
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBODIPY-based regioisomers and a donor–acceptor rotor as organic photosensitizers for maximizing singlet oxygen quantum yields and for the photooxidation of thioanisole†
Sushil
Sharma
,
Sakshi
Chawla
,
Vidushi
Gupta
,
Arijit K.
De
 * and
Sanchita
Sengupta
*
* and
Sanchita
Sengupta
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Knowledge City, Sector 81, P.O. Manauli, Mohali, Punjab 140306, India. E-mail: sanchita@iisermohali.ac.in; akde@iisermohali.ac.in
First published on 20th February 2024
Abstract
The efficient production of singlet oxygen using organic photosensitizers (PSs) is highly attractive for photocatalytic applications. Herein, we designed and synthesized regioisomeric biphenyl-BODIPYs such as pp-BODIPY, mp-BODIPY, mm-BODIPY and T-BODIPY (thiophene at the meso-position) and a triad T-ADA (based on benzodithiophene as the donor and BODIPY as the acceptor) as organic PSs. Singlet oxygen quantum yields reached up to 77% in T-BODIPY due to the heavy atom effect (bromine substitution in the thiophene ring) and charge transfer (CT)-mediated intersystem crossing (ISC) and ∼35% in T-ADA due to CT-mediated ISC. The variation in connectivities of spacers in regioisomeric BODIPYs and the type of spacer in T-BODIPY significantly altered the photophysical properties. Among all the PSs, T-ADA showed a charge transfer band at ∼650 nm. Detailed insights into the ultrafast dynamics and excited state processes were obtained using femtosecond transient absorption spectroscopy (fs-TAS). The technique allowed for a thorough investigation of these systems, indicating the participation of charge-transfer (CT) states in the observed dynamics of triplet state formation. Owing to the efficient generation of 1O2, T-BODIPY and T-ADA as well as regioisomers pp-BODIPY, mp-BODIPY and mm-BODIPY were utilized in the aerobic photooxidation of thioanisole to methyl phenyl sulfoxide with high selectivity towards sulfoxide formation.
Introduction
The field of photocatalysis where organic transformations are performed under mild conditions using light activable catalysts has attracted great attention among synthetic chemists in the last few decades.1–4 Photocatalysis offers a powerful strategy to activate organic molecules by transferring electrons or energy between an excited photocatalyst and substrate that leads to the formation of new chemical bonds or breakage of old bonds. There are several reports on transition metal-based photocatalysts containing Ru, Ir, Pt and Pd that have demonstrated their effectiveness in a variety of reactions.5–8 However, these photocatalysts suffer from limitations, such as high cost, toxicity, and difficulty in their disposal, which have directed attention towards the development of earth-abundant, non-toxic, metal-free, sustainable photocatalysts.9 In recent years, there have been various reports on the synthesis of metal-free photocatalysts and their utilization in organic transformations such as oxidation, cycloaddition, C–O bond formation, C–C bond formation, and C–S bond formation.1,2 Generally, highly efficient triplet photosensitizers (energy transfer photocatalysis) are based on fast intersystem crossing (ISC) occurring from singlet to triplet states and are preferred for organic photocatalysis.10 Among the various organic photocatalysts, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY)-based organic photocatalysts have been employed in recent years because of their outstanding thermal and photochemical stabilities, strong visible light absorption, high fluorescence quantum yields, good solubility and facile accessibility of their triplet excited states.11–14 Moreover, few chemical modifications in the skeleton of BODIPY, such as substitution by heavy atoms (i.e., bromine and iodine), enhance spin–orbit coupling, resulting in the increase in the population of triplet states.12 Furthermore, BODIPYs have been widely investigated for a variety of applications, such as photodynamic therapy, chemosensing, biological labelling, organic photocatalysis, dye-sensitized solar cells and laser dyes.12,15 In iodo-BODIPY derivative-based photocatalysts, the I atom attached on the π-core of the BODIPY chromophore drives efficient ISC, resulting in high yields of singlet oxygen (1O2).16 These photocatalysts were utilized for the singlet oxygen-mediated aerobic oxidative coupling of amines and the photooxidation of dihydroxynaphthalene, followed by aniline addition to a naphthoquinone intermediate. The visible light-driven hydroxylation of aryl halides in water was achieved by the utilization of a BODIPY photocatalyst and Ni as a co-catalyst in the presence of a base.17 An iodo-BODIPY and quinine-based photocatalyst, where iodo-BODIPY serves as the 1O2 producer and quinine serves as both chiral catalyst and quencher of 1O2 in the absence of a substrate, was synthesized and utilized for the asymmetric hydroxylation of β-dicarbonyl compounds.18 Recently, porous organic polymers containing halogenated BODIPY were reported for the singlet oxygen-mediated photo-degradation of 2-chloroethyl ethyl sulfide (chemical warfare agent) to its corresponding non-toxic 2-chloroethyl ethyl sulfoxide.13 Conversion of 1,5-dihydroxynaphthalene to juglone was achieved using BODIPY and a copper-based photocatalyst.19 It was reported that substitution with I at the meso-position results in a lower singlet oxygen quantum yield (0.01) compared to the 2- and 6-positions (0.83) during the oxidation of sulphides.12,20,21 All these reports achieved photocatalyzed organic transformations by introducing heavy atoms (Br or I) directly into the BODIPY skeleton. Filatov et al. reported on heavy-atom free BODIPY and anthracene-based dyads as triplet photosensitizers, in which the triplet state was generated by photoinduced electron transfer.22 An anthracene-based dyad was converted into a highly fluorescent species using 1O2, and was further utilized for imaging applications. Donor–acceptor (D–A) systems based on phenothiazine as the donor and BODIPY as the acceptor were synthesized, and triplet excited state formation was achieved by photoinduced electron transfer and charge recombination.23 In the D–A system, charge transfer (CT) occurs from the donor to acceptor upon photoexcitation, resulting in the formation of a singlet excited CT state (1CT). In the absence of heavy atoms, 1CT states can undergo efficient ISC by CT-mediated ISC involving either radical pair ISC (RP-ISC), or by spin–orbit charge transfer ISC (SOCT-ISC).10,12,14In this work, we have synthesized the biphenyl-BODIPY regioisomers pp-BODIPY, mp-BODIPY and mm-BODIPY with Br attached at different positions of the biphenyl, and thiophene BODIPY (T-BODIPY) where a thiophene spacer functionalized with Br is attached at the meso-position of BODIPY (Fig. 1). We have studied the effect of the regioisomerism and spacer on the photophysical properties, as well as on photocatalysis. Compound T-BODIPY showed a red shift in absorption and in emission compared to the other biphenyl-BODIPY regioisomers due to the attachment of thiophene at the meso-position. Furthermore, we have synthesized a heavy atom-free T-ADA rotor that consists of a T-BODIPY acceptor and benzodithiophene (BDT) as the donor, and the BDT was connected through a thiophene spacer at the meso-position of T-BODIPY. Previously, we reported the synthesis of tetramethyl-substituted BODIPY and unsubstituted BODIPY, along with their triads (A–D–A). We observed that due to the hindered rotation of tetramethyl-substituted BODIPY, the triad was twisted and intramolecular charge transfer (TICT)-was inactive.24 However, it exhibited energy transfer from the benzodithiophene (BDT) donor to the BODIPY acceptor. Therefore, in this work, we have replaced two of the methyl groups with hydrogen and a new spacer to investigate the effects on the charge transfer properties and triplet state population. Rotor T-ADA showed a pronounced charge transfer (CT) emission band at 650 nm.
 | ||
| Fig. 1 Chemical structures of pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA synthesized and investigated in this study. | ||
Additionally, T-ADA showed efficient 1O2 generation that was most likely due to CT from BDT to BODIPY-mediated ISC, which populated the triplet state. Due to the generation of 1O2 for T-BODIPY and T-ADA, these compounds as well as the three regioisomers were efficiently utilized in the aerobic photooxidation of thioanisole to methyl phenyl sulfoxide.
The alteration of the spacer from biphenyl to thiophene and the introduction of a heavy atom in BODIPYs lead to significant changes in the photophysical characterization, manifesting in remarkably high singlet oxygen quantum yields of these molecules. The observed outcomes are attributed to the structural modifications at the meso-position of BODIPY.
Results and discussion
Synthesis
The syntheses of the BODIPYs pp-BODIPY, new regioisomers mp-BODIPY, mm-BODIPY, and T-BODIPY, and triad T-ADA were accomplished following our earlier reported procedures,24 as shown in Schemes S1–S9 in the ESI.† Before the synthesis of BODIPYs, the spacer biphenyl was synthesized by the Suzuki coupling reaction between the suitably substituted halogenated benzene and formyl phenyl boronic acid (Schemes S1–S3, ESI†).25,26 Subsequently, the regioisomeric BODIPYs were synthesized by the reaction between different spacers (bromo biphenyl carbaldehyde) and 2,4-dimethyl pyrrole in a catalytic amount of trifluoroacetic acid (TFA), followed by subsequent addition of chloranil, triethylamine, and boron trifluoride diethyl etherate (Schemes S4–S6, ESI†).27 Meanwhile, for the synthesis of T-BODIPY, 2-methyl pyrrole was synthesized by Wolff–Kishner reduction of pyrrole-2-carboxaldehyde, and then it was used for the synthesis of T-BODIPY using 5-bromo-2-thiophenecarboxaldehyde, as shown in Scheme S8 in ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 The synthesis of the new triad T-ADA was accomplished by Stille coupling reaction between T-BODIPY and distannylated benzodithiophene precursor (BDT), as shown Scheme S9 in ESI.†24 All compounds were purified via column chromatography (TLC), and characterized using 1H NMR, 13C NMR and high-resolution mass spectrometry (HRMS).
28 The synthesis of the new triad T-ADA was accomplished by Stille coupling reaction between T-BODIPY and distannylated benzodithiophene precursor (BDT), as shown Scheme S9 in ESI.†24 All compounds were purified via column chromatography (TLC), and characterized using 1H NMR, 13C NMR and high-resolution mass spectrometry (HRMS).
Absorption and emission
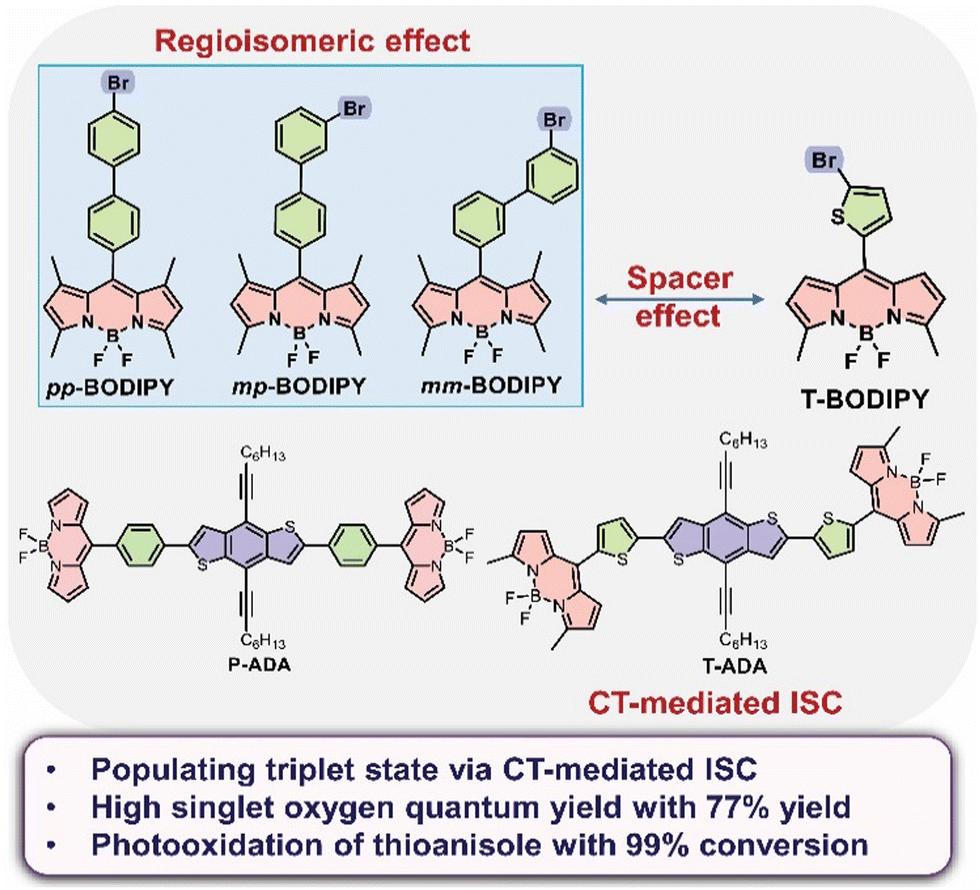
UV/vis absorption and emission spectra of pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA were recorded in CHCl3 at a concentration of ∼10−6 M. In the case of pp-BODIPY, mp-BODIPY and mm-BODIPY, the major absorption peak was observed at 504 nm, corresponding to the S0–S1 transition. Upon excitation at 504 nm, the emission peak was observed at 513 nm. Meanwhile, in the case of T-BODIPY, apart from a minor absorption peak at 390 nm, the major absorption peak was observed at 527 nm (Fig. 2a). Upon excitation at 527 nm, the emission was observed at 548 nm, as shown in Fig. 2b. Due to the thiophene spacer in T-BODIPY, the emission was red shifted by 35 nm in comparison to regioisomeric biphenyl-BODIPYs. A similar shift due to the thiophene spacer was suggested earlier in the literature.29–31 In the case of T-ADA, apart from minor absorption peaks at 327 nm and 395 nm, the major absorption band was observed at 529 nm. The observed smaller peak at 327 nm is attributed to the BDT part, while the bands at 398 nm and 529 nm are attributed to the BODIPY part in T-ADA. Upon excitation at 327 nm, apart from emission at 407 nm and 430 nm, a highly intense peak was observed at 650 nm, as shown in Fig. 2b. Upon excitation at 529 nm, the peak at 650 nm was obtained, while the BODIPY emission disappeared (Fig. S1, ESI†). Compared to our earlier reported rotor p-ADA that contained a phenyl spacer,32T-ADA contains a thiophene spacer and it showed significantly altered photophysics compared to p-ADA. Upon excitation at 380 nm for p-ADA, apart from the emission at 405 nm (BDT donor) and ∼512 nm (acceptor), a broad and intense peak at ∼698 nm (twisted intramolecular charge transfer) was observed.32 However, in the case of T-ADA, emission bands were observed only at 407 nm and 430 nm (BDT donor) and at 650 nm, and no separate emission of BODIPY (or local excited (LE) emission) was observed. Thus, for T-ADA, it can be presumed that the energy barrier between LE and CT is very low, such that the population immediately reaches the CT state from the LE state upon photoexcitation. The molar absorption coefficients (ε) of pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA were 6.62 × 104 M−1 cm−1, 5.96 × 104 M−1 cm−1, 7.67 × 104 M−1 cm−1, 6 × 104 M−1 cm−1 and 1.28 × 105 M−1 cm−1, respectively. The fluorescence quantum yields were determined by relative method using Rhodamine B and 4′,6-diamidino-2-phenylindole (DAPI) as reference compounds, and the fluorescence quantum yields were obtained as 0.70, 0.65, 0.74, 0.14 and 0.19 for pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA, respectively (Table S1, ESI†). In the case of regioisomeric BODIPYs (such as pp-BODIPY, mp-BODIPY, mm-BODIPY), the tetramethyl substitution at BODIPY hindered the rotations, which leads to the enhancement of fluorescence quantum yields. However, in the case of T-BODIPY and T-ADA, due to the possibility of ISC and non-radiative deactivation, the fluorescence quantum yields were lower compared to regioisomeric BODIPYs.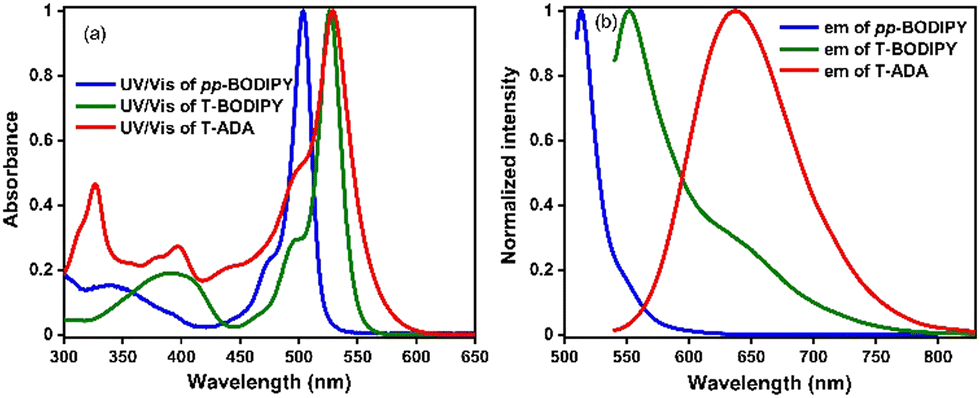 | ||
| Fig. 2 (a) UV/vis absorption and (b) emission spectra of pp-BODIPY (λex = 504 nm), T-BODIPY (λex = 527 nm) and T-ADA (λex = 529 nm). | ||
Solvatochromism of T-ADA
Among molecules pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA, a longer wavelength emission band at ∼650 nm was observed for T-ADA that can be attributed to charge transfer (CT). In the case of CT, due to the change in the dipole moment of the excited state, the molecule shows positive solvatochromic shifts in the emission spectra.33 Hence, a solvatochromism study was performed in solvents of different polarities for T-ADA (Fig. 3). In non-polar solvents such as methyl cyclohexane (MCH) and toluene (Tol), the emission of T-ADA was observed at 569 nm and 594 nm, respectively (Fig. 3a). Meanwhile, in polar solvents such as dichloromethane (DCM), tetrahydrofuran (THF) and chloroform (CHCl3), T-ADA showed emission bands at 662 nm, 633 nm, and 638 nm, respectively. Hence, the bathochromic shift in emission from 569 nm (MCH) to 662 nm (DCM) for T-ADA confirmed the occurrence of the CT state. Furthermore, a solvatochromic study was performed for T-ADA in a binary mixture of polar and nonpolar (DCM/Tol) solvents (Fig. 3b). As the percentage of DCM was increased from 0% to 100% (v/v), the emission was bathochromically shifted from 594 nm to 660 nm with a bathochromic shift of 66 nm, as shown in Fig. 3. Hence, an increase in the solvent polarities resulted in a bathochromic shift in the emission due to the higher dipole moment in the excited state being preferentially stabilized by polar solvents, thereby confirming a CT state. Also, T-ADA showed aggregation-induced emission (AIE) with an increase in the water percentage in the tetrahydrofuran (THF) solution (Fig. S2, ESI†). Additionally, the rotor T-ADA showed a decrease in emission intensity by ∼4-fold with a hypsochromic shift of ∼23 nm with an increase in the temperature, as shown in Fig. S3 (ESI†).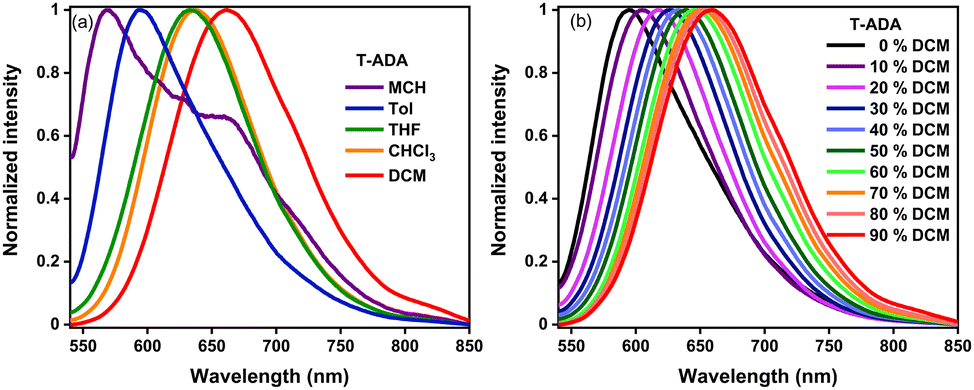 | ||
| Fig. 3 Emission of T-ADA (λex = 529 nm) (a) in solvents of different polarities and (b) in different percentages of DCM/Tol (c = ∼10−6 M) (inside the graph, 0–100% indicate the percentage of DCM in solutions). | ||
Cyclic voltammetry and spectroelectrochemistry
In order to assess the redox properties of pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA, cyclic voltammetry (CV) measurements were performed in deoxygenated DCM containing (0.1 M) tetrabutylammonium hexafluorophosphate (TBAHPF) as a supporting electrolyte (Fig. S4 and Table S2 in ESI†).The internal calibration was performed using a ferrocene/ferrocenium redox couple prior to all measurements. All of the regioisomeric BODIPYs showed reduction potentials of ∼−0.99 V to ∼−0.98 V and oxidation potentials at ∼1.35 V to ∼1.43 V, indicating the ease of reduction due to the boron attached to the core. Similarly, T-BODIPY and T-ADA showed reduction potentials at −0.94 V and −0.96 V, respectively, suggesting a facile reduction facilitated by the donor attached to the chromophores. Based on the first oxidation potential onsets (Eonsetox) and first reduction potential onsets (Eonsetred), the HOMO and LUMO levels were calculated as shown in ESI.† Accordingly, the calculated HOMO energies for pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA were −5.93 eV, −5.95 eV, −5.88 eV, −6.01 eV and −5.74 eV, respectively, and LUMO energies were −4.06 eV, −4.11 eV, −4.22 eV, −3.98 eV and −4.12 eV, respectively.
In order to understand the spectral profiles of radical anions or cations of T-BODIPY and T-ADA, spectroelectrochemical measurements were performed in DCM containing 0.1 M TBAHPF as a supporting electrolyte, as shown in Fig. S5 in ESI.† Upon applying a positive potential (0 to +2 V) for T-BODIPY, a shoulder was observed in the UV/vis absorption spectrum at ∼600 nm due to the formation of a radical cation, as shown in Fig. S5 (ESI†), while no significant spectral change was observed in T-ADA upon applying a positive potential. Upon application of a negative potential for T-BODIPY (0 to −2 V), the depletion of the absorption bands at ∼390 nm and 527 nm and the emergence of a new absorption band at ∼680 nm were observed due to the formation of the monoanion and dianion radical. Similarly, in T-ADA, the depletion of the absorption bands at ∼327 nm and 529 nm and the emergence of an absorption band at ∼398 nm were observed due to the formation of an anion radical, as shown in Fig. S5 (ESI†). The corresponding changes in color while applying the positive potential and negative potentials for both compounds can be observed in the inset of Fig. S5 (ESI†). Based on spectroelectrochemistry, both compounds are considered to be electrochromic.
Density functional theory calculations
Density functional theory (DFT) calculations were performed for all molecules in the ground state using the Gaussian 09 package at the B3LYP/6-31G(d,p) level to calculate the HOMO and LUMO energy levels, as well as torsion angles, in geometry optimized structures (Fig. S6–S9 and Tables S3–S7, ESI†). The frontier molecular orbital (FMO) composition analysis revealed no possibility of charge transfer from the biphenyl part to the BODIPY part in regioisomeric BODIPYs through HOMO to LUMO transitions. However, for T-BODIPY, FMO composition analysis suggested a lower likelihood of charge transfer from the thiophene part to the BODIPY part. For T-ADA, a higher possibility of charge transfer from the BDT part to the BODIPY part was observed (Table S7, ESI†). The feasibility of charge transfer was further calculated for T-ADA using the Rehm–Weller equation, and the negative values of the photoinduced charge separation and charge recombination indicate that both photoinduced forward and backward electron transfer processes are exergonic and thermodynamically feasible, as shown in ESI† (Section 10). Time-dependent DFT (TD-DFT) calculations of T-BODIPY and T-ADA were performed to calculate the singlet and triplet energy levels, and their energies are provided in Table S4 (ESI†). Natural transition orbital (NTO) calculations were performed for the first four excited states of T-BODIPY and T-ADA. Both compounds T-BODIPY and T-ADA showed charge transfer features in the S1 state where electrons and holes are completely separated on different parts of the molecules (i.e., on acceptor and donor, respectively), as shown in Fig. S7–S8 in ESI.† The calculated dipole moments and oscillator strengths of various singlet states are provided in Table S6 in ESI.† The larger oscillator strengths of the first singlet excited states for both compounds (S1) are indicative of larger molar absorptivities (strong absorption). Also, the dipole moments of the S1 states of T-BODIPY and T-ADA were 9.76 D and 16.6 D, respectively. The large dipole moments are attributed to the CT character of these states. The above results indicate that the S1 states for both molecules have CT character.Furthermore, the calculated singlet and triplet energy gaps of T-BODIPY (between the S1 and T3 states) and T-ADA (between the S1 and T4 states) were 0.02 eV and 0.10 eV, respectively, which is well-suited for an efficient ISC process. Based on these calculated energy levels, ISC could therefore occur from S1 to T3 in the case of T-BODIPY, and from S1 to T4 in the case of T-ADA. The NTO transitions of triplet states T3 and T4 of T-BODIPY and T-ADA, respectively, showed the possibility of CT character, as shown in Fig. S8–S9 (ESI†). The singlet and triplet energies were also calculated in the polarization continuum model (PCM) model using CHCl3 and heptane as solvents. In the case of CHCl3, the energy levels were slightly stabilized for T-ADA compared to that in heptane, which indicates that the states under consideration involve CT characters, as provided in Table S5 (ESI†). Hence, all results based on TD-DFT calculations are indicative of the fact that the S1 state is most likely involved in the ISC process through a CT-mediated ISC and populates the triplet state, which further assists in the generation of singlet oxygen (vide infra singlet oxygen quantum yields).
Fluorescence lifetime
The fluorescence lifetimes of pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA were calculated in CHCl3, as well as in Tol, using the time-correlated single photon counting (TCSPC) technique. The fluorescence lifetimes of all compounds were measured upon excitation by a 375 nm laser diode (Fig. 4 and S10, Table S8, ESI†). All the data were fitted by bi-exponential functions, and the τavg values of regioisomeric BODIPYs pp-BODIPY, mp-BODIPY and mm-BODIPY at the emission wavelength of 513 nm were 3.1 ns, 2.9 ns and 4.4 ns in CHCl3 and 2.8 ns, 2.7 ns and 4.1 ns in Tol, respectively. The τavg values of T-BODIPY at the emission wavelength of 554 nm were 0.44 ns in CHCl3 and 0.45 ns in Tol. The τavg value of T-ADA at the emission wavelength of 650 nm was ∼0.45 ns in CHCl3. | ||
| Fig. 4 Collected fluorescence lifetime decays of (a) T-BODIPY and (b) T-ADA using TCSPC in CHCl3 and Tol. | ||
Singlet oxygen quantum yields
To confirm the ability of pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA to generate 1O2, singlet oxygen quantum yields (SOQYs) (ΦΔ) were calculated by relative method in THF and MeOH as a solvent. The singlet oxygen quantum yield (ΦΔ) was determined by monitoring the photooxidation of 1,3-diphenylisobenzofuran (DPBF), which is a well-known 1O2 scavenger that rapidly gives a colourless oxidation product.34,35 It absorbs in the visible region (∼410 nm) and due to the scavenging of 1O2, the absorbance decreases with time. Singlet oxygen quantum yields were calculated at low concentrations (5 μM of dyes and 50 μM of DPBF) to minimize the possibility of quenching of 1O2 by dyes. A solution of dyes and DPBF in THF was irradiated with monochromatic light at specific wavelengths (ESI†), and the change in absorbance of DPBF was monitored with time through recording the absorbance of the solution at different time intervals, as shown in Fig. S11–S14 in ESI.† The SOQYs were calculated by plotting the change in absorbance at the initial time and at variable times against the irradiation time, as shown in ESI† (Section 11). The calculated ΦΔ values for pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA were 5.7%, 1.9%, 12.2%, 77% and 35% in THF and 0.4%, 0.3%, 0.5%, 17% and 6.4% in MeOH, respectively (Table S9, ESI†). In the case of the regioisomeric BODIPYs pp-BODIPY, mp-BODIPY and mm-BODIPY, the singlet oxygen quantum yields are significantly lower compared to those of T-BODIPY and T-ADA (Table S9, ESI†). This reduction in the singlet oxygen quantum yield is attributed to the inefficient ISC, which is a result of the bromine atom being not directly attached to the BODIPY core. In the case of T-BODIPY, Br is linked to the meso-position of BODIPY with a thiophene ring as a spacer, which minimizes the distance between bromine and the spin density surface of BODIPY, as compared to regioisomeric biphenyl-BODIPYs. Hence, a higher SOQY was observed due to the population of the triplet state by the higher rate of ISC and possibility of CT-mediated ISC. In T-ADA, however, there is no heavy atom attached to the core. Therefore, ISC can occur by CT-mediated intersystem crossing, involving either RP-ISC or SOCT-ISC. As discussed before, the small energy gaps between the singlet and triplet energy levels are suggestive of the presence of a strong spin–orbit coupling, leading to the generation of singlet oxygen. To further elucidate the specific mechanism, we conducted steady state EPR studies in the presence and absence of 5,5-dimethyl-1-pyrroline 1-oxide (DMPO). We did not find any signal corresponding to the radical species. Therefore, it is likely that an SOCT-type mechanism is dominant over an RP-ISC mechanism. However, the involvement of the RP-ISC pathway is not completely ruled out as the radical species, generated in situ in the presence of light, are short-lived. Time-resolved EPR measurements are necessary to confirm the absence of the RP-ISC pathway.Transient absorption studies
Furthermore, to elucidate the underlying mechanism, we meticulously examined the dynamics of the excited states using femtosecond transient absorption spectroscopy (fs-TAS). Details of the experimental setup can be found elsewhere.36,37 A pump pulse centered at 400 nm was chosen to photoexcite the sample, followed by a broadband white light probe pulse with a varying time delay. This delay allows us to measure the pump-induced changes in absorbance (ΔOD) in the visible wavelength range, providing information about the properties of the excited state. For transient absorption spectra, negative ΔOD represents ground state bleaching (GSB)/stimulated emission (SE), and positive ΔOD represents excited state absorption (ESA). The two-dimensional fs-TAS contour plots are shown in Fig. 5a. Spectral traces at different pump–probe delays for pp-BODIPY in Tol are shown in Fig. 5b. Excitation at 400 nm populates the higher lying S2 state of pp-BODIPY, which undergoes fast internal conversion (IC) to the S1 state within 500 fs.38 The TA spectra of pp-BODIPY show an intense negative signal at 500 nm, which matches well with the S1 ← S0 absorption maximum (Fig. 5b), and is assigned to GSB. A less intense negative signal at 520–600 nm appears in the region of the steady state fluorescence emission (Fig. 2b) and is attributed to SE, which spectrally overlaps with the GSB signal. Both of these characteristic features exhibit a slow gradual decay over a nanosecond timescale, a consequence of the radiative decay of the singlet excited state. A positive absorption ΔA signal rising within the pulse width was observed, which spans from 420 nm to 460 nm. This ESA band is plausibly ascribed to the photoexcited 1pp-BODIPY* state, representing singlet–singlet absorption, rather than arising from triplet excited-states (triplet–triplet absorption). This rationale finds support from the high quantum yield of the singlet emission from pp-BODIPY (Φf ∼ 74%, as reported in Table S1 in the ESI†) and the negligible formation of the singlet oxygen (1O2) quantum yield. | ||
| Fig. 5 (a) A 2D contour plot for fs-TA of pp-BODIPY in Tol following pump excitation at 400 nm. (b) Spectral traces at the indicated probe delay times. (c) Evolution-associated difference spectra (EADS) reconstructed via singular value decomposition (SVD) and global fitting of A. The transient data used sequential models A to B to GS (ground state), where A is 1pp-BODIPY* and B is relaxed 1pp-BODIPY. (d) Population dynamics for species A and B. (e) Kinetic traces along with corresponding global fits at indicated probe wavelengths. (f) Kinetic model employed to perform global analysis. | ||
Furthermore, to extricate the timescales of ultrafast relaxation processes, a global analysis of the fs-TAS data was carried out in which the transients at all detection wavelengths were analysed simultaneously with a single set of exponentials (i.e., the principal components) using Glotaran programming.39,40 Details of the analysis procedure may be found in section 12 in ESI.† For pp-BODIPY, a two-step consecutive sequential kinetic model (Fig. 5f), A → B → GS, is employed to obtain the evolution-associated difference spectra (EADS) and associated time constants related to the different excited state processes. The EADS thus obtained for pp-BODIPY in Tol are shown in Fig. 5c. The first EADS (Fig. 5c) corresponds to the initially populated, vibrationally hot 1pp-BODIPY, which relaxes into the local excited state 1pp-BODIPY (1LE) with a timescale of 46.9 ps (τ1) to give rise to the second EADS. This second EADS subsequently decays to the ground state with a timescale of 2.3 ns (τ2), which closely matches with the fluorescence lifetime studies from the TCSPC measurements (Table S8, ESI†). Kinetic traces along with the fit at different probe wavelengths are shown in Fig. 5e. Relative population profiles of the excited state fitted by kinetic models are represented in Fig. 5f. The first component matches with (within an order of magnitude) the previously reported timescale associated with the BODIPY-core structural changes in the excited state, and was attributed to vibrational relaxation41 or/and conformational evolution42 in the excited state. Additional measurements were performed in the more polar solvent THF (Fig. S16, Section S12, ESI†). It should be noted that the solvent polarity does not have a significant impact on the spectral position, and the shape of the transient absorption spectra looks very similar. Results of the global analysis in THF are shown in Fig. S16 (ESI†). The EADS are found to be very similar to those obtained in Tol, although there are small differences in the extracted kinetic parameters. The first-time component, τ1, is comparatively faster in THF (25.8 ps) compared to that in Tol (46.9 ps), as shown in Table 1.
| Compound | Solvent | Dielectric constant | τ 1 (ps) | τ 2 (ps) | τ 3 (ns) | τ 4* (ns) | τ 5* (ns) | RMSE |
|---|---|---|---|---|---|---|---|---|
| pp-BODIPY | Tol | 2.38 | 46.9 | — | — | — | 2.9 | 0.0003 |
| THF | 7.6 | 25.8 | — | — | — | 2.8 | 0.0002 | |
| T-BODIPY | Tol | 2.38 | 4.02 | 235.1 | 7.5 | 0.3 | 4.4 | 0.0002 |
| THF | 7.6 | 3.54 | 218.8 | 5.9 | 0.3 | 4.7 | 0.0004 | |
| T-ADA | Tol | 2.38 | 6.32 | 147.1 | 5.1 | — | 0.61 | 0.0005 |
| THF | 7.6 | 1.73 | 146.1 | 3.2 | — | 0.64 | 0.0003 |
In the T-BODIPY and T-ADA compounds, the BODIPY part, when combined with a bromo thiophene unit and BDT unit respectively, acts as an electron accepting component. A significant reduction in the fluorescence quantum yields was observed in these compounds (around 85% in T-BODIPY and 73% in T-ADA) compared to the regioisomeric BODIPYs. This reduction indicates that the separation of charges competes with the emission from the excited state of BODIPY. Moreover, the lifetime of the BODIPY component is found to decrease in both T-BODIPY and T-ADA structures (Table S8 in ESI†). In the case of T-BODIPY, the lifetime becomes biexponential, indicating the presence of two emissive states: the locally excited (LE) state and the CT state.43 However, in T-ADA, the emission from the LE state emission is quenched, and only emission from the 1CT state is observed (Fig. 2b). The presence of a significant population of triplets suggests that the charge separation process facilitates the generation of the triplet state, contributing to the high singlet oxygen quantum yield observed in both T-BODIPY and T-ADA compounds. This phenomenon underscores the importance of understanding the role of charge separation in the overall photophysical properties and potential applications of these compounds. The two-dimensional fs-TAS contour plot for T-BODIPY in Tol is shown in Fig. 6a. The fs-TA spectra at various probe delays are shown in Fig. 6b. Clearly, at a very short delay (within 1 ps), the transient absorption spectra show an ESA band centred on 440 nm, a strong GSB band on 520 nm, and an SE band spanning from 550 to 650 nm. All these signals are in line with the initial population of the locally excited singlet state of the BODIPY moiety 1(T-BODIPY*). After 1 ps, the SE signal vanishes with the concomitant appearance of a new ESA band in the 555 nm to 645 nm range with the maximum at 580 nm. This band corresponds to the formation of the BODIPY˙− anion (which is the reduced BODIPY moiety). Additionally, the appearance of a negative signal (SE) in the region of 650 nm to 750 nm corresponds to the formation of the singlet CT state 1(T˙+-BODIPY˙−), which is spectrally overlapped with the ESA band of BODIPY˙−. Notably, one could also observe the blue shift in ESA from 440 nm (originating from the local excited state) to 425 nm, which might be due to triplet–triplet (Tn ← T1) absorption in 3BODIPY. A clear distinction between the two can be clearly seen in the kinetics plotted at two different wavelengths, 425 nm and 440 nm, shown in Fig. 6e. Now, in order to reconstruct the spectra-associated difference spectra (SADS) associated with the individual species, we modeled the kinetics using a target analysis of the fs-TAS matrix by assuming a kinetic model, as shown in Fig. 6f. It should be noted that two components from the fluorescence obtained from TCSPC are fixed to generate the SADS spectrum (τ4 and τ5). The first SADS corresponds to the 1T-BODIPY* state, which decays in τ1 = 4 ps, as a second transient species is formed after charge transfer, 1(T˙+-BODIPY˙−) or 1CT. This is shown by the second SADS, which is characterized by a sharp absorption at 580 nm that grows in over the same timescale and decays with τ2 = 235.1 ps (Fig. 6c). This 1CT undergoes CT-mediated ISC to generate another new species 3(T˙+-BODIPY˙−)/3CT, which finally undergoes charge recombination (CR) with time constant τ3 = 7.5 ns via internal conversion (IC) to form lower lying (T-3BODIPY*), where the triplet is localized over the BODIPY unit and responsible for the formation of singlet oxygen. Kinetic traces at selected wavelengths along with the fits from target analysis are shown in Fig. 6e. Population profiles of different species involved in T-BODIPY are shown in Fig. 6d. Furthermore, to track the effects of the solvent polarity on the ISC rate, additional studies were carried out in THF. The time constants obtained from the target analysis of the TA data are tabulated in Table 1. As already mentioned earlier, for these systems, the IC from S2 is fast enough and therefore not taken into account while fitting the data into the target model. The corresponding SADS and population dynamics for T-BODIPY in THF are shown in Fig. S17 in ESI.† As the solvent polarity shifts from Tol to THF, it becomes evident that the rate of the 1CT state formation remains relatively consistent. However, the acceleration of the ISC rate is notably pronounced in the strongly polar solvent THF (τ2 = 218.8 ps) as compared to Tol (235.1 ps). This emphasizes the considerable influence of the solvent polarity on the 3CT state, rendering the CR rates (τ3) equally sensitive to variations in solvent polarity, as detailed in Table 1. It should be noted that the presence of the heavy atom Br with thiophene may additionally help in increasing the singlet oxygen quantum yield via spin–orbit coupling ISC apart from the CT-mediated ISC, but the latter is believed to be the overall dominating process.
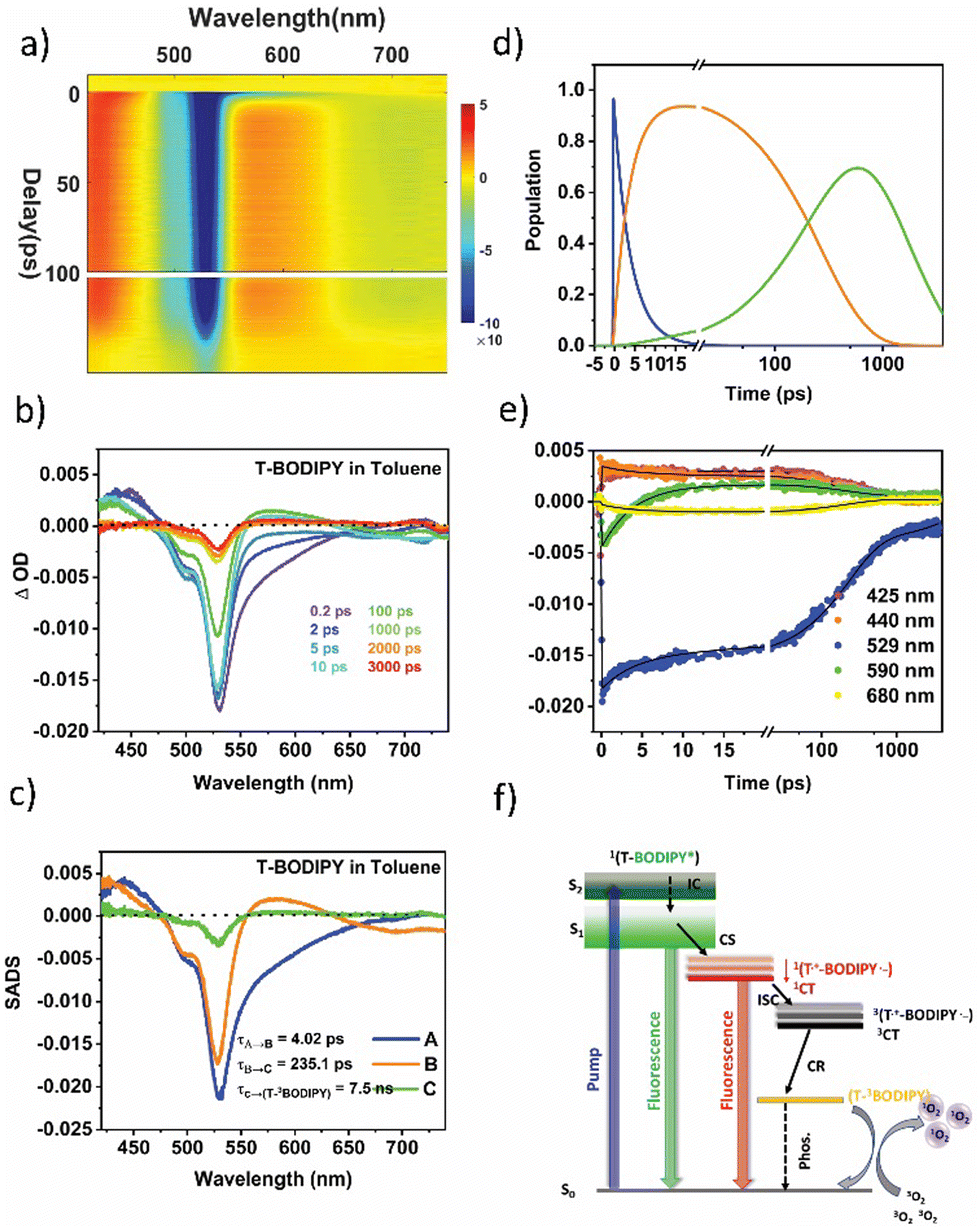 | ||
| Fig. 6 (a) A 2D contour plot for fs-TA of T-BODIPY in Tol following pump excitation at 400 nm. (b) Spectral traces at the indicated probe delay times. (c) Species-associated difference spectra (SADS) reconstructed via singular value decomposition (SVD) and global fitting of the transient data (global analysis) using a target model (f), where A is 1T-BODIPY*, B is 1(T˙+-BODIPY˙−)/1CT and C is 3(T˙+-BODIPY˙−)/3CT. (d) Population kinetics, (e) kinetic traces along with corresponding global fits at indicated probe wavelengths and (f) the kinetic model employed to perform target analysis. | ||
Likewise, the dynamics of excited states within the T-ADA triad was investigated by changing the polarity of the solvent. The fs-TA contour plots for T-ADA in Tol, along with the spectral traces corresponding to the indicated probe delays, are illustrated in Fig. 7a and b, respectively. A close examination of the spectral trace reveals that during the initial probe delay, no discernible excited state absorption (ESA) band is observed in the blue region of the probe spectrum (around 450 nm), despite the presence of the ground state bleach (GSB) band. This occurrence could potentially stem from a robust electronic coupling between the BDT donor and BODIPY acceptor. Following the initial photoexcitation, this coupling could lead to the absence of any spectral feature associated with the parent BODIPY component. Furthermore, a broad absorption extending past 700 nm appears, which follows similar kinetics as that of reduced BODIPY. We, therefore, assign the second spectral component to the charge-separated state. Notably, a distinct isosbestic point around 620 nm is evident, signifying the conversion from the initially excited state to the charge-separated state.
 | ||
| Fig. 7 (a) A 2D contour plot for fs-TA of T-ADA in Tol following pump excitation at 400 nm. (b) Spectral traces at indicated probe delay times. (c) Species associated difference spectra (SADS) reconstructed via singular value decomposition (SVD) and global fitting of the transient data (global analysis) using a target model (f), where A is 1(BDT-BODIPY*), B is 1(T˙+-BODIPY˙−)/1CT and C is 3(BDT˙+A˙−)/3CT. (d) Population kinetics, (e) kinetic traces with the corresponding global fits at the indicated probe wavelengths and (f) the kinetic model employed to perform target analysis. | ||
To extract the time components for various excited state processes in T-ADA after photoexcitation, the data were subjected to target analysis employing the branched kinetic model shown in Fig. 7f. The emission lifetime of the CT state, ascertained from TCSPC measurements (Table S8 in ESI†), is kept fixed during this analysis. The reconstructed SADS spectra are displayed in Fig. 7c. The initial SADS associated with the formation of 1(BDT-BODIPY)* displays a strong ground state bleach (GSB) signal coupled with an extensive excited state absorption (ESA) spanning from 620 nm to 740 nm. This particular species undergoes decay within a time constant of τ1 = 6.32 ps, leading to the emergence of another species characterized by charge separation, denoted as 1(BDT˙+-BODIPY˙−)/1CT. The subsequent SADS reveals a distinct and sharp new ESA centered at 580 nm, accompanied by an isosbestic point at 665 nm. Subsequently, this state undergoes CT-mediated ISC with the time scale of τ2 = 147.1 ps to form the 3(BDT˙+BODIPY˙−)/3CT state. This state is long-lived and characterized by a broad triplet–triplet (Tn ← T1) absorption feature in the range of 550 nm to 740 nm at later probe delays, which is seen in the third SADS. To show the quality of the fitting results, the kinetics at several typical selected wavelengths are shown in Fig. 7e, and the time-dependent population dynamics is shown in Fig. 7d. In contrast to T-BODIPY, in the case of T-ADA, a faster rate of charge separation was observed on increasing the solvent polarity from 6.32 ps to 1.73 ps. This results from the significant stabilization of the CT state in THF. Furthermore, the rates of ISC were found to be nearly identical in both solvents, indicating an equivalent degree of stabilization for the 1CT state. Moreover, this 3CT state recombines to the ground state in timescales of 5.1 ns and 3.2 ns, respectively. It should be noted that changing the solvent polarity does not change the rate of ISC significantly, which is most likely due to the fact that both 1CT and 3CT states are stabilized in more polar solvent (THF), but to the same extent.
Photocatalysis
Having understood the origin of triplet state formation in the compounds by fs-TAS studies and efficient generation of singlet oxygen, their photocatalytic abilities were investigated. Methylphenyl sulfoxides and sulfones are important compounds that are found in a variety of natural products and pharmaceuticals. Hence, their synthesis has been widely investigated.44 We chose the oxidation of thioanisole to methyl phenyl sulfoxide as a model reaction to be performed in the presence of O2 and 0.5 mol% of the photocatalyst in MeOH under irradiation (green light) at room temperature, as shown in Scheme 1 and ESI.† The reaction was screened with pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA as photocatalysts, and the conversion percentages of sulfoxide and sulfones were calculated as shown in Table 2 and ESI† (Section 13). Thioanisole was taken in MeOH (2 mL) and 0.5 mol% of catalyst was added to the mixture, and then irradiated by green light at room temperature. In the case of the pp-BODIPY photocatalyst, only 11% conversion was obtained after 6 h. After 12 h, 36% conversion was obtained, as shown in Table 2 and through 1H-NMR analysis in ESI,† Section 13. However, after 24 h, the highest conversion of 95% of sulfoxide and 1% of sulfones was obtained, and ∼4% thioanisole still remained unreacted after 24 h. | ||
| Scheme 1 Oxidation of thioanisole using pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and T-ADA as photocatalysts in MeOH in the presence of oxygen. | ||
| Catalyst | Conversion (%) | Selectivity (sulfoxide:sulfone) |
||
|---|---|---|---|---|
| 6 h | 12 h | 24 h | ||
| pp-BODIPY | 11 | 36 | 95 | 94:1 |
| mp-BODIPY | 86 | 99 | 99 | 99:1 |
| mm-BODIPY | 98 | 99 | 92 | 92:8 |
| T-BODIPY | 95 | 95 | 91 | 91:9 |
| T-ADA | 77 | 92 | 99 | 99:1 |
In the case of mp-BODIPY and mm-BODIPY, high conversion percentages of 86% and 98%, respectively, were observed after 6 h, and both rotors showed good selectivity of sulfoxide formation after 24 h. Similarly, in the case of T-BODIPY, 95% of the sulfoxide product was observed after 6 h. Compounds mm-BODIPY and T-BODIPY showed over-oxidation of sulfoxide to sulfone after 12 h, resulting in a decrease in the sulfoxide yield. A similar conversion of sulfide to sulfoxide in 6 h was observed in the case of T-ADA. After 24 h, 99% of sulfoxide and 1% of sulfone were observed, as shown in Table 2. The photooxidation of thioanisole was also performed using PDI as PC, as outlined in a previously reported study using green LED in an oxygen atmosphere. Comparable results44 were observed, as shown in ESI† (Table S13).
To gain further insight into the photocatalytic reaction mechanism, control experiments under various conditions were performed. When the reaction was performed in the absence of green light and without a photocatalyst, no trace of product was obtained even after 24 h of reaction, as shown in Scheme S11 and Tables S10–S12 in ESI.† Hence, light and photocatalyst are essential for the oxidation of sulfides, as revealed by the control experiments. The photocatalytic oxidation of sulfides occurs by two well-established mechanistic pathways: either through the formation of 1O2 due to energy transfer, or through superoxide anion (O2˙−) formation by electron transfer.44–46 In order to confirm the mechanism of the photooxidation of sulfides, additional reactions for mp-BODIPY, T-BODIPY and T-ADA, were performed in the presence of 1,4-diazabicyclo [2.2.2]-octane (DABCO), which is a singlet oxygen inhibitor, and the radical inhibitor 1,4-benzoquinone. In the case of mp-BODIPY, in the presence of DABCO and 1,4-benzoquinone, the photocatalysis reaction was suppressed and the conversion was only 3% and 8%, respectively, after 12 h, as shown in 1H NMR and Tables S10–S12 (ESI†). Similarly, in the case of T-BODIPY, the conversion was only 38% and 4% using DABCO and 1,4-benzoquinone, respectively, after 24 h. Similarly, in the case of T-ADA, the reaction was suppressed using DABCO and 1,4-benzoquinone, and the conversion was 51% and 11%, respectively, after 24 h. The detailed plausible mechanism for the photooxidation of sulfides using all compounds involve the participation of both 1O2 and O2˙−,44–46 as shown in Scheme S12 in ESI.†
Conclusions
We have synthesized BODIPY-based organic photosensitizers pp-BODIPY, mp-BODIPY, mm-BODIPY, T-BODIPY and triad T-ADA, in which BODIPY and BDT are connected through a thiophene spacer at the meso-position of BODIPY, where BDT behaves as a donor and T-BODIPY behaves as an acceptor. The regioisomeric effect and type of spacer significantly alter the photophysical properties, such as absorption, emission, fluorescence quantum yields, and singlet oxygen quantum yields of pp-BODIPY, mp-BODIPY, mm-BODIPY and T-BODIPY. The rotor triad T-ADA showed an emission band at ∼650 nm that originated from CT. This emission band was responsive to variations in solvent polarity and solubility in solvents (water in THF solution, i.e., AIE effect). Among all molecules, T-BODIPY and T-ADA showed high singlet oxygen quantum yields of 77% and 35% in THF, respectively. Regioisomers pp-BODIPY, mp-BODIPY, mm-BODIPY, and T-BODIPY and T-ADA showed excellent selectivities in the aerobic photooxidation of thioanisole to methyl phenyl sulfoxide. Femtosecond transient absorption studies were employed to shed light on the underlying mechanisms for the triplet state formation and involvement of the CT state. Additionally, density functional DFT calculations revealed a smaller energy gap between the singlet and triplet states, coupled with a rapid charge separation in T-BODIPY and T-ADA. This suggests a multifaceted yet efficient pathway for the generation of triplet states in this compound. T-BODIPY involves a combination of spin–orbit coupling and CT-mediated ISC mechanism, whereas T-ADA relies solely on CT-mediated ISC for the generation of triplet states. These intricate processes likely contribute to the notably high yields of singlet oxygen (1O2) observed in both T-BODIPY and T-ADA systems. The results presented here underscores the involvement of the CT state in these donor–acceptor systems to be utilized as triplet photosensitizers. Grasping these mechanisms is crucial in crafting resilient triplet photosensitizers. It is anticipated that as our understanding deepens, more complex and multifaceted systems will be developed, resulting in enhanced efficiency for their applications in photocatalysis, photodynamic therapy, and photon upconversion.Author contributions
S. S. G. designed the project and supervised the synthesis, optical characterization and photocatalysis studies of the project. A. K. D. supervised the TA measurements and analysis of the TA data. S. S. performed the synthesis, and carried out the structural, steady-state photophysical and redox characterization, as well as the DFT calculations of all molecules. S. C. performed TA measurements and analysed TA data of all the compounds. S. S. and V. G. performed photocatalysis studies of all compounds. S. S., S. C., V. G., A. K. D. and S. S. G. wrote the manuscript. All authors discussed the results and commented on the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. and S. C. thank IISER Mohali for PhD fellowships and V. G. thanks PMRF for PhD fellowship. S. S. G. thanks DST-SERB (SPG/2021/003570) and A. K. D. thanks DST-SERB (CRG/2021/003981) for funding. The authors sincerely thank Dr Sugumar Venkataramani for providing access to monochromatic light sources and for helpful discussions. The authors sincerely acknowledge the central facilities of IISER Mohali, such as NMR and mass spectrometry, and departmental facilities for the various measurements.References
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L. A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100–103 CrossRef CAS PubMed.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726–7727 CrossRef CAS PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- P. De Bonfils, L. Péault, P. Nun and V. Coeffard, Eur. J. Org. Chem., 2021, 1809–1824 CrossRef CAS.
- E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607–6628 RSC.
- A. Atilgan, M. M. Cetin, J. Yu, Y. Beldjoudi, J. Liu, C. L. Stern, F. M. Cetin, T. Islamoglu, O. K. Farha, P. Deria, J. F. Stoddart and J. T. Hupp, J. Am. Chem. Soc., 2020, 142, 18554–18564 CrossRef CAS PubMed.
- M. A. Filatov, Org. Biomol. Chem., 2019, 18, 10–27 RSC.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS PubMed.
- L. Yang, Z. Huang, G. Li, W. Zhang, R. Cao, C. Wang, J. Xiao and D. Xue, Angew. Chem., 2018, 130, 1986–1990 CrossRef.
- J. Fischer, L. Mele, H. Serier-Brault, P. Nun and V. Coeffard, Eur. J. Org. Chem., 2019, 6352–6358 CrossRef CAS.
- K. Chen, S. Guo, H. Liu, X. Li, Z. Zhang and T. Lu, Angew. Chem., 2020, 132, 13051–13057 CrossRef.
- W. Li, L. Li, H. Xiao, R. Qi, Y. Huang, Z. Xie, X. Jing and H. Zhang, RSC Adv., 2013, 3, 13417–13421 RSC.
- P. Rana, N. Singh, P. Majumdar and S. Prakash Singh, Coord. Chem. Rev., 2022, 470, 214698 CrossRef CAS.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed.
- K. Chen, W. Yang, Z. Wang, A. Iagatti, L. Bussotti, P. Foggi, W. Ji, J. Zhao and M. Di Donato, J. Phys. Chem. A, 2017, 121, 7550–7564 CrossRef CAS PubMed.
- S. Sharma, Z. Wei, F. C. Grozema and S. Sengupta, Phys. Chem. Chem. Phys., 2020, 22, 25514–25521 RSC.
- D. Simoni, G. Giannini, M. Roberti, R. Rondanin, R. Baruchello, M. Rossi, G. Grisolia, F. P. Invidiata, S. Aiello, S. Marino, S. Cavallini, A. Siniscalchi, N. Gebbia, L. Crosta, S. Grimaudo, V. Abbadessa, A. Di Cristina and M. Tolomeo, J. Med. Chem., 2005, 48, 4293–4299 CrossRef CAS PubMed.
- H. Bonin, D. Delbrayelle, P. Demonchaux and E. Gras, Chem. Commun., 2010, 46, 2677–2679 RSC.
- C. Thivierge, A. Loudet and K. Burgess, Macromolecules, 2011, 44, 4012–4015 CrossRef CAS PubMed.
- M. Yilmaz, M. Erkartal, M. Ozdemir, U. Sen, H. Usta and G. Demirel, ACS Appl. Mater. Interfaces, 2017, 9, 18199–18206 CrossRef CAS PubMed.
- T. Gayathri, A. K. Barui, S. Prashanthi, C. R. Patra and S. P. Singh, RSC Adv., 2014, 4, 47409–47413 RSC.
- E. X. Flores, A. J. Sánchez, H. G. Ortega, N. S. Puig, M. R. Ávila, R. Santillan and N. Farfán, New J. Chem., 2016, 40, 4500–4512 RSC.
- X. Liu, W. Chi, Q. Qiao, S. V. Kokate, E. P. Cabrera, Z. Xu, X. Liu and Y. T. Chang, ACS Sens., 2020, 5, 731–739 CrossRef CAS PubMed.
- S. Sengupta and U. K. Pandey, Org. Biomol. Chem., 2018, 16, 2033–2038 RSC.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4031 CrossRef PubMed.
- N. Adarsh, R. R. Avirah and D. Ramaiah, Org. Lett., 2010, 12, 5720–5723 CrossRef CAS PubMed.
- S. G. Awuah, J. Polreis, V. Biradar and Y. You, Org. Lett., 2011, 13, 3884–3887 CrossRef CAS PubMed.
- G. Bhutani, P. Verma, A. Jayachandran, S. Paul, K. Chattopadhyay and A. K. De, J. Phys. Chem. B, 2023, 127, 3197–3207 CrossRef CAS PubMed.
- Y. Silori, S. Chawla and A. K. De, ChemPhysChem, 2020, 21, 1908–1917 CrossRef CAS PubMed.
- D. W. Cho, M. Fujitsuka, J. H. Ryu, M. H. Lee, H. K. Kim, T. Majima and C. Im, Chem. Commun., 2012, 48, 3424–3426 RSC.
- I. H. M. Van Stokkum, D. S. Larsen and R. Van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Software, 2012, 49, 1–22 Search PubMed.
- P. Toele, H. Zhang, C. Trieflinger, J. Daub and M. Glasbeek, Chem. Phys. Lett., 2003, 368, 66–75 CrossRef CAS.
- H. L. Kee, C. Kirmaiery, L. Yu, P. Thamyongkit, W. J. Youngblood, M. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheldt, R. R. Birge, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 109, 20433–20443 CrossRef CAS PubMed.
- J. Kong, W. Zhang, G. Li, D. Huo, Y. Guo, X. Niu, Y. Wan, B. Tang and A. Xia, J. Phys. Chem. Lett., 2020, 11, 10329–10339 CrossRef CAS PubMed.
- Y. Gao, H. Xu, S. Zhang, Y. Zhang, C. Tang and W. Fan, Org. Biomol. Chem., 2019, 17, 7144–7149 RSC.
- E. Baciocchi, T. Del Giacco, F. Elisei, M. F. Gerini, M. Guerra, A. Lapi and P. Liberali, J. Am. Chem. Soc., 2003, 125, 16444–16454 CrossRef CAS PubMed.
- S. M. Bonesi, M. Fagnoni and A. Albini, Eur. J. Org. Chem., 2008, 2612–2620 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00117f |
| This journal is © The Royal Society of Chemistry 2024 |