
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalized graphene oxide–antibody conjugate-based electrochemical immunosensors to detect Opisthorchis viverrini antigen in urine†
Nang Noon Shean
Aye
ab,
Pornsuda
Maraming
b,
Patcharaporn
Tippayawat
b,
Sakda
Daduang
c,
Anchalee
Techasen
bd,
Wassana
Jamnongkan
d,
Paiboon
Sithithaworn
de and
Jureerut
Daduang
 *b
*b
aBiomedical Sciences Program, Graduate School, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: nangnoonshean.aye@kkumail.com
bCentre for Research and Development of Medical Diagnostic Laboratories, Faculty of Associated Medical Sciences, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: pornsma@kku.ac.th; patchatip@kku.ac.th; jurpoo@kku.ac.th
cDivision of Pharmacognosy and Toxicology, Faculty of Pharmaceutical Sciences, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: sakdad@kku.ac.th
dCholangiocarcinoma Research Institute, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: anchte@kku.ac.th; wassana_jk@hotmail.co.th
eDepartment of Parasitology, Faculty of Medicine, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: paib_sit@kku.ac.th
First published on 23rd March 2024
Abstract
The liver fluke, Opisthorchis viverrini (OV), is the most important risk factor associated with cholangiocarcinoma (CCA) and its infection is a major public health burden in the Greater Mekong Subregion. The limitations of conventional diagnostic tools such as prolonged detection time and multistep procedures have led to an upsurge in demand for point-of-care testing that can facilitate timely and early action for better treatment. Herein, a novel, yet simple, electrochemical immunosensor for the onsite detection of OV antigen (Ag) in urine samples was developed. The modification of graphene-oxide (GO) surfaces with monoclonal anti-OV antibodies (mAb) through covalent conjugation not only shortened the time of antibody immobilization but also enhanced the sensitivity of the immunosensor. The successful conjugation of GO–mAb was characterized using functional and structural analysis. Furthermore, the electrochemical properties of the modified screen-printed sensor electrodes were verified by electrochemical and surface characterization. The developed electrochemical immunosensor achieved a low limit detection of OV Ag of 0.08 ng mL−1 in standard buffer and 1.50 ng mL−1 in urine. Moreover, it exhibited a broad linear range (0.2–200 μg mL−1) of OV Ag detection and demonstrated excellent diagnostic potential with 93% sensitivity and 69% specificity compared to the traditional ELISA, and 92% sensitivity and 90% specificity compared to the commercial Rapid Diagnostic Test kit for urine OV Ag detection (OV-RDT). The highest AUC of 0.950 with 96% sensitivity and 90% specificity was obtained in comparison with the combined ELISA and RDT, without requiring the sample pre-treatment step. The results of the application of the developed immunosensor in the field-collected urine samples (n = 530) have further emphasized its potential utility as a prototype for point-of-care OV Ag detection.
1. Introduction
Cholangiocarcinoma (CCA) is a malignant tumour originating from the epithelial lining of the biliary tract.1 The highest incidence of CCA has been reported in Northeast Thailand and the Greater Mekong Subregion (GMS) of Southeast Asia.2 The liver fluke, Opisthorchis viverrini (OV), which is attained by consumption of raw or undercooked (salted, pickled, or smoked) infected cyprinid fish carrying the larval stage of the parasite called metacercaria, is the most important risk factor associated with CCA.3 The risk factors for opisthorchiasis-related CCA include chronic inflammation and associated injury of the biliary epithelium due to the persistent parasitic infection.1 OV has been classified as a group 1 biological carcinogen because the highest incidence of CCA was primarily due to OV infection.4,5 Approximately 40 million people are infected with OV in the GMS region alone6,7 and more than 600 million people are at risk,5 indicating that rapid diagnostic detection methods are in demand.The gold standard to diagnose OV infection is to detect OV eggs in faecal samples quantitatively using the formalin-ethyl acetate concentration technique (FECT).8 However, this technique has numerous shortcomings, such as limited sensitivity and specificity, extensive faecal sampling, and the need for a qualified microscopist to distinguish the OV eggs from eggs of other similar helminths.9 Moreover, light infection was unable to be detected and biliary tract obstruction leads to impaired flow of OV eggs into faeces, which would be a limitation for the detection of OV eggs using the FECT, resulting in false-negative cases.
Recently, a variety of immunological and molecular diagnostic methods have been reported to detect OV infection.10–14 OV Ag can be generally classified into somatic and excretory-secretory (ES) antigens. The crude somatic extract of adult OV worms was the most common antigen used to detect antibody in the sera by ELISA.15,16 Antibody response to both somatic and ES antigens was detected.17,18 IgG, IgA and IgE antibodies in serum, urine and bile have been reported in patients with opisthorchiasis.18–21 Even though these techniques exhibited better diagnostic sensitivity and specificity than the FECT to detect fecal eggs, these assays still have limitations in clinical application in terms of analytical sensitivity, cost of testing, prolonged and complicated procedures and the requirement of sophisticated instruments. Moreover, the serological diagnosis by OV antibody detection has drawbacks such as the cross interaction of the antigen used and the persistence of antibody after treatment that cannot reflect active infection with the parasite.22 The ELISA techniques have been developed to detect urine OV-ES antigen using a specific monoclonal antibody (IgG1 murine mAb, clone KKU 505) and sandwich OV Ag with protein A-purified rabbit IgG against OV Ag. The urine OV ELISA assay proved that urine antigen detection yielded higher diagnostic sensitivity and specificity (81% and 71%) compared to the FECT technique. However, these methods still have shortcomings such as detection limit, multistep procedures, and requirement of well-trained technicians. These challenges encourage the development of simple and cost-effective point-of-care biosensors for OV antigen detection in urine.
Amongst numerous immunosensors constructed, electrochemical immunosensors are promising breakthroughs because of their benefits such as high sensitivity, specificity, reproducibility, portability, and reduced environmental noise. Three-electrode combined screen-printed carbon electrodes (SPCEs) have been widely applied in electrochemical sensors due to their large surface area, cost-effectiveness, high reproducibility, and ease of mass production.23 Moreover, modification of SPCEs can easily be performed for better analytical performances. In electrochemical biosensing, the voltammetric signal of the target analyte can easily be extracted from interfering substances by appropriate modification of electrodes with analyte specific micro/nanomaterials. Graphene oxide (GO), produced by chemical exfoliation of graphite using strong oxidizing agents, has been widely applied for electrode modification. GO comprises numerous oxygen-containing functional groups such as carboxyl, carbonyl, phenol, lactone and quinone at the edges of GO, hydroxyl and epoxy groups on the basal plane of GO.24 Due to these functional groups, GO provides a broad surface area, great optical, electronic, mechanical, and electrochemical properties.25,26 GO is a promising material for applications in the field of bioelectrochemistry due to its advantageous properties.
Besides, the utilization of antibodies as bioreceptors together with GO can enhance the sensitivity and specificity of the electrochemical biosensor through specific antibody–antigen interaction. Monoclonal antibodies are more commonly used in biomolecular interactions than other bioreceptors such as aptamers or enzymes, as antibodies possess high specificity and unique in vivo features, while enzyme reactions are more complicated and aptamers can easily be degraded by the action of nucleases.27 For the construction of an immunosensor, the immobilization of antibodies on the sensor surface without decreasing their binding affinities and capacities is of particular importance.
In this study, the functionalized GO conjugated with OV-specific monoclonal antibody (mAb) was employed for the sensitive and reliable detection of OV Ag in urine samples. The monoclonal antibody specific to OV somatic and ES antigens provided successful diagnosis of opisthorchiasis. Since the biomolecules can be coated on GO surfaces via non-specific physical adsorption and π–π stacking, any proteins with an aromatic ring could be attached to GO. To circumvent this, surface modification of GO with monoclonal antibodies was proposed due to the higher specific binding of GO and mAb through covalent bonding. Antibody-conjugated GO-based sensors were primarily reported for fluorescent quenching28,29 but were rarely applied for electrochemical immunosensing. To the best of our knowledge, this is the first study reported for the fabrication of a point-of-care electrochemical detection system of urine OV Ag. The proposed immunosensor has been validated for analytical and diagnostic performance by using real urine samples. The developed electrochemical immunosensor will contribute to improve diagnostics and applications for mass screening of OV infection within the community.
2. Experimental section
2.1. Chemicals
The crude somatic antigen of Opisthorchis viverrini (OV Ag) and a monoclonal antibody specific to OV somatic Ag (mAb, KKU 505) were prepared by one of the co-authors (PS). The commercial screen-printed carbon electrodes were purchased from Quasense, Thailand. Graphene oxide (GO), N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 4-morpholinoethanesulfonic acid (MES) free acid and bovine serum albumin (BSA) were obtained from Sigma-Aldrich (Singapore). Potassium ferricyanide (K3[Fe (CN)6], 5 mM) with 0.1 M KCl in phosphate-buffered saline (PBS) (1×, pH 7.4) was used as a redox indicator. Washing buffer (1× PBS, pH 7.4) was used between each coating procedure.2.2. Instruments and characterization
The voltammograms were performed on the PalmSens4 potentiostat (PalmSens BV Co., Ltd, Netherlands) using PS Trace 5.6 software. The disposable screen-printed electrode with a three-electrode system, including a 2.5 mm working carbon electrode, a carbon counter electrode and a silver/silver chloride reference electrode, was used. Cyclic voltammetry (CV) was performed by applying a potential of −0.5 V to 0.9 V at a scan rate of 100 mV s−1 and the electrochemical impedance spectra (EIS) were recorded using the frequency range (100 mHz to 100 kHz) in 0.01 M PBS containing 5 mM [Fe (CN)6]3−/4− and 0.1 M KCl. The square wave voltammetry (SWV) measurements were performed by applying a potential range of −0.6 to 0.6 V with a frequency of 8 Hz, a step potential of 5 mV, and an amplitude of 2 mV. The characterization of GO and GO–mAb conjugates was conducted using a Bruker TENSOR II attenuated total reflectance (ATR)-Fourier transform infrared (FTIR) spectrometer (Bruker, Germany), an Eppendorf BioSpectrometer® fluorescence (Hamburg, Germany), a field-emission scanning electron microscope (FE-SEM, Jeol, JSM-IT200 InTouchScope™, Tokyo, Japan), an atomic force microscope (AFM, Park systems, NX-10, Korea), an XploRA plus Raman spectrometer (Horbita, Kyoto, Japan) and an X-ray diffractometer (XRD) (SmartLab, Rigaku, Japan) with Cu Kα radiation by collecting the data at 2.4° per min.2.3. Production of monoclonal Ab
For the isolation of monoclonal Ab specific to OV Ag, mice were immunized with an OV somatic Ag preparation. Then, antibody-secreting cells from the immunized mice were fused with the myeloma cells. After fusion, the cells were diluted in selective culture medium (HAT medium). Hybridoma cells survived in the selective medium and were recognized by their ability to grow indefinitely in the medium. They were then tested for the presence of the desired antibody. Suitable monoclonal antibody-producing cells were cloned and expanded in serum-free RPMI medium. The culture media was collected and centrifuged at 5000g at 4 °C for 1 h. The protein concentration in the supernatant was measured using a Pierce™ _BCA Protein Assay Kit. The secreted-monoclonal antibody (IgM mAb, clone KKU505) was kept frozen at −20 °C for subsequent use.2.4. Crude somatic OV Ag extraction
Crude somatic OV Ag was extracted from OV adult worms after washing three times with sterile PBS at pH 7.4. Adult worms in the extraction buffer were homogenized and sonicated three times (30 s per time) by an ultrasonic disintegrator (MISONIC sonicator 3000, US). The sonicated worm was kept at 4 °C overnight followed by centrifugation at 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 913g at 4 °C for 30 min. The protein concentration of the supernatant was measured using the Pierce™ _BCA Protein Assay Kit. The crude somatic OvAg in the supernatant was collected and stored at −20 °C prior to use.
913g at 4 °C for 30 min. The protein concentration of the supernatant was measured using the Pierce™ _BCA Protein Assay Kit. The crude somatic OvAg in the supernatant was collected and stored at −20 °C prior to use.
2.5. Functionalization of GO and preparation of GO–mAb conjugates
The functionalization of GO was performed as reported in our previous study with slight modifications.30 Briefly, the GO powder (0.05 g) was suspended in 10 mL of 100 μM EDC and 100 μM NHS that was prepared in MES buffer (0.1 M, pH 5.5). The suspension was stirred for 4 h at room temperature, followed by centrifugation of the EDC/NHS-activated GO suspension at 16100g for 25 min, and washed twice with deionized water (DI) to remove excess EDC, and NHS was attached. The deposited pellets were collected and dried at 60 °C overnight. Activated GO (1 mg mL−1) was resuspended in DI by magnetic stirring for 30 min at room temperature followed by sonication for 10 min. The GO–mAb conjugation was simply obtained by covalent interaction of carboxyl groups of functionalized GO and amine groups on the surface of mAb. The monoclonal Ab specific to OV antigen (100 μg mL−1) was added to the GO suspension (1:9 v/v) and stirred for 20 min at room temperature.
2.6. Fabrication of electrochemical immunosensors
The layer-by-layer immobilization of GO and mAb has been omitted in this study as the GO–mAb conjugate was drop-coated onto the SPCE surface. Seven microliters of the GO–mAb conjugate were dropped onto the working area of SPCEs and allowed to dry in air for 90 min. The modified electrode surfaces were further blocked by 0.1% BSA solution for 30 min. The fabrication of an electrochemical immunosensor was accomplished through a simple modification and the modified sensor was applied for the detection of OV Ag.2.7. Electrochemical analysis
The electrochemical determination of OV Ag was conducted by introducing various concentrations of somatic OV Ag onto the developed immunosensor surface for 60 min. A redox indicator of 5 mM [Fe(CN)6]3−/4− in PBS containing 0.1 M KCl was applied. To study the fabrication process, CV and EIS measurements were performed using the parameter mentioned before. The detection of OV Ag was performed using SWV measurements with an applied potential of −0.6 to 0.6 V, a step potential of 5 mV, an amplitude of 2 mV, and a frequency of 8 Hz in the presence of 5 mM [Fe (CN)6]3−/4−. The peak currents before (I0) and after (I1) OV Ag detection were recorded. The peak current changes (ΔI = I0 − I1) were observed to correlate with OV Ag concentration present in the sample. A schematic diagram of the proposed electrochemical immunosensor is illustrated in Scheme 1.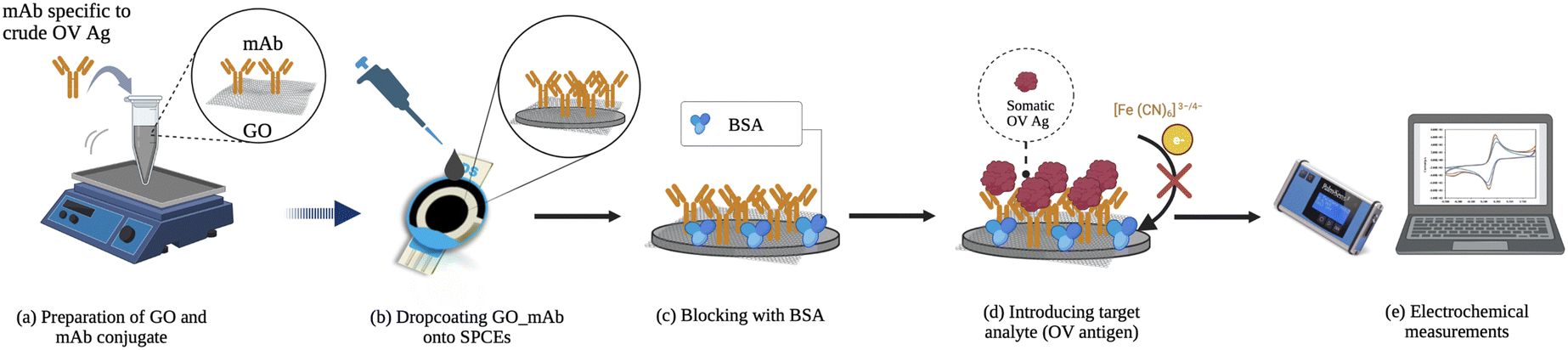 | ||
| Scheme 1 Schematic diagram for the construction of the electrochemical immunosensor. (a) Preparation of the GO–mAb conjugate, (b) dropcoating GO–mAb onto SPCEs, (c) blocking with 0.1% BSA, (d) incubation with OV antigen and (e) electrochemical measurements using 5 mM [Fe (CN)6]3−/4− as a redox probe. | ||
2.8. Evaluation of clinical utility and performance of the developed immunosensor in field testing
To evaluate the proficiency of the developed immunosensor for OV Ag detection, the urine samples were analysed as the real samples. To equilibrate the interference of other substances in urine on OV Ag detection, the standard curve was calibrated using different concentrations of OV Ag spiked into urine samples. The normal urine sample collected was diluted (1:10 dilution) prior to the experiment. The detection of OV Ag in urine samples was carried out using the same procedure as mentioned earlier except that 20 μg mL−1 mAb and 0.5% BSA were used for better sensitivity. A total of 530 urine samples from OV positive and OV negative individuals were collected from the endemic areas of opisthorchiasis in Khon Kaen Province, Thailand. The samples were further divided into three groups depending on method comparison. Group 1 consisted of 98 OV positive cases and 49 OV negative cases as confirmed by ELISA, and Group 2 involved 50 OV positive and 80 OV negative individuals as screened by the OV-rapid diagnostic test (OV-RDT) kit. The OV-RDT kit is a chromatographic assay that has been applied for mass screening of OV Ag, and indirect ELISA has been widely utilized for the determination of OV Ag concentrations in urine samples. Finally, 55 OV positive and 198 OV negative cases were included in Group 3 which were screened for OV positivity by OV-RDT and further confirmed by ELISA. Seven microliters of urine samples were dropped onto SPCEs and the SWV measurements were carried out as described in the previous section. Diagnostic sensitivity and specificity of the developed immunosensor were compared against the standard indirect ELISA technique.
2.9. Comparison of the diagnostic methods
The quantitative comparison of urine OV Ag concentration was conducted by the conventional ELISA and the developed electrochemical immunosensor was evaluated. The comparison of qualitative measurements was also performed between the ELISA, OV-RDT and the immunosensor. The indirect sandwich ELISA assay was performed using mAb and polyclonal anti-rabbit IgG antibody to capture OV Ag present in urine samples. The procedures for the ELISA assay were used as described previously11 and the detailed protocol for the ELISA assay can be found in the ESI.† Urine samples were centrifuged at 5000g for 15 min at 4 °C and the supernatant was collected and stored at −20 °C for ELISA detection. The urine OV-RDT assay was performed by applying 120 μL of urine sample followed by 40 μL of buffer and allowing the reaction to occur for 10 min. The results were observed by naked eyes and scored as OV-positive and OV-negative.2.10. Data analysis and statistical methods
Statistical analyses were performed using SPSS program, version 27.0 (SPSS Inc., Chicago, IL, USA) and MedCal (Med-Calc Software, Ostend, Belgium). Differences between two groups were analyzed using the Mann–Whitney U test for non-parametric data. The correlation between OV Ag detection by the proposed immunosensor and the reference method (ELISA) was evaluated using multivariate linear regression. The performance of the immunosensor in terms of diagnostic sensitivity, specificity, positive and negative predictive values was evaluated using the receiver operating curve (ROC) analysis.3. Results and discussion
3.1. Characterization of electrochemical immunosensors
Field emission scanning electron microscopy (FE-SEM) was used to observe morphological structures of the modified SPCE surfaces. Fig. 1 illustrates the surface morphology of bare SPCEs, GO/SPCEs, GO–mAb/SPCEs, BSA/GO–mAb/SPCEs and OV Ag/BSA/GO–mAb/SPCEs at 20000 × magnification (Fig. 1(A)–(E)) and the OV Ag immobilized immunosensor surface at 5000× magnification (Fig. 1(F)). The surface of bare SPCEs exhibited a smooth and uniform layer of carbon-based materials (Fig. 1(A)). In contrast, the SEM images of GO–mAb/SPCEs (Fig. 1(B)) and GO–mAb/SPCEs (Fig. 1(C)) showed that GO appeared with several layers of flakes with distinguishable edges of individual sheets and small number of wrinkled textures giving rough morphology.31 The surface of the GO–mAb conjugate showed a larger and rougher surface area formed by the aggregation of GO layers by mAb, indicating the increase of surface roughness. FE-SEM analysis confirmed the uniform modification of the GO–mAb conjugate. Subsequent blocking with BSA and immobilization with target OV Ag expressed a smooth web-like structure coating on the GO–mAb surfaces, showing the formation of a new layer coated on the sensor surface (Fig. 1(D)–(F)).
 | ||
| Fig. 1 FE-SEM images of (A) bare SPCEs, (B) GO-modified SPCEs, (C) GO–mAb-modified SPCEs, (D) BSA blocked SPCEs, (E) OV Ag immobilized BSA/GO–mAb/SPCEs at 20000× and (F) 5000× magnifications. | ||
The three-dimensional surface topology of bare SPCEs, GO-modified SPCEs and GO–mAb SPCEs was investigated using atomic force microscopy (AFM) (Fig. 2(A)–(C)). The root mean square (Rq) and the average (Ra) surface roughness of the modified electrode surface were increased from 26 ± 1.07 nm and 20.2 ± 2.04 nm in bare SPCEs to 38.3 ± 2.3 nm and 33.9 ± 3.5 nm in GO-modified SPCEs and 45.8 ± 3.05 nm and 35.8 ± 3.29 nm in GO–mAb SPCEs. The surface roughness was slightly decreased after blocking with BSA representing Rq of 43.8 ± 1.05 nm and Ra of 34.4 ± 2.96 nm (Fig. 2(D)). Finally, the immobilization of OV Ag indicated a smooth morphology due to the formation of the GO–mAb-OV Ag layer covering the electrode surface. The lowest Rq values of 19.7 ± 4.1 nm and Ra 16.3 ± 2.96 nm were observed for OV Ag/GO–mAb modified surfaces (Fig. 2(E)). AFM images of the sensor surfaces showed resemblance with those of FE-SEM images, indicating complete immobilization of the GO–mAb conjugate on SPCEs as well as the binding of OV Ag onto the GO–mAb modified sensor surface.
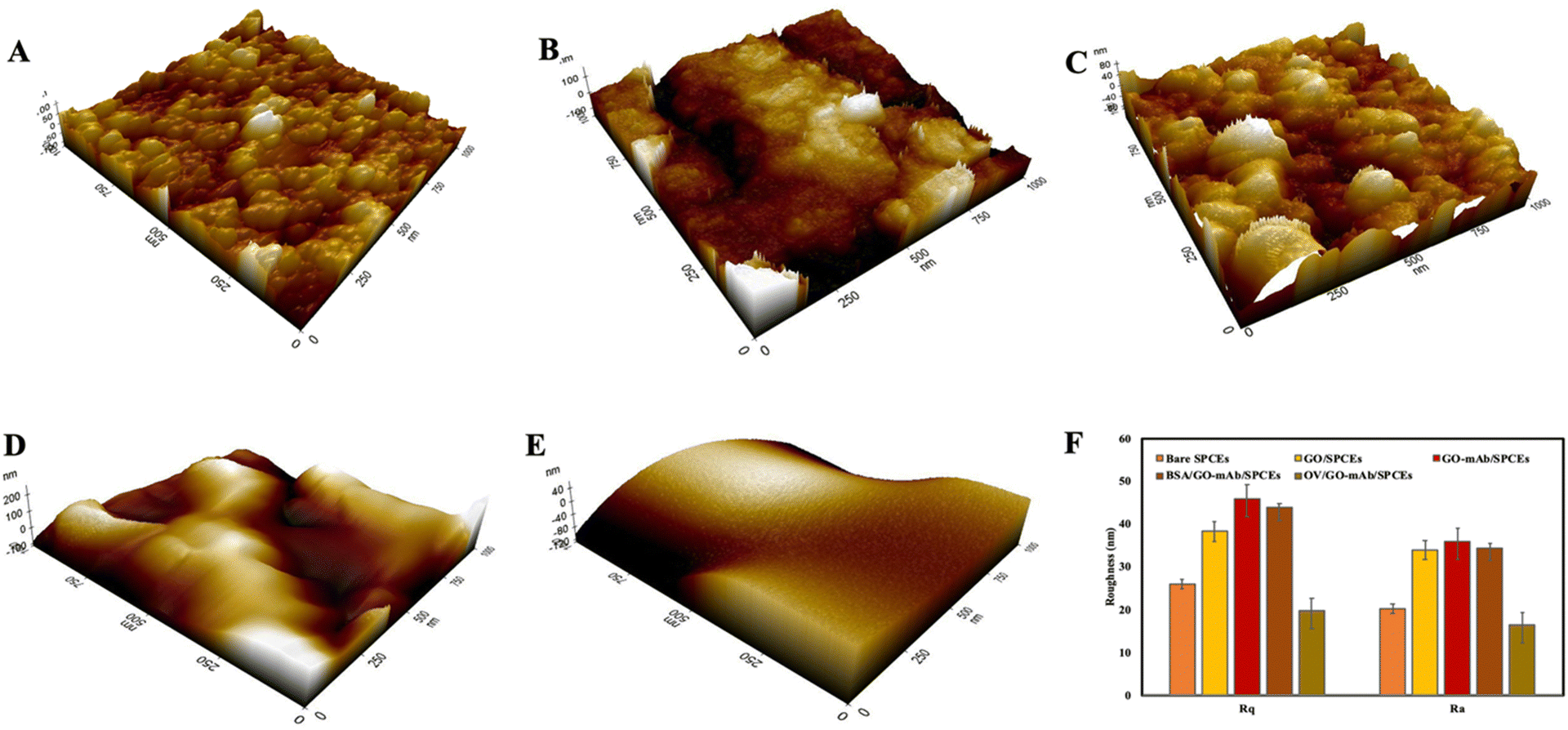 | ||
| Fig. 2 AFM images of (A) bare SPCEs, (B) GO-modified SPCEs, (C) GO–mAb-modified SPCEs, (D) BSA blocked SPCEs, (E) OV Ag immobilized GO–mAb/SPCEs and (F) root mean square (Rq) and average (Ra) surface roughness of each modification. | ||
Fourier-transformed infrared (FTIR) characterization was carried out for the functionalization of GO with mAb. Fig. 3(A) illustrates the FTIR spectra of the original GO and GO–mAb conjugates. The peak at 1098 cm−1 was attributed to the C–O stretching (alcohol) of GO.32 C–H bending (aromatic) functional groups were observed in the 1804 and 1800 cm−1 peak positions for original GO and GO–mAb, respectively. The peak at 1696 cm−1 represented the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching carboxyl group of the original GO33 which later disappeared upon GO–mAb conjugation as the carboxyl group was consumed by the amine group of the monoclonal antibody. The strong peak at 1572 cm−1 represented the amide I bonding while stretching.34 The band at 2665 cm−1 was attributed to the O–H stretching of GO and GO–mAb. C–C stretching and CH2 stretching groups of GO were observed at 2380 and 2945 cm−1, respectively. The C–H stretching alkene group and the C–N stretching amine group were attributed to the 654 cm−1 and 573 cm−1 wavenumbers of GO, but were later expressed as less intensive peaks in the GO–mAb complex. These factors contribute to the characteristics of GO, as reported in previous studies.35–38 The appearance of characteristic peaks of bending of the amide II band at 1528 cm−1 indicates successful immobilization of the antibodies onto GO. Moreover, the broader peak between 1600 and 1800 cm−1 is the spectral region most sensitive to antibody structural components.39 X-ray diffraction (XRD) patterns of original GO and EDC/NHS functionalized GO are illustrated in Fig. S1 (ESI†). XRD patterns showed the characteristics of pristine GO and carboxylated GO.
O stretching carboxyl group of the original GO33 which later disappeared upon GO–mAb conjugation as the carboxyl group was consumed by the amine group of the monoclonal antibody. The strong peak at 1572 cm−1 represented the amide I bonding while stretching.34 The band at 2665 cm−1 was attributed to the O–H stretching of GO and GO–mAb. C–C stretching and CH2 stretching groups of GO were observed at 2380 and 2945 cm−1, respectively. The C–H stretching alkene group and the C–N stretching amine group were attributed to the 654 cm−1 and 573 cm−1 wavenumbers of GO, but were later expressed as less intensive peaks in the GO–mAb complex. These factors contribute to the characteristics of GO, as reported in previous studies.35–38 The appearance of characteristic peaks of bending of the amide II band at 1528 cm−1 indicates successful immobilization of the antibodies onto GO. Moreover, the broader peak between 1600 and 1800 cm−1 is the spectral region most sensitive to antibody structural components.39 X-ray diffraction (XRD) patterns of original GO and EDC/NHS functionalized GO are illustrated in Fig. S1 (ESI†). XRD patterns showed the characteristics of pristine GO and carboxylated GO.
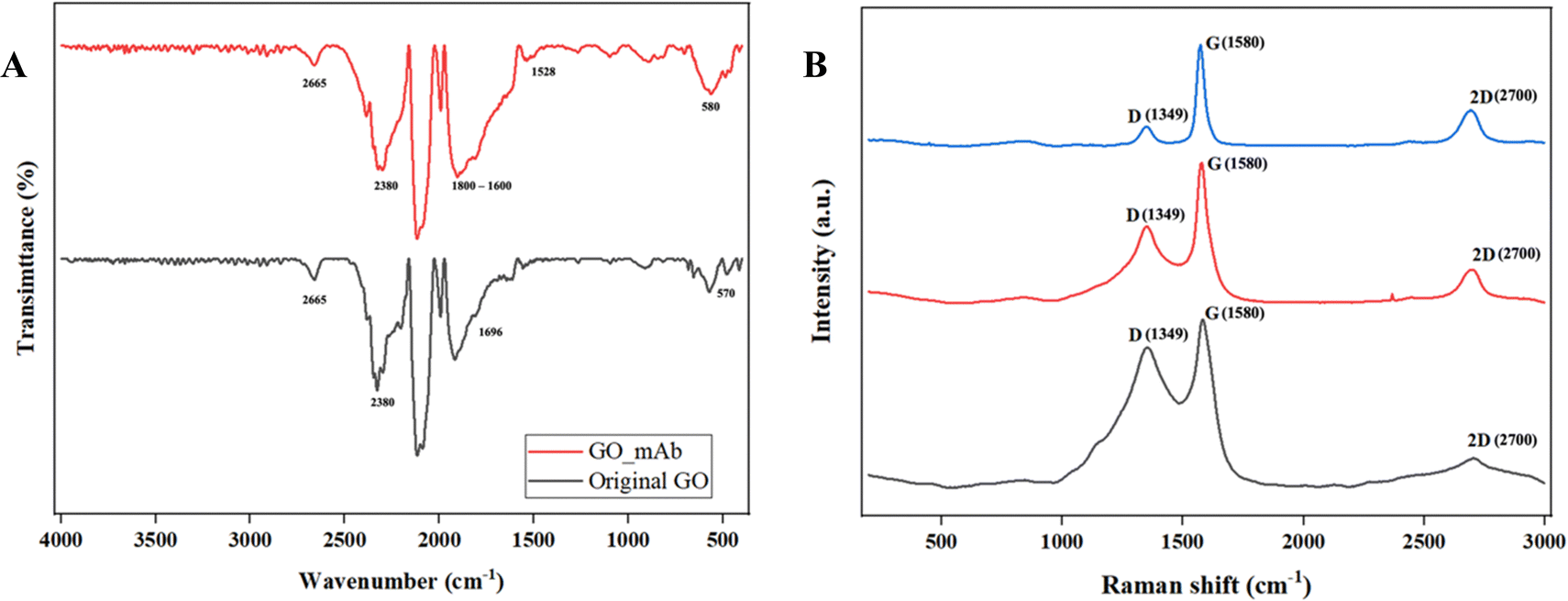 | ||
| Fig. 3 (A) FTIR original GO (black) and GO–mAb (red) and (B) Raman spectra of original GO (black), activated GO (red) and GO–mAb (blue). | ||
Raman spectra of bare SPCEs, GO/SPCEs and GO_mAb/SPCEs are illustrated in Fig. 3(B). Two significant peaks were observed at ∼1350 cm−1 and 1580 cm−1 that corresponded to the D and G bands of graphene. The additional 2D peak which is sensitive to the number of the graphene layer due to the second-order Raman scattering of the 2D band was observed around 2700 cm−1. The D band is associated with breaking the sp2 symmetry of carbon by the structural disorder and defects due to the presence of heteroatom and grain boundaries.40 Furthermore, the G band represents the stretching vibration mode (E2g) observed for sp2 carbon domains, relative to the first-order scattering.41 In bare SPCEs, the D and G bands were observed to have equal intensity though the pristine graphite usually exhibits a predominant sharp peak for the G band. The ID/IG ratio of bare SPCEs was 0.83 which is directly proportional to the disorder in its hexagonal lattice.42 The ratio of the integrated intensities of the D and G bands (ID/IG) is taken as an indicator of the relative disorder present in graphitic structures. The decrease in the intensities of the D band and ID/IG ratio was observed in GO/SPCEs and GO–mAb/SPCEs with the respective ratio of 0.55 and 0.19. The decrease in the ID/IG intensity was due to the aggregation of mAb on the GO layer and the increase in number of graphitic domains, which led to the second amorphization stage beyond the critical defect density.43 Furthermore, the increase in the intensity of the 2D-band indicates that the layer of the GO sheet has multiplied after the immobilization of mAb, which is accompanied by the decrease in the ID/IG intensity.44 The results of ATR-FTIR and Raman spectra validated the functionalization of GO with monoclonal antibody and immobilization on SPCE surfaces.
| Ip = 2.69 × 105A × D1/2n3/2v1/2C |
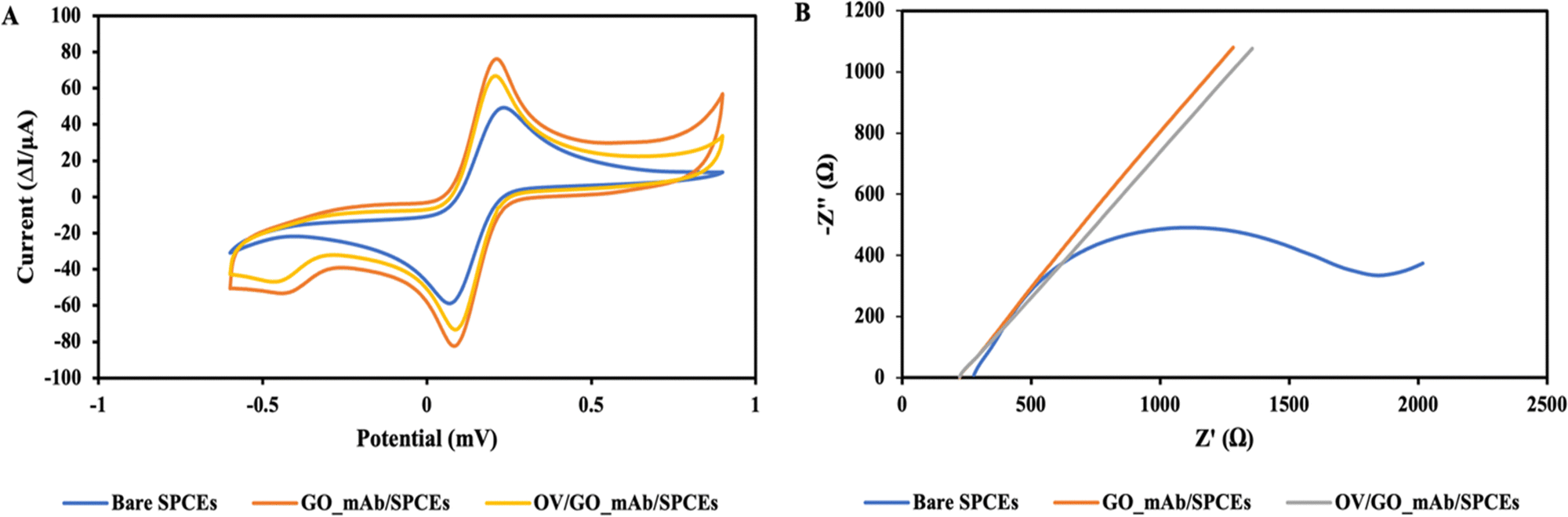 | ||
| Fig. 4 (A) Cyclic voltammograms of bare SPCEs, GO–mAb/SPCEs and OV/GO–mAb/SPCEs, showing a successful immobilization of each layer, (B) impedance spectra of the stepwise modification of the immunosensor using 5 mM [Fe (CN)6]3−/4− containing 0.1 M KCl in PBS as a redox indicator. | ||
Fig. 4(B) illustrates the Nyquist plots of each step and changes of charge transfer resistance (Rct) upon stepwise modification of the immunosensor surface. The impedance spectra of the GO–mAb modified surface showed the corresponding decrease in charge transfer resistance (Rct) due to the increase of electron-transfer capacity and enhanced electroactive sites on the electrode surface. Consequently, Rct was increased due to the steric hinderance onto the electrode surface after the specific interaction of OV Ag with immobilized mAb on the sensor surface. The EIS data supported the CV result showing the successful immobilization of the GO–mAb conjugate and the detection of OV Ag.
Fig. 4(B) illustrates the Nyquist plots in each step and changes in charge transfer resistance (Rct) upon the stepwise modification of the immunosensor surface. The impedance spectra of the GO_mAb modified surface also showed the corresponding decrease in charge transfer resistance (Rct) due to the increased electron-transfer capacity and enhanced electroactive sites on the electrode surface. Consequently, Rct was increased due to the steric hinderance onto the electrode surface after the specific interaction of OV antigen with immobilized mAb on the sensor surface. The EIS data supported the CV results, showing the successful immobilization of the GO_mAb conjugate and the detection of OV Ag.
3.2. Optimization of electrochemical immunosensors
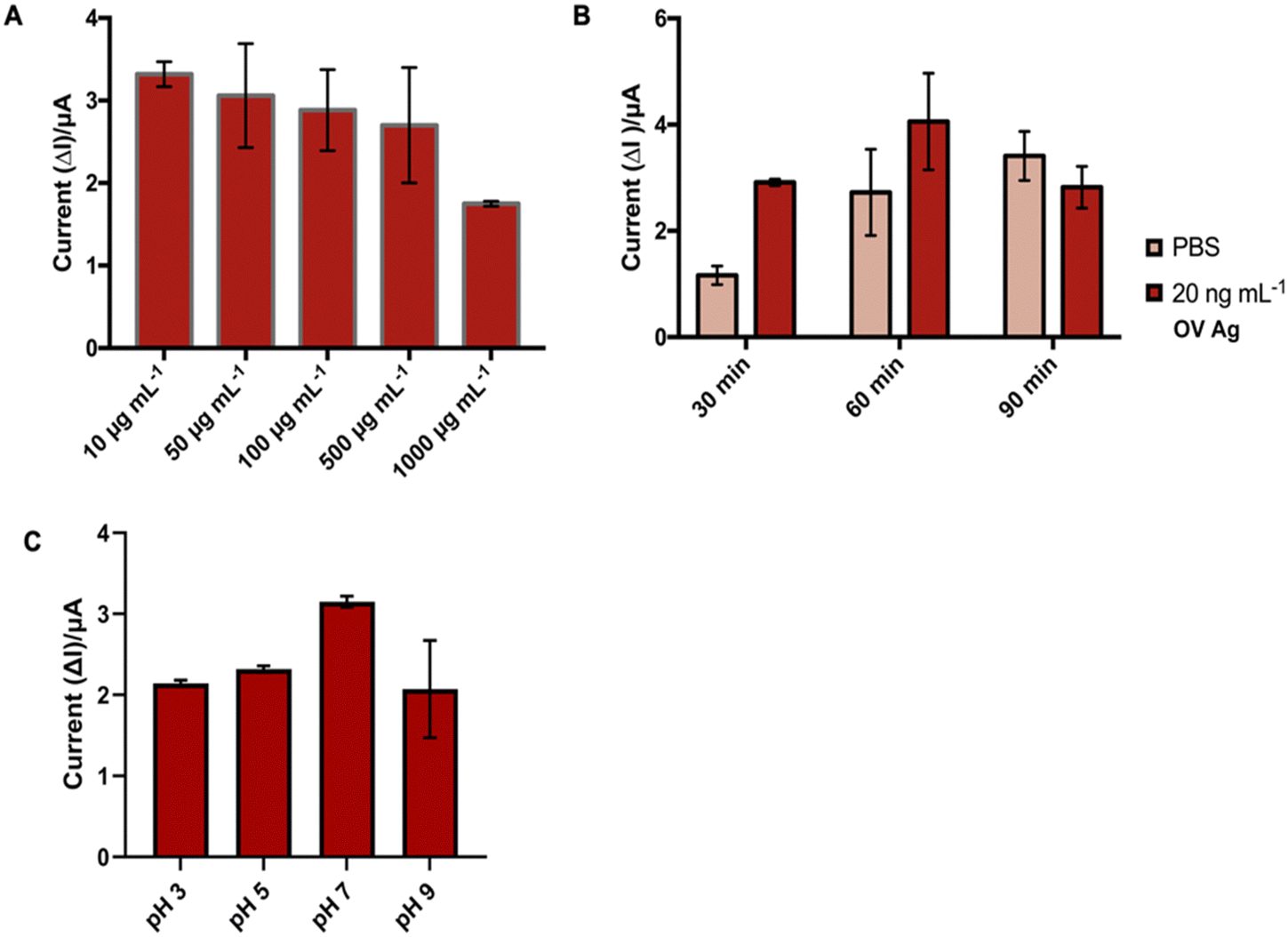 | ||
| Fig. 5 Optimization of (A) mAb concentration, (B) blocking time with 0.1% BSA, and (C) pH of the buffer. | ||
3.3. Analytical performance of immunosensors to detect OV Ag
Under the optimal experimental conditions, the applicability of the immunosensor developed to detect OV Ag was examined. The SWV voltammogram was recorded before incubating the sensor surface with target antigen and the peak current was extracted as the baseline (I0) signal. During the electrochemical measurement, the immobilized GO–mAb conjugate exhibited a free and stable assembly on the electrode surface, indicating a significant peak current of the redox solution. As the concentration of OV Ag bound to immobilized antibody increases, the antigen–antibody complex will act as a hindrance on the electrode surface and less redox species can penetrate it, leading to relative signal reduction (Fig. 6(A)). The peak current signal before and after target recognition was calculated as peak current changes (ΔI). As the concentration of OV Ag increased, there was a linear relationship between the peak current changes and logarithm of OV Ag concentration (I = 3.4179 log(c) − 5.844) with a correlation coefficient (R2) of 0.98 (Fig. 6(B)). The limit of detection (LOD) was calculated using the formula, LOD = 3σ/S, where σ is the standard deviation obtained from the average measurement of the blank and S is the slope of a linear graph. The calculated LOD of the developed immunosensor for OV Ag detection is 0.08 ng mL−1 with a linear response range of 0.2–200 μg mL−1. Table 1 shows the LOD and linear range of various other detection methods for OV Ag compared to our study. The proposed electrochemical immunosensor exhibited a relatively low LOD with a larger detection range and higher sensitivity.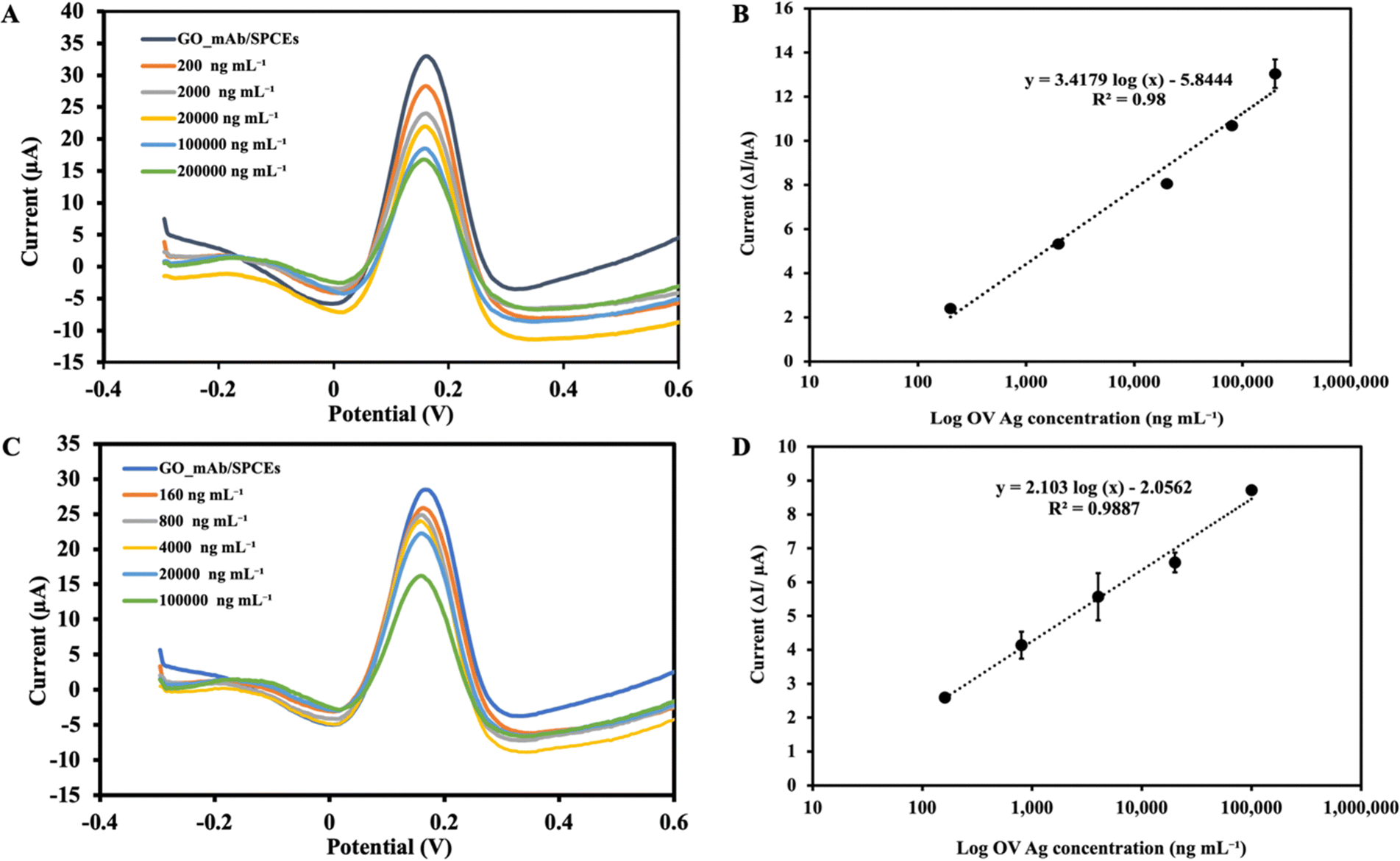 | ||
| Fig. 6 (A) SWV responses of the GO–mAb modified sensor (baseline) and different concentrations of OV in PBS, (B) linear relationship between log OV Ag concentration (0.2–200 μg mL−1) in PBS and peak current changes (ΔI). (C) SWV responses of the GO–mAb modified sensor (baseline) and different concentrations of OV Ag in urine, (D) standard curve of the immunosensor showing the linear relationship between the logarithm of OV Ag concentration (0.16–100 μg mL−1) spiked in urine and the corresponding maximum current (ΔI) from SWV. | ||
| Method | Limit of detection | Linearity | Reference |
|---|---|---|---|
| OV excretory-secretory (OV-ES) ELISA | 39 ng mL−1 | — | 11 |
| AuNPs-LISA assay | 23.9 ng mL−1 | 5.9–12000 ng mL−1 |
12 |
| Enzyme-linked immunosorbent assay | 93.8 ng mL−1 | — | 46 |
| Smartphone-based fluorescent ELISA | 34 ng mL−1 | 1.95–125 ng mL−1 | 47 |
| OV-RDT | 28.5 ng mL−1 | 28.5–5000 ng mL−1 | 48 |
| Electrochemical immunosensor | 0.08 ng mL−1 | 0.2–200 μg mL−1 | This study |
| 1.5 ng mL−1 | 0.16–100 μg mL−1 (Spiked urine) |
3.4. Analytical performance of the immunosensor in real clinical samples
To equilibrate the interference of other substances in urine on OV Ag detection, a standard curve was calibrated using different concentrations of OV Ag spiked into urine samples. A normal urine sample was diluted (1:10 dilution) prior to the experiment. The SWV peak current was observed to decrease with increasing concentration of OV Ag in urine (Fig. 6(C)). A calibration plot was constructed as described in the previous section. SWV with increasing concentration of OV Ag showed that the peak current changes were increased linearly (I = 2.10 log(c) − 2.0562) with the R2 of 0.988 (see Fig. 6(D)). Each point of the calibration graph represents the mean value obtained from three independent measurements. LOD calculated from the linear graph was 1.5 ng mL−1 which was higher than that obtained from the linear graph using the standard antigen spiked in PBS. It also showed a narrower linear detection range of 0.16–100 μg mL−1. The application of the developed immunosensor to the real clinical samples supported its significance for the clinical diagnosis of OV infection. Serological methods suffer from drawbacks such as invasive sample collection, necessity of cold chains, and persistence of antibodies for prolonged periods after treatment. Therefore, the use of non-invasive urine samples for OV Ag detection facilitated the applicability of the immunosensor.
3.5. Reproducibility and stability
The reproducibility of the proposed immunosensor was evaluated by the analysis of three different concentrations of OV Ag (0.02, 2, 20 μg mL−1) across ten independent studies performed by a different operator. The results are given in Table 2. The relative standard deviations (RSDs) obtained were 5.78%, 5.06%, and 4.4%, respectively, suggesting that the proposed immunosensor has acceptable reproducibility which was less than 10%.49 Therefore, the data provided that the immunosensor can generate highly reproducible signals along with high consistency of electrode surface modification and precision. The stability of the immunosensor was investigated by storing the developed immunosensor at 4 °C for various intervals up to 30 days and used for the detection of 500 ng mL−1 of OV Ag. There was no obvious change during the storage of 30 days, retaining about 99% of the initial current response (Fig. 7). The proposed immunosensor exhibited great stability for potential utility.| OV Ag concentration (μg mL−1) | Peak current (μA) | RSD (%) (n = 10) |
|---|---|---|
| 0.02 | 2.08 ± 0.12 | 5.78% |
| 2 | 5.78 ± 0.29 | 5.06% |
| 20 | 10.69 ± 0.47 | 4.40% |
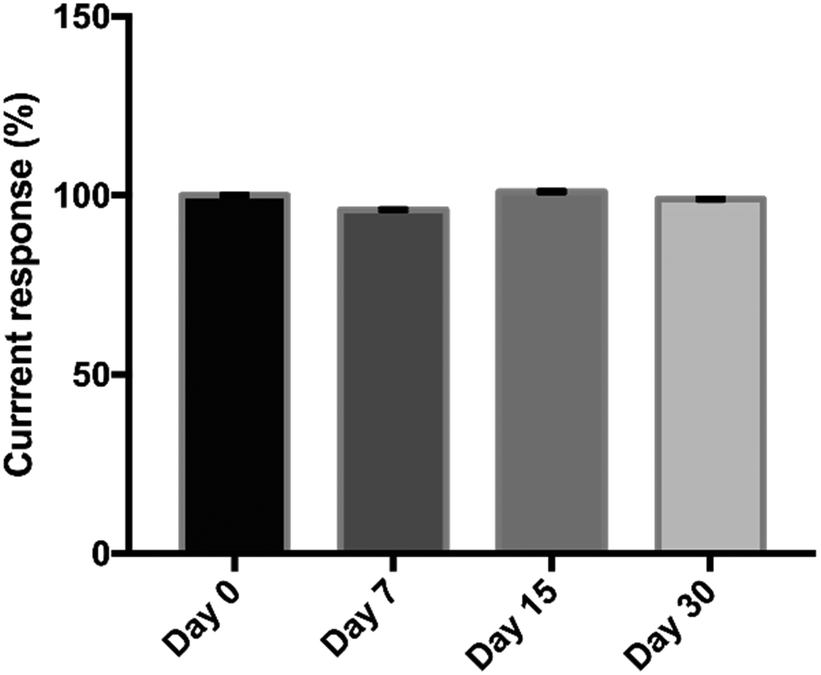 | ||
| Fig. 7 Stability of the immunosensor after its storage at 4 °C for 0, 7, 15 and 30 days for the detection of 500 ng mL−1 of OV Ag. | ||
3.6. Evaluation of field utility of the developed electrochemical immunosensor
As shown in Table S1 (ESI†), the collected 530 samples were separated into ELISA tested, OV-RDT tested and ELISA & OV-RDT tested groups. The concentrations of OV Ag detected by the immunosensor in OV infected urine samples were significantly higher than those in normal urine samples at a 95% confident interval with a p-value of <0.001 (Fig. 8(A)–(C)). The mean concentrations of OV Ag in OV-positive and OV-negative urine in each sample group were 211.41 ± 30.77 ng mL−1 and 26.27 ± 5.72 ng mL−1 (Fig. 8(A)), 220.34 ± 62.63 ng mL−1 and 30.50 ± 14.52 ng mL−1 (Fig. 8(B)), and 202.29 ± 31.68 ng mL−1 and 15.33 ± 3.58 ng mL−1 (Fig. 8(C)), respectively. These data indicated that the proposed immunosensor can be used as a promising tool to detect OV infection.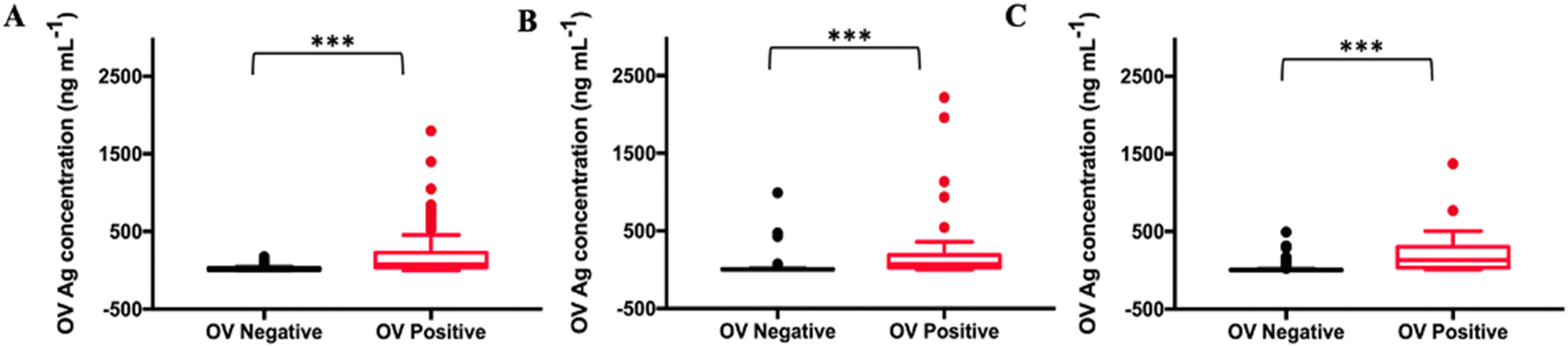 | ||
| Fig. 8 Statistical comparison of OV antigen detection in OV positive and OV negative individuals in (A) Group 1 by ELISA (n = 148), (B) Group 2 by OV-RDT (n = 130) and (C) Group 3 by ELISA & OV-RDT (n = 253). The plot shows a statistically significant difference in OV Ag concentration in urine between OV positive and negative individuals (p <0.001). | ||
Fig. 9(A)–(C) show the ROC curve analysis to compare the performance of the developed method to the conventional ELISA test, OV-RDT test kit and ELISA and OV-RDT tests. The electrochemical detection technique showed high AUC values of 0.846, 0.923 and 0.950 respectively, indicating that using the developed sensor is a superior method to classify OV positive and OV negative individuals correctly. Using the diagnostic cut-off (12.06 ng mL−1) obtained from the ROC curve, the diagnostic sensitivity and specificity were 93% and 69%, respectively (Fig. 9(A)). To further evaluate the assay performance, positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (LR+) and negative likelihood ratio (LR−) were estimated (Table 3). The diagnostic performance of the assay was verified by the PPV of 86%, the NPV of 82%, the LR+ of 3.0, the LR− of 0.10, the DP of 67.6 and the accuracy of 85%. False positive results from sensor detection were correlated with proteinuria (p < 0.05) probably because the high concentration of protein in urine interfered with biosensing. Compared to the ELISA, the developed immunosensor exhibited better sensitivity and specificity to detect OV infection with the advantage of not requiring urine pre-treatment.
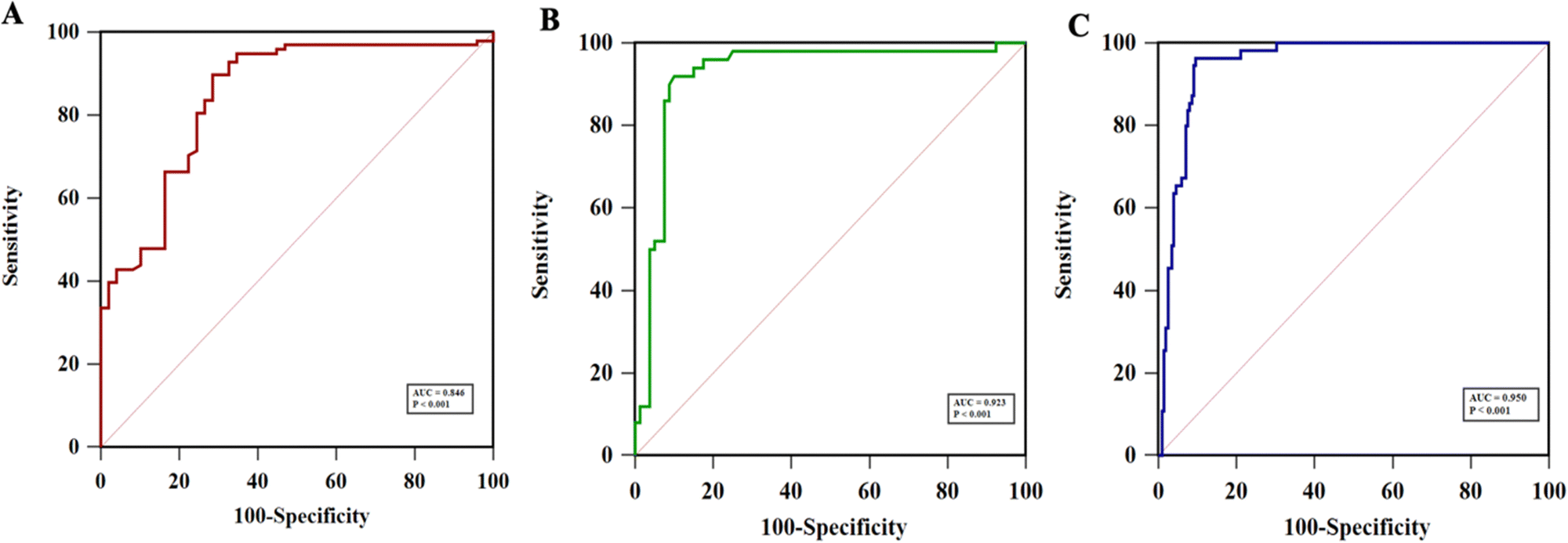 | ||
| Fig. 9 Receiver operating characteristics (ROC) analyses of the developed immunosensor for the detection of OV antigen in urine samples compared to (A) ELISA (AUC = 0.846), (B) OV-RDT (AUC = 0.923), and (C) ELISA and OV-RDT techniques (AUC = 0.950). | ||
| Comparator | Diagnostic performance of electrochemical immunosensors | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC | Cutoff | Sensitivity | Specificity | PPV | NPV | LR+ | LR− | Disease prevalence | |
| ELISA (n = 147) | 0.865 | 10.85 | 0.93 | 0.69 | 0.86 | 0.82 | 3.0 | 0.10 | 67.6 |
| OV-RDT (n = 130) | 0.923 | 11.94 | 0.92 | 0.90 | 0.85 | 0.96 | 9.2 | 0.08 | 38.5 |
| ELISA & OV-RDT (n = 253) | 0.950 | 12.06 | 0.96 | 0.90 | 0.75 | 0.99 | 9.6 | 0.04 | 21.7 |
The diagnostic performance of the developed electrochemical immunosensor was further validated through comparison with the commercialized rapid OV Ag test kit (OV-RDT). Urine OV Ag RDT has been used for the on-site detection of OV Ag. The ROC curve analysis indicated a high AUC of 0.923 (Fig. 9(B)), providing excellent sensitivity and specificity to OV Ag detection. The diagnostic performance of the proposed electrochemical immunosensor compared to OV-RDT presented 92% sensitivity, 90% specificity, 85% PPV, 96% NPV, 9.2 LR+, 0.08 LR−, 38.5% DP and 92% accuracy (see Table 3). Further comparison with the ELISA and OV-RDT exhibited the highest AUC of 0.950 with 96% sensitivity and 90% specificity (Fig. 9(C)). The diagnostic performance of the proposed electrochemical immunosensor compared to ELISA and OV-RDT presented 75% PPV, 99% NPV, 9.6 LR+, 0.04 LR−, 21.7% DP and 92% accuracy. The developed electrochemical immunosensor showed the highest accuracy when compared to RDT or a combination of ELISA and RDT. The prevalence of OV infection detected by the developed immunosensor (45%) was higher than that of ELISA (38%). The diagnostic cut-off to diagnose OV-positive individuals by the immunosensor was 12.06 ng mL−1 and that by the ELISA was 32 ng mL−1. Therefore, the lower cut-off of LOD for OV Ag by the immunosensor may lead to higher sensitivity and prevalence of OV infection. However, the reference range for OV Ag in the urine sample is yet to be identified which may evaluate the relevance of the proposed diagnostic method compared to the traditional techniques. Besides, correlation analysis between the concentration of OV Ag in urine detected by the immunosensor and the intensity of OV eggs by the gold standard FECT techniques should be conducted in the future to verify the clinical utility and diagnostic performance of the immunosensor.
The gold standard diagnosis FECT is sometimes inaccurate in the case of the presence of bile duct obstruction due to periductal fibrosis or in the case of the low intensity infection where the eggs cannot be detected in faeces. Even in those cases, the metabolic products can be secreted from adult worms and OV Ag can readily be detected in urine. Moreover, urine OV Ag has been reported to be significantly correlated with the intensity of OV infection.11 Therefore, the quantitative measurements of urine OV Ag by the proposed immunosensor can estimate the intensity of the infection as well as the progression after the treatment. The developed electrochemical immunosensor can detect urine OV Ag with high accuracy via a specific monoclonal antibody and antigen reaction coupled by the electrochemical transducing activity of graphene. Therefore, the electrochemical immunosensor is highly promising to detect OV Ag in urine samples for the diagnosis of opisthorchiasis in mass screening in the field.
3.7. Comparison of the diagnostic methods
Urine OV Ag concentrations detected by the developed immunosensor were compared with those detected by the ELISA as the reference. In this study, the comparison with a gold standard FECT was omitted due to the impracticability of collecting intensive fecal samples, particularly in field-collected samples. At the 95% confident interval, the calculated values of the paired t-test and F-test implied that there was no noticeable difference between the performances of the two methods compared. The Bland–Altman plot exhibited the concordance between the sensor and the ELISA measurements (Fig. S2, ESI†). The results obtained with both methods showed an overall agreement with some outliers. Moreover, the weighted Cohens kappa analysis showed moderate agreement between the immunosensor and the ELISA with the Kappa value of 0.72 and strong agreement when compared to OV-RDT with a Kappa value of 0.80. The discrepancy in the OV Ag concentration was particularly evident at the lower OV Ag concentration. The lowest OV Ag concentration detected by the immunosensor was 0.002 ng mL−1, whereas that by the ELISA was 16.02 ng mL−1, resulting in deviation from linear results. The frequency distribution of OV Ag concentration measured by the immunosensor indicated a wider detection range compared to the ELISA (Fig. S3A and B, ESI†). Moreover, sample collection, storage and detection time affected urine OV Ag concentration because of the short lifespan of OV Ag in urine and bacterial multiplication in the urine sample over prolonged storage. Overall, the validation of the newly developed immunosensor method using the conventional ELISA as reference has highlighted the reliability of the immunosensor.4. Conclusion
In this study, the newly developed electrochemical immunosensor introduces a novel concept for the detection of OV Ag in urine samples. The functionalization of GO with monoclonal antibody provides antibody-mediated specific binding sites for antigen detection and enhanced sensitivity of the immunosensor. The quantitative measurements of OV Ag in infected patients can play a crucial role in monitoring the diagnosis and prognosis of the disease. The developed immunosensor exhibited low LOD (1.5 ng mL−1) for OV Ag in urine samples, which is the lowest reported LOD for OV Ag detection to date. Furthermore, the proposed immunosensor exhibited excellent diagnostic potential with 96% sensitivity and 90% specificity without requiring a sample pre-treatment step. The analytical validation and clinical performance of the immunosensor outperformed traditional measurement techniques. The developed electrochemical immunosensor is a promising tool for point-of-care detection of OV Ag in field application as well as for monitoring disease progression with high accuracy.Institutional review board statement
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Centre for Ethics in Human Research, Khon Kaen University (HE664025).Author contributions
Conceptualization, Nang Noon Shean Aye, Pornsuda Maraming, and Jureerut Daduang; formal analysis, Nang Noon Shean Aye; investigation, Nang Noon Shean Aye; methodology, Nang Noon Shean Aye; supervision, Jureerut Daduang; writing – original draft, Nang Noon Shean Aye; writing – review & editing, Nang Noon Shean Aye, Pornsuda Maraming, Patcharaporn Tippayawat, Sakda Daduang, Anchalee Techasen. Resources – Wassana Jamnongkan, Paiboon Sithithaworn.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research was supported by the NSRF under the Basic Research Fund of Khon Kaen University through the Cholangiocarcinoma Research Institute: CARI-BRF64-47. The authors would like to acknowledge the Khon Kaen University ASEAN-GMS scholarship and the grant support from the NSRF under the Basic Research Fund of Khon Kaen University through the Cholangiocarcinoma Research Institute, Thailand (CARI-BRF64-47). Furthermore, the authors would like to acknowledge Professor Yukifumi Nawa and KKU publication clinic for editing the manuscript and providing insightful comments.References
- P. J. Brindley, M. Bachini, S. I. Ilyas, S. A. Khan, A. Loukas, A. E. Sirica, B. T. Teh, S. Wongkham and G. J. Gores, Nat. Rev. Dis. Primer, 2021, 7, 1–17 CrossRef PubMed.
- R. H. Andrews, P. Sithithaworn and T. N. Petney, Trends Parasitol., 2008, 24, 497–501 CrossRef PubMed.
- P. Sithithaworn, P. Yongvanit, K. Duenngai, N. Kiatsopit and C. Pairojkul, J. Hepato-Biliary-Pancreat. Sci., 2014, 21, 301–308 CrossRef PubMed.
- N. Khuntikeo, N. Chamadol, P. Yongvanit, W. Loilome, N. Namwat, P. Sithithaworn, R. H. Andrews, T. N. Petney, S. Promthet, K. Thinkhamrop, C. Tawarungruang, B. Thinkhamrop and on behalf of the CASCAP investigator, s, BMC Cancer, 2015, 15, 459 CrossRef.
- B. Sripa, J. M. Bethony, P. Sithithaworn, S. Kaewkes, E. Mairiang, A. Loukas, J. Mulvenna, T. Laha, P. J. Hotez and P. J. Brindley, Acta Trop., 2011, 120(Suppl 1), S158–S168 CrossRef PubMed.
- D. M. Parkin, Int. J. Cancer, 2006, 118, 3030–3044 CrossRef CAS PubMed.
- Z.-R. Lun, R. B. Gasser, D.-H. Lai, A.-X. Li, X.-Q. Zhu, X.-B. Yu and Y.-Y. Fang, Lancet Infect. Dis., 2005, 5, 31–41 CrossRef PubMed.
- M.-B. Qian, P. Yap, Y.-C. Yang, H. Liang, Z.-H. Jiang, W. Li, J. Utzinger, X.-N. Zhou and J. Keiser, Parasit. Vectors, 2013, 6, 314 CrossRef PubMed.
- M. V. Johansen, P. Sithithaworn, R. Bergquist and J. Utzinger, Adv. Parasitol., 2010, 73, 171–195 Search PubMed.
- X.-Q. Cai, H.-Q. Yu, R. Li, Q.-Y. Yue, G.-H. Liu, J.-S. Bai, Y. Deng, D.-Y. Qiu and X.-Q. Zhu, Sci. World J., 2014, 2014, 893981 Search PubMed.
- C. Worasith, C. Kamamia, A. Yakovleva, K. Duenngai, C. Wangboon, J. Sithithaworn, N. Watwiengkam, N. Namwat, A. Techasen, W. Loilome, P. Yongvanit, A. Loukas, P. Sithithaworn and J. M. Bethony, PLoS Negl. Trop. Dis., 2015, 9, e0004157 CrossRef.
- W. Taron, W. Jamnongkan, A. Techasen, J. Phetcharaburanin, N. Namwat, P. Sithithaworn, N. Khuntikeo, S. Mukdasai, S. Sayasone, W. Loilome and W. Ngeontae, Talanta, 2020, 209, 120592 CrossRef CAS.
- Y. Arimatsu, S. Kaewkes, T. Laha, S.-J. Hong and B. Sripa, Parasitol. Int., 2012, 61, 178–182 CrossRef CAS.
- L. Sadaow, R. Rodpai, P. Janwan, P. Boonroumkaew, O. Sanpool, T. Thanchomnang, H. Yamasaki, W. Ittiprasert, V. H. Mann, P. J. Brindley, W. Maleewong and P. M. Intapan, Trop. Med. Infect. Dis., 2022, 7, 308 CrossRef.
- S. Wongratanacheewin, R. W. Sermswan and S. Sirisinha, Acta Trop., 2003, 88, 195–207 CrossRef CAS.
- M. R. Haswell-Elkins, P. Sithithaworn, E. Mairiang, D. B. Elkins, S. Wongratanacheewin, S. Kaewkes and P. Mairiang, Clin. Exp. Immunol., 1991, 84, 213–218 CrossRef CAS.
- P. S. Akai, S. Pungpak, W. Chaicumpa, V. Kitikoon, Y. Ruangkunaporn, D. Bunnag and A. D. Befus, Int. J. Parasitol., 1995, 25, 971–973 CrossRef CAS.
- S. Tanvanich, G. Doungchawee, S. Boonpucknavig and W. Thamavit, Clin. Exp. Immunol., 1988, 74, 355–358 CAS.
- S. Wongratanacheewin, D. Bunnag, N. Vaeusorn and S. Sirisinha, Am. J. Trop. Med. Hyg., 1988, 38, 356–362 CrossRef CAS PubMed.
- S. Wongratanacheewin, R. Chawengkirttikul, D. Bunnag and S. Sirisinha, Parasitology, 1988, 96, 119–128 CrossRef CAS.
- S. Wongratanacheewin and S. Sirisinha, Southeast Asian J. Trop. Med. Public Health, 1987, 18, 511–520 CAS.
- C. Worasith, C. Wangboon, K. Duenngai, N. Kiatsopit, K. Kopolrat, A. Techasen, J. Sithithaworn, N. Khuntikeo, W. Loilome, N. Namwat, P. Yongvanit, E. J. Carlton and P. Sithithaworn, PLoS Negl. Trop. Dis., 2019, 13, e0007186 CrossRef CAS.
- R. A. D. Faria, Y. Messaddeq and L. G. D. Heneine, et al. , Int. J. Biosens. Bioelectron., 2019, 5(1), 1–2 Search PubMed.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef PubMed.
- Q. Zheng, Z. Li, J. Yang and J.-K. Kim, Prog. Mater. Sci., 2014, 64, 200–247 CrossRef CAS.
- M. McKeague and M. C. DeRosa, J. Nucleic Acids, 2012, 2012, e748913 CAS.
- A. Huang, L. Zhang, W. Li, Z. Ma, S. Shuo and T. Yao, R. Soc. Open Sci., 2018, 5, 171808 CrossRef PubMed.
- A. Huang, W. Li, S. Shi and T. Yao, Sci. Rep., 2017, 7, 40772 CrossRef CAS.
- N. N. S. Aye, P. Maraming, R. Tavichakorntrakool, A. Chaibunruang, P. Boonsiri, S. Daduang, N. Teawtrakul, P. Prasongdee, V. Amornkitbamrung and J. Daduang, Appl. Sci., 2021, 11, 10315 CrossRef CAS.
- C. Thunkhamrak, P. Chuntib, K. Ounnunkad, P. Banet, P.-H. Aubert, G. Saianand, A.-I. Gopalan and J. Jakmunee, Talanta, 2020, 208, 120389 CrossRef CAS PubMed.
- Z. Çiplak, N. Yildiz and A. Çalimli, Fullerenes, Nanotubes Carbon Nanostruct., 2015, 23, 361–370 CrossRef.
- Sudesh, N. Kumar, S. Das, C. Bernhard and G. D. Varma, Supercond. Sci. Technol., 2013, 26, 095008 CrossRef CAS.
- C. Sign and G. Sumana, J. Phys. Conf. Ser., 2016, 704, 012014 CrossRef.
- C. Shan, H. Yang, J. Song, D. Han, A. Ivaska and L. Niu, Anal. Chem., 2009, 81, 2378–2382 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- C. Shan, H. Yang, D. Han, Q. Zhang, A. Ivaska and L. Niu, Langmuir, 2009, 25, 12030–12033 CrossRef CAS PubMed.
- M. Tertiş, O. Hosu, L. Fritea, C. Farcau, A. Cernat, R. Săndulescu and C. Cristea, Electroanalysis, 2015, 27, 638–647 CrossRef.
- H. C. Lai, S. F. Chin, S. C. Pang, M. S. Henry Sum and D. Perera, J. Nanomater., 2017, 2017, e3615707 Search PubMed.
- K. Ghosh, M. Kumar, T. Maruyama and Y. Ando, Carbon, 2010, 48, 191–200 CrossRef CAS.
- P. Han, Y. Yue, L. Zhang, H. Xu, Z. Liu, K. Zhang, C. Zhang, S. Dong, W. Ma and G. Cui, Carbon, 2012, 50, 1355–1362 CrossRef CAS.
- N. Karikalan, R. Karthik, S.-M. Chen, M. Velmurugan and C. Karuppiah, J. Colloid Interface Sci., 2016, 483, 109–117 CrossRef CAS.
- S. C. Jun, Graphene-based Energy Devices, John Wiley & Sons, Ltd, 2015, pp.1–48 Search PubMed.
- Z. Luo, T. Yu, K. Kim, Z. Ni, Y. You, S. Lim, Z. Shen, S. Wang and J. Lin, ACS Nano, 2009, 3, 1781–1788 CrossRef CAS.
- J.-Z. Tsai, C.-J. Chen, K. Settu, Y.-F. Lin, C.-L. Chen and J.-T. Liu, Biosens. Bioelectron., 2016, 77, 1175–1182 CrossRef CAS.
- S. Sirisinha, R. Chawengkirttikul, M. R. Haswell-Elkins, D. B. Elkins, S. Kaewkes and P. Sithithaworn, Am. J. Trop. Med. Hyg., 1995, 52, 521–524 CrossRef CAS PubMed.
- W. Taron, K. Phooplub, S. Sanchimplee, K. Piyanamvanich, W. Jamnongkan, A. Techasen, J. Phetcharaburanin, P. Klanrit, N. Namwat, N. Khuntikeo, T. Boonmars, P. Sithithaworn, S. Ouiganon, P. Kanatharana, P. Thavarungkul, C. Buranachai, W. Loilome and W. Ngeontae, Sens. Actuators, B, 2021, 348, 130705 CrossRef CAS.
- C. Worasith, J. Sithithaworn, P. Wongphutorn, C. Homwong, K. Khongsukwiwat, A. Techasen, K. Y. Kopolrat, W. Loilome, N. Namwat, B. Thinkamrop, C. Tawarungruang, A. Titapun, T. Laha, R. H. Andrews, S. D. Taylor-Robinson and P. Sithithaworn, Infect. Dis. Poverty, 2023, 12, 102 CrossRef PubMed.
- H. T. Karnes and C. March, Pharm. Res., 1993, 10, 1420–1426 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: XRD analysis of GO and GO-EDC, Fig. S2: Bland–Altman plot for concordance analysis between the immunosensor and ELISA, Fig. S3: frequency distribution histogram. See DOI: https://doi.org/10.1039/d3ma01075a |
| This journal is © The Royal Society of Chemistry 2024 |