
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrafast magnetisation dynamics in a chromium-based Prussian blue analogue†
Harry A.
Lewis
 a,
Thomas M.
Stonelake
a,
Oscar
Céspedes
b and
J. Olof
Johansson
*a
a,
Thomas M.
Stonelake
a,
Oscar
Céspedes
b and
J. Olof
Johansson
*a
aEaStCHEM School of Chemistry, University of Edinburgh, David Brewster Road, EH9 3FJ, Edinburgh, UK. E-mail: olof.johansson@ed.ac.uk
bSchool of Physics and Astronomy, University of Leeds, LS2 9JT, Leeds, UK
First published on 7th August 2024
Abstract
Ultrafast transient transmittance (TT) and time-resolved magnetic circular dichroism (TR-MCD) measurements were used to investigate the electronic and magnetisation dynamics in thin films of a mixed-valence chromium Prussian blue analogue. Measurements performed for 800 nm photoexcitation unveil a TT spectrum that consists of a broad, complex, bleach-like feature with some additional excited-state absorption contributions, while the spectral dependence of the TR-MCD closely matched the shape of the static MCD spectrum. Both the TT and TR-MCD response evolved on different timescales (2.5 and 1.5 ps, respectively) and superimposed on this is a slow growth in both the TT and TR-MCD response (220 and 90 ps, respectively). Decomposition analyses of the broadband transient spectrum using temperature-dependent transmittance data indicate that the slow growth in pump–probe response is a result of sample heating following lattice thermalisation. These thermal effects have a much more profound impact on the magnetisation dynamics, particularly at high pump fluences, where the >70% loss in magneto-optical response observed after several hundred ps vastly exceeds the corresponding 4% change in transmittance. These conclusions could only be reached through the determination of both static and dynamic responses of the sample, thus illustrating the power of using complementary spectroscopic methods.
1 Introduction
The observation of demagnetisation in ferromagnetic Ni thin films on femtosecond timescales upon excitation by an ultrashort laser pulse1 prompted the emergence of ultrafast magnetism as an area of active research. Currently at the forefront of the field is the all-optical perturbation of long-range magnetic order,2 which has been extensively demonstrated in dielectrics3–5 and more recently in 2D ferromagnetic materials.6–10 This opportunity should be extended to molecular-based systems, given their desirable characteristics: they possess chemical flexibility that can give rise to tuneable electronic and magnetic properties, and carry localised sources of magnetic moment. The dynamics after photoexcitation in such systems were first reported in 1995 although with no direct spin-sensitivity.11The aforementioned ultrafast studies were only possible through the use of magneto-optical techniques to infer the sample's magnetic properties. Magnetic circular dichroism (MCD) is one such method, whereby the Zeeman splitting of normally degenerate electronic states corresponding to non-zero magnetic moment leads to differential absorption of the right- and left-circular polarisation components of light incident on a sample in the presence of a magnetic field. In transmission (i.e. Faraday) geometry, this induces ellipticity in the polarisation state of the transmitted light, a measurable quantity that changes as the magnetisation of the sample is varied. As such, MCD is suitable for use as a probe in an all-optical pump–probe scheme and can be used in conjunction with transient spectroscopy to ascertain both the electronic and magnetisation dynamics that ensue upon photoexcitation.8,12 Since molecular-based materials may possess intense and well-defined features in their electronic and MCD spectra,13–15 this should render these systems amenable to ultrafast studies using an optical probe to observe their magnetisation dynamics. However, such investigations are currently underreported, with only one published example to date.16
One notable subset of molecule-based magnetic materials is an array of polynuclear transition metal cyanide systems that have been shown to undergo sustained, yet reversible, magnetic phase transitions upon light irradiation at different wavelengths.17–23 Of these, the Prussian blue analogue (PBA) family of cyanide-bridged coordination polymers are the simplest representatives: they possess a face-centred cubic crystal structure and conform to the formula MxAy[B(CN)6]z·nH2O, where A and B are transition metal cations, M is a monovalent alkali cation inserted into the lattice for charge balance, and H2O is present in both coordinated and non-coordinated form.24 The identities and stoichiometry of A, B and M may be readily changed to give rise to materials with highly tuneable physical properties.25–27 These traits make PBAs useful model systems for elucidating the dynamical processes that underpin magnetisation changes in response to the photoexcitation of molecule-based systems. Indeed, the photophysics of Prussian blue itself and the lattice/localised spin dynamics associated with photoinduced phase transitions in CoII–FeIII/CoIII–FeII and MnIII–FeII PBAs have already been studied extensively using transient absorption, time-resolved X-ray diffraction and time-resolved X-ray absorption near-edge structure measurements.28–33 Nevertheless, the magnetisation dynamics themselves have yet to be investigated directly using a magneto-optical probe in any of these materials.
Our group previously investigated the ultrafast dynamics observed in thin films of a mixed-valence VII/III–CrIII PBA upon optical pumping of a charge-transfer transition.16 Complementary transient electronic absorption and time-resolved magneto-optical measurements revealed that the charge and magnetisation dynamics were inextricably linked, with the kinetics for both conforming to similar timescales. The interpretation was that spin-flips on CrIII-sites within 250 fs of photoexcitation were accompanied by a change in the bulk magnetic properties on sub-ps timescales, which was rationalised by perturbation of the superexchange interactions that give rise to long-range magnetic ordering. In this present work, we report a continuation of our investigations on Cr-based PBAs using our recently commissioned broadband pump–probe experiment.12 We have studied Cr–Cr PBA as it possesses greater resilience to air-oxidation and more consistent magnetic behaviour than V–Cr PBA, yet still magnetically orders at temperatures that are accessible with liquid nitrogen.
2 Materials and methods
Cr–Cr PBA is a mixed-valence material where Cr occupies both A- and B-sites in either its +2 or +3 oxidation states. This results in highly variable stoichiometry and sensitivity to air-oxidation. Previous research has shown that the prevalence of oxidation within the material can have profound effects on its electronic spectrum, coercivity, and magnetic-ordering temperature, Tc.34,35 In our samples, we observed that reduced materials exhibit increased absorption across the spectrum, an additional absorption feature in the NIR, and wider hysteresis loops than their oxidised counterparts (see Fig. S1, ESI†), albeit at the expense of a lower Tc. As the enhancements to the electronic spectrum provided more viable targets for photoexcitation and the Tc was still easily accessible with liquid nitrogen, the reduced material was used in this study.2.1 Sample preparation and characterisation
Samples of Cr–Cr PBA were prepared in the form of thin films through electrodeposition from solution, which has previously been described.36–40 In summary, an electrochemical cell was charged with a degassed aqueous solution containing Cr3+, [Cr(CN)6]3− and K+ of concentration 15, 10 and 530 mM, respectively. All reagents were commercially produced and used without further purification. A transparent conductive substrate comprising a layer of fluorine-doped tin oxide on 1 mm-thick soda–lime glass (Ossila S302) was used as a working electrode, and two Pt rods were employed as counter and reference electrodes. Films were deposited onto the substrates using a potentiostat (Metrohm μAUTOLAB III) at a constant potential of E = −1.2 V under a nitrogen atmosphere at room temperature, with film thickness controlled by varying the deposition time. Following characterisation, the films were encapsulated with a UV-cured adhesive (Norland optical adhesive 61) and a glass coverslip using a previously outlined procedure40 to improve sample transmittance and stave off oxidation.The composition of the films was confirmed through vibrational spectroscopy to assess the frequencies and intensities of characteristic C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch bands. For this, a Raman microscope (Renishaw inVia) with a reflectance IR attachment (Smiths IlluminatIR) was used. Two characteristic bands were observed in reflectance IR spectra at 2188 and 2075 cm−1 (Fig. S2, ESI†), corresponding to CrIII–CN and low-spin (LS) CrIILS–CN moieties, respectively.34–37,39 This indicated that the films contained a mixture of both CrIII and CrIILS in B-sites. In complementary Raman spectra obtained using a 514 nm laser line on the same sample, only a single peak at 2194 cm−1 was observed, which roughly coincides with the CrIII–CN frequency. The lack of a second band suggests that the CrIILS–CN moiety is not Raman active.
N stretch bands. For this, a Raman microscope (Renishaw inVia) with a reflectance IR attachment (Smiths IlluminatIR) was used. Two characteristic bands were observed in reflectance IR spectra at 2188 and 2075 cm−1 (Fig. S2, ESI†), corresponding to CrIII–CN and low-spin (LS) CrIILS–CN moieties, respectively.34–37,39 This indicated that the films contained a mixture of both CrIII and CrIILS in B-sites. In complementary Raman spectra obtained using a 514 nm laser line on the same sample, only a single peak at 2194 cm−1 was observed, which roughly coincides with the CrIII–CN frequency. The lack of a second band suggests that the CrIILS–CN moiety is not Raman active.
Film thickness was determined using an atomic force microscope (Veeco Nanoman VS) in tapping-mode configuration to assess the height profile across the boundary between substrate and film at various positions. Height data were exported to a routine data analysis package for assessment using the native software (NanoScope Analysis v1.5). For the film subject to pump–probe measurements in this study, the determined thickness, d, was 900(40) nm.
UV-visible-NIR transmittance of encapsulated Cr–Cr PBA films was measured at ambient conditions using a commercial double-beam spectrophotometer (Shimadzu UV1800). The absorption coefficient, α, was calculated using film transmittance and the path length through the material (i.e. d) while assuming negligible reflection at normal incidence. The resultant absorption spectrum (Fig. 1(a)) possessed multiple features: a strong absorption edge in the UV (λmax < 350 nm) with a large absorption coefficient, a peak at 380 nm, several poorly resolved transitions between 500 and 750 nm, and a broad absorption band in the NIR (λmax > 1000 nm). All these features are consistent with previous reports.15,34,37,41
 | ||
| Fig. 1 (a) Room-temperature electronic spectrum for the Cr–Cr PBA sample used in this study. The region pumped in all measurements is highlighted in red, the monochromatic probe in blue, and the region covered by the supercontinuum probe in broadband measurements is denoted by the rainbow. (b) Schematic of the sample geometry used in the pump–probe experiments, which determine pump-induced changes in the intensity, I*, and polarisation state, P*, of transmitted light. | ||
Magnetic hysteresis data for Cr–Cr PBA were acquired by measuring the field-dependent Faraday ellipticity, ηF, using a linearly polarised, 400 nm beam and a balanced polarisation bridge scheme (see below). The wavelength-dependence of ηF (i.e. the MCD spectrum; see Fig. S3, ESI†) was obtained by measuring the differential absorption of circularly polarised light upon reversing the applied field direction. Both methodologies have been previously described.12 From these measurements, the coercivity, μ0Hc, was determined to be 14.2 mT at 80 K. Tc was estimated to be ∼160 K, as indicated by the onset of observable coercivity and MCD features.
2.2 Optical setup
A schematic illustrating the geometry of the pump–probe experiment is provided in Fig. 1(b). The optical setup used to investigate the dynamics of Cr–Cr PBA has been outlined previously.42 In short, 90% (0.36 mJ) of the fundamental output (1028 nm, 270 fs) from an amplified Yb:KGW-based femtosecond laser system (Light Conversion PHAROS) was used to pump an optical parametric amplifier (OPA) (Light Conversion ORPHEUS-F) that generated ultrashort (∼40 fs-duration) signal pulses of variable wavelength to serve as a pump for experiments. These pulses were subject to mechanical modulation by an optical chopper operating at half the repetition rate of the laser (1 or 20 kHz, see below), with its phase set to block every second pulse for the purpose of calculating pump-induced differential transmittance/ellipticity. A variable delay line was employed to stagger the pump with respect to the probe, enabling pump–probe delays, tdelay, of up to 3 ns. A fraction (2.5 μJ) of the remaining fundamental from the femtosecond laser was used to generate a supercontinuum in CaF2, spanning a detectable range of 350–750 nm. This was split into two arms: the minor arm functioned as a reference beam, while the major arm served as a broadband probe. The pump and probe were individually focussed into the sample, with incident beam diameters on the order of 100 and 50 μm, respectively. The probe transmitted through the sample at normal incidence, while the pump was near-collinear (∼1°) with the probe. After the sample, changes in probe intensity were detected using a homebuilt dispersive multi-channel spectrometer employing a CCD camera (Entwicklungsbuero Stresing) with a 512×58-pixel sensor (Hamamatsu S7030). A second camera was dedicated for referencing to reduce shot-to-shot noise.Samples were held under high vacuum (<10−5 mbar) in an optical cryostat (Oxford Instruments MicrostatHe2) and cooled using liquid N2. The cryostat itself was positioned between the poles of a water-cooled electromagnet (GMW 3470), which could sustain magnetic fields of up to 407 mT. The external field was applied normal to the sample plane and parallel to the probe propagation direction. The cryostat was supported by a custom-built mount fixed to a mechanical translation stage (Zaber X-LSM025A-E03/X-VSR20A) capable of 2-dimensional movement transverse to the probe propagation direction. This enabled the sample to be translated to new positions as required.
Broadband transient transmittance (TT) and time-resolved magnetic circular dichroism (TR-MCD) spectra were both measured simultaneously using a procedure previously described by our group.12 To summarise, TT measurements were performed pairwise at a 1 kHz repetition rate having circularly polarised the supercontinuum probe before the sample using an achromatic quarter-wave plate (Thorlabs SAQWP05M-700): each measurement pair consisted of spectra recorded with an applied magnetic field in either the positive or negative direction, with 1000 laser shots accumulated for each time point. The total pump exposure time for a single measurement pair was ∼14 minutes; for reference, sample integrity measurements performed using maximum pump fluence indicated that the material was photostable for at least double this time (Fig. S4(a), ESI†). For each new pair of measurements, the sequence of the applied field directions was reversed, and the sample was translated to a fresh position with a transmittance matching that of a marked reference point. This was to prevent unwanted artefacts resulting from long-term fluctuations in laser power, sample inhomogeneity, or photodegradation/heating from prolonged pump exposure. All scans for a given field direction were corrected and averaged to yield the photoinduced response for that specific field. The overall TT spectrum was calculated by averaging the spectra for the two field directions and is expressed as the relative transmittance change, ΔT/T, in percent. Conversely, TR-MCD spectra were obtained by computing the difference between the two datasets and are expressed as a change in ellipticity angle, ΔηF, in mdeg. A 750 nm shortpass filter situated after the sample was used to remove any scattered pump light before detection.
To enhance the signal-to-noise ratio in magneto-optical kinetic traces, single-wavelength TR-MCD (also known as time-resolved Faraday ellipticity) measurements were conducted at 20 kHz repetition rate using balanced polarisation bridge detection. For this, a small portion of the 800 nm signal output from the OPA was frequency-doubled to yield a 400 nm linearly polarised probe with a fraction (<1%) of the pump beam's power. To accommodate the higher modulation speed, two lenses were inserted before and after the chopper to focus the pump beam through the smallest wheel slots and subsequently collimate. Consequently, the pump pulse durations were significantly longer compared to the broadband pump–probe measurements. Consecutive ellipticity traces were collected pairwise for a given sample position—twice for each field direction—and the difference between the two measurements was calculated to yield only the magnetic response of the sample, i.e. ΔηF. This scheme was also used to measure time-resolved hysteresis within the sample, where ΔηF was measured as the magnetic field was varied for a series of fixed tdelay. To maintain consistency across different measurements, the sample was positioned such that the transmitted beam intensity matched that of the marked reference point, as indicated by the static photodiode detector voltage. The same position was used for all single-wavelength kinetic traces, while a different position was used for time-resolved hysteresis. The static response (i.e. with no pump present) was reassessed ahead of each measurement to ensure that there was no significant change in sample transmittance that would suggest photodegradation. Likewise, assessment of TR-MCD data collected using maximum fluence revealed that consecutive traces were reproducible and did not show any significant change in shape or intensity (Fig. S4(b), ESI†), thereby indicating the sample is photostable throughout these measurements.
3 Results and discussion
From inspection of the static (or steady-state) electronic spectrum (Fig. 1(a)), there are two clear targets for photoexcitation in the pump–probe studies: the strong peak observed in the UV (λmax = 380 nm) and the broad feature observed in the NIR (λmax > 1000 nm). These have previously been attributed to metal-to-metal charge transfer (MM′CT).34,37 However, it is notable that the physical properties of Cr–Cr PBA are consistent with those exhibited by “class II” mixed-valence systems, as described by Robin and Day.43 The material is magnetically dilute with a low Tc (∼160 K), and possesses coordination environments of similar symmetry that contain different valencies (octahedral CrIII and CrII). In such systems, electronic spectra generally consist of a superposition of the localised transitions observed within the coordination environments (e.g. d–d), as well as more intense intervalence transitions between the different environments (i.e. MM′CT). Indeed, this description also accounts for the features observed in the spectrum for Cr–Cr PBA, in which the transition energies of the aforementioned peaks appear to match those of isolated [Cr(CN)6]3− and [Cr(CN)6]4− chromophores.44–48 Therefore, it is reasonable to assume that these absorption features may possess contributions from localised B-site transitions.Broadband TT and TR-MCD were recorded for Cr–Cr PBA using an 800 nm pump to excite the transition in the NIR at 80 K under an applied magnetic field strong enough to saturate the sample (μ0H > |65| mT). The resultant spectra are shown in Fig. 2. In the TT difference spectra, the UV is dominated by a broad positive signal that corresponds to the onset of a strong absorption band observed in the material, indicating that there is a sizeable ground state bleach (GSB) contribution. The asymmetry towards the blue end of the largest peak (λmax = 422 nm) and the trough (λ = 470 nm) in the middle of the GSB band are inconsistent with the overlaid static transmittance spectrum, which may instead arise from overlap with at least two excited state absorption (ESA) components. Two distinct negative signals are observed in the TT that correspond to additional ESA bands: a broad feature is observed in the middle of the visible region (λ = 580 nm), and a second feature in the red comprising two peaks (λmax = 708 and 730 nm, respectively). Notably, this latter feature is unique to low temperatures, being completely absent from spectra recorded at 295 K (see Fig. S5(a), ESI†).
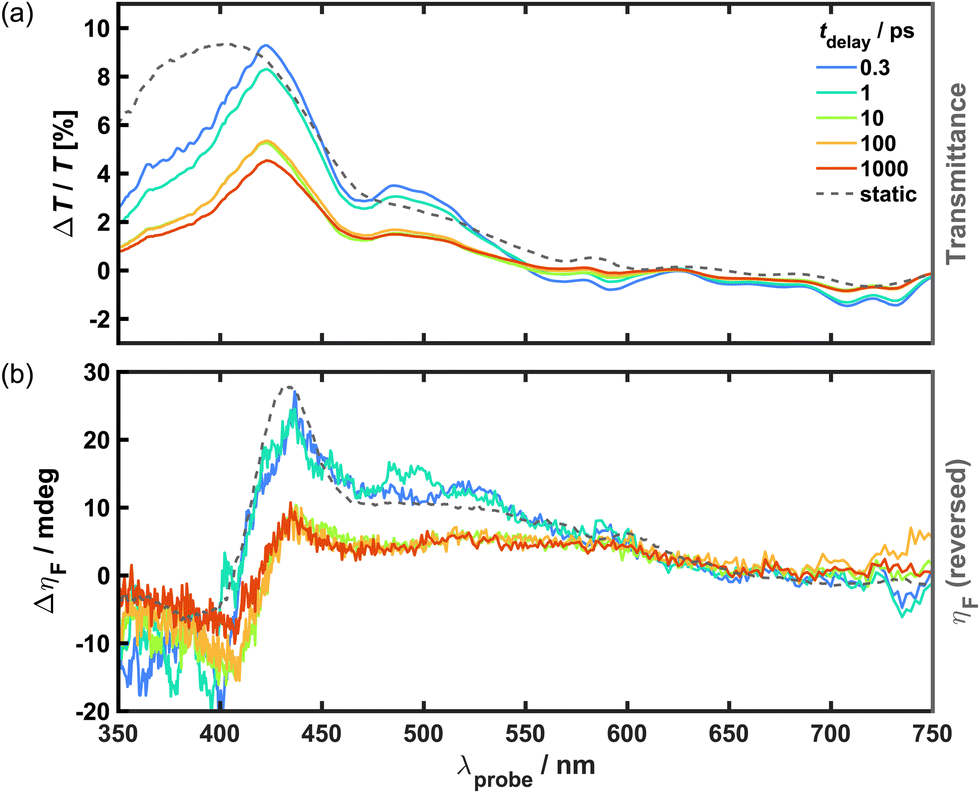 | ||
| Fig. 2 Spectral-dependence of electronic and magnetisation dynamics at discrete tdelay in Cr–Cr PBA upon photoexcitation with an 800 nm pump of fluence 4.2 mJ cm−2 at 80 K under an applied field of 407 mT. (a) TT difference spectra (coloured solid lines, left axis) with overlaid static transmittance spectrum (grey dotted line, right axis). (b) TR-MCD difference spectra (coloured solid lines, left axis) with overlaid static MCD spectrum (grey dotted line, right axis). The static MCD is reversed along the y-direction to aid comparison to the TR-MCD. | ||
The TR-MCD from the outset closely resembles the shape of the corresponding static MCD spectrum but with opposite sign. This indicates an overall decrease of the MCD is observed across the spectrum upon photoexcitation, which is consistent with demagnetisation and could be viewed as a GSB-like effect. While it might be expected that excited state MCD is observed, analogous to the ESA seen in the TT, the differences between the spectral profiles of the TR-MCD and static MCD are subtle and are difficult to analyse quantitatively given the noise in the data. This suggests that any potential excited-state MCD features are either very weak compared to the GSB contribution or lie outside of the detectable wavelength range, i.e. in the UV or NIR. In equivalent measurements performed at 295 K (see Fig. S5(b), ESI†), no TR-MCD response is observed that is discernible from baseline noise at any time delay. Likewise, the static MCD spectrum also exhibits no strong features.
The kinetics for the magnetisation dynamics were observed through polarimetric measurements performed at a higher repetition rate. Hence, TR-MCD was measured for a 400 nm probe at 20 kHz, and the data compared to TT acquired under similar excitation conditions (800 nm pump of fluence 2 mJ cm−2) at 1 kHz. To provide a closer match to the polarimetric experiment, the supercontinuum probe for this TT measurement was linearly polarised, and the 400 nm kinetic trace was generated through the Gaussian-weighted average of the spectrally resolved data with a bandwidth (5.89 nm) selected to match those expected of the probe in the TR-MCD experiment. The resultant TT and TR-MCD kinetic traces are shown in Fig. 3. Both traces conformed well to a triexponential phenomenological model convoluted with an experiment-specific instrument response function (IRF) to account for the temporal widths of the pump and probe pulses. The IRF was determined through the fitting of non-linear artefacts around time-zero, with values of 50 and 200 fs for TT and TR-MCD, respectively. This is a result of the difference in the pulse durations between the two experiments, as previously outlined. For both experiments, maximum change is observed within 200 fs, followed by a rapid decay within a few ps (τ1 = 2.45(2) and 1.5(1) ps for TT and TR-MCD, respectively), signal growth on intermediate timescales (τ2 = 220(40) and 90(30) ps), and long-term decay on protracted timescales that surpass the maximum tdelay (τ3 ≫ 1000 ps). The significant differences in the early/intermediate kinetic timescales for TT and TR-MCD suggest that the two signals evolve independently, and that the latter response is not merely the manifestation of an optical effect induced by the pump. From the observed time constants, it can be surmised that τ1 likely describes the relaxation of an excited state populated within 130 fs of photoexcitation to some long-lived intermediate state, while the long-term decay described by τ3 pertains to ground-state recovery. Conversely, the origin of τ2 is less intuitive: this slow signal growth is observed across much of the TT spectrum and is shown to possess a much larger amplitude in the TR-MCD trace, suggesting the underlying physical process has a greater influence on the magnetisation dynamics. Notably, the dissimilarity in the early kinetics observed for TT and TR-MCD does not reflect previous findings in V–Cr PBA,16 and the slow growth is likewise unique to the measurements on Cr–Cr PBA.16,49
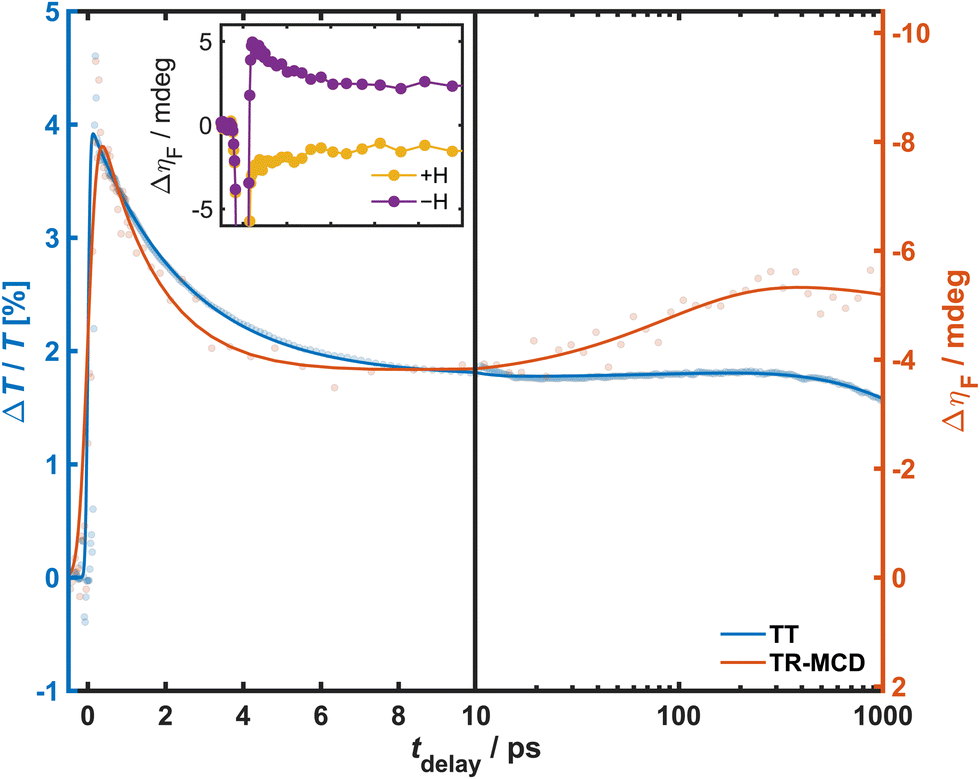 | ||
| Fig. 3 TT and TR-MCD kinetics observed in 400 nm probe upon photoexcitation with an 800 nm pump of fluence 2 mJ cm−2 at 80 K under an applied field of 65 mT. TT data were acquired at 1 kHz repetition rate, while TR-MCD data were acquired at 20 kHz. Recorded values are represented by filled circles, fits represented by solid lines. Note that the TR-MCD y-scale is reversed for easier visual comparison of kinetic timescales. Inset: TR-MCD traces obtained for each applied field direction. | ||
To test the field-dependence of the magnetisation dynamics and determine the effect of photoexcitation on material coercivity and remanence, time-resolved hysteresis measurements were carried out under the same excitation conditions as the TR-MCD trace. ΔηF was measured as a function of μ0H for various tdelay of interest, and the total ellipticity of the photoexcited sample,  , calculated using this and static hysteresis data via
, calculated using this and static hysteresis data via . The resultant loops are provided in Fig. 4, alongside μ0Hc, remanence
. The resultant loops are provided in Fig. 4, alongside μ0Hc, remanence 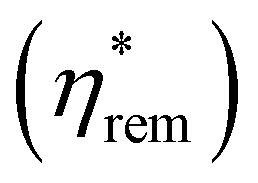 and saturation
and saturation  observed at different tdelay. The evolution of
observed at different tdelay. The evolution of 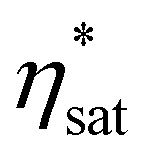 closely matches observations of the TR-MCD kinetics shown in Fig. 3, as would be expected given that the previous measurements were performed on a magnetically saturated sample. Likewise,
closely matches observations of the TR-MCD kinetics shown in Fig. 3, as would be expected given that the previous measurements were performed on a magnetically saturated sample. Likewise,  also conforms to the same trend: an initially strong decrease, which shows partial recovery within a few ps before decreasing again at protracted timescales. μ0Hc is observed to obey the same trend, but inverted: this is instead enhanced upon photoexcitation and grows again on protracted timescales. This implies that there may be an increase in anisotropy in the excited state. The same triexponential function used to fit the TR-MCD traces was also applied to the time-dependence of μ0Hc,
also conforms to the same trend: an initially strong decrease, which shows partial recovery within a few ps before decreasing again at protracted timescales. μ0Hc is observed to obey the same trend, but inverted: this is instead enhanced upon photoexcitation and grows again on protracted timescales. This implies that there may be an increase in anisotropy in the excited state. The same triexponential function used to fit the TR-MCD traces was also applied to the time-dependence of μ0Hc, 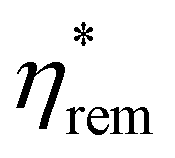 and
and  , in which the IRF and τ values from before were retained, but amplitudes were varied and a constant offset added. This crude model provided a reasonable description of the evolution of the three quantities upon photoexcitation, indicating that all changes in the shape of the hysteresis likely conform to the same kinetic timescales.
, in which the IRF and τ values from before were retained, but amplitudes were varied and a constant offset added. This crude model provided a reasonable description of the evolution of the three quantities upon photoexcitation, indicating that all changes in the shape of the hysteresis likely conform to the same kinetic timescales.
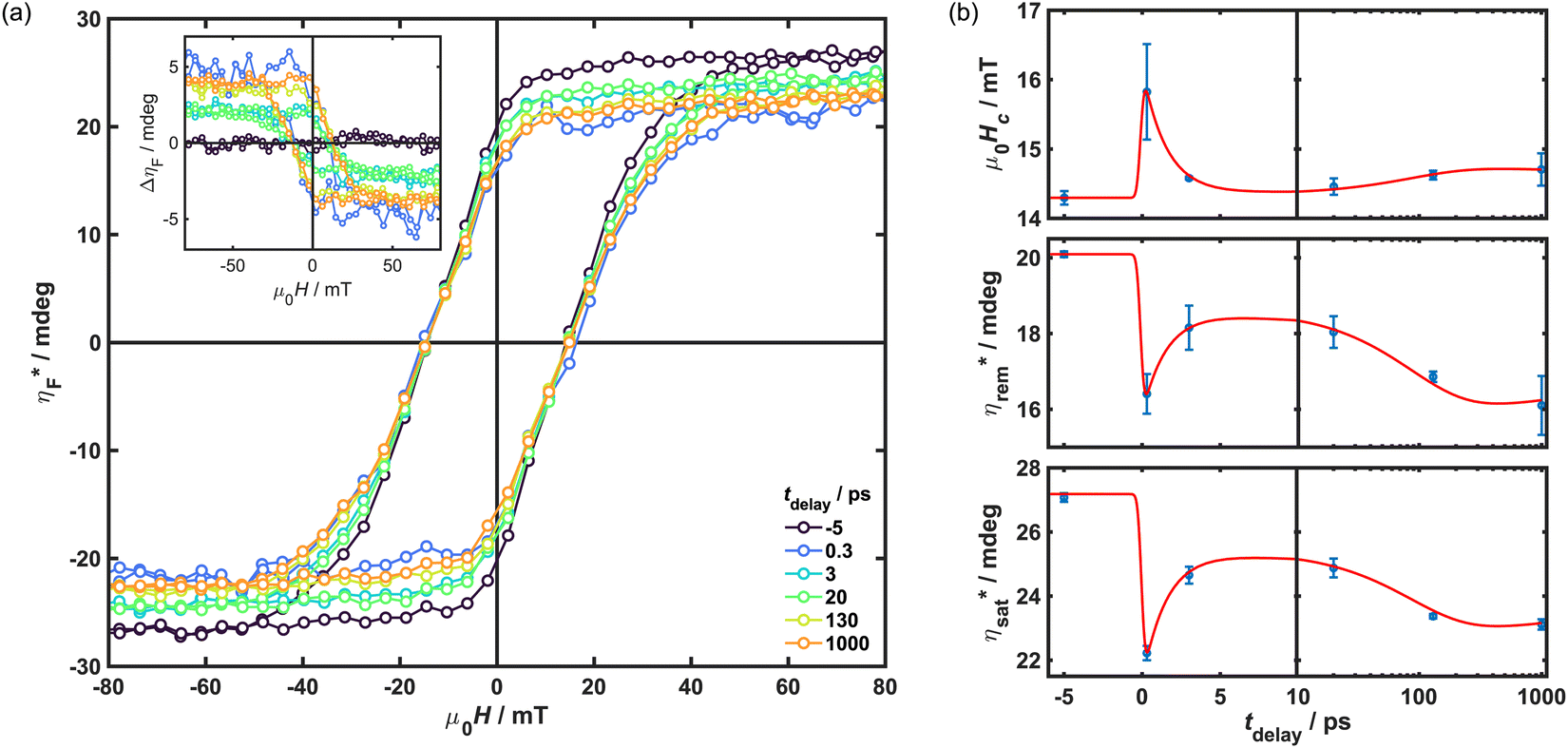 | ||
Fig. 4 Time-resolved hysteresis recorded upon photoexcitation with an 800 nm pump of fluence 2 mJ cm−2 at 80 K. Data were obtained through TR-MCD using a 400 nm probe beam at a repetition rate of 20 kHz. (a) Total ellipticity, 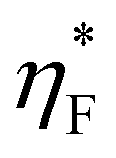 , calculated at fixed tdelay from ΔηF (shown inset). (b) Evolution of coercivity, remanence and saturation as a function of tdelay. Overlaid are fits possessing the same τ values as those determined from the kinetic modelling shown in Fig. 3. , calculated at fixed tdelay from ΔηF (shown inset). (b) Evolution of coercivity, remanence and saturation as a function of tdelay. Overlaid are fits possessing the same τ values as those determined from the kinetic modelling shown in Fig. 3. | ||
The apparent growth in the TT response echoes findings from previous pump–probe investigations on solid thin films, where authors reported primarily thermal contributions to transient spectra 100 ps after photoexcitation in haematite50 and BiVO4.51 In the latter study, the effects of sample heating were first observed after 10 ps, which is roughly consistent with the onset of the signal growth observed in the data presented here. Thus, these kinetics could be ascribed to slower heating effects that occur following the redistribution of energy from photoexcited electrons to the lattice of the sample. To test this hypothesis, static transmittance spectra were measured for various sample temperatures above 80 K and the difference between them calculated to determine the effect of macroscopic sample heating on transmittance (see Fig. S6(a), ESI†). These thermal difference spectra were then used as a basis function alongside Gaussians representing the various ESA components to decompose broadband TT difference spectra acquired at protracted time delays using a dedicated software package (KOALA v1.152). Notably, inclusion of the steady-state transmittance spectrum to specifically represent the GSB in addition to the thermal difference contribution did not provide any improvement to the fit; hence, this was omitted from the analysis. The resultant lineshape fit, shown in Fig. 5, provides a reasonable approximation of the experimental data. This indicates there is a significant thermal contribution to the difference spectrum alongside at least two overlapping ESA components: one band in the UV (λmax < 350 nm) and one broad band in the middle of the visible (λmax = 550 nm), as predicted. While the trough at 470 nm and the negative features beyond 700 nm were previously thought to have arisen from ESA, these are well described by the thermal difference component, suggesting that they are instead signals introduced by sample heating.
 | ||
| Fig. 5 Spectral decomposition analysis for TT data recorded at 80 K. The difference spectrum observed at tdelay = 100 ps was fitted using the KOALA52 software package. Overlaid are experimental data, the total fit, and the three component basis functions that comprise the total fit: the thermal difference spectrum obtained by sample heating and two Gaussians for ESA contributions. | ||
As MCD and TR-MCD could only be observed when the sample was magnetically ordered, it would be expected that the TR-MCD trace exhibits a greater sensitivity to changes in sample temperature than the TT, particularly if increased towards the Tc. The greater growth amplitude in the TR-MCD than the TT upon photoexcitation with the same pump fluence gives credence to this: the response observed after 300 ps (ΔηF = −5.3 mdeg) corresponds to 19% loss in MCD signal (cf. ηF = 27.9 mdeg), an order of magnitude higher than the 1.8% increase in transmittance observed at the same tdelay. In principle, the amount of heating observed will be determined by the pump fluence, as higher fluences will excite more sites within the sample and provide more point sources of heat. Hence, the pump fluence-dependence of the material's dynamic response is shown in Fig. 6. Both the initial (tdelay = 0.3 ps) and longer-term (tdelay = 300 ps) TT response increase linearly with fluence up to 4.2 mJ cm−2. In comparison, while the fluence-dependence of ΔηF for early timescales also remains linear, a non-linear dependence is observed at protracted timescales. At the highest fluence, which should correspond to the greatest amount of heat deposited into the sample, the growth is shown to dominate the long-term TR-MCD response (ΔηF = −20 mdeg at 1 ns) and is equivalent to >72% loss in MCD signal. The reasons for this could be the shape of the temperature-dependence of the magnetisation close to Tc, which is non-linear, and the change in temperature due to the pump laser which will also be influenced by the temperature-dependence of the heat capacity. Note that no trend could be discerned for the kinetic timescales with respect to increasing fluence for either the TT or TR-MCD data. This is unlike the outcome of ultrafast studies on Co–Fe and Mn–Fe PBA, where photoexcitation using pump fluences exceeding a particular threshold has been observed to affect the lifetime of photoinduced phase transitions.53–55
 | ||
| Fig. 6 Fluence-dependent kinetics observed for TT and TR-MCD measurements. (a) Kinetic traces for a 400 nm probe upon photoexcitation with an 800 nm pump of varied fluence. Recorded values are represented by filled circles, fits are represented by solid lines. The scale on the lower right axis indicates the relative change in TR-MCD observed with respect to the static ηF value. (b) Response observed at selected tdelay with respect to fluence, with fits represented by solid lines (quadratic fit to the 300 ps TR-MCD signal). | ||
As some ambiguity remains as to the nature of the transition excited by the 800 nm pump, the identities of the states involved in the early-time kinetics observed in the TT and TR-MCD are not immediately intuitive, nor are the origins of the two ESA features. As previously indicated, the NIR absorption band that is being photoexcited arises only in reduced materials with B = CrIILS content, so it can be assumed that optical pumping initially populates either an interconfigurational (i.e. d–d) state centred on B = CrIILS or a charge-transfer state involving B = CrIILS and neighbouring A-sites. In the latter event, the occupation of A-sites by either high-spin (HS) CrIIHS or CrIII would give rise to several possible MM′CT transitions, each of which would be a candidate for assignment of the NIR absorption. Interestingly, broadband TT/TR-MCD measurements performed having photoexcited the energetically distinct shoulder at 514 nm (Fig. S7 and S8, ESI†) yield difference spectra which possess identical features to those recorded for the 800 nm pump. Likewise, the TT kinetics observed are also similar for the two pump wavelengths. This would indicate that some common intermediate state is populated within the experimental time resolution for the two measurements, irrespective of pump photon energy, and that decay of this into some number of trapped exciton states and thermal energy is described by τ1.
Given the inhomogeneity of the material (mixture of oxidation states on both A- and B-sites, vacancies, crystal boundaries, etc.), one interpretation for the electronic component of the dynamics is that a series of excitons, whether they be charge-transfer or Frenkel (i.e. d–d), in different environments are generated by the pump pulse. These would give rise to the initial change in transmittance and the ESA features observed. While some may decay quickly (τ1), other excitons may be long-lived or trapped, hence the observation of a non-zero signal that does not appreciably decay within the tdelay accessible within the measurements (τ3). Assuming that the decay is non-radiative, the energy released will be distributed to the lattice through phonon excitation. The slow growth (τ2) observed could then be the result of lattice thermalisation following phonon–phonon coupling, thus explaining why the TT difference spectra resemble the thermal difference spectra associated with macroscopically heating the sample. Within this interpretation of the dynamics, the initial TR-MCD response would result from the disruption of magnetic order following the formation of the excitons, as well as any additional optical component (i.e. change in magneto-optical constant). As these decay, the sample response would also decrease as the magnetisation recovers. On longer timescales, the growth in the TR-MCD could again be attributed to the quenching of magnetisation by sample heating as lattice thermalisation takes place. Beyond the endpoint of the measurement, this would again be expected to recover and the TR-MCD response decay towards zero.
4 Conclusion
TT and TR-MCD measurements have been used to elucidate the ultrafast dynamics that ensue in photoexcited mixed-valence Cr–Cr PBA at 80 K. Optical pumping of the NIR absorption band at 800 nm gives rise to a complex broadband TT response characterised by distinct ESA and a significant thermal component, while the broadband TR-MCD response only appears to consist of a bleach feature that aligns well with the static MCD. The evolution of the TT and TR-MCD response for a 400 nm probe conformed well to a triexponential model consisting of a fast decay (τTT = 2.45(2) and τTR-MCD = 1.5(1) ps), slower growth from the thermal contribution (τTT = 220(40) and τTR-MCD = 90(30) ps) and ground state recovery on ns timescales, which is also reflected in time-resolved hysteresis measurements. Pump fluence is indicated to play a crucial role in determining the magnitude of the thermal contribution, which is more apparent for the TR-MCD. The early dynamics are attributed to the generation of a series of excitons corresponding to different chemical environments within the material. Late dynamics are ascribed to thermally induced effects that occur upon the decay of said excitons and redistribution of their energy to the lattice. The work reported here has demonstrated how an experimental framework comprising complementary transient absorption and magneto-optical techniques has the potential to uncover a myriad of information in investigations of both the electronic and magnetisation dynamics in molecular-based solids.Author contributions
H. A. L. and T. M. S. performed the synthesis and ultrafast measurements. H. A. L. analysed the ultrafast data, performed the material characterisation, and wrote the manuscript. O. C. performed SQUID magnetometric characterisation of the oxidised material. J. O. J. conceptualised the project, acquired funding, and edited the manuscript.Data availability
Data for this article, including transient absorption data and magneto-optical spectra for various wavelengths are available from the authors on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded in whole, or in part, by EPSRC (EP/S018824/1 and EP/V010573/1). The authors thank N. Robertson for the use of the potentiostat for making films and A. V. Gromov for Raman/reflectance IR measurements.References
- E. Beaurepaire, J.-C. Merle, A. Daunois and J.-Y. Bigot, Phys. Rev. Lett., 1996, 76, 4250–4253 CrossRef CAS PubMed.
- A. V. Kimel and M. Li, Nat. Rev. Mater., 2019, 4, 189–200 CrossRef.
- R. Mikhaylovskiy, E. Hendry, A. Secchi, J. Mentink, M. Eckstein, A. Wu, R. Pisarev, V. Kruglyak, M. Katsnelson, T. Rasing and A. Kimel, Nat. Commun., 2015, 6, 8190 CrossRef CAS PubMed.
- A. Stupakiewicz, K. Szerenos, D. Afanasiev, A. Kirilyuk and A. V. Kimel, Nature, 2017, 542, 71–74 CrossRef CAS PubMed.
- R. Mikhaylovskiy, T. Huisman, V. Gavrichkov, S. Polukeev, S. Ovchinnikov, D. Afanasiev, R. Pisarev, T. Rasing and A. Kimel, Phys. Rev. Lett., 2020, 125, 157201 CrossRef CAS PubMed.
- M. Dąbrowski, S. Guo, M. Strungaru, P. S. Keatley, F. Withers, E. J. G. Santos and R. J. Hicken, Nat. Commun., 2022, 13, 5976 CrossRef PubMed.
- M. Khela, M. Dąbrowski, S. Khan, P. S. Keatley, I. Verzhbitskiy, G. Eda, R. J. Hicken, H. Kurebayashi and E. J. G. Santos, Nat. Commun., 2023, 14, 1378 CrossRef CAS PubMed.
- E. Sutcliffe, X. Sun, I. Verzhbitskiy, T. Griepe, U. Atxitia, G. Eda, E. J. G. Santos and J. O. Johansson, Phys. Rev. B, 2023, 107, 174432 CrossRef CAS.
- D. Khusyainov, T. Gareev, V. Radovskaia, K. Sampathkumar, S. Acharya, M. Šiškins, S. Mañas-Valero, B. A. Ivanov, E. Coronado, T. Rasing, A. V. Kimel and D. Afanasiev, APL Mater., 2023, 11, 071104 CrossRef CAS.
- F. Hendriks, R. R. Rojas-Lopez, B. Koopmans and M. H. D. Guimarães, Nat. Commun., 2024, 15, 1298 CrossRef CAS PubMed.
- P. J. Reid, C. Silva, P. F. Barbara, L. Karki and J. T. Hupp, J. Phys. Chem., 1995, 99, 2609–2616 CrossRef CAS.
- J. Sutcliffe and J. O. Johansson, Rev. Sci. Instrum., 2021, 92, 113001 CrossRef CAS PubMed.
- S. Ohkoshi, M. Mizuno, G. Hung and K. Hashimoto, J. Phys. Chem. B, 2000, 104, 9365–9367 CrossRef CAS.
- Y. Sato, S. Ohkoshi and K. Hashimoto, J. Appl. Phys., 2002, 92, 4834–4836 CrossRef CAS.
- S. Ohkoshi, J. Shimura, K. Ikeda and K. Hashimoto, J. Opt. Soc. Am. B, 2005, 22, 196–203 CrossRef CAS.
- J. O. Johansson, J.-W. Kim, E. Allwright, D. M. Rogers, N. Robertson and J.-Y. Bigot, Chem. Sci., 2016, 7, 7061–7067 RSC.
- O. Sato, Y. Einaga, T. Iyoda, A. Fujishima and K. Hashimoto, J. Electrochem. Soc., 1997, 144, L11–L13 CrossRef CAS.
- O. Sato, Y. Einaga, A. Fujishima and K. Hashimoto, Inorg. Chem., 1999, 38, 4405–4412 CrossRef CAS PubMed.
- A. Bleuzen, C. Lomenech, V. Escax, F. Villain, F. Varret, C. C. Dit Moulin and M. Verdaguer, J. Am. Chem. Soc., 2000, 122, 6648–6652 CrossRef CAS.
- H. Tokoro, T. Matsuda, T. Nuida, Y. Moritomo, K. Ohoyama, E. D. L. Dangui, K. Boukheddaden and S. Ohkoshi, Chem. Mater., 2007, 20, 423–428 CrossRef.
- S. Ohkoshi, K. Imoto, Y. Tsunobuchi, S. Takano and H. Tokoro, Nat. Chem., 2011, 3, 564–569 CrossRef CAS PubMed.
- S. Ohkoshi, K. Shiraishi, K. Nakagawa, Y. Ikeda, O. Stefanczyk, H. Tokoro and A. Namai, J. Mater. Chem. C, 2021, 9, 3081–3087 RSC.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704–705 CrossRef CAS PubMed.
- M. Verdaguer and G. S. Girolami, Magnetism: Molecules to Materials V, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 283–346 Search PubMed.
- K. Hashimoto and S. Ohkoshi, Philos. Trans. R. Soc., A, 1999, 357, 2977–3003 CrossRef CAS.
- H. Tokoro and S. Ohkoshi, Dalton Trans., 2011, 40, 6825–6833 RSC.
- M. Reczyński, K. Nakabayashi and S. Ohkoshi, Eur. J. Inorg. Chem., 2020, 2669–2678 CrossRef.
- D. C. Arnett, P. Voehringer and N. F. Scherer, J. Am. Chem. Soc., 1995, 117, 12262–12272 CrossRef CAS.
- D. Weidinger, D. J. Brown and J. C. Owrutsky, J. Chem. Phys., 2011, 134, 124510 CrossRef CAS PubMed.
- S. Zerdane, M. Cammarata, L. Balducci, R. Bertoni, L. Catala, S. Mazerat, T. Mallah, M. N. Pedersen, M. Wulff, K. Nakagawa, H. Tokoro, S. Ohkoshi and E. Collet, Eur. J. Inorg. Chem., 2017, 272–277 Search PubMed.
- M. Cammarata, S. Zerdane, L. Balducci, G. Azzolina, S. Mazerat, C. Exertier, M. Trabuco, M. Levantino, R. Alonso-Mori, J. M. Glownia, S. Song, L. Catala, T. Mallah, S. F. Matar and E. Collet, Nat. Chem., 2020, 13, 10–14 CrossRef PubMed.
- A. Asahara, M. Nakajima, R. Fukaya, H. Tokoro, S. Ohkoshi and T. Suemoto, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195138 CrossRef.
- G. Azzolina, H. Tokoro, K. Imoto, M. Yoshikiyo, S. Ohkoshi and E. Collet, Angew. Chem., Int. Ed., 2021, 60, 23267–23273 CrossRef CAS PubMed.
- T. Mallah, S. Thiebaut, M. Verdaguer and P. Veillet, Science, 1993, 262, 1554–1557 CrossRef CAS PubMed.
- W. E. Buschmann, S. C. Paulson, C. M. Wynn, M. A. Girtu, A. J. Epstein, H. S. White and J. S. Miller, Chem. Mater., 1998, 10, 1386–1395 CrossRef CAS.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 271, 49–51 CrossRef CAS.
- E. Coronado, M. Makarewicz, J. P. Prieto-Ruiz, H. Prima-García and F. M. Romero, Adv. Mater., 2011, 23, 4323–4326 CrossRef CAS PubMed.
- H. Prima-Garcia, E. Coronado, J. P. Prieto-Ruiz and F. M. Romero, Nanoscale Res. Lett., 2012, 7, 232 CrossRef PubMed.
- P. Bhatt, S. M. Yusuf, R. Bhatt and G. Schütz, J. Solid State Electrochem., 2013, 17, 1285–1293 CrossRef CAS.
- H. A. Lewis, J. Kirkpatrick and J. O. Johansson, Thin Solid Films, 2021, 732, 138767 CrossRef CAS.
- S. Ohkoshi, A. Fujishima and K. Hashimoto, J. Am. Chem. Soc., 1998, 120, 5349–5350 CrossRef CAS.
- K. Barlow, J. Eng, I. Ivalo, M. Coletta, E. K. Brechin, T. J. Penfold and J. O. Johansson, Dalton Trans., 2022, 51, 10751–10757 RSC.
- M. B. Robin and P. Day, Advances in Inorganic Chemistry and Radiochemistry, Elsevier, 1968, pp. 247–422 Search PubMed.
- R. Krishnamurthy and W. B. Schaap, Inorg. Chem., 1963, 2, 605–608 CrossRef CAS.
- J. J. Alexander and H. B. Gray, J. Am. Chem. Soc., 1968, 90, 4260–4271 CrossRef CAS.
- A. Chiang and A. W. Adamson, J. Phys. Chem., 1968, 72, 3827–3831 CrossRef CAS.
- M. F. A. Hendrickx, V. S. Mironov, L. F. Chibotaru and A. Ceulemans, J. Am. Chem. Soc., 2003, 125, 3694–3695 CrossRef CAS PubMed.
- J. P. Eaton and D. Nicholls, Transit. Metal Chem., 1981, 6, 203–206 CrossRef CAS.
- L. Hedley, M. D. Horbury, F. Liedy and J. O. Johansson, Chem. Phys. Lett., 2017, 687, 125–130 CrossRef CAS.
- D. Hayes, R. G. Hadt, J. D. Emery, A. A. Cordones, A. B. F. Martinson, M. L. Shelby, K. A. Fransted, P. D. Dahlberg, J. Hong, X. Zhang, Q. Kong, R. W. Schoenlein and L. X. Chen, Energy Environ. Sci., 2016, 9, 3754–3769 RSC.
- J. K. Cooper, S. E. Reyes-Lillo, L. H. Hess, C.-M. Jiang, J. B. Neaton and I. D. Sharp, J. Phys. Chem. C, 2018, 122, 20642–20652 CrossRef CAS.
- M. P. Grubb, A. J. Orr-Ewing and M. N. R. Ashfold, Rev. Sci. Instrum., 2014, 85, 064104 CrossRef PubMed.
- H. Kamioka, Y. Moritomo, W. Kosaka and S. Ohkoshi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 180301 CrossRef.
- T. Suemoto, R. Fukaya, A. Asahara, H. Watanabe, H. Tokoro and S. Ohkoshi, Curr. Inorg. Chem., 2016, 6, 10–25 CrossRef CAS.
- H. Tokoro, S. Ohkoshi and K. Hashimoto, Appl. Phys. Lett., 2003, 82, 1245–1247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01153d |
| This journal is © The Royal Society of Chemistry 2024 |