
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trimetallic MOF1@MOF2 heterostructure derived Co–NC@CuM–C for enhanced photothermal catalysis in styrene epoxidation†
Xinliu
Zhao
,
Fusheng
Liu
,
Yu-Zhen
Chen
 * and
Zhibo
Li
*
* and
Zhibo
Li
*
College of Chemical Engineering, Qingdao University of Science and Technology, Qingdao 266042, Shandong, P. R. China
First published on 16th January 2024
Abstract
Constructing hierarchical MOF1-on-MOF2 composites using different binary MOFs is a promising strategy to develop multifunctional MOF precursors and their derivatives. However, the orientated growth of the heteroid MOF shell requires overcoming the mismatched cell lattices. Therefore, the desired assembly of MOF1@MOF2 with regular morphologies and sizes remains a significant challenge. Herein, a novel tri-metallic BMZIF@HKUST-1 with good dodecahedral morphology was successfully fabricated and pyrolyzed to obtain porous Co–NC@Cu–C, in which Cu and Co species were highly dispersed in the carbon skeleton and no agminated metal particles were observed. Interestingly, the carbon materials exert a good photothermal effect and can raise the system temperature from 25 to ∼50 °C under visible light irradiation. In addition, a metal ion redox exchange strategy was adopted to avoid runoff of unstable Cu species in Co–NC@Cu–C during catalysis under harsh conditions. The optimal Co–NC@CuOPd–C achieved excellent activity (99%) and high selectivity (92.9%) in styrene epoxidation by photothermal catalysis benefitting from its multiple active sites and highest stability. The novel synthetic strategy can be further extended to more MOF types and provides plentiful metal sites for producing various fine chemicals.
1 Introduction
The catalytic epoxidation of styrene to produce styrene oxide (SO) has attracted considerable attention because SO is an essential raw material for industrial synthesis of spices, pesticides and pharmaceuticals.1–5 Of these, spice intermediates account for 90.78% of the total SO volume utilized. The global market size for styrene oxide was $70.0 million in 2021, with estimates surpassing $109 million in 2028.6 Therefore, it is urgent to produce industrially SO with good yield and high purity. Three crucial factors, namely, catalyst, oxidant, and reaction temperature, significantly influence the styrene epoxidation efficiency. Generally, noble-metal-based catalysts like Ag, Pt, and Au show superb catalytic activity toward styrene epoxidation;7–9 however, SO selectivity above 90% remains a great challenge.10,11 Traditional oxidants such as organic peracids, KMnO4 and chlorohydrin are usually added to help achieve good activity.12–14 But the large amount of chemical waste produced has caused a serious pollution.15,16 From economical, green, and long-life vantage points, molecular oxygen as an oxidant has been the optimal choice. For example, Fe2O3@mSiO2-SH–Au gave a 65% conversion with 58% SO selectivity using O2 as the oxidant.17 Au/TiO2-EC afforded a 56% conversion, yet only 17.5% SO selectivity.18 Compared to precious metal catalysts, low-cost cobalt and copper species are also active toward styrene epoxidation.19–21 Especially, copper species (Cu, CuxO) can activate molecular O2 and enhance effectively the SO selectivity. Unfortunately, unstable copper species are readily oxidized and then leaching into some solvents. For example, as the most commonly used solvent for styrene epoxidation, acetonitrile readily coordinates with copper. Therefore, designing a method to stabilize Cu species in Cu-based catalysts would be more significant for good cost-efficiency and high SO selectivity.Porous materials have been commonly utilized as supports to stabilize small metal NPs. Typically, metal–organic frameworks (MOFs) are well-known crystalline porous materials, constructed via organic ligands with metal ions (or clusters).22–29 MOFs have been applied extensively in various fields such as heterogeneous catalysis,30–32 drug delivery and imaging,33,34 sensors,35,36 and gas adsorption/separation.37–40 More recently, MOFs served as excellent precursors or templates for the fabrication of various carbon-based materials and porous metals or metallic oxides.41–48 These hybrid materials provided more possibilities in electrochemical reactions and heterogeneous catalysis.49,50 Nonetheless, monometallic MOFs are subject to the limited metals in the periodic table, which hampers their utility, such as the Cu-MOF (HKUST-1) and Co-MOF (ZIF-67).51 To enrich the metal sites, isostructural MOF-on-MOF materials (with the same ligand) have been developed,52–58 and they include ZIF-67@ZIF-8, Fe-MIL-88B@Fe-MIL-88C, and UiO-67@UiO-66. In particular, the conjugation of two or more isomerous MOFs with different metals and ligands yields greater structural flexibility and more active species. For instance, HKUST-1-on-ZIF can provide both Cu and Co active sites, but such assembly is very challenging due to their unmatched cell lattices. Currently, similar assembly architectures like core–shell, hollow multi-shell and film-on-film MOFs (e.g., UiO-66@ZIF-8, MIL-88B@ZIF-8, PCN-222@NU-1000, MIL-125@ZIF-67) and ternary MOF-on-MOF hybrids (e.g., MIL-125@ZIF-67@ZIF-8) have been designed for various applications since 2009.59–63 Anisotropic growth often results in abnormal morphology or poor assembly of the final composite (like sandwich-type hybrid MOFs and In-MIL-88B/Fe-MIL-88B/In-MIL-88B).64 Research findings indicate that the morphology of MOFs and MOF derived materials may impact directly the intrinsic properties and their catalytic performance.65 Furthermore, the well-defined crystalline structures of core–shell MOFs could help the establishment of the structure–activity relationship. To date, directional growth of the guest HKUST-1 on the host ZIF with well-matched cell lattices and regular morphology has not been studied.
Herein, two different MOFs (BMZIF (ZnCo-ZIF) and HKUST-1 (Cu-MOF)) with distinct crystal structures and morphologies were directionally assembled. The dodecahedral BMZIF core provided active Co sites,66 and the HKUST-1 shell afforded Cu species to improve SO selectivity. This core–shell hierarchical BMZIF@HKUST-1, namely, HKZIF, is fleetly in situ reduced through procedural calcination under 20% H2/Ar to obtain a porous core–shell Co–NC@Cu–C. Due to rapid reduction, no distinct Co and Cu NPs were observed; they were uniformly dispersed throughout the core or shell carbon framework. Copper readily oxidizes in an oxidizing atmosphere and washes into solution. To stabilize and preserve the copper sites, a metal ion (Mn+, M = Ag, Pt, Pd) exchange based on a spontaneous redox reaction yielded Co–NC@CuAg–C, Co–NC@CuOPd–C and Co–NC@Cu2OPt–C (Scheme 1). Among them, Co–NC@CuOPd–C showed excellent catalytic activity (99% conversion and 92.9% selectivity) and high stability for styrene epoxidation using O2 by photothermal catalysis. The trimetallic isostructural MOFs and the intricate metal ion exchange will open up novel pathways toward the rational design of MOF-based multifunctional catalysts for more widespread applications.
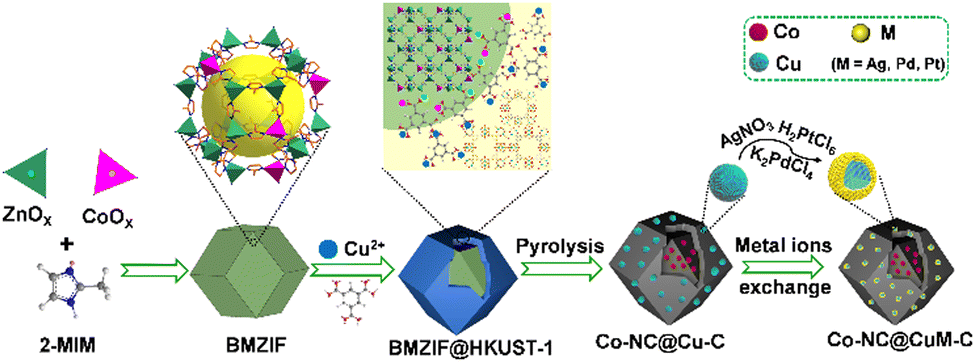 | ||
| Scheme 1 Schematic illustration of the synthesis of isomerous BMZIF@HKUST-1, porous Co–NC@Cu–C and Co–NC@CuM–C. | ||
2 Results and discussion
Cobalt species are active in oxidation reactions and copper species generally improve the chemoselectivity, so we selected the Co-ZIF and HKUST-1 (Cu-MOF) as MOF precursors. Given the presence of Zn could improve the surface area and porosity of the final porous carbon, Co-ZIF-67 was replaced by the ZnCo-BMZIF. The BMZIF and HKUST-1 are two completely different MOFs in terms of metal species, ligands, crystal structures, morphologies and properties. Therefore, the orientated growth of the heteroid HKUST-1 on the BMZIF needs to overcome the mismatched cell lattices. First, the nanosized BMZIF dodecahedron core was prepared by a typical coordination approach, in which Zn2+ and Co2+ (Zn/Co molar ratio, 5/1) were used as metal nodes to coordinate with 2-methylimidazole. The BMZIF was added into a mixed solution of DMSO, Cu(NO3)2, and 1,3,5-H3BTC to grow HKUST-1. To construct the core–shell HKZIF, instead of growing separately, reasonable control of the core/shell molar ratio and growth time is a crucial factor. Increasing the feed ratio of HKUST-1 gradually altered the corresponding composite colours from purple to aqua blue (Fig. 1(a)), indicating their successful recombination. Subsequently, the components in these composites were confirmed by powder X-ray diffraction (PXRD). Fig. 1(b) shows that the characteristic diffraction peaks of HKUST-1 began to appear at a core/shell molar ratio of 10/1; their peak intensities gradually increased as the ratios decreased, while it is opposite to the BMZIF. From 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1, XRD patterns showed coexisting diffraction peaks for both MOFs that matched their simulation patterns well. N2 sorption curves suggested the composite (using a 2/1 core/shell molar ratio) has combined the microporous features of the BMZIF and HKUST-1. BET surface areas for the BMZIF-5, HKUST-1, and HKZIF (2/1) were 1647.5, 1673.0 and 863.3 m2 g−1 (Fig. 1(c) and Fig. S1, ESI†), respectively. The decreased surface area of the HKZIF (2/1) was observed. We guess the partial pore windows of the BMZIF particle surface were covered and blocked by the growing HKUST-1.
:1 to 1:1, XRD patterns showed coexisting diffraction peaks for both MOFs that matched their simulation patterns well. N2 sorption curves suggested the composite (using a 2/1 core/shell molar ratio) has combined the microporous features of the BMZIF and HKUST-1. BET surface areas for the BMZIF-5, HKUST-1, and HKZIF (2/1) were 1647.5, 1673.0 and 863.3 m2 g−1 (Fig. 1(c) and Fig. S1, ESI†), respectively. The decreased surface area of the HKZIF (2/1) was observed. We guess the partial pore windows of the BMZIF particle surface were covered and blocked by the growing HKUST-1.
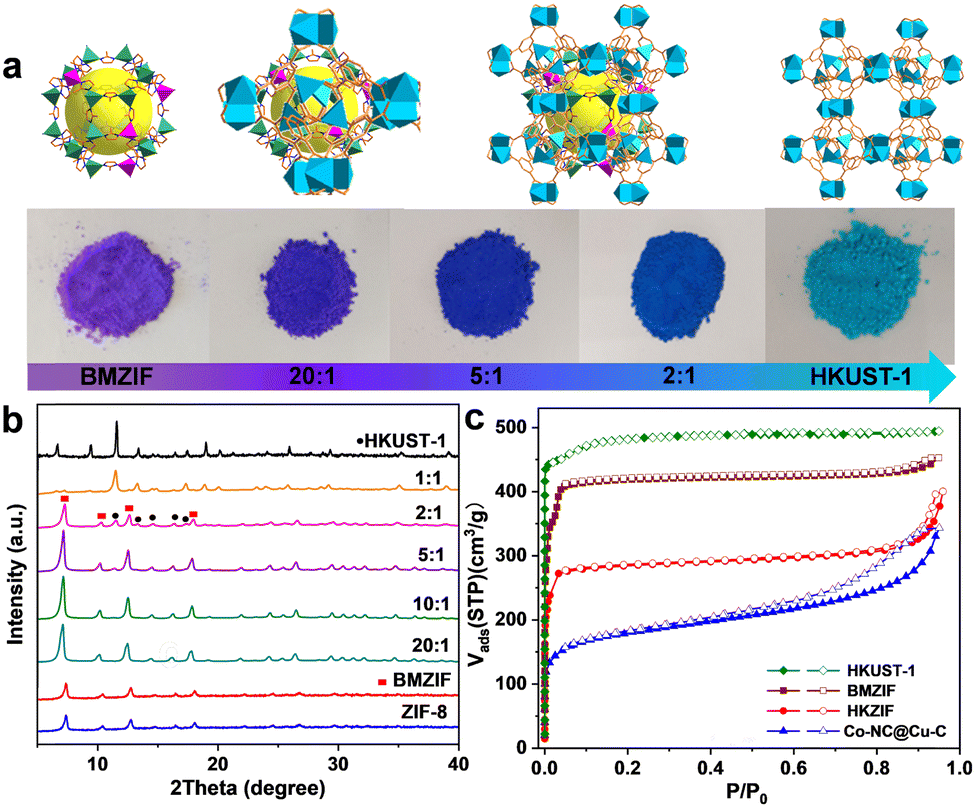 | ||
| Fig. 1 (a) The sample color and the corresponding structure of the as-synthesized BMZIF, HKUST-1 and HKZIF. (b) PXRD patterns of a series of as-synthesized MOFs. (c) N2 sorption isotherms of MOF precursors and Co–NC@Cu–C. | ||
A series of measurements confirmed the detailed BMZIF@HKUST-1 structure. The morphologies of the BMZIF and HKZIF (2:1) were measured by scanning electron microscopy (SEM). Fig. 2(a) shows BMZIF particles (150 nm) uniformly dispersed with well-defined dodecahedron morphology. After coating HKUST-1, the core–shell HKZIF particle retains a regular dodecahedron morphology and a size of ∼200 nm (Fig. 2(b)). The detailed structure of the hybrid was further analyzed by transmission electron microscopy (TEM), elemental mapping and other measurements. The TEM image shows the dispersed core–shell NPs with the dark BMZIF core and gray HKUST-1 shell in each particle (Fig. 2(c)). The enlarged TEM image of one HKZIF particle clearly shows the core–shell structure and regular dodecahedron shape. The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image of the HKZIF in Fig. 2(d) confirmed the regular morphology. Elemental mappings of Zn, Co, and Cu verify their uniform dodecahedral distribution throughout the carbon skeleton (Fig. 2(e)–(g) and Fig. S2, ESI†). In addition, the smaller distribution area of Zn and Co relative to Cu illustrates that Zn and Co concentrate at the centre and Cu species distribute mainly on the outside of the particle. The HAADF-STEM image in Fig. 2(d) and the corresponding line-scan data confirm this structure assessment (Fig. 2(i)). These results clearly prove the well-directed growth of HKUST-1 on the BMZIF and successful synthesis of the core–shell HKZIF. Actually, there is some difficulty for the successful assembly of HKUST-1 and the BMZIF. We also attempted to grow the BMZIF on HKUST-1, but it failed. This was because the HKUST-1 surface was easily etched by alkalescent 2-methylimidazole. In addition, the growth time of HKUST-1 on the BMZIF was also one tricky point and affected greatly the final morphology of the HKZIF. When the core/shell molar ratio was 2/1, the optimal growth time was 10 min (Fig. S3, ESI†). There is not enough time for coordinative growth of HKUST-1 on the ZIF in a short time (5 min), while it would spontaneously nucleate and grow individual HKUST-1 particles when the time was extended (≥20 min).
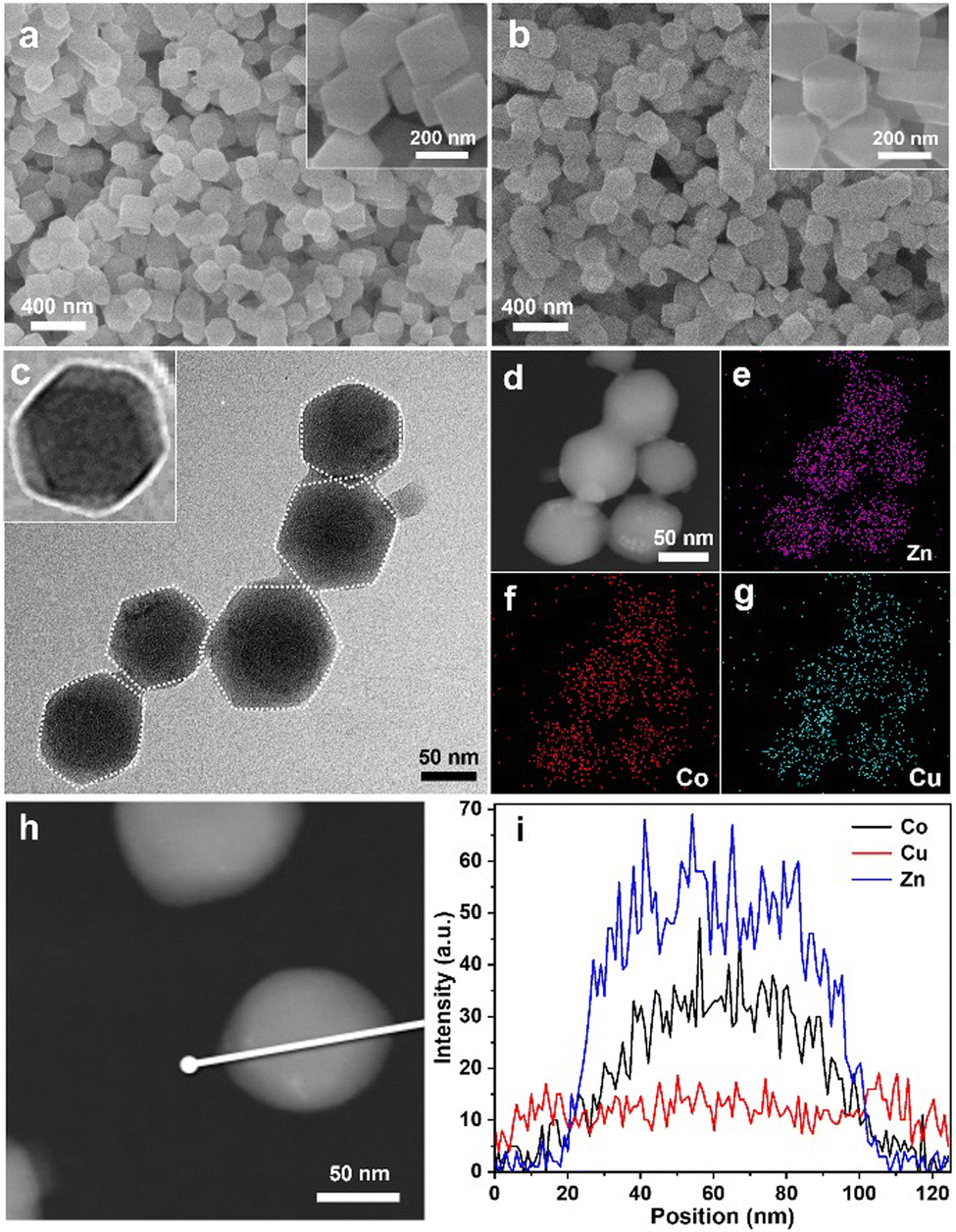 | ||
| Fig. 2 SEM images of the (a) BMZIF and (b) HKZIF. (c) TEM image of the HKZIF. (d)–(g) HAADF-STEM images and the corresponding EDS elemental (Zn, Co, Cu) mapping of the HKZIF. (h) and (i) Line scanning spectra of the HKZIF. | ||
Subsequently, the HKZIF precursor underwent pyrolysis under a 20% H2/Ar atmosphere. During pyrolysis, the metal centers (Zn(II), Cu(II) and Co(II)) are reduced to their corresponding zero-valent states. Zn species readily evaporated along with the carrier gas due to its low boiling point. As a result, the BMZIF core produced Co encapsulated by porous N-doped C (named Co@NC) and the HKUST-1 shell converted into porous Cu@C. Their powder X-ray diffraction peaks are shown in Fig. 3(a). The diffraction peaks at ∼44.21°, 51.52°, and 75.85° were assigned to metallic Co (JCPDS no. 15-0806), and the three peaks at around 43.29°, 50.43°, and 74.13° were indexed to metallic Cu (JCPDS no. 04-0836). No characteristic peak for the CoCu alloy was detected, which confirmed the successful generation of Co–NC@Cu–C NPs. The decreased surface areas and newly generated mesopores indicate the pore structure transformation (Fig. 1(c) and Fig. S1, ESI†). The SEM images (Fig. 3(b)) show that Co–NC@Cu–C particles were well dispersed and retained the sizes and morphologies of their precursors. From the TEM images, most Co–NC@Cu–C particles retained the core–shell structure with the black core and dark shell (Fig. 3(c)). Generally, Co tends to have a deeper defocus contrast compared with Cu in TEM. The corresponding elemental mapping of C, Co, and Cu showed that the C element uniformly dispersed throughout the particle, Co species occupied the central position, and the Cu element distributed over a wider area with a darker centre than Co (Fig. 3(d)–(g)). These results further demonstrate the formation of the HKZIF and its successful transition to Co–NC@Cu–C.
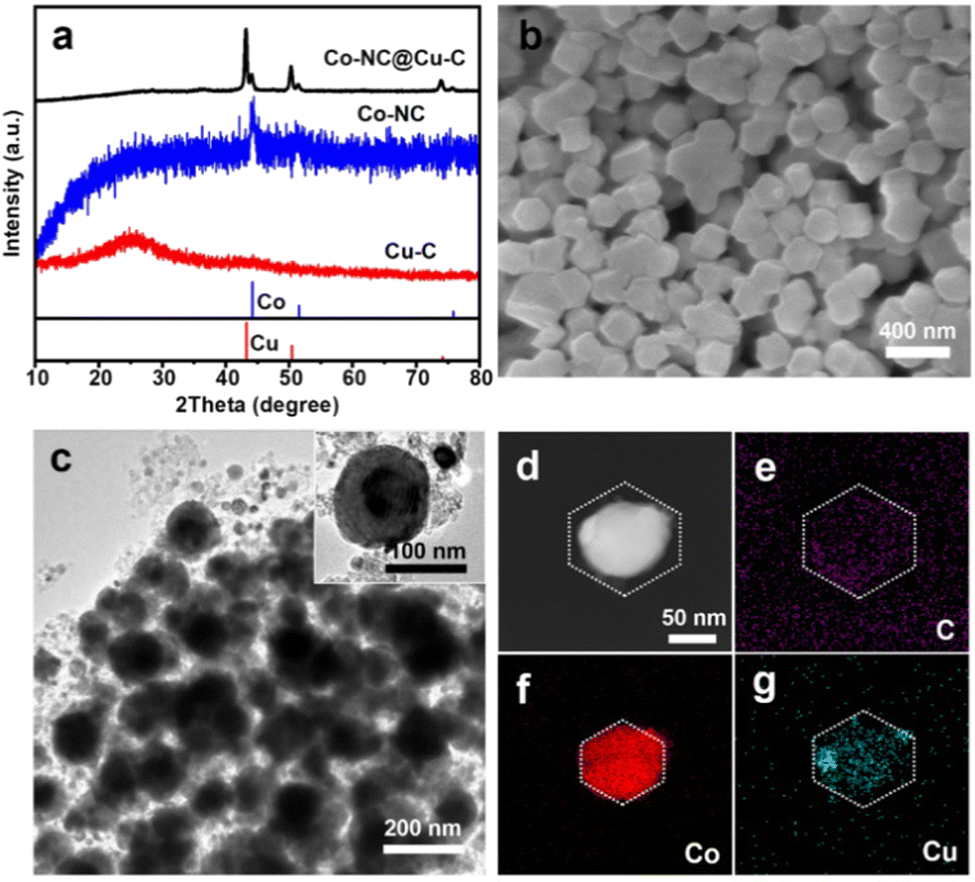 | ||
| Fig. 3 (a) PXRD patterns of Cu–C, Co–NC, and Co–NC@Cu–C. (b) SEM, (c) TEM, and (d)–(g) HAADF-STEM images as well as the corresponding EDS elemental (C, Co, Cu) mapping of Co–NC@Cu–C. | ||
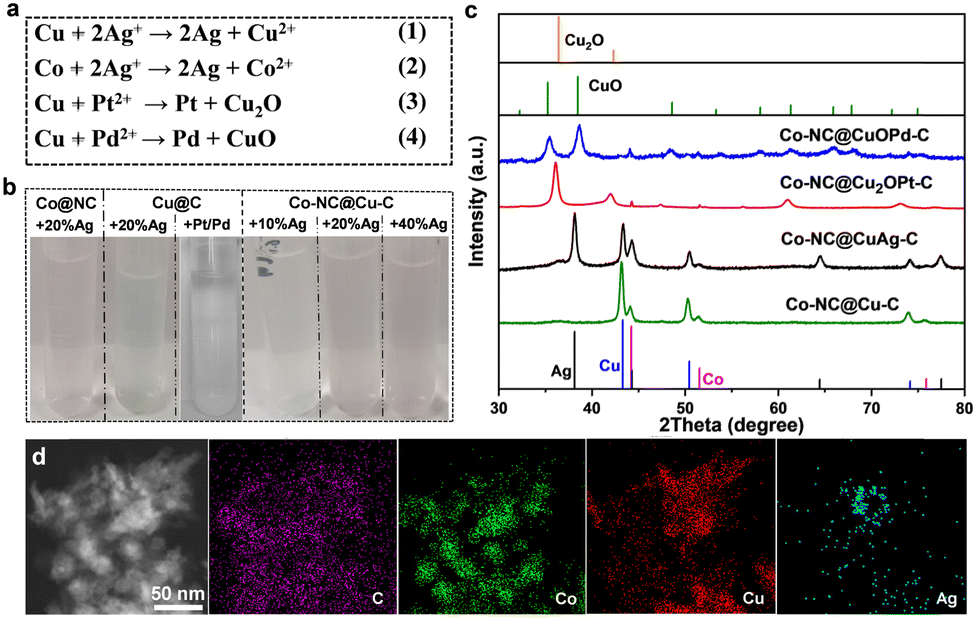
X-ray photoelectron spectroscopy (XPS) characterization further confirmed the forms of Co and Cu in the samples. The Co 2p spectrum presents three forms of Co species at 778.35, 780.35 and 782.1 eV; these were metallic Co, CoxOy or CoCxNy, and Co–Nx, respectively (Fig. S4a, ESI†). The Cu 2p peaks at 934.2 eV and 932.7 eV were attributed to divalent Cu and zero-valent Cu, respectively (Fig. S4b, ESI†). Cu(0) species were oxidized readily to generate dissociative Cu(II) ions in most oxidation reactions, especially using coordinating solvents (e.g., acetonitrile). Therefore, some changes should be made to avoid oxidation and prolong the lifetimes of Cu-based catalysts. Ion exchange by a spontaneous oxidation–reduction reaction effectively replaces a metal with another metal in solution. The standard reduction potentials of the Co/Co2+ (−0.28 V vs. the standard hydrogen electrode, SHE) and Cu/Cu2+ (0.34 V vs. SHE) pairs are much lower than those of noble metals, such as the Ag+/Ag pair (0.799 V vs. SHE), Pd2+/Pd pair (0.8 V vs. SHE) and the Pt2+/Pt pair (1.2 V vs. SHE). Therefore, Cu species in Co–NC@Cu–C can be oxidized or replaced by the noble metal ions (Ag+, Pt2+, Pd2+) (Fig. 4(a)). The solution colour changed during this replacement, which preliminarily confirmed successful ion exchange (Fig. 4(b)). For Co–NC@Cu–C, the solution colour gradually turned from blue to pink with increasing Ag+ content, indicating that the copper shell is replaced first and then the cobalt core by Ag+. Differently, the solution colour remained unchanged when adding PtCl62− or PdCl42− into the Cu–C sample, indicating no dissociative Cu2+ was released. According to the XRD analysis in Fig. 4(c), the emerging characteristic peaks of Ag and decreased peak intensity of Cu compared with those of Co–NC@Cu–C confirm the partial substitution of Cu by Ag. The surface valence state of various elements in Co–NC@CuAg–C was analyzed using XPS (Fig. S5, ESI†). The Cu 2p3/2 peaks of both divalent Cu and zero-valent Cu remained unchanged (Fig. S5a, ESI†). The sample displayed two obvious peaks centred at 368.33 and 374.33 eV, which corresponded to metallic Ag (Fig. S5b, ESI†).
 | ||
| Fig. 4 (a) Ion exchange reactions. (b) Solution color changes during exchange using a series of catalysts. (c) PXRD patterns of Co–NC@CuM (M = Ag, Pt, Pd)–C. (d) HAADF-STEM images and the corresponding EDS elemental (C, Co, Cu, Ag) mapping of Co–NC@CuAg–C. | ||
Different from the commutative product by Ag+, Cu(0) was mainly oxidized to Cu2O by Pt2+, i.e., to Cu2O at low Pd2+ content and CuO at high Pd2+ content (Fig. 4(c) and Fig. S6, ESI†). As the amount of Pd2+ increased, the peak intensities of Cu2O gradually decreased until they disappeared. Finally, only the characteristic peaks of CuO were observed when using 10 wt% Pd. This is the same as that observed for Cu@C derived from HKUST-1: the Cu NPs were oxidized and transformed completely to CuO by 10 wt% Pd (Fig. S6, ESI†). Very weak diffractions of Pt or Pd species are detected from powder XRD patterns, presumably due to their low concentrations or small sizes. The presence of Pd(0) can be demonstrated from the XPS spectrum of Co–NC@CuOPd–C. The Pd 3d spectrum shows two signals of Pd 3d5/2 and Pd 3d3/2 electrons at 336.53 eV and 342.25 eV (Fig. S7, ESI†). The Cu 2p XPS spectrum shows two main signals of Cu 2p3/2 and Cu 2p1/2 at 934.2 eV and 954.3 eV, respectively (Fig. S7, ESI†), demonstrating that Cu0 was oxidized completely to CuO by Pd2+. Fig. S8 (ESI†) shows the XPS spectral comparison of the Co 2p of Co–NC and Co–NC@CuOPd–C. It is worth noting that the Co 3d peaks in Co–NC@CuPd–C shifted to higher binding energies by ∼1.22 eV compared to Co–NC. The obvious blue shift of Co 3d suggested the existence of an interaction between the Co surface at the core–shell interface and the shell layer CuOPd–C. The elemental mapping in Fig. 4(d) confirmed the existence of C, Co, Cu, and Ag elements. Ag disperses primarily at the edges of the particle like hollow NPs, while the externally distributed Cu decreased declined due to their exchange. The actual amounts of Ag, Pd, and Pt in the Co–NC@CuAg–C, Co–NC@CuOPd–C and Co–NC@Cu2OPt–C samples were measured by inductively coupled plasma atomic emission spectrometry (ICP-AES). The weight percentages (wt%) of Ag, Pd, and Pt were lower than their nominal values (Table S1, ESI†).
The performance of Co–NC@Cu–C and Co–NC@CuM (M = Ag, Pt, Pd)–C towards epoxidation of styrene employing O2 as the oxidant, isobutyraldehyde as the co-catalyst and acetonitrile as the solvent was investigated. Table 1 shows a comparison of various catalysts for styrene oxidation. Co–NC@Cu–C had an 84.7% conversion with a selectivity of 81.8% to styrene oxide within 2 h at 80 °C (entry 1). For comparison, the CoCu-ZIF was synthesized and then pyrolyzed to give CoCu alloy NPs stabilized by N-doped C. The lattice fringe with an interplanar spacing of 0.207 nm in the high resolution transmission electron microscopy (HRTEM) image was assigned to the (111) plane of CuCo alloy (Fig. S9, ESI†). The CoCu@NC presented relatively low conversion (58%) with a 74.5% selectivity of styrene epoxide under the same reaction conditions (entry 2). The result proved the advantage of constructing multilayer MOF structures. However, the solution colour after the above two reactions turned blue (Fig. S10, ESI†), which meant that most Cu was oxidized to Cu(II) by O2, then dissolved upon coordination to acetonitrile. Although Co–NC@CuAg(20%)–C had a higher conversion (94.0%) and selectivity (81.5%) (entry 4), the light blue solution after the reaction was also observed (Fig. S10, ESI†). Silver also readily coordinates with acetonitrile and cannot protect copper from leaching. The Co–NC@Cu2OPt(10%)–C had a conversion of 94.3% and a 71.6% selectivity (entry 5). However, the blue solution after the reaction indicated the dissolution of Cu2O in acetonitrile. Interestingly, the nearly colourless solution after catalysis for Co–NC@CuOPd(10%)–C confirmed the good stability of CuO in acetonitrile solvent (Fig. S10, ESI†). Introducing a small amount of Pd not only stabilized Cu, but increased the reaction conversion (95.0%) and SO selectivity (87.0%) (entry 6). For comparison, monometallic Co–NC and Cu–C had poor yields due to their few metal active sites (entries 7 and 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time | T (°C) | Conv. (%) | Select. of a (%) |
| a Reaction conditions: catalyst (20 mg), acetonitrile (10 mL), styrene (1.1 mmol), isobutyraldehyde (2.5 mmol), O2 bubbling. b Reaction conditions: catalyst (20 mg), acetonitrile (10 mL), styrene (0.1 mmol), isobutyraldehyde (2 mmol), O2 bubbling, visible light (λ ≥ 420 nm). | |||||
| 1 | Co–NC@Cu–C | 2 h | 80 | 84.7 | 81.8 |
| 2 | CoCu@NC | 2 h | 80 | 58.0 | 74.5 |
| 3 | Cu@Ag@C(20%) | 2 h | 80 | 66.0 | 79.8 |
| 4 | Co–NC@CuAg(20%)–C | 2 h | 80 | 94.0 | 81.5 |
| 5 | Co–NC@Cu2OPt(10%)–C | 2 h | 80 | 94.3 | 71.6 |
| 6 | Co–NC@CuOPd(10%)–C | 2 h | 80 | 95.0 | 87.0 |
| 7 | Co–NC | 2 h | 80 | 41.0 | 72.0 |
| 8 | Cu–C | 2 h | 80 | 53.5 | 73.1 |
| 9b | Co–NC@Cu2OPt(10%)–C | 30 min | 47 | >99.0 | 94.5 |
| 10b | Co–NC@CuOPd(10%)–C | 30 min | 42 | 99.0 | 92.9 |
| 11b | Co–NC@Cu–C | 30 min | 53 | 52.3 | 49.4 |
| 12b | Co–NC | 30 min | 45 | 15.5 | 20.0 |
| 13b | Cu–C | 30 min | 46 | 21.6 | 25.2 |
Interestingly, we found the photothermal effects of these carbon samples. Fig. 5(a) shows the photothermal conversion efficiencies of Co–NC@Cu2OPt(10%)–C and Co–NC@CuOPd(10%)–C under visible light irradiation. The solution temperature went up approximately to 45–55 °C upon the Co–NC@CuPd–C, Co–NC@Cu2OPt–C and Co–NC@Cu–C. This can be explained by their good light absorption ability. According to UV-Vis diffuse reflectance spectra, all the samples have a visible light absorption band at 330–400 nm (Fig. 5(b)). Therefore, the epoxidation of styrene was further investigated using visible light instead of external heating. The Co–NC@CuOPd–C exhibited high activity (>99% conversion in 30 min) and selectivity (92.9%). Co–NC@Cu2OPt–C also had excellent activity and SO selectivity compared with monometallic Co–NC and Cu–C (entries 9–13). Their catalytic performance exceeded most reported catalysts for styrene epoxidation (Table S2, ESI†). The PXRD patterns of Co–NC@Cu–C and Co–NC@CuM–C (M = Ag, Pt, Pd) catalysts before and after the reaction are shown in Fig. 5(c). According to the data, the diffraction peaks of Cu decreased clearly for the Co–NC@Cu–C catalyst after the reaction. The characteristic peaks of both Cu and Ag had a clear intensity decline, accompanied by the emerging peaks of AgCN for Co–NC@CuAg–C after the 1st run. This result combined with the blue solution confirmed our speculation that both Cu and Ag initially oxidized and coordinated with acetonitrile. Similarly, the strong peaks of Cu2O almost disappeared for Co–NC@Cu2OPt–C after the reaction, indicating the same instability of Cu2O in acetonitrile. Interestingly, the diffraction peaks of used Co–NC@CuOPd–C remained virtually unchanged, which confirmed the superior stability of CuO. In addition, the ICP analysis of Co–NC@CuOPd–C after catalysis shown almost the same metal contents as those of fresh Co–NC@CuOPd–C (Table S1, ESI†). This result further demonstrated the stability of Co–NC@CuOPd–C. In order to investigate the mechanism of epoxidation of styrene, the quenchers of different reactive oxygen species (1O2, ˙OH, ˙O2−) were introduced into the reaction system. In the presence of sodium amide (NaN3, quenching for 1O2) and IPA (quenching for ˙OH), there was almost no obvious decrease in the conversion of styrene (Fig. S11, ESI†). No products were detected when adding superoxide dismutase (SOD), indicating that ˙O2− has an important contribution to the epoxidation of styrene. According to the above experimental results and related literature,67,68 a possible mechanism for the epoxidation of styrene with O2 is proposed. As shown in Scheme S1a (ESI†), the CuOPd shell with abundant charge transferred electrons to O2 to produce ˙O2−. The obtained ˙O2− and isobutyryl radical combined with H+ to generate isobutyl peroxyacid. The isobutyl peroxyacid produced finally reacted with styrene to form styrene oxide. The oxidation pathway of benzaldehyde is shown in Scheme S1b (ESI†). The isobutyryl radical reacted with O2 to produce an isobutylperoxy radical, which reacted with the olefinic bond of styrene to form a complex radical. Then the intermediate reacted with O2 to generate a new radical intermediate, which further decomposed to give benzaldehyde.
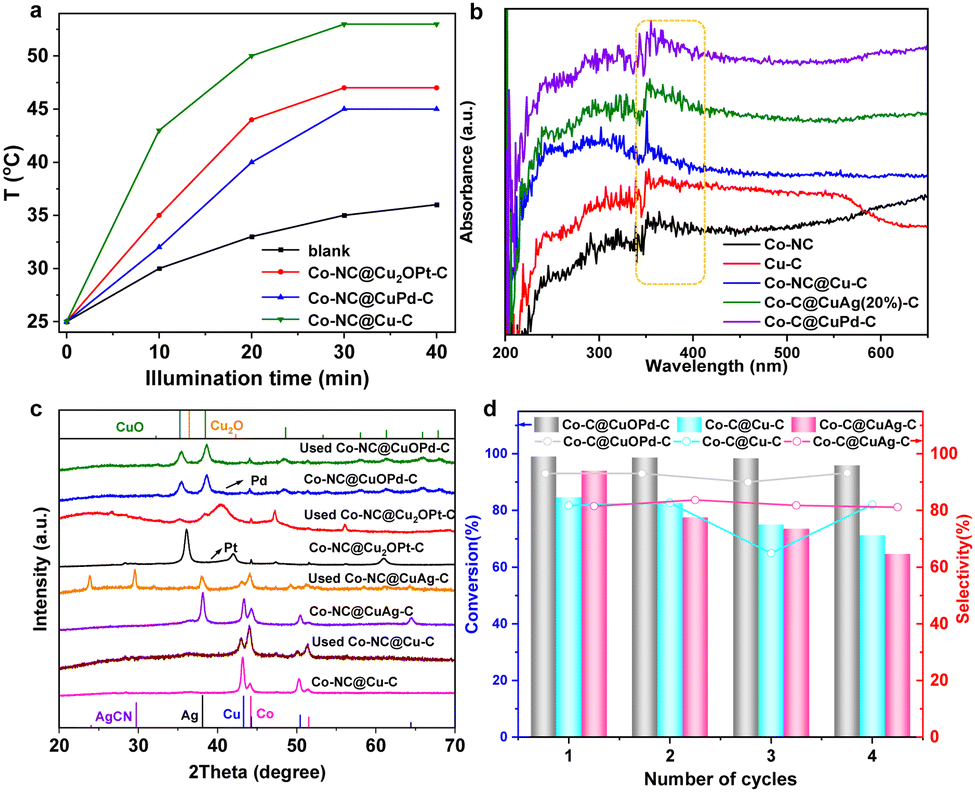 | ||
| Fig. 5 (a) Photothermal conversion efficiency based on different materials. (b) UV-Vis absorption spectra of Co–NC, Cu–C, Co–NC@Cu–C and Co–NC@CuM–C. (c) XRD patterns of the catalysts Co–NC@Cu–C and Co–NC@CuM–C before and after the reaction. (d) Recyclability of Co–NC@CuOPd(10%)–C, Co–NC@Cu–C, and Co–NC@CuAg(20%)–C for styrene epoxidation. | ||
Subsequently, the recyclability of several position exchanged samples was investigated under the same reaction conditions, which is a very significant practical consideration for large-scale applications. It is worth noting that the Co–NC@CuOPd–C catalyst can be reused at least four times without an obvious activity change (Fig. 5(d)). For comparison, the obvious activity decline during four reuse cycles for both Co–NC@CuAg–C and Co–NC@Cu–C was mainly due to the massive leaching of Cu and Ag species (Fig. 5(d)). Although all exchanged products (Cu, Cu2O, CuO) have good catalytic activity, only CuO has the best stability under harsh reaction conditions. In addition, the alloyed Pd may protect the CuO from dissolution to some extent. The Co–NC@CuAg–C and Co–NC@Cu2OPt–C may be suitable for other reaction conditions. Therefore, this ion exchange strategy provides a novel and effective method to prevent the leaching of Cu species under an oxidizing atmosphere.
3 Conclusions
In this work, we have successfully realized the directed epitaxial growth of hierarchical HKUST-1 on the BMZIF to obtain a novel trimetallic BMZIF@HKUST-1 heterostructure. During assembly, the regular morphology, size and composition of the hybrids were effectively controllable. After direct carbonization, the core–shell Co–NC@Cu–C material containing both Cu and Co active sites was successfully obtained. To stabilize Cu species in Co–NC@Cu–C for styrene epoxidation under harsh conditions, ion exchange with noble metal ions (M = Ag, Pt, Pd) was adopted. Among the exchanged products, Co–NC@CuOPd–C optimized the stability in catalytic styrene epoxidation by photothermal catalysis under moderate conditions. Moreover, the good SO yield with >90% selectivity exceeds those of most reported catalysts, even some noble metals. The excellent catalytic performance of Co–NC@CuOPd–C stems mainly from multiple active sites (Co, CuO, Pd), good stability, and its porous structure. This work provided a novel method to prevent Cu NPs in Cu-based catalysts from coordinative leaching. In addition, the design strategy of hierarchical core–shell MOFs would explore more active sites for the synthesis of various fine chemicals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22275108) and the Excellent Youth Foundation of Shandong Natural Science Foundation (ZR2020YQ08).Notes and references
- Y. Xiong, W. Sun, P. Xin, W. Chen, X. Zheng, W. Yan, L. Zheng, J. Dong, J. Zhang, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2000896 CrossRef CAS PubMed.
- M. Li, L. Ma, L. Luo, Y. Liu, M. Xu, H. Zhou, Y. Wang, Z. Li, X. Kong and H. Duan, Appl. Catal., B, 2022, 309, 121268 CrossRef CAS.
- H. Fu, K. Huang, G. Yang, Y. Cao, H. Wang, F. Peng, X. Cai, H. Gao, Y. Liao and H. Yu, ACS Catal., 2021, 11, 8872–8880 CrossRef CAS.
- K. Kamata, K. Yonehara, Y. Sumida, K. Yamaguchi, S. Hikichi and N. Mizuno, Science, 2003, 300, 964–966 CrossRef CAS PubMed.
- D. Banerjee, R. V. Jagadeesh, K. Junge, M.-M. Pohl, J. Radnik, A. Bruckner and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 4359–4363 CrossRef CAS PubMed.
- B. Jia, L. Bai, Z. Han, R. Li, G. Huangfu, C. Li, J. Zheng, Y. Qu, K. Leng, Y. Wang and J. Bai, ACS Appl. Mater. Interfaces, 2022, 14, 10337–10343 CrossRef CAS PubMed.
- F. Zhang, X. Zhao, C. Feng, B. Li, T. Chen, W. Lu, X. Lei and S. Xu, ACS Catal., 2011, 1, 232–237 CrossRef CAS.
- S. Tian, B. Wang, W. Gong, Z. He, Q. Xu, W. Chen, Q. Zhang, Y. Zhu, J. Yang, Q. Fu, C. Chen, Y. Bu, L. Gu, X. Sun, H. Zhao, D. Wang and Y. Li, Nat. Commun., 2021, 12, 3181 CrossRef CAS PubMed.
- B. Liu, P. Wang, A. Lopes, L. Jin, W. Zhong, Y. Pei, S. L. Suib and J. He, ACS Catal., 2017, 7, 3483–3488 CrossRef CAS.
- J. Liu, R. Meng, J. Li, P. Jian, L. Wang and R. Jian, Appl. Catal., B, 2019, 254, 214–222 CrossRef CAS.
- F. Li, J. Tang, Q. Ke, Y. Guo, M. N. Ha, C. Wan, Z. Lei, J. Gu, Q. Ling, V. N. Nguyen and W. Zhan, ACS Catal., 2021, 11, 11855–11866 CrossRef CAS.
- A. S. Shaarma, V. S. Sharma, H. Kaur and R. S. Varma, Green Chem., 2020, 22, 5902–5936 RSC.
- J. Sebastian, K. M. Jinka and R. V. Jasra, J. Catal., 2006, 244, 208–218 CrossRef CAS.
- G. Xu, Q.-H. Xia, X.-H. Lu, Q. Zhang and H.-J. Zhan, J. Mol. Catal. A: Chem., 2007, 266, 180–187 CrossRef CAS.
- X. Chen, Y. Zou, M. Zhang, W. Gou, S. Zhang and Y. Qu, J. Mater. Chem. A, 2022, 10, 6016 RSC.
- B. Paul, S. K. Sharma, R. Khatun, S. Adak, G. Singh, V. Joshi, M. K. Poddar, A. Bordoloi, T. Sasaki and R. Bal, Ind. Eng. Chem. Res., 2019, 58, 18168–18177 CrossRef CAS.
- X. Li, X.-H. Zhu, Y. Fang, H.-L. Yang, X. Zhou, W. Chen, L. Jiao, H. Huo and R. Li, J. Mater. Chem. A, 2014, 2, 10485–10491 RSC.
- L. Wang, H. Wang, A. E. Rice, W. Zhang, X. Li, M. Chen, X. Meng, J. P. Lewis and F.-S. Xiao, J. Phys. Chem. Lett., 2015, 6, 2345–2349 CrossRef CAS PubMed.
- Y. Fu, L. Xu, H. Shen, H. Yang, F. Zhang, W. Zhu and M. Fan, Chem. Eng. J., 2016, 299, 135–141 CrossRef CAS.
- Y. Huang, Z. Liu, G. Gao, G. Xiao, A. Du, S. E. Bottle, S. Sarina and H. Zhu, ACS Catal., 2017, 7, 4975–4985 CrossRef CAS.
- M. R. Nabid, Y. Bide and M. Abuali, RSC Adv., 2014, 4, 35844 RSC.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC.
- R.-B. Lin, L. Li, H.-L. Zhou, H. Wu, C. He, S. Li, R. Krishna, J. Li, W. Zhou and B. Chen, Nat. Mater., 2018, 17, 1128–1133 CrossRef CAS PubMed.
- Z. Lu, J. Duan, H. Tan, L. Du, X. Zhao, R. Wang, S. Kato and S. Yang, J. Am. Chem. Soc., 2023, 145, 4150–4157 CrossRef CAS PubMed.
- O. M. Yaghi and Z. Rong, Science, 2023, 379, 330–331 CrossRef CAS PubMed.
- L. Jiao, J. Wang and H.-L. Jiang, Acc. Mater. Res., 2021, 2, 327–339 CrossRef CAS.
- H. Liu, W. Liu, G. Xue, T. Tan, C. Yang, P. An, W. Chen, W. Zhao, T. Fan, C. Cui, Z. Tang and G. Li, J. Am. Chem. Soc., 2023, 145, 11085–11096 CrossRef CAS PubMed.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- Q. Huang, Q. Niu, X.-F. Li, J. Liu, S.-N. Sun, L.-Z. Dong, S.-L. Li, Y.-P. Cai and Y.-Q. Lan, Sci. Adv., 2022, 8, eadd5598 CrossRef CAS PubMed.
- C.-C. Cao, C.-X. Chen, Z.-W. Wei, Q.-F. Qiu, N.-X. Zhu, Y.-Y. Xiong, J.-J. Jiang, D. Wang and C.-Y. Su, J. Am. Chem. Soc., 2019, 141, 2589–2593 CrossRef CAS PubMed.
- M. A. El-Bindary, M. G. El-Desouky and A. A. El-Bindary, Appl. Organomet. Chem., 2022, 36, e6660 CrossRef CAS.
- M. Ding, W. Liu and R. Gref, Adv. Drug Delivery Rev., 2022, 190, 114496 CrossRef CAS PubMed.
- H. Yuan, N. Li, W. Fan, H. Cai and D. Zhao, Adv. Sci., 2022, 9, 2104374 CrossRef CAS PubMed.
- W.-H. Deng, L. He, E.-X. Chen, G.-E. Wang, X.-L. Ye, Z.-H. Fu, Q. Lin and G. Xu, J. Mater. Chem. A, 2022, 10, 12977 RSC.
- M. A. Gaidimas, F. A. Son, M. R. Mian, T. Islamoglu and O. K. Farha, ACS Appl. Mater. Interfaces, 2022, 14, 47222–47229 CrossRef CAS PubMed.
- Y. Ye, Y. Xie, Y. Shi, L. Gong, J. Phipps, A. M. Al-Enizi, A. Nafady, B. Chen and S. Ma, Angew. Chem., Int. Ed., 2023, 62, e202302564 CrossRef CAS PubMed.
- O. T. Qazvini and S. G. Telfer, J. Mater. Chem. A, 2020, 8, 12028–12034 RSC.
- X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed.
- L. Yang, X. Zeng, W. Wang and D. Cao, Adv. Funct. Mater., 2018, 28, 1704537 CrossRef.
- H.-L. Jiang, B. Liu, Y.-Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- S.-N. Zhao, X.-Z. Song, S.-Y. Song and H.-J. Zhang, Coord. Chem. Rev., 2017, 337, 80–96 CrossRef CAS.
- X. Xie, L. Shang, X. Xiong, R. Shi and T. Zhang, Adv. Energy Mater., 2022, 12, 2102688 CrossRef CAS.
- Y. Zhou, B. Tang, S. Wang and J. Long, Int. J. Hydrogen Energy, 2020, 45, 15785–15795 CrossRef CAS.
- H.-Q. Yin, Z.-M. Zhang and T.-B. Lu, Acc. Chem. Res., 2023, 56, 2676–2687 CrossRef CAS PubMed.
- J. Li, Y. Chen, Y. Tang, S. Li, H. Dong, K. Li, M. Han, Y.-Q. Lan, J. Bao and Z. Dai, J. Mater. Chem. A, 2014, 2, 6316 RSC.
- L. Hu, W. Li, L. Wang and B. Wang, EnergyChem, 2021, 3, 100056 CrossRef CAS.
- J. Gao, Q. Huang, Y. Wu, Y.-Q. Lan and B. Chen, Adv. Energy Sustainability Res., 2021, 2, 2100033 CrossRef CAS.
- Y. Gu, Y. Wu, L.-N. Li, W. Chen, F. Li and S. Kitagawa, Angew. Chem., Int. Ed., 2017, 56, 15658–15662 CrossRef CAS PubMed.
- S. Choi, T. Kim, H. Ji, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2016, 138, 14434–14440 CrossRef CAS PubMed.
- X.-G. Wang, L. Xu, M.-J. Li and X.-Z. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18078–18086 CrossRef CAS PubMed.
- T. Li, J. E. Sullivan and N. L. Rosi, J. Am. Chem. Soc., 2013, 135, 9984–9987 CrossRef CAS PubMed.
- J. Yang, F. Zhang, H. Lu, X. Hong, H. Jiang, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 10889–10893 CrossRef CAS PubMed.
- G. Lee, S. Lee, S. Oh, D. Kim and M. Oh, J. Am. Chem. Soc., 2020, 142, 3042–3049 CrossRef CAS PubMed.
- Y. Gong, Y. Yuan, C. Chen, P. Zhang, J. Wang, A. Khan, S. Zhuiykov, S. Chaemchuen and F. Verpoort, J. Catal., 2019, 375, 371–379 CrossRef CAS.
- S. Furukawa, K. Hirai, K. Nakagawa, Y. Takashima, R. Matsuda, T. Tsuruoka, M. Kondo, R. Haruki, D. Tanaka, H. Hirotoshi Sakamoto, S. Shimomura, O. Sakata and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 1766–1770 CrossRef CAS PubMed.
- M.-S. Yao, J.-W. Xiu, Q.-Q. Huang, W.-H. Li, W.-W. Wu, A.-Q. Wu, L.-A. Cao, W.-H. Deng, G.-E. Wang and G. Xu, Angew. Chem., Int. Ed., 2019, 58, 14915–14919 CrossRef CAS PubMed.
- W. Liu, J. Huang, Q. Yang, S. Wang, X. Sun, W. Zhang, J. Liu and F. Huo, Angew. Chem., Int. Ed., 2017, 56, 5512–5516 CrossRef CAS PubMed.
- S. Choi, T. Kim, H. Ji, H.-J. Lee and M. Oh, J. Am. Chem. Soc., 2016, 138, 14434–14440 CrossRef CAS PubMed.
- J. Zhuang, L.-Y. Chou, B. T. Sneed, Y. Cao, P. Hu, L. Feng and C.-K. Tsung, Small, 2015, 11, 5551–5555 CrossRef CAS PubMed.
- M. Zhao, J. Chen, B. Chen, X. Zhang, Z. Shi, Z. Liu, Q. Ma, Y. Peng, C. Tan, X.-J. Wu and H. Zhang, J. Am. Chem. Soc., 2020, 142, 8953–8961 CrossRef CAS PubMed.
- C. Liu, Q. Sun, L. Lin, J. Wang, C. Zhang, C. Xia, T. Bao, J. Wan, R. Huang, J. Zou and C. Yu, Nat. Commun., 2020, 11, 4971 CrossRef CAS PubMed.
- H. J. Lee, Y. J. Cho, W. Cho and M. Oh, ACS Nano., 2013, 7, 491–499 CrossRef PubMed.
- H. Guo, A. Che, W. Mi, Y. Zhang and X. Shi, J. Mater. Chem. A, 2023, 11, 19145–19152 RSC.
- Y.-Z. Chen, C. Wang, Z.-Y. Wu, Y. Xiong, Q. Xu, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- Q. Yue, Y. Lu, Z. Zhang, H. Tian, H. Wang, X. Li and S. Liu, New J. Chem., 2020, 44, 16913–16920 RSC.
- B.-B. Wentzel, P.-A. Gosling, M.-C. Feiters and R. J. M. Nolte, J. Chem. Soc., Dalton Trans., 1998, 2241–2246 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01024d |
| This journal is © The Royal Society of Chemistry 2024 |