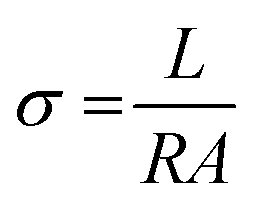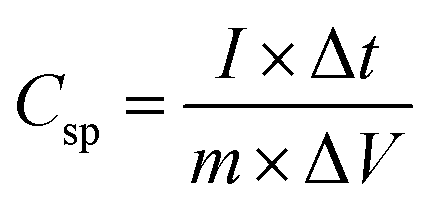DOI:
10.1039/D4MA00118D
(Paper)
Mater. Adv., 2024,
5, 6170-6184
Electrochemical performance enhancement of MnO2 nanowires through silver incorporation for next-generation supercapacitors
Received
5th February 2024
, Accepted 7th June 2024
First published on 27th June 2024
Abstract
Increased demand for effective energy storage systems emphasizes the urgency to overcome the bottlenecks of existing technology. Supercapacitors (SCs), owing to their high specific power and fast charging/discharging capabilities, are perfect candidates for future energy applications but their low energy density makes them impractical for commercial applications. Because of their high energy density and variable oxidation states, transition metal oxides (TMOs) have great potential as supercapacitor electrode materials. But for practical applications, their poor intrinsic conductivity needs to be improved. Noble metal doping offers a compelling method to raise the conductivity and structural stability of TMOs. Herein, we have prepared AgxMnO2 (x = 0.05, 0.10, and 0.15) to improve the conductivity and structural stability of the electro-active material. FESEM micrographs exhibit cracks on the nanowire (NW) surface by Ag doping, proposing less dead volume. Ag doping also fortified electrode pulverization during charging/discharging cycles by imparting structural stability. These properties enabled Ag0.05MnO2 NWs to demonstrate a specific capacitance of 1027 F g−1 at a current density of 1 A g−1. The electrode also retained a capacitance of 93.16% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 GCD cycles@12 A g−1 along with 86% rate capability at 9 A g−1. By tackling critical difficulties such as poor conductivity and structural stability, this study advances energy storage technologies and lays the groundwork for the creation of high-performance supercapacitors for future energy applications.
000 GCD cycles@12 A g−1 along with 86% rate capability at 9 A g−1. By tackling critical difficulties such as poor conductivity and structural stability, this study advances energy storage technologies and lays the groundwork for the creation of high-performance supercapacitors for future energy applications.
1. Introduction
Addressing the escalating environmental pollution concerns resulting from increased fossil fuel consumption highlights the growing importance of renewable energy storage devices to fulfill next-generation energy demands.1–4 Fossil fuel burning produces almost 35 billion tons of CO2 per annum, which is adversely affecting the air quality index.5,6 The combustion of fossil fuels is also damaging soil and water sources. Materials scientists and researchers are trying to develop green and sustainable energy production systems such as hydro-power, solar cells, and windmills.7,8 However, these energy sources are climate dependent. Therefore, it is challenging to construct energy storage systems to meet the ever-increasing energy demands. Modern devices, such as portable electronics, medical equipment, smartphones, load-managing systems, and hybrid vehicles, necessitate immediate access to high power. This requirement presents a barrier that rechargeable batteries cannot adequately meet.
Supercapacitors are excellent rechargeable energy storage systems that can power up modern electronics and electrical gadgets.9,10 They have superior power density than rechargeable batteries.11 Supercapacitors have received a lot of interest for their potential as replacements to conventional capacitors and rechargeable batteries. They have remarkable characteristics such as high specific power, extended cycle life, outstanding rate capability, and quick charging/discharging capabilities. These distinct properties make supercapacitors ideal candidates for applications in new energy systems such as wearable electronic gadgets, load management systems, and powering off-grid homes. Furthermore, supercapacitors provide energy to power up electric vehicles through increased specific power. However, supercapacitors have low energy density as compared to rechargeable batteries. The energy density is related to the specific capacitance and potential window of a supercapacitor by the equation 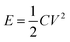 . So, the energy density could be improved either by increasing specific capacitance or by extending the voltage window. Recent research in the domain of supercapacitors has witnessed substantial advances in the use of transition metal oxides and sulfides as electrode materials. Studies by Vilhal et al., Prabhu et al., Jiang et al., Sun et al., Pappu et al., and Van et al. have shown that NiO, CuO, and Cu doped ceria, manganese oxide/carbon composite cobalt manganese oxide, and Ni–Co binary sulfides have potential electrochemical characteristics for improving energy storage performance in supercapacitors.12–17 Furthermore, comprehensive studies by Melkiyur et al. and Imran et al. have summarized the existing state-of-the-art and future prospects of transition metal oxides/sulfides and manganese oxide/sulfide in supercapacitor technology respectively.18,19
. So, the energy density could be improved either by increasing specific capacitance or by extending the voltage window. Recent research in the domain of supercapacitors has witnessed substantial advances in the use of transition metal oxides and sulfides as electrode materials. Studies by Vilhal et al., Prabhu et al., Jiang et al., Sun et al., Pappu et al., and Van et al. have shown that NiO, CuO, and Cu doped ceria, manganese oxide/carbon composite cobalt manganese oxide, and Ni–Co binary sulfides have potential electrochemical characteristics for improving energy storage performance in supercapacitors.12–17 Furthermore, comprehensive studies by Melkiyur et al. and Imran et al. have summarized the existing state-of-the-art and future prospects of transition metal oxides/sulfides and manganese oxide/sulfide in supercapacitor technology respectively.18,19
Especially, transition metal oxides have gained substantial interest as supercapacitor electrode materials because of their faradaic redox nature to store charge, which improves the energy density of the supercapacitor. Sana et al. demonstrated that Ag–V2O5 exhibited a specific capacitance of 516.6 F g−1@1 A g−1 current density.20 Wang et al. reported that Co3O4 nanowires demonstrated a specific capacitance of 880.2 F g−1@1 A g−1 current density with a rate capability of 70%@20 A g−1.21 Liu et al. reported that the Ag–MnO2/rGO composite exhibited a specific capacitance of 804.6@1 A g−1.22 Ozoemena et al. demonstrated that K+/Na+ pre-intercalated MnO2 NWs exhibited a specific capacitance of 124 F g−1 in KOH electrolyte.23 Among TMOs, manganese dioxide (MnO2) nanostructures have attracted great attraction as electrode materials for supercapacitors, thanks to their economic and environmentally friendly nature, theoretical capacitance (1370 F g−1), and non-toxicity. MnO2 nanostructures exist in a variety of crystallographic forms (α, β, γ, and δ).24 These crystallographic forms are distinct from each other by the linkage of basic [MnO6] octahedra. In α, β, and γ MnO2, [MnO6] octahedron links in such a way that it forms (2 × 2 and 1 × 1), (1 × 1), and (2 × 1) tunnels respectively. α-MnO2 is regarded as a potential candidate for supercapacitor applications due to a larger tunnel structure of 4.62 Å.25 The larger tunnel structure makes electrolyte ion insertion and de-insertion quite easy resulting in fast charging/discharging rates. However, the α-phase of MnO2 suffers from stability issues and transforms into other phases of MnO2.26,27 Previous studies have shown that the α phase of MnO2 is stabilized by doping with larger ions.28–30 MnO2 also has poor electronic conductivity like other transition metal oxides.31 Ag doping in MnO2 has been proved to be beneficial due to excellent conductivity of silver which contributes to the overall conductivity enhancement of the electrode material. Ag+ ions have larger ionic radii (1.15 Å) due to which they are able to stabilize α-MnO2. Additionally, hybridization between Ag 4d and O 2p induces new states between conduction and valence bands. The p–d repulsion is so high that it moves the Fermi level towards the conduction band resulting in an ease in electron transfer to the conduction band. Oxygen and interstitial defects produced by silver doping also induce new occupied states between conduction and valence band resulting in an upward movement of the Fermi level. Silver doping also induced n-type characteristics in p-type MnO2.32 Consequently, silver is expected to induce structural stability to maintain the lifespan of SCs over longer charging/discharging cycles along with improvement in conductivity. Structural stability could also be obtained by doping alkali metal ions but Ag 4d and O 2p hybridization which contributes towards conductivity improvement makes Ag preferable over alkali metal ions and transition metal ions. Moreover, silver is the most conductive metal in the periodic table.33 Additionally, silver being a redox active material could introduce more redox active sites in the electrode material contributing to improved pseudocapacitance, leading to enhanced energy density. Owing to these characteristics, silver is considered as an excellent dopant for MnO2. The synergistic effect of silver's excellent conductivity and α-MnO2's pseudocapacitance and large tunnel structure could improve energy density along with power density, enabling supercapacitors applicable where quick and longer energy discharge is needed. Rahman et al. prepared silver doped MnO2@carbon cloth for supercapacitor applications. The electrode demonstrated a capacitance of 350 F g−1@1 A g−1 current density. They also reported the performance of a Ag–MnO2/activated carbon asymmetric supercapacitor device. The device demonstrated an energy density of 54 W h kg−1 in a 0–2 V potential window.34 Zhang et al. assessed the supercapacitor performance of silver nanoparticle loaded MnO2 nanosheets. Ag–MnO2 exhibited a capacitance of 272 F g−1@10 mV s−1, which is three times higher than that of the undoped MnO2 (90 F g−1) in 1 M Na2SO4 electrolyte.35 Dai et al. prepared silver nanoparticles embedded in MnO2 nanowires and studied their electrochemical supercapacitor properties. The electrode material demonstrated a specific capacitance of 1230 F g−1@1 mV s−1 scan rate along with a capacitance retention of almost 91% after 10 K charging/discharging cycles.36
In this research, we have studied the consequences of varying the amount of silver on the structural and supercapacitor performance of MnO2. Ag doping in MnO2 is believed to demonstrate better structural stability and electrochemical performance. Ag doping has tuned the structure of MnO2 which is confirmed through characterization techniques like XRD, XPS, and FTIR. FESEM micrographs have shown that the NW surface becomes rough after Ag doping, which is expected to provide more redox active sites for electrolyte ions. Ag doping in MnO2 NWs showed an increase in surface area as confirmed by BET analysis. It was observed that the electrochemical performance of MnO2 NWs showed an improvement with an optimum silver concentration of 0.05, therefore, MnO2 doping with an optimum dopant concentration can improve electrochemical performance.
2. Experimental
2.1. Materials
Potassium permanganate (KMnO4, Daejung, 98%), ammonium persulfate [(NH4)2S2O8, Merck, 98%], nitric acid (HNO3, Merck, 70%), silver nitrate (AgNO3, Merck, 98%), absolute ethanol (C2H5OH, Merck, 98%) and deionized (DI) water.
2.2. Synthesis of AgxMnO2 nanowires
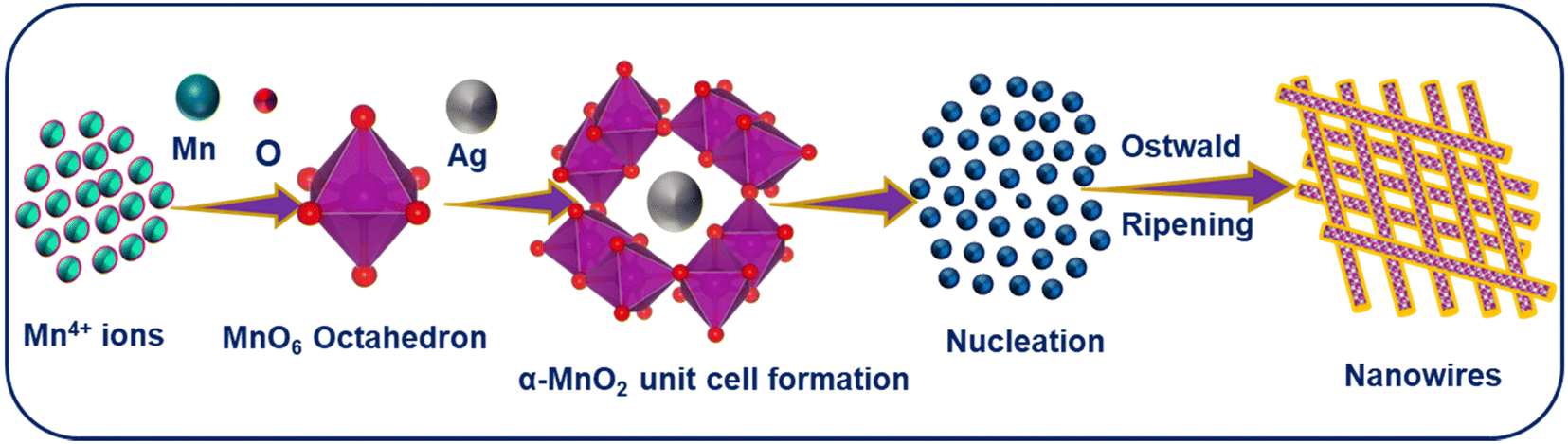
MnO2 NWs were synthesized through a low temperature hydrothermal treatment. A uniform aqueous solution of KMnO4 was prepared by dissolving it (4 g) in DI water with continuous stirring. Subsequently, aqueous solution of ammonium persulfate was prepared by dissolving it (2 g) in a separate beaker. This solution was slowly added to the aqueous solution of KMnO4. After that, 1 cm3 of nitric acid was added to the above mixture. The total volume of the mixture was made up to 80 cm3 by the addition of some more DI water. The resulting mixture was then transferred to a Teflon-lined autoclave for heat treatment at 180 °C for 16 h. Afterwards, the mixture was washed multiple times with DI water and ethanol to eliminate impurities and neutralize the solution. Finally, the precipitates were dried overnight at 100 °C. The resulting nanowires were finely ground into powder and stored for further characterization and potential application. To introduce Ag into MnO2, the same protocol was employed with different Mn:Ag molar ratios (1:0.05, 1:0.1, 1:0.15). AgxMnO2 NWs grow via Ostwald ripening.37,38 A schematic illustration for the formation mechanism of AgxMnO2 NWs is depicted in Fig. 1.
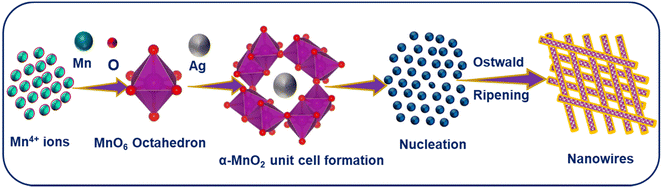 |
| | Fig. 1 A schematic illustration for the formation mechanism of AgxMnO2 NWs. | |
2.3. Electrode preparation
Sonication was used to prepare binder-free electrodes of MnO2 NWs and AgxMnO2 NWs on the nickel foam. In a typical synthesis procedure, MnO2 NW paste in DI water was created by sonication for 60 min. The homogenized paste was then transferred on HCl treated and washed nickel foam using a micropipette. The electroactive material-loaded nickel foam was then placed in an oven at 50 °C for complete removal of DI water. Subsequent evaluation of electrochemical supercapacitor performance was done using the prepared electrode.
2.4. Electrochemical studies
Electrochemical performance of MnO2 NWs and AgxMnO2 NWs was tested in a half-cell configuration using 1 M Na2SO4. Cyclic voltammetric (CV) response was assessed in a potential range 0–0.6 V. GCD measurements were performed in order to assess the charging/discharging behavior of the electrodes. Additionally, the effect of various current densities on Ag0.05MnO2 was also studied. Measurements using electrochemical impedance spectroscopy (EIS) were conducted between 0.1 Hz and 100 kHz frequency.
2.5. Characterization
Structural investigations of AgxMnO2 NWs were conducted on a LabX XRD-6100 X-ray diffractometer. Functional group identification was performed using an IR Affnity-1S spectrophotometer. Studies on morphology were executed using a ZEISS LEO SUPRA 55 field emission scanning electron microscope. Elemental presence was evaluated using a JEOL JCM-6000Plus SEM. Transmission electron microscopy (TEM) was performed using a JEOL JSM 2100 transmission electron microscope operating at 200 kV. The XPS measurements were carried out using a Kratos Amicus/ESCA 3400 instrument. A DuPont type analyzer was used to examine the energy of photoelectrons that were emitted at 0° from the surface normal after the sample was exposed to 240 W unmonochromated Mg Kα X-rays at a pass energy of 150 eV. Using the Micromeritics ASAP 2020 Physisorption analyzer, the BET surface area of the samples was determined. Electrochemical performance of the as synthesized electrodes was tested on GAMRY interface 5000E.
3. Results and discussion
3.1. X-ray diffraction (XRD)
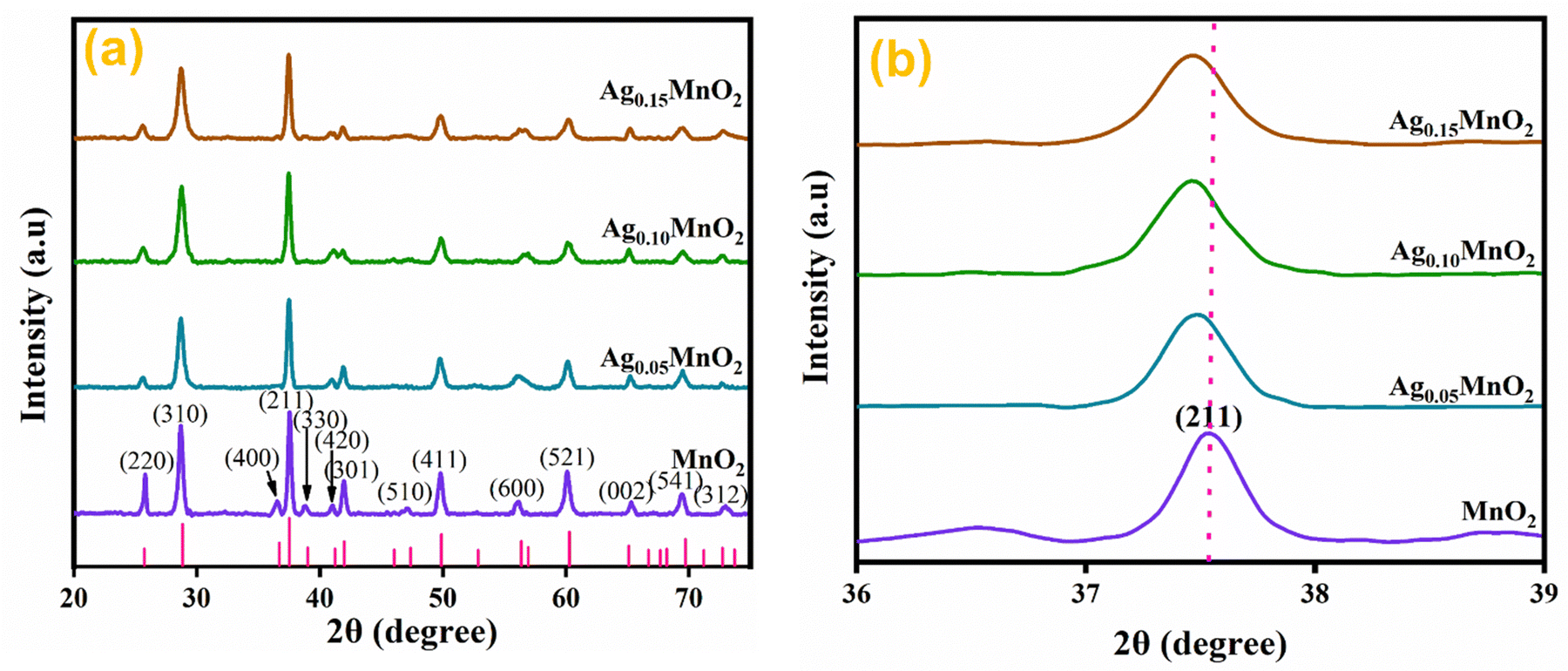
Structural investigations and crystallite size determination of bare and doped MnO2 NWs have been carried out by XRD analysis.39 The XRD spectra of AgxMnO2 NWs are shown in Fig. 2(a). All the diffraction peaks are generated by (220), (310), (400), (211), (330), (420), (301), (510), (411), (600), (521), (002), (541), and (312) diffraction planes of tetragonal MnO2 according to ICDD card # 00-044-0141.40 The intense diffraction peaks indicated good crystallinity and the intensity was observed to decrease through Ag doping. This decrease in intensity showed that the crystallinity is disturbed by doping. This could be attributed to the insertion of Ag+ ions into the MnO2 lattice.41 This intensity reduction may be associated with the fact that doping can induce defects and dislocations in the crystal lattice. Defects, such as vacancies or interstitials, introduce local atomic rearrangements, while dislocations are line defects caused by lattice mismatch. Both defects and dislocations scatter X-rays, leading to a decrease in peak intensity. Moreover, doping can lead to the formation of amorphous or poorly crystalline regions within the material. Fig. 2(b) shows the magnified view of the diffraction peak associated with the (211) plane. With silver doping, the diffraction peak was shifted towards a lower angle. This shifting is attributed to ionic radii difference between Ag+ (1.15 Å) and Mn4+ (0.53 Å).42 When Ag atoms substitute Mn atoms in the MnO2 lattice, the average atomic size increases. This increase in atomic size can cause lattice expansion, leading to a shift in the XRD peaks towards lower diffraction angles.43–45 The average crystallite size “D” was calculated using the Debye Scherrer equation.46,47| | | D = Kλ/βcosθ | (1) |
The calculated crystallite sizes for AgxMnO2 NWs (x = 0, 0.05, 0.10, and 0.15) were found to be 13.65 nm, 13.20 nm, 12.80 nm, and 12.50 nm, respectively. Upon increasing the Ag concentration, the crystallite size decreased, and is well consistent with the previous literature.48 The surface area of all the samples was calculated from XRD data using the eqn (2).49,50The calculated surface area of MnO2, Ag0.05MnO2, Ag0.10MnO2, and Ag0.15MnO2 NWs was found to be 57.2 m2 g−1, 73.1 m2 g−1, 68.3 m2 g−1, and 63.7 m2 g−1, respectively.
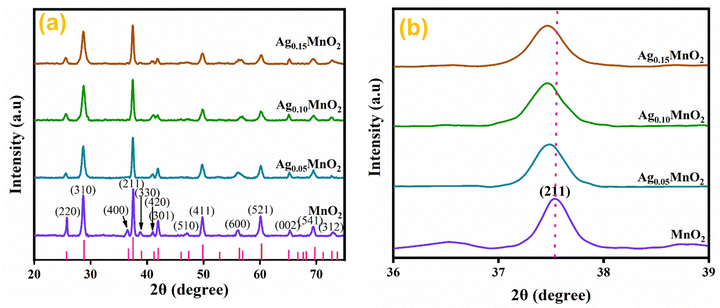 |
| | Fig. 2 (a) XRD patterns of bare and Ag doped MnO2 NWs, (b) enlarged view of diffraction peak from (211) diffraction plane. | |
3.2. Fourier transform infrared (FTIR) spectroscopy
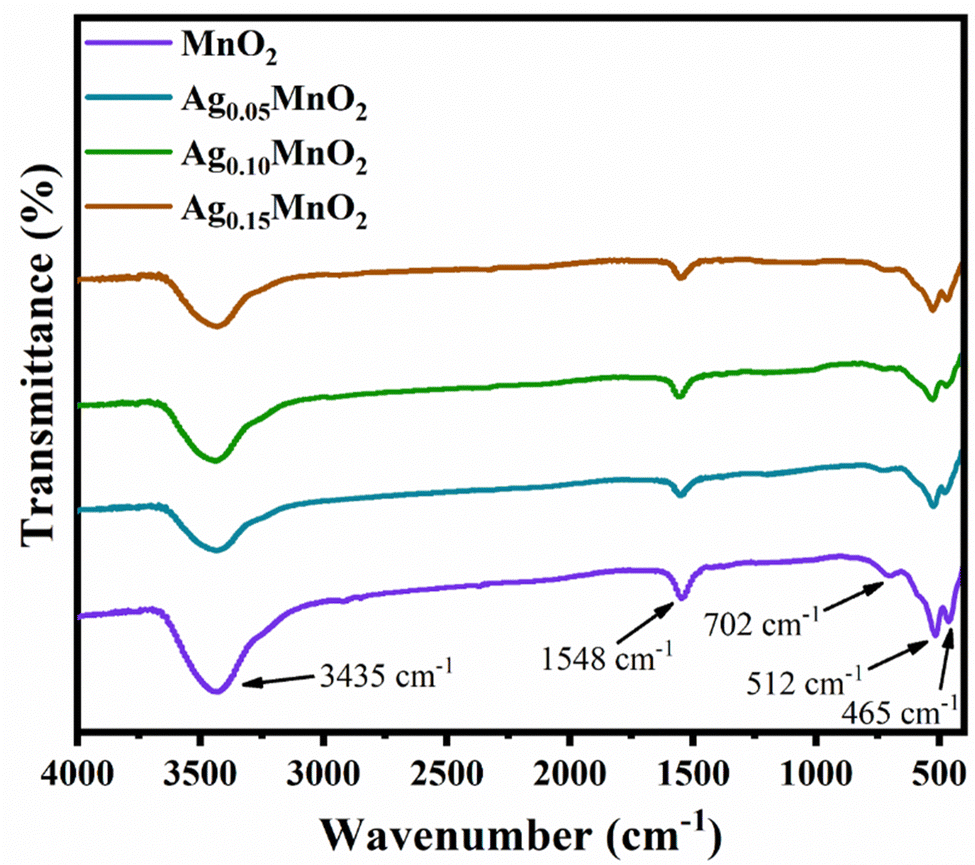
Functional groups of pristine, and AgxMnO2 NWs were confirmed by FTIR technique.51 FTIR spectra of AgxMnO2 NWs are given in Fig. 3. All the absorption bands are in good agreement with the literature.28 The absorption bands below 1000 cm−1 (low frequency region) confirmed the successful synthesis of MnO6 octahedra.52 Briefly, the absorption bands located at 465 cm−1 and 512 cm−1 are ascribed to Mn–O stretching vibration. The absorption band located at 702 cm−1 is attributed to the stretching vibration of Mn–O–Mn, suggesting the formation of MnO6 octahedra. These absorption bands were located at slightly different positions for the Ag doped samples. This illustrates that Ag has been successfully doped into MnO2. The band at 1548 cm−1 is due to Mn–O–H vibration.53 The intensity decrease with doping indicated that oxygen vacancies are produced. In addition, the band positioned at 3435 cm−1 is due to the stretching vibration of the O–H bond.
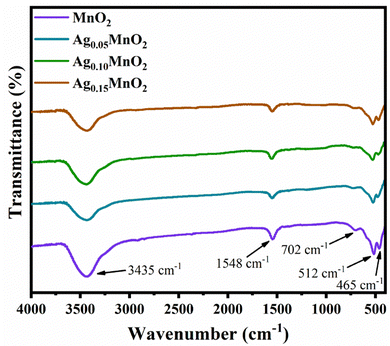 |
| | Fig. 3 FTIR spectra of AgxMnO2 NWs. | |
3.3. Field emission scanning electron microscopy (FESEM)
Morphology of AgxMnO2 NWs was evaluated by FESEM analysis.54 The FESEM micrograph of pristine MnO2 showed the nanowires morphology as depicted in Fig. 4(a). Pristine nanowires are ultrathin as well as ultralong having a diameter below 50 nm. Fig. 4(b)–(d) are the FESEM micrographs for MnO2 NWs doped with different concentrations of silver. Silver doping has made the surface rough, and aggregation has also taken place. The aggregation is resulted from high surface charge.55,56 The augmented surface charge could be attributed to greater ion concentration and reduced pH, as AgNO3 aqueous solution is acidic. It is generally understood that a change in pH value has a significant impact on surface charge (negative or positive). Augmented ionic strength will decrease or remove the repulsive energy barrier, resulting in fast aggregation.57 Diameters of pristine nanowires are below 50 nm. For Ag doped MnO2 nanowires, the diameter ranges between 50 and 70 nm.
 |
| | Fig. 4 FESEM images of (a) MnO2, (b) Ag0.05MnO2, (c) Ag0.10MnO2, (d) Ag0.15MnO2. | |
3.4. High resolution transmission electron microscopy (HRTEM)
HRTEM analysis was performed to study the in-depth morphology and diameter of nanowires.58 HRTEM images of MnO2 and Ag0.05MnO2 are illustrated in Fig. 5(a) and (b). MnO2 and Ag0.05MnO2 nanowires average diameter measured using ImageJ software was found to be 31 nm and 38 nm respectively. TEM images of MnO2 and Ag0.05MnO2 demonstrate that both the samples have grown nanowires. The slight increase in diameter for Ag0.05MnO2 is credited to the stronger diffusion of Ag+ ions in the MnO2 structure.59 This diffusion is also confirmed by the non-existence of silver particles on the nanowire surface. Fig. 5(c) and (d) demonstrate the selected area diffraction (SAD) pattern of MnO2 and Ag0.05MnO2. It is clear that both the samples are crystalline and are consistent with the XRD diffractogram. Fig. 5(c) and (d) (inset) illustrate the lattice fringes showing the interplanar spacing (d). MnO2 has well resolved lattice fringes with the interplanar distance (d = 0.245 nm). While the inter-planar distance for Ag0.05MnO2 is 0.249 nm. This increase is credited to the stress and lattice expansion caused by the diffusion of Ag+ ions in MnO2 crystal as confirmed by XRD peak shifting towards lower angle.60,61 The lattice fringes of Ag0.05MnO2 (Fig. 5(d)) shows that crystallinity of MnO2 NWs has reduced with Ag doping, which is also confirmed by XRD diffractogram.
 |
| | Fig. 5 (a) HRTEM image of MnO2, (b) HRTEM image of Ag0.05MnO2, (c) SAD pattern of MnO2, (d) SAD pattern of Ag0.05MnO2. | |
3.5. Energy dispersive X-ray (EDX) spectroscopy
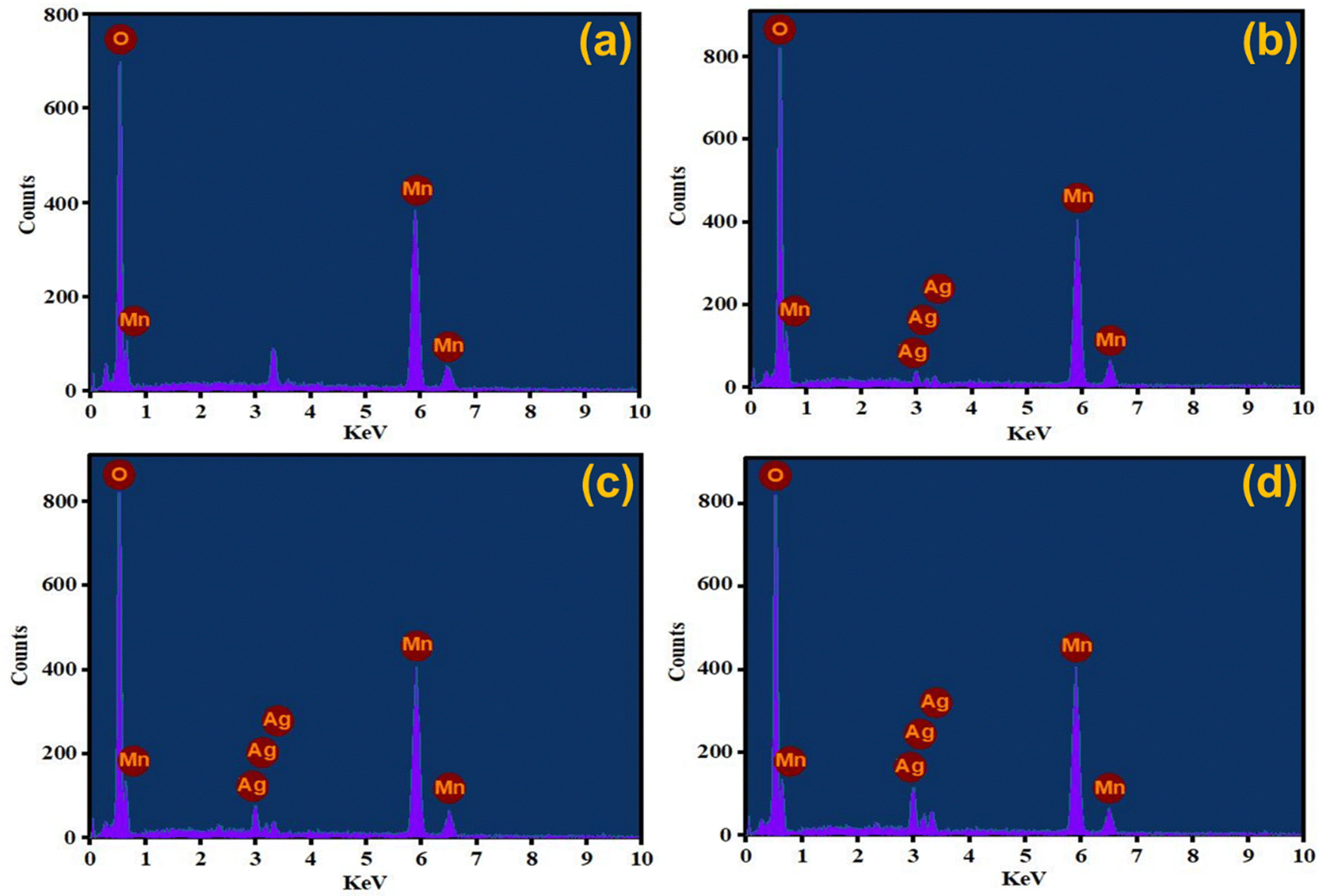
Elemental detection of AgxMnO2 NWs was done by EDX technique62 and the results are presented in Fig. 6. The EDX analysis of MnO2 NWs revealed de-excitation peaks corresponding to the manganese and oxygen atoms. This confirms that the pristine MnO2 sample is free from any impurities since the detected peaks exclusively correspond to the elemental composition of Mn and O. However, an unlabeled peak is observed in the spectrum positioned between 3–4 keV. It is attributed to the de-excitation of potassium atoms. The potassium peak originated from the precursor salt used in the synthesis of the MnO2 NWs. These atoms are believed to reside within the tunnel structure of the nanowires, indicating that they are incorporated into the material during the synthesis process to stabilize the α-phase of MnO2. The EDX analysis of Ag-doped MnO2 NWs samples presented in Fig. 6(b)–(d) show the presence of both manganese and oxygen, similar to the pristine sample. However, in addition to peaks associated with manganese and oxygen, de-excitation peaks corresponding to silver atoms are also observed. These peaks indicate the successful incorporation of silver as a dopant into the MnO2 matrix during the synthesis process. The presence of varied intensity de-excitation peaks for Ag in the EDX spectra was observed by varying the dopant concentration. This confirms that the amount of silver incorporated in the MnO2 matrix can be varied and controlled by adjusting the dopant concentration during the synthesis process.
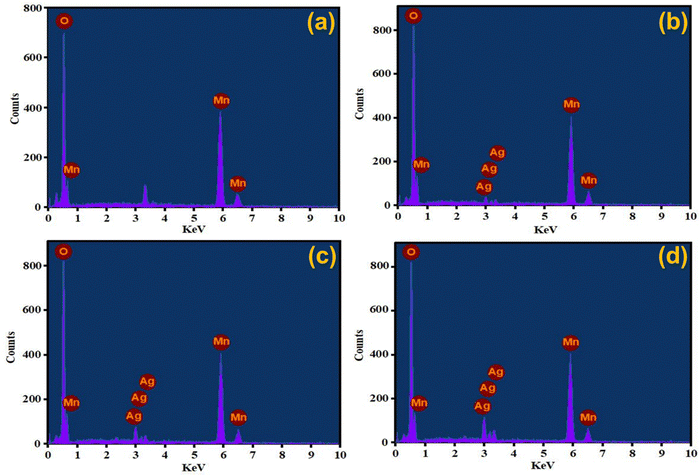 |
| | Fig. 6 EDX spectra of (a) MnO2, (b) Ag0.05MnO2, (c) Ag0.10MnO2, (d) Ag0.15MnO2. | |
3.6. X-ray photoelectron spectroscopy (XPS)
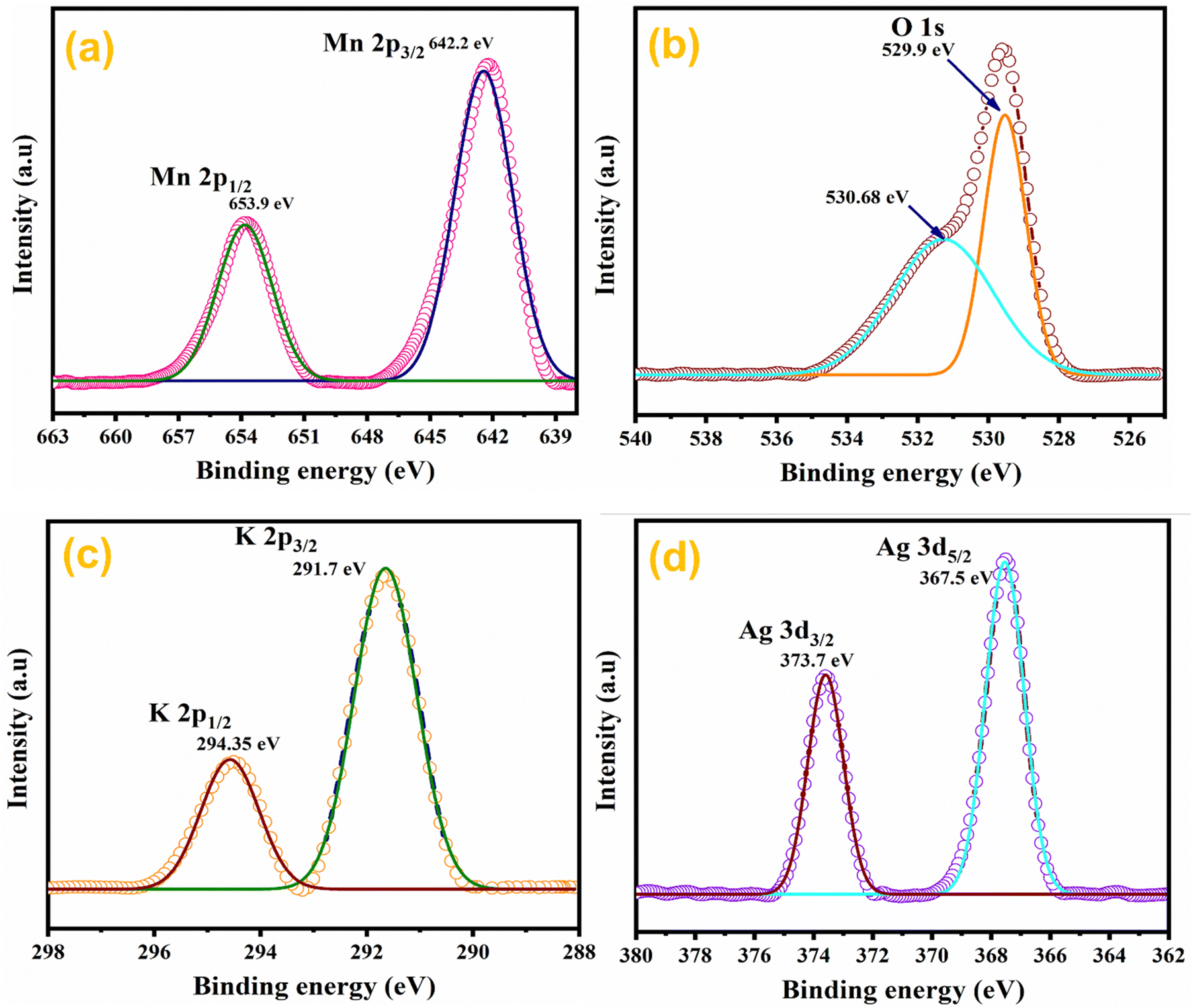
The elemental composition of Ag0.05MnO2 NWs was further confirmed by XPS studies.63 XPS spectra of the sample are depicted in Fig. 7. Fig. 7(a) shows the XPS spectrum of Mn 2p in Ag0.05MnO2. There has been observed a peak at 642.2 eV that is associated with Mn 2p3/2. The other peak located at 653.9 eV is associated with Mn 2p1/2. Spin energy separation of almost 11.7 eV between spin–orbit doublet suggests that Mn exists in +4 oxidation state.64–66Fig. 7(b) and (c) show the XPS spectra of O 1s and K 2p in Ag0.05 MnO2. The XPS spectrum of O 1s showed a peak at 529.9 eV along with a shoulder peak around 530.7 eV. The peak at 529.9 eV is ascribed to the structural oxygen while the peak around 530.68 eV is associated with the oxygen of the hydroxyl group.67Fig. 7(c) reveals that K atoms from precursor KMnO4 residing in α-MnO2 tunnels were also detected by the XPS spectrum of K 2p.68Fig. 7(d) displays the XPS spectrum of Ag 3d. There has been observed a peak located at 367.5 eV and is associated with Ag 3d5/2, which clearly suggests that Ag exists as an Ag+ ion.69 The other peak located at 373.7 eV is ascribed to Ag 3d3/2.70 This shows that Ag has been successfully doped in MnO2 NWs. The theoretical doping concentration of Ag for Ag0.05MnO2 was 9.58 wt%, while the actual doping concentration calculated by XPS and EDX spectra was found to be 8.71 wt%.
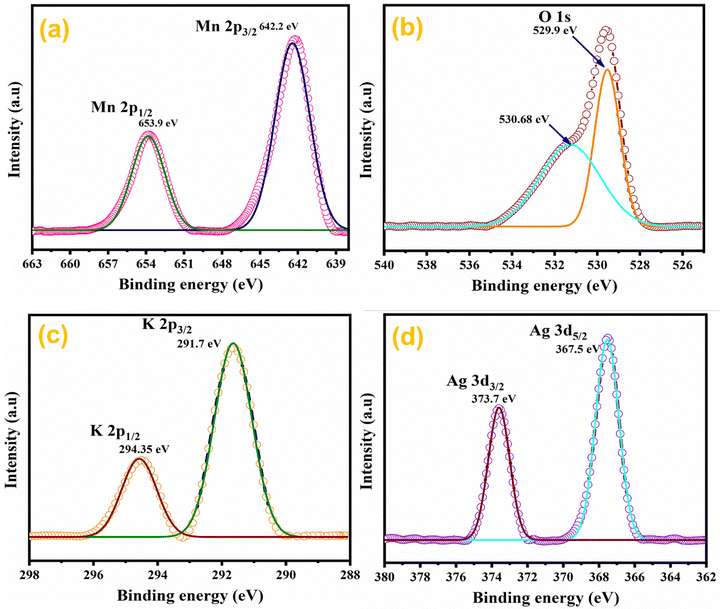 |
| | Fig. 7 XPS spectra of Ag0.05MnO2 nanowires. | |
3.7. Current–voltage (I–V) studies
By applying voltage (V) we observed the current (I) response of bare and doped MnO2 NWs. I–V profiles are depicted in Fig. 8. Conductivity of bare and doped MnO2 NWs has been calculated using eqn (3).| | 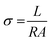 | (3) |
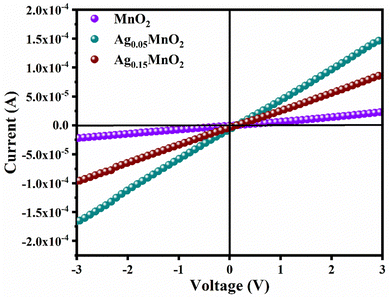 |
| | Fig. 8
I–V profiles of bare and doped MnO2 NWs. | |
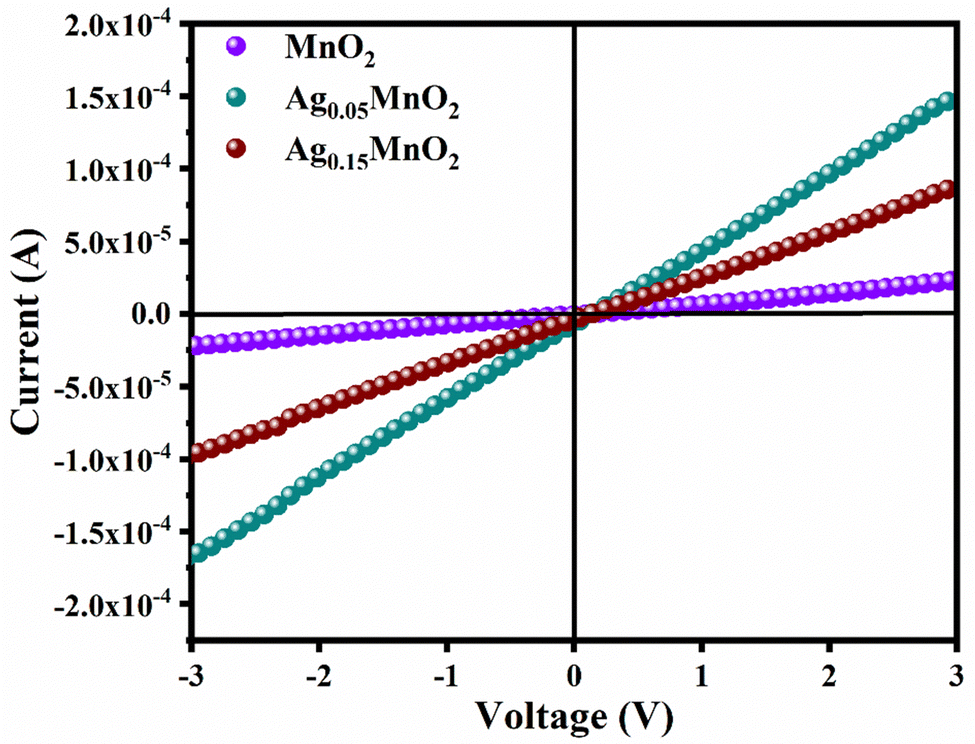
It is also clear from I–V profile that with Ag doping, the I–V curve becomes more vertical suggesting an increased current under applied voltage. Conductivity values of bare and doped samples are shown in Table 1. An increase in the conductivity of MnO2 is observed with Ag doping. This increase is ascribed to defects (oxygen vacancies) which are generated to balance the charge. Generation of oxygen vacancies left behind electrons. Consequently, charge carrier concentration increases which is confirmed by Mott–Schottky analysis, giving rise to an increase in the conductivity of MnO2 NWs. However, at higher doping concentrations, a large number of defects are generated which leads to increased charge scattering. Thus, giving rise to a decrease in conductivity.71
Table 1 Electrical conductivity and charge carrier density of AgxMnO2 NWs
| S. no. |
Sample name |
Electrical conductivity (S cm−1) |
Charge carrier density (cm−3) |
| 1 |
MnO2 |
2.90 × 10−5 |
1.48 × 1026 |
| 2 |
Ag0.05MnO2 |
1.5 × 10−3 |
1.20 × 1027 |
| 3 |
Ag0.15MnO2 |
5.2 × 10−4 |
2.01 × 1028 |
3.8. Physisorption analysis
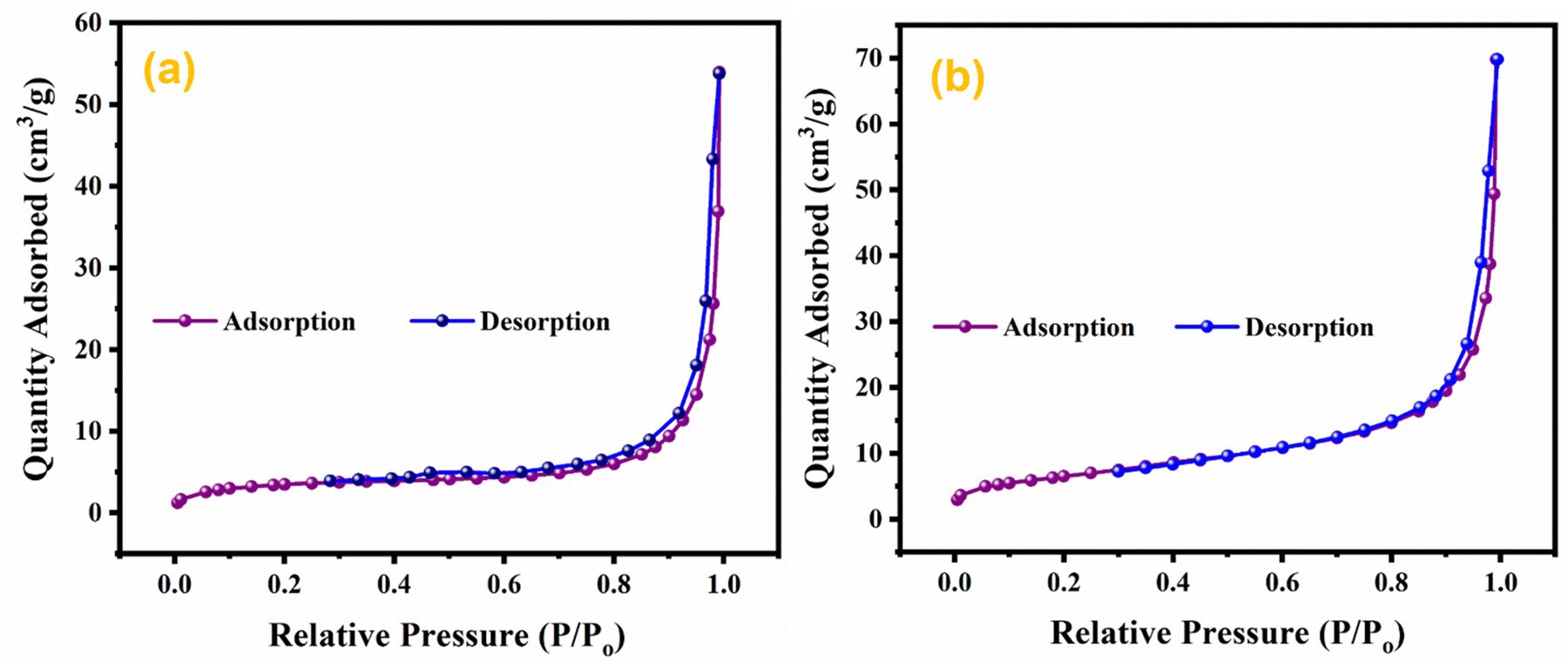
The effect of doping and composite formation on the specific surface area was investigated through BET analysis.72 BET measurements were carried out with nitrogen at 77 K. BET isotherms of pristine MnO2 NWs, and Ag0.05MnO2 NWs are shown in Fig. 9. Pristine and doped samples exhibited the isotherms that resemble the type III isotherm of IUPAC having a H3 hysteresis loop. Rapid higher adsorption at higher relative pressure indicates the presence of small pores in the samples. Both the samples displayed the same type of adsorption/desorption trend, however, the amount of gas adsorbed in Ag doped MnO2 sample is higher than the pristine MnO2 NWs. This showed that doping MnO2 with noble metal is a good way to improve the surface area. The BET surface area of Ag0.05MnO2 NWs is 74 m2 g−1, which is superior to the surface area of pristine MnO2 NWs. During the synthesis process, the introduction of noble metal (Ag) can influence the growth and arrangement of the material's microstructure, resulting in the formation of pores or voids. These pores contribute to the increased surface area as they provide additional sites for reactant molecules to interact with the material.
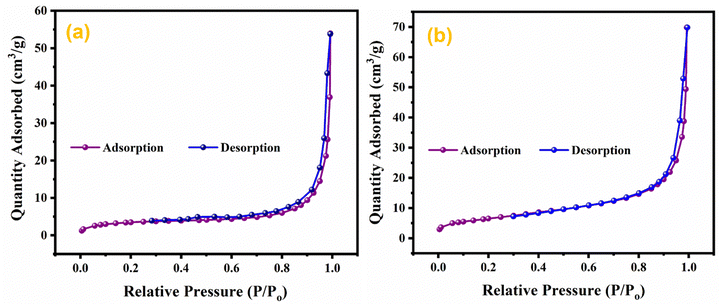 |
| | Fig. 9 BET isotherms of (a) MnO2, (b) Ag0.05MnO2. | |
3.9. Electrochemical measurements
Electrochemical measurements of AgxMnO2 NWs were executed to assess the supercapacitor performance of the as-prepared samples. For electrochemical measurements the electro-active material was deposited on 3-D nickel foam (NF). Cyclic voltammetry (CV), galvanostatic charge/discharge (GCD), and electrochemical impedance spectroscopy (EIS) were performed to evaluate the supercapacitor performance of the electrodes. CV measurements for AgxMnO2 (x = 0, 0.05, 0.10, 0.15) were performed to get a better understanding of the electron transfer rate, and redox reactions involved at electrode–electrolyte interface. CV is also used to get an estimation of the appropriate potential window. CV measurements were performed in a potential window 0–0.6 V in 1 M Na2SO4 using platinum as the counter electrode, and Ag/AgCl as the reference electrode in a half-cell configuration.
CV profiles of AgxMnO2 are shown in Fig. 10. An anodic peak is observed in the positive current region while the cathodic peak is observed in the negative current region. The bumps in CV curves confirmed the reversible faradaic reaction. The possible electrode reaction may be explained by two different pathways.
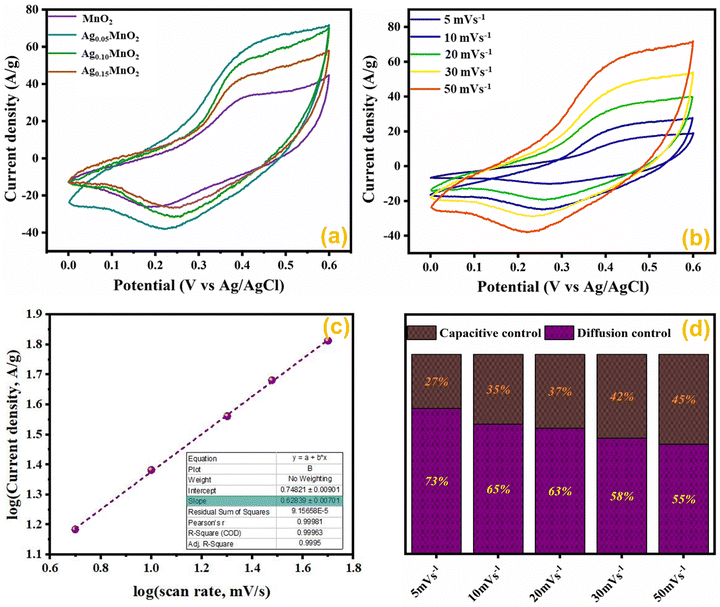 |
| | Fig. 10 (a) CV profile of AgxMnO2 NWs, (b) CV profile of Ag0.05MnO2 at various scan rates, (c) logi vs. logν graph of Ag0.05MnO2, and (d) impact of the scan rate on the prominent charge storage process of Ag0.05MnO2. | |
The first pathway is the adsorption/desorption of electrolyte ions (Na+) at the electrode–electrolyte interface and is represented below.
| MnO2 surface + Na+ + e− → (MnO2−Na+) surface |
The second pathway involves the insertion/disinsertion of electrolyte ions (Na
+) and protons (H
+) in the bulk involving a redox reaction and is represented below.
| MnO2 bulk + Na+ + e− → (MnOONa) bulk |
Comparative CV measurements for AgxMnO2 NWs were performed at 50 mV s−1 scan rate and are displayed in Fig. 10(a). The Ag0.05MnO2 electrode showed the greatest integral area among all the samples. The amplified area under the curve is credited to the improved conductivity, thanks to the silver. The presence of silver imparts a new channel for rapid electron transfer by lowering the intrinsic resistivity. In addition, the amplified loop area may be credited to the increased surface area, which will offer more active sites for electrolyte ions to be adsorbed. It is evident from Fig. 10(a) that as the silver concentration increases beyond 0.05 molar, the current response of the electro-active material decreases at 50 mV s−1 scan rate and is well consistent with the previous literature.1 Excessive doping can result in a higher concentration of charge carriers. These carriers can screen the electric field, reducing the effective electric field experienced by the remaining charges and, consequently, the amount of charge that can be stored. This phenomenon is known as charge screening and can lead to a decrease in specific capacitance.
The effect of different scan rates on CV profile of Ag0.05MnO2 NWs was also studied. Fig. 10(b) displays the CV curves of Ag0.05MnO2 at different scan rates. With an increase in scan rate, loop area also increases resulting in an increase in specific capacitance. The enlarged loop area is attributed to the fact that increasing the sweep rate enables more current to flow through the electrode material.
To conclude whether the prominent charge storage process is a pseudocapacitive or electric double layer, the power law relation was used.
By taking logν on the x-axis, and logi on the y-axis, and plotting its linear fit gives the value of the slope and intercept. The slope value ranges between 0.5 and 1. For a perfect pseudocapacitor, the slope is 0.5, and for an ideal EDLC it is 1. The linear fit plot of logν vs. logi is given in Fig. 10(c). The inset of Fig. 10(c) illustrates the slope value, which is 0.62. It shows that the prominent charge storage process is pseudocapacitive.
Impact of scan rate on the charge storage mechanism was also investigated by Dunn's method given in eqn (5) and (6).73
| | | i(ν)/ν1/2 = K1ν1/2 + K2 | (6) |
The results obtained by using the above relations are shown in Fig. 10(d). By increasing the scan rate, the capacitive contribution towards total capacitance increased from 27% to 45%. This increase is due to the reduced time accessible for electrolyte ions to permeate the electrode material at a high scan rate. However, the diffusion-controlled charge storage process is prominent and its contribution towards total capacitance at all the scan rates is dominant suggesting the pseudocapacitive nature of the electro-active material.
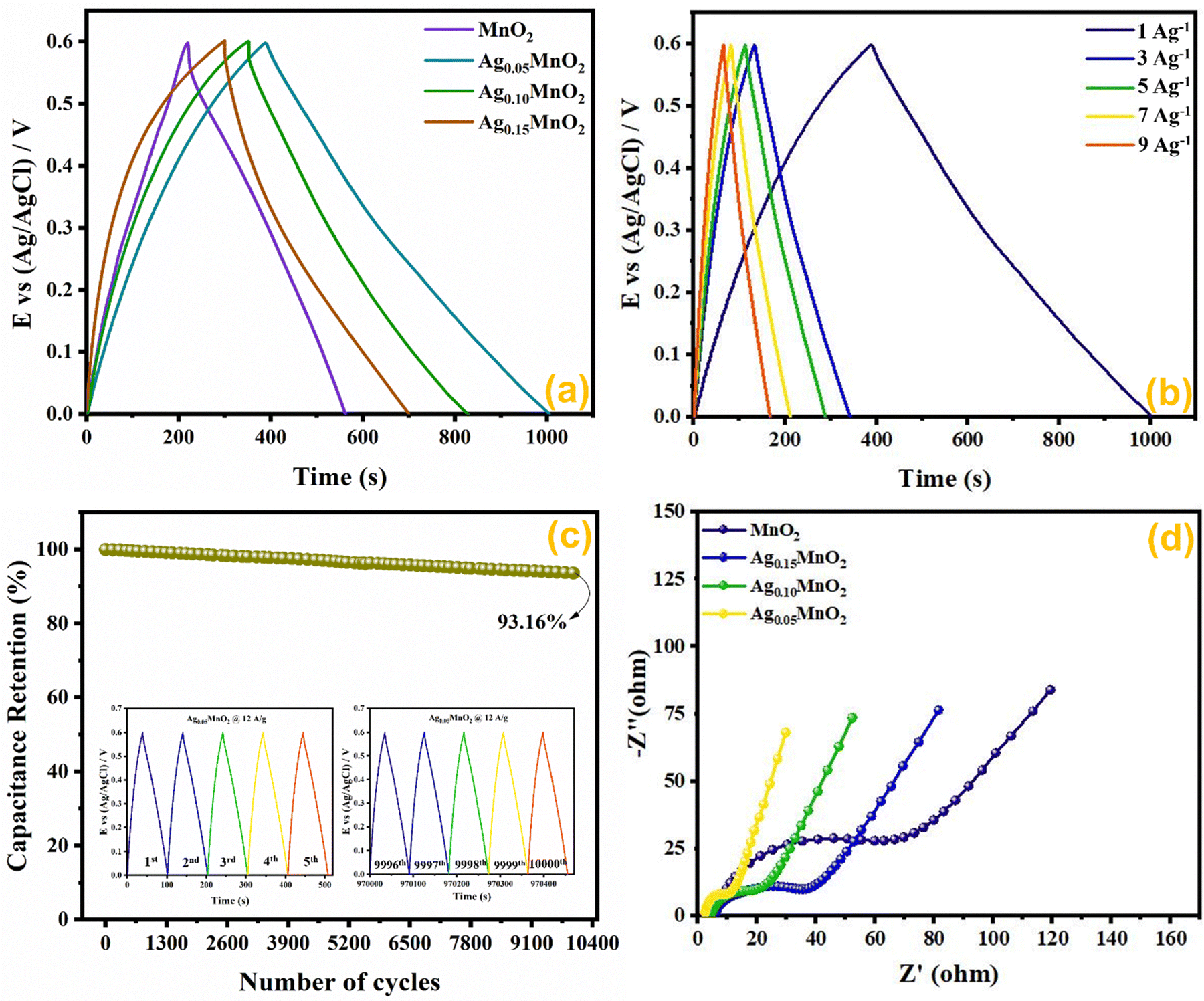
The capacitance value for pristine and Ag doped MnO2 NWs was calculated from the galvanostatic charge discharge (GCD) profile, which is obtained in the same range as that of CV. Comparative GCD measurements of all the samples performed at 1 A g−1 current density are shown in Fig. 11(a). The highest discharging time of 616 s was shown by Ag0.05MnO2 among all the samples. This suggests that the optimum concentration of Ag into MnO2 is 0.05. The discharging time showed by other samples is 342 s, 400 s, and 480 s for MnO2 NWs, Ag0.15MnO2, and Ag0.10MnO2 NWs respectively. The specific capacitance was determined from GCD profiles using eqn (7).
| | 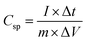 | (7) |
 |
| | Fig. 11 (a) GCD curves of AgxMnO2, (b) impact of varying current densities on GCD profile of Ag0.05MnO2, (c) capacitance retention after 10000 GCD cycles (inset: first and last 5 cycles during cyclic test of Ag0.05MnO2), and (d) EIS profile of AgxMnO2. | |
Ag0.05MnO2 exhibited the highest specific capacitance of 1027 F g−1@1 A g−1 current density. This improvement is ascribed to the introduction of new gap states between the conduction and the valence band induced by the hybridization between Ag 4d with O 2p. Large size of Ag+ ions and high energy of Ag 4d orbitals result in a strong repulsion between Ag 4d and O 2p which pushes energy levels upward causing fermi level to shift closer to conduction band. Moreover, oxygen and interstitial defects produced by silver doping also induced new occupied states between the conduction and the valence band resulting in an upward movement of the Fermi level. Thus, silver doping induced n-type characteristics in MnO2.74 The order of specific capacitance for different MnO2 samples follows the order: Ag0.05MnO2 > Ag0.10MnO2 > Ag0.15MnO2 > MnO2. The specific capacitance of all the electro-active materials is given in Table 2. It is observed that as the silver concentration exceeds 0.05 molar, the specific capacitance decreases by increasing the dopant concentration. Doping introduces impurities into the material, which can create defects or dislocations in the crystal lattice. At low doping concentrations, these defects can enhance the electrochemical surface area, thereby increasing the specific capacitance. However, at high doping concentrations, the excessive defects can lead to increased charge scattering, decreased electrolyte penetration, and increased resistance, resulting in a decrease in specific capacitance. It is inferred from the GCD profile of all the samples that MnO2 showed an IR drop in the beginning of the discharge curve, which almost disappeared for Ag0.05MnO2. However, this IR drop again increased for Ag0.10MnO2 and Ag0.15MnO2. This decrease in IR drop for Ag0.05MnO2 is ascribed to the decreased resistivity of the electrode material.75,76 GCD curve of MnO2 is the characteristic curve of α-MnO2.77 With Ag doping, the GCD curve has become steeper and consistent with the already reported GCD curves of α-MnO2.67,78 It is due to the improved reaction kinetics, increased conductivity and shorter diffusion path of ions.
Table 2 Specific capacitance and resistance values for all the prepared samples
| S. no. |
Sample |
Specific capacitance (F g−1) |
Charge transfer resistance (Rct/Ω) |
Solution resistance (Rs/Ω) |
| 1 |
MnO2 |
570 |
70 |
5.97 |
| 2 |
Ag0.05MnO2 |
1027 |
10 |
2.35 |
| 3 |
Ag0.10MnO2 |
800 |
20 |
4.16 |
| 4 |
Ag0.15MnO2 |
667 |
40 |
5.58 |
The impact of current densities on Ag0.05MnO2 NWs was also investigated. It can be assessed from Fig. 11(b) that by increasing the current density specific capacitance was decreased. However, the shape of the charging/discharging curve remains virtually the same.
It is inferred from Table 3 that an increase in current density resulted in a decrease in specific capacitance. It might be ascribed to slow redox reaction at elevated current densities. Moreover, at lower current densities electrolyte ions not only adsorb on the surface but also penetrate the bulk. Consequently, more active sites are involved giving rise to an improved capacitance. Specific capacitance decreases when the current density is high because electrolyte ions are confined to the surface and do not have enough time to permeate the bulk. We also studied the cyclic stability of Ag0.05MnO2 NWs by applying 10,000 GCD cycles@12 A g−1. The cyclic stability test findings are given in Fig. 11(c). The Ag0.05MnO2 electrode retained 93.16% of its initial capacitance. Inset of Fig. 11(c) shows the first five and last five GCD cycles. The excellent cyclic stability proved that doping a noble metal into the MnO2 matrix is a good way to boost electrochemical performance of MnO2. Table 4 compares the electrochemical performance of the current electrode material with the recent literature.
Table 3 Impact of varying current density on the specific capacitance of Ag0.05MnO2 NWs
| S. no. |
Current density (A g−1) |
Specific capacitance (F g−1) |
Retention (%) |
| 1 |
1 |
1027 |
100 |
| 2 |
3 |
998 |
97 |
| 3 |
5 |
950 |
93 |
| 4 |
7 |
920 |
89 |
| 5 |
9 |
890 |
86 |
Table 4 Comparison of the current electrode material with previously reported studies
| Electroactive material |
Specific capacitance (F g−1) |
Retention (%) |
Cyclic activity (%)/cycles |
Ref. |
| F–MnO2 |
148/0.5 A g−1 |
100@10 A g−1 |
82.80/10000 |
79
|
| Fe–MnO2 |
196/1 A g−1 |
70@30 A g−1 |
83.3/20000 |
80
|
| K–MnO2 |
283.7/1 A g−1 |
66.9@20 A g−1 |
90.7/10000 |
81
|
| Na–MnO2 |
324.7/0.5 A g−1 |
56.1@50 A g−1 |
91/10000 |
82
|
| MnO2/MXene |
457/1 A g−1 |
— |
102.5/1000 |
83
|
| Eu–MnO2 |
362.1/1 A g−1 |
63@10 A g−1 |
100/10000 |
84
|
| Zn–MnO2 |
392/1 A g−1 |
70.4@20 A g−1 |
94.3/10000 |
85
|
| Ag–MnO2 |
520.8/0.5 A g−1 |
— |
90.6/2000 |
86
|
| Ag–MnO2/GO |
868/2 A g−1 |
49.1@5 A g−1 |
94.5/5000 |
87
|
| Ag0.05MnO2 |
1027/1 A g−1 |
86%@9 A g−1 |
93.16%/10000 |
This work |
EIS measurements provide information about the mass transfer resistance, internal resistance, and resistance present at the electrode–electrolyte interface. EIS measurements for AgxMnO2 NWs were performed in the frequency ranging between 0.1 Hz and 100 kHz, and are illustrated in Fig. 11(d). A typical EIS spectrum entails an x-intercept followed by a semicircle and a linear line in a low frequency region representing electrode–electrolyte interfacial resistance, charge transfer resistance (Rct), and mass transfer resistance respectively. Ag0.05MnO2 NWs showed the smallest diameter indicating the smallest charge transfer resistance. It can be credited to Ag doping, which provided a new pathway for rapid electron transfer. The introduction of silver provide ions in the tunnels of MnO2 leading to an improvement in conductivity. The resistance values are depicted in Table 2. It is observed that when doping concentration exceeds x = 0.05 the Rct increases. It is ascribed to the fact that heavy doping causes pinning of the Fermi level near the surface of the electro-active material, resulting in energy barrier change across the electrode–electrolyte interface, thus increasing resistance in charge transfer.88,89 Additionally, high doping leads to more scattering centers which also resists the charge transfer at the electrode–electrolyte interface.90
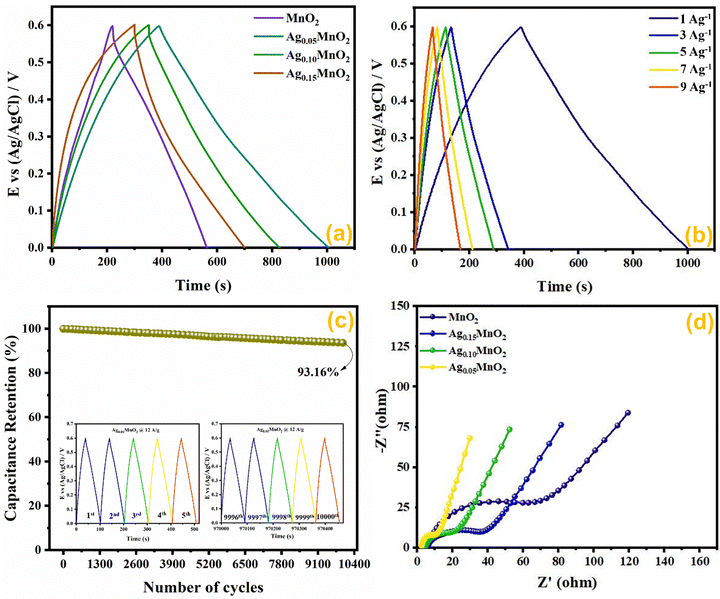
3.10. Mott–Schottky (M–S) analysis
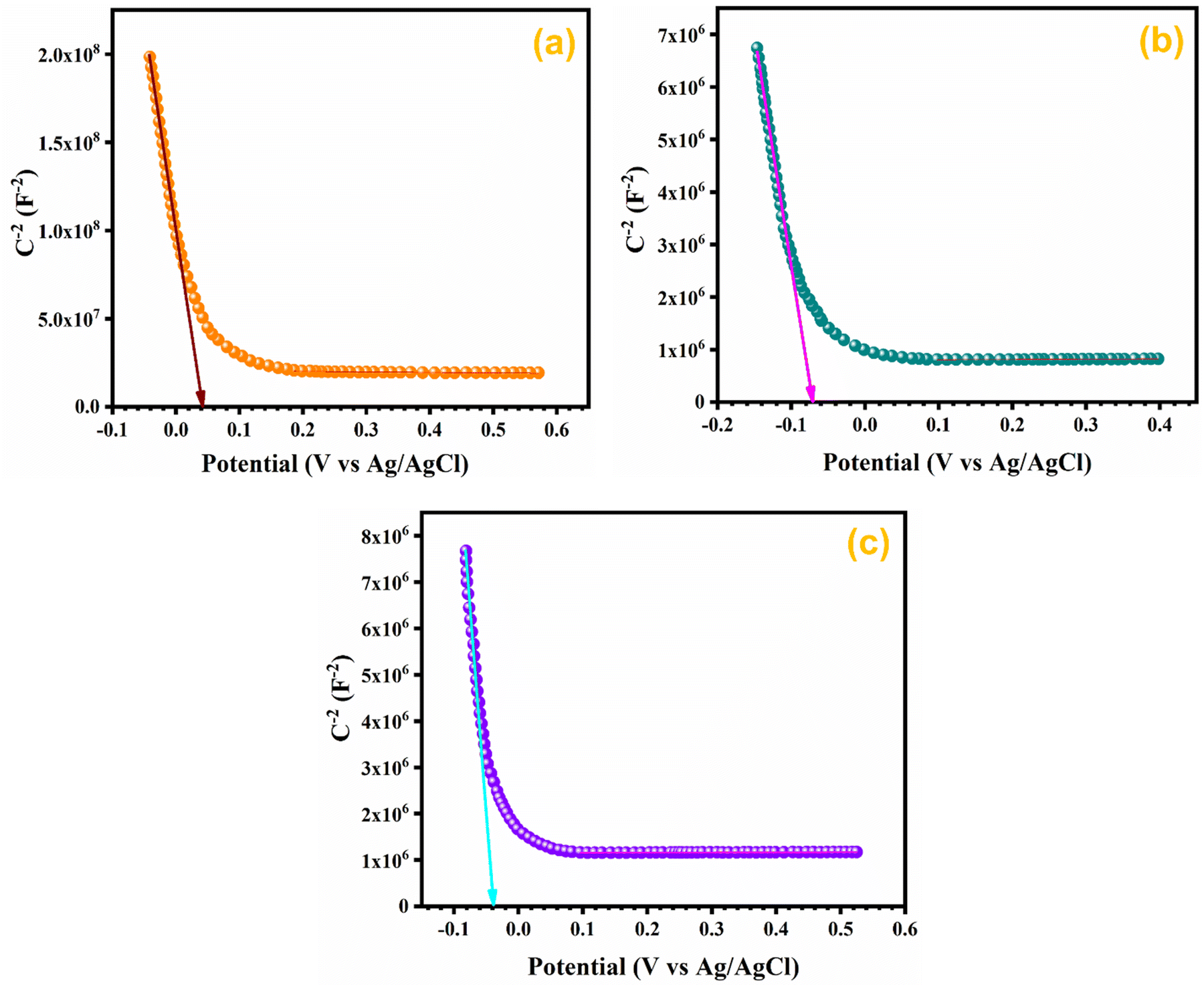
In order to assess the charge carrier density and flat band potential M–S studies were performed. M–S profiles of Ag0.05MnO2 and Ag0.15MnO2 are illustrated in Fig. 12. The slope of the M–S plot for MnO2 is negative, which suggests the p-type nature of the material,91 while the slope of Ag0.05MnO2 and Ag0.15MnO2 is positive, indicating that doping has induced n-type character in MnO2.92–94 This transformation from p-type to n-type is associated with the production of oxygen vacancies. Production of oxygen vacancies is the result of charge balance which is caused by doping silver in MnO2 due to variation in oxidation state of Ag+ and Mn4+. As a result of oxygen vacancy generation, electrons (charge carriers) will be left behind. Thus, the concentration of electrons will increase.95 The flat band potentials of MnO2, Ag0.05MnO2, and Ag0.15MnO2 are 0.042 V, −0.07 V, and −0.04 V respectively. The charge carrier densities of MnO2, Ag0.05MnO2 and Ag0.15MnO2 are given in Table 1. Charge carrier density calculated from the M–S plot was found to increase with an increase in the Ag content. Bappi et al. have reported that doping induces scattering centers.71 Heavy doping induced more scattering centers (increased charge carrier density) which increased the energy barrier at the electrode–electrolyte interface giving rise to an increase in Rct and reduced mobility of charge carriers which is also confirmed by EIS and I–V measurements respectively.
 |
| | Fig. 12 M–S profile of (a) MnO2, (b) Ag0.05MnO2, and (c) Ag0.15MnO2. | |
4. Conclusions
In summary, a facile and low-temperature hydrothermal process has been used to synthesize AgxMnO2 NWs. The effect of Ag doping on the structure and morphology has been validated by physico-chemical techniques. The influence of the dopant amount on electrochemical performance was also studied. XRD analysis showed a shift in the diffraction angle towards a lower 2θ value. FESEM analysis displayed cracks on the NWs’ surface by Ag doping in MnO2 NWs. BET studies revealed an increase in the surface area. Electrochemical investigations were carried out using porous 3D nickel foam as a current collector. Cyclic voltammetry investigations showed typical redox peaks in positive and negative current regions suggesting a prominent pseudocapacitive charge storage process. Ag0.05MnO2 NWs had a significantly higher specific capacitance of 1027 F g−1 in comparison to bare MnO2@1 A g−1. Furthermore, Ag0.05MnO2 showed lower resistance compared to other samples and excellent capacitance maintenance (93.16% at 12 A g−1 after 10000 cycles). Ag0.05MnO2's superior electrochemical performance is ascribed to increased conductivity, Ag intercalation into (2 × 2) tunnels and a shorter diffusion path length for electrolyte ions.
Author contributions
Muhammad Usman Khalid: investigation, data curation, and writing – review & editing; Sonia Zulfiqar: formal analysis, validation, and writing – review & editing; Imran Shakir: formal analysis; M. Nazirduddin Khan: resources; Muhammad Farooq Warsi: supervision and project administration; Eric W. Cochran: resources and project administration.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The researchers wish to extend their sincere gratitude to the Deanship of Scientific Research at the Islamic University of Madinah for the support provided to the Post-Publishing Program. Prof. Dr Sonia Zulfiqar is highly thankful for the support provided by the Statutory City of Ostrava, Czechia through Research Grant “Global Experts” and to the American University in Cairo, Egypt for using STRC facilities. Profs Cochran and Zulfiqar are grateful for the TEM imaging provided by Dr Tracey Stewart of the Roy J. Carver High Resolution Microscopy Facility and for the XPS measurements provided by Dr Dapeng Jing of the Materials Analysis and Research Laboratory of the Iowa State University Office of Biotechnology. E. W. C. and S. Z. are also thankful to the National Science Foundation for financial support through research grants NSF-2113695, NSF-2218070 and NSF-2242763.
References
- A. K. Worku, D. W. Ayele, N. G. Habtu and M. D. Ambaw, Heliyon, 2022, 8, e10960 CrossRef CAS PubMed.
- H. R. Barai, A. N. Banerjee and S. W. Joo, J. Ind. Eng. Chem., 2017, 56, 212–224 CrossRef CAS.
- Q. Liu, S. Ji, J. Yang, H. Wang, B. G. Pollet and R. J. M. Wang, Materials, 2017, 10, 988 CrossRef PubMed.
- J. Li, S. Yi, R. Rajagopalan, Z. Zhang, Y. Tang and H. Wang, Green Energy Environ., 2023, 8, 308–317 CrossRef CAS.
- V. J. Mane, S. B. Kale, S. B. Ubale, V. C. Lokhande and C. D. Lokhande, Mater. Today Chem., 2021, 20, 100473 CrossRef CAS.
- P. Simon, Y. Gogotsi and B. J. S. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- Y. Xiang, S. Lu and S. P. Jiang, Chem. Soc. Rev., 2012, 41, 7291–7321 RSC.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CAS.
- D. P. Dubal, N. R. Chodankar, D.-H. Kim and P. Gomez-Romero, Chem. Soc. Rev., 2018, 47, 2065–2129 RSC.
- T. Xiong, T. L. Tan, L. Lu, W. S. V. Lee and J. J. A. E. M. Xue, Adv. Energy Mater., 2018, 8, 1702630 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- N. B. Velhal, N. C. Maile, C. Paeng, H. Lee, T. Kim, J. Kim and C. Yim, J. Energy Storage, 2024, 90, 111764 CrossRef.
- K. Kalaiselvi, M. Mubarak Ali, S. Prabhu, P. Muthu Mareeswaran and R. Ramesh, Inorg. Chem. Commun., 2024, 162, 112145 CrossRef CAS.
- J.-Z. Jiang, Y.-J. Gu, W. Wen, Z.-Z. Ye and J.-M. Wu, J. Energy Storage, 2024, 84, 110957 CrossRef.
- H. Li, H. Yang, H. Sun, Y. Huang, P. An, Y. Yunhua and H. Zhao, Electrochim. Acta, 2024, 473, 143514 CrossRef CAS.
- H. A. Hamouda, S. Cui, X. Dai, X. Xie, H. Peng, G. Ma and Z. Lei, J. Energy Storage, 2022, 47, 103616 CrossRef.
- T. P. Manh, N. Nguyen Van, V. B. T. Phung, L. Ngo Thi, Q. Ngo Quy, S. Le The, P. Doan Tien, D. Tran Quang, T. Nguyen Van and N. To Van, Ceram. Int., 2024, 50, 22757–22770 CrossRef CAS.
- M. Imran, A. M. Afzal, M. W. Iqbal, H. H. Hegazy, M. Z. Iqbal, S. Mumtaz and R. Qureshi, Mater. Sci. Semicond. Process., 2023, 158, 107366 CrossRef CAS.
- I. Melkiyur, Y. Rathinam, P. S. Kumar, A. Sankaiya, S. Pitchaiya, R. Ganesan and D. Velauthapillai, Renewable Sustainable Energy Rev., 2023, 173, 113106 CrossRef CAS.
- S. Munir, M. Aadil, M. F. Warsi, H. H. Somaily, N. U. Ain and M. Shahid, Ceram. Int., 2022, 48, 33306–33314 CrossRef CAS.
- W. Guo, X. Lian, Y. Tian, T. Yang and S. Wang, J. Energy Storage, 2021, 38, 102586 CrossRef.
- S. Sun, G. Jiang, Y. Liu, B. Yu and U. Evariste, J. Energy Storage, 2018, 18, 256–258 CrossRef.
- T. P. Mofokeng, S. Shabalala, A. B. Haruna, P. V. Mwonga, Z. N. Tetana and K. I. Ozoemena, J. Electroanal. Chem., 2023, 948, 117809 CrossRef CAS.
- K. Li, C. Chen, H. Zhang, X. Hu, T. Sun and J. Jia, Appl. Surf. Sci., 2019, 496, 143662 CrossRef CAS.
- M. U. Khalid, K. M. Katubi, S. Zulfiqar, Z. A. Alrowaili, M. Aadil, M. S. Al-Buriahi, M. Shahid and M. F. Warsi, Fuel, 2023, 343, 127946 CrossRef CAS.
- D. Gangwar and C. Rath, Appl. Surf. Sci., 2021, 557, 149693 CrossRef CAS.
- C. Ling, R. Zhang, T. S. Arthur and F. Mizuno, Chem. Mater., 2015, 27, 5799–5807 CrossRef CAS.
- E. Hastuti, A. Subhan, P. Amonpattaratkit, M. Zainuri and S. Suasmoro, RSC Adv., 2021, 11, 7808–7823 RSC.
- K. Chen, W. Pan and D. Xue, J. Phys. Chem. C, 2016, 120, 20077–20081 CrossRef CAS.
- X. Duan, J. Yang, H. Gao, J. Ma, L. Jiao and W. Zheng, CrystEngComm, 2012, 14, 4196–4204 RSC.
- J. Liu, J. Bao, X. Zhang, Y. Gao, Y. Zhang, L. Liu and Z. Cao, RSC Adv., 2022, 12, 35556–35578 RSC.
- M. Thomas, W. Sun and J. Cui, J. Phys. Chem. C, 2012, 116, 6383–6391 CrossRef CAS.
- S. G. Hassan and E. J. Mohammad, Mater. Today: Proc., 2021, 42, 2749–2751 CAS.
- A. U. Rahman, N. Zarshad, J. Wu, F. Faiz, F. Raziq, A. Ali, G. Li and H. Ni, Mater. Sci. Eng., B, 2021, 269, 115150 CrossRef CAS.
- G. Zhang, L. Zheng, M. Zhang, S. Guo, Z.-H. Liu, Z. Yang and Z. Wang, Energy Fuels, 2012, 26, 618–623 CrossRef CAS.
- Y. Dai, S. Tang, S. Vongehr and X. Meng, ACS Sustainable Chem. Eng., 2014, 2, 692–698 CrossRef CAS.
- E. Umeshbabu, M. Satyanarayana, G. Karkera, A. Pullamsetty and P. J. M. A. Justin, Mater. Adv., 2022, 3, 1642–1651 RSC.
- X. Wang and Y. J. C. A. E. J. Li, Chem. – Eur. J., 2003, 9, 300–306 CrossRef CAS PubMed.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- A. Alalawi, U. E. Romman, K. M. Katubi, S. T. Shafa, M. Usman Khalid, Z. A. Alrowaili, M. S. Al-Buriahi, M. I. Din and I. Shakir, Curr. Appl. Phys., 2024, 60, 32–42 CrossRef.
- B. Rajesh Kumar and B. Hymavathi, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2018, 9, 035018 Search PubMed.
- B. Bashir, M. U. Khalid, M. Aadil, S. Zulfiqar, M. F. Warsi, P. O. Agboola and I. J. C. I. Shakir, Ceram. Int., 2021, 47, 3603–3613 CrossRef CAS.
- R. Karmakar, S. K. Neogi, A. Banerjee and S. Bandyopadhyay, Appl. Surf. Sci., 2012, 263, 671–677 CrossRef CAS.
- Y. Gu, Y. Min, L. Li, Y. Lian, H. Sun, D. Wang, M. H. Rummeli, J. Guo, J. Zhong and L. Xu, Chem. Mater., 2021, 33, 4135–4145 CrossRef CAS.
- H. Usui, S. Suzuki, Y. Domi and H. Sakaguchi, ACS Sustainable Chem. Eng., 2020, 8, 9165–9173 CrossRef CAS.
- M. U. Khalid, M. F. Warsi, M. I. Sarwar, P. O. Agboola, I. Shakir and S. Zulfiqar, Ceram. Int., 2020, 46, 14287–14298 CrossRef CAS.
- M. F. Warsi, B. Bashir, S. Zulfiqar, M. Aadil, M. U. Khalid, P. O. Agboola, I. Shakir, M. A. Yousuf and M. J. C. I. Shahid, Ceram. Int., 2021, 47, 5044–5053 CrossRef CAS.
- D. Prieur, W. Bonani, K. Popa, O. Walter, K. W. Kriegsman, M. H. Engelhard, X. Guo, R. Eloirdi, T. Gouder, A. Beck, T. Vitova, A. C. Scheinost, K. Kvashnina and P. Martin, Inorg. Chem., 2020, 59, 5760–5767 CrossRef CAS PubMed.
- M. Siddiq, A. G. Taki, M. Aadil, S. Mubarik, E. W. Cochran, S. Zulfiqar, A. A. A. Mohammed and S. Ijaz, Ceram. Int., 2023, 49, 36590–36599 CrossRef CAS.
- M. Rashid, W. Hassan, M. Aadil, H. H. Somaily, N. M. Mahdi, R. Lataef, A. G. Taki, K. Srithilat, D. F. Baamer, S. M. Albukhari, M. A. Salam and A. Llyas, Opt. Mater., 2023, 135, 113192 CrossRef CAS.
- H. S. N. Jayawardena, S. H. Liyanage, K. Rathnayake, U. Patel and M. Yan, Anal. Chem., 2021, 93, 1889–1911 CrossRef CAS PubMed.
- M. V. Ananth, S. Pethkar and K. Dakshinamurthi, J. Power Sources, 1998, 75, 278–282 CrossRef CAS.
- D. P. Dubal, W. B. Kim and C. D. Lokhande, J. Phys. Chem. Solids, 2012, 73, 18–24 CrossRef CAS.
- Y. Khan, H. Sadia, S. Z. Ali Shah, M. N. Khan, A. A. Shah, N. Ullah, M. F. Ullah, H. Bibi, O. T. Bafakeeh and N. B. J. C. Khedher, Catalysts, 2022, 12, 1386 CrossRef CAS.
- S. Yousaf, S. Zulfiqar, M. Shahid, A. Jamil, I. Shakir, P. O. Agboola and M. F. Warsi, Ceram. Int., 2020, 46, 14304–14310 CrossRef CAS.
- M. F. Warsi, M. Bilal, S. Zulfiqar, M. U. Khalid, P. O. Agboola and I. Shakir, Mater. Res. Express, 2020, 7, 105015 CrossRef CAS.
- W. Zhang, Nanomaterial, 2014, 19–43 Search PubMed.
- W. Zhou and H. Greer, Eur. J. Inorg. Chem., 2016, 941–950 CrossRef CAS.
- S. M. Jadhav, R. S. Kalubarme, N. Suzuki, C. Terashima, J. Mun, B. B. Kale, S. W. Gosavi and A. Fujishima, ACS Omega, 2021, 6, 5717–5729 CrossRef CAS PubMed.
- S. Liu, L. Kang, J. Zhang, S. C. Jun and Y. J. N. A. M. Yamauchi, NPG Asia Mater., 2023, 15, 9 CrossRef CAS.
- L.-T. Tseng, Y. Lu, H. M. Fan, Y. Wang, X. Luo, T. Liu, P. Munroe, S. Li and J. Yi, Sci. Rep., 2015, 5, 9094 CrossRef CAS PubMed.
- A. d’Alfonso, B. Freitag, D. Klenov and L. J. P. R. B. Allen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 100101 CrossRef.
- F. A. Stevie and C. Donley, J. Vac. Sci. Technol., A, 2020, 38, 063204 CrossRef CAS.
- J. Kim, Y. H. Min, N. Lee, E. Cho, K. Y. Kim, G. Jeong, S. K. Moon, M. Joo, D. B. Kim and J. Kim, ACS Omega, 2017, 2, 7424–7432 CrossRef CAS PubMed.
- B. Wei, L. Wang, Q. Miao, Y. Yuan, P. Dong, R. Vajtai and W. J. C. Fei, Carbon, 2015, 85, 249–260 CrossRef CAS.
- M. Li, W. Lei, Y. Yu, W. Yang, J. Li, D. Chen, S. Xu, M. Feng and H. J. N. Li, Nanoscale, 2018, 10, 15926–15931 RSC.
- J. Cremonezzi, D. Tiba and S. Domingues, SN Appl. Sci., 2020, 2, 1689 CrossRef CAS.
- K. Rajkumari, D. Das, G. Pathak and S. Rokhum, New J. Chem., 2019, 43, 2134–2140 RSC.
- Y. Pei, X. Chen, D. Xiong, S. Liao and G. J. P. O. Wang, PLoS One, 2013, 8, e81627 CrossRef PubMed.
- W. Zhang, X. Li, S. Liu, J. Qiu, J. An, J. Yao, S. Zuo, B. Zhang, H. Xia and C. J. C. Li, ChemSusChem, 2022, 15, e202102158 CrossRef CAS PubMed.
- M. B. Pramanik, M. A. Al Rakib, M. A. Siddik, S. Bhuiyan and T. Research, Eur. J. Eng. Technol. Res., 2024, 9, 10–15 CrossRef.
-
S. Thomas, R. Thomas, A. K. Zachariah and R. Kumar, Thermal and rheological measurement techniques for nanomaterials characterization, Elsevier, 2017 Search PubMed.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS PubMed.
- M. A. Thomas, W. W. Sun and J. B. Cui, J. Phys. Chem. C, 2012, 116, 6383–6391 CrossRef CAS.
- S. Dalvand, Z. Khoushab, S. M. Mousavi-Khoshdel, H. Ghafuri, H. R. E. Zand and M. Omidvar, Int. J. Hydrogen Energy, 2023, 48, 10098–10107 CrossRef CAS.
- S. Yousaf, M. Aadil, S. Zulfiqar, M. F. Warsi, P. O. Agboola, M. F. Aly Aboud and I. Shakir, J. Mater. Res. Technol., 2020, 9, 14158–14167 CrossRef CAS.
- E. A. Arkhipova, A. S. Ivanov, K. I. Maslakov, R. Y. Novotortsev, S. V. Savilov, H. Xia, A. V. Desyatov and S. M. J. I. Aldoshin, Ionics, 2022, 28, 3501–3509 CrossRef CAS.
- M. Mahmood, A. Rasheed, I. Ayman, T. Rasheed, S. Munir, S. Ajmal, P. O. Agboola, M. F. Warsi and M. Shahid, Energy Fuels, 2021, 35, 3469–3478 CrossRef CAS.
- A. Xia, J. Li, X. Zeng, J. Chen and G. Tan, Ceram. Int., 2024, 50, 13061–13069 CrossRef CAS.
- L. Zhang, X. Li, L. Li, X. Cheng, H. Wu and J. Zheng, J. Energy Storage, 2024, 85, 111136 CrossRef.
- X. Zhang, F. Zhang, D. Wei, Z. Cai, Y. Song and X. Wang, J. Energy Storage, 2023, 72, 108468 CrossRef.
- H. Bai, S. Liang, T. Wei, Q. Zhou, M. Shi, Z. Jiang, J. Feng, M. Zhang and Z. Fan, J. Power Sources, 2022, 523, 231032 CrossRef CAS.
- H. Peçenek, S. Yetiman, F. K. Dokan, M. S. Onses, E. Yılmaz and E. Sahmetlioglu, Ceram. Int., 2022, 48, 7253–7260 CrossRef.
- Y. Liang, D. Zhu, S. Chao, M. Hu, D. Li, W. Zhou, J. Xu, X. Duan and P. Liu, J. Energy Storage, 2023, 60, 106673 CrossRef.
- J. Wu, W. Raza, P. Wang, A. Hussain, Y. Ding, J. Yu, Y. Wu and J. Zhao, Electrochim. Acta, 2022, 418, 140339 CrossRef CAS.
- Y. Feng, H. Qu, Y. Wang, L. Wang, Y. Wang, D. Yang, B. Ding, Y. Sun, J. Guo and S. J. M. Dai, Materials, 2024, 17, 1312 CrossRef CAS PubMed.
- V. Mane, S. Kale, S. Ubale, V. Lokhande and C. J. M. T. C. Lokhande, Mater. Today Chem., 2021, 20, 100473 CrossRef CAS.
- S. Wu, X. Yang, H. Huang, Z. Shen, Y. Xue, H. Yang, L. Wang, F. Xu, X. Wang and W. Ge, Jpn. J. Appl. Phys., 2022, 61, 090901 CrossRef CAS.
- J. M. d O. Cremonezzi, D. Y. Tiba and S. H. J. S. A. S. Domingues, SN Appl. Sci., 2020, 2, 1–9 Search PubMed.
- M. B. Pramanik, M. A. Al Rakib, M. A. Siddik, S. Bhuiyan and T. Research, Eur. J. Eng. Technol. Res., 2024, 9, 10–15 CrossRef.
- S. Yousaf, K. M. Katubi, S. Zulfiqar, M. F. Warsi, Z. A. Alrowaili, M. S. Al-Buriahi and I. Ahmad, Int. J. Hydrogen Energy, 2023, 48, 27201–27214 CrossRef CAS.
- S. Yousaf, S. Zulfiqar, H. H. Somaily, M. F. Warsi, A. Rasheed, M. Shahid and I. Ahmad, RSC Adv., 2022, 12, 23454–23465 RSC.
- J. Jia, W. Yang, P. Zhang and J. Zhang, Appl. Catal., A, 2017, 546, 79–86 CrossRef CAS.
- J. Ma, C. Wang and H. He, Appl. Catal., B, 2017, 201, 503–510 CrossRef CAS.
-
W. Du, X. Zhang, L. Wu, Z. Wang, W. Liu, J. Liu and F. Wang, in Metal Oxide Defects, ed. V. Kumar, S. Som, V. Sharma and H. C. Swart, Elsevier, 2023, pp. 491–531 DOI:10.1016/B978-0-323-85588-4.00021-0.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sonia
Zulfiqar
a,
Sonia
Zulfiqar
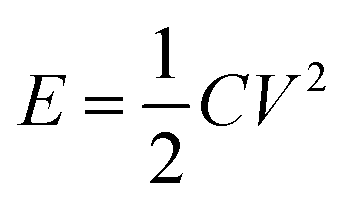 . So, the energy density could be improved either by increasing specific capacitance or by extending the voltage window. Recent research in the domain of supercapacitors has witnessed substantial advances in the use of transition metal oxides and sulfides as electrode materials. Studies by Vilhal et al., Prabhu et al., Jiang et al., Sun et al., Pappu et al., and Van et al. have shown that NiO, CuO, and Cu doped ceria, manganese oxide/carbon composite cobalt manganese oxide, and Ni–Co binary sulfides have potential electrochemical characteristics for improving energy storage performance in supercapacitors.12–17 Furthermore, comprehensive studies by Melkiyur et al. and Imran et al. have summarized the existing state-of-the-art and future prospects of transition metal oxides/sulfides and manganese oxide/sulfide in supercapacitor technology respectively.18,19
. So, the energy density could be improved either by increasing specific capacitance or by extending the voltage window. Recent research in the domain of supercapacitors has witnessed substantial advances in the use of transition metal oxides and sulfides as electrode materials. Studies by Vilhal et al., Prabhu et al., Jiang et al., Sun et al., Pappu et al., and Van et al. have shown that NiO, CuO, and Cu doped ceria, manganese oxide/carbon composite cobalt manganese oxide, and Ni–Co binary sulfides have potential electrochemical characteristics for improving energy storage performance in supercapacitors.12–17 Furthermore, comprehensive studies by Melkiyur et al. and Imran et al. have summarized the existing state-of-the-art and future prospects of transition metal oxides/sulfides and manganese oxide/sulfide in supercapacitor technology respectively.18,19