
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInnovative strategies for nitrogen-incorporating silicon oxycarbide-based preceramic polymer synthesis†
B.
Pérez-Román
 *a,
A.
Merchán del Real
b,
J.
Rubio
a,
M. A.
Mazo
a and
F.
Rubio-Marcos
a
*a,
A.
Merchán del Real
b,
J.
Rubio
a,
M. A.
Mazo
a and
F.
Rubio-Marcos
a
aCeramic and Glass Institute (ICV-CSIC), Kelsen St. 5, 28049, Madrid, Spain. E-mail: berta.perez@icv.csic.es; Fax: +34 91 735 58 43; Tel: +34 91 735 58 40
bOrganic Chemistry Department, Faculty of Science, Autonomous University of Madrid, Francisco Tomás y Valiente St. 7, 28049, Madrid, Spain
First published on 18th January 2024
Abstract
In this study, we introduce a pioneering methodology for crafting silicon oxycarbonitride materials (SiOCN) by harnessing the intricate synergy between allyl-substituted hydrido polycarbosilane (AHPCS) and a novel triazine-based dendron, serving as a nitrogen-containing polymeric precursor. The synthetic journey involves meticulous hydrosilylation reactions between AHPCS's Si–H moieties and the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 groups embedded within the triazine dendritic architecture, followed by controlled pyrolysis under an argon atmosphere at temperatures up to 900 °C. Through systematic variations in reaction durations and pyrolytic temperatures, we uncover the prevalence of SiC4−xOx motifs within the material matrix, with oxygen content modulation observed in samples under extended reaction times and heightened pyrolysis temperatures. Nitrogen-based bonding's paramount importance within the N-containing polymeric precursor is also established, revealing a preference for retaining N sp2–C over N sp3–C bonds due to intricate nitrogen–AHPCS interactions that yield robust Si–N linkages. This inquiry not only advances our fundamental understanding but also charts a course for tailoring silicon oxycarbonitride –SiOCN– material properties. Promisingly, these advancements position such materials as prospective candidates for high-energy silicon oxycarbonitride-based supercapacitors, bridging pioneering materials science with sustainable energy storage technology.
CH2 groups embedded within the triazine dendritic architecture, followed by controlled pyrolysis under an argon atmosphere at temperatures up to 900 °C. Through systematic variations in reaction durations and pyrolytic temperatures, we uncover the prevalence of SiC4−xOx motifs within the material matrix, with oxygen content modulation observed in samples under extended reaction times and heightened pyrolysis temperatures. Nitrogen-based bonding's paramount importance within the N-containing polymeric precursor is also established, revealing a preference for retaining N sp2–C over N sp3–C bonds due to intricate nitrogen–AHPCS interactions that yield robust Si–N linkages. This inquiry not only advances our fundamental understanding but also charts a course for tailoring silicon oxycarbonitride –SiOCN– material properties. Promisingly, these advancements position such materials as prospective candidates for high-energy silicon oxycarbonitride-based supercapacitors, bridging pioneering materials science with sustainable energy storage technology.
Introduction
Undoubtedly, the realm of carbosilane chemistry has witnessed burgeoning interest as a means to procure precursor molecules, fostering the development of advanced silicon carbide (SiC) materials, and novel polymer-derived compounds with the capacity to finely control ultimate ceramic chemistry and nanostructure.1 Preceding polymers undergo transformation from their polymeric state into inorganic entities via elevated-temperature heat treatment, enabling the creation of ceramic materials with intricacies surpassing those attainable through conventional ceramic powder processing methodologies.2,3In the journey of polymer-derived ceramics (PDCs), the architecture of the polymeric precursor has demonstrated a pivotal role in steering both the microstructure of the resultant material and its resilience at elevated temperatures.4 The allure of highly branched polymers, inclusive of hyperbranched polymers and dendrons, has magnetized substantial research endeavors, aiming to yield new ceramic materials endowed with tailored properties.5 These molecular constructs, boasting substantial molecular weights, mitigate volatilization of low-mass species and potential reactivity linked to the abundance of reactive or functional groups.
In this context, various cross-linking methodologies have been explored to fabricate novel preceramic precursors, thereby targeting the attainment of ceramic materials marked by optimal ceramic yields. Typically encompassing ring-opening polymerization of metallocenes,6,7 hydrosilylation reactions,8 dehydrocoupling,9 dehalocoupling of chlorosilanes,10 and other redistribution reactions, these synthetic routes culminate in preceramic structures that, during the polymer-to-ceramic conversion, undergo intricate processes involving covalent bond reorganization and the liberation of organic species in gaseous form. Herein, the molecular weight of the precursor, its bonding strength, and degree of branching substantively sway the ensuing ceramic yield.11,12
Yu et al.13 conducted a systematic examination of functionality distribution within preceramic polymers, revealing that hyperbranched polymers containing outer-layer allyl units underwent redistribution and decomposition reactions at lower temperatures compared to those housing allyl functionalities in both outer and inner layers. Notably, a mere hyperbranched polymer construct does not suffice for high ceramic yield; apt crosslinking is imperative to achieve ceramic material with commendable yield.14 Correspondingly, a heavily crosslinked preceramic polymer exhibits superior ceramic yield compared to its loosely bonded counterpart.15
An avenue brimming with promise for enhancing PDCs involves introducing distinct elements into the structure, yielding ternary, quaternary ceramic systems, and nanostructured ceramic materials. Among these, nitrogen and boron have garnered substantial attention. Boron's role in enhancing SiC sintering and crystallization is long-established,16 while nitrogen incorporation yields amorphous silicon nitride and silicon carbonitride ceramics (SiCN).17 Notably, the type of heteroatom–Si bonds in the preceramic structure dictates its retentiveness during the polymer-to-ceramic transition. N–Si bonds exhibit greater resilience than N–C bonds,18 potentially giving rise to Si3N4-type domains or N-doped carbon. Incorporating N into a C-rich phase can be adeptly accomplished via pyrolysis of melamine precursors or pyridine.19,20 Hydrosilylation and click-chemistry reactions offer suitable strategies for producing hyperbranched polymers containing diverse heteroatoms.21,22 Linear or hyperbranched silanes, synthesized via Si–H-containing molecules and alkenes or alkynes, facilitate tailored preceramic polymer creation with predetermined compositions, densities, and characteristics.
In this endeavour, we capitalize on click chemistry and hydrosilylation reactions to synthesize a novel hyperbranched preceramic precursor from vinylaniline and cyanuric acid. Additionally, we introduce a novel dendron inspired by Vidya et al.,23 featuring a 1,3,5-triazine derivative incorporating styrene rings through –NH bonding at positions 2, 4, and 6 of the triazine core. This synthesized hyperbranched dendron coalesces with a commercial silicon carbide polymer precursor, forming a preceramic polymer construct that undergoes controlled pyrolysis within the temperature range of 700 to 900 °C. This orchestrated transformation seeks to yield a silicon oxycarbonitride (SiOCN)-based material embedding N within both its glassy phase and Cfree nanodomains, marking a pioneering achievement where N is incorporated into the carbon phase and the ceramic backbone of a preceramic dendritic structure.
Experimental details
Synthesis of the materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) as the eluent. The final product materialized as an off-white solid, with a commendable reaction yield of 80%.
:10 wt/wt ratio (AHPCS/TRIAZ-3) with vigorous magnetic stirring in anhydrous THF. Additionally, 1 wt% of platinum-1,3-divinyl-1,1,3,3-tetramethyldisiloxane (3–3.5% Pt), abcr GmbH (Germany) was incorporated to initiate crosslinking. The entire reaction transpired under an argon atmosphere using a Schlenk line. This intricate reaction progression was meticulously tracked through Fourier transform infrared spectroscopy (FTIR) at distinct time intervals, ensuring comprehensive polymerization was attained. Subsequently, synthesized materials were obtained after 20 hours and 48 hours of reaction time. Both resultant products were subjected to pyrolysis under a flowing argon atmosphere (150 cm3 min−1) at temperatures of 700, 800, and 900 °C for a duration of 2 hours and a prior dwell period at 280 °C for 5 hours were produced to ensure the crosslinking. The synthesized samples were appropriately designated, such as TZPC20-700, where “TZ” and “PC” denote the triazine-based dendron and polycarbosilane, respectively. The initial number signifies the reaction time, while the subsequent number indicates the pyrolysis temperature. Following pyrolysis, the SiOCN samples were subjected to milling in an agate mortar and sieved to attain particle sizes below 60 μm.
:4) as the eluent. The final product materialized as an off-white solid, with a commendable reaction yield of 80%.
:10 wt/wt ratio (AHPCS/TRIAZ-3) with vigorous magnetic stirring in anhydrous THF. Additionally, 1 wt% of platinum-1,3-divinyl-1,1,3,3-tetramethyldisiloxane (3–3.5% Pt), abcr GmbH (Germany) was incorporated to initiate crosslinking. The entire reaction transpired under an argon atmosphere using a Schlenk line. This intricate reaction progression was meticulously tracked through Fourier transform infrared spectroscopy (FTIR) at distinct time intervals, ensuring comprehensive polymerization was attained. Subsequently, synthesized materials were obtained after 20 hours and 48 hours of reaction time. Both resultant products were subjected to pyrolysis under a flowing argon atmosphere (150 cm3 min−1) at temperatures of 700, 800, and 900 °C for a duration of 2 hours and a prior dwell period at 280 °C for 5 hours were produced to ensure the crosslinking. The synthesized samples were appropriately designated, such as TZPC20-700, where “TZ” and “PC” denote the triazine-based dendron and polycarbosilane, respectively. The initial number signifies the reaction time, while the subsequent number indicates the pyrolysis temperature. Following pyrolysis, the SiOCN samples were subjected to milling in an agate mortar and sieved to attain particle sizes below 60 μm.
Characterization methods
The comprehensive characterization of TRIAZ-3 involved liquid-state nuclear magnetic resonance (NMR) analysis using a Bruker (USA) Advance Neo instrument in acetone-D6. This encompassed one-dimensional 1H (500 MHz) and 13C NMR (125 MHz) and 13C NMR Distortionless Enhancement by Polarization Transfer 135° (DEPT-135) analyses. We have employed the conventional notation where dd means doublet of doublet and s-br means broad singlet. J means coupling constant related to cis, trans or geminal position of alkenyl hydrogens (Jcis, Jtrans and Jgem). In addition, also ortho, meta and para are related to coupling constants of aromatic hydrogens (Jortho, Jmeta and Jpara). Additionally, the molecular weight of TRIAZ-3 was ascertained through Mass Spectrometry (MS), with the sample being suitably diluted in acetone, and analyzed using an Agilent (USA) 6520 model-Accurate-Mass LC/MS Q-TOF.SiOCN-based materials underwent thorough characterization employing elemental analysis for quantifying carbon, oxygen, hydrogen and nitrogen content. Leco (USA) instruments were used to perform the measurements, employing CS-200 and TC-500 and RC-412 analyzers for this purpose. Notice that silicon content was calculated by difference. FTIR characterization was executed using the KBr pellet technique for solid samples, while the attenuated total reflectance mode (ATR) was employed for liquid samples. Both analyses were performed using a PerkinElmer (USA) model BX spectrophotometer, with IR spectra being generated from an average of at least 32 scans.
To investigate the polymer-to-ceramic conversion process, thermogravimetric (TG) analysis was conducted to examine the thermal behavior of the synthesized polymeric precursors. This analysis was performed using an SDT Q600 TA instrument (USA). The experimental procedure involved heating the samples from room temperature to 1000 °C while maintaining a constant flow of argon. The heating rate was set at 10 °C min−1.
Furthermore, the materials underwent examination via solid-state NMR technique. The magic-angle spinning (MAS) method was utilized to explore the chemical environments of 29Si and 13C nuclei. The 13C NMR and 29Si spectra were obtained through direct irradiation employing a Bruker (USA) AV-400-WB 4 mm probe head, with a frequency of 100.61 MHz, π/3 pulse length at 65 kHz, and a relaxation time of 20 seconds.
Surface analysis was conducted utilizing X-ray photoelectron spectroscopy (XPS) on a SPECS GmbH (Germany) instrument equipped with an ultra-high vacuum (UHV) system and PHOIBOS 150 9MCD as an energy analyzer. A non-monochromatic Mg served as the energy source (200 W – 12 kV), with a sampling area of 500 × 500 μm2. The obtained curves were calibrated based on peak positions adjusted to 284.6 eV (C 1s), followed by baseline correction through a Shirley background. Gaussian-shaped functions were employed to fit the curves, thus enabling the estimation of surface analysis composition. Data analysis was done employing CASA XPS software.
Raman spectra were acquired using a Renishaw (United Kingdom) InVia spectrophotometer, employing an Ar+ ion laser with an excitation wavelength of 514 nm. Each spectrum was recorded with 10 accumulations and a duration of 10 seconds, with a minimum of 5 measurements conducted for each sample.
Results and discussion
Characterization of TRIAZ-3 and its cross-linking with AHPCS
The initial step in our exploration involves the meticulous synthesis of TRIAZ-3, followed by a thorough investigation of its properties through a combination of advanced analytical techniques. Employing a systematic approach, we used NMR (Section S1 and Fig. S1–S3, ESI†) and MS to unravel the molecular intricacies of TRIAZ-3, leading to the acquisition of the subsequent data:In the realm of NMR, the potent 1H NMR spectroscopy, furnished proton chemical shifts (δH (ppm)) of significance assigned in Fig. S1 (ESI†) as follows: H-1 (5.74 (dd, Jtrans = 17.6 Hz and Jgem = 1.0 Hz, 3H)), H-2 (5.16 (dd, Jcis = 10.9 Hz and Jgem = 1.0 Hz, 3H)), H-3 (6.76 (dd, Jtrans = 17.6 and Jgem = 10.9 Hz, 3H)), H-4, H-7 (7.44 (dd, Jortho = 6.6 Hz and Jmeta = 2.0 Hz, 6H)), H-5, H-6 (7.83 (dd, Jortho = 6.6 Hz and Jmeta = 2.0 Hz, 6H)), and H-8 (8.65 (s-br, 3H)). Furthermore, the intricate structural landscape was further illuminated through 13C NMR analyses revealing carbon resonances (δC (ppm)) assigned in Fig. S2 (ESI†) as follows: C-1 (111.3), C-2 (136.6), C-3 (131.9), C-4, C-8 (126.3), C-5, C-7 (120.3), C-6 (139.7), and C-9 (164.6). Finally, 13C NMR DEPT-135 spectra plays a crucial role in identifying and characterizing the resonance signals showcased in Fig. S3 (ESI†). It provides a clear distinction between primary, secondary, and tertiary carbons within the TRIAZ-3 dendron. Notice, CH3 and CH-related resonances are presented in an upright orientation, while CH2 resonances appear inverted and quaternary carbons do not appear. The assignation of carbon resonances (δC (ppm)) is the following: C-1 (113.3, CH2), C-5, C-7 (120.3, CH), C-4, C-8 (126.3, CH) and C-2 (136.6, CH).
In the realm of mass spectrometry, our calculated molecular mass for C27H24N6 was established at 432 g mol−1. This theoretical framework was fortified through empirical observations, culminating in measured values of 433.21 [M + H]+ and 455.20 [M + Na]+. This comprehensive characterization endeavour not only provides a profound understanding of TRIAZ-3's intricate structure but also sets the stage for unraveling its subsequent interactions, applications, and transformative potentials.
Having successfully synthesized TRIAZ-3, our investigation now shifts towards a fundamental exploration: understanding the intricate mechanism underpinning the cross-linking process between TRIAZ-3 and AHPCS, employing FTIR-ATR technique. In Fig. 1a, we present a comprehensive overview, showcasing the FTIR-ATR spectra of both precursor materials, AHPCS and TRIAZ-3, along with the profiles of the resultant preceramic polymers obtained at distinct reaction times, denoted as TZPC20 and TZPC48. In the AHPCS spectra, distinct high-intensity bands emerge prominently: one centered at 934 cm−1, indicative of the bending vibration of Si–H groups, and another at 2120 cm−1, corresponding to the stretching vibration of Si–H bonds.24 The allyl groups within the spectrum exhibit their own distinct footprints, with absorption bands at 1631 cm−1 and 3076 cm−1, attributed to the CC and C–H stretching vibrations, respectively. Additionally, the landscape of Si–C stretching vibrations manifests itself in absorption bands around 750 cm−1 and 832 cm−1. Delving deeper into the backbone of the polymer, the Si–CH2–Si groups bring forth two bands at 1036 cm−1 and 1354 cm−1, aligning with stretching and deformation vibrations, respectively.25 A telltale sign of Si-CH3 groups is discernible at 1253 cm−1.26–28
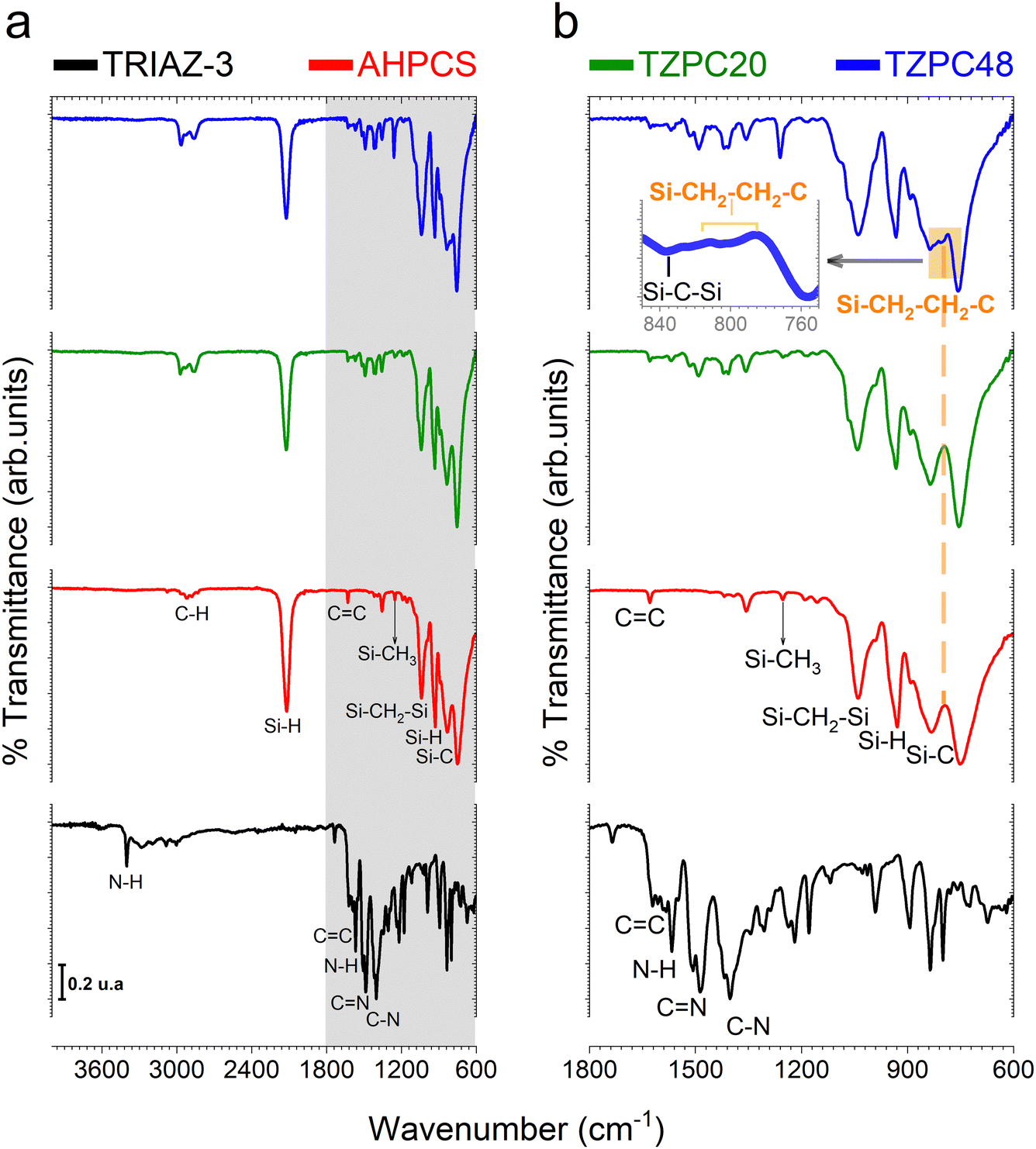 | ||
| Fig. 1 Investigating the impact of reaction time on cross-linking mechanism between TRIAZ-3 and AHPCS. (a) Presenting FTIR-ATR spectra that portray the distinct profiles of the ongoing as-synthesized preceramic polymers (TZPC20 and TZPC48), AHPCS, and the TRIAZ-3 dendron. (b) Providing a magnified view into the IR region spanning from 1800 to 600 cm−1 (region delineated in grey on panel a), revealing intricate molecular interactions and evolving vibrational resonances as a function of reaction time. The orange-shaded box highlights the establishment of Si–CH2–CH2–C bonds (via hydrosilylation reaction) on the TZPC48 preceramic polymers. | ||
Shifting our gaze to the TRIAZ-3 spectrum, its complexity is undeniable. Yet, it unveils distinct vibrational signatures: the tri-substituted triazine rings exhibit a breathing mode resonance at 835 cm−1. Notably, the vibrations associated with C–N bonds (at 1218 cm−1, 1303 cm−1, and 1401 cm−1) and CN bonds (at 1487 cm−1 and 1624 cm−1) emerge, further enriching our understanding of this intricate structure.29,30 Delving deeper, the aromatic C–H and in-plane bending modes of the amine-styrene rings reveal their presence in the 1000–1300 cm−1 range, with an overarching breathing mode attributed to the 895 cm−1 band. A dual representation of N–H bond vibrations materializes through peaks centered at 1565 cm−1 and 3405 cm−1, representing bending and stretching vibrations respectively. Meanwhile, the stretching vibration of conjugated CC vinyl groups finds resonance around 1621 cm−1. In addition, we also found the stretching vibrations of CC aryl groups that usually appears as doublets depending on symmetric factors and can be assigned to 1593 and 1582 cm−1 peaks.31 Finally, C–H stretching in –CHCH2 vibrations appear at 3085 cm−1.
In Fig. 1b, we gain enhanced visibility into the spectra of the reaction products (TZPC20 and TZPC48) within the range of 1800–600 cm−1. Additionally, an initial theoretical structure of the synthesized polymeric precursors is proposed in Scheme 1 of the interaction between AHPCS and TRIAZ-3. The FTIR-ATR spectra, as showcased in Fig. 1b, reveal noteworthy alterations. Specifically, a reduction in intensity is observed at approximately 1621 and 1631 cm−1, corresponding to the CC bonds of vinyl and allyl groups, respectively, which are inherent to both AHPCS and TRIAZ-3 precursors. Concurrently, a diminishment of the Si–H band at 2120 cm−1, exclusive to AHPCS, signifies the interaction between Si–H and the –CHCH2 and –CH2–CHCH2 moieties within the precursors. This reactivity leads to an elevation in the population of –CH2– groups and the emergence of some –CH3 groups. This evolution is confirmed by the heightened intensity of several bands, encompassing the band at 1036 cm−1 indicative of Si–CH2–Si bonds (via hydrosilylation reaction), and the band at 1257 cm−1 linked to Si–CH3 groups. Strikingly, within both TZPC20 and TZPC48 spectra, the presence of C–N and CN stretching vibration bands persists, implying the retention of triazine rings post-reaction. Notably, the emergence of a novel band closed to 790 cm−1 within TZPC48 signifies the establishment of Si–CH2–CH2–C bonds (via hydrosilylation reaction), see the orange-shaded box of the Fig. 1b. The broadening of the 832 cm−1 band associated with Si–C bonds suggests the nascent formation of a three-dimensional architecture within the as-synthesized polymeric precursors.
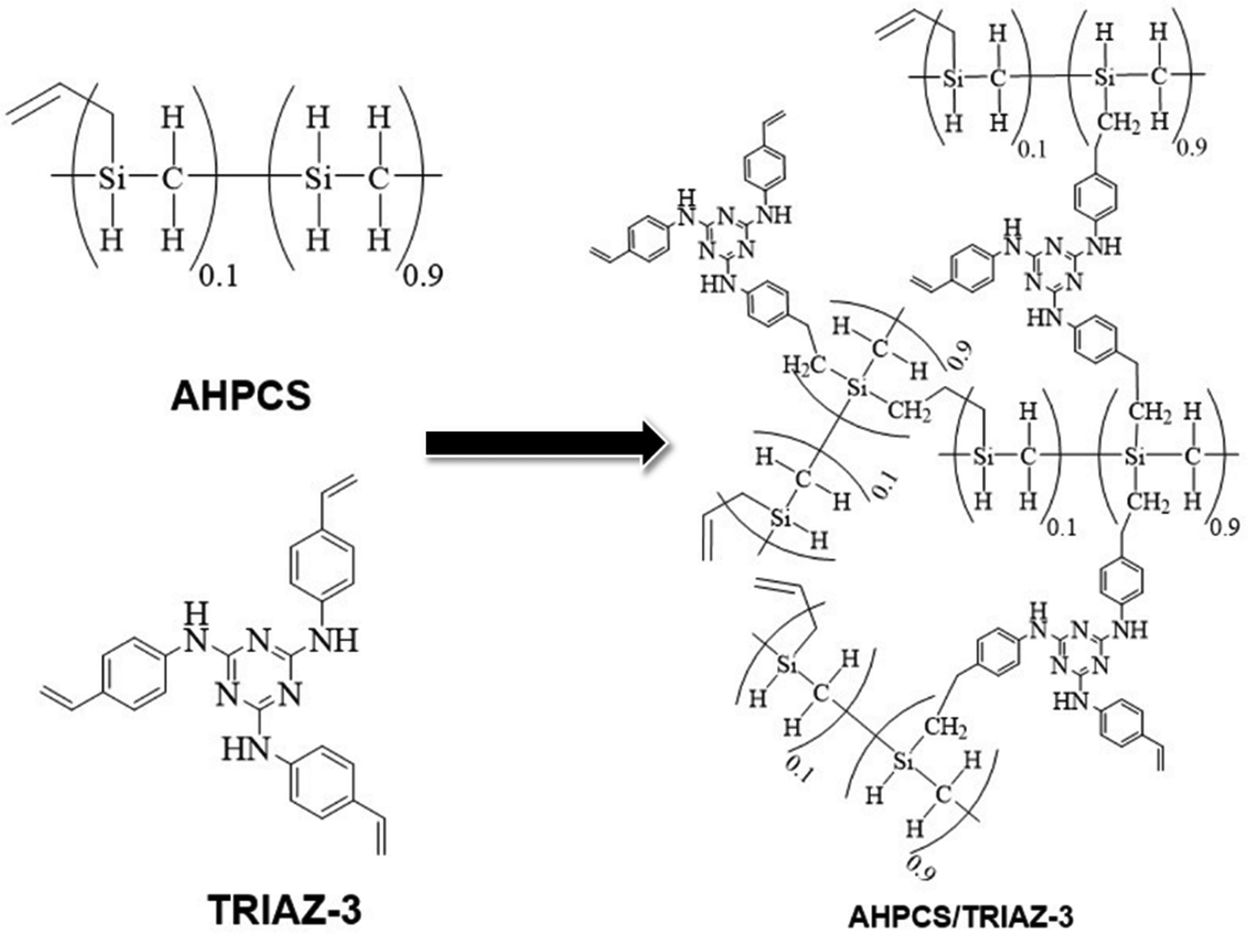 | ||
| Scheme 1 Theoretical configuration of the reaction product following hydrosilylation reactions at room temperature between vinyl functional groups of TRIAZ-3 and Si–H of AHPCS, and self-crosslinking of AHPCS. | ||
In summation, the intricate interplay between the TRIAZ-3 dendron and AHPCS unfolds via hydrosilylation reactions (as depicted in Scheme 1). In this mechanistic endeavor, Si–H bonds within AHPCS engage in a responsive interaction with the vinyl groups (–CHCH2) of TRIAZ-3, giving rise to a dynamic network of covalent linkages and molecular rearrangements. This phenomenon captures the cross-linking process between both starting material (TRIAZ-3 and AHPCS), underscored by the evolving vibrational signatures observed through FTIR-ATR analysis (Fig. 1).
TG and DTG analysis for polymer-to-ceramic conversion
With the intricate interplay between the TRIAZ-3 dendron and AHPCS having been established, the subsequent step embarked upon was the evaluation of the impact of pyrolysis on the resulting reaction products. For this purpose, TG analysis was employed (Fig. 2). In a succinct overview, the initial stages of AHPCS crosslinking begin with a weight loss occurring at 180 °C (marked as 1 in Fig. 2), attributed to the removal of H2 and the release of minor molecular species, stemming from thermal crosslinking reactions involving dehydrocoupling and hydrosilylation. Subsequently, at 450 °C, a further weight loss ensues, correlating to the disintegration of the organic side groups (signalled as 2 in Fig. 2). This process entails the emission of gaseous byproducts such as CH4 and H2, among others.32 On the other hand, the TRIAZ-3 dendron demonstrates a distinct decomposition profile, characterized by a single-step weight loss peak occurring sharply at 470 °C (marked as 4 in Fig. 2), alongside an additional decomposition close to 120 °C associated with traces of solvent (marked as process 3 in Fig. 2).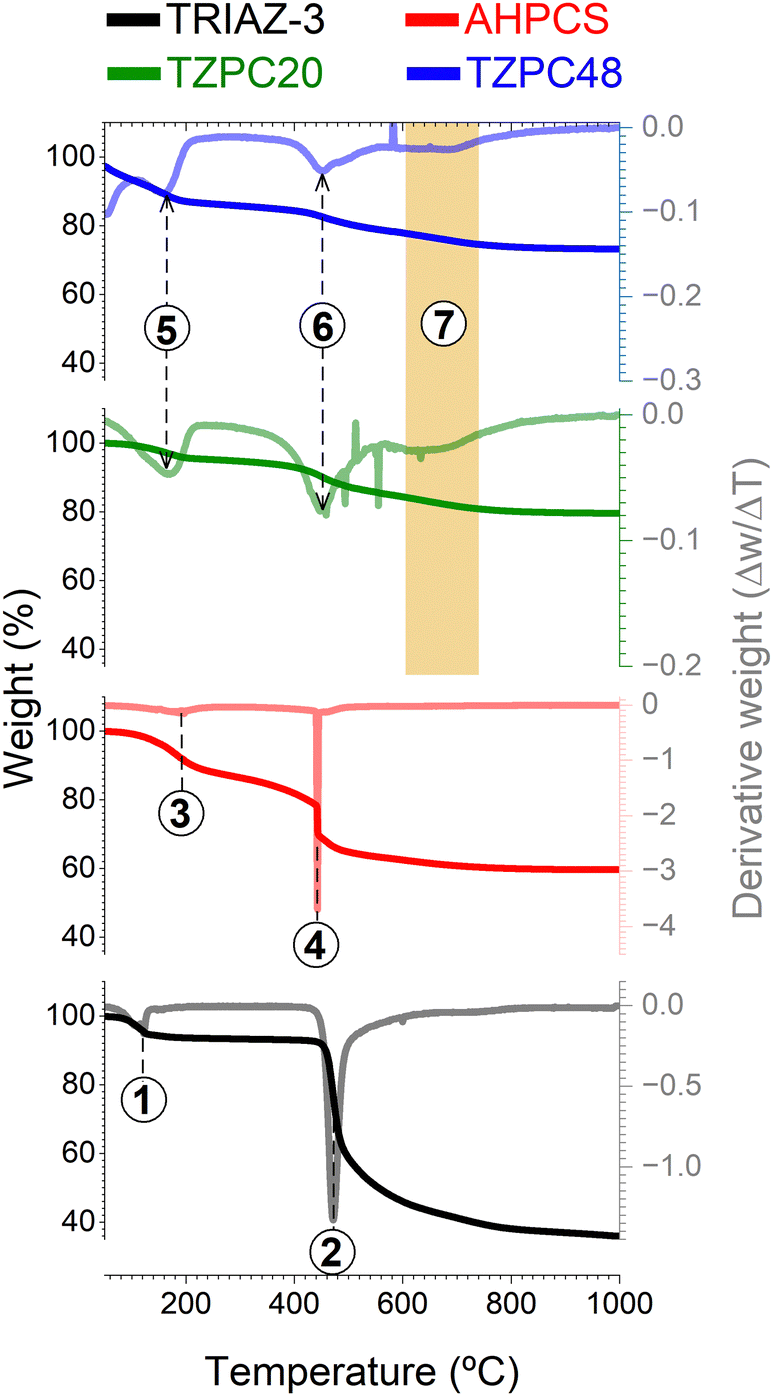 | ||
| Fig. 2 TG and DTG analysis for polymer-to-ceramic conversion. TG curves illustrating the thermal behavior of the precursors AHPCS and TRIAZ-3, as well as the as-synthesized materials TZPC20 and TZPC48. Additionally, DTG have been incorporated into each panel, offering insights into the decomposition profiles of these materials and highlighting distinct thermal degradation stages. Notably, the region delineated in orange corresponds to a novel decomposition peak, attributed to the presence of the crosslinked AHPCS/TRIAZ-3 structure. | ||
Delving into the synthesized materials, TZPC20 and TZPC48 manifest an elevated ceramic yield, achieving values of 75 and 79% respectively, as compared with the 60% achieved by the AHPCS. TZPC20 and TZPC48 exhibit ceramic yields of around 75 and 79% compared with the 60% achieved by the AHPCS. This enhancement is attributed to the augmented crosslinked structure that evolves during the reaction conducted at room temperature, a trend that is in agreement with the findings from FTIR-ATR spectroscopic analyses (Fig. 1 and Scheme 1). Within the lower temperature range of the thermogram (below 200 °C), AHPCS displays a more pronounced weight loss compared to TZPC20 and TZPC48. This divergence can be attributed to the reduced availability of Si–H groups for self-crosslinking within the as-synthesized materials, owing to the consumption of these bonds during hydrosilylation reactions with TRIAZ-3 dendron. Notably, this reaction transpires at a lower temperature, approximately 165 °C (signalled as 5 in Fig. 2). Subsequent weight loss peaks, observed at 450 °C for AHPCS, shift to 454 °C for TZPC20 and 456 °C for TZPC48, respectively. This weight loss phase is ascribed to the vaporization of molecular oligomers engendered by the crosslinking interactions involving Si–H within the allyl and vinyl groups of AHPCS and TRIAZ-3, respectively. The derivative thermograms (DTG) (Fig. 2) for TZPC20 and TZPC48 samples also reveal an additional shoulder at around 500 °C, directly attributed to the decomposition of the TRIAZ-3 dendron (signalled as 6 in Fig. 2). Significantly, only within the TZPC20 and TZPC48 samples is a novel decomposition peak observed, absent in both AHPCS and TRIAZ-3. This peak emerges between 600 °C and 690 °C, potentially linked to the crosslinked AHPCS/TRIAZ-3 structure (process marked as 7 in the region delineated in orange of the Fig. 2). Motivated by this observation, these materials were subjected to pyrolysis at higher temperatures (700, 800, and 900 °C), a stage at which the aforementioned reactions are presumed to have concluded.
Effect of pyrolysis temperature on elemental composition of ceramified materials
Once the temperatures for pyrolysis were determined, the subsequent investigation delved into assessing the impact of these temperatures on the composition of the resulting materials. The elemental analysis of the ceramic materials pyrolyzed between 700–900 °C, is detailed in Table 1, along with the corresponding empirical formulas derived from this analysis. As anticipated, the highest carbon (C) content is observed in samples obtained through extended reaction times and higher pyrolysis temperatures, reaching a maximum of 35% for specimens pyrolyzed at 900 °C. Conversely, the concentration of oxygen (O) experienced a marginal decrease as both synthesis parameters were elevated. Given that neither of the precursor materials inherently contains oxygen within their structures, it is postulated that its presence could be attributed to factors such as the solvent utilized during polymeric precursor preparation, the crosslinking and pyrolysis processes, wherein more extensively condensed structures exhibit reduced oxygen incorporation, or even potential contamination during handling. The presence of oxygen in these samples aligns with observations reported in various studies and has been ascribed to heightened reactivity with moisture subsequent to pyrolysis within the 700–900 °C range.33 Similar results have been demonstrated by Schitco et al.,34 and Kaur et al.,26 wherein oxygen concentrations of up to 10% (wt) were found for SMP-10® polycarbosilane pyrolyzed under argon atmosphere.26,34| Sample | Weight% | Empirical formula | ||||
|---|---|---|---|---|---|---|
| Si | O | C | N | H | ||
| TZPC20-700 | 53.23 | 12.68 ± 0.22 | 31.80 ± 0.22 | 1.32 ± 0.06 | 0.91 ± 0.05 | SiO0.41C1.39N0.05H0.47 |
| TZPC48-700 | 53.71 | 10.75 ± 0.58 | 33.53 ± 0.13 | 1.65 ± 0.12 | 0.36 ± 0.05 | SiO0.35C1.45N0.06H0.19 |
| TZPC20-800 | 54.89 | 10.19 ± 0.26 | 33.38 ± 0.50 | 1.53 ± 0.09 | 0.01 ± 0.01 | SiO0.32C1.41N0.05H0.007 |
| TZPC48-800 | 52.66 | 11.27 ± 0.40 | 34.40 ± 0.50 | 1.66 ± 0.07 | 0.01 ± 0.01 | SiO0.37C1.52N0.06H0.005 |
| TZPC20-900 | 54.18 | 9.19 ± 0.35 | 35.00 ± 0.19 | 1.62 ± 0.07 | — | SiO0.29C1.50N0.06 |
| TZPC48-900 | 53.22 | 10.92 ± 0.39 | 34.10 ± 0.10 | 1.46 ± 0.16 | — | SiO0.35C1.49N0.05 |
Exploring evolution of structure through comprehensive FTIR analysis
The intricate interplay between the TRIAZ-3 dendron and AHPCS, as deciphered via hydrosilylation reactions, has set the stage for the exploration of the impact of pyrolysis temperatures on the composition of the resulting ceramic materials. In pursuit of this understanding, we turn our attention to the structural characterization of the synthesized products. In Fig. 3a, the FTIR spectra of the materials subjected to pyrolysis at 700, 800, and 900 °C are presented. A distinguishing feature across these spectra resides in the prominence of two broad and high-intensity bands around 1000 cm−1 and 790 cm−1. The former is attributed to Si–CH2–Si bonds, while the latter corresponds to Si–C bonds within the SiC4 units.35–37 These bands exhibit substantial overlap, with their broad widths consistent with similar material systems.35,38–40 Examination of the spectra in Fig. 3a reveals that the band around 1000 cm−1 experiences a decrease in intensity and evolves into a wide shoulder when subjected to treatment at 900 °C. This shift confirms the removal of hydrogen from Si–CH2–Si bonds, as corroborated by Table 1. With most of the hydrogen extracted at 900 °C, the shoulder shifts to 990 cm−1, corresponding to Si–C bonds that exist in a non-organized or amorphous structure, indicative of SiC4 units. Conversely, the band centered at 790 cm−1 remains consistent in both position and width across all pyrolysis temperatures. This persistence underscores its association with a three-dimensional SiC4 network characteristic of the precursor of β-SiC.41,42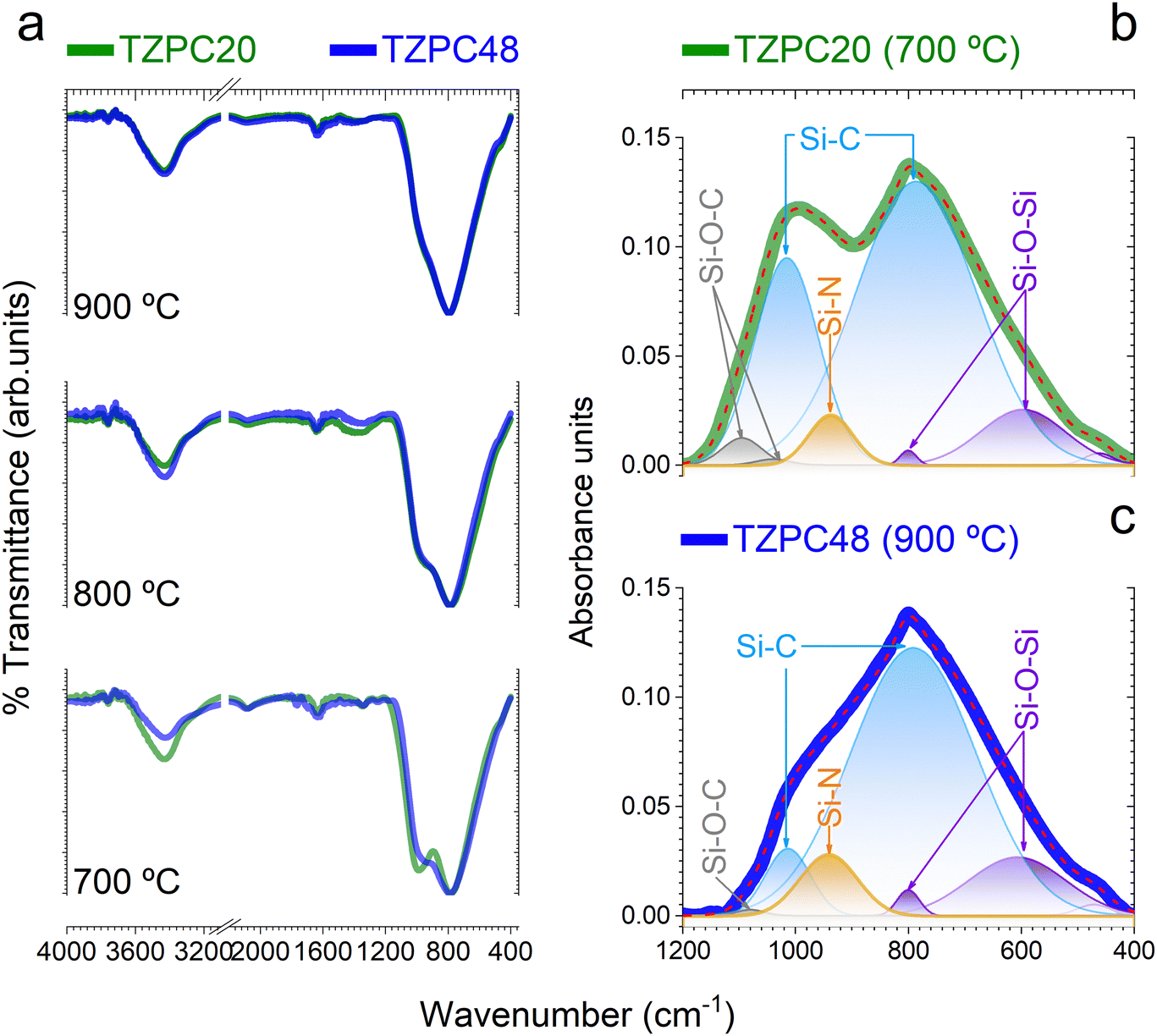 | ||
| Fig. 3 Probing vibrational modes via FTIR spectroscopy. (a) FTIR spectra of distinct samples, delineating their evolution in response to varying pyrolysis times and reaction durations. In green color is represented the TZPC20 sample, while in blue color is plotted the TZPC48-900 sample. Deconvoluted FTIR spectra of (b) TZPC20-700 and (c) TZPC48-900 samples representing the extreme scenarios explored offering insights into the intricate vibrational modes associated with Si–O–C bonds (gray), Si–C-bonds (blue), Si–O–Si bonds (purple), and Si–N bonds (orange). | ||
Each spectrum also features a faint band at 800 cm−1, which is overlapped with the 790 cm−1 peak and attributed to the formation of Si–O–Si bonds. Aligned with this observation, a subtle new shoulder appears near 460 cm−1, attributed to the bending vibration of O–Si–O bonds, consistent with the oxygen content obtained from elemental analysis (Table 1).38,43 The presence of oxygen, as discussed earlier (Table 1), likely arises from moisture adsorption and reaction, leading to the generation of Si–OH groups.33,44 Furthermore, spectra of materials pyrolyzed at 700 °C exhibit two low-intensity bands around 1260 and 1350 cm−1, linked to Si–CH2–Si and Si–CH3 bonds, respectively.45,46 These bands diminish as pyrolysis temperature exceeds 800 °C, a consequence of hydrogen removal due to the ceramicization of AHPCS-based materials.47
For an in-depth analysis of these FTIR spectra, Gaussian bands were employed through a deconvolution process. The spectra of AHPCS pyrolyzed at various temperatures were initially deconvoluted, and the resultant Gaussian bands were used as a preliminary approximation for the TZPC20 and TZPC48 samples. Additional bands were included if required to achieve optimal agreement between experimental and deconvoluted spectra. An illustrative example of this process can be found in Sections S2 and Fig. S4 (ESI†). In Fig. 3b and c, two instances of deconvoluted FTIR curves are provided, corresponding to the materials labelled as TZPC20-700 and TZPC48-900 representing the lower and upper extremes in terms of reaction times and pyrolysis temperatures. The positions, Full Width at Half Maximum (FWHM), and areas of the different bands are documented in Section S2 and Table S1 (ESI†). From this comprehensive data, it becomes evident that AHPCS exhibits two principal bands at 790 and 990 cm−1, along with a medium band centered at 605 cm−1. Additionally, small bands around 920, 1045, and 1070 cm−1 are noted, accompanied by two even smaller bands at 470 and 800 cm−1. The latter two bands slightly intensify in the sample pyrolyzed at 900 °C, associated with Si–O bonds (bending deformation δ(O–Si–O) at 475 cm−1 and Si–O–Si stretching in SiO4 units at 800 cm−1). The 920 cm−1 band is attributed to Si–OH bonds,48,49 a consequence of oxide layer formation against moisture. Bands at 1045 and 1070 cm−1, indicative of Si–O bonds, align with silica materials;50 however, their lower frequencies suggest the presence of Si–O–C and/or Si–O–N bonds.51,52 While these bands are prevalent across the synthesized materials, a new band at approximately 940 cm−1 emerges and increases in intensity due to the presence of TRIAZ-3. Literature suggests this band can be unambiguously assigned to the stretching of Si–N bonds,36,38,53,54 indicating the reaction between Si–H groups of AHPCS and NH–C sp2 groups of TRIAZ-3—signifying dendritic molecule rupture during thermal treatment.
In Fig. 3b and c, bands attributed to SiOC and SiON species are prominently displayed. For the TZPC20-700 sample, two bands at 1040 and 1095 cm−1 indicate the formation of oxygen-rich structural units, whereas TZPC48-900 displays a single and less intense band at a similar frequency, signifying lower oxygen content. The data in Section S2 and Table S1 (ESI†) highlight a decline in the intensity of bands at 1045 and 1070 cm−1 with increasing pyrolysis temperature, while those at 605 and 940 cm−1 intensify. This trend indicates the continuous formation of Si–C and Si–N bonds at the expense of Si–CH2–Si and Si–O bonds. Additionally, the 990 cm−1 band persists and is attributed to the presence of Si–C bonds, possibly in a linear structure at the studied pyrolysis temperatures. The Si–OH vibration band exhibits a slight increase with higher pyrolysis temperatures, indicating enhanced reaction with moisture.55
NMR insight into structural transformations: 29Si-MAS NMR and 13C-MAS NMR analysis
After conducting an extensive FTIR analysis on the ceramic products, the investigation progresses to a verification of the gathered data through NMR spectroscopy. Both 29Si-MAS NMR and 13C-MAS NMR techniques have been employed to provide a more comprehensive understanding of the evolving molecular arrangements. In Fig. 4a, the 29Si-MAS NMR spectra for all pyrolyzed samples are presented, revealing a characteristic broad band that signifies the amorphous nature of the resulting structures. Notably, the asymmetric shapes of these curves, especially apparent in the TZPC20-treated samples, strongly suggest a mixture of various silicon tetrahedral units. To elucidate these units’ presence, the spectra underwent fitting through multiple Gaussian bands, employing the approach proposed by Schitco et al.34 where the chemical shift values related to Si (δSi) were attributed to specific SiX4 groups (X = C, N, O). Furthermore, the main band at δSi = −8 ppm, assigned by Schitco et al.34 to 29SiC4 tetrahedron, served as the starting point for the deconvolution. Subsequent fitting of the asymmetrical experimental 29Si-MAS NMR profiles with additional Gaussian curves led to the addition of new bands at 6 ppm and −27 ppm. These newly introduced bands are attributed to SiCmOn and SiCmNp groups, where m + n + p = 4,34,56 respectively. Notably, these new bands correlate both with the chemical composition analyzed for the materials (Table 1) and with the presence of Si–C, Si–O, and Si–N bonds observed via FTIR spectroscopy (Fig. 3). Meanwhile, the presence of SiCmNp groups indicates the breaking of some NH–C sp2 of TRIAZ-3, reacting with the Si–H groups of AHPCS to form SiC3N groups, as it was elucidated by FTIR technique (Fig. 3).57,58 Moreover, the absence of a band at δSi = −48 ppm indicates the expected lack of Si3N4 groups, given the pyrolysis temperature employed for the production of the pyrolyzed materials.34,54 | ||
| Fig. 4 Structural characterization via NMR analysis. (a) 29Si-MAS NMR spectra of the various as-synthesized materials and (b). 13C-MAS NMR spectra of the different samples. The as-synthesized materials prepared for 20 hours are represented in green, while those obtained for 48 hours are depicted in blue. | ||
The evolution of Gaussian band areas from the deconvoluted spectra is presented in Table 2. These data suggest a higher concentration of SiC4 units with elevated pyrolysis temperatures. Conversely, SiC3O and SiC3N groups decrease with rising pyrolysis temperatures (due to the formation of more SiC4 units). Even with this decrease in both structural units, the data from Table 2 indicate that TZPC48 samples contain more SiC3N units at the expense of SiC3O units, consistent with the oxygen content observed in the chemical analysis (Table 1). Based on these simulated data, the samples primarily consist of SiC4 groups (53–77%), SiC3O groups (8–19%), and SiC3N groups (10–28%). These findings strongly suggest a reaction between AHPCS and TRIAZ-3 molecules, with Si–H bonds of AHPCS reacting with –CHCH2 and NH–C sp2 groups of TRIAZ-3, leading to the formation of Si–C and Si–N bonds, confirming the FTIR spectra results. Moreover, the presence of SiC3N and SiC3O groups, as well as SiCmOn and SiCmNp units, further highlights the intricate interplay between these compounds and the structural evolution observed.
| Si species | δ si (ppm) | TZPC20 | TZPC48 | ||||
|---|---|---|---|---|---|---|---|
| Calculated percentage (%) of different Si species | |||||||
| 700 | 800 | 900 | 700 | 800 | 900 | ||
| SiC3O | 6 ± 1.0 | 19.8 | 17.0 | 12.8 | 18.5 | 14.9 | 8.4 |
| SiC4 | −7 ± 0.8 | 57.9 | 64.3 | 76.9 | 53.4 | 59.3 | 77.0 |
| SiC3N | −27.2 ± 2.0 | 22.3 | 18.7 | 10.3 | 28.1 | 25.8 | 14.6 |
Turning to the 13C-MAS NMR analysis, Fig. 4b showcases the obtained curves. In general, these spectra exhibit two prominent bands centered around 135 ppm and 10 ppm, respectively. The former corresponds to the amorphous Cfree phase or sp2 hybridized carbon, while the latter is attributed to C sites bonded primarily to Si-based and/or N-based networks, forming diverse SiCxOy and SiCxNy structural units via sp3 hybridization.34,59 A closer inspection of the 13C MAS NMR spectra reveals a shift to a more positive δsi in the band assigned to C sp2 environments for materials prepared with the shortest reaction time. This shift is associated with C environments with one or more nitrogen nearest neighbors.60 Notably, small and narrow peaks can be observed over the main bands, particularly pronounced in the TZPC20-700 sample. These peaks at 150 ppm, 120 ppm, 27 ppm, and 19 ppm (indicated by an asterisk in Fig. 4b) correspond to nitrogen-containing three-ring aromatic groups with amino terminations61,62 aliphatic carbon segments,63 and Si–CH2–Si units,64 respectively. While these spectra have not been subjected to deconvolution due to the challenges outlined by Schitco et al.,34 it is evident that these aforementioned small peaks tend to decrease or vanish with increasing pyrolysis temperature. Moreover, the presence of a band at 135 ppm, showing an upfield asymmetry similar to that found in nitrogen-doped reduced graphene oxide, confirms the presence of CN bonds in nitrogen-containing functionalities.65 This finding further substantiates the existence of triazine aromatic rings within these materials.
XPS analysis: revealing surface dynamics of pyrolyzed materials
In this section, we delve into the exploration of the surface properties of the pyrolyzed materials through XPS analysis. This technique offers valuable insights into the electronic core levels (C 1s, O 1s, N 1s, Si 2s, and Si 2p) as well as the Auger bands of the various elements within the pyrolyzed materials (refer to Section S3 and Fig. S5, ESI†). Presented in Table 3 is the surface analysis composition, determined by atomic percentage derived from the survey spectra. While there are subtle differences compared to the chemical composition in Table 1, these discrepancies are attributed to a slight surface oxidation. This oxidation phenomenon might stem from the presence of adventitious carbon or the materials’ heightened reactivity with moisture, resulting in the formation of Si–OH groups subsequent to pyrolysis at 700–900 °C—a phenomenon discussed extensively in prior studies.33,44 It is important to note that the chemical composition calculations in Table 1 include H content, an aspect omitted from XPS calculations. The most important information extracted from these data is the confirmation of the presence of N in all samples.| Samples | wt% C | wt% Si | wt% O | wt% N |
|---|---|---|---|---|
| TZPC20-700 | 31.6 | 49.2 | 18.0 | 1.2 |
| TZPC48-700 | 26.8 | 46.4 | 25.4 | 1.4 |
| TZPC20-800 | 29.8 | 49.8 | 19.2 | 1.2 |
| TZPC48-800 | 25.8 | 46.6 | 26.4 | 1.2 |
| TZPC20-900 | 24.1 | 47.0 | 27.5 | 1.4 |
| TZPC48-900 | 26.5 | 50.7 | 21.4 | 1.4 |
Given these data, it is reasonable to assume that the composition remains relatively uniform between the bulk and the surface, suggesting that the bonding types identified within these analyses are representative of the entire material. Deconvoluted Gaussian bands corresponding to XPS spectra of the as-synthesized dendron TRIAZ-3 and the AHPCS treated at 900 °C are displayed in Section S3 and Fig. S6, S7 (ESI†), respectively. The XPS spectra of both reactants serve as a foundation for the deconvolution of the TZPC20 and TZPC48 materials. In the XPS N 1s spectrum (Fig. S6, ESI†), two contributions at 398.31 eV and 401.05 eV are observed for the TRIAZ-3 dendron, linked to NC bonds of triazine aromatic rings and –NH– bonding in C sp2 with alkenyl and triazine motifs (NH–C sp2), respectively.66–68 These contributions demonstrate similar intensities, aligned with the dendron's molecular structure depicted in Scheme 1. However, in the pyrolyzed materials shown in Fig. 5a, which also retain these two contributions, the intensity of the 401.05 eV peak is notably lower than in TRIAZ-3 spectrum. This result implies the disappearance of a portion of the NH–C sp2 bonds during the pyrolysis treatment. Employing the CASA® software, we analysed these contributions using the same peaks (position and FWHM) in both TRIAZ-3 and pyrolyzed samples. This analysis leads to the introduction of a new peak at 399.8 eV to accurately fit the spectra. This additional peak can be attributed to N–Si sp3 bonds, as supported by FTIR (Fig. 3) and 29Si MAS NMR (Fig. 4a) spectra. Table 4 captures the areas of these three peaks for TRIAZ-3 and the pyrolyzed samples, demonstrating an increase in N–Si sp3 bonds (399.8 eV) with rising treatment temperatures, consistent with findings from Nguyen et al.54 This rise coincides with a decrease in NH–C sp2 bonds at 401.05 eV, while NC within triazine at 398.31 eV bonds remain intact in comparison with the starting TRIAZ-3. This suggests that NH–C sp2 bonds within the TRIAZ-3 dendron react with the Si–H of the AHPCS to form Si–N bonds, preserving NC bonds of the triazine aromatic rings. Notably, the peak area of NH–C sp2 bonds accounts for about 25% of the NC bonds within the triazine aromatic ring, in contrast to the 50–50% contribution observed in TRIAZ-3. This discrepancy suggests that approximately 50% of the N–H bonds undergo fragmentation during thermal treatment, leading to the formation of two distinct fragments capable of reacting with the Si–H of the AHPCS. A conceptual illustration of the potential breakdown of TRIAZ-3 is proposed in the Scheme 2, where N–H may remain attached to either the triazine core or to the aromatic ring of the aniline. The provided scheme depicts the fracture of TRIAZ-3 detected during the pyrolysis treatment. However, it does not capture the complexity of the reactions involved in this process, primarily due to the challenge of illustrating the intricate 3D structure obtained after thermal treatment. The reactions occurring during pyrolysis include bonding rearrangements, crosslinking processes, and the removal of H, allyl and vinyl groups in the final structure.
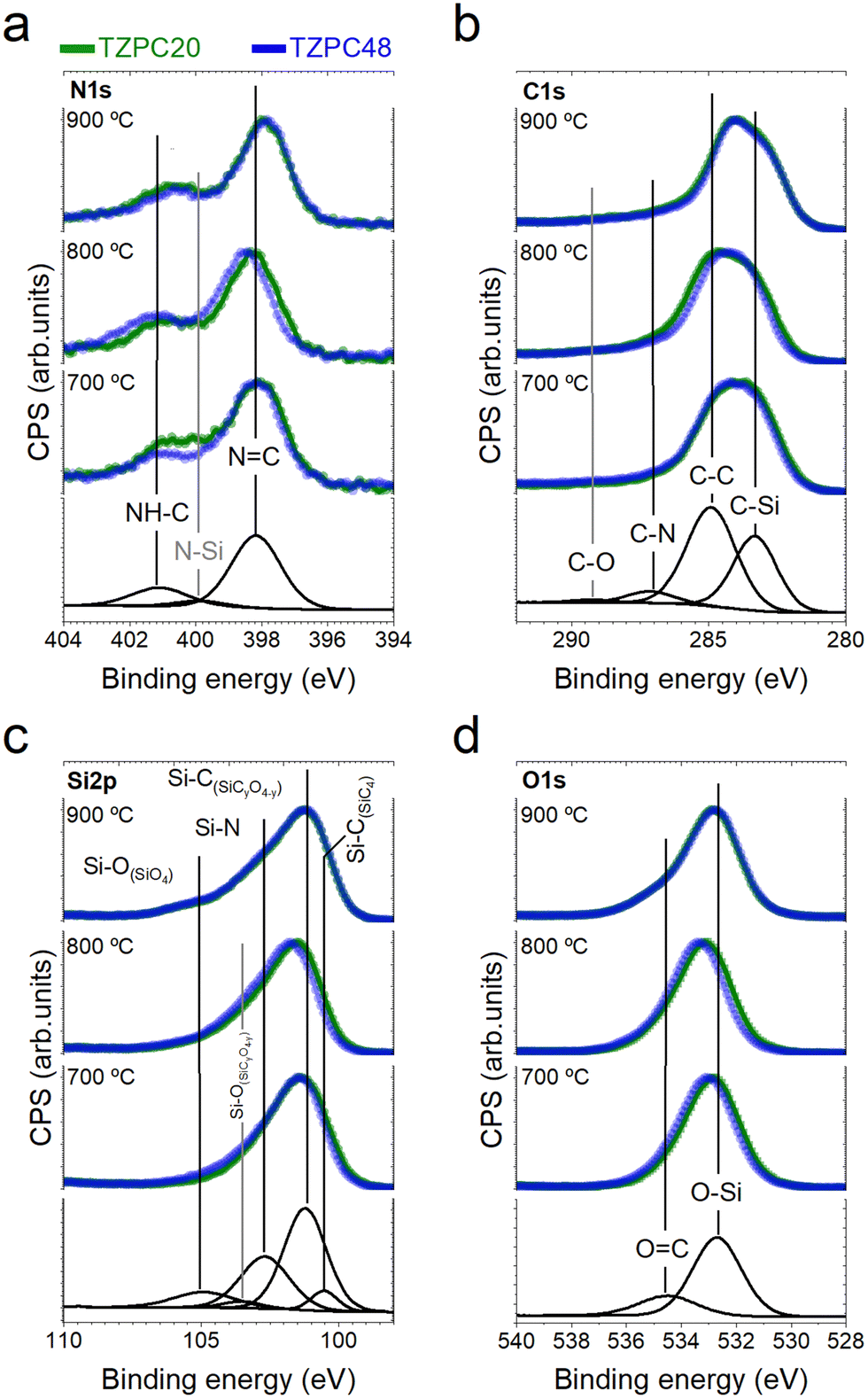 | ||
| Fig. 5 Revealing surface dynamics of pyrolyzed materials by XPS spectra. (a) N 1s XPS, (b) C 1s XPS, (c) Si 2p XPS, (d) O 1s XPS. The as-synthesized materials prepared for 20 hours are distinguished by the color green, while those obtained over a 48-hour duration are depicted in blue. Additionally, each panel includes individual fittings for the different signal components, which are represented by concise black lines positioned at the base of each graph. | ||
| Relative concentration of the N-containing bond unit (%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Bond units | BE (eV) | TRIAZ-3 | TZPC20 | TZPC48 | ||||
| 700 °C | 800 °C | 900 °C | 700 °C | 800 °C | 900 °C | |||
| NC |
398.31 | 49.3 | 70.2 | 69.7 | 70.3 | 76.7 | 70.9 | 71.3 |
| N–Si sp3 | 399.86 | — | 5.3 | 5.7 | 5.5 | 3.0 | 2.4 | 5.2 |
| NH–C sp2 | 401.05 | 50.7 | 24.5 | 24.6 | 24.2 | 20.3 | 26.7 | 23.5 |
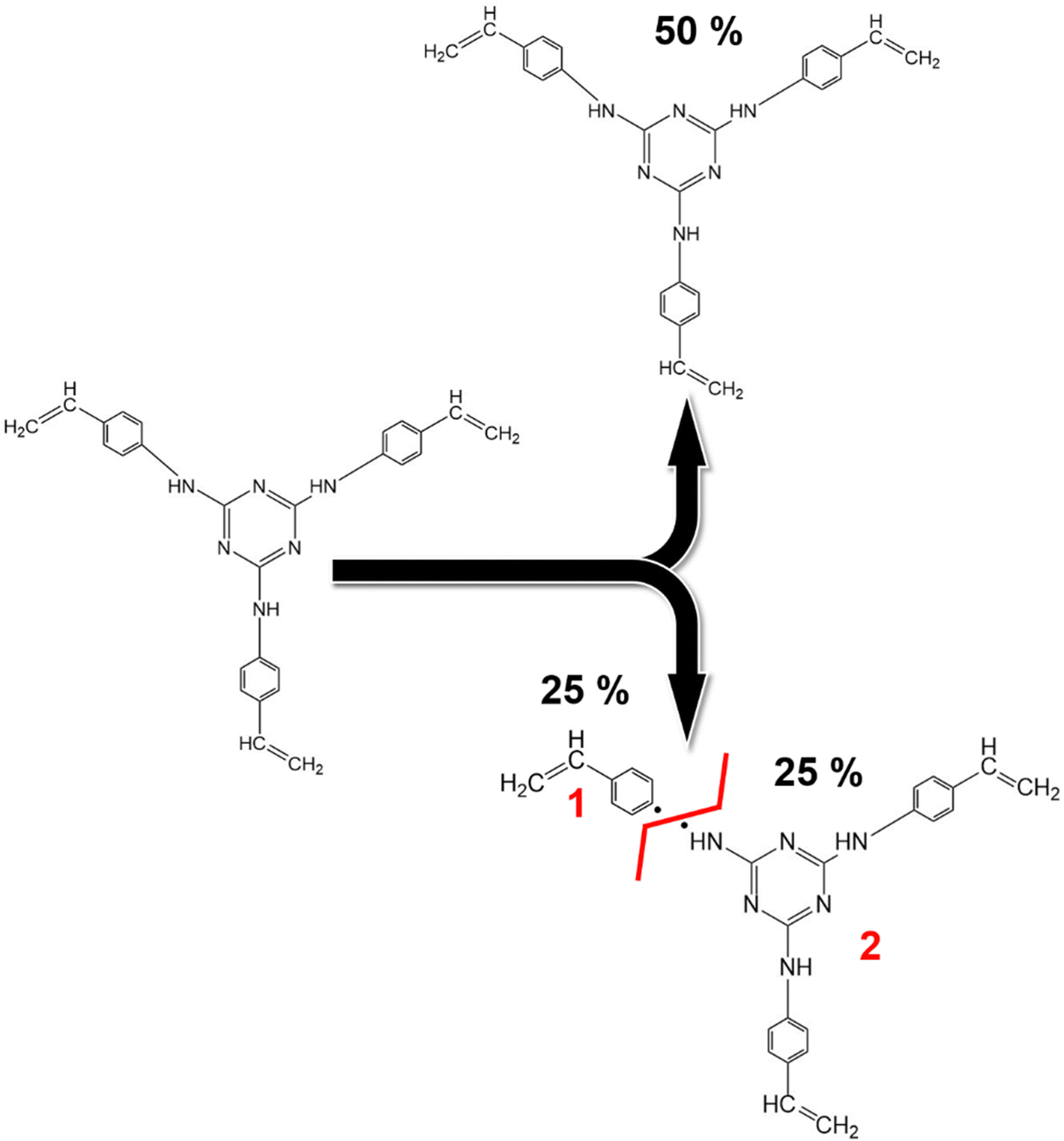 | ||
| Scheme 2 Proposed mechanism for the fragmentation of the dendritic molecule TRIAZ-3. The schematic representation above illustrates the proposed mechanism for the fracture of the dendritic molecule TRIAZ-3, neglecting the complex reactions entailed in the heat treatment of the preceramic material. This fracture primarily occurs through the cleavage of the –NH– bonding, which exists between the vinylaniline and the active center of the molecule. This cleavage results in the emergence of two distinct reactive fragments. The portion where only the aniline ring and vinyl group remain is designated as fragment 1, while the fragment containing the triazine active center is denoted as fragment 2. It is important to note that the XPS spectrum of the TRIAZ-3 dendron without fragmentation is provided in Section S3 and Fig. S6 (ESI†). | ||
Transitioning to the XPS C 1s spectra (Fig. 5b), the high-resolution profiles reveal a dominant and asymmetric peak at 284.9 eV. Deconvolution of this peak yields four distinct contributions at 283.9 eV, 285.1 eV, 286.5 eV, and 288.8 eV, assigned to C–Si sp3, CC sp2, CN, and C–O sp3 bonds, respectively.26,67,69 In contrast, the XPS spectrum of TRIAZ-3 dendron (Section S3 and Fig. S6, ESI†) presents only three peaks at 284.6 eV, 285.9 eV, and 287.8 eV, associated with CC sp2, CC vinyl, and CN bonds, respectively. The absence of the CC vinyl signal in the XPS spectra of pyrolyzed samples suggests that these groups have reacted with Si–H bonds of the AHPCS (undergoing hydrosilylation reaction), leaving behind CC sp2 and CN bonds in the material. This corroborates the N 1s XPS spectra findings mentioned earlier. Table 5 highlights the concentration of various peaks within the C 1s XPS spectra of the treated samples. These data indicate the sustained concentration of CN sp2 bonds within TRIAZ-3 at varying temperatures. Once again, the oxidation phenomenon amplifies with increased pyrolysis temperature, resulting in more C–O bonds in TZPC20-900 and TZPC48-900 samples. Additionally, the decline in C–Si sp3 bonds, possibly attributed to the formation of new Si–N bonds (as evidenced by the N 1s spectra), contributes to the rise in CC sp2 bonds, as evident in Table 5.
| Relative concentration of the C-containing bond unit (%) | |||||
|---|---|---|---|---|---|
| Samples | C–Si sp3 | CC sp2 |
CN sp2 |
CC vinyl |
C–O sp3 |
| 283.9 eV | 285.1 eV | 286.5 eV | 287.7 eV | 288.8 eV | |
| TRIAZ-3 | — | 78.8 | 10.7 | 10.5 | — |
| TZPC20-700 | 53.9 | 32.4 | 11.6 | — | 2.1 |
| TZPC48-700 | 55.7 | 28.4 | 13.1 | — | 2.8 |
| TZPC20-800 | 47.2 | 37.3 | 12.7 | — | 2.8 |
| TZPC48-800 | 49.1 | 34.1 | 12.8 | — | 4.0 |
| TZPC20-900 | 41.1 | 42.2 | 12.7 | — | 4.0 |
| TZPC48-900 | 41.9 | 42.9 | 11.6 | — | 3.6 |
The Si 2p core level is explored in XPS spectra of TZPC20 and TZPC48 pyrolyzed materials at varying temperatures (Fig. 5c). Analysis of the Si 2p spectra reveals an increasing asymmetry towards higher binding energies, indicating evolving bonding interactions between Si and other atoms. This asymmetry amplifies with escalating treatment temperatures, highlighting the ongoing reactions between AHPCS and TRIAZ-3. The spectra of TZPC20 and TZPC48 are further deconvoluted into five bands, building upon the insights gleaned from AHPCS pyrolyzed samples. The assignment of these bands is consistent with 13C-MAS NMR and 29Si-MAS NMR findings (Fig. 4), as well as prior literature reports.26,70Table 6 encapsulates the outcomes of this deconvolution process. The principal peak at 101.60 eV corresponds to Si–C bonds, while peaks at 103.26 eV and 105.20 eV correspond to Si–O bonds of SiC3O and SiO4 structural units, respectively. Notably, in TZPC20 and TZPC48 materials, two novel bands emerge at 100.80 and 102.68 eV, attributed to Si–C bonds in SiO(C)28 and Si–N in SiN(C)71,72 bonds, respectively. These additions are attributed to interactions between AHPCS, TRIAZ-3, and oxygen contamination. Refer to Scheme 3 depicting the chemical reaction between Si–H of the AHPCS and NH–C sp2 bonds of TRIAZ-3. Parallel to Scheme 2, this figure simplistically portrays the reaction mechanism involving the bonding of AHPCS and TRIAZ-3 fragments. However, they overlook the various phenomena involved during pyrolysis treatment, previously explained along the manuscript. Following the rupture of TRIAZ-3 along –NH– linkages, AHPCS engages with both dendritic structure fragments, establishing new Si–C linkages via fragment 1, which subsequently leads to Cfree phases. Moreover, new Si–NH–C linkages (fragment 2) form, with H atoms lost as pyrolysis temperature increases, while new linkages with Si or C atoms emerge during redistribution reactions. Notably, the band representing Si(O)C bonds (103.26 eV) shifts towards higher binding energies with rising temperature pyrolysis, indicating a higher prevalence of O atoms due to the oxidation phenomenon, corroborating the findings in Table 3.
| Relative concentration of the Si-containing bond unit (%) | |||||
|---|---|---|---|---|---|
| Samples | Si–C | SiN(C) | SiC3O | SiO4 | |
| 100.80 eV | 101.60 eV | 102.68 eV | 103.26 eV | 105.20 eV | |
| TZPC20-700 | 13.4 | 58.6 | 3.6 | 22.4 | 2.0 |
| TZPC48-700 | 4.9 | 60.4 | 4.4 | 22.3 | 8.0 |
| TZPC20-800 | 17.9 | 51.1 | 4.2 | 22.9 | 3.9 |
| TZPC48-800 | 10.0 | 58.5 | 3.5 | 23.1 | 4.9 |
| TZPC20-900 | 9.9 | 51.5 | 9.1 | 19.3 | 10.2 |
| TZPC48-900 | 5.1 | 52.7 | 4.8 | 28.9 | 8.5 |
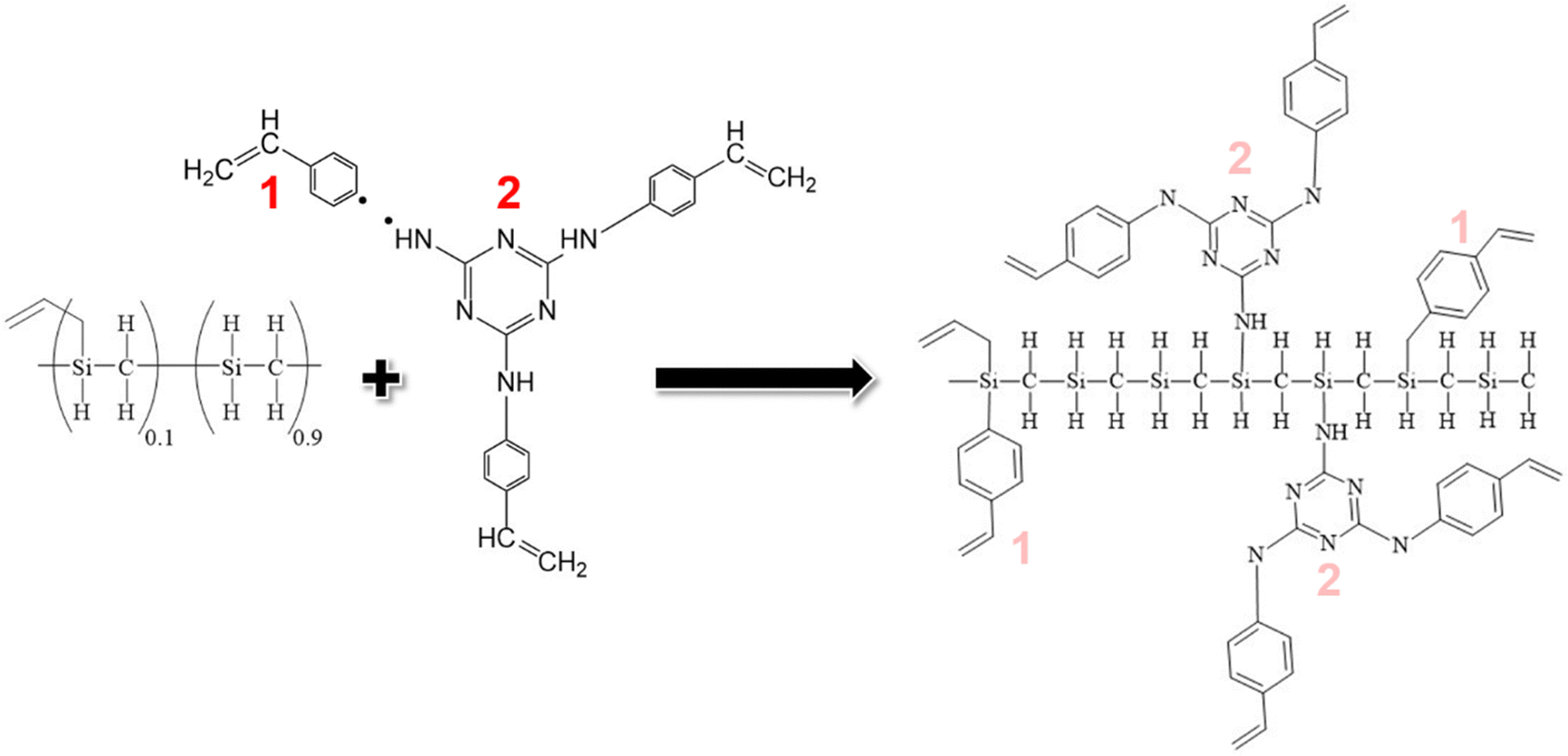 | ||
| Scheme 3 Theoretical structure of the starting material. The schematic representation illustrates the theoretical structure of the starting material obtained following the fracture of the dendron TRIAZ-3. This fracture results in the establishment of new Si–C bonds and Si–NH–C bonds through the reaction of AHPCS with fragments 1 and 2, respectively. It is essential to note that this fragmentation of the dendron occurs during the pyrolysis process, and concurrently, hydrogen loss occurs at temperatures exceeding 450 °C (Fig. 2). This depiction provides a theoretical framework to visualize the structural transformations that transpire during the synthesis of SiOCN materials, elucidating the critical role played by the fracture of the dendritic molecule and subsequent bonding reactions in shaping the final material composition. | ||
Finally, the high-resolution O 1s spectra are presented in Fig. 5d. The dominant peak at approximately 533.00 eV is deconvoluted into two bands, with the main peak situated at 532.80 eV attributed to O–Si bonds from the structural network of the SiOC. A smaller band at 534.80 eV likely arises from OC bonds26,73 resulting from surface oxidation. The data in Table 7 demonstrate the presence of these components in both TRIAZ-3 and AHPCS treated at varying temperatures. Interestingly, OC bonds increase relative to O–Si with escalating treatment temperatures in both samples TZPC20 and TZPC48. Despite this, elemental analysis (Table 1) indicates consistent oxygen concentration across samples. This suggests the possible oxidation of CO bonds to C–OH or the formation of new Si–OH groups. These findings underscore the preference for surface oxidation in C atoms rather than Si atoms,68 further illuminating the intricate surface dynamics of the pyrolyzed materials.
| Relative concentration of the O-containing bond unit (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BE (eV) | TRIAZ-3 | AHPCS | TZPC20 | TZPC48 | |||||||
| 700 | 800 | 900 | 700 | 800 | 900 | 700 | 800 | 900 | |||
| O–Si | 532.80 | 94.82 | 84.6 | 84.9 | 75.1 | 94.7 | 90.6 | 79.3 | 91.8 | 85.6 | 79.1 |
| OC |
534.80 | 5.18 | 15.4 | 15.1 | 24.9 | 5.3 | 9.4 | 20.7 | 8.2 | 14.4 | 20.9 |
Raman analysis: investigating the state of Cfree phases in pyrolyzed materials
To further delve into the characterization of the pyrolyzed materials, Raman spectroscopy was employed to elucidate the state of the Cfree phase, which was previously detected through 13C-MAS NMR and XPS analyses (Fig. 4b and 5b). Moreover, the presence of Cfree phase was also discerned through X-ray diffraction (XRD), included in Section S4 and Fig. S8 (ESI†). This comprehensive approach aimed to provide a deeper understanding of the carbonaceous phases present in the samples. The experimental and deconvoluted Raman spectra of the pyrolyzed materials are presented in Fig. 6a and b, respectively. The Raman spectra exhibit characteristic peaks, notably the D and G peaks at approximately 1350 cm−1 and 1590 cm−1, respectively. Particularly, the TZPC20-700 sample displayed a distinctive feature – an intense peak centered around 2909 cm−1 (marked with an asterisk in Fig. 6a) corresponding to the symmetric stretching vibration of C–H bonds.74 This peak suggests the presence of remaining groups that did not undergo full mineralization during the pyrolysis process,75 revealing that conversion from inorganic polymer to ceramic structure remain incomplete within the shortest reaction time and the lowest temperature pyrolysis. This phenomenon indicates that synthesis of 20 hours duration and heat treatments at 700 °C are insufficient to complete the mineralization process. The D band, located around 1350 cm−1, signifies the breathing mode of disordered sp2-hybridized rings within the carbon structures.76,77 The G band, appearing around 1590 cm−1,78,79 is linked to the electron–phonon coupling interaction and reflects the E2g symmetry mode. The deconvolution analysis discerns additional bands, including the D* (also known as T) band at around 1150 cm−1 attributed to sp2 and sp3 carbon atoms at the carbon network's edges (Fig. 6b). This band is indicative of impurities or symmetry breakage due to nitrogen incorporation,80 corresponding to the rupture of the TRIAZ-3 (Scheme 2).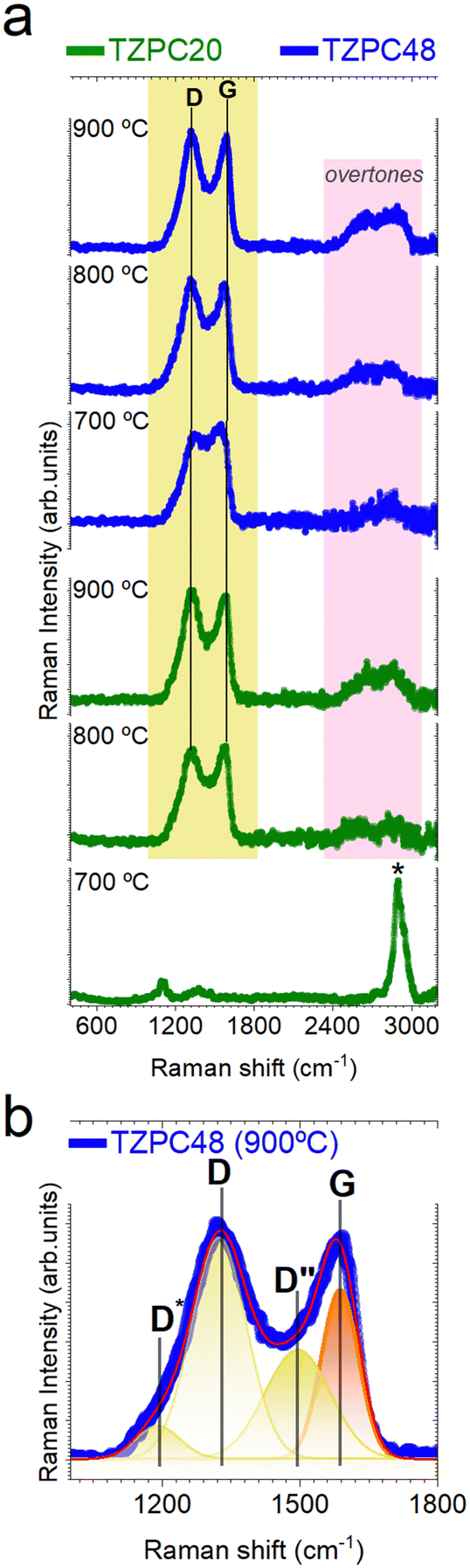 | ||
| Fig. 6 Exploring the nature of Cfree phases in pyrolyzed materials through Raman spectroscopy. (a) Raman spectra series of TZPC20 and TZPC48 materials subjected to varying temperatures. The yellow shaded region corresponds to characteristic modes associated with the carbonaceous phases present in the samples, while the pink highlighted region corresponds to overtones of different vibration modes. (b) Deconvolution analysis of the TZPC48-900 spectrum, focusing on the spectral range between 1000–2000 cm−1. The Raman vibration modes associated with the D band are represented in yellow, while the G band mode is highlighted in orange. | ||
Additionally, the D′′ band is detected, associated with amorphous carbon,77,81 yet its attribution remains debated.76,82 These bands collectively provide insights into the composition and structure of the carbonaceous materials. The second-order Raman spectra reveal overtones of different vibration modes, such as the 2D (also known as G′) and D + G bands, further characterizing the samples. The 2D band's intensity and shape indicate the stacking layers of graphite. Although the 2D band is less defined due to defects in the segregated carbon phase, the D and G bands become more well-defined with increased reaction time and pyrolysis temperature.83
By employing the equation proposed by A. C. Ferrari,77 for disordered and amorphous carbons, the obtained fitting data were used to determine the lateral crystallite size (La) of the free carbon nanoclusters. The ID/IG ratio increases with higher pyrolysis temperatures, denoting an augmentation of defects in the carbon sheets. Interestingly, samples subjected to longer reaction times show a red shift in the D band with increasing pyrolysis temperature, attributed to disorder-induced lattice defects. The G band, sensitive to nitrogen doping, experiences an upshift due to increased nitrogen content. In our materials, the Raman G band shifts from 1567 to 1582 cm−1 as a result of nitrogen doping.81 The change in the position of the G band corresponds to the transition from disordered carbon to nanocrystalline carbon,77 while the shift in the D band position signifies alterations in the number of ordered threefold aromatic rings. The obtained La values, approximately 1.3–1.6 nm, increase with higher pyrolysis temperatures, especially evident in samples with longer reaction times. Notably, the sample obtained at the lowest pyrolysis temperature and shorter reaction time did not exhibit D and G bands in the spectra, hence no La data is presented for this sample in Table 8.
| Treatment temperature (°C) | TZPC20 | TZPC48 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D* | D | D′′ | G | I D/IG | L a | D* | D | D′′ | G | I D/IG | L a | ||
| 700 | Position | — | — | — | — | — | — | 1156 | 1343 | 1486 | 1567 | 1.07 | 1.39 |
| FWHM | — | — | — | — | — | 207 | 181 | 142 | 116 | ||||
| 800 | Position | 1218 | 1326 | 1456 | 1567 | 1.02 | 1.36 | 1159 | 1320 | 1469 | 1569 | 1.11 | 1.42 |
| FWHM | 117 | 130 | 138 | 110 | 181 | 168 | 112 | 101 | |||||
| 900 | Position | 1194 | 1325 | 1450 | 1568 | 1.11 | 1.42 | 1181 | 1324 | 1498 | 1582 | 1.34 | 1.56 |
| FWHM | 161 | 144 | 116 | 121 | 116 | 155 | 35 | 80 | |||||
In brief, our study has successfully demonstrated the feasibility of incorporating nitrogen into polymeric precursors, leading to the formation of N-doped Si(O)C materials. Crucially, our findings emphasize the pivotal role of reaction time and pyrolysis temperature in tailoring the properties of SiOCN materials. Specifically, these parameters influence the extent of doping in the glassy phase and the concentration of SiNC3 within the material. Longer reaction times tend to preserve the triazine rings and result in a more defective carbon structure, primarily due to the incorporation of nitrogen heteroatoms into the carbonaceous phase. This investigation unveils a novel synthesis pathway for N-doped SiOC structures through an in situ synthesis method, underscoring the significance of precise control over the synthesis parameters of polymer-derived ceramics (PDCs). The outcomes of this research hold great promise for the development of innovative functional materials with potential applications in diverse fields, including electrochemical devices such as the production of nitrogen-doped carbide-derived carbons (CDCs). As we continue to explore the possibilities offered by this synthesis strategy, it becomes evident that our work contributes to the ongoing pursuit of tailored materials designed to address evolving technological and environmental challenges.
Conclusions
In this study, we have successfully designed and synthesized a novel N-containing dendritic structure to produce N-doped SiOC materials. The synthesis involved well-defined steps of hydrosilylation reactions between a polymer-derived carbide and the newly designed dendron, followed by controlled pyrolysis. Notably, we not only established the synthesis pathway but also proposed a comprehensive reaction mechanism underlying SiOCN formation. By chemically modifying a commercially available allyl-hydrido polycarbosilane with the dendritic structure, we incorporated nitrogen into the SiOCN material while preserving the integrity of the triazine aromatic rings. During pyrolysis, a fraction of NH-C sp2 linkages fractured, indicating greater retention of N sp2–C over N sp3–C. The rupture of N sp3–C bonds facilitated Si–N bond formation, resulting in SiNC3, predominantly SiC4, along with SiC3O, SiO4, and a segregated carbon phase (Cfree) coexisting with triazine rings. This investigation confirms the effective incorporation of nitrogen into SiOC's carbon phase and ceramic backbone. Our work's broader impact lies in advancing high-energy supercapacitor, stablishing a new route for the synthesis of N-doped SiOC material for the future production of CDC with N heteroatoms, bridging materials science with sustainable energy storage technology. By tailoring materials at a molecular level, we enhance supercapacitor electrochemical performance. As the demand for efficient energy storage grows, our study paves the way for high-performance supercapacitors applicable across diverse fields. This holistic approach fosters sustainable progress in materials science and energy technology, driving us towards a more energy-efficient and environmentally conscious future.Author contributions
J. R., and F. R.-M. conceived the idea. B. P.-R., A. M. R., J. R., M. A. M. and F. R.-M. planed the research and experiments. B. P.-R. and A. M. R prepared the samples. B. P.-R. performed structural characterizations. B. P.-R. carried out Raman measurements and the microscopic strain characterization. B. P.-R. wrote the manuscript draft with inputs from J. R., and F. R.-M. All authors contribute to the discussion and to the final version of the manuscript. The work was supervised by J. R., M. A. M. and F. R.-M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation (MCIN/AEI/10.13039/501100011033), through the PID2020-114192RB-C41 project program and Comunidad de Madrid for the financial support through the Doctorados Industriales project (IND2020/IND-17375), which is co-financed by the European Social Fund. We also acknowledge projects TED2021-130957B-C51 and TED2021-132800B-I00, funded by the Spanish Ministry of Science and Innovation.References
- C. G. Pantano, A. K. Singh and H. Zhang, Silicon Oxycarbide Glasses, J. Sol-Gel Sci. Technol., 1999, 14, 7–25 CrossRef CAS.
- C. Stabler, E. Ionescu, M. Graczyk-Zajac, I. Gonzalo-Juan and R. Riedel, Silicon oxycarbide glasses and glass-ceramics: “All-Rounder” materials for advanced structural and functional applications, J. Am. Ceram. Soc., 2018, 101, 4817–4856 CrossRef CAS.
- P. Colombo, G. Mera, R. Riedel and G. D. Sorarù, Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics, J. Am. Ceram. Soc., 2010, 93, 1805–1837 CrossRef CAS.
- G. Mera, M. Gallei, S. Bernard and E. Ionescu, Ceramic nanocomposites from tailor-made preceramic polymers, Nanomaterials, 2015, 5, 468–540 CrossRef CAS PubMed.
- M. Douloudi, E. Nikoli, T. Katsika, M. Vardavoulias and M. Arkas, Dendritic polymers as promising additives for the manufacturing of hybrid organoceramic nanocomposites with ameliorated properties suitable for an extensive diversity of applications, Nanomaterials, 2021, 11, 1–38 Search PubMed.
- Y. Tian, M. Ge, W. Zhang, X. Lv and S. Yu, Metallocene Catalytic Insertion Polymerization of 1-Silene to Polycarbosilanes, Sci. Rep., 2015, 5, 1–6 CAS.
- Z. Yu, H. Min, J. Zhan and L. Yang, Preparation and dielectric properties of polymer-derived SiCTi ceramics, Ceram. Int., 2013, 39, 3999–4007 CrossRef CAS.
- G. Liu, J. Kaspar, L. M. Reinold, M. Graczyk-Zajac and R. Riedel, Electrochemical performance of DVB-modified SiOC and SiCN polymer-derived negative electrodes for lithium-ion batteries, Electrochim. Acta, 2013, 106, 101–108 CrossRef CAS.
- S. C. K. Yive, R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, Silicon Carbonitride from Polymeric Precursors: Thermal Cross-Linking and Pyrolysis of Oligosilazane Model Compounds, Chem. Mater., 1992, 4, 141–146 CrossRef.
- S. Yajima, K. Okamura, J. Hayashi and M. Omori, Synthesis of continuous SiC fibers with high tensile strength, J. Am. Ceram. Soc., 1976, 59, 324–327 CrossRef CAS.
- H. Li, et al., Effect of the polycarbosilane structure on its final ceramic yield, J. Eur. Ceram. Soc., 2008, 28, 887–891 CrossRef CAS.
- J. Lücke, J. Hacker, D. Suttor and G. Ziegler, Synthesis and characterization of silazane-based polymers as precursors for ceramic matrix composites, Appl. Organomet. Chem., 1997, 11, 181–194 CrossRef.
- Z. Yu, et al., Influence of the architecture of dendritic-like polycarbosilanes on the ceramic yield, J. Eur. Ceram. Soc., 2015, 35, 1161–1171 CrossRef CAS.
- L. A. Baldwin, et al., Synthesis of a Two-Component Carbosilane System for the Advanced Manufacturing of Polymer-Derived Ceramics, Chem. Mater., 2018, 30, 7527–7534 CrossRef CAS.
- Z. D. Apostolov, E. P. Heckman, T. S. Key and M. K. Cinibulk, Effects of low-temperature treatment on the properties of commercial preceramic polymers, J. Eur. Ceram. Soc., 2020, 40, 2887–2895 CrossRef CAS.
- A. R. Puerta, E. E. Remsen, M. G. Bradley, W. Sherwood and L. G. Sneddon, Synthesis and ceramic conversion reactions of 9-BBN-modified allylhydridopolycarbosilane: A new single-source precursor to boron-modified silicon carbide, Chem. Mater., 2003, 15, 478–485 CrossRef CAS.
- E. Kroke, et al., Silazane derived ceramics and related materials, Mater. Sci. Eng., R, 2000, 26, 97–199 CrossRef.
- N. B. Laidani and G. D. Sorarù, N-doped polymer-derived Si (N) OC: The role of the N-containing precursor, J. Mater. Res., 2015, 30, 770–781 CrossRef.
- H. F. Gorgulho, F. Gonçalves, M. F. R. Pereira and J. L. Figueiredo, Synthesis and characterization of nitrogen-doped carbon xerogels, Carbon, 2009, 47, 2032–2039 CrossRef CAS.
- B. Cao, et al., Preparation of Nitrogen-Doped Carbon Spheres by Injecting Pyrolysis of Pyridine, ACS Sustainable Chem. Eng., 2015, 3, 1786–1793 CrossRef CAS.
- F. Bouzat, S. Foucaud, Y. Leconte, A. Maître and R. Lucas, From click to ceramic: An efficient way to generate multielement Si/Zr/C clicked-polymer-derived ceramics (cPDC), Mater. Lett., 2013, 106, 337–340 CrossRef CAS.
- F. Bouzat, A.-R. Graff, R. Lucas and S. Foucaud, Preparation of C/SiC ceramics using a preceramic polycarbosilane synthesized via hydrosilylation, J. Eur. Ceram. Soc., 2016, 36, 2913–2921 CrossRef CAS.
- V. M. Vidya, S. Pola and P. Chetti, Optoelectronic and charge transport properties of D-n-A type 1,3,5-triazine derivatives: A combined experimental and DFT study, Spectrochim. Acta, Part A, 2021, 245, 118940 CrossRef PubMed.
- R. Sreeja, B. Swaminathan, A. Painuly, T. V. Sebastian and S. Packirisamy, Allylhydridopolycarbosilane (AHPCS) as matrix resin for C/SiC ceramic matrix composites, Mater. Sci. Eng., B, 2010, 168, 204–207 CrossRef CAS.
- L. J. Print, J. J. Liggat, S. Moug, H. Seaton and D. C. Apperley, A Study of the Ceramicisation of Allylhydridopolycarbosilane by Thermal Volatilisation Analysis and Solid-State Nuclear Magnetic Resonance, Silicon, 2023, 15, 1355–1379 CrossRef CAS.
- S. Kaur, G. Mera, R. Riedel and E. Ionescu, Effect of boron incorporation on the phase composition and high-temperature behavior of polymer-derived silicon carbide, J. Eur. Ceram. Soc., 2016, 36, 967–977 CrossRef CAS.
- H. Q. Ly, R. Taylor, R. J. Day and F. Heatley, Conversion of polycarbosilane (PCS) to SiC-based ceramic: Part 1. Characterisation of PCS and curing products, J. Mater. Sci., 2001, 36, 4037–4043 CrossRef CAS.
- S. Kaur, et al., Single-source-precursor synthesis of novel V8C7/SiC(O)-based ceramic nanocomposites, J. Eur. Ceram. Soc., 2016, 36, 3553–3563 CrossRef CAS.
- S. P. Lee, N. Mellon, A. M. Shariff and J. M. Leveque, Adsorption of CO2 and Methane on Covalent Organic Polymer, E3S Web Conf., 2018, 43, 01001 CrossRef.
- D. S. Kumar, W. Ximbo and L. Zhiping, Facile synthesis of triazine-triphenylamine-based microporous covalent polymer adsorbent for flue gas CO2 capture, Microporous Mesoporous Mater., 2018, 255, 76–83 CrossRef.
- W. Simon and T. Clerc, Tablas de elucidación estrructural de compuestos orgánicos por métodos espectroscópicos, 1980 Search PubMed.
- H. Li, et al., Polymer–ceramic conversion of a highly branched liquid polycarbosilane for SiC-based ceramics, J. Mater. Sci., 2008, 43, 2806–2811 CrossRef CAS.
- A. K. Singh and C. G. Pantano, Surface Chemistry and Structure of Silicon Oxycarbide Gels and Glasses, J. Sol-Gel Sci. Technol., 1997, 8, 371–376 CAS.
- C. Schiţco, et al., Silicon oxycarbonitrides synthesized by ammonia-assisted thermolysis route from polymers: A total X-ray scattering, solid-state NMR, and TEM structural study, J. Eur. Ceram. Soc., 2016, 36, 979–989 CrossRef.
- Q. Wen, et al., Single-source-precursor synthesis of dense SiC/HfCxN1−x-based ultrahigh-temperature ceramic nanocomposites, Nanoscale, 2014, 6, 13678–13689 RSC.
- V. L. Nguyen, N. B. Laidani and G. D. Sorarù, N-doped polymer derived Si(N)OC The role of the N-containing, J. Mater. Res., 2015, 30(6), 770–781 CrossRef CAS.
- Z. Yu, et al., Single-source-precursor synthesis, microstructure and high temperature behavior of TiC-TiB2-SiC ceramic nanocomposites, Ceram. Int., 2017, 43, 5949–5956 CrossRef CAS.
- M. A. Schiavon, K. J. Ciuffi and I. V. P. Yoshida, Glasses in the SiOCN system produced by pyrolysis of polycyclic silazane/siloxane networks, J. Non. Cryst. Solids, 2007, 353, 2280–2288 CrossRef CAS.
- S. Rangarajan and P. B. Aswath, Role of precursor chemistry on synthesis of Si-O-C and Si-O-C-N ceramics by polymer pyrolysis, J. Mater. Sci., 2011, 46, 2201–2211 CrossRef CAS.
- R. Yao, Z. Feng, L. Chen and Y. Zhang, Effects of oxidation curing and Al atoms on the formation of near-stoichiometric freestanding SiC(Al) films derived from polyaluminocarbosilane (PACS), J. Eur. Ceram. Soc., 2013, 33, 1675–1683 CrossRef CAS.
- G. Ramis, P. Quintard, M. Cauchetier, G. Busca and V. Lorenzelli, Surface Chemistry and Structure of Ultrafine Silicon Carbide: An FT-IR Study, J. Am. Ceram. Soc., 1989, 72, 1692–1697 CrossRef CAS.
- C. Vix-Guterl, I. Alix, P. Gibot and P. Ehrburger, Formation of tubular silicon carbide from a carbon–silica material by using a reactive replica technique: infra-red characterisation, Appl. Surf. Sci., 2003, 210, 329–337 CrossRef CAS.
- Y. de Hazan and D. Penner, SiC and SiOC ceramic articles produced by stereolithography of acrylate modified polycarbosilane systems, J. Eur. Ceram. Soc., 2017, 37, 5205–5212 CrossRef CAS.
- L. Bois, J. Maquet, F. Babonneau, H. Mutin and D. Bahloul, Structural Characterization of Sol-Gel Derived Oxycarbide Glasses. 1. Study of the Pyrolysis Process, Chem. Mater., 1994, 6, 796–802 CrossRef CAS.
- B. Perez-Roman, et al., Insights into the structural and surface characteristics of microporous carbide derived carbons obtained through single and double halogen etching, Microporous Mesoporous Mater., 2021, 310, 110675 CrossRef CAS.
- S. Otoishi and Y. Tange, Preparation of Si3N4 Whiskers from Polycarbosilane, J. Eur. Ceram. Soc., 1997, 105, 1072–1078 CrossRef CAS.
- L. J. Print, J. J. Liggat, S. Moug, H. Seaton and D. C. Apperley, A Study of the Ceramicisation of Allylhydridopolycarbosilane by Thermal Volatilisation Analysis and Solid - State Nuclear Magnetic Resonance, Silicon, 2023, 15, 1355–1379 CrossRef CAS.
- J. R. Ramos, C. Morales and Y. Matsumoto, Oxygen concentration effect on properties of SiOC thin films obtained by HWCVD, in 2017 14th International Conference on Electrical Engineering, Computing Science and Automatic Control (CCE), 2017, pp. 22–25.
- C. Y. Kim, et al., Ultraviolet irradiation effect on the properties of leakage current and dielectric breakdown of low-dielectric-constant SiOC(H) films using comb capacitor structure, Thin Solid Films, 2011, 519, 6732–6736 CrossRef CAS.
- R. Ellerbrock, M. Stein and J. Schaller, Comparing amorphous silica, short-range-ordered silicates and silicic acid species by FTIR, Sci. Rep., 2022, 12, 1–8 CrossRef PubMed.
- R. Coustel, et al., Vibrational frequencies of hydrogenated silicon carbonitride: A DFT study, Surf. Coatings Technol., 2017, 325, 437–444 CrossRef CAS.
- D. D. S. Meneses, et al., Investigation of medium range order in silicate glasses by infrared spectroscopy, Vib. Spectrosc., 2013, 65, 50–57 CrossRef.
- M. C. Bechelany, et al., Preparation of polymer-derived Si-B-C-N monoliths by spark plasma sintering technique, J. Eur. Ceram. Soc., 2015, 35, 1361–1374 CrossRef CAS.
- V. L. Nguyen, N. B. Laidani and G. D. Sorarù, N-doped polymer-derived Si(N)OC: The role of the N-containing precursor, J. Mater. Res., 2015, 30, 770–781 CrossRef CAS.
- M. A. Mazo, et al., Evaluation of thermal shock resistance of silicon oxycarbide materials for high-temperature receiver applications, Sol. Energy, 2018, 173, 256–267 CrossRef CAS.
- C. Gérardin, F. Taulelle and D. Bahloul, Pyrolysis chemistry of polysilazane precursors to silicon carbonitride: Part 2. - Solid-state NMR of the pyrolytic residues, J. Mater. Chem., 1997, 7, 117–126 RSC.
- J. L. Oteo, M. A. Mazo, C. Palencia, F. Rubio and J. Rubio, Synthesis and Characterization of Silicon Oxycarbide Derived Nanocomposites Obtained through Ceramic Processing of TEOS/PDMS Preceramic Materials, J. Nano Res., 2011, 14, 27–38 CAS.
- M. Alejandra Mazo, A. Tamayo and J. Rubio, Stable highly porous silicon oxycarbide glasses from pre-ceramic hybrids, J. Mater. Chem. A, 2015, 3, 23220–23229 RSC.
- D. Mocaer, G. Chollon, R. Pailler and L. Filipuzzi, & Naslain, R. Si-C-N ceramics with a high microstructural stability elaborated from the pyrolysis of new polycarbosilazane precursors - Part V Oxidation kinetics of model filaments, J. Mater. Sci., 1993, 28, 3059–3068 CrossRef CAS.
- S. Widgeon, et al., Nanostructure and Energetics of Carbon-Rich SiCN Ceramics Derived from Polysilylcarbodiimides: Role of the Nanodomain Interfaces, Chem. Mater., 2012, 24, 1181–1191 CrossRef CAS.
- J. Lamanna, J. Braddock-Wilking, S. H. Lin and B. J. Feldman, 13C NMR spectroscopy of amorphous hydrogenated carbon nitride, Solid State Commun., 1999, 109, 573–576 CrossRef CAS.
- E. G. Gillan, Synthesis of nitrogen-rich carbon nitride networks from an energetic molecular azide precursor, Chem. Mater., 2000, 12, 3906–3912 CrossRef CAS.
- M. A. Schiavon, G. Domenico Sorarù and I. V. P. Yoshida, Synthesis of a polycyclic silazane network and its evolution to silicon carbonitride glass, J. Non. Cryst. Solids, 2002, 304, 76–83 CrossRef CAS.
- Q. D. Nghiem, A. Asthana, I. K. Sung and D. P. Kim, Fabrication of porous SiC-based ceramic microchannels via pyrolysis of templated preceramic polymers, J. Mater. Res., 2006, 21, 1543–1549 CrossRef CAS.
- H. W. Kim, et al., Carbon Defect Characterization of Nitrogen-Doped Reduced Graphene Oxide Electrocatalysts for the Two-Electron Oxygen Reduction Reaction, Chem. Mater., 2019, 31, 3967–3973 CrossRef CAS.
- K. Yamamoto, Y. Koga and S. Fujiwara, XPS studies of amorphous SiCN thin films prepared by nitrogen ion-assisted pulsed-laser deposition of SiC target, Diam. Relat. Mater., 2001, 10, 1921–1926 CrossRef CAS.
- T. S. Miller, et al., Carbon nitrides: Synthesis and characterization of a new class of functional materials, Phys. Chem. Chem. Phys., 2017, 19, 15613–15638 RSC.
- M. Ismael, Y. Wu, D. H. Taffa, P. Bottke and M. Wark, Graphitic carbon nitride synthesized by simple pyrolysis: Role of precursor in photocatalytic hydrogen production, New J. Chem., 2019, 43, 6909–6920 RSC.
- K. Schwinghammer, et al., Crystalline carbon nitride nanosheets for improved visible-light hydrogen evolution, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed.
- G. J. Leonel, X. Guo, G. Singh, C. Gervais and A. Navrotsky, Energetics and structure of SiC(N)(O) polymer-derived ceramics, J. Am. Ceram. Soc., 2023, 5086–5101 CrossRef CAS.
- Y. Feng, N. Feng, Y. Wei and Y. Bai, Preparation and improved electrochemical performance of SiCN-graphene composite derived from poly(silylcarbondiimide) as Li-ion battery anode, J. Mater. Chem. A, 2014, 2, 4168–4177 RSC.
- Q. Hanniet, et al., Investigation of polymer-derived Si–(B)–C–N ceramic/reduced graphene oxide composite systems as active catalysts towards the hydrogen evolution reaction, Sci. Rep., 2020, 10, 1–15 CrossRef PubMed.
- Z. Ren, C. Gervais and G. Singh, Preparation and structure of SiOCN fibres derived from cyclic silazane/poly-acrylic acid hybrid precursor, R. Soc. open Sci., 2019, 6, 190690 CrossRef CAS PubMed.
- J. H. Hibben, Raman spectra in organic chemistry, Chem. Rev., 1936, 18(1), 1–232 CrossRef.
- D. Hourlier, S. Venkatachalam, M. R. Ammar and Y. Blum, Pyrolytic Conversion of organopolysiloxanes, J. Anal. Appl. Pyrolysis, 2017, 123, 296–306 CrossRef CAS.
- D. Hu, G. Shao, J. Wang, A. Gurlo and M. F. Bekheet, Revealing nanodomain structures of bottom-up fabricated graphene embedded silicon oxycarbide ceramics, Polymers, 2022, 14, 3675 CrossRef CAS PubMed.
- A. C. F. Robertson, and J. Interpretation of Raman spectra of disordered and amorphous carbon, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef.
- R. Vidano and D. B. Fischbach, New Lines in the Raman Spectra of Carbons and Graphite, J. Am. Ceram. Soc., 1978, 61, 13–17 CrossRef CAS.
- L. G. Cançado, et al., Quantifying defects in graphene via Raman spectroscopy at different excitation energies, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martinez-Alonso and J. M. D. Tascón, Raman microprobe studies on carbon materials, Carbon, 1994, 32, 1523–1532 CrossRef CAS.
- S. Claramunt, et al., The importance of interbands on the interpretation of the Raman spectrum of graphene oxide, J. Phys. Chem. C, 2015, 119, 10123–10129 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- G. Mera, A. Navrotsky, S. Sen, H. J. Kleebe and R. Riedel, Polymer-derived SiCN and SiOC ceramics-structure and energetics at the nanoscale, J. Mater. Chem. A, 2013, 1, 3826–3836 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00898c |
| This journal is © The Royal Society of Chemistry 2024 |