
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiple length-scale control of Boc-protected diphenylalanine aggregates through solvent composition†
Sara
Catalini
 *abc,
Francesco
Bagni
cd,
Stefano
Cicchi
d,
Mariangela
Di Donato
ce,
Alessandro
Iagatti
bc,
Andrea
Lapini
cf,
Paolo
Foggi
bcg,
Caterina
Petrillo
a,
Alessandro
Di Michele
a,
Marco
Paolantoni
g,
Giorgio
Schirò
h,
Lucia
Comez
*i and
Alessandro
Paciaroni
*a
*abc,
Francesco
Bagni
cd,
Stefano
Cicchi
d,
Mariangela
Di Donato
ce,
Alessandro
Iagatti
bc,
Andrea
Lapini
cf,
Paolo
Foggi
bcg,
Caterina
Petrillo
a,
Alessandro
Di Michele
a,
Marco
Paolantoni
g,
Giorgio
Schirò
h,
Lucia
Comez
*i and
Alessandro
Paciaroni
*a
aDipartimento di Fisica e Geologia, Università di Perugia, Via Pascoli, 06123 PG, Italy. E-mail: sara.catalini@unipg.it; alessandro.paciaroni@unipg.it
bCNR-INO, Largo Fermi 6, 50125 FI, Italy
cEuropean Laboratory for Non-Linear Spectroscopy, Via Nello Carrara 1, 50019 Sesto Fiorentino, FI, Italy
dDipartimento di Chimica “Ugo Schiff”, Università di Firenze, via della Lastruccia, 3-13, 50019 Sesto Fiorentino, FI, Italy
eCNR-ICCOM, via Madonna del Piano 10, I-50019 Sesto Fiorentino, FI, Italy
fDipartimento di Scienze Chimiche, della Vita e della Sostenibilità Ambientale, Università di Parma, Parco Area delle Scienze, 17/A, 43124 Parma, PR, Italy
gDipartimento di Chimica, Biologia e Biotecnologie, Università di Perugia, Via Elce di sotto 8, 06123 PG, Italy
hUniv. Grenoble Alpes, CNRS, CEA, IBS, F-38000 Grenoble, France
iCNR - Istituto Officina dei Materiali (IOM), c/o Dipartimento di Fisica e Geologia, Università di Perugia, Via Pascoli, 06123 PG, Italy. E-mail: comez@iom.cnr.it
First published on 20th March 2024
Abstract
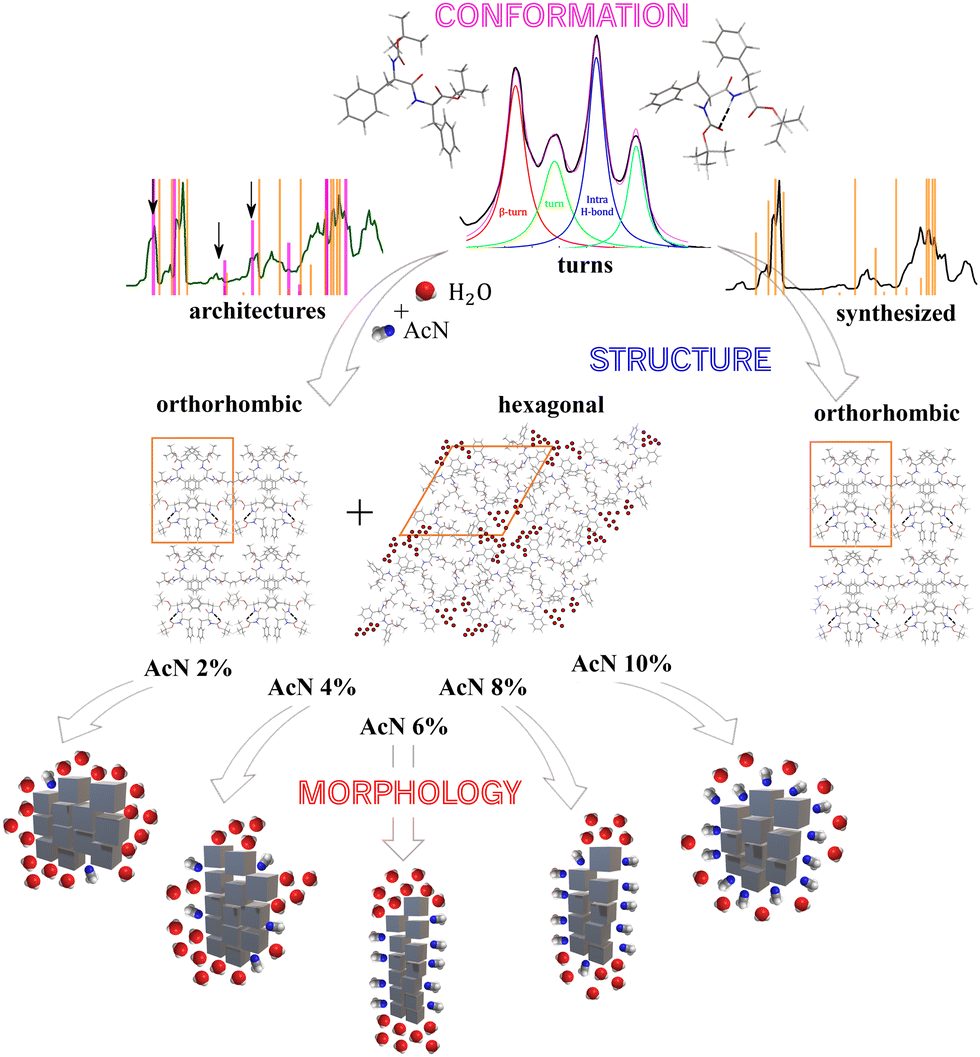
Diphenylalanine and its variants are among the most studied building blocks in materials science due to their ability to form extended architectures with a wide range of morphologies. Given this propensity to self-assemble into very different shapes, controlling the aggregation process of the basic units is very challenging. Using a selection of techniques, this work investigates the critical role of solvation in influencing the association of Boc-protected diphenylalanine. The results indicate that the balance of solvents, specifically acetonitrile-water, significantly impacts the self-assembly process. Indeed, a change in acetonitrile content from 2% to 10% in water solutions drives the formation of different morphologies of the aggregates, spanning from spheres to plates. In a pure organic solvent a single orthorhombic crystalline phase is observed, whereas the presence of water reveals the coexistence of two phases: orthorhombic and hexagonal. The fraction of hexagonal phase relies on the solvent composition, while the peptides always adopt turn conformations, promoting the plane-to-plane stacking of aromatic rings. The architectural growth is driven by aromatic stacking and hydrophobic interactions; the development of different morphologies is dependent on the extent of the aggregate–solvent interface. Interestingly, we find a morphology re-entrant behaviour induced by finely tuning the acetonitrile content, thereby linking aggregate morphology with molecular characteristics. The selection of suitable building blocks and solvation conditions is crucial in steering the nature of aggregates toward creating smart biomaterials with enhanced properties.
Introduction
The design of biomaterials at the molecular level, utilizing natural biomolecules and their self-assembly processes, is widely used in materials science and several strategies have been implemented to control the morphology of final architectures.1 The latter can be used as building blocks to develop dry or soft materials and, depending on the application they are intended for, some morphologies are preferred over others. For example, if the material destination is drug delivery, aggregates with spherical or cubic shapes are preferred such as those of lipidic liposomes2 or cubosomes.3 Nanomaterials for photothermal or photodynamic therapy applications4,5 usually consist of spherical objects with functionalized surfaces. Instead, for the development of a three-dimensional network for cellular scaffolding, morphologies with axial preferential extension are desirable, such as tubular or fibrillar structures.6 The latter can interact with each other, forming branched structures leading to system percolation and making soft matrices able to retain a large amount of solvent.7–9 Among biomolecules, short peptides are attractive due to their effortless availability, programmable primary structure, and widely tunable self-assembly architecture.10–14 Self-assembling of these systems depends on multiple factors, making it challenging to have a precise control over the process. The formation of peptide aggregates indeed occurs through weak interactions among monomers, including hydrogen bonding, electrostatic, hydrophobic,15 and π–π aromatic stacking interactions.10 Short peptides offer the possibility to form versatile supramolecular architectures displaying unique physical and chemical properties, often presenting intriguing optical features.4 Peptide- and protein-based amyloid-like fibrils are examples of robust and stable structures, which can act as scaffolds for developing light-harvesting arrays.16 Diphenylalanine (FF), which is the key structural motif of the Aβ amyloid polypeptide, has a strong propensity to self-assemble in an aqueous environment as demonstrated by a wide number of recent experimental17–22 and numerical23–25 studies. For example, coarse-grained molecular dynamics simulations found that FF spontaneously assembles into nano- and micro-aggregates, forming ordered and dimensionally extended structures. One of the predominant factors determining the final morphology of the aggregates is the medium where the aggregation takes place. Energetic and structural analysis suggests that the interplay among peptide–peptide and peptide–solvent interactions results in the formation of different structures.23 In particular, an important role in self-assembly is played by aromatic side chains, which strongly interact with each other. The first nucleation seeds are the precursors of the final nano- and micro-structures and are involved in substantial hydrophobic clustering. Therefore, to control the morphologies of the resultant architectures, the microenvironment surrounding hydrophobic aromatic side chains should be ad hoc determined.26 In addition, the presence of protecting tertbutyl groups reduces the hydrophilic points of the dipeptide and opens new aggregation pathways. Previous studies, on natural, Boc-27 and Fmoc-protected28 FF, found a morphological tunability of the aggregates, depending both on the protecting group and the employed mixture of solvents. The most utilized organic solvents are hexafluoro isopropanol,18,29,30 tetrahydrofuran and dichloromethane,22,31,32 while ethanol, methanol and water are commonly used as cosolvents triggering the peptide aggregation.22,31,32 Depending on the mixtures, the aggregate architectures can span from spheres to tubes.33 An appealing alternative to aforementioned organic solvents is acetonitrile (AcN), which is widely used in several chemical processes, proving to be a key organic solvent in pharmaceutical manufacture.34 Furthermore, AcN's strong polarity enables it to stay miscible with water at all concentrations and to dissolve various inorganic and organic molecules.35 Because peptide aggregation relies on nucleation events and is common in highly organized self-assembly processes,25,36 it is essential to combine molecular structural characterizations with nano/microscale morphological analysis for a comprehensive understanding across different scales of hierarchy.On these grounds, we decided to adopt an integrated approach to investigate the architectures of Boc-protected FF (Boc-FF) in the presence of AcN–water mixtures.37 By combining optical, diffraction, and absorption techniques with electronic and light microscopy, we discover that the self-assembly and morphology of the resulting aggregates are under thermodynamic and kinetic38,39 control and are strongly modulated by the AcN–water solvent ratio.
Experimental section
Boc-FF synthesis
All the reagents were commercially available and have been used without any further purification. The short peptide Boc-FF (chemical structure shown in Fig. S1, ESI†) was synthesized according to literary procedure;40 HRMS-ESI (m/z): [M + H]+ calculated for C27H36N2O5, 469.2702; found, 469.2696. El. anal. calculated for C27H36N2O5: C, 69.21; H, 7.74; N, 5.98. Found: C, 69.32; H, 7.54; N, 5.91.Aggregates preparation
The preparation of the Boc-FF aggregated structures relies on a protocol based on fine-tuning the mixed solvents ratio. According to this preparation methodology, the synthesized compounds are dissolved in AcN organic solvent, and subsequently H2O is introduced to induce the aggregation process. After H2O addition the aggregation immediately occurs, giving rise to a colloidal suspension. A concentrated stock solution (43 mM) of Boc-FF dissolved in pure AcN is prepared. Solvent mixtures are prepared with the following AcN percentage: 2%, 4%, 6%, 8% and 10% v/v. The Boc-FF aggregates are formed at room temperature by introducing the concentrated peptide solution into the AcN–H2O mixture, resulting in a final peptide concentration of 1.1 mM (0.5 mg ml−1). This concentration falls within the commonly used range in the literature, which spans from 0.2 mg ml−1 to 10 mg ml−1.41,42 The preparation of the Boc-FF aggregated structures at high temperature has been performed directly at 85 °C. Solvent mixtures are prepared with the same AcN percentage as for the room temperature experiments. All the mixtures and the concentrated stock solution of Boc-FF in pure AcN are preheated for 30 minutes at 85 °C. A volume of Boc-FF stock solution is added to the solvent mixtures to reach the final concentration of 1.1 mM. Samples are kept at 85 °C for 5 h and then cooled at room temperature. After cooling, all the samples display the presence of a small amount of precipitate along with a transparent supernatant solution.Optical microscopy images
The dry samples for XRD experiments, are prepared in glass Petri dishes, and observed through a HRX-01 Hirox digital microscope equipped with a high-range triple zoom lens HR-5000.SEM images
The morphology of the samples is analysed by field emission scanning electron microscopy FE SEM LEO 1525 (ZEISS). A drop of solution is deposited with a glass Pasteur pipette on a silicon substrate and is left to dry at room temperature for 15 h. Afterward, samples are metalized with 8 nm of chromium and the section is observed by SEM. Measurements are carried out using an In-lens detector at 15 kV. The dimensional distribution of spherical aggregates has been determined with ImageJ software.43XRD patterns
X-ray diffraction (XRD) experiments are carried out at the FIP2-BM07 beamline of European Synchrotron Radiation Facility (ESRF) (Grenoble) equipped with a Pilatus 6 M detector. Its optics deliver a focused beam on a fixed sample position, with an energy resolution of about 10−3 to 10−4 and a large accessible energy range (5–25 keV). The X-ray beam profile has a Gaussian shape, with a beam size that can be adjusted between 50 × 50 and 250 × 250 μm2. For our experiment we used the biggest beam size to account for the different orientations of the crystals. The beam energy is 12.65 keV, corresponding to a wavelength λ = 0.98 Å. A 2D diffraction patterns have been processed using PyFAI software package44 to obtain 1D plots (azimuthal integration).45 We performed a pixel binning to have a final array of 600 points. For each sample we recorded the XRD pattern of three different positions of the powder and the final profiles are obtained from the average of the three points and the subtraction of the mylar capillary curve.FTIR spectra
Fourier transform infrared (FTIR) measurements in attenuated total reflection (ATR) configuration are performed with an Alpha (Bruker Optics) spectrometer on sample powders. The software Opus 7.5 is employed for spectral acquisition and analysis. Each spectrum is recorded in the 300–5000 cm−1 spectral region with a resolution of 1 cm−1 by averaging 30 scans acquisition.CD spectra
Circular dichroism (CD) measurements are performed using a Jasco J810 spectropolarimeter. Each spectrum of the colloidal suspensions is collected in the range from 200 to 500 nm, with a scan speed of 50 nm min−1. Quartz cuvettes with a path-length of 1 mm are used to obtain the optimal signal-to-noise ratio for the Boc-FF samples with concentrations of 1.1 mM. Spectra of the depositions are collected in the 190–300 nm spectral range with a scan speed of 50 nm min−1 and each spectrum is the average of 4 measurements. All spectra are acquired at room temperature and subtracted for the cuvette contribution. CD data are expressed as the difference in the CD intensity of the right- and left-handed circularly polarized light.UV-Vis absorption spectra
UV-Visible (UV-Vis) absorption measurements are performed using a Jasco V-570 spectrophotometer. Each spectrum of the colloidal suspensions is collected in the range from 200 to 700 nm, with a 1 nm resolution. Quartz cuvettes with path-length of 1 mm are used and the temperature is controlled by means of an integrated Perliter system. Spectra of the depositions are collected in 200–450 nm spectral range. All spectra are subtracted for the cuvette contribution.Emission spectra
Fluorescence measurements are performed using a PerkinElmer LS 55 spectrofluorometer. Samples are excited at 250 nm and the emission is collected from 270 to 550 nm. A cut-off filter at 270 nm are used to remove the second-order diffraction of the grating. The excitation and emission slits are set at 10 and 5 nm, respectively. All the spectra are acquired with an integration time of 200 nm min−1, using a quartz cuvette with a 1 cm optical path.Results
Interestingly, optical microscopy (OM) and SEM imaging show that a significant morphological differentiation is generated by the weak variation in the AcN–H2O ratio within the composition of the cost-effective and safe solvents used for preparing the Boc-FF aggregates, as displayed in Fig. 1. Fig. S2 and S3 in the ESI† present additional images taken at different magnifications and in different regions of the sample. | ||
| Fig. 1 OM (a, d, g, l and o) and SEM (b, e, h, m, p, c, f, i, n and q) images of Boc-FF 1.1 mM aggregates formed from the colloidal suspensions at 25 °C in the different solvent mixtures. Optical microscopy images are acquired with 1k magnification (a, d, g, l and o); the scale bar is 50 μm. SEM images are acquired with 5k magnification (b, e, h, m and p) the scale bar is 5 μm, and 50k magnification (c, f, i, n and q); the scale bar is 500 nm. | ||
By using 2% AcN solutions, principally spherical particles with an average diameter of 650 nm are produced. 4% AcN results in plate-like particles, which become longer at 6% AcN. In 8% AcN solutions, the plates shorten back and appear to be more spaced apart compared to the aforementioned samples. Lastly, the use of 10% AcN yields spherical-like particles with an average diameter of 600 nm and a narrower dimensional distribution compared to the spheres formed with 2% AcN, as reported in Fig. S4 (ESI†). To shed light on the structure and conformation of the Boc-FF aggregates, we used XRD, FTIR, and CD measurements to probe the supramolecular organization on different length scales. To directly compare the results from such measurements to those from imaging techniques, samples have been dried from colloidal suspensions.
The crystalline structure of the Boc-FF aggregates is confirmed by XRD characterization. The wavevector, q, the distance, d, and the intensities, I(q), of all the observed lines of the powder diffraction data are listed in sequence in the ESI† (Tables T1–T6). As shown in Fig. 2, the diffraction patterns of all the aggregates are quite similar in the wavevector range going from 2.6 to 3.6 Å−1. This indicates that the characteristic distances in the range 2.4–1.7 Å, compatible with typical peptide–peptide hydrogen bond lengths, remain unaltered in all the systems.46,48 Such a similarity can be also observed in the intermediate q range from 1.0 Å−1 to 2.2 Å−1, corresponding to the typical distances between π–π stacked ordered aromatic rings on the order of 4.0–5.0 Å.17,46,48–52 Quite interestingly, in the lowest investigated q range (0.4–1.0 Å−1), which reports on the size of the crystalline cell, three main peaks at the 7.0 Å, 8.0 Å and 12.1 Å distances are detected only in the diffraction patterns of the architectures formed by the AcN–H2O mixtures. The intensity of these three peaks is strongly dependent on the amount of AcN in the initial suspensions, being stronger in the 2% and 6% AcN mixtures, decreasing in the 4% and 8% AcN mixtures, and almost disappearing in the architectures prepared at the 10% AcN mixture. To provide new insights on the structural features of the polycrystalline phase of the measured samples, we compared the low-q XRD experimental patterns with single crystal XRD data obtained from online databases for FF-like molecules. We find out that the ensemble of the peaks of the formed architectures is qualitatively well reproduced by the combination of an orthorhombic and a hexagonal phase (Fig. 2a and b). More in detail, in the presence of a large amount of organic solvent (Fig. 2a and f) the structure appears to be predominantly orthorhombic, with a likely more compressed unit cell compared to the reference single crystal (COD: 4510160).46 On the other hand, for a water percentage higher than 90%, an hexagonal phase seems to emerge in the XRD patterns, as demonstrated by the superposition of the additional peaks with the corresponding reference structure (CSD: 163340).47 The FTIR spectra of the synthesized Boc-FF peptides and the formed architectures are reported in Fig. 3 (1200–3400 cm−1 region).
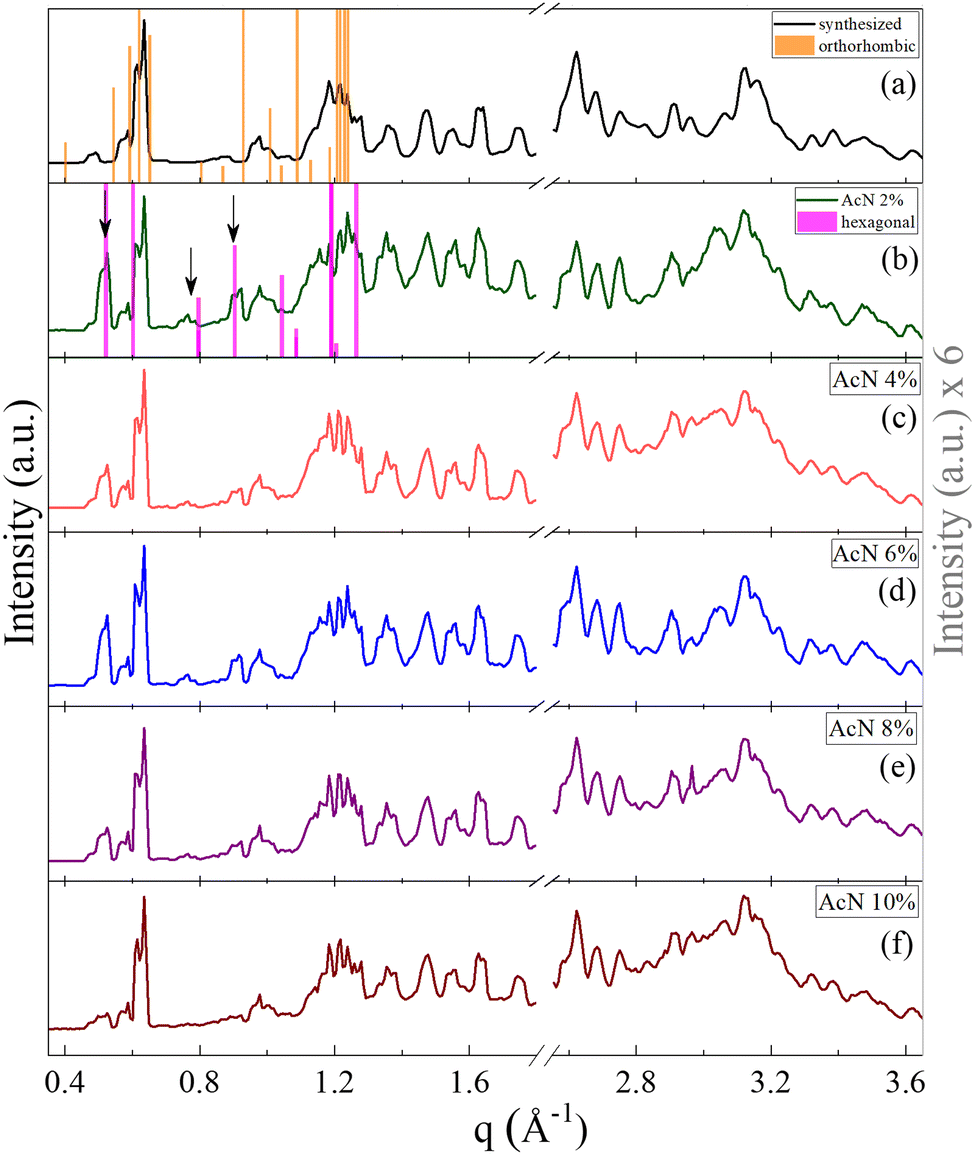 | ||
| Fig. 2 Powder XRD patterns of the synthesized Boc-FF (a) and of Boc-FF dried samples formed from the colloidal suspensions (b, c, d, e and f), acquired over a q range of 0.4–3.6 Å−1. XRD pattern at low q values of the orthorhombic (orange line COD: 4510160)46 and the hexagonal (pink line CSD: 163340)47 single crystal structures of FF-like molecules downloaded from Crystallography Open Database and Cambridge Structural Database, respectively. | ||
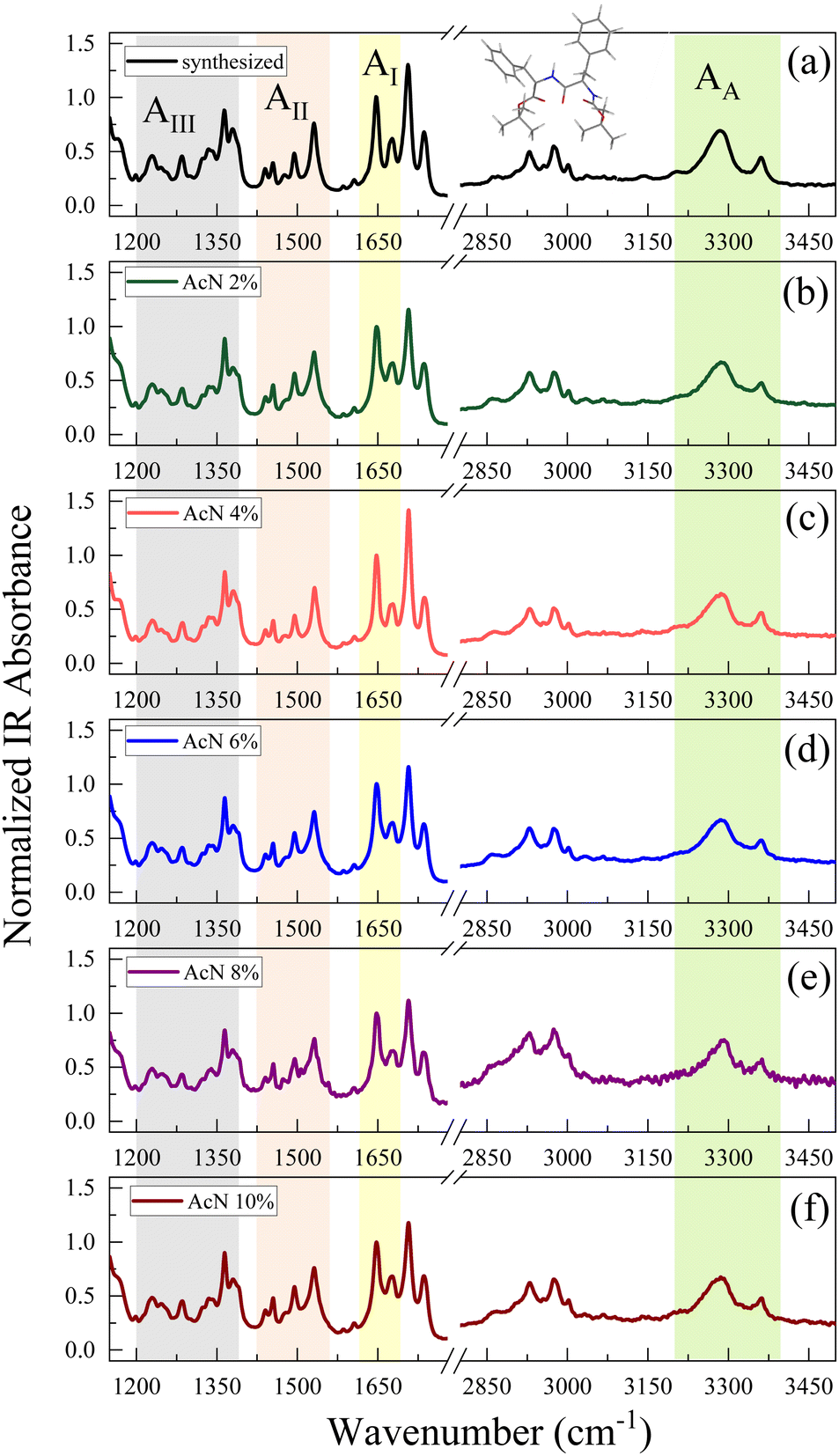 | ||
| Fig. 3 FTIR spectra of Boc-FF synthetized compound (a) and dried samples formed from the colloidal suspensions (b–f) that are dried for 15 h, recorded in ATR configuration. The coloured bands highlight the characteristic absorption bands of the amide groups. | ||
The amide spectral regions provide precious information on intra- and inter-molecular hydrogen bonding interactions. In particular, the characteristic absorption bands of amide III (AIII) 1200–1400 cm−1, amide II (AII) 1430–1570 cm−1, amide I (AI) 1600–1680 cm−1 and amide A (AA) 3200–3400 cm−1 can be identified.53,54 Compared to natural FF, the Boc-FF peptide presents a tert-butyloxycarbonyl group linked to the terminal parts (see chemical structure in the insert of Fig. 3). Four different C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peaks related to the three distinct amide, carbamate and ester carbonyl groups, contribute to the signal in the 1600 cm−1 to 1800 cm−1 region.54,55 The FTIR spectra of Boc-FF aggregates obtained by drying the colloidal suspensions for 15 h show identical peak positions and relative intensities as the synthesized Boc-FF compound. Their signals are narrow and well resolved, thus confirming that the samples are in a crystalline state.56 The AI band consists of two peaks at 1647 and 1676 cm−1, being compatible with structures with a significant β-turn character.54,57 Indeed, the peak at 1676 cm−1 is a marker of turn conformations, while the feature at 1647 cm−1 suggests an aperiodic secondary structure involving different types of β-turns.58 To obtain additional information on the way the drying procedure affects the molecular properties of the aggregates, FTIR measurements are performed also on samples left drying for a shorter time interval (2 h instead of 15 h Fig. S5). In this case, the spectra show broader signals, indicating a more amorphous structure and are centred at 1653 cm−1 and 1683 cm−1. These higher vibrational frequencies correspond to a weaker H-bonded β-turn conformation compared to the crystalline samples. Interestingly, spectra from samples obtained with the same drying time closely resemble each other, irrespective of the initial AcN–H2O ratio, suggesting that Boc-FF peptides consistently adopt a similar conformation. The intensity of the carbamate peak at about 1700 cm−1 is dominant in the crystalline samples, while it undergoes a strong decrease in amorphous samples. Even the AA spectral region is characterized by two NH populations, forming stronger and weaker H-bonds corresponding to a low- and a high-frequency peak respectively. As a general trend, all the signals arising from NH and CO modes display significant changes when passing from the amorphous to the crystalline form, while the CH2 and CH3 bands (2800–3100 cm−1) do not undergo any variation. The structural properties of the Boc-FF peptide are further analysed through CD spectroscopy as reported in Fig. 4.
O peaks related to the three distinct amide, carbamate and ester carbonyl groups, contribute to the signal in the 1600 cm−1 to 1800 cm−1 region.54,55 The FTIR spectra of Boc-FF aggregates obtained by drying the colloidal suspensions for 15 h show identical peak positions and relative intensities as the synthesized Boc-FF compound. Their signals are narrow and well resolved, thus confirming that the samples are in a crystalline state.56 The AI band consists of two peaks at 1647 and 1676 cm−1, being compatible with structures with a significant β-turn character.54,57 Indeed, the peak at 1676 cm−1 is a marker of turn conformations, while the feature at 1647 cm−1 suggests an aperiodic secondary structure involving different types of β-turns.58 To obtain additional information on the way the drying procedure affects the molecular properties of the aggregates, FTIR measurements are performed also on samples left drying for a shorter time interval (2 h instead of 15 h Fig. S5). In this case, the spectra show broader signals, indicating a more amorphous structure and are centred at 1653 cm−1 and 1683 cm−1. These higher vibrational frequencies correspond to a weaker H-bonded β-turn conformation compared to the crystalline samples. Interestingly, spectra from samples obtained with the same drying time closely resemble each other, irrespective of the initial AcN–H2O ratio, suggesting that Boc-FF peptides consistently adopt a similar conformation. The intensity of the carbamate peak at about 1700 cm−1 is dominant in the crystalline samples, while it undergoes a strong decrease in amorphous samples. Even the AA spectral region is characterized by two NH populations, forming stronger and weaker H-bonds corresponding to a low- and a high-frequency peak respectively. As a general trend, all the signals arising from NH and CO modes display significant changes when passing from the amorphous to the crystalline form, while the CH2 and CH3 bands (2800–3100 cm−1) do not undergo any variation. The structural properties of the Boc-FF peptide are further analysed through CD spectroscopy as reported in Fig. 4.
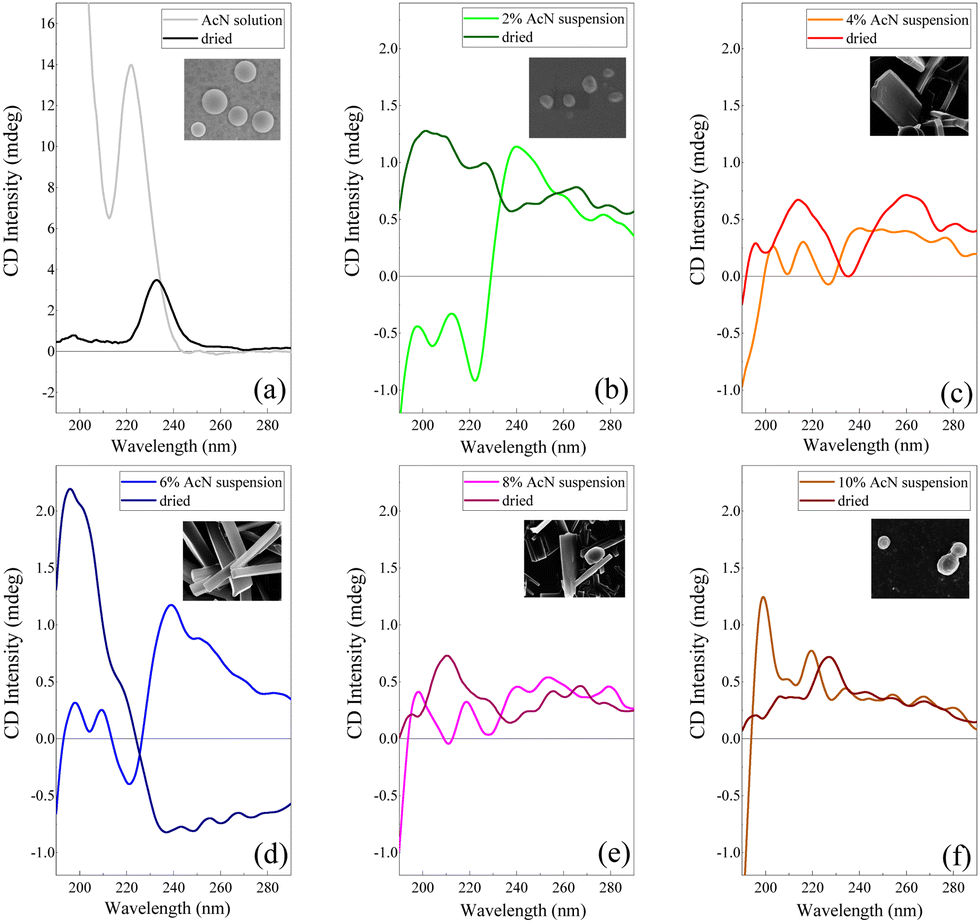 | ||
| Fig. 4 CD spectrum of the Boc-FF AcN solution (a) and Boc-FF colloidal suspensions in AcN–H2O mixtures (b–f) compared with the respective CD spectra of the dried samples. The inserts report the SEM images of the dried samples with 50k magnification. | ||
Electronic CD spectroscopy is widely used to investigate the secondary structure of peptides and proteins, analysing the two bands of the peptide bond at 200 nm and 220 nm assigned to π–π* and n–π* transitions, respectively.59 In the case of short peptides, like Boc-FF, secondary structures are classified according to the values of the peptide bond torsion angle, in analogy with more extended structures like the α-helix or β-sheet motifs found in proteins. On the other hand, the π–π* transitions due to phenylalanine aromatic rings, falling at 260 nm (Lb) and at 210 nm (La), also contribute to the CD spectra.59 The CD signal of the peptide in its monomeric form (Fig. 4a), i.e. Boc-FF in AcN solution, is characterized by an intense positive maximum at 220 nm and a strong upturn of the ellipticity below 200 nm.29,48 A major change of the spectral features is visible in the CD profiles of the colloidal suspensions (Fig. 4b–f) compared to the spectrum of the monomer solution, including a strong decrease of the dichroic signal itself. Such a change reflects both the variation of peptide bonds dihedral angles and the coexistence of different Boc-FF conformations.29 A weak negative minimum centred in the 225–230 nm interval characterizes the spectra of the colloidal suspensions formed with 2%, 4%, 6% and 8% AcN (Fig. 4b–e). On the other hand, the CD spectrum of the colloidal suspension formed using 10% AcN (Fig. 4f) qualitatively resembles that of Boc-FF AcN solution. The CD profiles of the colloidal suspension have been compared with those of the dried powder, for consistency of sample preparation with other techniques (SEM, OM, XRD and FTIR). Regarding the latter, it is worth of note that the 10% AcN sample displays spectral features that are qualitatively very similar to those of FF dried from pure AcN solutions. On the other hand, the 4% and 8% AcN samples exhibit comparable spectral trends, with two positive structured bands located in the 190–230 nm and 230–280 nm regions. Lastly, the dried sample obtained from the 6% AcN solution shows markedly distinct features.
Finally, optical absorption and fluorescence measurements, shown in Fig. 5, provide insights into the electronic properties of the monomeric and aggregated forms of Boc-FF.
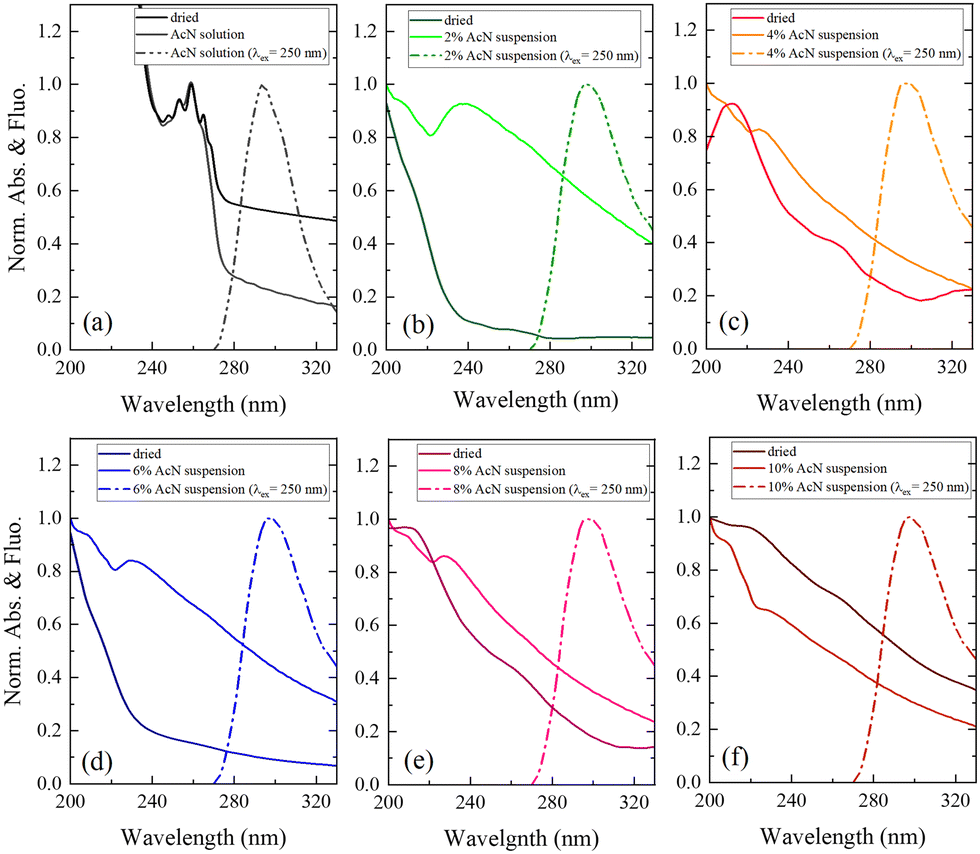 | ||
| Fig. 5 UV-Vis absorption spectra (solid lines) of the Boc-FF AcN solution and dried sample (a) and Boc-FF colloidal suspensions in AcN–H2O mixtures and dried samples (b–f) compared with the respective emission spectra (dashed lines) of the solution and colloidal suspensions, excited at 250 nm. | ||
The optical absorption spectrum confirms that a monomeric form of the peptide exists in pure AcN solution, as both a strong absorption feature below 240 nm, assigned to the aromatic ring of phenylalanine, and a smaller contribution from the peptide link are observed. This is also supported by the characteristic structured band at about 250 nm, due to the vibronic transitions of the phenylalanine aromatic ring.29 All the Boc-FF colloidal suspensions share similar absorption spectral features (Fig. 5b–f), with a high background arising from the scattering of particles in suspension. The absorption spectra show two main bands, at about 210 nm and 250 nm. The latter is broader than that of Boc-FF AcN solution, extending to both higher and lower wavelengths. Conversely, in the dried samples the band at 250 nm almost disappears while the prevalent contribution from the band at 210 nm suggests that the aromatic rings may establish face-to-face interactions typical of H-aggregates.60 The emission spectra recorded by exciting Boc-FF molecules in the AcN solution and in the colloidal suspensions are different, making it possible to identify the self-assembling phenomenon even through fluorescence spectral features (Fig. 5a–f). Upon excitation at 250 nm, Boc-FF in the monomeric state emits at 290 nm while in the aggregate state its emission shifts to 300 nm due to excimer formation.61–64 As a final consideration, it is noteworthy that the generation of peptide architectures is a process under both thermodynamic and kinetic control.65–67 Indeed, self-assembly processes relying on physical interactions are significantly influenced by various factors, including evaporation time, as well as environmental conditions like humidity and temperature.68 Architectures formed at higher temperatures, 85 °C instead of 25 °C, indeed exhibit distinct morphologies.69 They are slightly larger in dimension and show reduced morphologic selectivity, yet they remain quite stable over time (see SI Fig. from S6 to S10, ESI†). The architectures prepared at 25 °C exhibit evolving structures over time, as depicted in Fig. S11 and S12 (ESI†). Interestingly, the re-entrant morphological behaviour remains consistent even with variations in evaporation time, the aggregates grow while retaining significant morphological differentiation, and the morphological recovery induced by the subtle variation of the AcN–H2O ratio is sustained, reinforcing our experimental findings.
Discussion
Our findings reveal that gradually changing the AcN–H2O solvent ratio allows for precise tuning of the morphology in Boc-FF peptide aggregates. As highlighted by OM and SEM images (Fig. 1), even a slight variation in the amount of AcN significantly affects the morphology of Boc-FF architectures, that change from the spherical shape at 2% AcN to long plates at 6% AcN, and finally recovers the spherical form at 10% AcN.AcN and water mixtures, depending on their molar ratio and temperature value, form interpenetrating clusters70 that generate microheterogeneity in solution.35 The latter have a role in determining the microscopic environment around hydrophobic molecules71 and affect molecular interactions such as aromatic stacking, which can be modulated by changing the AcN–H2O ratio.72 However, the structure and composition of the solvation layer around a peptide does not only depend on the microheterogeneities present in the mixture, but also on the molecular groups of the peptide backbone that can generate preferential solvation in such mixed solvents.73,74 As a result, variations in the AcN–H2O ratio have an impact on various levels, including changes in the Boc-FF solubility. Unlike H2O, which can establish H-bonds with both CO and NH groups, AcN can interact with NH and tertbutyl and phenyl hydrophobic groups, possibly leading to the preferential solvation of Boc-FF molecules.75 As for the hierarchical self-assembly of Boc-FF molecules, the interactions most likely responsible of the process are the hydrophobic forces of nonpolar groups and π–π stacking interactions.28 The balancing of all these interactions determines either an isotropic or a preferential growth of the self-assembled architectures, providing both morphological tuning through solvent composition variation and morphological recovery moving from the lowest to the highest AcN concentration. For the smallest amounts of AcN, the hydrophobic effect predominates, resulting in the association of Boc-FF monomers. This is consistent with the spherical shape formed at 2% AcN. On the other hand, when increasing the AcN fraction in the range from 4% to 8%, the preferential solvation around hydrophobic groups favours the anisotropic growth along a specific direction through hydrophobic and π–π stacking interactions.75 Finally, a further increase of the AcN amount to 10%, may interfere with π–π stacking due to the availability of enough AcN molecules, interacting even with phenyl rings, thus producing the recovery of the isotropic morphology.75
Powder XRD measurements reveal the presence of the hexagonal phase only in the presence of water, and the persistence of the orthorhombic phase in all the studied solvation environments. Additionally, the XRD peaks influenced by the varying solvent compositions exclusively belong to the hexagonal phase, wherein solvent molecules are part of the crystalline structure. In this picture, the development of different morphologies depends on the extent of the interface between aggregates and solvent, and on the solvent properties, which promote either the isotropic or preferential growth65,67 of the architectures. Our findings agree with previous works of He et al.,65,67 wherein both microtubes and nanofibers of natural FF have the same X-ray diffraction peaks, which are also very similar to those of the FF single crystal.65,67 This suggests that different morphologies can be generated starting from units having the same peptide conformation.
As for the structural properties of the different aggregates, the FTIR spectra of all the Boc-FF samples show narrow peaks typical of the crystalline form, whose position is consistent with turn conformations of the peptide. A key determinant for the structural properties of the aggregates is the drying time. When samples are dried over 2 h instead of 15 h, the FTIR spectra are characterized by red shifted broader peaks typical of an amorphous state, indicating less ordered turn conformations. During the evolution of the peptide architecture from the amorphous to the crystalline state, Boc-FF molecules undergo intra- and inter-molecular conformational rearrangements and establish strong H-bonds among CO and NH molecular groups. Indeed, in the crystalline form, the FTIR amides signals show a marked change, with the carbamate signal becoming dominant in the spectrum, indicating the formation of an intramolecular H-bond with the amidic NH. Regardless of whether the sample is in an amorphous or crystalline form, all the FTIR spectra exhibit qualitative similarities, suggesting that Boc-FF peptides maintain a consistent conformation, regardless of the aggregate morphology.
As described in previous literature on a comparable system,41 the supramolecular assembly of Boc-FF peptides can be described as a multi-step phase transition process. It has been observed that initially the spheres form rapidly through condensation in the solution phase. Subsequently, they undergo Ostwald ripening over an extended period, ultimately transforming into tubular structures, which represent the thermodynamically most stable form. Considering that, in our case, architectures also tend to lengthen over time while exhibiting re-entrant behaviour, it is plausible that a secondary self-assembly event also occurs in our system.
Unravelling the secondary structure of peptides forming aggregates in both colloidal suspensions and dried samples is not a trivial task. As a cue, Marchesan et al.76 hypothesize that CD signatures are related to a statistical coil, whereby an ensemble of peptides conformational states coexist in equilibrium.76 Based on previous theoretical and experimental studies from Woody et al.77–79 on peptide conformations, the CD spectral features of our colloidal suspensions can be assigned to a distribution of β-conformations.48,77,78 This distribution encompasses both β-sheet and β-turns, with an additional variability influenced by the peptide backbone twist.80,81 The maximum near 200 nm corresponds to the β-turn π–π* transition, and the second maximum at 218 nm is indicative of a n–π* transition. In contrast, the minimum at 226 nm (n–π* transition) can be interpreted as a signature of β-sheet arrangements of Boc-FF molecules.82 The CD spectral features of colloidal suspensions with 2% AcN (Fig. 4b) and 10% AcN (Fig. 4f) exhibit significant differences. This divergence is possibly attributed to the distinct solvation environments surrounding the first aggregate nuclei, namely H2O and AcN, respectively. Indeed, as a confirmation of the influence of the solvation environment, the CD spectra of the 10% AcN colloidal suspension (Fig. 4f) and the solution in AcN (Fig. 4a) reveal identical spectral features, with the only discrepancy lying in intensity, attributable to scattering phenomena that occur in the colloidal suspension. In the case of the 2% (Fig. 4b) and 6% (Fig. 4d) AcN samples, the CD spectra in the colloidal suspensions appear similar, though with substantial differences at low wavelengths. The 2% AcN suspension exhibits a negative Cotton effect at 195 nm and 210 nm, while the 6% AcN suspension displays a positive one. This dissimilarity suggests variations in π–π* and n–π* orbital interactions between the two types of aggregates.83 Furthermore, when comparing the CD spectra of the same samples after drying, the similarity persists between samples dried from pure AcN and those dried from 10% AcN. However, a distinction remains between samples dried from 2% AcN and 10% AcN suspensions. Our data highlight that the crucial factor influencing morphology is the hydration shell around the surface of the aggregates. Conversely, in intermediate phases (4% AcN and 8% AcN), where solvation is preferential but less ordered, the architectures grow in a preferential direction but to a lesser extent compared to the conditions observed in structures formed at 6% AcN. The 6% AcN dried sample demonstrates a stronger dichroic signal, possibly linked to the extension of the architectural ordering in space. These observations support the idea that surface solvation profoundly influences morphology.
 | ||
| Fig. 6 Schematic representation of the mechanism of Boc-FF architecture formation from the molecular to the microscopic length scale. | ||
The interactions among Boc-FF monomers are also responsible for the optical properties of aggregate structures. In particular, the absorption band centred at 250 nm undergoes a significant broadening in the aggregated form. Indeed, aromatic rings can assemble in a parallel orientation, i.e. plane-to-plane stacking (H-aggregates), associated to an hypsochromic shifted H-band, or in a head-to-tail arrangement (J-aggregates), generating a bathochromic shifted J-band.60 The optical properties of H- and J-aggregates are determined by their distinct structural packing, specifically the variation in slip angles among the stacked molecules.60 In colloidal suspensions, where the aggregates do not have a crystalline order, the aromatic rings interact to form both H- and J-aggregates. In dried samples, the hypsochromic shift of the absorption band indicates a preference for H-aggregates. A schematic representation outlining our interpretation of the development of Boc-FF architectures from the molecular to the micrometric level is shown in Fig. 6.
Our findings also indicate that temperature affects the Boc-FF self-assembly process by influencing several possibly competing interactions, such as H-bond interactions and aromatic rings stacking.67 Specifically, the morphology of the aggregates prepared by the incubation of the colloidal suspensions at 85 °C is different from those prepared at 25 °C for the same solvent compositions. Boc-FF peptides adopt turn conformations in all the aggregated forms and these structures remain stable after one day even without solvent removal, indicating that the aggregates have attained their stable form. The fact that temperature is a key parameter in controlling the self-assembly of FF aggregates has been already observed in the irreversible phase transition from the hexagonal to the orthorhombic crystalline phase of FF nanotubes at 140 °C.84 In addition, FF nanowires are aligned in a vertical direction starting from the peptide amorphous films, aging them at 150 °C under aniline vapor.85 Moreover, a thermo-responsive organogel, undergoing a structural transition to a crystalline nanowire at 95 °C, has developed through a temperature-induced reversible self-assembly of FF peptide in isopropanol.67 In conclusion, morphological control of structures formed by weak variations of the AcN–H2O ratio can be achieved either through temperature regulation or by inducing solvent evaporation at different times. In our work, a control over the length of the plates can be reached, and the Boc-FF molecule with 6% AcN and 8% AcN demonstrates the ability to form branched structures, which can be used as a scaffold for the systematic introduction of fluorescent dyes for energy and/or biomedical applications.
Conclusions
In this work, Boc-FF peptide aggregates are prepared using a mixed-solvents protocol, with AcN as the organic solvent and water as the solvent inducing aggregation. The self-assembly process of Boc-FF peptide is studied at different solvent ratios ranging from 2% to 10% v/v of AcN. We find that peptides spontaneously assemble into either spheres or plates depending on the solvent ratio, showcasing a capability for morphological re-entrant behaviour. Colloidal suspensions quickly form upon water addition, and Boc-FF peptides arrange themselves in a distribution of turn and β-sheet conformations. Here, aromatic rings interact in both parallel and head-to-tail arrangements depending on the peptide dihedral angles. The first nuclei formed in the colloidal suspension grow over time until the solvent is removed,38,39 while retaining memories of the first morphological differentiation. After solvent withdrawn, only the turn conformation remains, and the Boc-FF peptide crystallizes, possibly forming an orthorhombic crystalline structure. This phase coexists with a hexagonal one that occurs when the architectures originate in the presence of over 90% water. Interestingly, we observe that the peptides adopt the same conformations and crystalline structures regardless of the morphology of the final architectures. We find that the isotropic or preferential growth of aggregates may be due to the preferential solvation of AcN along the peptide backbone, and that the aggregation process is dominated by hydrophobic interactions and π–π stacking. The latter interaction is responsible for the growth along a preferential axis, but when AcN content exceeds the threshold of 10% the isotropic growth is recovered. Our findings offer the potential for precise control over the morphology of the Boc-FF architectures, driven by hydrophobic and aromatic interactions, through the slight variation of the AcN content. Temperature control and evaporation time parameters can also be employed to influence the final properties of peptide architectures. This simple bottom-up approach could open additional routes for the growth of peptide-based materials with different building block morphologies. This may provide a novel platform for designing a variety of versatile peptide structures for new biomaterial fabrications.Author contributions
Conceptualization S. Catalini, M. Di Donato, S. Cicchi, P. Foggi, L. Comez and A. Paciaroni; methodology S. Catalini, validation S. Catalini and F. Bagni; formal analysis S. Catalini; investigation S. Catalini, F. Bagni, A. Di Michele, G. Schirò, A. Lapini, A. Iagatti and M. Paolantoni; resources S. Cicchi, P. Foggi, A. Di Michele, G. Schirò, M. Paolantoni, L. Comez and A. Paciaroni; data curation S. Catalini; writing original draft S. Catalini, L. Comez and A. Paciaroni; writing review & editing all authors contributed to the revision of the final version of this manuscript; visualization S. Catalini, L. Comez and A. Paciaroni; supervision L. Comez and A. Paciaroni; project administration A. Paciaroni and C. Petrillo funding acquisition A. Paciaroni, C. Petrillo, L. Comez, A. Iagatti and A. Lapini.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. C. and A. P. thank the research project “FSE-REACT EU” financed by National Social Fund–National Operative Research Program and Innovation 2014–2020 (D. M. 1062/2021), personal Grant number 23-G-15445-3. S. C., P. F., C. P., A. D. M., M. P., L. C. and A. P. thank the European Union-NextGenerationEU project under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041-VITALITY-CUP: J97G22000170005 and B43C22000470005. S. C. and G. S. thank A. Royant and S. Engilberge of the FIP2-BM07 beamline of the European Synchrotron Radiation Facility (ESRF) (Grenoble) for providing their internal beamtime and support for XRD measurements. L. C., A. I., and A. L. thank Ministero dell’Istruzione dell’Università e della Ricerca Italiano LANTERN (PRIN2022-20225NPY8P).References
- L. Wang, R. Lai, L. Zhang, M. Zeng and L. Fu, Adv. Mater., 2022, 34, 2201956 CrossRef CAS PubMed.
- J. Wang, J. Gong and Z. Wei, AAPS PharmSciTech, 2022, 23, 1–14 CrossRef PubMed.
- Y. Yao, S. Catalini, B. Kutus, J. Hunger, P. Foggi and R. Mezzenga, Angew. Chem., Int. Ed., 2021, 60, 25274–25280 CrossRef CAS PubMed.
- B. Sun, K. Tao, Y. Jia, X. Yan, Q. Zou, E. Gazit and J. Li, Chem. Soc. Rev., 2019, 48, 4387–4400 RSC.
- T. P. Pivetta, C. E. A. Botteon, P. A. Ribeiro, P. D. Marcato and M. Raposo, Nanomaterials, 2021, 11, 3132 CrossRef CAS PubMed.
- M. Diener, J. Adamcik, J. Bergfreund, S. Catalini, P. Fischer and R. Mezzenga, ACS Macro Lett., 2020, 9, 115–121 CrossRef CAS PubMed.
- S. Catalini, A. Taschin, P. Bartolini, P. Foggi and R. Torre, Appl. Sci., 2019, 9 Search PubMed.
- S. Catalini, V. Lutz-Bueno, M. Usuelli, M. Diener, A. Taschin, P. Bartolini, P. Foggi, M. Paolantoni, R. Mezzenga and R. Torre, iScience, 2022, 25, 1–15 CrossRef PubMed.
- S. Catalini, D. R. Perinelli, P. Sassi, L. Comez, G. F. Palmieri, A. Morresi, G. Bonacucina, P. Foggi, S. Pucciarelli and M. Paolantoni, Biomacromolecules, 2021, 22, 1147–1158 CrossRef CAS PubMed.
- Q. Zou, K. Liu, M. Abbas and X. Yan, Adv. Mater., 2016, 28, 1031–1043 CrossRef CAS PubMed.
- Z. C. Adams, E. J. Olson, T. L. Lopez-Silva, Z. Lian, A. Y. Kim, M. Holcomb, J. Zimmermann, R. Adhikary and P. E. Dawson, Chem. Sci., 2022, 13, 10020–10028 RSC.
- R. Chang, C. Yuan, P. Zhou, R. Xing and X. Yan, Acc. Chem. Res., 2024, 57, 289–301 CrossRef CAS PubMed.
- C. Yuan, Q. Li, R. Xing, J. Li and X. Yan, Chem, 2023, 9, 2425–2445 CAS.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS.
- I. Insua, A. Cardellini, S. Díaz, J. Bergueiro, R. Capelli, G. M. Pavan and J. Montenegro, Chem. Sci., 2023, 14, 14074–14081 RSC.
- A. Nandy and S. Mukherjee, J. Phys. Chem. Lett., 2022, 13, 6701–6710 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- N. Amdursky, M. Molotskii, E. Gazit and G. Rosenman, J. Am. Chem. Soc., 2010, 132, 15632–15636 CrossRef CAS PubMed.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- J. H. Kim, M. Lee, J. S. Lee and C. B. Park, Angew. Chem., 2012, 124, 532–535 CrossRef.
- M. I. Souza, T. Prieto, T. Rodrigues, F. F. Ferreira, F. B. Nascimento, A. O. Ribeiro, E. R. Silva, F. Giuntini and W. A. Alves, Sci. Rep., 2017, 7, 13166 CrossRef PubMed.
- K. Karikis, A. Butkiewicz, F. Folias, G. Charalambidis, C. Kokotidou, A. Charisiadis, V. Nikolaou, E. Nikoloudakis, J. Frelek, A. Mitraki and A. G. Coutsolelos, Nanoscale, 2018, 10, 1735–1741 RSC.
- C. Guo, Y. Luo, R. Zhou and G. Wei, ACS Nano, 2012, 6, 3907–3918 CrossRef CAS PubMed.
- C. Guo, Z. A. Arnon, R. Qi, Q. Zhang, L. Adler-Abramovich, E. Gazit and G. Wei, ACS Nano, 2016, 10, 8316–8324 CrossRef CAS PubMed.
- Q. Xiong, Y. Jiang, X. Cai, F. Yang, Z. Li and W. Han, ACS Nano, 2019, 13, 4455–4468 CrossRef CAS PubMed.
- Y. Pang, Q. Zhang, X. Sun, J. Ji, F. Pi and X. Shen, Int. J. Electrochem. Sci., 2018, 13, 8518–8529 CrossRef CAS.
- R. C. G. Creasey, I. Louzao, Z. A. Arnon, P. Marco, L. Adler-Abramovich, C. J. Roberts, E. Gazit and S. J. B. Tendler, Soft Matter, 2016, 12, 9451–9457 RSC.
- C. Diaferia, E. Rosa, G. Morelli and A. Accardo, Pharmaceuticals, 2022, 15, 2–14 CrossRef PubMed.
- A. Handelman, N. Kuritz, A. Natan and G. Rosenman, Langmuir, 2016, 32, 2847–2862 CrossRef CAS PubMed.
- M. P. Conte, N. Singh, I. R. Sasselli, B. Escuder and R. V. Ulijn, Chem. Commun., 2016, 52, 13889–13892 RSC.
- E. Nikoloudakis, K. Mitropoulou, G. Landrou, G. Charalambidis, V. Nikolaou, A. Mitraki and A. G. Coutsolelos, Chem. Commun., 2019, 55, 14103–14106 RSC.
- A. Arul, P. Rana, K. Das, I. Pan, D. Mandal, A. Stewart, B. Maity, S. Ghosh and P. Das, Nanoscale Adv., 2021, 3, 6176–6190 RSC.
- Q. Li, Y. Jia, L. Dai, Y. Yang and J. Li, ACS Nano, 2015, 9, 2689–2695 CrossRef CAS PubMed.
- S. Chaudhari, Y. Kwon and M. Y. Shon, Bull. Korean Chem. Soc., 2019, 40, 220–229 CrossRef CAS.
- T. Takamuku, M. Tabata, A. Yamaguchi, J. Nishimoto, M. Kumamoto, H. Wakita and T. Yamaguchi, J. Phys. Chem. B, 1998, 102, 8880–8888 CrossRef CAS.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- Y. Zhao, L. Deng, J. Wang, H. Xu and J. R. Lu, Langmuir, 2015, 31, 12975–12983 CrossRef CAS PubMed.
- C. Yuan, R. Xing, J. Cui, W. Fan, J. Li and X. Yan, CCS Chem., 2023, 1–11 Search PubMed.
- C. Yuan, M. Yang, X. Ren, Q. Zou and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 17456–17460 CrossRef CAS PubMed.
- L. Pérez-Picaso, J. Escalante, H. F. Olivo and M. Y. Rios, Molecules, 2009, 14, 2836–2849 CrossRef PubMed.
- A. Levin, T. O. Mason, L. Adler-Abramovich, A. K. Buell, G. Meisl, C. Galvagnion, Y. Bram, S. A. Stratford, C. M. Dobson, T. P. J. Knowles and E. Gazit, Nat. Commun., 2014, 5(1), 5219 CrossRef CAS PubMed.
- Q. Li, Y. Jia, L. Dai, Y. Yang and J. Li, ACS Nano, 2015, 9, 2689–2695 CrossRef CAS PubMed.
- C. Igathinathane, L. O. Pordesimo, E. P. Columbus, W. D. Batchelor and S. R. Methuku, Comput. Electron. Agric., 2008, 63, 168–182 CrossRef.
- J. Kieffer and D. Karkoulis, J. Phys.: Conf. Ser, 2013, 425 Search PubMed.
- J. Kieffer and J. Wright, Powder Diffr., 2013, 28, 339–35038 CrossRef.
- S. Bera, P. Jana, S. K. Maity and D. Haldar, Cryst. Growth Des., 2014, 14, 1032–1038 CrossRef CAS.
- C. H. Görbitz, Chem. – Eur. J., 2001, 7, 5153–5159 CrossRef.
- S. Kralj, O. Bellotto, E. Parisi, A. M. Garcia, D. Iglesias, S. Semeraro, C. Deganutti, P. D’Andrea, A. V. Vargiu, S. Geremia, R. De Zorzi and S. Marchesan, ACS Nano, 2020, 14, 16951–16961 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Nat. Nanotechnol., 2006, 1, 195–200 CrossRef CAS PubMed.
- D. Haldar, M. G. B. Drew and A. Banerjee, Tetrahedron, 2006, 62, 6370–6378 CrossRef CAS.
- S. Maity, P. Kumar and D. Haldar, Org. Biomol. Chem., 2011, 9, 3787–3791 RSC.
- C. H. Görbitz, Chem. – Eur. J., 2007, 13, 1022–1031 CrossRef PubMed.
- Y. Ji, X. Yang, Z. Ji, L. Zhu, N. Ma, D. Chen, X. Jia, J. Tang and Y. Cao, ACS Omega, 2020, 5, 8572–8578 CrossRef CAS PubMed.
- N. Wilkosz, M. Czaja, S. Seweryn, K. Skirlinska-Nosek, M. Szymonski, E. Lipiec and K. Sofinska, Molecules, 2020, 25, 2–26 CrossRef PubMed.
- G. Ramer, F. S. Ruggeri, A. Levin, T. P. J. Knowles and A. Centrone, ACS Nano, 2018, 12, 6612–6619 CrossRef CAS PubMed.
- J. Xia, B. Sun, C. Wang, N. Sun, H. Cao, Y. Jia, Y. Yang and J. Li, J. Colloid Interface Sci., 2021, 599, 661–666 CrossRef CAS PubMed.
- A. Adochitei and G. Drochioiu, Roumanian J. Chem., 2011, 56, 783–791 CAS.
- M. Gupta, A. Bagaria, A. Mishra, P. Mathur, A. Basu, S. Ramakumar and V. S. Chauhan, Adv. Mater., 2007, 19, 858–861 CrossRef CAS.
- D. M. Rogers, S. B. Jasim, N. T. Dyer, F. Auvray, M. Réfrégiers and J. D. Hirst, Chem, 2019, 5, 2751–2774 CAS.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- S. M. Hsu, F. Y. Wu, T. S. Lai, Y. C. Lin and H. C. Lin, RSC Adv., 2015, 5, 22943–22946 RSC.
- X. Liu, J. Fei, A. Wang, W. Cui, P. Zhu and J. Li, Angew. Chem., 2017, 129, 2704–2707 CrossRef.
- Z. Gan and H. Xu, Macromol. Rapid Commun., 2017, 38 Search PubMed.
- M. Pellach, S. Mondal, L. J. W. Shimon, L. Adler-Abramovich, L. Buzhansky and E. Gazit, Chem. Mater., 2016, 28, 4341–4348 CrossRef CAS.
- R. Huang, W. Qi, R. Su, J. Zhao and Z. He, Soft Matter, 2011, 7, 6418–6421 RSC.
- J. Anderson, P. T. Lake and M. McCullagh, J. Phys. Chem. B, 2018, 122, 12331–12341 CrossRef CAS PubMed.
- R. Huang, Y. Wang, W. Qi, R. Su and Z. He, Nanoscale Res. Lett., 2014, 9, 1–9 CrossRef CAS PubMed.
- J. Chen, S. Qin, X. Wu and P. K. Chu, ACS Nano, 2016, 10, 832–838 CrossRef CAS PubMed.
- Y. Su, X. Yan, A. Wang, J. Fei, Y. Cui, Q. He and J. Li, J. Mater. Chem., 2010, 20, 6734–6740 RSC.
- R. D. Mountain, J. Phys. Chem. A, 1999, 103, 10744–10748 CrossRef CAS.
- A. Wakisaka, Y. Shimizu, N. Nishi, K. Tokumaru and H. Sakuragi, J. Chem. Soc., Faraday Trans., 1992, 88, 1129–1135 RSC.
- Y. Lin, M. Penna, M. R. Thomas, J. P. Wojciechowski, V. Leonardo, Y. Wang, E. T. Pashuck, I. Yarovsky and M. M. Stevens, ACS Nano, 2019, 13, 1900–1909 CAS.
- A. Wakisaka, J. S. Takahashi and N. Nishi, J. Chem. Soc., Faraday Trans., 1995, 91, 4063–4069 RSC.
- A. Wakisaka, S. Komatsu and Y. Usui, J. Mol. Liq., 2001, 90, 175–184 CrossRef CAS.
- I. Rosbottom, C. Y. Ma, T. D. Turner, R. A. O’Connell, J. Loughrey, G. Sadiq, R. J. Davey and K. J. Roberts, Cryst. Growth Des., 2017, 17, 4151–4161 CrossRef CAS.
- A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862–1876 CAS.
- Z. Shi, K. Chen, Z. Liu, A. Ng, W. Clay Bracken and N. R. Kallenbach, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17964–17968 CrossRef CAS PubMed.
- L. He, A. E. Navarro, Z. Shi and N. R. Kallenbach, J. Am. Chem. Soc., 2012, 134, 1571–1576 CrossRef CAS PubMed.
- C. Toniolo, F. Formaggio and R. W. Woody, Electron. Circ. Dichroism Pept., 2012, 2, 499–544 CAS.
- C. A. Bush, S. K. Sarkar and K. D. Kopple, Biochemistry, 1978, 17, 4951–4954 CrossRef CAS PubMed.
- J. Bandekar, D. J. Evans, S. Krimm, S. J. Leach, S. Lee, J. R. McQuie, E. Minasian, G. Némethy, M. S. Pottle, H. A. Scheraga, E. R. Stimson and R. W. Woody, Int. J. Pept. Prot. Res., 1982, 19, 187–205 CrossRef CAS PubMed.
- X. Yan, Y. Cui, Q. He, K. Wang and J. Li, Chem. Mater., 2008, 20, 1522–1526 CrossRef CAS.
- D. M. Rogers, S. B. Jasim, N. T. Dyer, F. Auvray, M. Réfrégiers and J. D. Hirst, Chem, 2019, 5, 2751–2774 CAS.
- A. Heredia, I. Bdikin, S. Kopyl, E. Mishina, S. Semin, A. Sigov, K. German, V. Bystrov, J. Gracio and A. L. Kholkin, J. Phys. D: Appl. Phys., 2010, 43, 462001 CrossRef.
- J. Ryu and C. B. Park, Adv. Mater., 2008, 20, 3754–3758 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00018h |
| This journal is © The Royal Society of Chemistry 2024 |