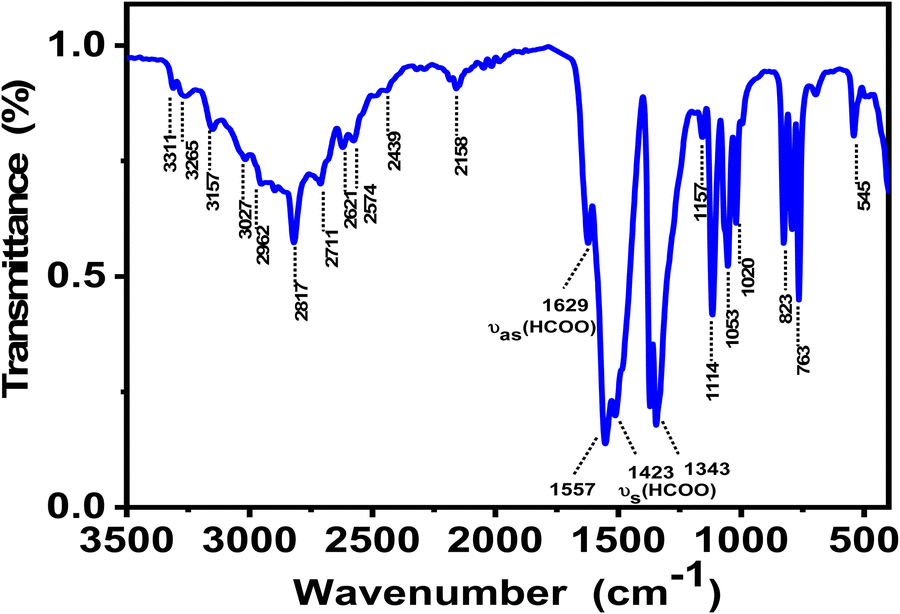 | ||
| Fig. 4 Infrared spectrum of (NH3(CH2)2NH3)[Cu(HCO2)4] polycrystals. | ||
3.3. Thermal properties
The thermal stabilities of 1 were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), with a heating rate of 283 K min−1 under nitrogen flow from RT to 1150 K. Fig. 5 shows that the compound is stable up to 373 K, and it decomposes in three degradation steps, as can be seen in the TGA result (blue curve). The first decomposition is observed between 373 and 423 K. The weight loss is around 19.84% (theoretical value, 19.65%) and corresponds to the loss of organic diamine (NH2(CH)2NH2) molecule per formula unit. This decomposition is accompanied by an intense endothermic peak of the DSC curve at 406 K (red curve). The last two steps of degradation between 423 and 553 K correspond to the decomposition of four formate linkers by releasing different species: HCOOH, CO, CO2, H2 and H2O with an experimental value of 59.08% (theoretical value, 60.22%). It is well known that formic acid (HCOOH) can decompose into H2O and CO (dehydration). It can also undergo a less favourable decomposition process into H2 and CO2 (decarboxylation).42 The degradation of the formate groups is coupled with two endothermic peaks in the DSC signal; the first one occurs at 441 K and second one at 520 K. These degradation processes of the formate linkers are consistent with proposed possible mechanisms of decomposition of formic acid.42 During the whole degradation process of the Cu-based formate, the resulting residue formed corresponds to pure Cu. Subsequently, the residue became nearly stable up to 573 K. A systematic study of the thermal decomposition of hybrid perovskites of formula [AH][M(HCOO)3] under an inert atmosphere was performed using thermogravimetry and simultaneous infrared spectroscopy of evolved gases, and the results show that the Cu-based formate decomposes to pure Cu, in agreement with the observed degradation process of 1, and the difference is that in our case the decomposition occurs in three steps instead of one in the reported decomposition.43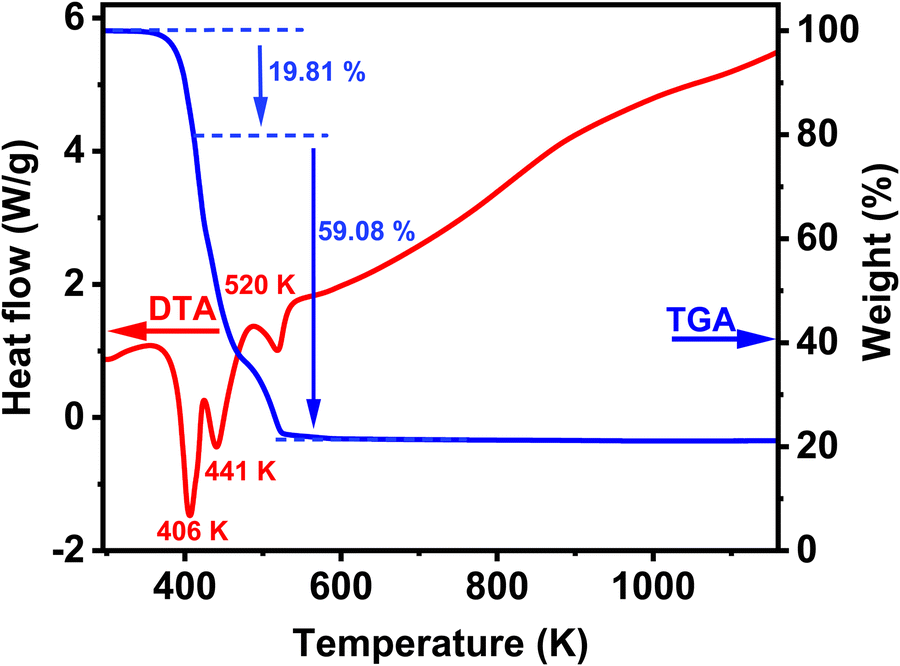 | ||
| Fig. 5 TGA and DSC traces of the synthesized compound (NH3(CH2)2NH3)[Cu(HCO2)4] at a rate of 283 K min−1 under air flow. | ||
3.4. Magnetic properties
Magnetic susceptibility measurements on ground crystals of 1 were carried out in the 5–300 K temperature range with applied fields between 40 and 10 kOe. Above 40 K, the compound shows a typical paramagnetic behavior. However, at lower temperatures, the susceptibility exhibits a strong field dependence. The high temperature data (T > 200 K) are well described by a Curie–Weiss law, with Cm = 0.416 cm3 K mol−1 and θ = −74 K (Fig. S1, ESI†). The effective magnetic moment (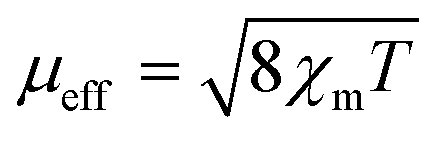 ) value per Cu(II) ion measured at room temperature is 1.63µB, slightly lower than the theoretical spin-only value for a S = 1/2 system (1.73µB), as generally occurs for antiferromagnetic compounds. The thermal evolution of the magnetic molar susceptibility is rather complex showing a broad maximum at about 60 K, followed by a sharp peak at 15 K (Fig. S2A, ESI†). The χmT value decreases with the temperature, reaching a minimum value at around 35 K. Then, the related curve suddenly increases reaching a maximum at 18 K, and at lower temperatures, it decreases again (Fig. 6A). In addition, Fig. S2B (ESI†) shows the M/H vs. temperature curves obtained between 5 and 300 K under applied fields of 100 Oe and 10 kOe, respectively. Since the magnetic behavior observed at temperatures above 30 K was similar in both cases, magnetization data under applied fields of 40 and 1000 Oe were only registered in the 5–30 K range.
) value per Cu(II) ion measured at room temperature is 1.63µB, slightly lower than the theoretical spin-only value for a S = 1/2 system (1.73µB), as generally occurs for antiferromagnetic compounds. The thermal evolution of the magnetic molar susceptibility is rather complex showing a broad maximum at about 60 K, followed by a sharp peak at 15 K (Fig. S2A, ESI†). The χmT value decreases with the temperature, reaching a minimum value at around 35 K. Then, the related curve suddenly increases reaching a maximum at 18 K, and at lower temperatures, it decreases again (Fig. 6A). In addition, Fig. S2B (ESI†) shows the M/H vs. temperature curves obtained between 5 and 300 K under applied fields of 100 Oe and 10 kOe, respectively. Since the magnetic behavior observed at temperatures above 30 K was similar in both cases, magnetization data under applied fields of 40 and 1000 Oe were only registered in the 5–30 K range.
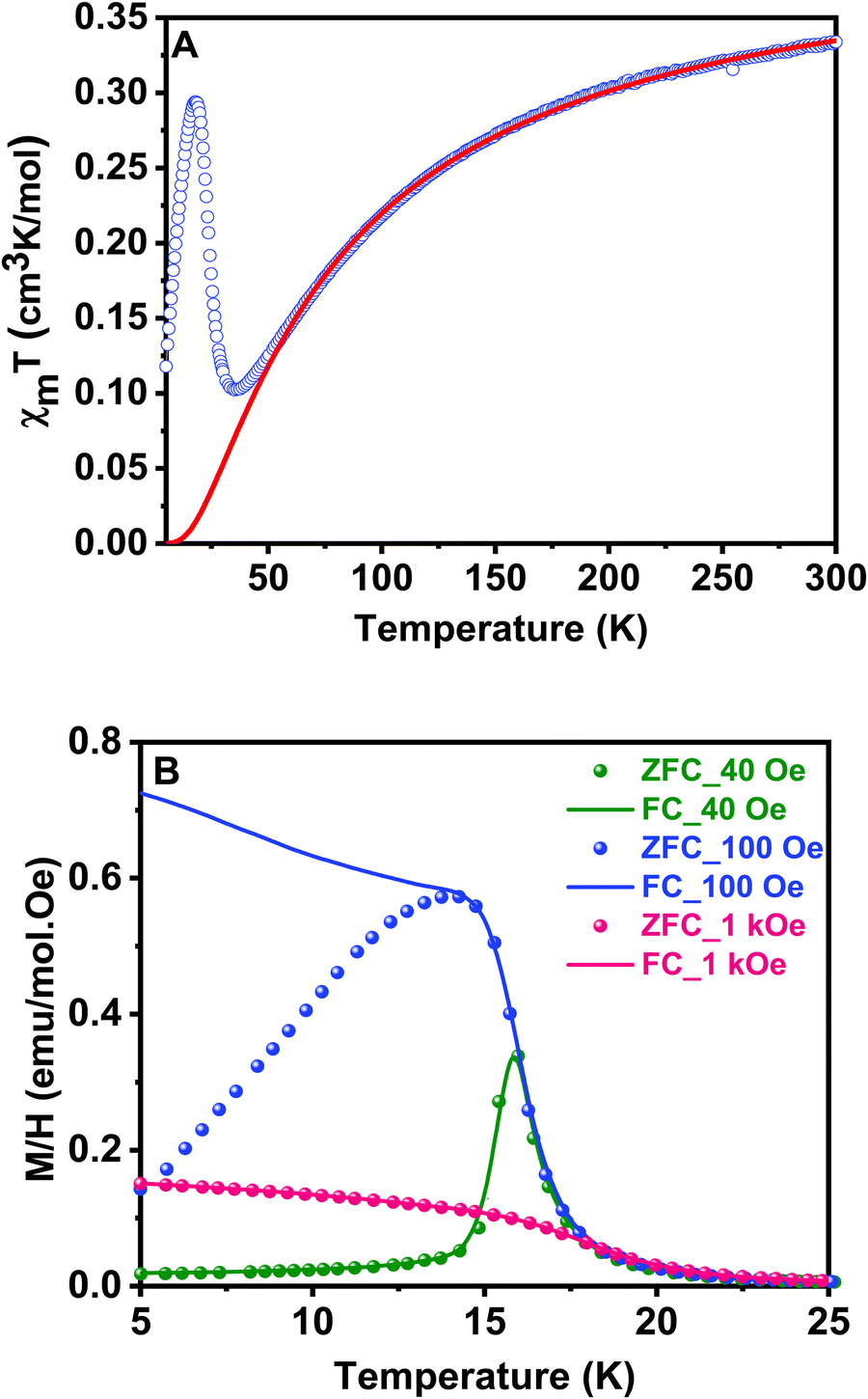 | ||
| Fig. 6 (A) Thermal variation of χmT for 1 at 10 kOe; the solid red line represents the theoretical curve for a 2D antiferromagnet. (B) Thermal evolution of M/H for field cooled (FC) and zero field cooled (ZFC) samples measured at 40 Oe, 100 Oe and 1 kOe. | ||
The negative Weiss temperature, together with the initial decrease of the χmT data, suggests that the predominant magnetic interactions in 1 are antiferromagnetic. Taking into account the structural features of this compound, we expect bidimensional couplings within the bc plane by a super exchange mechanism via the formate groups between Cu2+ ions. Therefore, we tried to fit the high temperature susceptibility data using the high temperature series expansion given by Rushbrooke and Wood for a quadratic layer Heisenberg antiferromagnet.44 We derived eqn (1) by taking into account S = 1/2 for the Cu(II) ion and based on the spin Hamiltonian 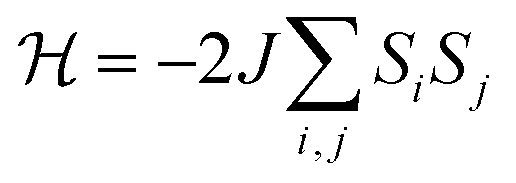 :
:
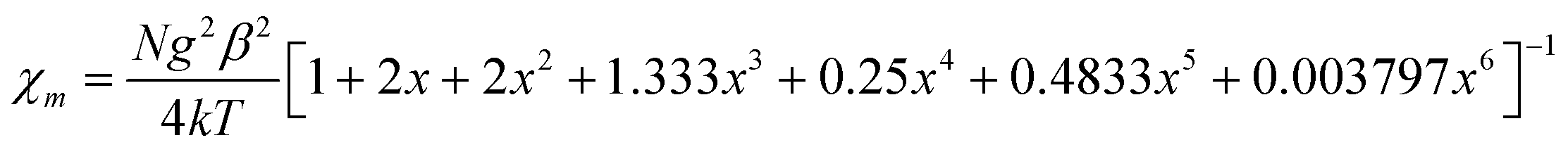 | (1) |
Fig. 6B shows the thermal evolution of M/H for field cooled (FC) and zero field cooled (ZFC) samples at different fields. FC and ZFC curves are similar above 20 K for all magnetic fields, but both curves diverge below this temperature for applied fields between 100 Oe and 1 kOe. In the ZFC curves, a maximum in M/H is observed at 16 K, which is indicative of predominant antiferromagnetic interactions. Moreover, this is a field-dependent effect: the less intense the applied field, the sharper the maximum observed. On the contrary, in FC measurements, the magnetization appears to saturate at low temperatures for applied fields higher than 100 Oe. This behavior is characteristic of a canting phenomenon, in which a weak remanent magnetization is provided by a not exactly collinear antiferromagnetic arrangement of the magnetic moments. The AC susceptibility measurements accurately show that the magnetic ordering transition occurs at 16 K (Fig. S3, ESI†). We observed a sharp peak in the real part of the susceptibility with no associated anomaly in the imaginary part, confirming the essentially antiferromagnetic nature of the arrangement and weakness of the remanent magnetization. The χm’ vs. T curve does not exhibit any evolution on changing the frequency. This fact allows to exclude a spin-glass-like behavior for the sample.49
Hysteresis loops from −50 to 50 kOe at different temperatures have also been measured for sample 1. Well above the magnetic ordering temperature, M is linear with H over the whole range of applied fields, as observed at T = 50 K in Fig. 7A. The magnetization values are also lower than predicted by the Brillouin function for a paramagnetic system with S = 1/2, in good agreement with previously proposed antiferromagnetic exchange couplings (Fig. S4, ESI†). On the contrary, at low temperatures, M vs. H curves are linear just at high values of the magnetic field, but at low fields a spontaneous magnetization is detected and magnetic hysteresis can be observed at H < 1 kOe and T < 16 K (Fig. 7B). A low saturation magnetization value is observed, 0.027 Nβ units at T = 3 K and H = 500 Oe, far below the saturation value expected for a S = 1/2 system with g = 2.10. This observation is consistent with a weak ferromagnetism arising from spin canting in an ordered antiferromagnetic material. It is remarkable that the remanent magnetization and coercive field are practically zero, despite the described magnetic hysteresis. Thus, the canting in different perovskite layers cancels out in zero-field. However, a small field is enough to align residual moments and a net magnetization is produced.50 The weak interplane interactions are responsible for the long range ordering achieved at low temperature by forcing the alignment of the canted magnetic moments of neighboring planes. The origin of spin canting is proposed to be an antisymmetric exchange term in the exchange Hamiltonian due to the tilting of CuO6 octahedra.49
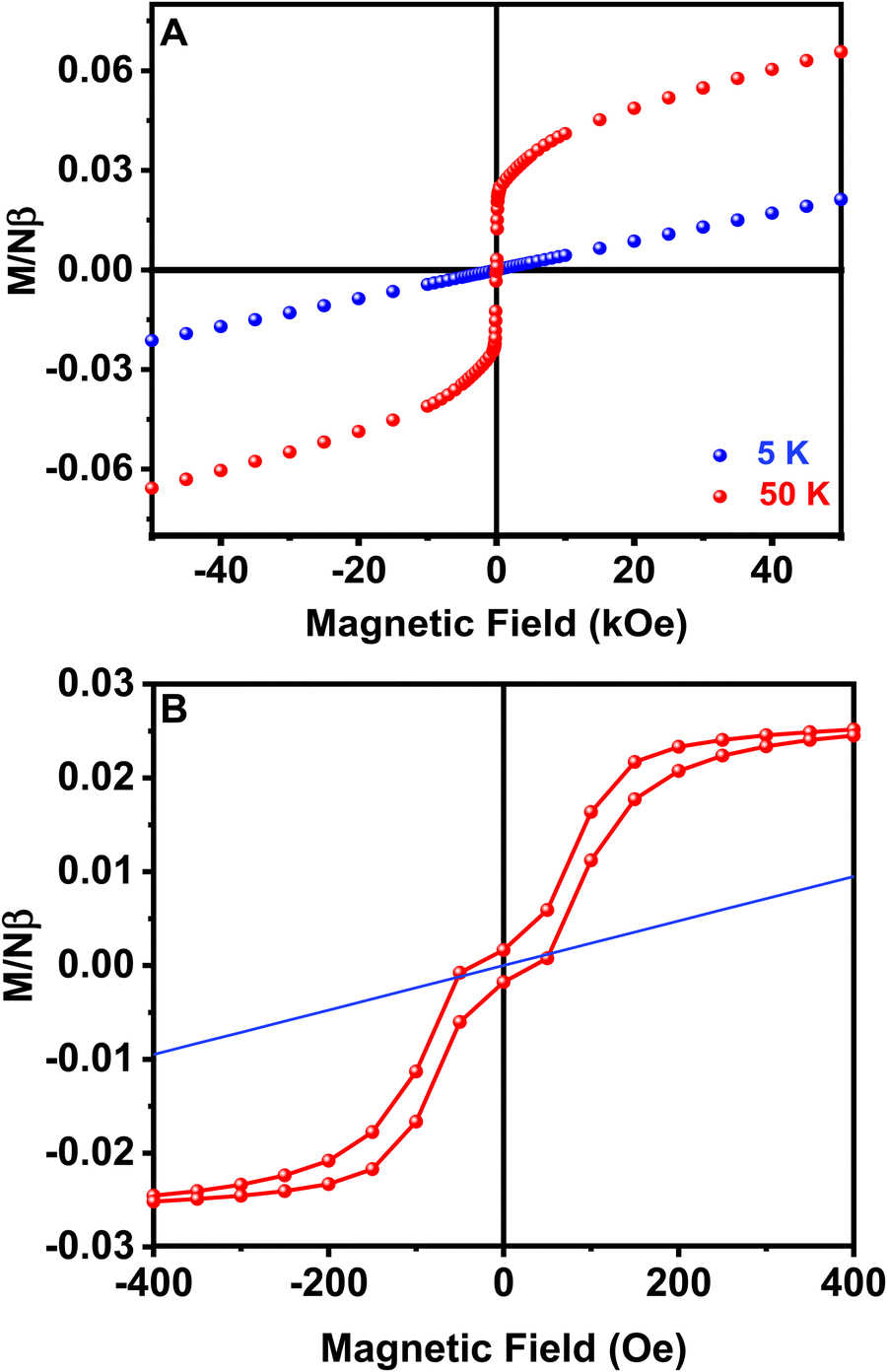 | ||
| Fig. 7 (A) Magnetization vs. field curves obtained above and below the ordering temperature. (B) Hysteresis loop registered at 3 K (the solid line corresponds to the Brillouin function at that temperature). | ||
Conclusion
To conclude, we have successfully prepared a novel perovskite-type copper (II) organic framework formulated as (NH3(CH2)2NH3)[Cu(HCO2)4], using a mild solution chemistry approach by a facile slow diffusion process. The compound was characterized by SCXRD, FT-IR TGA and DSC techniques. The compound crystallizes in the monoclinic system with P21/c space group, and its structure consists of 2D squared layers of formato-bridged [Cu(HCOO)4]2− and an interlayer of [NH3(CH2)2NH3]2+ organic dications, which is sandwiched between the inorganic layers and extended in a zigzag structure with inorganic anions. The infrared spectrum of the compound shows the characteristic vibrational modes of the [NH3(CH2)2NH3]2+ organic cation and HCOO− groups. The TGA results indicate that this compound is stable until about 373 K. In addition to that, magnetic studies reveal that this compound shows a 2D Heisenberg antiferromagnetic behavior with moderate interactions between the Cu(II) ions through the O–C–O bridges below TN = 16 K. Additionally, compound 1 exhibits weak ferromagnetic behavior arising from spin canting; the origin of spin canting in an ordered antiferromagnetic material is proposed to be an antisymmetric exchange term in the exchange Hamiltonian due to the tilting of the CuO6 octahedra.51Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the following grants: PID2020-116519RB-I00 funded by MCIN/AEI/10.13039/501100011033 and the European Union (EU) and EU through “Fondo Europeo de Desarrollo Regional” (FEDER): 2022-GRIN-343259 and 2022-GRIN-34313 funded by UCLM (FEDER, EU). A.B.A. acknowledges the grant from the Spanish Service for the Internationalization of Education (SEPIE) for her stay at the UCLM, through the EU Erasmus+ key action program (2020-1-ES01-KA107-079868). We are thankful for the technical assistance of Interface Regional University Center at the University Sidi Mohammed Ben Abdellah (USMBA, Fez, Morocco) and thank the USMBA for the financial support.References
- S. Gonzalez-Carrero, R. E. Galian and J. Pérez-Prieto, Opt. Express, 2016, 24, A285–A301 CrossRef CAS PubMed.
- S.-T. Ha, R. Su, J. Xing, Q. Zhang and Q. Xiong, Chem. Sci., 2017, 8, 2522–2536 RSC.
- S. Ma, M. Cai, T. Cheng, X. Ding, X. Shi, A. Alsaedi, T. Hayat, Y. Ding, Z. a Tan and S. Dai, Science China Materials, 2018, 61, 1257–1277 CrossRef CAS.
- S. Shimizu, M. Yoshizawa-Fujita, Y. Takeoka and M. Rikukawa, ACS Omega, 2019, 4, 13260–13264 CrossRef CAS PubMed.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- A. Kaiba, M. H. Geesi, Y. Riadi, E. O. Ibnouf, T. A. Aljohani and P. Guionneau, J. Solid State Chem., 2021, 304, 122587 CrossRef CAS.
- W. Zhang and R.-G. Xiong, Chem. Rev., 2012, 112, 1163–1195 CrossRef CAS PubMed.
- S.-T. Ha, R. Su, J. Xing, Q. Zhang and Q. Xiong, Chem. Sci., 2017, 8, 2522–2536 RSC.
- Y. Wang, H. Xu, F. Wang, D. Liu, H. Chen, H. Zheng, L. Ji, P. Zhang, T. Zhang, Z. D. Chen, J. Wu, L. Chen and S. Li, Chem. Eng. J., 2020, 391, 123589 CrossRef CAS.
- Y. H. Kim and N. Hur, J. Korean Phys. Soc., 2020, 77, 1026–1030 CrossRef CAS.
- Z.-C. Zhang, T. Zhang, C.-Y. Su, M.-M. Lun, Y. Zhang, D.-W. Fu and Q. Wu, Inorg. Chem., 2022, 61, 13322–13329 CrossRef CAS PubMed.
- L. C. Gómez-Aguirre, B. Pato-Doldán, A. Stroppa, S. Yáñez-Vilar, L. Bayarjargal, B. Winkler, S. Castro-García, J. Mira, M. Sánchez-Andújar and M. A. Señarís-Rodríguez, Inorg. Chem., 2015, 54, 2109–2116 CrossRef PubMed.
- L. C. Gómez-Aguirre, B. Pato-Doldán, A. Stroppa, L.-M. Yang, T. Frauenheim, J. Mira, S. Yáñez-Vilar, R. Artiaga, S. Castro-García, M. Sánchez-Andújar and M. A. Señarís-Rodríguez, Chem. – Eur. J., 2016, 22, 7863–7870 CrossRef PubMed.
- Y. Wu, S. Shaker, F. Brivio, R. Murugavel, P. D. Bristowe and A. K. Cheetham, J. Am. Chem. Soc., 2017, 139, 16999–17002 CrossRef CAS PubMed.
- Y. Jie, H. Yuan, Z. YouQuan, F. Ting, H. Fan, K. Qian and Y.-H. Ye, Z. Naturforsch. B, 2018, 73, 571–575 CrossRef CAS.
- J. García Ben, J. Salgado Beceiro, I. Delgado Ferreiro, P. Dafonte-Rodríguez, J. López Beceiro, R. Artiaga, S. Castro-Garcia, M. Sánchez-Andújar, J. Bermúdez-García and M. Senaris-Rodriguez, Crystals, 2022, 12, 860 CrossRef.
- C.-Q. Jing, X. Yin, P.-C. Xiao, Y.-J. Gao, X.-M. Wu, C.-Y. Yue and X.-W. Lei, Chem. – Eur. J., 2022, 28, e202103043 CrossRef CAS PubMed.
- J. Zhou and J. Huang, Adv. Sci., 2018, 5, 1700256 CrossRef PubMed.
- A. Cuquejo-Cid, A. García-Fernández, C. Popescu, J. M. Bermúdez-García, M. A. Señarís-Rodríguez, S. Castro-García, D. Vázquez-García and M. Sánchez-Andújar, iScience, 2022, 25, 104450 CrossRef CAS PubMed.
- K. D. Hughey, A. J. Clune, M. O. Yokosuk, J. Li, N. Abhyankar, X. Ding, N. S. Dalal, H. Xiang, D. Smirnov, J. Singleton and J. L. Musfeldt, Inorg. Chem., 2018, 57, 11569–11577 CrossRef CAS PubMed.
- L. C. Gómez-Aguirre, B. Pato-Doldán, J. Mira, S. Castro-García, M. A. Señarís-Rodríguez, M. Sánchez-Andújar, J. Singleton and V. S. Zapf, J. Am. Chem. Soc., 2016, 138, 1122–1125 CrossRef.
- B. Pato-Doldán, L. Cañadillas-Delgado, L. C. Gómez-Aguirre, M. A. Señarís-Rodríguez, M. Sánchez-Andújar, Ó. Fabelo and J. Mira, J. Phys. Chem. C, 2023, 127, 3330–3338 CrossRef.
- X.-Y. Wang, Z.-M. Wang and S. Gao, Chem. Commun., 2008, 281–294, 10.1039/B708122G.
- Z. Yang, G. Cai, C. L. Bull, M. G. Tucker, M. T. Dove, A. Friedrich and A. E. Phillips, Philos. Trans. R. Soc., A, 2019, 377, 20180227 CrossRef CAS PubMed.
- M. Mączka, A. Ciupa, A. Gągor, A. Sieradzki, A. Pikul, B. Macalik and M. Drozd, Inorg. Chem., 2014, 53, 5260–5268 CrossRef.
- P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625–13627 CrossRef CAS PubMed.
- M. Mączka, J. Janczak, M. Trzebiatowska, A. Sieradzki, S. Pawlus and A. Pikul, Dalton Trans., 2017, 46, 8476–8485 RSC.
- S. Liu, B.-W. Wang, Z.-M. Wang and S. Gao, Dalton Trans., 2018, 47, 11925–11933 RSC.
- J. Luo, Q. Fu, H. Wang, D. Zhao, L. Luo and W. Li, AIP Adv., 2017, 7, 105119 CrossRef.
- P. Peksa, J. K. Zarȩba, M. Ptak, M. a M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) czka, A. Ggor, S. Pawlus and A. Sieradzki, J. Phys. Chem. C, 2020, 124, 18714–18723 CrossRef CAS.
czka, A. Ggor, S. Pawlus and A. Sieradzki, J. Phys. Chem. C, 2020, 124, 18714–18723 CrossRef CAS. - Z. Wang, K. Hu, S. Gao and H. Kobayashi, Adv. Mater., 2010, 22, 1526–1533 CrossRef CAS PubMed.
- B. Pato-Doldán, L. C. Gómez-Aguirre, J. M. Bermúdez-García, M. Sánchez-Andújar, A. Fondado, J. Mira, S. Castro-García and M. A. Señarís-Rodríguez, RSC Adv., 2013, 3, 22404–22411 RSC.
- G.-C. Xu, X.-M. Ma, L. Zhang, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2010, 132, 9588–9590 CrossRef CAS PubMed.
- P. J. Calderone, P. M. Forster, L. A. Borkowski, S. J. Teat, M. Feygenson, M. C. Aronson and J. B. Parise, Inorg. Chem., 2011, 50, 2159–2167 CrossRef CAS PubMed.
- S. Chen, R. Shang, B.-W. Wang, Z.-M. Wang and S. Gao, APL Mater., 2018, 6, 114205 CrossRef.
- P. Danylchenko, R. Tarasenko, A. Orendáčová, J. Strecka, E. Cizmar, V. Tkáč and M. Orendáč, J. Phys. Chem. Solids, 2023, 182, 111613 CrossRef CAS.
- M. Mączka, M. Ptak, S. Pawlus, W. Paraguassu, A. Sieradzki, S. Balciunas, M. Simenas and J. Banys, Phys. Chem. Chem. Phys., 2016, 18, 27613–27622 RSC.
- R. Shang, Z.-M. Wang and S. Gao, Angew. Chem., Int. Ed., 2015, 54, 2534–2537 CrossRef CAS PubMed.
- M. Trzebiatowska, B. Zarychta, A. Pikul, M. Mączka, P. Peksa and R. Poprawski, Phys. Chem. Chem. Phys., 2017, 19, 16749–16757 RSC.
- R. Scatena, R. D. Johnson, P. Manuel and P. Macchi, J. Mater. Chem. C, 2020, 8, 12840–12847 RSC.
- P. Šenjug, J. Dragović, F. Torić, I. Lončarić, V. Despoja, K. Smokrović, E. Topić, I. Đilović, M. Rubčić and D. Pajić, Materials, 2021, 14, 1730 CrossRef PubMed.
- P. H. McBreen, S. Serghini-Monim, D. Roy and A. Adnot, Surf. Sci., 1988, 195, L208–L216 CrossRef CAS.
- S. Castro-Garcia, Solids, 2021, 2, 2 Search PubMed.
- G. S. Rushbrooke and P. J. Wood, Mol. Phys., 1963, 6, 409–421 CrossRef CAS.
- R. L. Carlin, K. Kopinga, O. Kahn and M. Verdaguer, Inorg. Chem., 1986, 25, 1786–1789 CrossRef CAS.
- A. Rodríguez-Fortea, P. Alemany, S. Alvarez and E. Ruiz, Chem. – Eur. J., 2001, 7, 627–637 CrossRef.
- H. Kobayashi and T. Haseda, Sci. Rep. Res. Inst., Tohoku Univ., Ser. A, 1963, 16, 168 Search PubMed.
- T. Tajiri, S. Matsumoto, H. Deguchi, M. Mito, S. Takagi, C. Moriyoshi, K. Itoh and K. Koyama, J. Magn. Magn. Mater., 2007, 310, e566–e568 CrossRef CAS.
- A. J. Van Duyneveldt and C. A. M. Mulder, Physica, 1982, 114, 82–100 CAS.
- N. Burger, H. Fuess and P. Burlet, Solid State Commun., 1980, 34, 883–886 CrossRef CAS.
- I. Dzyaloshinsky, J. Phys. Chem. Solids, 1958, 4, 241–255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2332899. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ma00222a |
| This journal is © The Royal Society of Chemistry 2024 |