
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe osteoconductive properties of graphene-based material surfaces are finely tuned by the conditioning layer and surface chemistry†
Federica
Tiberio‡
 a,
Francesco
Amato‡
b,
Claudia
Desiderio
c,
Federica
Vincenzoni
de,
Giordano
Perini
f,
Irene
Moretti
f,
Alberto
Augello
e,
Ginevra
Friggeri
e,
Lishan
Cui
f,
Leonardo
Giaccari
b,
Martina
Salvati
a,
Luca
Polito
a,
Ornella
Parolini
ae,
Marco
De Spirito
ef,
Andrea Giacomo
Marrani
b,
Wanda
Lattanzi
*ae,
Massimiliano
Papi
*efg,
Lorena
Di Pietro‡
ae and
Valentina
Palmieri‡
eg
a,
Francesco
Amato‡
b,
Claudia
Desiderio
c,
Federica
Vincenzoni
de,
Giordano
Perini
f,
Irene
Moretti
f,
Alberto
Augello
e,
Ginevra
Friggeri
e,
Lishan
Cui
f,
Leonardo
Giaccari
b,
Martina
Salvati
a,
Luca
Polito
a,
Ornella
Parolini
ae,
Marco
De Spirito
ef,
Andrea Giacomo
Marrani
b,
Wanda
Lattanzi
*ae,
Massimiliano
Papi
*efg,
Lorena
Di Pietro‡
ae and
Valentina
Palmieri‡
eg
aDipartimento di Scienze della Vita e Sanità Pubblica, Università Cattolica del Sacro Cuore, Rome, Italy. E-mail: wanda.lattanzi@unicatt.it
bDipartimento di Chimica, Università di Roma “La Sapienza”, Rome, Italy
cIstituto di Scienze e Tecnologie Chimiche (SCITEC) “Giulio Natta”, Consiglio Nazionale delle Ricerche, Rome, Italy
dDipartimento di Scienze Biotecnologiche di Base, Cliniche Intensivologiche e Perioperatorie, Università Cattolica del Sacro Cuore, Rome, Italy
eFondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy. E-mail: Massimiliano.papi@unicatt.it
fDipartimento di Neuroscienze, Università Cattolica del Sacro Cuore, Rome, Italy
gIstituto dei Sistemi Complessi, Consiglio Nazionale Ricerche, Rome, Italy
First published on 17th April 2024
Abstract
The molecular layer that adsorbs on the biomaterial surface upon contacting body tissues and fluids, termed the conditioning layer (CL), influences cell behavior regulating scaffold integration and resilience in a patient-specific fashion. To predict and improve the clinical outcome of 3D-printed scaffolds, graphene coatings are employed in bone tissue engineering, due to the possibility to functionalize its chemical/physical properties. In this study, we investigated the composition and the influence of the CL on three different graphene oxide-based coatings of 3D-printed polycaprolactone (PCL) implants: graphene oxide (–GO), carboxylated GO (–GO–COOH) and reduced GO (–rGO). The effects of surface features and CL were evaluated in vitro using bone marrow-derived mesenchymal stromal cells (hBM-MSC). Our results showed that the CL formed on negatively charged PCL–GO–COOH and PCL–rGO scaffolds reduced cell adhesion, while simultaneously enhancing cell cluster formation and proliferation by a fivefold increase. The quantification of bone mineralized matrix highlighted that CL on both PCL–GO–COOH and PCL–rGO coatings sustained the osteogenic potential of these two types of GO. The analysis of CL components adsorbed on the scaffolds revealed that the PCL–GO–COOH and PCL–rGO coatings tend to entrap specific patterns of serum proteins (e.g. anti-adhesive and osteogenic modulators) and ions (carbonate and phosphate), suggesting a correlation between these enriched components and the observed biological outcomes of conditioned scaffolds. Lastly, PCL–rGO coatings maintained unique antibacterial properties after in vitro simulated CL formation, representing a suitable promising strategy to improve bone grafting capable of shaping CL formation while preserving the favorable osteoinductive properties of scaffolds.
Introduction
Bone grafting represents the second most common transplation worldwide, following blood transfusions,1,2 and there is an increasily growing demand for bone substitutes for reconstructive surgery.Bone is a highly dynamic tissue characterized by a remarkable self-renewal ability to remodel, regenerate and repair its structure and function upon injury.3,4 Nevertheless, spontaneous complete bone healing is usually limited to small defects.5 Bone defects larger than 2 cm in length (i.e. non-union defects) usually exceed the intrinsic regenerative capacity, thus representing a relevant clinical challenge that requires surgical grafting procedures.5,6 Non-union bone defects may result from multiple clinical conditions, including complex traumas, degenerative diseases, tumor resection and congenital malformations. A functionally effective bone regeneration mandatorily requires an efficient restoration of the mechanical strength and support (osteoconduction), along with the appropriate biochemical environment of osteoinductive stimuli for resident progenitor cells. Also, the designed strategy needs to minimize the risk of implant-site infections, which still represent the main adverse event affecting the outcome of bone grafting surgery and often require implant removal, with increased morbidities.7–10 Autologous bone grafting remains the gold standard in reconstructive orthopedic surgery, although its widespread clinical application is limited by several drawbacks, including the restricted availability of donor bone, pain and morbidity in grafting sites, risk of hematoma, and resorption.2,11,12
The recent advancements in 3D printing provide suitable alternatives to autologous bone grafts.13–16 3D printed scaffolds can meticulously mimic the microarchitecture and the anatomical structure of bone acting as osteoconductive supports by ensuring host cell adhesion, proliferation and differentiation.17,18 Among all, polycaprolactone (PCL) is a synthetic polyester polymer widely exploited for the fabrication of 3D scaffolds for bone tissue engineering, due to its good biocompatibility, its moldability, slow degradation rate, and loadbearing properties. Nonetheless, the clinical application of PCL is still limited owing to its low cell adhesion properties causing poor osteoconductive performance, and due to the limited ability to control its in vivo osteointegration.19–22 The success and clinical translation of biomimetic constructs indeed also depend on the interaction of the biomaterial with the in vivo biological microenvironment upon implantation, that it is now well known can largely affects its properties. The in vivo controlling of scaffolds remains a considerable hurdle to be considered to accurately predict individual responses and a successful bone grafting.
Specifically, a molecular layer initially adheres to the biomaterial surface upon contact with bodily tissues and fluids, namely the conditioning layer (CL), significantly influencing scaffold integration and its pro-regenerative capabilities.23–26 The CL alters the physicochemical properties of the scaffold surface playing a crucial role in biomaterial-tissue interactions and serving as the primary interface at the implantation site, thus providing more relevant than the material composition itself in triggering the initial cellular responses.27–29 The CL affects biomaterial integration also by modulating the colonization by microorganisms as well as the inflammation process, which altogether amount to the successful outcome of the implant.30 On the other hand, the chemical composition, charge, topography, and roughness of the biomaterial influence the CL dynamics,31 thus modifying the biomaterial surface chemistry can help in controlling its formation.
In this context, to predict the in vivo capabilities of biomaterials, there is a need for effective and diversified surface modification strategies to shape the CL and boost scaffold integration at the bone defect site.
Graphene oxide (GO) has stood out as an innovative and low-cost material for bone reconstruction.32 GO is derived from the oxidation of graphene, a carbon allotrope where carbon atoms are densely arranged in a two-dimensional honeycomb pattern. It is characterized by oxygen-containing functional groups bound to the carbon backbone along both its surfaces and edges. There is a large variety of functionalities on the GO surface, such as hydroxyl and epoxy groups on the basal plane and carbonyl, carboxylic acid ethers, quinones, lactones, and ketones at the edges.33–36 The functional groups can be selectively removed from GO by reduction methods in a spatially controlled fashion.34,37,38 These methods also allow the modulation of GO topography and roughness. The reactivity of these groups on GO, as their balance and distribution, also influence the chemical interaction and conjugation of GO-based scaffolds and thus their CL composition.34,39 In fact, the reduction of the oxygen content of GO inhibits complement activation, and the reduction along geometric patterns can cause selective binding of growth factors and act as patterned topological cues, altering cell polarization and growth patterns, while inducing the osteogenic differentiation of stem cells and progenitors.40 When used as coatings on titanium or magnesium devices or calcium phosphate scaffolds, graphenic materials have been observed to enhance cell attachment and differentiation. This bone-promoting effect is attributed to their high concentration of potential attachment points, diverse modes of binding (such as electrostatic interactions, hydrogen bonding, and π–π stacking), and large surface area. These characteristics enable graphenic materials to rapidly gather and focus essential signaling proteins that are crucial for bone formation and blood vessel development.41 Then, while graphene oxide, with its abundant oxygen-containing functional groups, offers enhanced biocompatibility and provides numerous sites for cellular attachment, which is crucial for the initial cell adhesion and subsequent differentiation, reduced graphene oxide (rGO), with its restored π-conjugated structure, tends to have better electrical conductivity. This feature can be particularly beneficial for bone differentiation.42 Also, an enhanced osteoconductivity of rGO has been explained due to the denaturation of insulin, an adipogenic differentiation stimulus. GO's oxygen-containing groups can provide multiple binding sites for proteins, which stabilize the protein structure. In contrast, the more hydrophobic surface of rGO leads to stronger π–π interactions, which induce structural changes due to strong interactions with the protein's aromatic residues.43 Controlling the GO surface chemistry and topography has the potential to meet the need for new strategies to personalize biomaterials for bone regeneration.
In this paper, we exploited recently developed GO synthesis methods44,45 to enable the chemical manipulation of GO coatings for 3D-printed PCL scaffolds. Specifically, GO, carboxylated GO (GO–COOH), and reduced GO (rGO) coatings were produced and the effects of the different GO surfaces on cell behaviour were evaluated in simulated in vivo conditions. To this aim, GO-coated scaffolds were conditioned with a control human serum, and cell adhesion and viability, growth, osteogenic differentiation, and bacterial proliferation have been studied and compared to the uncoated PCL. We have demonstrated that the surface chemistry of GO not only influenced protein adsorption and concentration but, more importantly, facilitate the formation of a biomimetic mineralized layer after exposure to simulated body fluid (SBF), conditioning the regenerative capacities of the GO-based surfaces. Overall, we propose an optimized approach to define an ideal bone graft coating strategy based on GO functionalities.
Experimental
Materials synthesis
Pristine GO (GO) was synthesized by following a reported procedure.44 Briefly, 69 mL of H2SO4 96 wt% were added to a mixture of synthetic graphite (3.0 g) and NaNO3 (1.5 g) and the mixture was cooled at 0 °C. Then, KMnO4 (9.0 g) was added in portions and the mixture was stirred at 35 °C for 30 min. Subsequently, Milli-Q water (138 mL) was added slowly, and the mixture was heated at 98 °C for 15 min. Next, additional distilled water (420 mL) and H2O2 (3 mL, 30% v/v) were added. For workup, filtration through a filter paper was performed and the solid obtained was centrifuged at 4000 rpm for 30 min in the presence of HCl 1.12 M and Milli-Q water (four times). During this step of purification, the supernatant was discarded and replaced with Milli-Q water. Next, the solid was transferred to a round-bottom flask, then Milli-Q water was added, and the mixture was stirred for a night to ensure proper exfoliation. Next, the dispersion was sonicated for 30 min and centrifuged at 3000 rpm for 40 min to recover the yellowish supernatant and discharge, as precipitate, the graphite oxide that may be present. This procedure was repeated until the obtainment of an almost colorless supernatant. Finally, the dispersion of GO was freeze-dried, and the solid powder was stored in a desiccator. Carboxyl-functionalized GO (GO–COOH) was prepared by following a reported procedure44 in which 25.5 mg of GO and 25.5 mg of K2CO3 (0.18 mmol) were added to 30 mL of dry DMF. Then, 130 mg of succinic anhydride (1.30 mmol) were added, and the mixture was sonicated for 30 min. Subsequently, the mixture was stirred at 50 °C for 24 h under an inert atmosphere (N2). After this time, the stirring was stopped and, subsequently, 160 mL of diethyl ether were added to precipitate the product. The mixture was then filtered through a filter paper and the solid collected was washed with an additional 40 mL of diethyl ether. Finally, after the complete solvent evaporation, 76 mg of a brown solid were obtained. Reduced GO (rGO) was synthesized from GO by follwing a reported procedure.45 Briefly, 10 mL of an aqueous dispersion of GO 4.25 mg mL−1 was diluted to 50 mL of volume using Milli-Q water. Then, 425 mg of sodium ascorbate (2.14 mmol) was added to the dispersion and the mixture was stirred at room temperature for 72 h. Subsequently, the mixture was centrifuged at 4000 rpm for 30 min in water (four times) discharging away the supernatant and keeping the solid black precipitate. Finally, the sample was freeze-dried, and the solid powder was stored in a desiccator.3D printing
3D printing of scaffolds was performed with a BIO X 3D bioprinter (Cellink) using a thermoplastic printhead (Cellink, heating capacity of up to 250 °C) and PCL pellets. The structure of scaffolds was designed using modeled 3D computer graphics and computer-aided design (CAD) software Rhinoceros software (Robert McNeel & Associates). The extrusion-based printing was done at a printhead temperature of 65 °C and a printed temperature of 25 °C. The extrusion pressure was set at 40 kPa, with a preflow of 20 ms and a speed of 22 mm s−1, and the nozzle diameter was 200 μm.Dynamic light scattering and Zeta potential
Dynamic light scattering measurements were performed with Zetasizer Nano ZS (Malvern), equipped with a 633 nm He–Ne laser and operating at an angle of 173°. UV-transparent cuvettes (Malvern) were used for experiments with a sample volume of 500 μL and a concentration of 100 μg mL−1. The measurements were performed at a fixed position (4.65 mm) with an automatic attenuator. For each sample, three measurements were averaged, and the diffusion coefficient D was retrieved through cumulant analysis from autocorrelation functions. The equivalent hydrodynamic radius (Z-Average size) was obtained by the Stokes–Einstein equation. Data analysis was performed by Malvern Zetasizer software. The zeta potential (ζ) was calculated from the electrophoretic mobility by means of the Henry correction to Smoluchowski's equation. Data analysis was performed by Malvern Zetasizer software.Atomic force microscopy
Atomic force microscopy (AFM) was performed with a NanoWizard II (JPK Instruments AG, Berlin, Germany). The images were acquired using silicon cantilevers with high aspect-ratio conical silicon tips (CSC37 Mikro-Masch, Tallinn, Estonia) characterized by an end radius of about 10 nm, a half conical angle of 20°, and a spring constant of 0.6 N m−1. Scan areas (10 × 10 μm) were imaged. The surface roughness of all samples was evaluated by using the software JPK SPM data processing. Briefly, 3 scan areas were imaged with AFM for each sample. Then, the roughness was measured in terms of both the arithmetical mean deviation of the assessed profile (Ra) and of root mean squared (Rq). Furthermore, in the ESI† the Rt, i.e. the height difference between the highest (Max) and lowest peak (Min) in the images were reported. Finally, Kurtosis was measured to describe the “sharpness” of the surface topography.Raman spectroscopy
Raman spectra were run at room temperature in backscattering geometry with an inVia Renishaw micro-Raman spectrometer equipped with an air-cooled CCD detector and super-Notch filters. An Ar+ ion laser (λlaser = 514 nm) was used, coupled to a Leica DLML microscope with a 20× objective. The resolution was 2 cm−1 and spectra were calibrated using the 520.5 cm−1 line of a silicon wafer. Raman spectra of GO and its derived materials were collected with 1% of power, 10 seconds of spectral acquisition and 20 scans.Fourier transform infrared spectroscopy (FTIR)
The chemical analysis of PCL and GO-coated PCL surfaces was carried out using attenuated total reflectance-Fourier transform infrared spectroscopy, using a Bruker ALPHA II FTIR Spectrometer, equipped with an attenuated total reflection module (Eco-ATR). The surfaces were directly laid upon the ATR crystal and the spectra were recorded in the wave number range of 4000–550 cm−1.X-ray photoelectron spectroscopy (XPS)
XPS measurements were carried out using a modified Omicron NanoTechnology MXPS system equipped with a monochromatic Al Kα (hν = 1486.7 eV) X-ray source (Omicron XM-1000), operating the anode at 14 kV and 16 mA. The C 1s photoionization region was acquired using an analyzer pass energy of 20 eV, while the survey spectra were acquired with a pass energy of 50 eV. A take-off angle of 21° with respect to the sample surface normal was adopted. The experimental spectra were theoretically reconstructed by fitting the secondary electrons background to a Shirley function and the elastic peaks to pseudo-Voigt functions described by a common set of parameters: position, full-width at half-maximum (FWHM), Gaussian–Lorentzian ratio. The relative amount of the different oxygenated functional groups was determined through the area of the peaks within the curve-fitting envelope of the C 1s region, with an uncertainty of ±10%. For the sake of comparison between different samples, all intensities (areas) of the peaks were normalized to the one due to the contribution from isolated sp2 C atoms (peak at 284.8 eV) in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C network, including also that related to extended π-delocalization (peak at 284.3 eV) in the case of rGO sample.
C network, including also that related to extended π-delocalization (peak at 284.3 eV) in the case of rGO sample.
Scanning electron microscopy
Scanning electron microscopy (SEM) was performed to evaluate the 3D printed scaffold morphology, simulated body fluid adhesion, cell adhesion and bone matrix formation using SEM Supra 25 microscope (Zeiss, Germany). For cell imaging samples were fixed for 30 minutes with glutaraldehyde (2.5%) and dehydrated in a series of ethanol solutions. All samples were sputter-coated with gold prior to imaging.Cell culture
Primary human bone marrow-derived mesenchymal stromal cells (hBM-MSC) were purchased from STEMCELL Technologies (Inc, Vancouver, Canada). hBM-MSC were cultured in growth medium [GM; MesenCult MSC Basal Medium (STEMCELL Technologies) supplemented with Mesencult MSC Stimulatory Supplement (STEMCELL Technologies)] and expanded for multiple passages (up to 4 passages) for subsequent experiments.Cell viability and proliferation assay
To evaluate the effects of GO-based biomaterials on hBM-MSC adhesion and proliferation, PCL, PCL–GO, PCL–GO–COOH and PCL–rGO scaffolds were placed into 48-well plates and sterilized by 70% ethanol (v/v) for 5 minutes. PCL was used as a control. A sample of the different biomaterials was also incubated with human serum (Sigma-Aldrich, St. Louis, MO, USA) for 1 h at 37 °C to allow the adsorption of its components (i.e. organic and inorganic species) on biomaterials’ surfaces. After this pretreatment, scaffolds were extensively washed with Dulbecco's phosphate buffered saline w/o calcium w/o magnesium (PBS; Aurogene, Rome, Italy) to remove loosely bound species. Thereafter, hBM-MSC were seeded on both untreated and pretreated surfaces, using a 7 × 103 cells-per-well density in fresh complete GM, and incubated in a humidified incubator at 37 °C with 5% CO2. After 48 hours of incubation, cell viability was measured using the CellTiter-Glo Cell viability assay (Promega, Madison, WI, USA). Briefly, the medium was carefully removed by the well and replaced with 120 μL of fresh GM containing 20 μL of CellTiter-Glo reagent and incubated at 37 °C in 5% CO2 for 2 hours. Fluorescence intensity was recorded with cytation 3 cell imaging multi-mode reader (Biotek, Winooski, VT, USA) by exciting at 550 nm and reading the emission at 600 nm. Cell growth was assessed after 24 and 72 hours of culture. For this purpose, cells on biomaterials were fixed in paraformaldehyde 4 wt% and cell nuclei stained with DAPI (Sigma-Aldrich). The experiment was repeated in triplicate and Aat least 3 nonoverlapping images representing each experimental condition were acquired using cytation 3 and the number of nuclei/field was counted.Osteogenic differentiation assay
To investigate the effect of the different GO-based biomaterials on osteogenic differentiation of hBM-MSC, cells were seeded on PCL, PCL–GO, PCL–GO–COOH and PCL–rGO scaffolds at a density of 7 × 103 cells per well and grown until confluence. The GM was then replaced with an osteoinductive medium (OM) composed as follow: DMEM with low glucose (1 g L−1) (Aurogene) supplemented with 1% L-glutamine (Aurogene), 1% antibiotics (penicillin 100 IU mL−1, streptomycin 100 mg mL−1; Aurogene), 10% fetal bovine serum (FBS; GIBCO, Thermo Fisher Scientific, Waltham, MA, USA), Dexamethasone (0,1 μM), ascorbic acid (10 μM) and β-glycerophosphate (50 μM) (Sigma Aldrich) as previously described.46 OM was changed every 3 days. After 7 days, osteogenic differentiation was assessed morphologically using Alizarin red S (ARS) staining (Sigma-Aldrich) to detect the in vitro matrix mineralization.46 Furthermore, ARS dye was extracted from the stained monolayer by dissolving each sample in 10% acetic acid and ARS adsorption spectrum at 405 nm was quantified47,48 using an automatic microplate photometer (ELx800; Biotek, Bad Friedrichshall, Germany). Since GO interacts with ARS, the concentration of each sample was calculated by subtracting the optical density (OD) of neat sample (blank). We replicated the experiment in triplicate.In addition, to analyze the effect of (+)-sodium L-ascorbate of PCL–rGO on osteogenic differentiation, hBM-MSC were seeded into 48-well plates and cultured in GM until confluence, when the GM was switched with OM supplemented with (+)-sodium L-ascorbate at the concentration of 17.5 μg mL−1 and 1.75 μg mL−1. Cells grown with standard OM were used as controls. ARS staining and quantification of stained monolayers were performed to investigate the osteogenic differentiation of cells as described above.
A modified-simulated body fluid (SBF; Biochemazone, Leduc, Alberta, Canada), a solution of apatite containing calcium ions and phosphate ions, was used to evaluate the binding of inorganic components, excluding the contribution of the organic fraction. To this aim, the different scaffolds were treated with SBF for 7 days; thereafter the chemical composition of the surfaces was analyzed by using FTIR.
Protein analysis and characterization
Serum proteins were detached from the scaffolds using a protocol optimized for phage display. Briefly, after 10× washing with PBS to remove loosely bonded proteins, 100 μL of 0.2 M glycline–HCl (pH = 2.2) were added to each well and incubated for 8 minutes upon gently rocking; then the pH of the eluate was neutralized with 15 μL 1 M Tris-HCl (pH = 9.1).49 Protein samples were quantified using the Pierce BCA protein assay kit (Thermo Fisher Scientific) and a UV spectrophotometer (NanoDrop™ One/OneC Microvolume UV-vis, Thermo Fisher Scientific). Sample quality was assessed through SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (Bio-Rad Laboratories GmbH, Feldkirchen, Germany). Protein samples were submitted to filter aided sample preparation (FASP) (10 kDa molecular cut-off) protocol for protein digestion before LC–MS analyses. Specifically, a volume of the protein sample corresponding to 50 μg of total protein content was mixed with urea buffer solution (8 M urea in 0.1 M Tris/HCl buffer pH = 8.5), transferred to the filter device, and centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min. After urea buffer solution washing (1× volume) and centrifugation, the supernatant was added with 8 mM 1,4-dithiothreitol (DTT) in urea buffer solution to allow disulfide bridges reduction for 15 min incubation at 37 °C. After centrifugation, any excess of DTT was eliminated by repeated cycles of urea buffer washings and centrifugations. The supernatant was then treated for thiols carboxamide methylation with 50 mM iodoacetamide (IAA) solution in urea buffer solution. The mixture was incubated at room temperature in the dark for 20 min. After centrifugation, any excess of IAA was removed by incubating the supernatant with DTT solution at 37 °C for 15 min, followed by urea buffer solution washings and filter buffer exchange with 50 mM ammonium bicarbonate filter buffer exchange. Sample digestion was carried out overnight at 37 °C using trypsin enzyme (1 μg/μL solution in 50 mM ammonium bicarbonate buffer) in ratio 1:50 (w/w). Enzymatic digestion was stopped by the addition of concentrated formic acid (FA) to the final level in the sample of 1% (v/v). The proteolytic peptides were collected by centrifugation of FASP device, then lyophilized and redissolved in 0.1% FA water solution (v/v) for LC–MS analysis.
000 rpm for 15 min. After urea buffer solution washing (1× volume) and centrifugation, the supernatant was added with 8 mM 1,4-dithiothreitol (DTT) in urea buffer solution to allow disulfide bridges reduction for 15 min incubation at 37 °C. After centrifugation, any excess of DTT was eliminated by repeated cycles of urea buffer washings and centrifugations. The supernatant was then treated for thiols carboxamide methylation with 50 mM iodoacetamide (IAA) solution in urea buffer solution. The mixture was incubated at room temperature in the dark for 20 min. After centrifugation, any excess of IAA was removed by incubating the supernatant with DTT solution at 37 °C for 15 min, followed by urea buffer solution washings and filter buffer exchange with 50 mM ammonium bicarbonate filter buffer exchange. Sample digestion was carried out overnight at 37 °C using trypsin enzyme (1 μg/μL solution in 50 mM ammonium bicarbonate buffer) in ratio 1:50 (w/w). Enzymatic digestion was stopped by the addition of concentrated formic acid (FA) to the final level in the sample of 1% (v/v). The proteolytic peptides were collected by centrifugation of FASP device, then lyophilized and redissolved in 0.1% FA water solution (v/v) for LC–MS analysis.
Ultra high-performance liquid chromatography-nanoESI mass spectrometry analysis (UHPLC-ESI-MS/MS)
UHPLC-ESI-MS/MS analyses of the digested protein samples were performed in triplicate on UltiMate 3000 RSLCnano System coupled to Orbitrap Elite MS detector with EASY-Spray nanoESI source (Thermo Fisher Scientific, Waltham, MA, USA) and equipped with Thermo Xcalibur 2.2 computer program (Thermo Fisher Scientific) for instrumental operations and data acquisition. Chromatographic separations were performed on PepMap C18 EASY-Spray column (2 μm particles, 100 Å pore size, 15 cm length, 50 μm I.D.) (Thermo Fisher Scientific) to an Acclaim PepMap100 nano-trap cartridge (C18, 5 μm, 100 Å, 300 μm, ID × 5 mm) (Thermo Fisher Scientific). The separation was performed under thermostated temperature (40 °C) and gradient elution program using 0.1% FA aqueous solution as Eluent A and acetonitrile/water/FA solution (99.9:0.1%v/v) as Eluent B as following: (i) 5% B for 7 min, (ii) 5% to 35% B for 113 min, (iii) 35% B to 99% for 2 min, (iv) 99% B for 3 min, (v) 99% to 1.6% B for 2 min, (vi) 1.6% B for 3 min, (vii) 1.6% to 78% B for 3 min, (viii) 78% B for 3 min, (ix) 78% to 1.6% B for 3 min, (x) 1.6% B for 3 min, (xi) 1.6% to 78% B for 3 min, (xii) 78% B for 3 min, (xiii) 78% B to 5% B for 2 min, and (xiv) 5% B for 20 min. The mobile phase flow rate was 0.3 μL min−1. The injection volume was 5 μL. The Orbitrap Elite operated in positive ionization mode with a 60000 full scan resolution, in 350–2000 m/z acquisition range. MS/MS fragmentation was obtained using collision-induced dissociation (CID) with a normalized collision energy of 35% and data-dependent scan (DDS) mode on the top 20 most intense signals of each MS spectrum. The minimum signal threshold set was 500.0, with an isolation width of 2 m/z and a default charge state to +2. MS/MS spectra acquisition was performed in the linear ion trap at a normal scan rate.
Data analysis
LC–MS raw data were processed using Proteome Discoverer 1.4 software (version 1.4.1.14, Thermo Fisher Scientific) for protein identification based on SEQUEST HT cluster as search engine against the Homo Sapiens databank (UniProtKB/Swiss-Prot protein knowledgebase released in February 2022). The signal-to-noise (S/N) threshold was set to 1.5. Trypsin was set as a cleavage enzyme with a maximum of 2 missed cleavage sites. Minimum and maximum peptide lengths were set to 6 and 144 residues, respectively. Mass tolerance was set at 10 ppm and fragment mass tolerance of 0.5 Da and 0.02 Da; both “use average precursor mass” and “use average fragment mass” were set to “False”. Methionine oxidation (+15.99 Da) and N-terminal acetylation (+42.011 Da) were set as dynamic modifications, while carbamidomethylation of cysteine (+57.02 Da) as a static modification. Validation of protein and peptide identifications was performed through a decoy database search and calculation of the false discovery rate (FDR) using the Percolator node, with strict and relaxed FDR target values set at 0.01 and 0.05, respectively.The resulting protein identification data were further filtered for high-confidence peptide identification, requiring a minimum of at least 2 peptides per protein, each with a minimum peptide length of 9 amino acids and peptide rank 1, according to the human proteome project (HPP) mass spectrometry data interpretation guidelines.50 Sample data grouping analysis was performed using the Venn diagram tool at https://bioinformatics.psb.ugent.be/webtools/Venn/.
Antibacterial effects
Samples were tested for their antibacterial performance. E. coli ATCC 25922 was chosen as the standard bacterium. Bacteria were inoculated in a Lennox LB Broth at 37 °C overnight. Afterwards, 250 μL of cell suspension was subcultured in 250 mL of LB and harvested at the exponential growth phase and diluted in PBS. Samples were incubated with 50 μL of E. coli suspension diluted in PBS to a concentration of 105 CFU mL−1 for 3 hours. At the end of the incubation, the samples were washed and vortexed in PBS to recover cells from the surface. The resulting solution was then plated on LB Agar plates at 37 °C overnight and the CFU were quantified. The experiment was repeated in triplicate.Results and discussion
Morphological and spectroscopical analysis of GO coatings on PCL scaffolds
PCL–GO coatings were prepared using GO samples with tailored surface chemistry and compared with uncoated PCL scaffolds used as controls. Precisely, coatings were made with GO, a GO enriched with additional carboxyl groups (GO–COOH) and a reduced graphene oxide (rGO) depleted in oxygen-based functional groups after the treatment with the mild and green reducing agent sodium ascorbate44,45 (Fig. S1A, ESI†). The characterization of GO flakes was obtained by dynamic light scattering and ζ potential measurements (Table S1, ESI†). The determined average size was 111 ± 35 nm for GO, 249 ± 62 nm for GO–COOH and 245 ± 31 nm for rGO and with the relative surface charges determined through ζ measurements of −24.8 ± 1.7 mV for GO, followed by −31 ± 0.5 mV for GO–COOH and −37 ± 2.2 mV for rGO.For the coating preparation, GO, GO–COOH or rGO aqueous dispersions at concentration 0.5 mg mL−1 were deposited by drop casting on PCL scaffolds and let dry overnight in sterile conditions. The AFM characterization of surface properties is reported in Fig. 1A where images show the typical rough surfaces, regardless of the type of GO employed. Values obtained for surface roughness are reported in Fig. S1B (ESI†), that shows a significantly higher roughness for PCL–GO–COOH followed by PCL–GO and PCL–rGO.
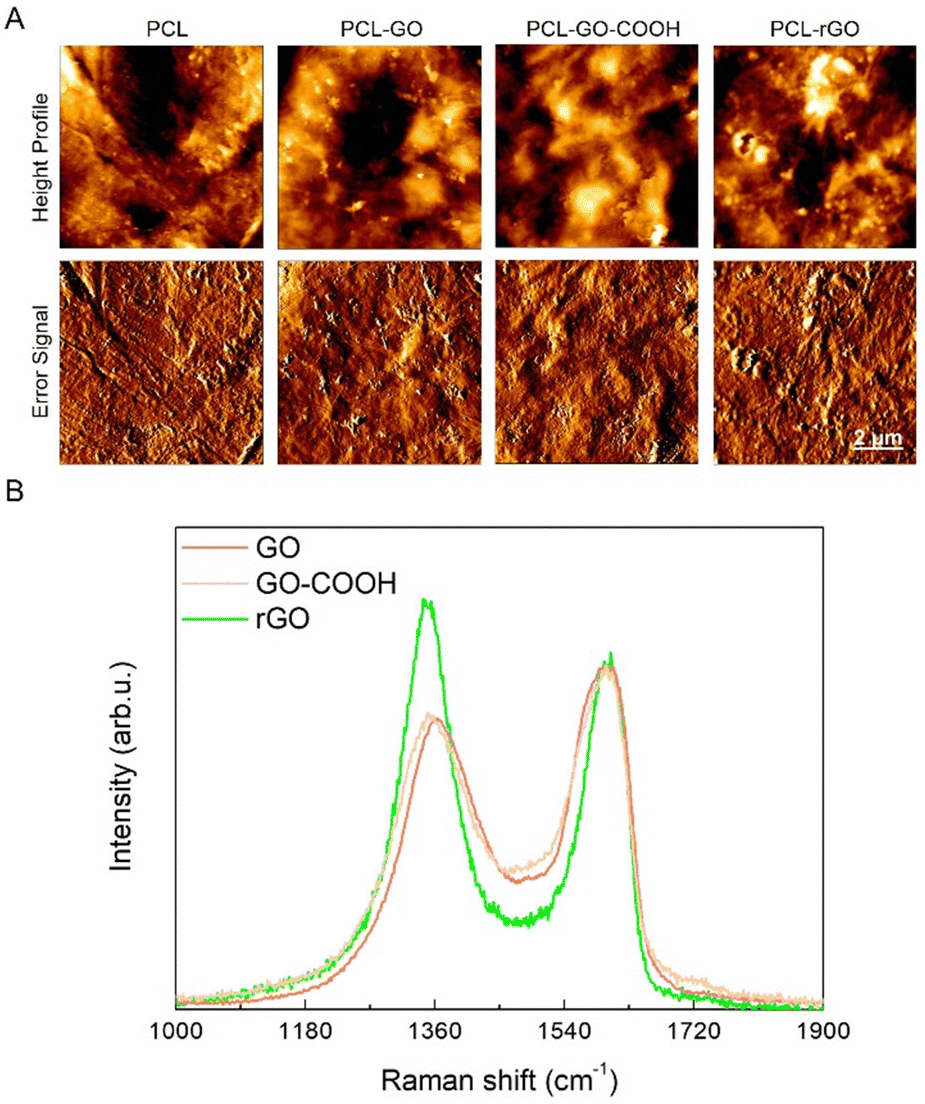 | ||
| Fig. 1 Morphological and spectroscopical characterization of GO coatings. (A) Representative images displaying surface characterization by AFM of different coatings on PCL. The size of the scanned area is 10 × 10 μm. (B) Normalized Raman spectra of GO (red line), GO–COOH (yellow line) and rGO (green line). | ||
The Raman spectrum of the pristine GO displayed the diagnostic D and G bands. The former, associated to the ring breathing mode from sp2 carbon-rings adjacent to an edge or defect, was localized at ≈1363 cm−1 while the latter was centered at ≈1600 cm−1 (Fig. 1B, black line) and due to the C–C stretching mode of the sp2 domains.51,52 It is well established that the variation of the ID/IG ratio is an important parameter to estimate the variation in the number of defects in the layers. In fact, this ratio slightly increased from 0.84 in GO to 0.86 in GO–COOH as a consequence of the carboxyl-functionalization (Fig. 1B, red line), which only marginally led to additional structural defects. Conversely, after the mild chemical reduction of GO with sodium ascorbate, the ID/IG ratio rose up to 1.18 (Fig. 1B, blue line) owing to the depletion of the epoxy-functionalities that were mainly localized in the basal plane of the layers, in accordance with the literature.44,45,53,54
In order to further investigate the surface functionalities, present in the layers of the GO-based materials, XPS measurements were performed. A comparison of the C 1s region after curve fitting of the various GO-based nanomaterials has been presented in Fig. 2. To obtain an accurate fitting, the XPS spectrum of GO (Fig. 2) was deconvoluted into five components (see Table S1, ESI†): Csp2 (red), hydroxyls (blue), epoxides (green), carbonyls/carboxylates (purple) and carboxyls (orange).55 By observing the spectrum of the carboxylated graphene oxide (GO–COOH) shown in Fig. 2, an extensive increase was evident in the peak associated to carbonyls/carboxylates (purple), whose area ratio versus Csp2 (Ax/Asp2) rose from 0.19 to 0.30 (Table S1, ESI†), while the component relative to epoxides considerably decreased from 0.75 to 0.28, due to their involvement in the carboxylation reaction of GO with succinic anhydride. Furthermore, to obtain an optimal fit, it was necessary to add a component associated to the methylene groups of the appended succinate residual. Regarding the rGO sample, it was evident that the chemical reduction of GO with sodium ascorbate led to a general drop of oxygen functionalities, as observable in the 285.5–289.0 eV BE range (Fig. 2). In the case of epoxide and carbonyls, this decrease was larger than for hydroxyls, whereas carboxyl amount was maintained similar to the pristine GO. Finally, to attain an accurate curve-fitting of the experimental data, the rGO spectrum required an additional low-energy asymmetrical component at 284.3 eV (Cgr), associated with an increased carbon sp2 network after reduction.45,56,57
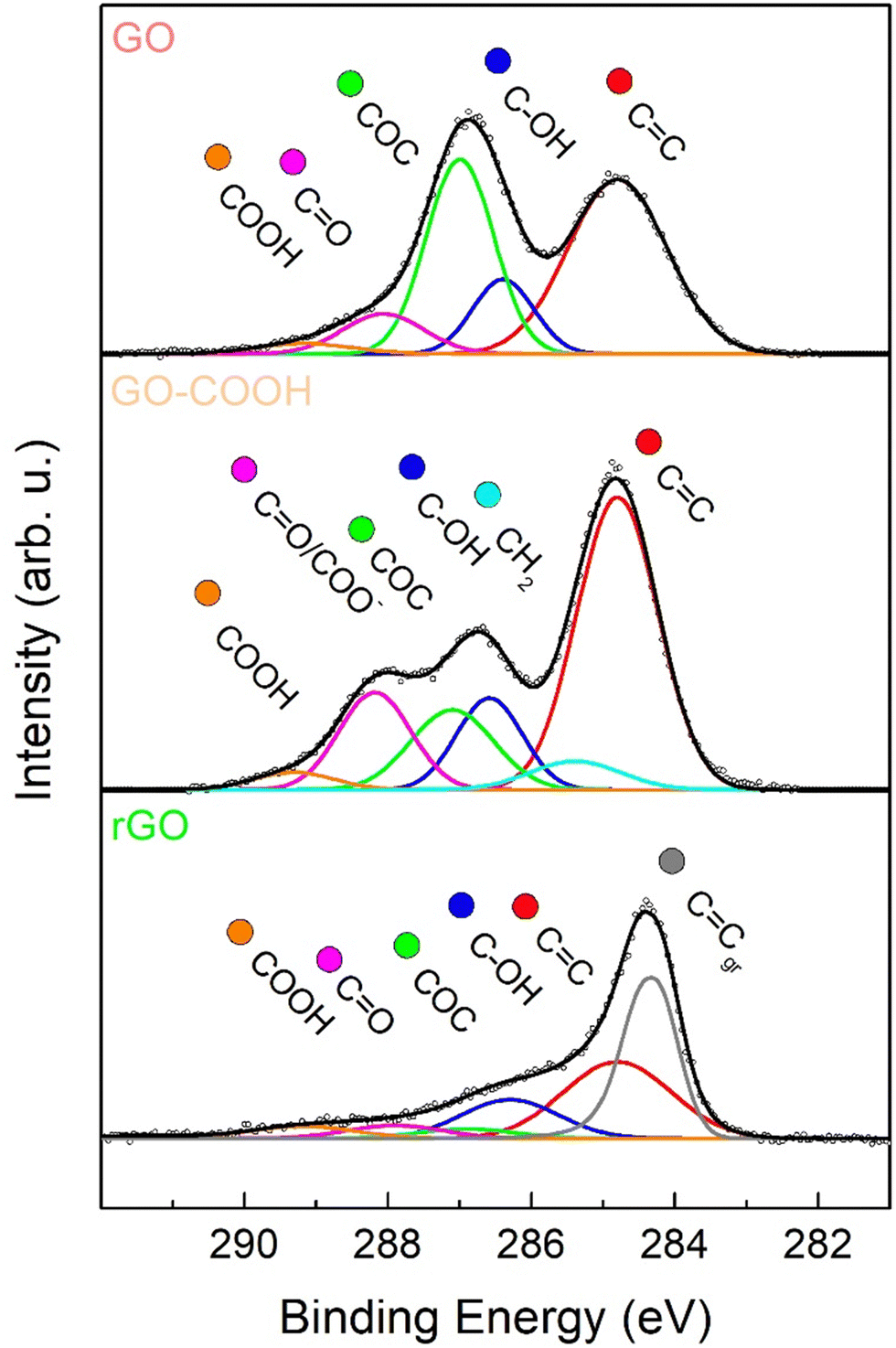 | ||
| Fig. 2 X-ray photoelectron spectroscopy (XPS) characterization of GO coatings. XPS spectra in the C 1s region of GO (upper panel), GO–COOH (center panel) and rGO (lower panel). Experimental data are reported in dots, while curve-fitting results are reported with continuous coloured lines, corresponding to different oxygenated functional groups. | ||
Cell adhesion and proliferation on GO coatings in simulated in vivo conditions
The biocompatibility of the different GO-based coatings and their capabilities to support cell growth were investigated using hBM-MSC grown on the different scaffolds. Cell adhesion, viability and proliferation were analyzed comparatively among the differently coated PCL scaffolds, with and without human serum preconditioning. Our results showed that, while serum pretreatment did not affect the viability of cells grown on PCL, the metabolic activity of hBM-MSC was slightly reduced on conditioned PCL–GO, PCL–GO–COOH and PCL–rGO already suggesting that a specific CL influenced surfaces’ biological capability (Fig. S2A, ESI†).We then analyzed the adhesion of cells at different time points, as well as the coating effect on cell proliferation (Fig. 3A). Our results displayed that hBM-MSC adhesion was comparable between cells on the different untreated scaffolds analyzed after 24 hours of culture, whereas the number of cells attached on scaffolds previously conditioned with serum decreased on GO-based biomaterials already after 1 day of growth (Fig. 3B and Fig. S2B, ESI†). To elaborate, pretreated PCL–GO–COOH and PCL–rGO showed the lowest adhesion capacity compared with all the untreated scaffolds and with both pretreated PCL and PCL–GO (Fig. 3B and Fig. S2B, ESI†). Conversely, the number of cells on untreated and serum-conditioned scaffolds showed no difference after 24 hours of culture (Fig. 3B and Fig. S2B, ESI†). After 72 hours, the number of cells remained low on conditioned GO-based biomaterials compared with the respective unconditioned scaffolds, whereas conditioned PCL scaffolds showed a significantly increased number of adherent hBM-MSC compared with the other pretreated conditions (Fig. 3B and Fig. S2C, ESI†).
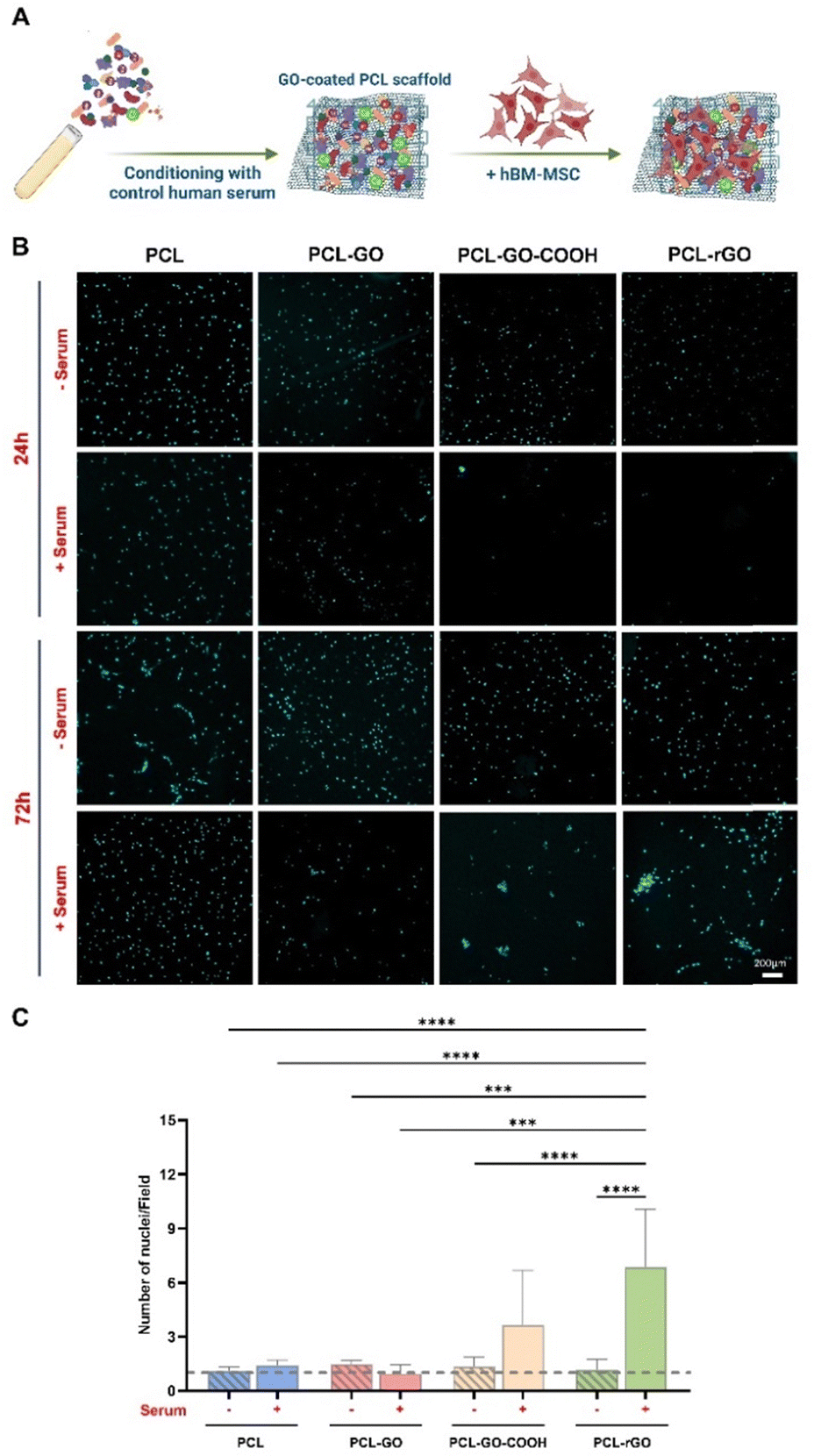 | ||
| Fig. 3 Cell adhesion and proliferation on GO coatings. (A) Schematic representation of the workflow for the analysis of hBM-MSC growth on GO-based scaffolds conditioned with human serum (created with https://Biorender.com). (B) Images of nuclei stained with DAPI after 24 and 72 hours of culture of cells cultured on both untreated (−) and serum pretreated (+) scaffolds. (C) The histogram summarizes the proliferation rate of cells on GO coatings and each bar indicates the ratio between the number of nuclei at 72 h and nuclei of cells at 24 h in each condition. The results are reported in the graph bar as mean ± standard deviation (SD). P values were assessed by ordinary one-way ANOVA followed by Tukey's multiple comparisons test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
The proliferation rate on each surface, calculated by considering the number of cells observed after 72 hours and 24 hours, indicated that hBM-MSC growth was fostered on PCL, PCL–GO–COOH and PCL–rGO pretreated with serum compared with the same experimental condition at 24 hours (Fig. 3C). Specifically, the highest growth of hBM-MSC has been observed on serum conditioned PCL–GO–COOH and PCL–rGO (Fig. 3C). The proliferation data might reflect the metabolism reported in Fig. S2A (ESI†), i.e. even if the cell number is reduced (DAPI in Fig. S2B and C, ESI†), there is an evident response to proteins on the surface.
Of note, the proliferation rate on the pretreated PCL–rGO resulted particularly interesting since a 5-fold increase in cell number was observed and this trend was significantly higher compared to the other conditions (Fig. 3C). Then, analyzing the distribution of cells on the different surfaces, we observed that in treated PCL–GO–COOH and PCL–rGO cells formed packed clusters, while on other biomaterials, the cells distributed and grew evenly over the whole area (Fig. 3B).
These results suggested that CL on GO surfaces influenced both cell adhesion and proliferation, reducing the number of cells that initially adhere to the scaffolds, whereas strongly promoting cells proliferation above all on PCL–rGO. Several studies demonstrated that the capability of graphene derivatives to enhance cell viability, adhesion and growth, partially resides in their strong proficiency to adsorb proteins through electrostatic interaction by oxygenated groups on their surface, thereby creating a layer between cells and material surfaces.58–60
Effects of GO biomaterials on osteogenic differentiation
The osteoinductive abilities of GO and rGO were largely described both in in vitro and in vivo studies,40,59,61 though insufficient insights are available regarding the correlation between GO chemistry, CL composition, and the osteogenic commitment of MSC. We thus analyzed if the CL could also influence the osteogenic differentiation of hBM-MSC (Fig. 4A).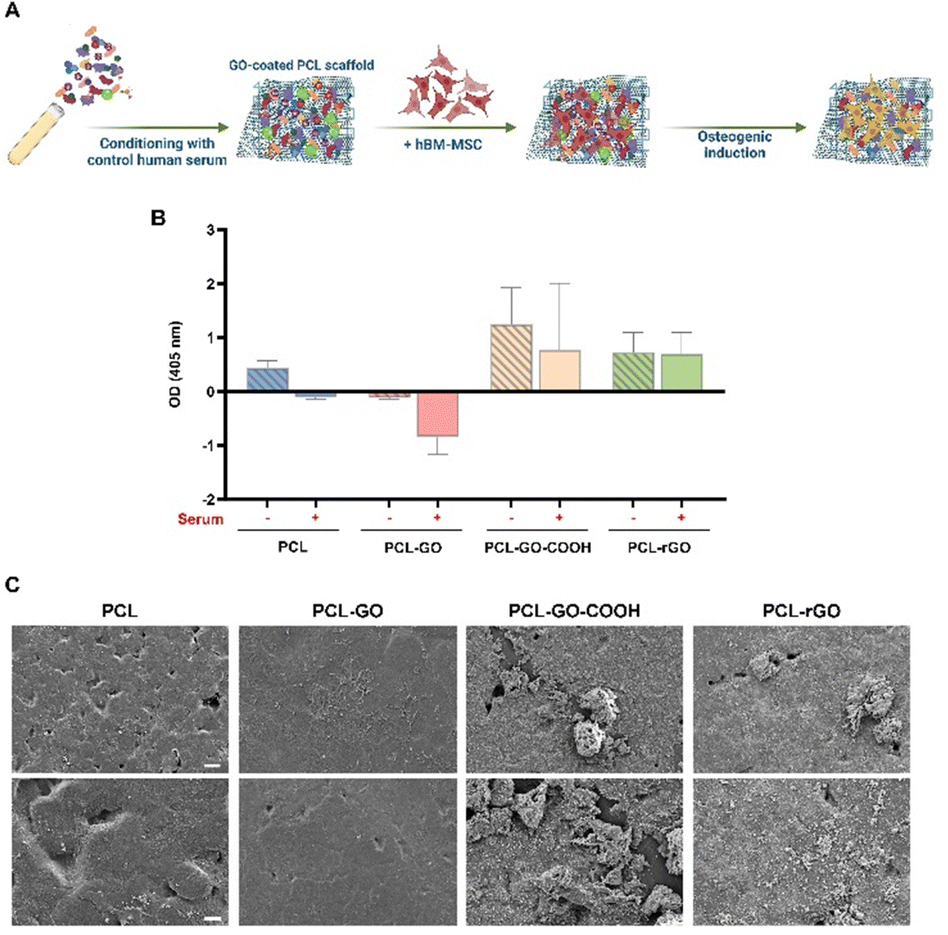 | ||
| Fig. 4 Osteogenic differentiation assays of hBM-MSC on GO coatings. (A) Schematic workflow for the culture and osteogenic induction of hBM-MSC on GO-coated scaffolds after pretreatment with serum (created with https://Biorender.com). (B) The graph displays the quantification of bone matrix deposited by hBM-MSC on the different scaffolds conditioned with human serum (+) or not (−). The results are reported in the graphs as mean ± SD and they represent the optical density of each sample, from which the corresponding blank values were subtracted, resulting in some negative values. These negative values were only observed in conditions where the mineralized matrix was slightly detected. (C) Representative images of the mineralized matrix deposits on serum-coated scaffolds after 7 days of osteogenic induction of hBM-MSC obtained by SEM [Scale bar: 20 μm (top images), 10 μm (bottom images)]. | ||
ARS staining and quantification showed that PCL–GO–COOH and PCL–rGO displayed the best osteoinductive capacity compared with both PCL and PCL–GO (Fig. 4B). Additionally, the conditioning with serum did not significantly affect the osteogenesis of hBM-MSC on PCL–GO–COOH and PCL–rGO, whereas the osteogenic differentiation of hBM-MSC cultured on serum conditioned PCL and PCL–GO was completely inhibited (Fig. 4B). To confirm CL effects on the osteogenic commitment of cells, SEM images of samples after 7 days of induction in serum coated scaffolds were analyzed. Our results demonstrated the presence of an evident mineralized matrix layer on both pretreated PCL–GO–COOH and PCL–rGO coatings (Fig. 4C).
Considering that the PCL–rGO chemical synthesis is based on sodium ascorbate, we verified whether part of its effect on osteogenic differentiation of hBM-MSC could be mediated by the presence of increased concentrations of sodium L-ascorbate in the culture medium. We evaluated the effect of a concentration of sodium ascorbate comparable to that used to synthesize PCL–rGO deposited on the scaffold (1.75 μg mL−1) and of a 10× higher concentration (17.5 μg mL−1). Our data showed that the treatment did not significantly affect the osteogenic differentiation of cells, when compared with cells grown for 7 days in standard OM (Fig. S3, ESI†).
Overall, our results confirmed the osteoinductive effects of rGO coatings.59,62 We hypothesize that the specific adsorption of factors present in the OM by this nanomaterial provides cells with a concentrated pool of substances that strongly support their differentiation.58,59 Interestingly, our data showed that the CL on GO substrates negatively influenced its osteoinductive properties, while PCL–GO–COOH and PCL–rGO maintained their potential to enhance osteogenesis even after CL adsorption. Also, GO coating is known to affect cellular mechanosensing and mechanotransduction through the integrin/FAK axis,63 hence the differential nanopatterned surface achieved with the different GO modifications may differentially affect cellular adhesion, growth, osteogenic commitment and matrix deposition.
To better delve into the origin of the CL over the analyzed surfaces, we first evaluated the inorganic component studying the adsorption of SBF. SBF replicates ions in human serum and can be used to study the formation of apatite layers on the surface of implants to predict the in vivo bone bioactivity.64,65 We incubated samples in SBF for 7 days and then analyzed the chemical composition of the surfaces by using FTIR. FTIR spectra of samples without SBF have been reported for reference in Fig. S4 (ESI†). As seen in peaks at 875 cm−1, 1420 cm−1 and 1460 cm−1, marked with red dashed lines and ascribable to CO3−2, and 605 cm−1 and 1030 cm−1, marked with blue dashed lines ascribable to PO4−3, in PCL–rGO and PCL–GO–COOH samples an apatite layer was formed after exposure to SBF (Fig. 5A).66,67 From SEM images in Fig. 5B, the deposition of the apatite layer was evident, while small sparsed crystals were visible on the surface of PCL and PCL–GO.
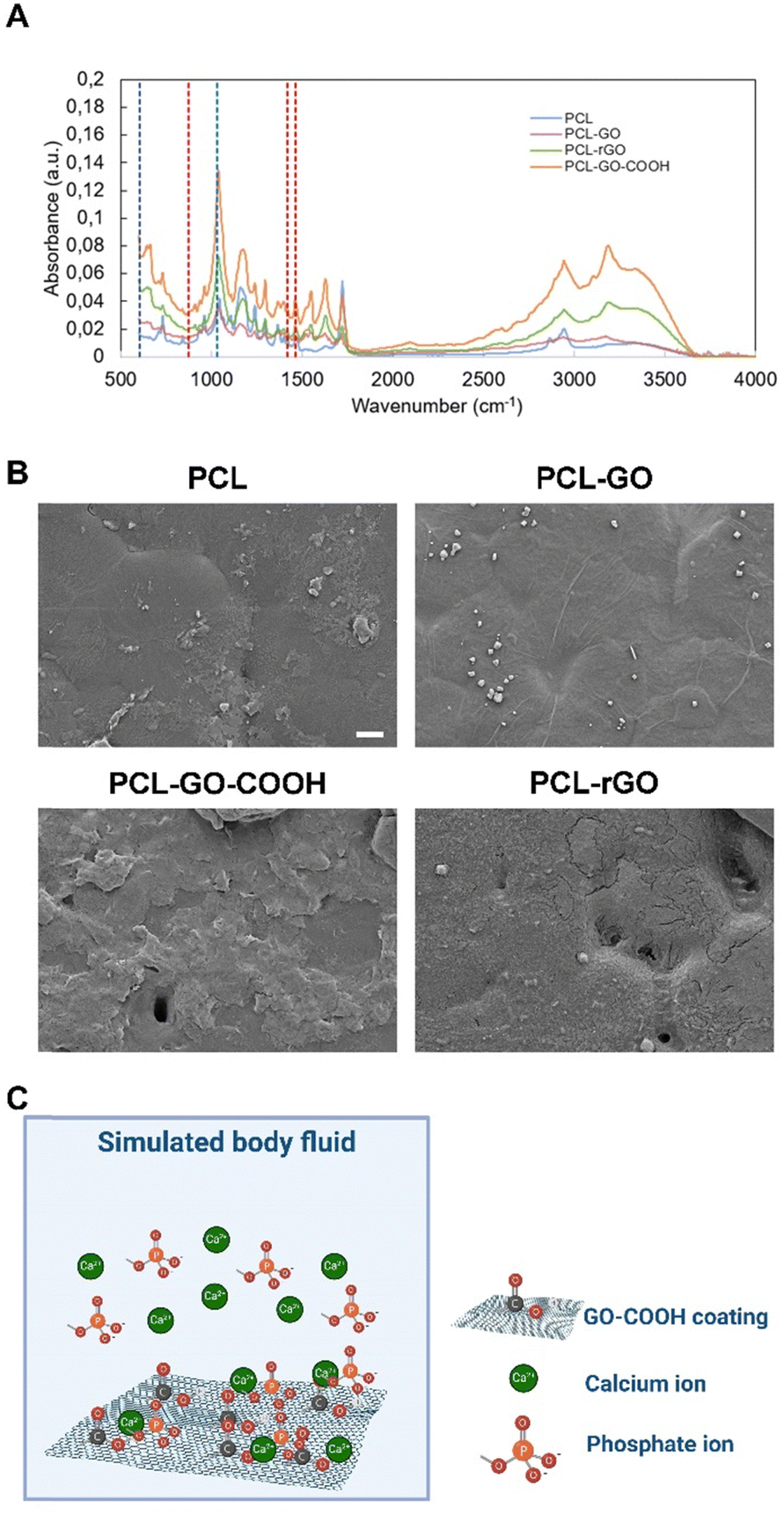 | ||
| Fig. 5 Analysis of the inorganic CL component. (A) FTIR analysis of SBF adsorbed on different coatings. (B) Representative SEM images of SBF treated coatings, scale bar is 10 μm. (C) Schematic representation of ions adsorption on GO-scaffolds surfaces put in contact with simulated body fluid (created with https://Biorender.com). | ||
The origin of this amorphous biomimetic hydroxyapatite layer on the GO–COOH surface can be explained by the charge of the functional groups, as previously described for COOH–carbon nanotubes.68–70 Specifically, negative electric charges were present as a consequence of the dissociation of the carboxyl groups, which electrostatically attract the positive calcium ions (Fig. 5C). After that, by exposing this unstable assemblage to phosphate and hydroxide ions dissolved in the SBF, an amorphous biomimetic apatite layer was formed (see schematic in Fig. 5C). Further supply of mineral ions could lead to the growth of this layer in different directions. Other probable functional sites may also have contributed simultaneously to local mineralized layer deposition. Similarly, rGO has a further negative zeta potential that could increase the binding of positive calcium ions.71,72
The mineralized layer on the analyzed scaffolds may also influence their capacities for both cell growth and differentiation. In this regard, it was reported that the amorphous apatite layer on the scaffold surface enhances bone-bonding ability of the biomaterial.66,73,74 Specifically, PCL and PCL–GO, despite the adhesion of cells, did not foster the deposition of SBF and consequently, the osteogenic properties were limited compared to PCL–rGO and PCL–GO–COOH. Conversely, the adsorption of carbonate and phosphate ions on PCL–rGO and PCL–GO–COOH resulting in the formation of a mineralized layer could be partially responsible for the increased osteogenic potential observed on these two coatings. Furthermore, cell communication via paracrine signals is affected by distance, as demonstrated recently by Raina and colleagues.75 Specifically, the range of cell–cell communication is mainly limited to the immediate proximity, influencing the differentiation of cells that is affected by direct cell–cell contacts.76 As reported in Fig. 5B, the surface becomes extremely irregular and therefore we hypothesize an uneven distribution of serum protein which reflects the cell adhesion and therefore clustering (refer to Fig. 3B).
Protein conditioning layer on GO coatings
Finally, we evaluated the adsorption of serum proteins comparing the different types of scaffolds to elucidate which proteins bind to the biomaterials and which may mediate their different in vitro properties. Serum proteins adsorbed on each scaffold were detached and analyzed by mass spectrometry coupled with liquid chromatography to study and compare the qualitative and quantitative composition of the protein CL (Fig. 6A). The protein identification data resulting from LC–MS raw data elaboration by Proteome Discoverer software were validated by FDR calculation and filtered to ensure high confidence identifications (see experimental section for details).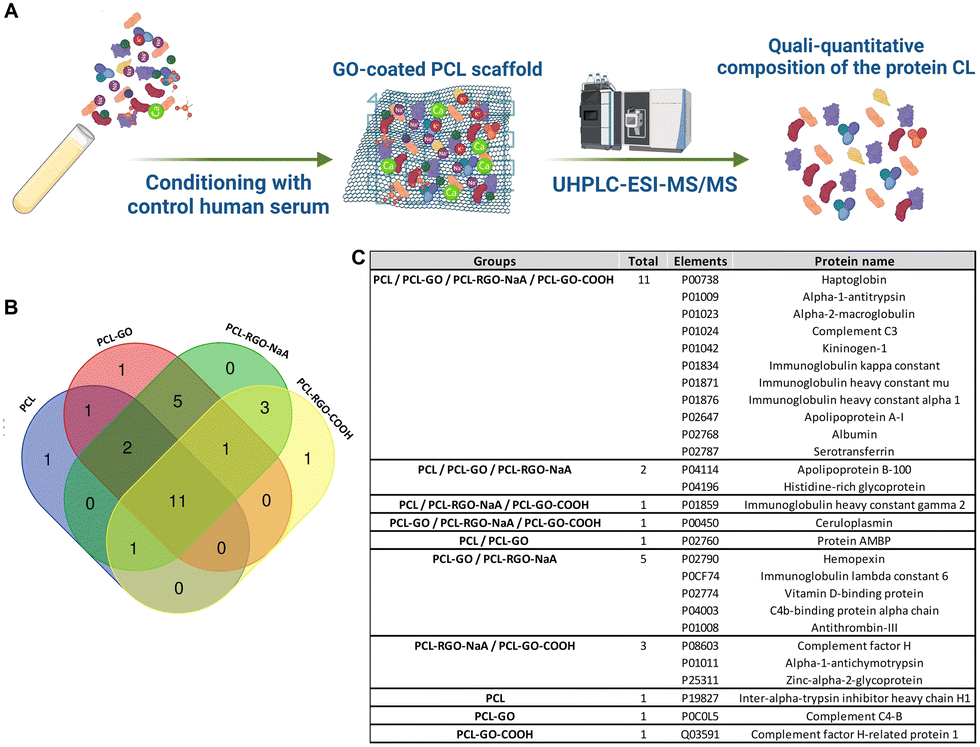 | ||
| Fig. 6 Analysis of the protein CL composition. (A) Schematic illustration of the experimental plan used to evaluate the human serum proteins adsorption on GO scaffolds (UHPLC-ESI-MS/MS: ultra high-performance liquid chromatography-nanoESI mass spectrometry analysis; created with https://Biorender.com) (B) the Venn diagram shows the intersection between the serum proteins adsorbed on PCL, PCL–GO, PCL–GO–COOH and PCL–rGO identified by means of mass spectrometry. (C) The table reports the unique UniProt identifier and full name of all the proteins exclusive of each scaffold and in common between the 4 different scaffolds analyzed. | ||
We detected 16, 21, 17 and 23 proteins on PCL, PCL–GO, PCL–GO–COOH and PCL–rGO, respectively (identification data in Supplementary file S1, ESI†). Of these, 11 elements were in common between the 4 different scaffolds, whereas 1 protein resulted exclusive of PCL (namely Inter-alpha-trypsin inhibitor heavy chain H1), 1 of PCL–GO (namely Complement C4-B) and 1 of PCL–GO–COOH (namely Complement factor H-related protein 1) (Fig. 6B and C). In addition, GO-based composites exclusively absorbed Ceruloplasmin (CP) compared with PCL scaffold, whereas Complement factor H, Alpha-1-antichymotrypsin and Zinc-alpha-2-glycoprotein bound solely PCL–GO–COOH and PCL–rGO (Fig. 6B and C). The results demonstrated that GO-based scaffolds used influenced the composition of protein coronas compared to uncoated PCL.
Considering only the common proteins identified, we also compared their levels on biomaterial surfaces by label-free relative quantification of the protein areas, since the amount of a specific protein could influence GO-scaffolds properties. These proteins showed statistically significant different levels between the PCL and each GO-coated sample analyzed. Differences were observed between PCL–GO–COOH and the other GO-based materials, often showing lower levels of these proteins, on some occasions similar to PCL (Fig. 7). Nevertheless, PCL–GO–COOH adsorbed more Complement C3 (C3), Immunoglobulin kappa constant and Serotransferrin compared with PCL alone (Fig. 7D, F and J). PCL–GO and PCL–rGO held a significantly higher quantity of all the common proteins adsorbed compared to both PCL and PCL–GO–COOH, except for C3 which showed comparable levels in PCL–GO materials. In addition, PCL–GO and PCL–rGO showed no significantly different levels of the common proteins except for alpha-2-macroglobulin (A2M), that exhibited a higher extent of adsorption on PCL–GO (Fig. 7C), and of apolipoprotein A1 (APOA1), that, on the contrary, was found higher on PCL–rGO compared to PCL–GO (Fig. 7K).
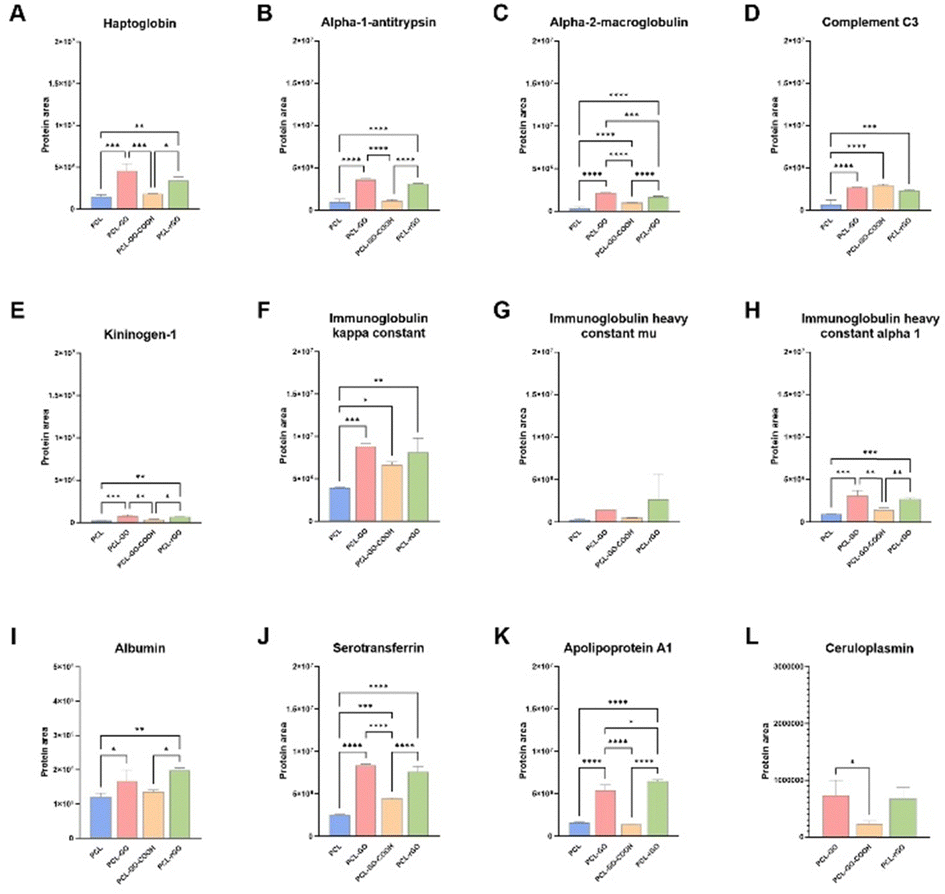 | ||
| Fig. 7 Quantification of serum proteins adsorbed on scaffolds surface. The histograms show levels of common serum proteins adsorbed on PCL, PCL–GO, PCL–GO–COOH and PCL–rGO (A)–(K) and on GO-based scaffolds (L). The results are reported in the graphs bar as mean ± SD. P values were assessed by ordinary one-way ANOVA followed by Tukey's multiple comparisons test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
It has been reported that some of these proteins can influence cell adhesion, proliferation, and differentiation and hereafter we have examined the most representative proteins suggesting a possible explanation for our results.
As already discussed above, serum proteins reduced hBM-MSC adhesion on GO coatings and mass spectrometry analysis suggested that this effect could be most likely associated to the greater adsorption on these coatings of anti-adhesive proteins, such as alpha-1-antitrypsin, alpha-2-macroglobulin and ceruloplasmin.77 Since it has been observed that C3 improved bone formation ability of MG63 osteoblast cells influencing the OPG/RANKL/RANK axis,78 we hypothesized that the enrichment of C3 on GO-based scaffolds could influence the osteogenic performance of serum-conditioned GO-biomaterials compared to the unconditioned ones. In addition, A2M is an extracellular macromolecule mainly known for its role as a broad-spectrum protease inhibitor, promoting osteoblastic differentiation through the TGF-β pathway.79,80 Moreover, APOA1, one of the major HDL protein constituents, has been described as a key modulator of osteoblast differentiation and bone mass production.81 Indeed, its deficiency in vivo has been associated with a change in the commitment of MSC towards adipocyte lineage rather than osteoblast progenitors.81–83 Similarly, serotransferrin is a protein involved in the iron balance which greatly affects MSC behavior and several studies demonstrated that iron overload reduced bone density, impairing osteoblast activity, and thus leading to decreased extracellular matrix (ECM) mineralization.84–87 Additionally, iron overload facilitates osteoclast differentiation and bone resorption.84–87 These processes may contribute to the diminished osteogenesis observed in conditioned PCL–GO scaffold. Of note, ceruloplasmin (CP), which resulted selectively adsorbed to GO-based biomaterials, showed a higher affinity for PCL–GO and PCL–rGO compared to PCL–GO–COOH (Fig. 7L). CP is a serum glycoprotein with ferroxidase activity responsible for 95% copper transport in the blood. It has been reported that CP can undergo oxidative modifications under oxidative conditions that promote a gain of function in integrin binding, signaling, and cell adhesion.88–90 In this context, we speculated that GO-based scaffolds could promote oxidative-induced structural change of serum proteins that interact with them, including CP, thus influencing cell adhesion and proliferation.
Overall, the different subsets of adsorbed proteins on each coating analyzed in this study may cause specific osteoinductive effects, thus affecting their biological performance. These results suggested that the serum-derived CL was not able to modify the low osteoinductive properties of PCL–GO. Conversely, the osteoinductive capacities of unconditioned PCL–GO–COOH and PCL–rGO, seemed to be strengthened or otherwise preserved by the serum conditioning.
Of note, in addition to the protein layer, as mentioned above, the ionic composition of serum was also an important co-stimulus, which strongly contributed to maintaining the osteoinductive abilities of PCL–GO–COOH and PCL–rGO. On the basis of our anlyses we hypothesized that in these GO-coatings the main stimulus for differentiation could be the biomimetic apatite layer formed on the scaffold surface rather than a specific protein pattern.91 In other words, we hypothesized that the higher the adsorption of mineralized matrix the lower the influence of the protein CL.
Antimicrobial properties of GO-based scaffold
Given the importance for the clinical translational of the interaction of the scaffolds with possible pathogens, the antimicrobial performance of the different biomaterials was analyzed and compared. Our results showed that PCL–rGO displayed excellent antibacterial effects significantly reducing the growth of E. coli (Fig. 8). Serum conditioning seemed to increase bacterial infection on PCL scaffolds, whereas it had no/fewer effects on GO coatings antimicrobial properties, thereby preserving the good antibacterial properties of PCL–rGO (Fig. 8).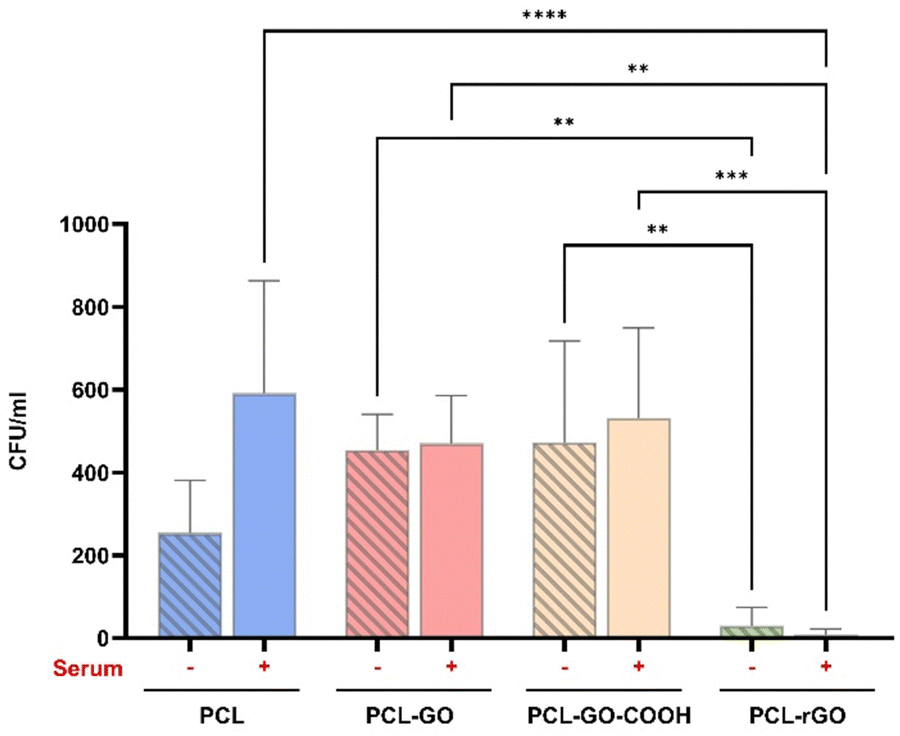 | ||
| Fig. 8 Antimicrobial test of GO coatings. The graph shows the number of colonies forming unit per ml on both pretreated and untreated scaffolds. The results are reported in the graph bar as mean ± SD. P values were assessed by ordinary one-way ANOVA followed by Tukey's multiple comparisons test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
A prerequisite for bacterial colonization of medical devices is the adsorption of adhesive serum proteins.92 We did not observe, in mass spectrometry analysis, a significant adsorption of adhesive proteins such as fibrinogen, fibronectin and vitronectin. However, all coatings showed a certain degree of binding of CP protein, a copper transportation protein that has antibacterial activity.93–95 We assumed that the bactericidal activity of CP was to some extent responsible for the lack of bacteria number increase when surfaces were treated with blood serum.96 However, a secondary mechanism not depending on the CL must be implicated in the high antibacterial efficacy measured for PCL–rGO coatings with or without serum conditioning.
As discussed previously, rGO had the lower zeta potential observed among the different coatings studied. We predicted that since this zeta potential increases the adsorption of divalent cations, the GO effect of bacteria trapping is enhanced.97 Further investigations will be conducted to elucidate the mechanism underlying the strong efficacy of PCL–rGO.
Conclusions
The improvement of bone cell proliferation and differentiation by graphene-based scaffolds was widely documented. Nevertheless, the formation of a conditioning layer on the surface of the scaffold once it comes into contact with biological fluids, alters the interaction between biomaterial and cells and influences the biological effects of the scaffold on cell growth, differentiation, and extracellular matrix deposition. In this work, we analyzed the effect of GO chemistry in coatings for 3D-printed PCL scaffolds and determined the influence of the CL on their regenerative capacities. We demonstrated that PCL–GO–COOH and PCL–rGO exhibited advantageous capabilities supporting hBM-MSC proliferation and osteogenic differentiation, maintaining these functionalities even when conditioned with serum (as summarized in Fig. 9). Our results also revealed that PCL–rGO presented excellent antimicrobial properties, and this effect persisted after the adsorption of CL on its surface, making it a highly promising coating for advancing the field of bone regeneration therapies given the possibility to better predict its properties once implanted in vivo (Fig. 9).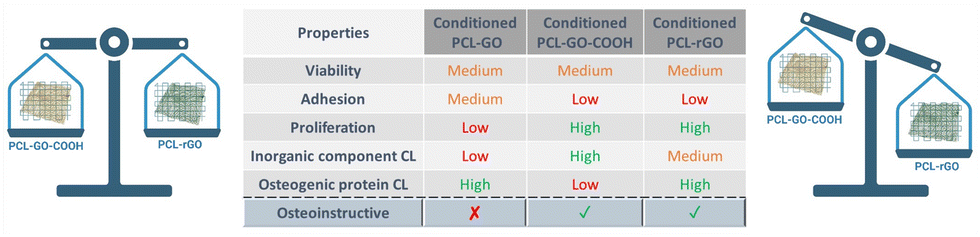 | ||
| Fig. 9 Regenerative properties of graphene-based material surfaces. In the image the regenerative properties of graphene-based material surfaces observed are summarized. The efficiency of each serum conditioned construct on cell viability, adhesion, proliferation, inorganic components, and proteins of CL composition as well as osteogenic differentiation has been classified as high, medium, or low. | ||
Overall, the qualitative and quantitative analyses of CL suggested that specific inorganic components combined with a definite protein pattern, including A2M, APOA1 and serotransferrin, adsorbed on GO-coatings may enhance and/or sustain the GO-related effects on cell behavior and improved bone grafting translational applications of 3D-printed PCL scaffolds.
Author contributions
VP, LDP, AGM conceptualization; FT, FA, VP, LDP, LG data curation; CD, FV, VP, FT, LDP, GP, IM, LC, LG, IM, AA formal analysis; VP, LDP, MP, AGM, MDS, OP, WL funding acquisition; VP, LDP, FT, FA, LP, MS investigation; FA, AGM, GP, AA, FT, LDP, VP, GF, LC, MS, LP methodology; LDP, VP, AGM project administration; VP, LDP, MP, AGM, MDS, OP, WP resources; FA, VP, CD, FV software; VP, LDP, MP, AGM, MDS, OP, WL supervision; MP, AGM, WL, VP, LDP validation; FA, FT, CD, FV visualization; VP, LDP, FT, FA writing – original draft; WL, AGM, FA, FV, CD, FT, MP, OP writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from AIRC under IG 2019 – ID. 23124 project Italian Ministry of Health, GR-2019-12370086, and under FLAGERA JTC2019 MARGO Project. We acknowledge the contribution of 3D Bioprinting Research Core Facility G-STeP of the Fondazione Policlinico Universitario “A. Gemelli” IRCCS for sample processing and the G-STeP Microscopy Facility of the Fondazione Policlinico Universitario “A. Gemelli” IRCCS for microscopy experiments. The authors also acknowledge the Regenerative Medicine Research Center (CROME) of the Università Cattolica del Sacro Cuore.Notes and references
- V. Campana, G. Milano, E. Pagano, M. Barba, C. Cicione, G. Salonna, W. Lattanzi and G. Logroscino, J. Mater. Sci. Mater. Med., 2014, 25, 2445–2461 CrossRef CAS PubMed.
- A. H. Schmidt, Injury, 2021, 52, S18–S22 CrossRef PubMed.
- I. L. Tsiklin, A. V. Shabunin, A. V. Kolsanov and L. T. Volova, Polymers, 2022, 14, 3222 CrossRef CAS PubMed.
- W. Lattanzi, R. Parolisi, M. Barba and L. Bonfanti, Front. Cell. Neurosci., 2015, 9 DOI:10.3389/fncel.2015.00455.
- M. Ansari, Prog. Biomater., 2019, 8, 223–237 CrossRef CAS PubMed.
- A. Stahl and Y. P. Yang, Tissue Eng., Part B, 2021, 27, 539–547 CrossRef PubMed.
- F. H. Lee, P. C. Shen, I. M. Jou, C. Y. Li and J. L. Hsieh, Medicine, 2015, 94, e2034 CrossRef PubMed.
- S. M. Kurtz, G. B. Higgs, E. Lau, R. R. Iorio, P. M. Courtney and J. Parvizi, J. Arthroplasty, 2022, 37, 205–212 CrossRef PubMed.
- T. Nisyrios, L. Karygianni, T. Fretwurst, K. Nelson, E. Hellwig, R. Schmelzeisen and A. Al-Ahmad, Materials, 2020, 13(9), 2102 CrossRef CAS PubMed.
- A. Peeters, G. Putzeys and L. Thorrez, J. Bone Jt. Infect., 2019, 4, 245–253 CrossRef PubMed.
- J. R. Peterson, F. Chen, E. Nwankwo, T. J. Dekker and S. B. Adams, Foot Ankle Orthop., 2019, 4, 1–9 Search PubMed.
- D. Offner, G. F. de Grado, I. Meisels, L. Pijnenburg, F. Fioretti, N. Benkirane-Jessel and A.-M. Musset, Cell Med., 2019, 11, 215517901985766 CrossRef PubMed.
- G. Battafarano, M. Rossi, V. De Martino, F. Marampon, L. Borro, A. Secinaro and A. Del Fattore, Int. J. Mol. Sci., 2021, 22, 1–22 Search PubMed.
- L. Vidal, C. Kampleitner, M. Brennan, A. Hoornaert and P. Layrolle, Front. Bioeng. Biotechnol., 2020, 8, 61 CrossRef PubMed.
- Z. Yazdanpanah, J. D. Johnston, D. M. L. Cooper and X. Chen, Front. Bioeng. Biotechnol., 2022, 10, 824156 CrossRef PubMed.
- D. S. M. Palmieri, V. Lattanzi, W. Perini, G. Augello and A. Papi, 2D Mater., 2020, 7, 022004 CrossRef.
- C. Wang, W. Huang, Y. Zhou, L. He, Z. He, Z. Chen, X. He, S. Tian, J. Liao and B. Lu, Bioact. Mater., 2020, 5, 82–91 Search PubMed.
- S. S. Lee, X. Du, I. Kim and S. J. Ferguson, Matter, 2022, 5, 2722–2759 CrossRef CAS.
- R. Dwivedi, S. Kumar, R. Pandey, A. Mahajan, D. Nandana, D. S. Katti and D. Mehrotra, J. Oral Biol. Craniofacial Res., 2020, 10, 381–388 CrossRef PubMed.
- Y. Xu, Y. Yang, Z. Hua, S. Li, Z. Yang, Q. Liu, G. Fu, P. Ji and Q. Wu, Biomaterials, 2021, 275, 120890 CrossRef CAS PubMed.
- M. Gharibshahian, M. Salehi, N. Beheshtizadeh, M. Kamalabadi-Farahani, A. Atashi, M. S. Nourbakhsh and M. Alizadeh, Front. Bioeng. Biotechnol., 2023, 11, 1168504 CrossRef PubMed.
- X. Zhao, S. Wang, F. Wang, Y. Zhu, R. Gu, F. Yang, Y. Xu, D. Xia and Y. Liu, J. Magnes. Alloy., 2022, 5, 042009 Search PubMed.
- W. Y. Zhu, P. Jiang, B. Luo, F. Lan and J. He, Nanoscale, 2019, 11, 6817–6827 RSC.
- W. Y. Zhu, Q. Yang, M. Yang, X. Zhan, F. Lan, J. He and Z. Gu, ACS Nano., 2017, 11, 3690–3704 CrossRef PubMed.
- R. García-álvarez and M. Vallet-Regí, Nanomaterials, 2021, 11, 888 CrossRef PubMed.
- Z. Othman, B. Cillero Pastor and S. van Rijt, Biomaterials, 2018, 167, 191–204 CrossRef CAS PubMed.
- K. N. Ekdahl, S. Huang, B. Nilsson and Y. Teramura, Seminars in immunology, Elsevier, 2016, vol. 28, p. 268–277 Search PubMed.
- L. Di Pietro, V. Palmieri, M. Papi and W. Lattanzi, Int. J. Mol. Sci., 2022, 23, 9493 CrossRef CAS PubMed.
- V. Serpooshan, M. Mahmoudi, M. Zhao, K. Wei, S. Sivanesan, K. Motamedchaboki, A. V. Malkovskiy, A. B. Goldstone, J. E. Cohen, P. C. Yang, J. Rajadas, D. Bernstein, Y. J. Woo and P. Ruiz-Lozano, Adv. Funct. Mater., 2015, 25, 4379–4389 CrossRef CAS PubMed.
- N. Groen, M. Guvendiren, H. Rabitz, W. J. Welsh, J. Kohn and J. De Boer, Acta Biomater., 2016, 34, 133–142 CrossRef CAS PubMed.
- A. E. Engberg, P. H. Nilsson, S. Huang, K. Fromell, O. A. Hamad, T. E. Mollnes, J. P. Rosengren-Holmberg, K. Sandholm, Y. Teramura, I. A. Nicholls, B. Nilsson and K. N. Ekdahl, Biomaterials, 2015, 36, 55–65 CrossRef CAS PubMed.
- V. Palmieri, W. Lattanzi, G. Perini, A. Augello and M. Papi, 2D Mater., 2020, 7, 022004 CrossRef CAS.
- A. Chouhan, H. P. Mungse and O. P. Khatri, Adv. Colloid Interface Sci., 2020, 283, 102215 CrossRef CAS PubMed.
- V. Palmieri, G. Perini, M. De Spirito and M. Papi, Nanoscale Horiz., 2019, 4, 464–471 RSC.
- P. P. Wibroe, S. V. Petersen, N. Bovet, B. W. Laursen and S. M. Moghimi, Biomaterials, 2016, 78, 20–26 CrossRef CAS PubMed.
- S. Syama and P. V. Mohanan, Comprehensive Application of Graphene: Emphasis on Biomedical Concerns, Springer Berlin Heidelberg, 2019, vol. 11, p. 6 Search PubMed.
- V. Palmieri, M. Barba, L. Di Pietro, S. Gentilini, M. C. Braidotti, C. Ciancico, F. Bugli, G. Ciasca, R. Larciprete, W. Lattanzi, C. Conti and M. Papi, 2D Mater., 2018, 19, 3336 Search PubMed.
- A. G. Marrani, A. Motta, R. Schrebler, R. Zanoni and E. A. Dalchiele, Electrochim. Acta, 2019, 304, 231–238 CrossRef CAS.
- P. P. Wibroe, S. V. Petersen, N. Bovet, B. W. Laursen and S. M. Moghimi, Biomaterials, 2016, 78, 20–26 CrossRef CAS PubMed.
- V. Palmieri, W. Lattanzi, G. Perini, A. Augello, M. Papi and M. De Spirito, 2D Mater., 2020, 7, 1–20 Search PubMed.
- Z. M. Wright, A. M. Arnold, B. D. Holt, K. E. Eckhart and S. A. Sydlik, Regen. Eng. Transl. Med., 2019, 5, 190–209 CrossRef CAS.
- S. Bahrami, N. Baheiraei and M. Shahrezaee, Sci. Rep., 2021, 11, 1–10 CrossRef PubMed.
- W. C. Lee, C. H. Lim, H. Shi, L. A. Tang, Y. Wang and C. T. Lim, ACS Nano., 2011, 5, 7334–7341 CrossRef CAS PubMed.
- F. Amato, A. Motta, L. Giaccari, R. Di Pasquale, F. A. Scaramuzzo, R. Zanoni and A. G. Marrani, Nanoscale Adv., 2023, 5, 893–906 RSC.
- F. Amato, G. Perini, G. Friggeri, A. Augello, A. Motta, L. Giaccari, R. Zanoni, M. De Spirito, V. Palmieri, A. G. Marrani and M. Papi, Adv. Mater. Interfaces, 2023, 10, 1–10 CrossRef.
- W. Lattanzi, M. Barba, F. Novegno, L. Massimi, V. Tesori, G. Tamburrini, S. Galgano, C. Bernardini, M. Caldarelli, F. Michetti and C. Di Rocco, Bone, 2013, 52, 474–484 CrossRef CAS PubMed.
- C. A. Gregory, W. G. Gunn, A. Peister and D. J. Prockop, Anal. Biochem., 2004, 329, 77–84 CrossRef CAS PubMed.
- V. Palmieri, M. Barba, L. Di Pietro, C. Conti, M. De Spirito, W. Lattanzi and M. Papi, Int. J. Mol. Sci., 2018, 19, 1–9 Search PubMed.
- S. G. Colombarolli and A. Vitali, Methods Mol. Biol., 2023, 2578, 237–247 CrossRef CAS PubMed.
- E. W. Deutsch, L. Lane, C. M. Overall, N. Bandeira, M. S. Baker, C. Pineau, R. L. Moritz, F. Corrales, S. Orchard, J. E. Van Eyk, Y. K. Paik, S. T. Weintraub, Y. Vandenbrouck and G. S. Omenn, J. Proteome Res., 2019, 18, 4108–4116 CrossRef PubMed.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud’homme and I. A. Aksay, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- Z. Li, L. Deng, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2023, 135, 101089 CrossRef CAS.
- S. Eigler, C. Dotzer and A. Hirsch, Carbon, 2012, 50, 3666–3673 CrossRef CAS.
- A. G. Marrani, A. Motta, F. Amato, R. Schrebler, R. Zanoni and E. A. Dalchiele, Nanomaterials, 2022, 12, 1–21 Search PubMed.
- I. Ferrari, A. Motta, R. Zanoni, F. A. Scaramuzzo, F. Amato, E. A. Dalchiele and A. G. Marrani, Carbon, 2023, 203, 29–38 CrossRef CAS.
- K. C. Prince, I. Ulrych, M. Peloi, B. Ressel, V. Cháb, C. Crotti and C. Comicioli, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 6866–6868 CrossRef CAS.
- C. C. K. C. Prince, I. Ulrych, M. Peloi, B. Ressel, V. Cháb and C. Crotti, J. Phys. Chem. C, 2012, 116, 9900–9908 Search PubMed.
- L. Daneshmandi, M. Barajaa, A. Tahmasbi Rad, S. A. Sydlik and C. T. Laurencin, Adv. Healthcare Mater., 2021, 10, e2001414 CrossRef PubMed.
- A. Ricci, A. Cataldi, S. Zara and M. Gallorini, Materials, 2022, 15, 2229 CrossRef CAS PubMed.
- J. T. Jeong, M. K. Choi, Y. Sim, J. T. Lim, G. S. Kim, M. J. Seong, J. H. Hyung, K. S. Kim, A. Umar and S. K. Lee, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- C. C. Shen, S. H. Hsu, K. B. Chang, C. A. Yeh, H. C. Chang, C. M. Tang, Y. C. Yang, H. H. Hsieh and H. S. Hung, Biomedicines, 2021, 9, 1632 CrossRef CAS PubMed.
- L. Ferroni, C. Gardin, F. Rigoni, E. Balliana, F. Zanotti, M. Scatto, P. Riello and B. Zavan, Coatings, 2022, 12, 799 CrossRef CAS.
- H. Xie, T. Cao, A. Franco-Obregón and V. Rosa, Int. J. Mol. Sci., 2019, 20, 574 CrossRef CAS PubMed.
- X. Chen, A. Nouri, Y. Li, J. Lin, P. D. Hodgson and C. Wen, Biotechnol. Bioeng., 2008, 101, 378–387 CrossRef CAS PubMed.
- D. Arcos and M. Vallet-regí, J. Mater. Chem. B, 2021, 8, 1781–1800 RSC.
- N. Vargas-Becerril, D. A. Sánchez-Téllez, L. Zarazúa-Villalobos, D. M. González-García, M. A. Álvarez-Pérez, C. de León-Escobedo and L. Téllez-Jurado, Ceram. Int., 2020, 46, 28806–28813 CrossRef CAS.
- A. L. B. Maçon, T. B. Kim, E. M. Valliant, K. Goetschius, R. K. Brow, D. E. Day, A. Hoppe, A. R. Boccaccini, I. Y. Kim, C. Ohtsuki, T. Kokubo, A. Osaka, M. Vallet-Regí, D. Arcos, L. Fraile, A. J. Salinas, A. V. Teixeira, Y. Vueva, R. M. Almeida, M. Miola, C. Vitale-Brovarone, E. Verné, W. Höland and J. R. Jones, J. Mater. Sci.: Mater. Med., 2015, 26, 1–10 CrossRef PubMed.
- K. PourAkbar, N. JamilPour and G. Rouhi, Biomed. Eng., 2009, 477–498 Search PubMed.
- R. R. Pandey and C. C. Chusuei, Molecules, 2021, 26, 6674 CrossRef CAS PubMed.
- Z. Peng, T. Zhao, Y. Zhou, S. Li, J. Li and R. M. Leblanc, Adv. Healthcare Mater., 2020, 9, 1–30 Search PubMed.
- G. Wei, C. Gong, K. Hu, Y. Wang and Y. Zhang, Nanomaterials, 2019, 9, 1–20 Search PubMed.
- S. Wang, Y. Yang, R. Wang, X. Kong and X. Wang, Bioact. Mater., 2020, 5, 387–397 Search PubMed.
- L. Zou, Y. Zhang, X. Liu, J. Chen and Q. Zhang, Colloids Surf., B, 2019, 178, 222–229 CrossRef CAS PubMed.
- X. Wu, K. Walsh, B. L. Hoff and G. Camci-Unal, Bioengineering, 2020, 7, 1–24 CrossRef PubMed.
- D. Raina, A. Bahadori, A. Stanoev, M. Protzek, A. Koseska and C. Schroter, Development, 2021, 148(21), dev199926 CrossRef CAS PubMed.
- H. Daneshpour, P. van den Bersselaar, C. H. Chao, T. G. Fazzio and H. Youk, Nat. Chem. Biol., 2023, 19, 596–606 CrossRef CAS PubMed.
- A. S. G. Curtis and J. V. Forrester, J. Cell Sci., 1984, 71, 17–35 CrossRef CAS PubMed.
- M. Li, X. Chu, D. Wang, L. Jian, L. Liu, M. Yao, D. Zhang, Y. Zheng, X. Liu, Y. Zhang and F. Peng, Biomaterials, 2022, 282, 121408 CrossRef CAS PubMed.
- F. Sadeghi, E. Vahednia, H. Naderi Meshkin and M. A. Kerachian, J. Cell. Mol. Med., 2020, 24, 4784–4790 CrossRef CAS PubMed.
- X. Qiu, S. Liu, H. Zhang, B. Zhu, Y. Su, C. Zheng, R. Tian, M. Wang, H. Kuang, X. Zhao and Y. Jin, Stem Cell Res. Ther., 2018, 9, 1–15 CrossRef PubMed.
- H. C. Blair, E. Kalyvioti, N. I. Papachristou, I. L. Tourkova, S. A. Syggelos, D. Deligianni, M. G. Orkoula, C. G. Kontoyannis, E. A. Karavia, K. E. Kypreos and D. J. Papachristou, Lab. Invest., 2016, 96, 763–772 CrossRef CAS PubMed.
- M. Mangaraj, R. Nanda and S. Panda, Indian J. Clin. Biochem., 2016, 31, 253–259 CrossRef CAS PubMed.
- A. Akhmetshina, D. Kratky and E. Rendina-Ruedy, Metabolites, 2023, 13, 578 CrossRef CAS PubMed.
- E. Balogh, E. Tolnai, B. Nagy, B. Nagy, G. Balla, J. Balla and V. Jeney, Biochim. Biophys. Acta, Mol. Basis Dis., 2016, 1862, 1640–1649 CrossRef CAS PubMed.
- A. Borriello, I. Caldarelli, M. C. Speranza, S. Scianguetta, A. Tramontano, D. Bencivenga, E. Stampone, A. Negri, B. Nobili, F. Locatelli, S. Perrotta and A. Oliva, Biochim. Biophys. Acta, Bioenerg., 2016, 1860, 1211–1223 CrossRef CAS PubMed.
- K. J. Mehta, J. Cell. Physiol., 2021, 236, 7266–7289 CrossRef CAS PubMed.
- B. K. Das, L. Wang, T. Fujiwara, J. Zhou, N. Aykin-Burns, K. J. Krager, R. Lan, S. G. Mackintosh, R. Edmondson, M. L. Jennings, X. Wang, J. Q. Feng, T. Barrientos, J. Gogoi, A. Kannan, L. Gao, W. Xing, S. Mohan and H. Zhao, eLife, 2022, 11, 1–32 Search PubMed.
- M. Barbariga, F. Curnis, A. Spitaleri, A. Andolfo, C. Zucchelli, M. Lazzaro, G. Magnani, G. Musco, A. Corti and M. Alessio, J. Biol. Chem., 2014, 289, 3736–3748 CrossRef CAS PubMed.
- M. Barbariga, A. Zanardi, F. Curnis, A. Conti, D. Boselli, S. Di Terlizzi and M. Alessio, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- A. Zanardi and M. Alessio, Int. J. Mol. Sci., 2021, 22, 1–13 Search PubMed.
- L. Lin, K. L. Chow and Y. Leng, J. Biomed. Mater. Res., Part A, 2009, 89, 326–335 CrossRef PubMed.
- G. Pietrocola, D. Campoccia, C. Motta, L. Montanaro, C. R. Arciola and P. Speziale, Int. J. Mol. Sci., 2022, 23, 5958 CrossRef CAS PubMed.
- K. SJ, Arch. Biochem. Biophys., 1992, 295, 302–308 CrossRef PubMed.
- I. Salah, I. P. Parkin and E. Allan, RSC Adv., 2021, 11, 18179–18186 RSC.
- A. M. Orta-Rivera, Y. Meléndez-Contés, N. Medina-Berríos, A. M. Gómez-Cardona, A. Ramos-Rodríguez, C. Cruz-Santiago, C. González-Dumeng, J. López, J. Escribano, J. J. Rivera-Otero, J. Díaz-Rivera, S. C. Díaz-Vélez, Z. Feliciano-Delgado and A. D. Tinoco, Inorganics, 2023, 11, 252 CrossRef CAS.
- B. Bakhautdin, M. Febbraio, E. Goksoy, C. A. de la Motte, M. F. Gulen, E. P. Childers, S. L. Hazen and X. Li, Gut, 2013, 62, 209–219 CrossRef CAS PubMed.
- V. Palmieri, L. Di Pietro, G. Perini, M. Barba, O. Parolini, M. De Spirito, W. Lattanzi and M. Papi, Front. Bioeng. Biotechnol., 2020, 8, 1–9 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00144c |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |