
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of polyethylene glycol and polydopamine coatings on the performance of camptothecin-loaded liposomes for localised treatment of colorectal cancer†
Anna Maria
Maurelli
 abc,
Bárbara
Ferreira
bc,
Sofia
Dias
bcd,
Helena
Almeida
bcd,
Vincenzo
De Leo
*a,
Bruno
Sarmento
bce,
Lucia
Catucci
*a and
José
das Neves
*bce
abc,
Bárbara
Ferreira
bc,
Sofia
Dias
bcd,
Helena
Almeida
bcd,
Vincenzo
De Leo
*a,
Bruno
Sarmento
bce,
Lucia
Catucci
*a and
José
das Neves
*bce
aDepartment of Chemistry, University of Bari Aldo Moro, Via Orabona 4, 70126 Bari, Italy. E-mail: vincenzo.deleo@uniba.it; lucia.catucci@uniba.it
bi3S – Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Rua Alfredo Allen 208, Porto, 4200-135, Portugal. E-mail: j.dasneves@i3s.up.pt
cINEB – Instituto de Engenharia Biomédica, Universidade do Porto, Rua Alfredo Allen 208, Porto, 4200-135, Portugal
dICBAS – Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, Rua Jorge de Viterbo Ferreira 228, 4050-313, Porto, Portugal
eIUCS – Instituto Universitário de Ciências da Saúde, Rua Central de Gandra 1317, Gandra, 4585-116, Portugal
First published on 4th April 2024
Abstract
Management of colorectal cancer (CRC) remains a global healthcare challenge as current therapies are unable to offer optimal clinical efficacy and meet patient expectations. The oral route is particularly appealing for managing CRC due to its low invasiveness and direct access to affected areas, making it possible for localised action. However, different barriers oppose achieving proper drug concentration at tumour sites. In this work, the drug candidate camptothecin was incorporated into liposomes to overcome its poor solubility and pH instability, as well as to enhance drug distribution in the gastrointestinal tract. Two polymeric coatings were tested to improve liposome performance, namely ‘standard’ polyethylene glycol (PEG) and ‘novel’ polydopamine (PDA). Liposomes were characterized for physicochemical properties, in vitro drug release, mucus transport, in vitro intestinal permeability and anticancer activity using HCT 116 cell-based 2D and 3D (spheroids) models. Drug-loaded liposomes (130–157 nm) were successfully prepared and featured sustained drug release in intestinal fluids in a pH-independent manner. Drug release was slower in the case of PDA-coating and consistent with long-reaching colorectal drug delivery. Both polymers conferred sub-diffusive behaviour to liposomes in a mucus surrogate and allowed a mild increase (up to roughly 2.5-fold) in epithelial drug permeability as compared to free camptothecin. Anticancer activity was maintained or even increased for liposomal formulations. For instance, camptothecin-loaded, PDA-coated liposomes were over 2-fold more cytotoxic to HCT 116 cell spheroids than the free drug. Overall, proposed formulations, namely PDA-modified liposomes, present the potential to be used for localised treatment of CRC.
1. Introduction
Colorectal cancer (CRC) remains a considerable healthcare burden, with increasing numbers in both global incidence and mortality.1 Advances in its treatment have been considerable over the last couple of decades, namely with the introduction of anti-epidermal growth factor receptor and anti-vascular endothelial growth factor monoclonal antibodies.2 However, the standard of care is still unable to achieve optimal clinical efficacy and safety or meet specific patient preferences and expectations. For example, patients at earlier stages of the disease (stage II and III) seem to prefer oral drug regimens to the detriment of injectable ones.3 Oral therapy also reduces the need for patients to resort to healthcare facilities for each administered dose. Moreover, the oral route appears quite attractive for therapy of localised CRC tumours due to its low invasiveness, and the possibility of targeted delivery to affected areas of the gastrointestinal tract (GIT), with a potential reduction in administered dose and side effects of injectable chemotherapy.4,5 Still, drugs and dosage forms with indication for oral treatment of CRC are limited in number and usage (typically only capecitabine and tegafur–uracil)6,7 and are intended to promote systemic drug absorption and distribution rather than promote localised treatment. Also, the frequent need for concomitant intravenous administration of other drugs (e.g., oxaliplatin) limits the benefits of currently available oral anticancer drugs.8,9 Camptothecin (CPT) is a natural alkaloid anticancer drug isolated from Camptotheca acuminata Decne.10 It presents the potential to serve as an alternative to thymidylate synthase inhibitors currently used for oral CRC therapy, acting through the inhibition of the DNA topoisomerase I via the establishment of non-covalent interactions.11 However, the drug presents some long known limitations, namely its poor water solubility and pH-dependent degradation.12,13 Above pH 5.5 the lactone form of CPT converts into the carboxylate one, which is characterized by reduced anticancer activity.14,15Several nanotechnology-based formulation approaches have been proposed to fully or partially overcome these issues, including the association of CPT to carriers such as liposomes,16,17 silicon nanoparticles (NPs),18 solid lipid NPs,19,20 polymeric micelles,21 poly(anhydride) NPs/cyclodextrins,22 and silica-based antioxidant NPs.23 Some of these systems, however, aimed at incrementing the solubility of CPT or at allowing targeted tumour delivery upon administration by parenteral routes.16–18,20 Others actually addressed the use of CPT-loaded nanocarriers for oral chemotherapy19,21–24 underlining, for instance, the need to boost drug performance at the gut, namely by tackling relevant chemical (e.g. pH, enzymes) and physical (e.g. mucus) barriers.
Bearing in mind the previous aims, we set out to further explore how differently coated liposomes could serve as carriers of CPT for localised treatment of CRC. Our choice for liposomes lies on the previous experience of our team with this type of carrier,25 as well as on reports that these systems can prevent the inactivation of CPT at neutral-alkaline conditions.26 Alongside size, surface properties are also known to determine the interactions and ultimately, the transport of nanocarriers in mucus.27 Mucus acts as a stringent physical barrier to most particulate systems, but the use of non-adhesive coatings can radically change transport behaviour. The most commonly used strategy involves dense surface modification of nanosystems with polyethylene glycol (PEG) of low molecular mass.25,28–30 Moreover, surface modification of simple liposomes made of phospholipids and cholesterol with PEG (as well as other polymers) can increase colloidal stability and improve the resistance of drug payloads to the action of gastric acids, bile salts and enzymes, as well as prevent premature drug release.31–36 One particular polymer of interest to our team has been polydopamine (PDA), a bioinspired and biocompatible material, which can be easily obtained in situ via self-polymerization of dopamine (DA) under weak-alkaline conditions.37,38 Recent works suggest that PDA-based nanocarriers are safe for mucosal use and do not trigger immune response upon single administration.39,40 Additionally, recent reports highlighted that PDA can also enhance the colloidal stability of coated nanosystems,41,42 while potentially facilitating mucus penetration due to its zwitterionic behaviour that is able to decrease interactions with mucins.43–45 However, to our best knowledge, this polymer has never been tested for coating liposomes intended for oral delivery.
In this work, we developed CPT-loaded liposomes with diameter in the range of 100–200 nm and coated with two different polymers, viz. PEG or PDA, and compared their performance as relevant to oral delivery and localised CRC therapy. In particular, we evaluated (i) the colloidal properties of liposomes; (ii) the in vitro release of CPT in simulated GIT fluids; (iii) the transport in an intestinal mucus surrogate; (iv) the drug permeability across two intestinal epithelial models; and (v) the anticancer activity using both 2D and 3D (tumour spheroids) cell models.
2. Materials and methods
2.1. Materials
Lipoid S 100 (≥94% soybean phosphatidylcholine) was purchased from Lipoid (Ludwigshafen, Germany); 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG-2000) from Avanti Polar Lipids (Alabaster, AL, USA); CPT (sodium salt) from Biosynth (Compton, UK); cholesterol, dopamine hydrochloride, sodium phosphate dibasic, potassium phosphate monobasic, potassium chloride, N,N-dimethyl-n-dodecylamine-N-oxide (LDAO), resazurin and purified type II mucin (from porcine stomach) from Merck (Darmstadt, Germany); sodium chloride, Triton X-100, Dulbecco's Modified Eagle Medium (DMEM; with GlutaMAX™), RPMI 1640, Hanks’ Balanced Salt Solution (HBSS) and non-essential amino acids solution 100X from Thermo Fisher Scientific (Waltham, MA, USA); agarose (SeaKem® LE) from Lonza (Norwest, Australia); DiOC18(3) from Biotium (San Francisco, CA, USA); and fetal bovine serum (FBS) and penicillin/streptomycin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U/10000 μg ml−1) from PAN-Biotech (Aidenbach, Germany). All other materials and reagents were of analytical grade or equivalent.
000 U/10000 μg ml−1) from PAN-Biotech (Aidenbach, Germany). All other materials and reagents were of analytical grade or equivalent.
2.2. Cell lines origin and maintenance
HCT 116 and Caco-2 (C2BBe1 clone) human colorectal carcinoma cell lines were obtained from ATCC (Manassas, VA, USA), and the mucus-producing HT29-MTX human colorectal carcinoma cell line was kindly provided by Dr T. Lesuffleur (INSERM U178, Villejuif, France). Caco-2 and HT29-MTX cells were maintained in DMEM, supplemented with 10% (v/v) heat-inactivated FBS, penicillin (100 U mL−1) and streptomycin (100 μg mL−1), and 1% (v/v) non-essential amino acids. HCT 116 cells were cultured in RPMI 1640, supplemented with 10% (v/v) heat-inactivated FBS, penicillin (100 U mL−1) and streptomycin (100 μg mL−1). Cells were maintained under standard culture conditions (37 °C, 5% CO2 and 95% RH).2.3. Preparation of liposomes
Non-coated CPT-loaded liposomes (CPT-L) were prepared using the thin-film hydration method followed by homogenization through extrusion, as previously detailed.32 In brief, 200 μL of Lipoid S 100 in chloroform (80 mg mL−1), 80 μL of cholesterol in chloroform (20 mg mL−1) and 150 μL of CPT in methanol (10 mg mL−1) were mixed. The blend was then dried under gentle nitrogen flux followed by vacuum. The lipid/drug film was dispersed in one millilitre of phosphate buffered saline solution (PBS, pH 7.4), vortexed and sonicated for three minutes to prepare multilamellar vesicles. The dispersion was further extruded across polycarbonate membranes (Avanti Polar Lipids) with nominal pore diameter of 200 nm and 100 nm, in a sequential manner, in order to obtain unilamellar liposomes.CPT-loaded liposomes coated with PEG (CPT-L-PEG) were similarly prepared by replacing one milligram of Lipoid S 100 with 3.68 mg of DSPE-PEG-2000, starting from a stock solution in chloroform at 40 mg mL−1 concentration, (PEG-2000 final molar ratio of 7%) and by dispersing the dried film in PBS (pH 7.4). CPT-loaded liposomes coated with PDA (CPT-L-PDA) were also prepared from non-coated CPT-L as previously described.38 Polymer formation and deposition at the surface of liposomes was achieved by incubating DA (4 mg mL−1) with CPT-L (16 mg mL−1) for one hour at 35 °C in PBS (pH 7.4). Excess DA and CPT were removed by overnight dialysis (10 kDa MWCO) against PBS (pH 7.4).
2.4. Physicochemical and technological characterization of liposomes
CPT content in liposomes was evaluated spectroscopically upon conversion of liposomes into mixed micelles using LDAO (4% v/v) to abbreviate the scattering contribution from the UV-Vis spectra. The drug was quantified by monitoring the absorbance at 420 nm. Results were used to calculate the percentages of encapsulation efficiency (EE%) and loading capacity (LC%) of the drug into liposomes as follows: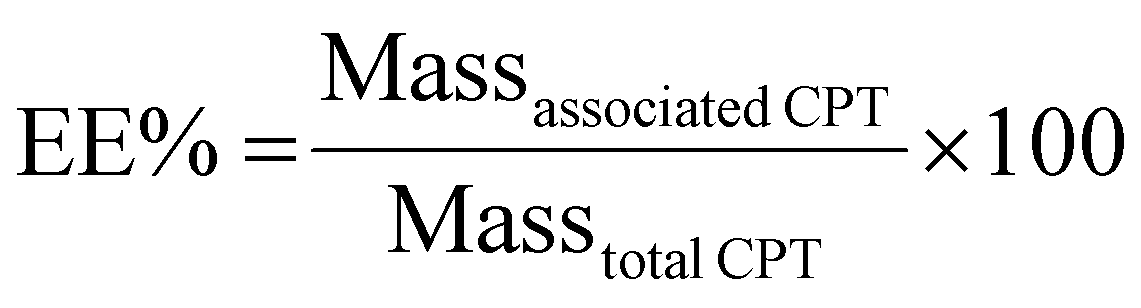 | (1) |
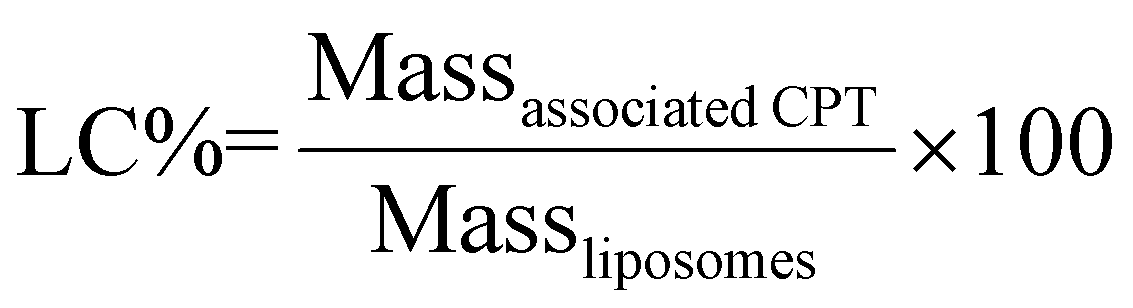 | (2) |
The hydrodynamic diameter and polydispersity index (PDI) of liposomes dispersed in PBS buffer were investigated by dynamic light scattering (DLS) analysis using a Zetasizer Nano ZS (Malvern Panalytical, Malvern, UK). The same equipment was used to assess the ζ-potential of liposomes diluted in ultrapure water by laser Doppler anemometry. In all cases, liposomes were dispersed at an approximate concentration of 0.8 mg mL−1 before conducting measurements. Size and morphology of liposomes were also assessed by transmission electron microscopy (TEM). Micrographs were acquired after negative staining of samples with uranyl acetate 1% using a JEM 1400 microscope (JEOL, Tokyo, Japan).
In vitro drug release experiments were performed using a dialysis-based method. First, the release was assessed sequentially in simulated gastric and intestinal fluids. To this end, liposome samples (one millilitre containing 130–230 μg mL−1 of CPT) were placed into dialysis tubes (12.4 kDa MWCO) and immersed in 14 mL of a gastric simulated fluid (sodium chloride 0.2% w/v, 1 M hydrochloric acid 8% v/v, pH 1.2). After two hours, the dialysis tube was transferred to 14 mL of an intestinal simulated fluid (potassium phosphate monobasic 0.68% w/v, sodium hydroxide 0.2 N 7.7%, pH 6.8) and the assay continued for an additional 4 h. To isolate and better understand the behaviour of liposomes in the colorectum, the release assay was further performed solely in intestinal simulated fluid (pH 6.8) over 24 h. In all cases, drug release was monitored throughout the experiment by collecting 400 μL of release medium at pre-defined time intervals and replacing it with fresh buffer. The amount of CPT released was monitored through fluorescence spectroscopy (370/450 nm). Cumulative drug release (Q%) was calculated according to the following equation:
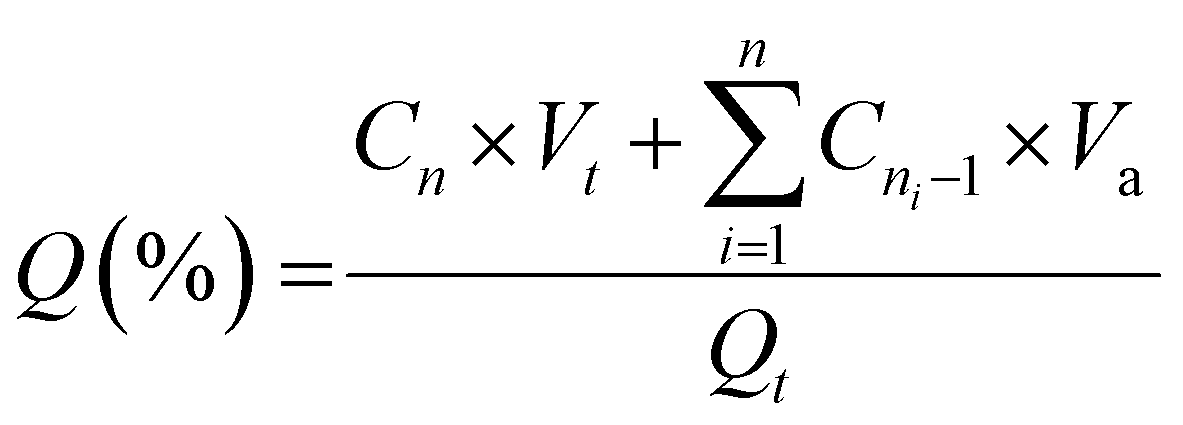 | (3) |
2.5. Transport of liposomes in intestinal mucus surrogate
An intestinal mucus surrogate was prepared by dissolving mucin in PBS (pH 7.4) at 50 mg mL−1 and leaving it to stabilize for 30 min at room temperature before use. Then, 65 μL of this fluid was mounted onto a glass microscope limited by 1.5 × 1.6 cm gene frames (Thermo Fisher Scientific). Two microlitres of a diluted dispersion of fluorescent liposomes (0.0025% w/v in PBS) were added on the top of the mucus surrogate, sealed with a coverslip, and left to equilibrate for at least 30 min before the analysis. Fluorescent liposomal formulations were obtained by simply adding one milligram of DiOC18(3) to the initial lipid blend, omitting the incorporation of CPT, and purifying the vesicles by dialysis (10 kDa MWCO).The transport of liposomes was assessed by multiple particle tracking (MPT) analysis as previously detailed.47 Briefly, real-time videos (1024 × 1024 pixels, 16-bit, 75 s duration, 250 msec temporal resolution) were captured using a ORCAFlash4.0 digital CMOS camera (Hamamatsu, Japan) mounted on a Leica DMI6000 inverted epifluorescence microscope (Wetzlar, Germany). Videos were then processed for background subtraction and extraction of individual liposome trajectories using Fiji software (v. 2.15.0)48 and the MosaicSuite 2D/3D single-particle tracking plugin.49 Data were then analysed by the in-house developed software MPTHub (v. 1.1.0)47 and trajectories containing at least 30 consecutive frames were considered for calculating values of time-averaged mean square displacements (MSD), effective diffusivity (Deff), as previously detailed.47 Ensemble MSD (<MSD>) values were further used to calculate the anomalous exponent (α). Values of α allow classifying liposomes as immobile (α < 0.2), sub-diffusive (0.2 ≤ α < 0.9) and diffusive (α ≥ 0.9). Results of Deff for each formulation were also compared with those of the theoretical diffusion coefficient of spherical particles with similar size in water (Dw), calculated according to the Stokes–Einstein equation. This comparison allows providing an idea of the level of transport impairment of liposomes in mucus.47
2.6. In vitro intestinal drug permeability
Colorectal permeability of CPT was assessed using Caco-2 and Caco-2/HT29-MTX cell monolayer models.50 Although routinely used for evaluating drug permeability at the small intestine, these models have also been shown useful for predicting drug behaviour at the colorectum.21,51 The Caco-2 cell monolayer model was established by seeding 105 cells per cm2 in CellQART® hanging cell culture inserts (PET, 12-well, 1 μm; SABEU, Northeim, Germany) for 21 days under standard cell culture conditions. The medium was renewed trice weekly and the transepithelial electrical resistance (TEER) was monitored throughout using a EVOM2 voltohmmeter (World Precision Instruments, Sarasota, FL, USA). The Caco-2/HT29-MTX cell monolayer model was established similarly by seeding both cell lines simultaneously at a cell ratio of 9:1.50
Just before proceeding with permeability experiments, the culture media were discarded, and the cell monolayers were washed twice with PBS. The system was then equilibrated in HBSS for 30 min at 37 °C under orbital shaking (100 rpm). Permeability of CPT was assessed by adding the free drug or CPL-loaded liposomes dispersed in 0.5 mL of HBSS at a drug concentration of 10 μg mL−1 in the apical compartment. This concentration was selected based on preliminary cell viability experiments using the resazurin reduction assay that showed no apparent toxicity of drug/liposomes to Caco-2 or HT29-MTX cells. Samples (200 μL) were collected at pre-determined time points from the basolateral compartment and the volume was replenished to 1.5 mL with fresh HBSS. TEER was monitored throughout time points. The amount of CPT that permeated cell monolayers was quantified by fluorescence spectroscopy and used for calculating the apparent permeability coefficient (Papp) as follows:
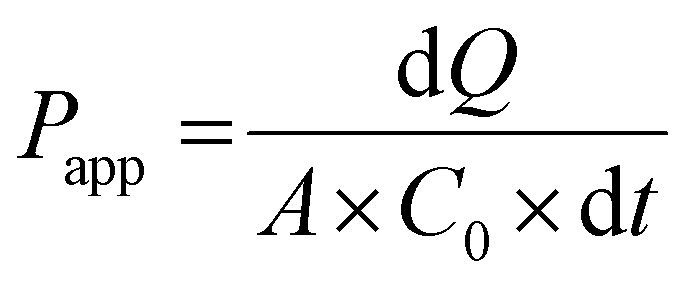 | (4) |
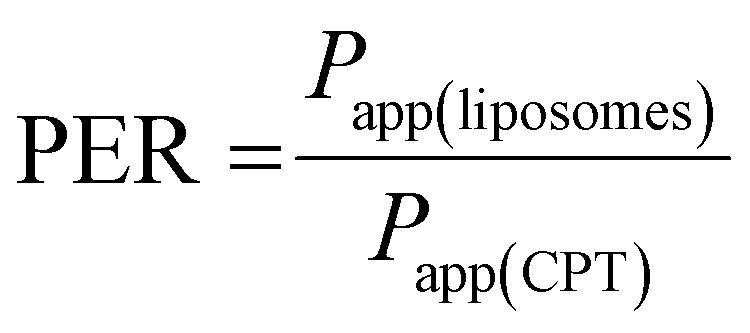 | (5) |
2.7. In vitro anticancer activity
The cytotoxicity potential of CPT-loaded liposomes was evaluated using the HCT 116 colorectal cancer cell line, both in 2D and 3D configurations. In the case of the former, HCT 116 cells were seeded in 96-well plates at a density of 2 × 104 cells per well and left under standard culture conditions for 24 h. Cells were then washed twice with PBS and incubated for an additional 24 h with different concentrations of free drug or CPT-loaded liposomes. Afterwards, cells were washed twice with PBS and incubated with 10% resazurin in supplemented RPMI 1640 for 2 h under standard cell culture conditions, before fluorescence signal (530/590 nm) being measured using a BioTek Synergy Mx multi-mode microplate reader (Winooski, VT, USA). Cells without treatment or incubated with 1% (v/v) Triton X-100 were used as negative and positive controls, respectively. Cell viability was expressed as percentage by comparing results of the fluorescence signal of samples with those of the negative control (100% viability).Spheroids of HCT 116 cells were prepared according to Bauleth-Ramos et al.52 and used as 3D colorectal tumour surrogates. Briefly, agarose round notch templates were generated by pouring 2% (w/v) agarose in normal saline into MicroTissues® 3D Petri Dish® micro-moulds (Merck). The agarose templates were placed in 12-well microplates, followed by the addition of 2 mL of supplemented RPMI 1640 and left to equilibrate for at least 2 h under standard cell culture conditions. The mould was then filled with a suspension of HCT 116 cells (190 μL; 5000 cells per spheroid) and, after 30 min, 2 mL of medium were added to each well. Spheroids were generated over 7 days (medium replaced once at day 4), transferred into 96-well plates (3 spheroids/well) and incubated for an additional 24 h period with 200 μL of free CPT or liposomal formulations in culture medium at different concentrations. The medium was then removed, and the spheroids were washed twice with PBS and mixed (5 min, 60 rpm) with 100 μL of medium and 100 μL of CellTiter-Glo® (Promega, Madison, WI, USA). After static incubation for 25 min at room temperature, supernatants were collected and assessed for luminescence (560 nm) using the BioTek Synergy Mx multi-mode microplate reader. The percentage of metabolic activity of spheroids was determined by comparing the results of the luminescence signal of samples with those of the negative control (spheroids without treatment).
Dose–response curves obtained for both 2D and 3D configurations were fitted by log-logistic regression and used to calculate half-maximal inhibitory concentration (IC50) values.
2.8. Statistical analysis
All experiments were performed in triplicate and data are presented as mean ± standard deviation (SD), unless otherwise noted. Comparison of results for mean size, zeta potential and MPT studies were performed by two-tailed Student's t-test, while one-way and two-way analysis of variance (ANOVA) with Tukey's multiple comparison post-hoc test was used for analysing data of permeability experiments using Prism® (v. 8, GraphPad Software, La Jolla, CA, USA). Values of p < 0.05 were considered as significant.3. Results and discussion
3.1. Production and physicochemical characterization of liposomes
Non-coated (naked) liposomes were successfully prepared through the thin-film hydration method, followed by a procedure of extrusion using membranes with a final pore diameter of 100 nm. PEGylated liposomes were prepared by adding DSPE-PEG-2000 at 7% molar ratio to the lipid blend, to provide a non-ionic, stable hydrophilic polymer coating grafted onto the surface of the liposomes,53,54 likely in the so-called “brush-like” configuration. This strategy has been long recognized as useful for providing enhance colloidal stability55 and, under specific conditions, mucus-penetrating properties.56,57 PDA-coated liposomes were further obtained by coating naked liposomes with PDA through the in-situ polymerization of DA around the vesicles under near-neutral pH conditions (pH 7.4). The feasibility of this technique has been previously demonstrated by our team,38 and it is clearly noticeable by a change in the visual appearance of the liposomal dispersion from translucid white to opaque brown at the end of the reaction (ESI,† Fig. S1).All CPT-loaded liposomes presented mean diameter in the range of 130–157 nm and typical low polydispersity (Table 1). Results for liposome formulations obtained without the incorporation of CPT were similar (ESI,† Table S1). While PEGylation did not seem to affect liposome size, PDA coating led to a slight but significative (p < 0.05) increase in mean values of diameter as compared to non-coated counterparts, likely denoting the formation of a polymeric shell with an average thickness of circa 10 nm. Mean diameter values of CPT-L-PDA were also significantly larger than for CPT-L-PEG. CPT-L were characterized by lower EE% and LC% values, contrasting with those for coated liposomes, particularly CPT-L-PDA (Table 1). The presence of a polymeric coating at the surface of liposomes appears to act as a barrier for CPT leakage across the phospholipid bilayer. The PDA coating may further provide a docking substrate to CPT, as related to its good adsorption properties.58
| Formulation | Mean diameter (nm) | PDI | ζ-potential (mV) | EE% | LC% | CPT (μg mL−1) |
|---|---|---|---|---|---|---|
| CPT-L (non-coated) | 138 ± 1 | 0.08 ± 0.02 | −5.7 ± 0.7 | 4 ± 2 | 0.4 ± 0.1 | 60 ± 4 |
| CPT-L-PEG | 130 ± 2 | 0.07 ± 0.01 | −19.2 ± 0.6 | 8 ± 2 | 0.8 ± 0.2 | 123 ± 4 |
| CPT-L-PDA | 157 ± 2 | 0.16 ± 0.02 | −13.2 ± 0.1 | 16 ± 2 | 1.5 ± 0.2 | 234 ± 9 |
Additionally, values of ζ-potential were significantly more negative for coated liposomes, presenting differences between CPT-L-PEG and CPT-L-PDA (Table 1). Overall, these data support that modifying liposomes with either PEG or PDA can not only provide an effective coating that reduces drug leakage from vesicles, but also has the potential to contribute to stability. Indeed, both CPT-L-PEG and CPT-L-PDA maintained colloidal properties (namely size) upon storage at 4 °C up to at least four weeks (ESI,† Fig. S2). The absence of polymeric coating led to a loss of the original colloidal features of liposomes when placed in biorelevant media, thus preventing their further development as a viable option for oral administration. This has been reported before and is particularly relevant for liposomes composed only of phospholipids and cholesterol in the GIT.31–33
We further analysed PEG- and PDA-coated liposomes for morphology and confirmed the results for hydrodynamic diameters using TEM (Fig. 1). Images attested the formation of round-shaped vesicles with a superficial lipid bilayer in the case of PEG-coated systems. However, PDA-modified liposomes appeared to have a denser contrast coating. Overall, the size of observed vesicles seemed to be in line with the values of hydrodynamic diameters measured by DLS.
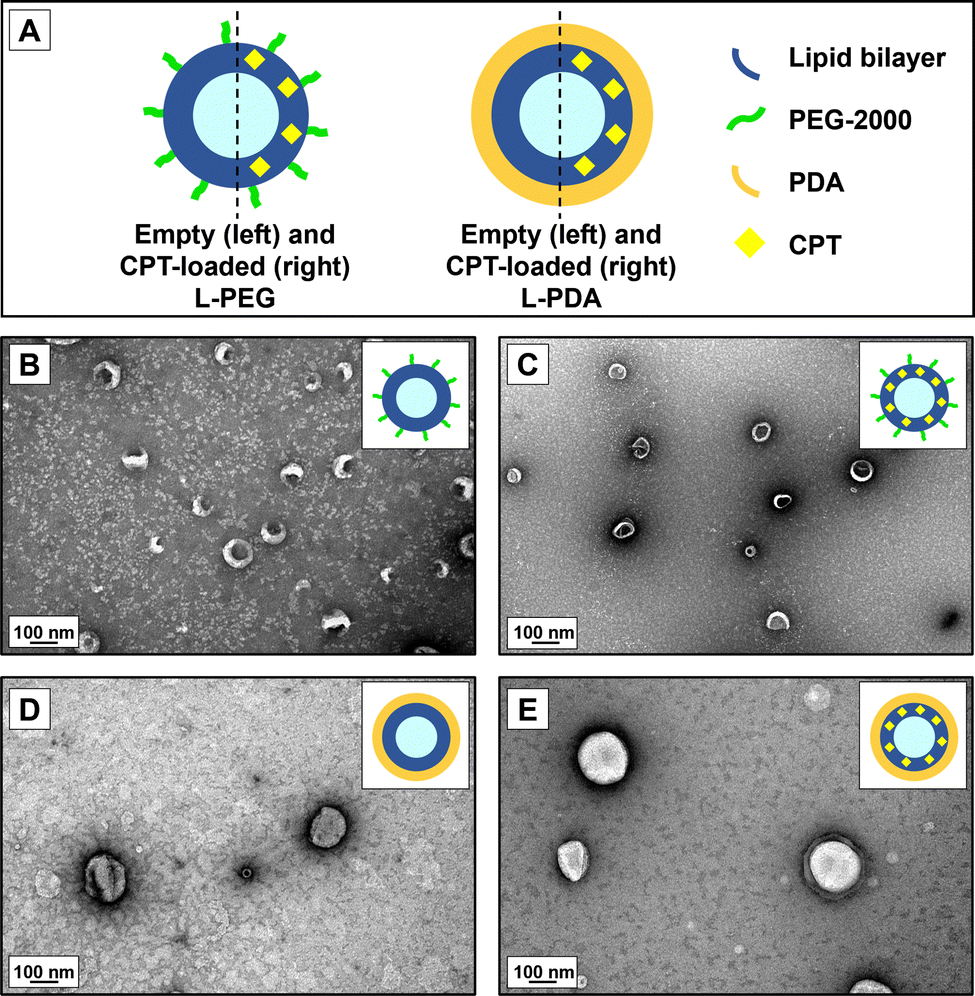 | ||
| Fig. 1 Organization and micromorphology of polymer-coated liposomes. (A) Schematic representation of the tentative structure of PEG- and PDA-modified liposomal formulations. Representative TEM images of (B) L-PEG, (C) CPT-L-PEG, (D) L-PDA and (E) CPT-L-PDA. | ||
3.2. In vitro drug release
We initially investigated the in vitro cumulative release of CPT from liposomes in a sequential fashion to simulate natural trafficking in the proximal GIT. Liposomes were first placed in simulated gastric fluid (pH 1.2) for 2 h, followed by 4 h incubation in intestinal fluids (pH 6.8). All polymer-coated liposome formulations allowed sustained release of the drug in acidic conditions, suggesting a good stability of both polymeric coatings in these conditions, as previously reported.43,59,60 The same trend was found for the “small intestine” phase (Fig. 2). CPT-L-PEG displayed faster drug release as compared to CPT-L-PDA. At the end of the “small intestinal phase” around 49% and 27% of total CPT were released from liposomes modified with PEG and PDA, respectively. The calculated value of f2 was 40, thus reinforcing that the release profiles were dissimilar.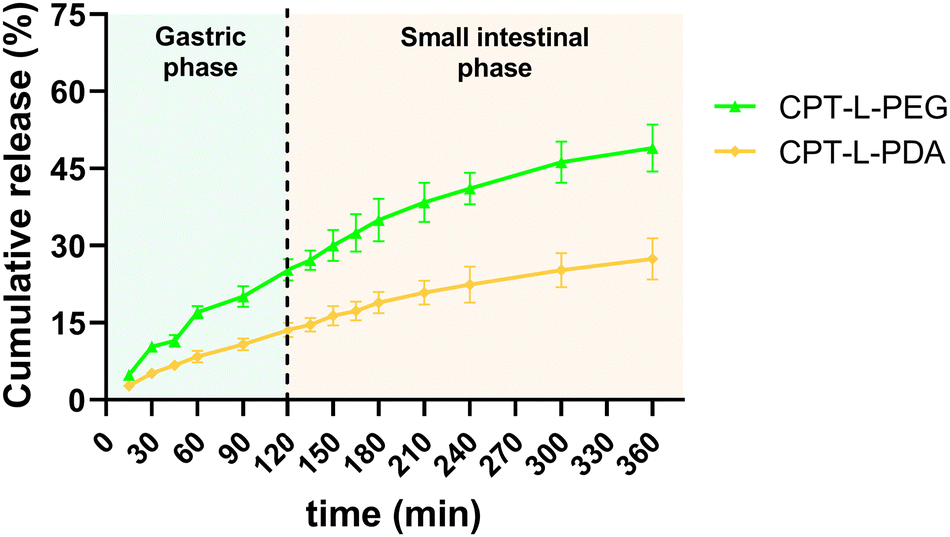 | ||
| Fig. 2 Sequential in vitro cumulative release of CPT from liposomes in gastric and intestinal simulated fluids. Results are expressed as mean ± SD (n = 3). | ||
Since the purpose of our liposomal formulations is to allow local action at the colorectum (often at quite distal locations),61 it would be preferable to avoid such pre-mature drug release at the stomach and small intestine. Assuming that developed liposomes could be further incorporated into dosage forms that allow contact with digestive fluids only at the colon (e.g., by using pH-sensitive capsules),62 we further tested the release of CPT only in simulated colorectal medium (pH 6.8) for 24 hours to study the “colorectal phase” behaviour of liposomes (Fig. 3). Again, drug release was faster from CPT-L-PEG as compared to CPT-L-PDA. The value of f2 was calculated as 35, thus reinforcing the relevance of these differences. Contrary to what happened under highly acid conditions, a burst effect was observed up to 2–4 h, followed by a nearly linear release stage until 24 h for all formulations. CPT-L-PDA and PEGylated liposomes were able to release around 34% and 61% at the end of the assay. Overall, all liposomes can provide sustained drug release irrespective of the pH conditions observed in the GIT.
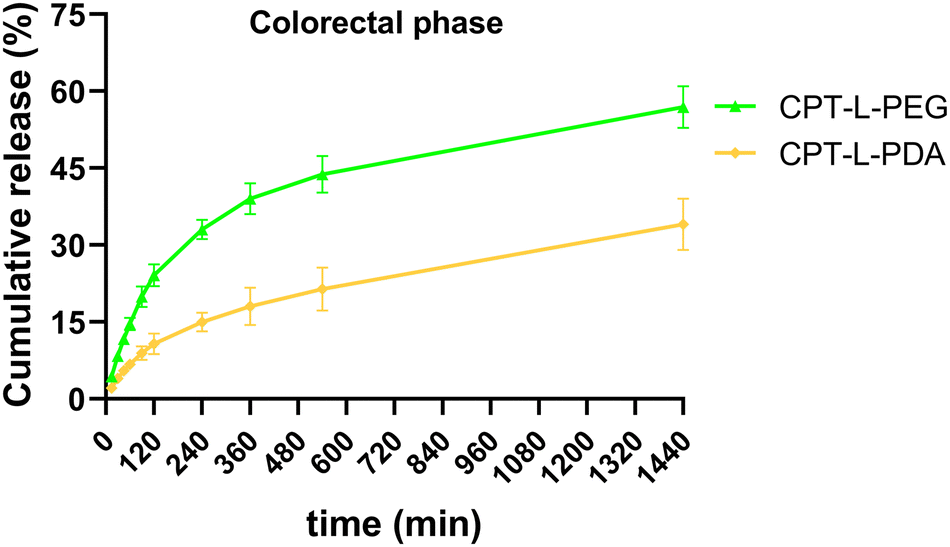 | ||
| Fig. 3 In vitro cumulative release of CPT from liposomes in simulated colorectal fluid. Results are expressed as mean ± SD (n = 3). | ||
3.3. Transport in intestinal mucus surrogate
Mucus represents a crucial barrier for the GIT transport of molecules and particles, including on their ability to translocate from the lumen to the mucosal tissue.27 In the particular case of oral delivery, drugs or nanocarriers need to cross the mucus layer to reach the intestinal epithelium. The development of mucus-penetrating systems is therefore seen as an interesting approach towards this goal. We performed MPT studies in an intestinal mucus surrogate (5% mucin in PBS) to understand the mobility of L-PEG and L-PDA. All formulations displayed a tandem linear dependence of <MSD> on time scale (τ), suggesting a homogeneous transport rate (Fig. 4A).63 The analysis of individual trajectories showed that liposomes were mainly sub-diffusive, though smaller fractions were also found to be immobile (around 10%) or diffusive (Fig. 4B). Interestingly, the relative amount of diffusive L-PDA (17%) was around 3-fold larger than diffusive L-PEG (6%). Average values of α were close (0.51 for L-PEG and 0.59 for L-PDA) but still significantly different (p = 0.039), thus suggesting higher mobility of L-PDA (Fig. 4C). Indeed, transport impairment in mucus surrogate was roughly 50% higher in the case of L-PEG, as denoted by values of the Dw/Deff ratio (Fig. 4D). These results are in line with the work of Wu et al.64 showing that low-density PEG-coating of liposomes yield mostly sub-diffusive behaviour in pig gastric mucus. To our best knowledge, the hereby presented study represents the first time PDA-modified liposomes are studied for their transport in mucus, but correlates with the findings of Poinard et al.45 These researchers reported that PDA-coated polystyrene NPs (≈100 nm) possessed sub-diffusive properties in reconstituted mucus (α = 0.82 at unspecified τ) due to the zwitterionic and hydrophilic properties of the polymer.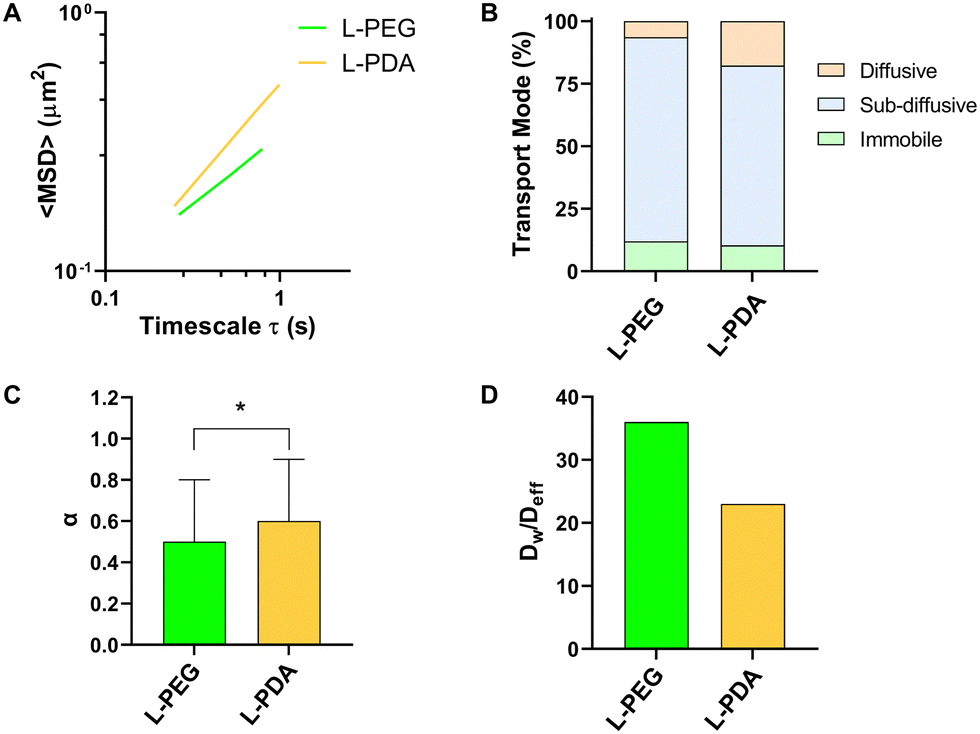 | ||
| Fig. 4 Transport behaviour of liposomes in intestinal mucus surrogate. (A) <MSD> as a function of timescale; (B) distribution percentage of liposomes according to transport mode; (C) values of α; and (D) Dw/Deff ratio. Results determined at τ = 1 s (≥96 valid trajectories per sample) and expressed in (C) as mean ± SD. (*) denotes p < 0.05. | ||
3.4. Intestinal permeability
The epithelium is easily recognized as the main barrier that orally delivered drugs and/or drug carriers must overcome before reaching systemic circulation. In addition – keeping in mind the objective of achieving localised action at the colorectum – drugs/carriers need to cross the epithelium in order to accumulate in the lamina propria (or even at the underlying muscle layers) where colorectal tumours may be present.65 In this regard, the intestinal permeability of CPT associated with liposomes was evaluated by using two in vitro models, i.e. Caco-2 and Caco-2/HT29-MTX cell monolayers. Although largely used as small intestinal epithelium surrogates, both models are also efficient in mimicking the colorectum and have been shown useful in characterizing drug permeability.51,66,67 The permeability assay was performed using a drug concentration of 10 μg mL−1 in all cases, which had no apparent detrimental effects on cell viability up to 4 h of incubation (ESI,† Fig. S3). TEER values monitored throughout permeability experiments also backup the maintenance of the integrity of cell monolayers (data not shown).The permeability profiles up to 4 h indicate that liposomal formulations provided higher CPT permeability across both cell monolayer models as compared to the free drug (Fig. 5A and B). Drug transport was apparently slower in the case of Caco-2/HT29-MTX cell monolayers, which suggests that the presence of the mucus-like secretions in this model may have had a mild impact on drug permeability. This seems to agree with the sub-diffusive nature of liposomes as determined by MPT. Overall, the enhancement in drug permeability associated with liposomes was moderate but well denoted by the significant increase in Papp and PER values as compared to free CPT (Fig. 5C and D). The exception was for CPT-L-PEG in the cell co-culture model, likely due to hindered transport in mucus. From a practical viewpoint, these results indicate that liposomes may lead to higher drug transport across the colorectal epithelium and promote CPT accumulation at the underlying intestinal tissues.
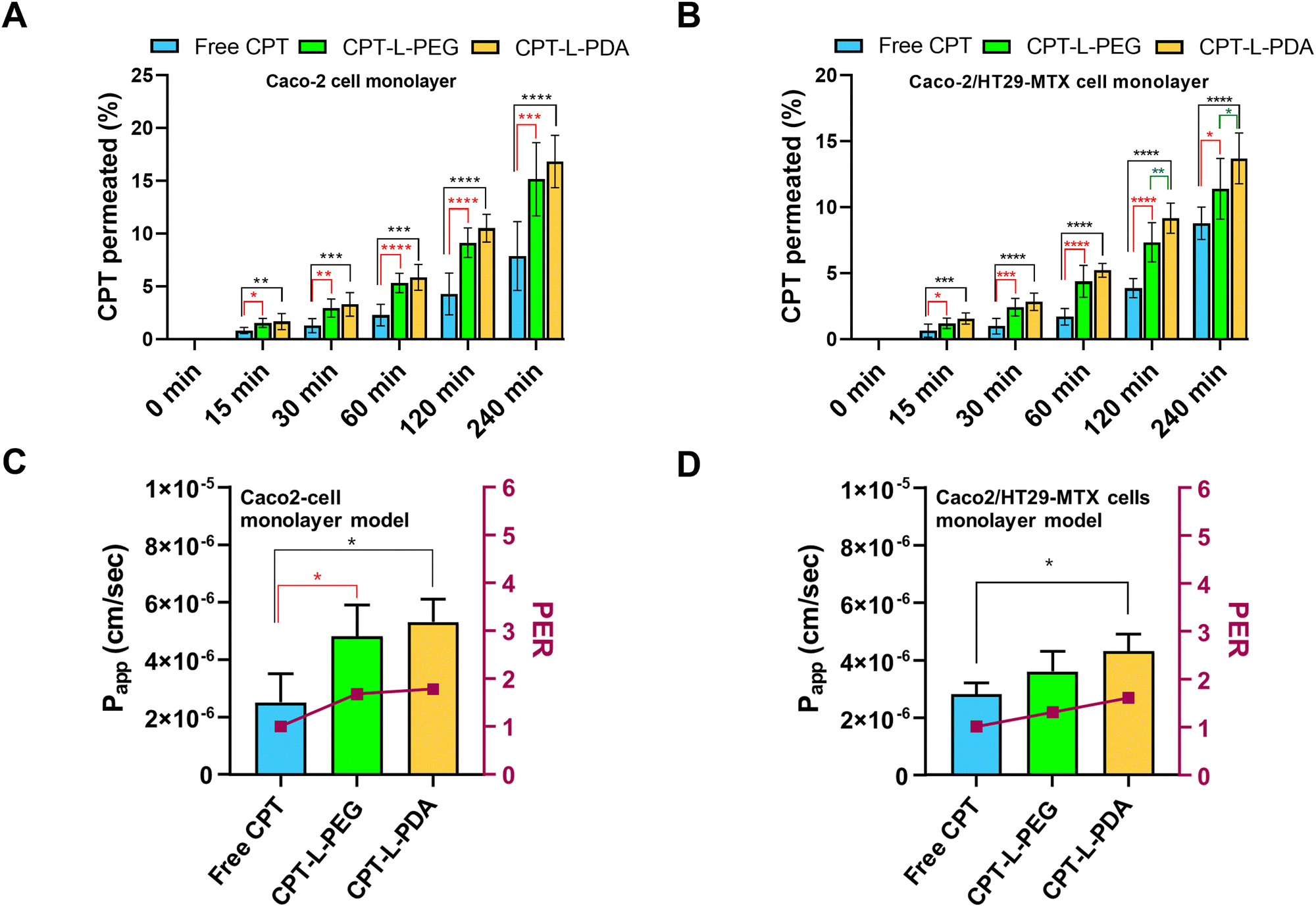 | ||
| Fig. 5 Permeability of CPT across intestinal cell monolayer models when in the free form or associated with liposomes. Percentage of CPT permeated across (A) Caco-2 and (B) Caco-2/HT29-MTX cell monolayer models and calculated Papp and PER values for (C–D) each of these models. Data are expressed as mean ± SD (n = 3). (*), (**), (***) and (****) denote p < 0.05, p < 0.01, p < 0.001 and p < 0.0001, respectively. | ||
3.5. Anticancer activity
The therapeutic efficacy of CPT, either in the free form or associated with liposomes, was assessed using 2D and 3D models based on the HCT 116 colorectal cancer cell line. These cells have been commonly used in 2D configuration (i.e. non-confluent single cell layer) for studying cancer biology and for screening potential anticancer compounds. However, 3D models such as spheroids may better represent the structural and pathophysiological properties of a solid tumour, thus providing an advanced tool for testing drugs and delivery systems.52,68,69HCT 116 spheroids were grown and monitored for morphology over seven days before usage (ESI,† Fig. S4). The metabolic activity of both 2D and 3D HCT 116 cell models was evaluated upon exposure to different concentrations of CPT in the free form or embedded in liposomes (Fig. 6) and results were used to calculate IC50 values (Table 2). Data indicate that HCT 116 cells were more resistant to treatment under 3D configuration, likely reflecting the more stringent barrier to drug/liposome transport posed by spheroids.70 Values of IC50 were around 2-log10 higher than those obtained using the 2D model. Moreover, no differences were observed when comparing liposomal formulations with the free drug under this last configuration (Table 2). In the case of HCT 116 spheroids, IC50 values seem to indicate that drug association to liposomes led to enhanced anticancer activity, namely in the case of CPT-L-PDA (over 2-fold improvement). The effects of CPT-L-PDA could be related to the ability of liposomes to protect CPT from premature degradation under cell culture conditions and/or to promote better spheroid penetration. PDA coating may further enhance uptake by cancer cells as compared with PEG.45
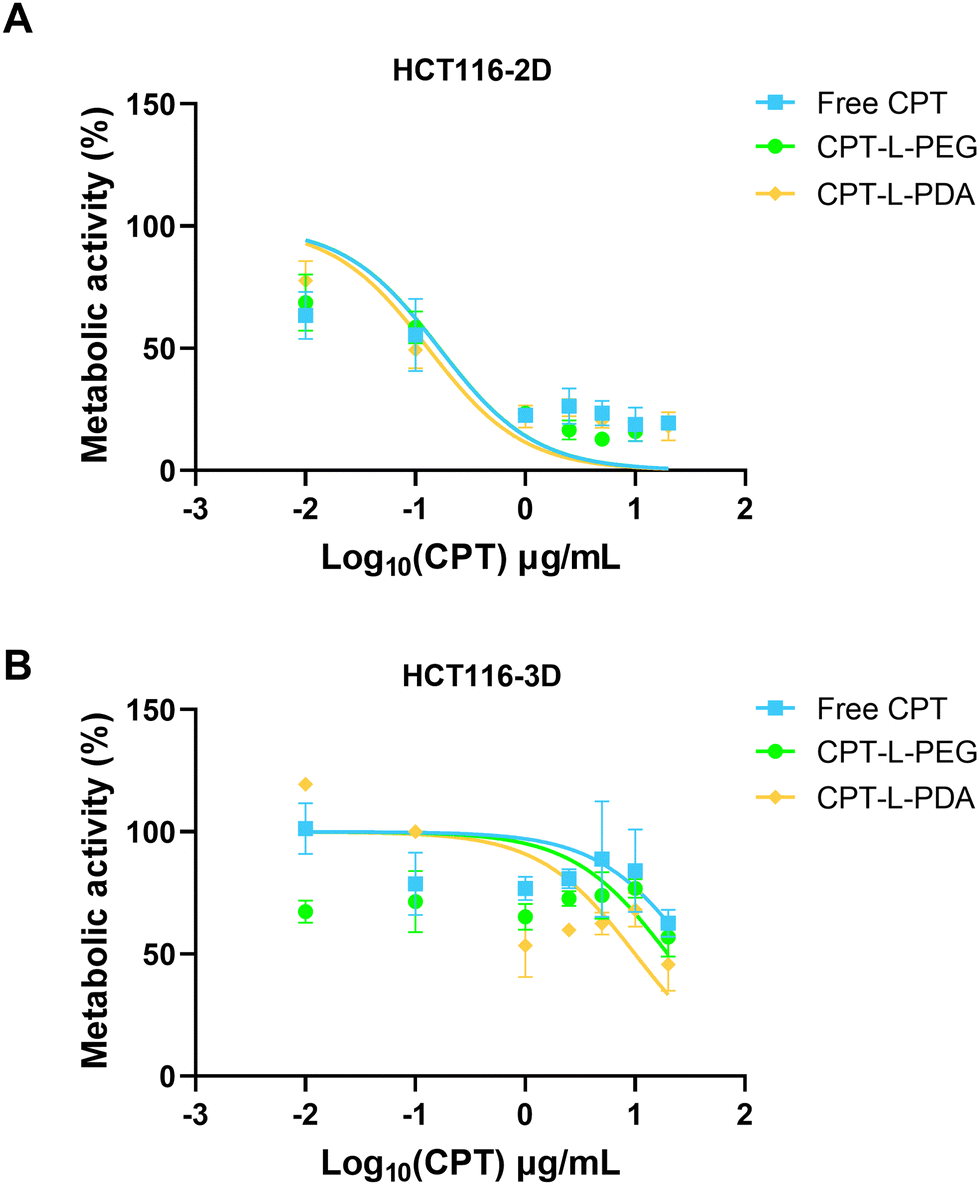 | ||
| Fig. 6 Dose–response curves of metabolic activity of HCT 116 cells under (A) 2D and (B) 3D configurations upon 24 h incubation with CPT or CPT-loaded liposomes. Experimental points are expressed as mean ± SD (n = 3). Lines represent curve fitting using log-logistic regression. | ||
| IC50 (μg mL−1) | ||
|---|---|---|
| 2D | 3D | |
| Free CPT | 0.16 | >20 |
| CPT-L-PEG | 0.17 | 19.8 |
| CPT-L-PDA | 0.13 | 10.0 |
We also tested liposomes containing no CPT to assess whether nanocarriers had any intrinsic anticancer activity (ESI,† Fig. S5). PEG-modified liposomes showed moderate activity in the 2D model, but only at around 10-fold higher levels of those observed for CPT-loaded counterparts. L-PDA did not affect cell viability in either 2D or 3D models.
4. Conclusions
We successfully prepared and characterized different polymer-coated, CPT-loaded liposomes for the localised treatment of CRC. Liposomes were modified with either PEG or PDA to solve the typical challenges presented to liposome-mediated oral drug delivery. Whereas PEGylation has been previously described for the stabilization of liposomes in the GIT, to the best of our knowledge PDA-coated liposomes have never been tested for such purpose. All CPT-loaded liposomes presented nanometric size and the addition of polymer coatings enhanced stability and EE%. Notably, CPT-L-PDA were able to provide slower and sustained drug release under different conditions simulating normal GIT transit, contrasting with PEGylated liposomes, which could be beneficial for drug delivery to tumoral sites located at distal areas of the colorectum. Modification with either PEG or PDA rendered sub-diffusive properties to liposomes and allowed enhancing the in vitro intestinal permeability of the CPT, inclusive in the presence of mucus-like secretions. Finally, the anticancer activity of CPT in both 2D or 3D tumour models was maintained or even enhanced when liposomes – namely those coated with PDA – were used. Overall, PDA-coating seems to be a promising strategy for improving the properties of liposomes intended for the oral administration of drugs, namely CPT for the localised treatment of CRC.Author contributions
A. M. Maurelli: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft. B. Ferreira: investigation, methodology, writing – review & editing. S. Dias: investigation, methodology, writing – review & editing. H. Almeida: data curation, formal analysis, writing – review & editing. V. De Leo: conceptualization, formal analysis, funding acquisition, methodology, resources, supervision, writing – review & editing. B. Sarmento: funding acquisition, resources, supervision, writing – review & editing. L. Catucci: Conceptualization, formal analysis, funding acquisition, methodology, resources, supervision, writing – review & editing. J. das Neves: conceptualization, formal analysis, methodology, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Dias, H. Almeida and J. das Neves gratefully acknowledges Fundação para a Ciência e a Tecnologia, Portugal for financial support (2021.07508.BD and 2020.06264.BD doctoral fellowships, and CEECIND/01280/2018 contract under the Individual CEEC Program, respectively). Vincenzo De Leo is a researcher at the University of Bari within the European Union program “FSE-REACT-EU, PON Research and Innovation 2014–2020”, DM1062/2021. Graphical abstract was created with BioRender.com.Notes and references
- GBD Colorectal Cancer Collaborators, Lancet Gastroenterol. Hepatol., 2022, 7, 627–647 CrossRef PubMed.
- L. Vecchione, S. Stintzing, G. Pentheroudakis, J. Y. Douillard and F. Lordick, ESMO Open, 2020, 5, e000826 CrossRef PubMed.
- K. Damm, A. Vogel and A. Prenzler, Eur. J. Cancer Care, 2014, 23, 762–772 CrossRef CAS PubMed.
- M. S. Alqahtani, M. Kazi, M. A. Alsenaidy and M. Z. Ahmad, Front. Pharmacol., 2021, 12, 618411 CrossRef CAS PubMed.
- Y. S. R. Krishnaiah and M. A. Khan, Pharm. Dev. Technol., 2012, 17, 521–540 CrossRef CAS PubMed.
- P. García-Alfonso, A. J. Muñoz Martín, L. Ortega Morán, J. Soto Alsar, G. Torres Pérez-Solero, M. Blanco Codesido, P. A. Calvo Ferrandiz and S. Grasso Cicala, Ther. Adv. Med. Oncol., 2021, 13, 17588359211009001 Search PubMed.
- S. Goyle and A. Maraveyas, Dig. Surg., 2006, 22, 401–414 CrossRef PubMed.
- G. Argilés, J. Tabernero, R. Labianca, D. Hochhauser, R. Salazar, T. Iveson, P. Laurent-Puig, P. Quirke, T. Yoshino, J. Taieb, E. Martinelli and D. Arnold, Ann. Oncol., 2020, 31, 1291–1305 CrossRef.
- S. Ünal, Y. Aktaş, J. M. Benito and E. Bilensoy, Int. J. Pharm., 2020, 584, 119468 CrossRef PubMed.
- M. Kang, R. Fu, P. Zhang, S. Lou, X. Yang, Y. Chen, T. Ma, Y. Zhang, Z. Xi and J. Liu, Nat. Commun., 2021, 12, 3531 CrossRef CAS PubMed.
- S. Jacob, M. l Aguado, D. Fallik and F. o Praz, Cancer Res., 2001, 61, 6555–6562 CAS.
- M. E. Wall, M. C. Wani, C. E. Cook, K. H. Palmer, A. T. McPhail and G. A. Sim, J. Am. Chem. Soc., 1966, 88, 3888–3890 CrossRef CAS.
- J. Fassberg and V. J. Stella, J. Pharm. Sci., 1992, 81, 676–684 CrossRef CAS PubMed.
- M. C. Wani, A. W. Nicholas, G. Manikumar and M. E. Wall, J. Med. Chem., 1987, 30, 1774–1779 CrossRef CAS.
- R. P. Hertzberg, M. J. Caranfa, K. G. Holden, D. R. Jakas, G. Gallagher, M. R. Mattern, S. M. Mong, J. O. L. Bartus, R. K. Johnson and W. D. Kingsbury, J. Med. Chem., 1989, 32, 715–720 CrossRef CAS PubMed.
- D. Sivadasan, M. H. Sultan, O. A. Madkhali, S. H. Alsabei and A. A. Alessa, Molecules, 2022, 27, 1086 CrossRef CAS PubMed.
- C.-H. Lin, S. A. Al-Suwayeh, C.-F. Hung, C.-C. Chen and J.-Y. Fang, J. Tradit. Complementary Med., 2013, 3, 102–109 CrossRef PubMed.
- M. Landgraf, C. A. Lahr, I. Kaur, A. Shafiee, A. Sanchez-Herrero, P. W. Janowicz, A. Ravichandran, C. B. Howard, A. Cifuentes-Rius, J. A. McGovern, N. H. Voelcker and D. W. Hutmacher, Biomaterials, 2020, 240, 119791 CrossRef CAS PubMed.
- S. Yang, J. Zhu, Y. Lu, B. Liang and C. Yang, Pharm. Res., 1999, 16, 751–757 CrossRef CAS.
- S. Martins, I. Tho, I. Reimold, G. Fricker, E. Souto, D. Ferreira and M. Brandl, Int. J. Pharm., 2012, 439, 49–62 CrossRef CAS.
- A. Almeida, F. Castro, C. Resende, M. Lúcio, S. Schwartz and B. Sarmento, J. Controlled Release, 2022, 349, 731–743 CrossRef CAS.
- J. Huarte, S. Espuelas, Y. Lai, B. He, J. Tang and J. M. Irache, Int. J. Pharm., 2016, 506, 116–128 CrossRef CAS PubMed.
- Q. N. Nguyen-Trinh, K. X. T. Trinh, N.-T. Trinh, V. T. Vo, N. Li, Y. Nagasaki and L. B. Vong, Acta Biomater., 2022, 143, 459–470 CrossRef CAS PubMed.
- M. Martínez-Martínez, G. Rodríguez-Berna, M. Bermejo, I. Gonzalez-Alvarez, M. Gonzalez-Alvarez and V. Merino, Eur. J. Pharm. Biopharm., 2019, 136, 174–183 CrossRef PubMed.
- V. De Leo, S. Ruscigno, A. Trapani, S. Di Gioia, F. Milano, D. Mandracchia, R. Comparelli, S. Castellani, A. Agostiano, G. Trapani, L. Catucci and M. Conese, Int. J. Pharm., 2018, 545, 378–388 CrossRef CAS.
- T. G. Burke, A. E. Staubus, A. K. Mishra and H. Malak, J. Am. Chem. Soc., 1992, 114, 8318–8319 CrossRef CAS.
- J. das Neves, R. Sverdlov Arzi and A. Sosnik, Chem. Soc. Rev., 2020, 49, 5058–5100 RSC.
- K. Maisel, L. Ensign, M. Reddy, R. Cone and J. Hanes, J. Controlled Release, 2015, 197, 48–57 CrossRef CAS PubMed.
- R. Nunes, F. Araújo, J. Tavares, B. Sarmento and J. das Neves, Eur. J. Pharm. Biopharm., 2018, 130, 200–206 CrossRef CAS PubMed.
- E. Yamazoe, J.-Y. Fang and K. Tahara, Int. J. Pharm., 2021, 593, 120148 CrossRef CAS PubMed.
- A. Jash, A. Ubeyitogullari and S. S. H. Rizvi, J. Mater. Chem. B, 2021, 9, 4773–4792 RSC.
- V. De Leo, A. M. Maurelli, L. Giotta, V. Daniello, S. Di Gioia, M. Conese, C. Ingrosso, F. Ciriaco and L. Catucci, Int. J. Mol. Sci., 2023, 24, 790 CrossRef CAS.
- H. He, Y. Lu, J. Qi, Q. Zhu, Z. Chen and W. Wu, Acta Pharm. Sin. B, 2019, 9, 36–48 CrossRef PubMed.
- K. Iwanaga, S. Ono, K. Narioka, K. Morimoto, M. Kakemi, S. Yamashita, M. Nango and N. Oku, Int. J. Pharm., 1997, 157, 73–80 CrossRef CAS.
- A. Singh, Y. R. Neupane, S. Shafi, B. Mangla and K. Kohli, J. Mol. Liq., 2020, 303, 112649 CrossRef CAS.
- G. R. Dakwar, E. Zagato, J. Delanghe, S. Hobel, A. Aigner, H. Denys, K. Braeckmans, W. Ceelen, S. C. De Smedt and K. Remaut, Acta Biomater., 2014, 10, 2965–2975 CrossRef CAS PubMed.
- Y. Tang, Y. Tan, K. Lin and M. Zhu, Front. Chem., 2021, 9, 727123 CrossRef CAS PubMed.
- V. De Leo, A. M. Maurelli, C. Ingrosso, F. Lupone and L. Catucci, Int. J. Mol. Sci., 2021, 22, 11916 CrossRef.
- A. M. Maurelli, V. De Leo, V. Daniello, C. D. Calvano, F. Ciriaco, F. Milano, C. Ingrosso, T. R. I. Cataldi, S. Di Gioia, M. Conese, A. Agostiano and L. Catucci, Mater. Today Chem., 2024, 37, 101994 CrossRef CAS.
- S. Sabzi, M. Habibi, F. Badmasti, S. Shahbazi, M. R. Asadi Karam and M. Farokhi, Int. J. Pharm., 2024, 654, 123961 CrossRef CAS PubMed.
- M. Farokhi, F. Mottaghitalab, M. R. Saeb and S. Thomas, J. Controlled Release, 2019, 309, 203–219 CrossRef CAS PubMed.
- L. Vannozzi, E. Catalano, M. Telkhozhayeva, E. Teblum, A. Yarmolenko, E. S. Avraham, R. Konar, G. D. Nessim and L. Ricotti, Nanomaterials, 2021, 11, 2105 CrossRef CAS PubMed.
- S. Hu, Z. Yang, S. Wang, L. Wang, Q. He, H. Tang, P. Ji and T. Chen, Chem. Eng. J., 2022, 428, 132107 CrossRef CAS.
- B. Poinard, S. A. E. Lam, K. G. Neoh and J. C. Y. Kah, J. Controlled Release, 2019, 300, 161–173 CrossRef CAS PubMed.
- B. Poinard, S. Kamaluddin, A. Q. Q. Tan, K. G. Neoh and J. C. Y. Kah, ACS Appl. Mater. Interfaces, 2019, 11, 4777–4789 CrossRef CAS PubMed.
- J. W. Moore and H. H. Flanner, Pharm. Technol., 1996, 20, 64–74 Search PubMed.
- L. Gabriel, H. Almeida, M. Avelar, B. Sarmento and J. das Neves, Nanomaterials, 2022, 12, 1899 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- I. F. Sbalzarini and P. Koumoutsakos, J. Struct. Biol., 2005, 151, 182–195 CrossRef CAS PubMed.
- I. Lozoya-Agullo, F. Araújo, I. González-Álvarez, M. Merino-Sanjuán, M. González-Álvarez, M. Bermejo and B. Sarmento, Mol. Pharmaceutics, 2017, 14, 1264–1270 CrossRef CAS PubMed.
- R. Nunes, F. Araújo, L. Barreiros, I. Bártolo, M. A. Segundo, N. Taveira, B. Sarmento and J. das Neves, ACS Appl. Mater. Interfaces, 2018, 10, 34942–34953 CrossRef CAS PubMed.
- T. Bauleth-Ramos, T. Feijão, A. Gonçalves, M.-A. Shahbazi, Z. Liu, C. Barrias, M. J. Oliveira, P. Granja, H. A. Santos and B. Sarmento, J. Controlled Release, 2020, 323, 398–411 CrossRef CAS PubMed.
- A. S. Nosova, O. O. Koloskova, A. A. Nikonova, V. A. Simonova, V. V. Smirnov, D. Kudlay and M. R. Khaitov, MedChemComm, 2019, 10, 369–377 RSC.
- Z. Amoozgar and Y. Yeo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 219–233 CAS.
- K. Nakamura, K. Yamashita, Y. Itoh, K. Yoshino, S. Nozawa and H. Kasukawa, Biochim. Biophys. Acta, 2012, 1818, 2801–2807 CrossRef CAS PubMed.
- Y. Y. Wang, S. K. Lai, J. S. Suk, A. Pace, R. Cone and J. Hanes, Angew. Chem., Int. Ed., 2008, 47, 9726–9729 CrossRef CAS PubMed.
- J. das Neves, M. F. Bahia, M. M. Amiji and B. Sarmento, Expert Opin. Drug Delivery, 2011, 8, 1085–1104 CrossRef CAS PubMed.
- Z. Wang, H.-C. Yang, F. He, S. Peng, Y. Li, L. Shao and S. B. Darling, Matter, 2019, 1, 115–155 CrossRef.
- W. Liu, A. Ye, C. Liu, W. Liu and H. Singh, Food Res. Int., 2012, 48, 499–506 CrossRef CAS.
- Y. Zhang, L. Wang, M. Xu, T. Zhao, L. Kuang and D. Hua, Adv. Healthcare Mater., 2020, 9, 1901778 CrossRef CAS PubMed.
- S. Stintzing, S. Tejpar, P. Gibbs, L. Thiebach and H. J. Lenz, Eur. J. Cancer, 2017, 84, 69–80 CrossRef PubMed.
- G. Van den Mooter, Expert Opin. Drug Delivery, 2006, 3, 111–125 CrossRef CAS PubMed.
- S. K. Lai, D. E. O'Hanlon, S. Harrold, S. T. Man, Y. Y. Wang, R. Cone and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1482–1487 CrossRef CAS PubMed.
- Y. Wu, W. Wang, Z. Yu, K. Yang, Z. Huang, Z. Chen, X. Yan, H. Hu and Z. Wang, Biomater. Adv., 2022, 136, 212798 CrossRef CAS.
- N. N. Mahmoud, Surg. Oncol. Clin. North Am., 2022, 31, 127–141 CrossRef.
- W. Rubas, M. E. Cromwell, Z. Shahrokh, J. Villagran, T. N. Nguyen, M. Wellton, T. H. Nguyen and R. J. Mrsny, J. Pharm. Sci., 1996, 85, 165–169 CrossRef CAS.
- L. Lohikangas, M. Wilen, M. Einarsson and P. Artursson, Eur. J. Pharm. Sci., 1994, 1, 297–305 CrossRef CAS.
- H. Karlsson, M. Fryknas, R. Larsson and P. Nygren, Exp. Cell Res., 2012, 318, 1577–1585 CrossRef CAS PubMed.
- A. Tchoryk, V. Taresco, R. H. Argent, M. Ashford, P. R. Gellert, S. Stolnik, A. Grabowska and M. C. Garnett, Bioconjugate Chem., 2019, 30, 1371–1384 CrossRef CAS.
- F. Castro, C. Leite Pereira, M. H. Macedo, A. Almeida, M. J. Silveira, S. Dias, A. P. Cardoso, M. J. Oliveira and B. Sarmento, Adv. Drug Delivery Rev., 2021, 175, 113824 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01158e |
| This journal is © The Royal Society of Chemistry 2024 |