
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking OER catalytic potential and chiral Fe3O4 film as a game-changer for electrochemical water oxidation pathway and by-product control†
Wenyan
Zhang
 *,
Chaoqun
Jiang
,
Hangmin
Guan
,
Yuanyuan
Wang
,
Yingfei
Hu
,
Wei
Wang
,
Wenjie
Tian
and
Lingyun
Hao
*,
Chaoqun
Jiang
,
Hangmin
Guan
,
Yuanyuan
Wang
,
Yingfei
Hu
,
Wei
Wang
,
Wenjie
Tian
and
Lingyun
Hao
College of Material Engineering, Jinling Institute of Technology, Nanjing 211169, China. E-mail: wiseyanyan@jit.edu.cn; Fax: +86-25-86188966; Tel: +86-25-86188966
First published on 5th January 2024
Abstract
Electrochemical water splitting is an attractive technique for hydrogen production, but its development is constrained due to the sluggish kinetics of the oxygen evolution reaction (OER), limited charge transmission efficiency, and the generation of harmful by-products (like hydrogen peroxide) accompanying the OER process. Herein, we constructed an Fe3O4 film with an inherent chiral structure to demonstrate its chiral induced spin selectivity (CISS) effect on modulating the OER pathway and facilitating the transfer of electrons extracted from the OER. Compared with normal Fe3O4, the chiral Fe3O4 film facilitated the OER pathway for 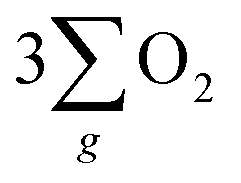 production via the CISS-induced spin alignment of electrons and radicals, while it suppressed the pathway for peroxide by-product generation. Meanwhile, as a half-metallic material, chiral Fe3O4 film promoted the transportation of spin-aligned electrons derived from water oxidation to an external circuit. Compared with normal Fe3O4 film which is not capable of aligning electrons, the potential at 10 mA cm−2 declined by 150 mV, the Tafel slope decreased by 104 mV dec−1, the charge transfer resistance decreased by 50%, the free charge carrier density was amplified by 10-fold, and the electron lifetime was prolonged within the chiral Fe3O4 film.
production via the CISS-induced spin alignment of electrons and radicals, while it suppressed the pathway for peroxide by-product generation. Meanwhile, as a half-metallic material, chiral Fe3O4 film promoted the transportation of spin-aligned electrons derived from water oxidation to an external circuit. Compared with normal Fe3O4 film which is not capable of aligning electrons, the potential at 10 mA cm−2 declined by 150 mV, the Tafel slope decreased by 104 mV dec−1, the charge transfer resistance decreased by 50%, the free charge carrier density was amplified by 10-fold, and the electron lifetime was prolonged within the chiral Fe3O4 film.
1. Introduction
For decades, the excessive consumption of fossil fuels has led to severe environmental pollution and a global energy crisis, posing a significant threat to the future development of human society.1–5 Consequently, there is an imperative to explore clean and sustainable energy sources capable of replacing fossil fuels. Among these alternatives, hydrogen has garnered considerable attention due to its status as an environmentally friendly energy carrier, characterized by high enthalpy and absence of carbon emissions (such as CO and CO2) during combustion.6–8Electrocatalytic water splitting has emerged as a cost-effective and promising technique for generating hydrogen from water, and it offers the advantage of not requiring extreme conditions like high temperature or pressure.9–13 A myriad of advanced catalysts for splitting water have been developed, such as MOF-derived catalysts,14 nickel oxide and nickel phosphide,15 NiFe selenide,16 CoNi hydrogen phosphate nanotubes,17 nickel nitride,18 and 2D-layered catalysts like NiCo hydroxide nanosheets.19
However, the efficiency of electrochemical water splitting falls short of expectations.20–23 The primary hurdle in this process lies in the sluggish oxygen evolution reaction (OER) that involves a complex four-electron process.24–28 Furthermore, an undesirable by-product, hydrogen peroxide, tends to form at the anodes when electrons derived from water oxidation are not spin-polarized. The presence of this by-product poses a detrimental impact on the OER reaction system, as it tends to poison the surface of electrocatalysts, thereby diminishing their catalytic activity and shortening their operational lifespan.29–31 To facilitate efficient water splitting, it has become imperative to accelerate OER kinetics while mitigating the production of hydrogen peroxide by-products during the OER process.
It is noted that the OER is inherently spin-sensitive, because prior to the formation of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, the active sites must facilitate the extraction of three out of four electrons with identical spin orientations. Consequently, electrocatalysts that are capable of ensuring spin selectivity at the catalyst/electrolyte interface and subsequently facilitating the seamless transport of these spin-aligned electrons to the external circuit have become highly desirable for enhancing OER efficiency.23–25,32,33
O, the active sites must facilitate the extraction of three out of four electrons with identical spin orientations. Consequently, electrocatalysts that are capable of ensuring spin selectivity at the catalyst/electrolyte interface and subsequently facilitating the seamless transport of these spin-aligned electrons to the external circuit have become highly desirable for enhancing OER efficiency.23–25,32,33
The chiral induced spin selectivity (CISS) effect represents a promising approach to spin control in the OER process. For example, the attachment of organic chiral molecules (such as proteins or DNA) to semiconductors like TiO2 or CdSe, induces a CISS effect, reducing the OER barrier while suppressing the formation of by-products.26,27,34 Similarly, Inorganic materials characterized by intrinsic chiral configurations, such as chiral CuO and chiral CoxO, have showcased the CISS effect on lowering the OER barrier and mitigating the generation of by-products. Notably, their current density can surpass that achieved with semiconductors decorated with organic chiral molecules by up to 1000-fold.35–37
As an inorganic material, magnetite (Fe3O4) is renowned for its exceptional catalytic activity resulting from its redox characteristics, narrow band gap, cost-effectiveness, low toxicity, and remarkable durability.38,39 It is plausible that incorporating Fe3O4 with an inherent chiral structure could lead to a CISS effect on spin control for the OER. Beyond these benefits, Fe3O4 stands out as a half-metallic material, exhibiting nearly 100% spin polarization in the electronic density of states (DOS) near the Fermi energy level. Consequently, the electrical conductivity of Fe3O4 is fundamentally dependent upon the spin states of electrons, with one spin channel exhibiting metallic behavior, while the other spin channel demonstrates insulating properties.40–45 Such a distinguished feature holds potential for enhancing the transfer of spin-aligned electrons from the catalyst to the external circuit.
To verify these hypotheses, we constructed an Fe3O4 film with an inherent chiral structure to investigate its CISS effect on spin alignment for the OER and its effectiveness in transferring spin-aligned electrons to the external circuit. We observed that, compared with normal Fe3O4 film without a chiral structure, the utilization of a chiral Fe3O4 film as an OER electrocatalyst resulted in notable catalytic improvements, encompassing the facilitation of the OER pathway while hindering hydrogen peroxide by-products, a 150 mV reduction in the applied potential required to achieve 10 mA cm−2, a 42% increase in current density at 1.8 V (vs. SCE), a decrease of 104 mV dec−1 in the Tafel slope, a 50% reduction in charge transfer resistance, and a 10-fold amplification in the free charge carrier density. The benefits brought about by the chiral Fe3O4 film are intricately intertwined with the CISS effect induced by its chiral structure and the capabilities of half-metallic materials to facilitate electron transfer.
2. Results and discussion
Fig. 1(a) shows the XRD patterns of the chiral Fe3O4 film coated on FTO by electrochemical deposition. (Details are provided in ESI†). The XRD patterns exhibit diffraction peaks corresponding to SnO2 (JCPDS 2-1337) and Fe3O4 (JCPDS 7-322). The intensity of the Fe3O4 diffraction peak is lower than that of SnO2, due to the relatively small amount of Fe3O4 on the FTO substrate. The XPS spectra were acquired to delve into the surface state of chiral Fe3O4 on FTO. In Fig. 1(b), the XPS survey spectra reveal that the surface of chiral Fe3O4 is primarily composed of Fe and O elements. The chiral Fe3O4 film has a regular laminate morphology, as demonstrated in Fig. 2(a). Furthermore, the laminate structure of chiral Fe3O4 is hierarchically constructed from nanoparticles, as Fig. 2(b) shows.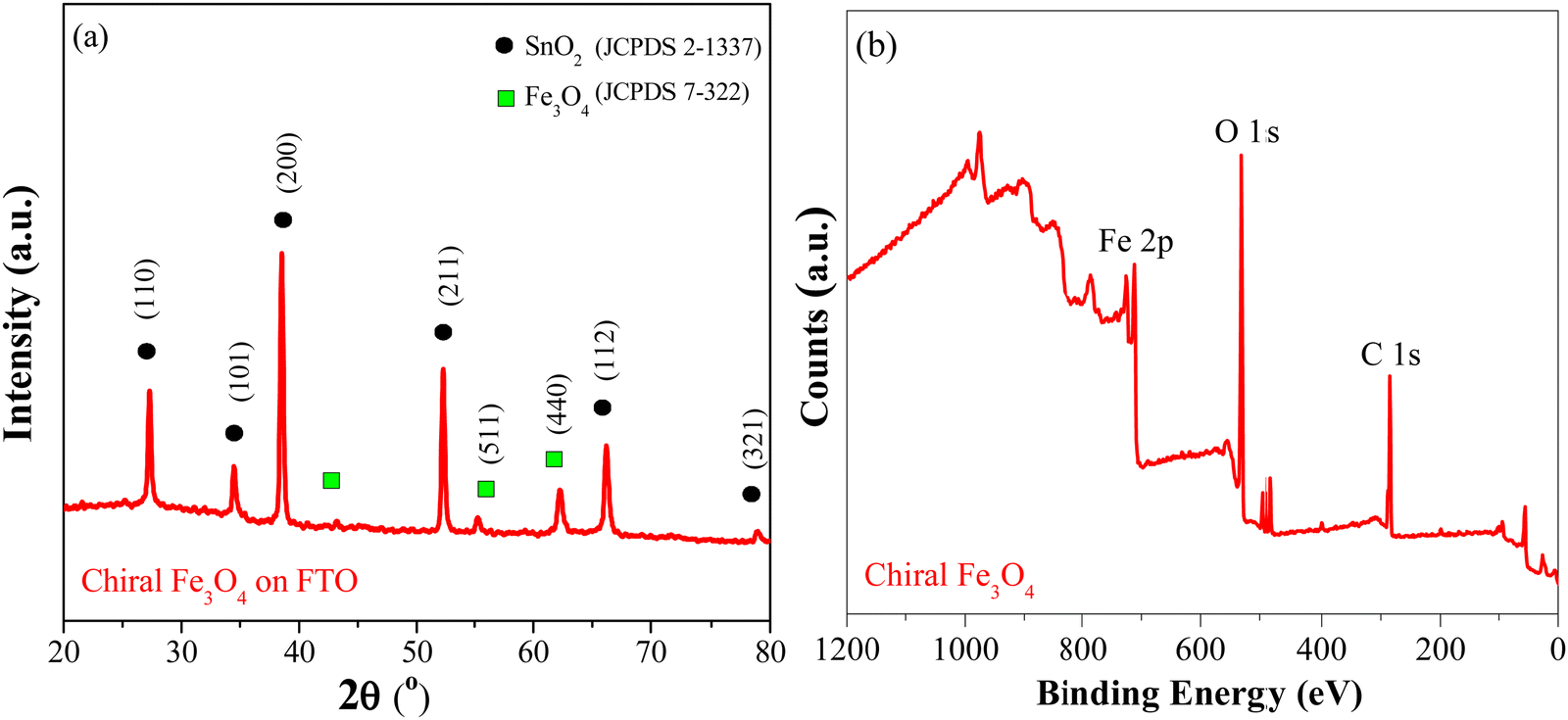 | ||
| Fig. 1 (a) XRD patterns and (b) XPS survey spectra (b) of chiral Fe3O4. | ||
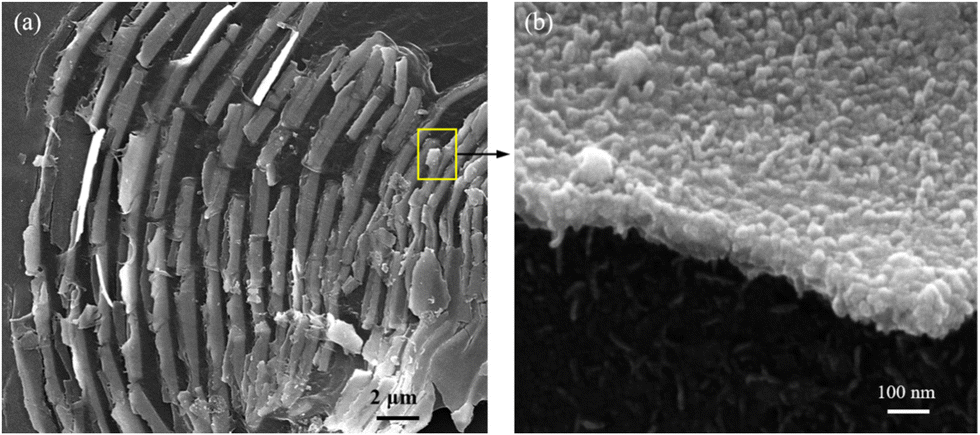 | ||
| Fig. 2 (a) SEM image of chiral Fe3O4; (b) magnification image of the rectangular region in (a). | ||
The Fe 2p XPS spectrum of the chiral Fe3O4 film is presented in Fig. 3. Within this spectrum, the binding energy peak centered at approximately 710.6 eV corresponds to the Fe 2p3/2 state, while the peak at 724.0 eV corresponds to the Fe 2p1/2 state.46–49 These peaks are fitted using a shakeup satellite and two spin–orbit doublets, which originate from the Fe3+ and Fe2+ states.46–48 The area ratio of Fe3+/Fe2+ in the spectrum is approximately 2, indicating that the material deposited on the FTO substrate is Fe3O4.
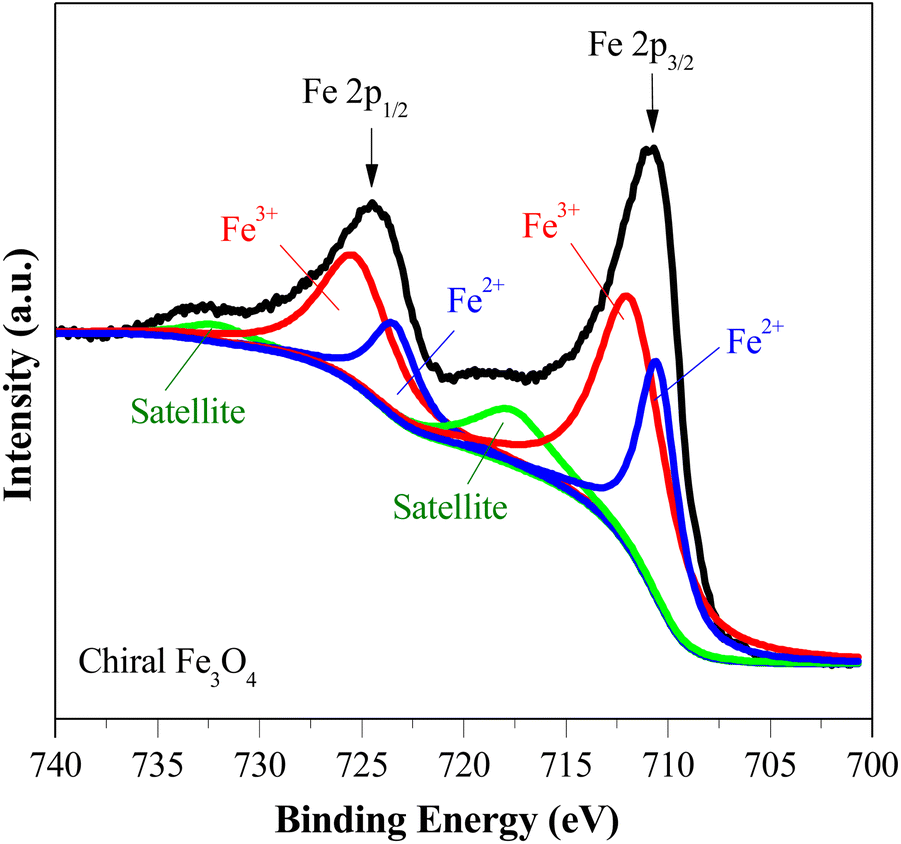 | ||
| Fig. 3 Fe 2p XPS spectra of chiral Fe3O4. | ||
The chirality of the catalyst surface was assessed through an electrochemical selectivity test, a sensitive technique for discerning the surface chirality of materials (details in ESI†).50,51 As shown in Fig. 4(a), chiral Fe3O4 exhibited a higher current density and smaller reduction potential when it was scanned in a chiral electrolyte comprising 10 mM L-tartaric acid than in a chiral electrolyte containing 10 mM D-tartaric acid. These results confirmed that chiral Fe3O4 exhibited contrasting chiral selectivity responses in electrolytes with opposite chirality, specifically preferring charge exchanges with L-tartaric acid.
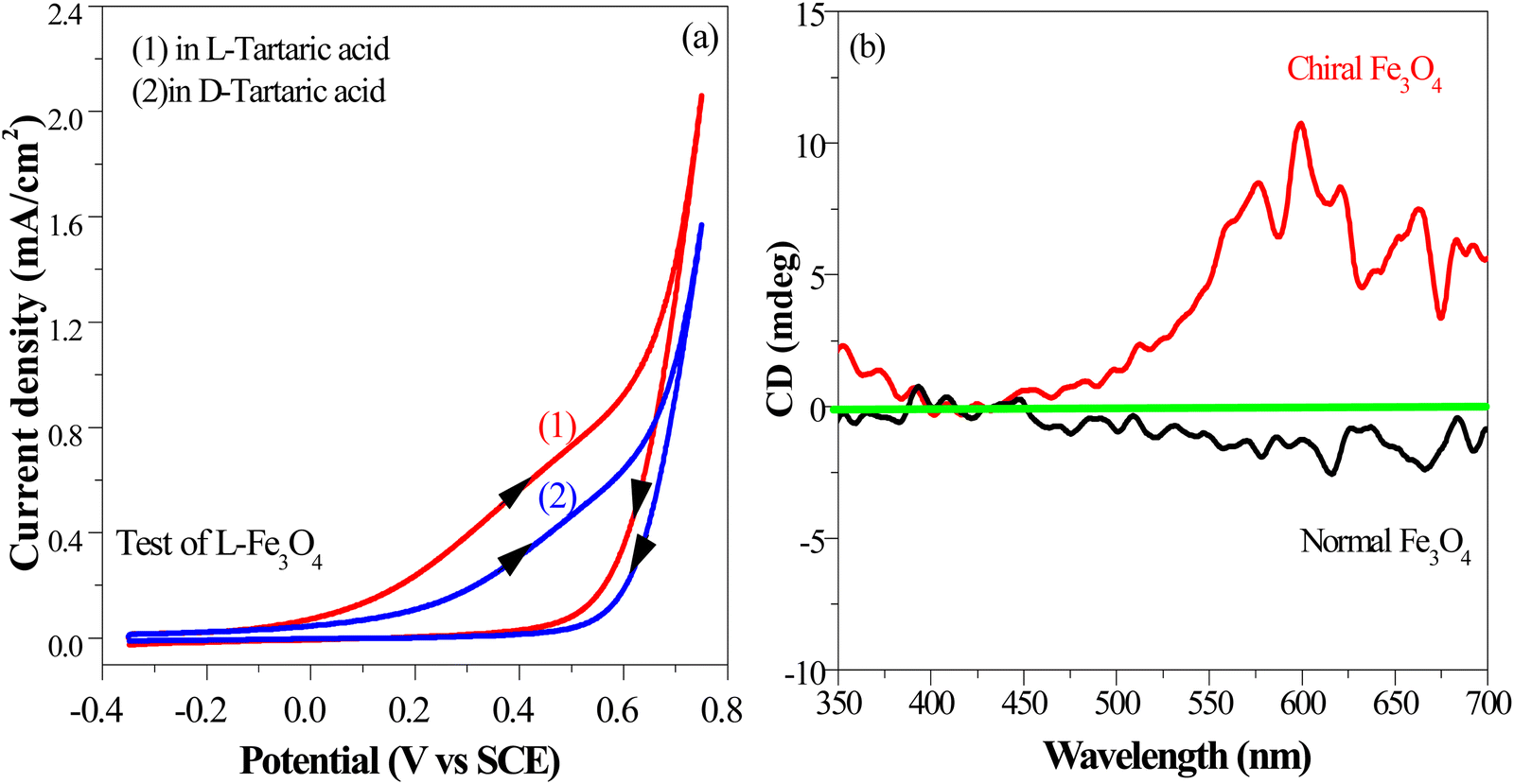 | ||
| Fig. 4 (a) Electrochemical selectivity test for chiral Fe3O4 (scanned in a chiral electrolyte involving 10 mM L-tartaric acid and in another chiral electrolyte involving 10 mM D-tartaric acid; arrows indicate scan direction). (b) CD spectra of the chiral Fe3O4 film and normal Fe3O4 film. | ||
To complement the findings from the electrochemical selectivity test, we further elucidated the surface chirality of the chiral Fe3O4 film using circular dichroism spectroscopy (CD) and polarized optical microscopy (POM) measurements performed in reflection mode. Fig. 4(b) demonstrates that the chiral Fe3O4 film prominently displays optical activity within the visible light region, whereas normal Fe3O4 exhibits no discernible optical activity within this region, suggesting inherent chirality with the chiral Fe3O4 film. This observation strongly implies the intrinsic chirality of the chiral Fe3O4 film. The results of POM are shown in Fig. S2 (ESI†), which also support the formation of a chiral film.
The chiral Fe3O4 film, which has inherent chiral structure, demonstrated a CISS effect on spin alignment during the OER. This was verified by monitoring the generation of the singlet-state by-product H2O2 throughout the OER process.32–34 The UV-vis spectra of the electrolyte in the water oxidation system are presented in Fig. 5(a), employing o-tolidine as an indicator to track the production of H2O2. When normal Fe3O4 was used as an electrocatalyst, an absorption peak emerged at approximately 436 nm, as H2O2 oxidized o-tolidine, leading to the formation of a yellow compound. However, this peak was substantially diminished when the chiral Fe3O4 film was utilized as an electrocatalyst, signifying the inhibition of H2O2 generation alongside OER.
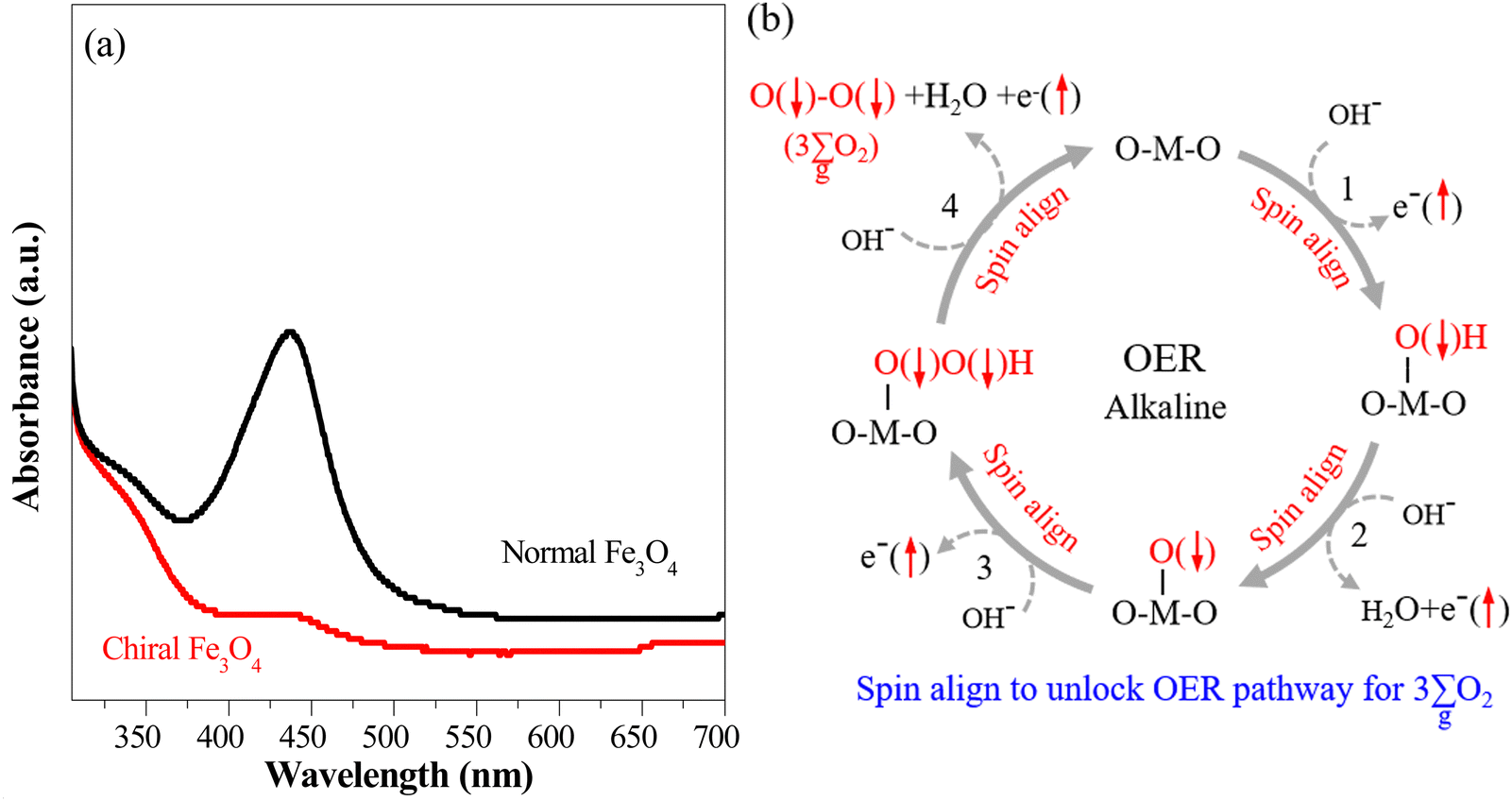 | ||
Fig. 5 “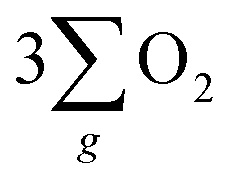 ”(a) Visible absorption spectra for the utilized electrolyte titrated with o-tolidine as an indicator to assess H2O2 by-product formation in systems employing the chiral Fe3O4 and normal Fe3O4 films as electrocatalysts. (b) Scheme for the CISS effect of chiral Fe3O4 on facilitating the water oxidation pathway of ”(a) Visible absorption spectra for the utilized electrolyte titrated with o-tolidine as an indicator to assess H2O2 by-product formation in systems employing the chiral Fe3O4 and normal Fe3O4 films as electrocatalysts. (b) Scheme for the CISS effect of chiral Fe3O4 on facilitating the water oxidation pathway of 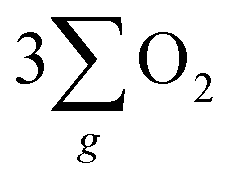 formation and forbidding the production of H2O2 during the OER in alkaline electrolyte. formation and forbidding the production of H2O2 during the OER in alkaline electrolyte. | ||
As depicted in Fig. 5(b), the OER pathway of Fe3O4 in alkaline media follows the adsorbate evolution mechanism (AEM), involving a sequential series of concerted four-proton-coupled electron transfer (PCET) processes that take place on active sites. These processes create multiple oxygen intermediates, including *OH, *O, and *OOH, to establish the pathway, *OH → *O → *OOH → O2 for the formation of O2. It is recognized that the OER process is spin sensitive, because the four electrons extracted from its four steps should have the same spin direction before the formation of the OO bond. Thereby, the spin orientations of electrons and radicals are beneficial for delivering 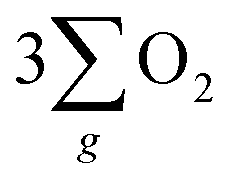 .
.
The inhibition of H2O2 by-product suggests that the chiral Fe3O4 film filtered the spin orientation of electrons acquired from water oxidation through the CISS effect,29,30,32,33 synchronously ensuring the alignment of spins in the newly generated ˙OH radicals that subsequently attached to the M sites of O–M–O species on the Fe3O4 surface during step 1 and step 3 of the AEM process, as demonstrated in Fig. 5(b). This facilitated the water oxidation pathway for the formation of the triplet-state product, 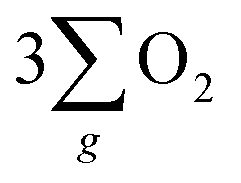 while promoting the alignment of electrons extracted from water oxidation throughout the four steps of AEM. Conversely, the formation of H2O2, which occurs on the singlet potential energy surface, was hindered.29,30,33
while promoting the alignment of electrons extracted from water oxidation throughout the four steps of AEM. Conversely, the formation of H2O2, which occurs on the singlet potential energy surface, was hindered.29,30,33
In accordance with the CISS-induced preference for the 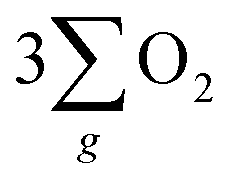 formation pathway during the OER, chiral Fe3O4 exhibited a lower water oxidation barrier than normal Fe3O4. As depicted in the inset of Fig. 6(a), the LSV onset potential was reduced in comparison with normal Fe3O4. Additionally, the potential required to achieve a current density of 10 mA cm−2 decreased by 150 mV when the chiral Fe3O4 film was used as an electrocatalyst.
formation pathway during the OER, chiral Fe3O4 exhibited a lower water oxidation barrier than normal Fe3O4. As depicted in the inset of Fig. 6(a), the LSV onset potential was reduced in comparison with normal Fe3O4. Additionally, the potential required to achieve a current density of 10 mA cm−2 decreased by 150 mV when the chiral Fe3O4 film was used as an electrocatalyst.
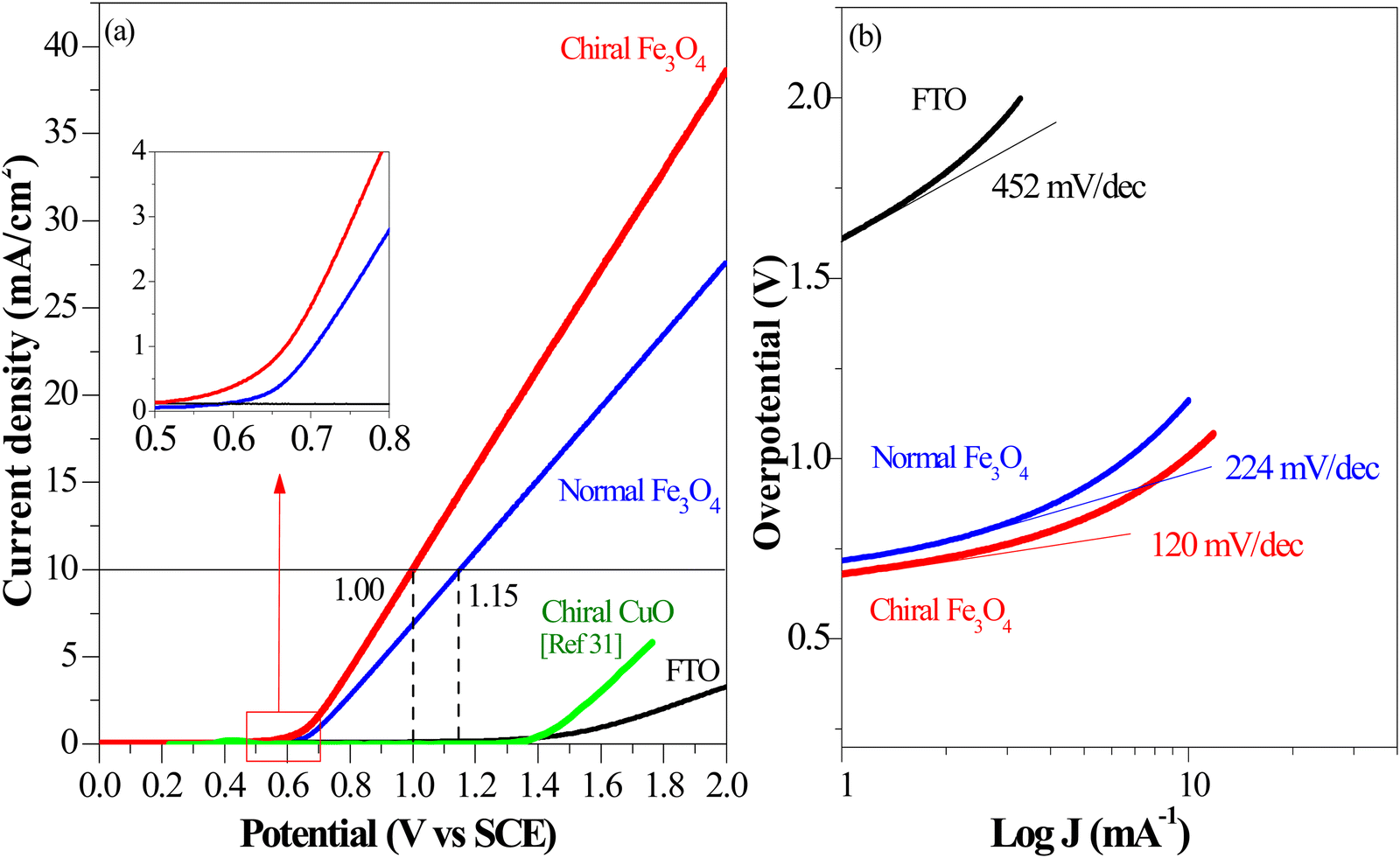 | ||
| Fig. 6 (a) Linear sweep voltammetry characterization of chiral Fe3O4, normal Fe3O4, FTO, and chiral CuO (the LSV of chiral Fe3O4, normal Fe3O4, and FTO were recorded in 0.1 M KOH as electrolyte at room temperature with calomel as reference; the LSV of chiral CuO refers to ref. 31). | ||
As mentioned above, being a half-metallic material, Fe3O4 should be fundamentally efficient for transferring spin-aligned electrons, even displaying metallic behavior. Here, it is noticed that the chiral Fe3O4 and normal Fe3O4 films demonstrated superior performance in OER current density compared to the chiral CuO film, which is not a half-metal, as shown in Fig. 6(a).
In addition, as seen in Fig. 6(a), the current density generated by the chiral Fe3O4 film at 1.8 V (vs. SCE) was 42% higher than that of normal Fe3O4, indicating that the chiral Fe3O4 film facilitated electron transfer compared to normal Fe3O4 film. Furthermore, as shown in Fig. 6(b), the Tafel slope of the chiral Fe3O4 film was 104 mV dec−1 smaller than that of normal Fe3O4, which suggests that the presence of chirality on the Fe3O4 film enhances OER kinetics and carrier transport. These results demonstrate that, in addition to its CISS effect in promoting the OER pathway for 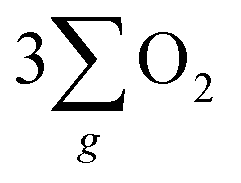 production, the chiral Fe3O4 film facilitates a more efficient transfer of spin-aligned electrons, derived from water oxidation, from the catalyst to the external circuit compared to the normal Fe3O4 film.
production, the chiral Fe3O4 film facilitates a more efficient transfer of spin-aligned electrons, derived from water oxidation, from the catalyst to the external circuit compared to the normal Fe3O4 film.
In line with the findings from LSV and Tafel characterization, the results from electrochemical impedance spectroscopy (EIS) reveal that the chiral Fe3O4 film exhibited smaller Nyquist circle diameters than normal Fe3O4 (see Fig. 7(a)). To gain further insights from these Nyquist plots, we employed an R(RQ) circuit model for fitting, and the fitting outcomes are summarized in Table 1. Herein, RΩ represents solution resistance, Qd denotes electrochemical double-layer capacitance, and Rct corresponds to charge transfer resistance. The Rct value for normal Fe3O4 was only 6% of that observed for FTO, suggesting the significance of Fe3O4 in promoting electron transfer for the OER. Notably, the chiral Fe3O4 film exhibits a remarkable 50% reduction in Rct compared to normal Fe3O4, suggesting that electron transport across Fe3O4 was enhanced due to its intrinsic chiral structure.
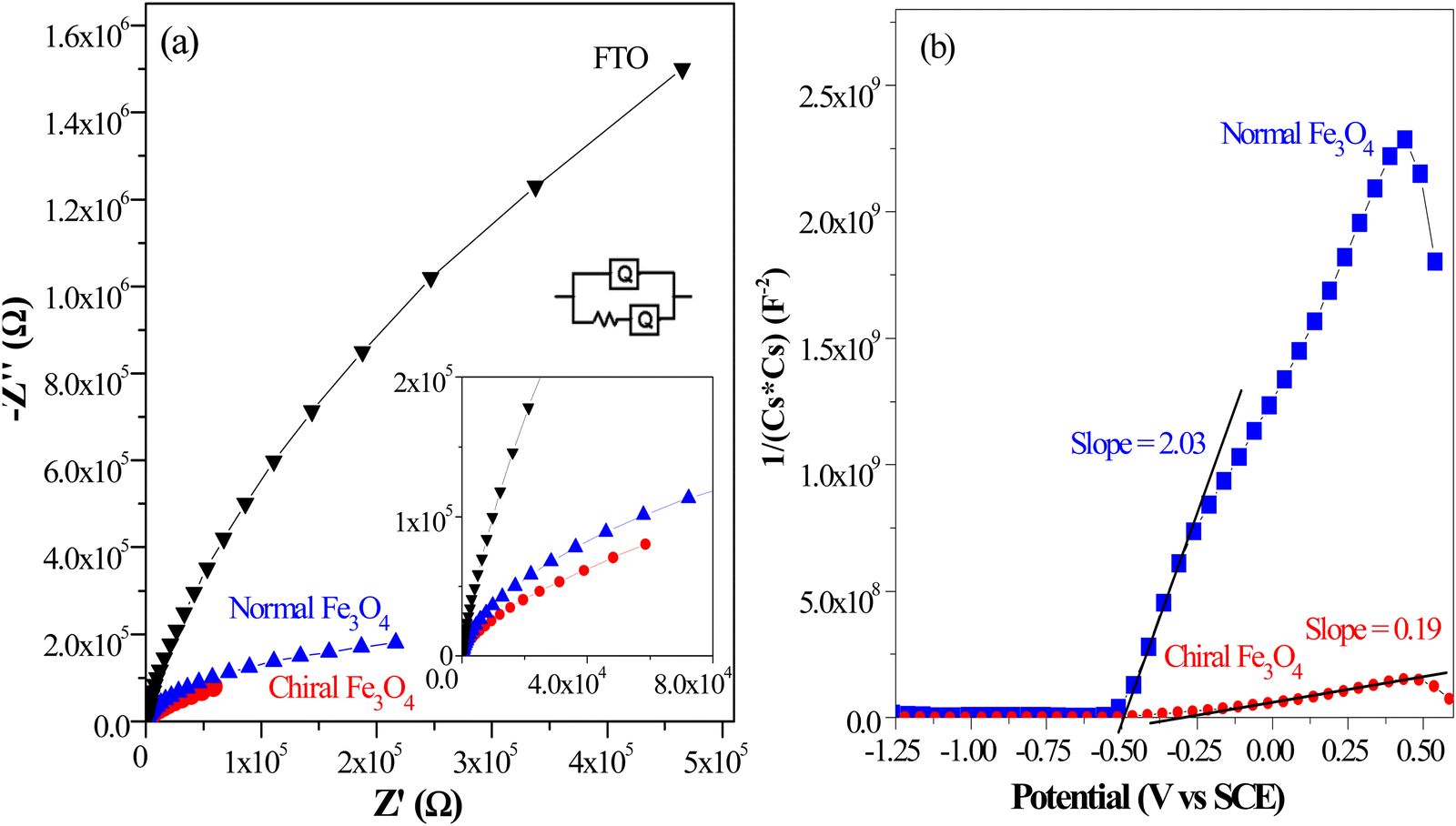 | ||
| Fig. 7 (a) Nyquist plots of L-Fe3O4, D-Fe3O4, normal Fe3O4 and FTO. (b) Mott–Schottky plots of L-Fe3O4, D-Fe3O4 and normal Fe3O4. | ||
| R Ω (Ω) | Q d (×10−4 F) | n | R ct (Ω) | |
|---|---|---|---|---|
| Fe3O4 | 42.95 | 0.2823 | 0.9434 | 3.314 × 105 |
| Chiral Fe3O4 | 33.48 | 1.122 | 0.95 | 1.369 × 105 |
| FTO | 36.45 | 0.08913 | 0.9642 | 5.135 × 106 |
Simultaneously, Mott–Schottky measurements provided further evidence of the pivotal role played by chirality in enhancing the catalytic activity of Fe3O4 film in the OER. The Mott–Schottky equation, expressed as (Csc)−2 = 2(E − Efb− kT/e)/(εε0A2eNd) (details in ESI†), dictates that the slope of the linear segments in the Mott–Schottky plot correspond to 2/(εε0A2eNd).52,53 These linear segments are associated with the depleted states of majority carriers located within the space charge region.54 As depicted in Fig. 7(b), the slope of the linear segment of normal Fe3O4 was approximately 2.03, while the slope of the chiral Fe3O4 film was around 0.19, merely 10% of that observed for normal Fe3O4. It is calculated that the charge carrier density (Nd) of normal Fe3O4 was about 3.5 × 1028 cm−3, while for the chiral Fe3O4 film, it was approximately 3.7 × 1029 cm−3. This result shows that the majority carrier density within the space charge region of Fe3O4 increased up to 10-fold due to the incorporation of chirality.
Furthermore, evidence from the Bode and phase angle plots serves to reinforce the enhanced electron transport capabilities observed in the chiral Fe3O4 film. In Fig. 8(a), the Bode plots illustrate that the chiral Fe3O4 film displays lower impedance modulus values compared to normal Fe3O4 and FTO. This observation signifies an increase in carrier mobility on the chiral Fe3O4 film.55 Moreover, as depicted in Fig. 8(b), the phase angle plot for chiral Fe3O4 shifts towards lower frequencies compared to normal Fe3O4. This red shift indicates accelerated carrier mobility across the chiral Fe3O4 film. Furthermore, Fig. 8(b) also reveals that the electron lifetime within the chiral Fe3O4 film is prolonged compared to that within the normal Fe3O4 film, taking into account that the electron lifetime (τe) can be calculated with the equation τe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = 1/2πfmax, where fmax represents the peak frequency.49,56 The elongated electron lifetime implies lower charge recombination probability in chiral Fe3O4 film than in normal Fe3O4 film.
= 1/2πfmax, where fmax represents the peak frequency.49,56 The elongated electron lifetime implies lower charge recombination probability in chiral Fe3O4 film than in normal Fe3O4 film.
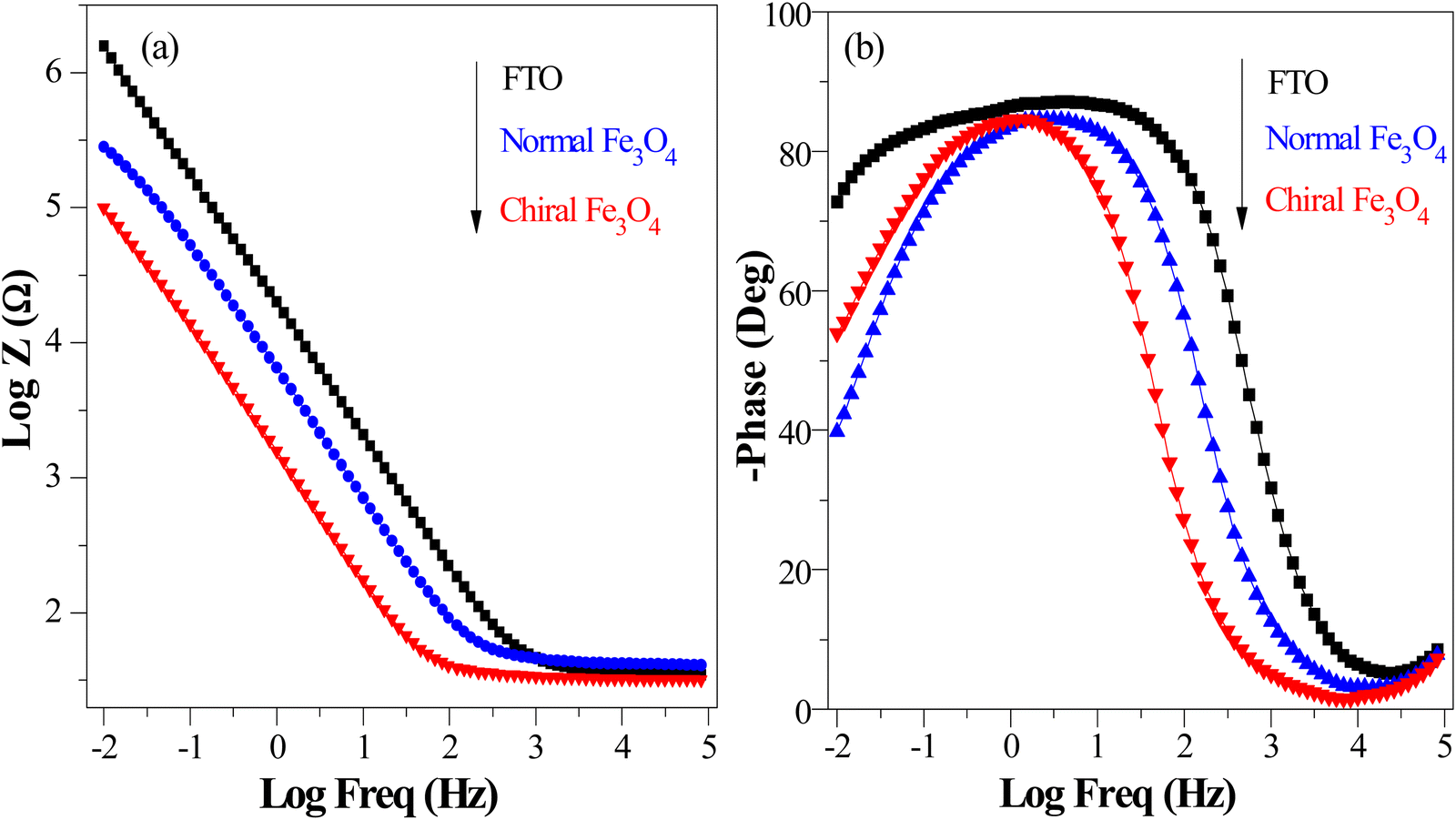 | ||
| Fig. 8 (a) Bode plots of chiral Fe3O4 film and normal Fe3O4 and FTO. (b) Phase angle plots of chiral Fe3O4 film and normal Fe3O4 and FTO. | ||
Additionally, hydrogen production in water splitting systems with different OER catalysts was demonstrated, as shown in Fig. 9(a) with chiral Fe3O4 film and normal Fe3O4 film as OER catalysts. The hydrogen production rate with chiral Fe3O4 film as OER catalyst was approximately 20% higher than that with normal Fe3O4 film as OER electrocatalyst and about 5 times of that with chiral CuO film as OER electrocatalyst. Furthermore, chiral Fe3O4 film exhibited robust electrocatalytic activity during the three experimental trials, as shown in Fig. S1 (ESI†). In each trial, the hydrogen production rate was enhanced by at least 20% when chiral Fe3O4 was employed as electrocatalyst rather than normal Fe3O4 as electrocatalyst. The growth of the hydrogen production rate is associated with the elevation of water oxidation kinetics and decline of the OER barrier, which was induced by the chiralization of the Fe3O4 catalyst. The stability also suggests the robustness of the chiral Fe3O4 film for the OER, as shown in Fig. S4 (ESI†).
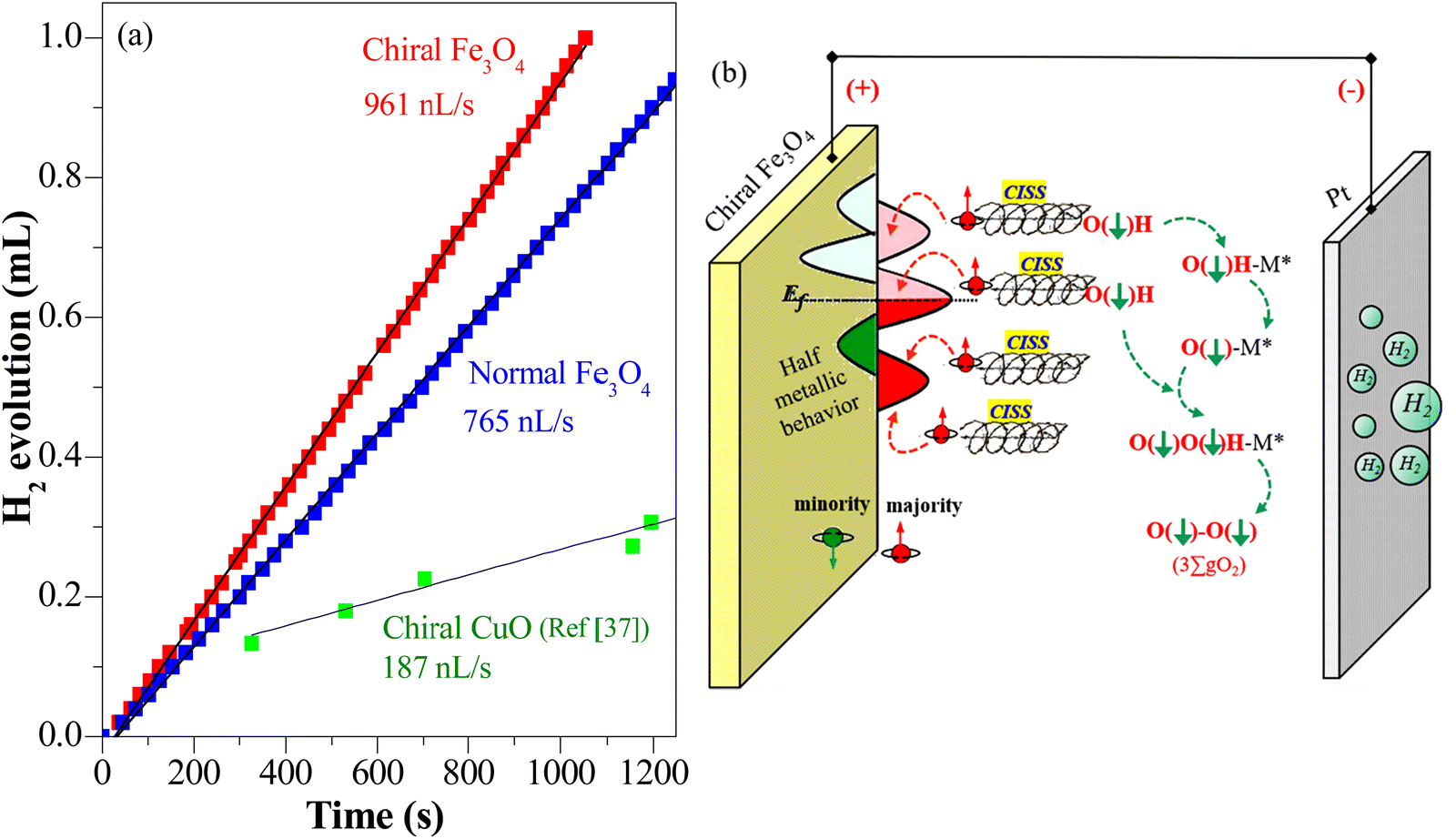 | ||
| Fig. 9 (a) Hydrogen production recorded in a water splitting system with chiral Fe3O4 and normal Fe3O4 as OER catalysts (conducted at a voltage of 1.5 V (vs. SCE) in 0.1 M KOH electrolyte, at room temperature, with calomel as reference, Pt wire as counter). The hydrogen production of chiral CuO refers to ref. 37. (b) Scheme of the CISS effect in conjunction with the half-metallic nature of chiral Fe3O4 film for aligning the spins of electrons and radicals for the OER and the transportation of spin-oriented electrons. | ||
The evidence presented above coherently demonstrates the advantages offered by the chiral Fe3O4 film compared to normal Fe3O4 film and chiral CuO. As illustrated in Fig. 9(b), during the OER, the CISS effect of the chiral Fe3O4 film aligned the spins of electrons and radicals. This spin-controllable process improved the possibility for the formation of the triplet-state product 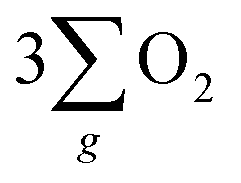 , promoted the AEM water oxidation pathway, and thereby reduced the barrier for the OER. Meanwhile, the half-metallic nature of Fe3O4 facilitated the transfer of spin-aligned electrons. The combined and synergistic influence of these factors plays a pivotal role in unlocking the catalytic potential of Fe3O4 film for the OER.
, promoted the AEM water oxidation pathway, and thereby reduced the barrier for the OER. Meanwhile, the half-metallic nature of Fe3O4 facilitated the transfer of spin-aligned electrons. The combined and synergistic influence of these factors plays a pivotal role in unlocking the catalytic potential of Fe3O4 film for the OER.
3. Conclusion
In summary, this work demonstrated a strategy that harnesses the synergy of chirality and half-metallic materials for unlocking the electrochemical catalytic potential of the OER. Though the current density enhancement of chiral Fe3O4 over normal Fe3O4 is not too high, it provides a promising way to facilitate the OER pathway towards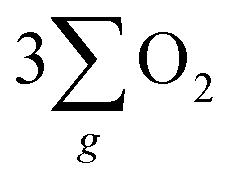 production through the CISS-induced spin control during AEM. When contrasted with normal Fe3O4 film which lacks the capability to align electrons, the chiral Fe3O4 film suppressed the pathway of H2O2 by-product formation, decreased the potential required to obtain 10 mA cm−2 current density by 150 mV, reduced the Tafel slope by 104 mV dec−1, elevated the current density by 42% at 1.8 V (vs. SCE), lowered the charge transfer resistance by 50%, achieved a 10-fold amplification of the free charge carrier density, and extended the electron lifetime within the catalyst. Furthermore, chiral Fe3O4 exhibited enhanced OER performance over chiral CuO as the transfer of spin-aligned electrons extracted from water oxidation to the external circuit was promoted due to the half-metallic nature of Fe3O4.
production through the CISS-induced spin control during AEM. When contrasted with normal Fe3O4 film which lacks the capability to align electrons, the chiral Fe3O4 film suppressed the pathway of H2O2 by-product formation, decreased the potential required to obtain 10 mA cm−2 current density by 150 mV, reduced the Tafel slope by 104 mV dec−1, elevated the current density by 42% at 1.8 V (vs. SCE), lowered the charge transfer resistance by 50%, achieved a 10-fold amplification of the free charge carrier density, and extended the electron lifetime within the catalyst. Furthermore, chiral Fe3O4 exhibited enhanced OER performance over chiral CuO as the transfer of spin-aligned electrons extracted from water oxidation to the external circuit was promoted due to the half-metallic nature of Fe3O4.
Notes
Experimental details and characterization details of this work are shown in our ESI.†Data availability
The data that supports the findings of this study are available within the article and its supplementary material. No additional data are available.Author contributions
Wenyan Zhang: conceptualization, supervision, writing – reviewing and editing, investigation, project administration, funding acquisition. Chaoqun Jiang: conceptualization, methodology, writing – reviewing and editing, investigation. Hangmin Guan: conceptualization, methodology, writing – reviewing and editing, investigation. Yuanyuan Wang: methodology, investigation, writing – original draft, funding acquisition. Yingfei Hu: methodology, investigation, writing – original draft, funding acquisition. Wei Wang: formal analysis, investigation. Wenjie Tian: visualization, formal analysis. Lingyun Hao: formal analysis, writing – original draft.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank the support from the Natural Science Foundation of China (22206065, 61804069, 51802130), Natural Science Foundation of Jiangsu Province (BK20221167), Jiangsu Overseas Visiting Scholar Program for University Prominent Young & Middle-aged Teachers and Presidents, fund of Nanjing Optometric New Materials and Application Technology Innovation Team, and QingLan project of Jiangsu Province. Dr Wenyan Zhang appreciates the spiritual support from her parents in heaven.References
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- W. L. Zhen, X. Yuan, X. F. Ning, X. Z. Gong and C. Xue, ACS Appl. Mater. Interfaces, 2020, 12, 868–876 CrossRef CAS PubMed.
- R. Zhang, Z. H. Wei, G. Y. Ye, G. J. Chen, J. J. Miao, X. H. Zhou, X. W. Zhu, X. Q. Cao and X. N. Sun, Adv. Energy Mater., 2021, 11, 2101758 CrossRef CAS.
- Y. F. Hu, H. T. Huang, J. Y. Feng, W. Wang, H. M. Guan, Z. S. Li and Z. G. Zou, Sol. RRL, 2021, 5, 2100100 CrossRef CAS.
- Y. Zeng, M. T. Zhao, Z. H. Huang, W. J. Zhu, J. X. Zheng, Q. Jiang, Z. C. Wang and H. F. Liang, Adv. Energy Mater., 2022, 12, 2201713 CrossRef CAS.
- N. Dean, Nat. Energy, 2022, 7, 785–787 CrossRef.
- N. Kittner, F. Lill and D. Kammen, Nat. Energy, 2017, 2, 17125 CrossRef.
- Y. Z. Zhang, Engineering, 2017, 3, 431 CrossRef CAS.
- J. Chen, Q. L. Zeng, X. P. Qi, B. J. Peng, L. Xu, C. Liu and T. X. Liang, Int. J. Hydrogen Energy, 2020, 45, 24828–24839 CrossRef CAS.
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- H. Dotan, A. Landman, S. W. Sheehan, K. D. Malviya, G. E. Shter, D. A. Grave, Z. Arzi, N. Yehudai, M. Halabi, N. Gal, N. Hadari, C. Cohen, A. Rothschild and G. S. Grader, Nat. Energy, 2019, 4, 786–795 CrossRef CAS.
- S. Anwar, F. Khan, Y. H. Zhang and A. Djire, Int. J. Hydrogen Energy, 2021, 46, 32284–32317 CrossRef CAS.
- M. Nemiwal, V. Gosu, T. C. Zhang and D. Kumar, Int. J. Hydrogen Energy, 2021, 46, 10216–10238 CrossRef CAS.
- K. L. Ao, Q. F. Wei and W. A. Daoud, ACS Appl. Mater. Interfaces, 2020, 12, 33595–33602 CrossRef CAS PubMed.
- P. Bhanja, Y. Kim, B. Paul, Y. V. Kaneti, A. A. Alothman, A. Bhaumik and Y. Yamauchi, Chem. Eng. J., 2021, 405, 126803 CrossRef CAS.
- Y. J. Guo, C. R. Zhang, J. Zhang, K. Dastafkan, K. Wang, C. Zhao and Z. Q. Shi, ACS Sustainable Chem. Eng., 2021, 9, 2047–2056 CrossRef CAS.
- N. L. W. Septiani, Y. Valentino Kaneti, K. B. Fathoni, Y. N. Guo, Y. Ide, B. Yuliarto, X. C. Jiang, N. Nugraha, H. K. Dipojono, D. Golberg and Y. Yamauchi, J. Mater. Chem. A, 2020, 8, 3035–3047 RSC.
- S. G. Hu, S. Q. Wang, C. Q. Feng, H. M. Wu, J. J. Zhang and H. Mei, ACS Sustainable Chem. Eng., 2020, 8, 7414–7422 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, Y. N. Guo, B. Yuliarto, X. C. Jiang, Y. Ide, N. Nugraha, H. K. Dipojono, A. B. Yu, Y. Sugahara, D. Golberg and Y. Yamauchi, ChemSusChem, 2020, 13, 1645–1655 CrossRef CAS PubMed.
- Y. Zuo, Y. P. Liu, J. S. Li, R. F. Du, X. Han, T. Zhang, J. Arbiol, N. J. Divins, J. Llorca, N. Guijarro, K. Sivula and A. Cabot, Chem. Mater., 2019, 31, 7732–7743 CrossRef CAS.
- D. Li, J. Y. Shi and C. Li, Small, 2018, 14, 1704179 CrossRef PubMed.
- R. R. Mohamed, G. A. E. I. Muhammad, H. A. Hosam, M. R. Wael, A. H. Mohamed, F. F. Hazem, M. Mohsen, A. S. Kamel and S. E. D. Mohamed, Int. J. Hydrogen Energy, 2022, 47, 32145–32157 CrossRef.
- J. Du, F. Li and L. C. Sun, Chem. Soc. Rev., 2021, 50, 2663–2695 RSC.
- F. A. Garcés-pineda, M. Blasco-ahicart, D. Nieto-castro, N. López and J. R. Galán-mascarós, Nat. Energy, 2019, 4, 519–525 CrossRef.
- X. Li, Z. Cheng and X. Wang, Electrochem. Energy Rev., 2021, 4, 136–145 CrossRef CAS.
- D. J. Zhou, P. S. Li, X. Lin, A. McKinley, Y. Kuang, W. Liu, W. F. Lin, X. M. Sun and X. Duan, Chem. Soc. Rev., 2021, 50, 8790–8817 RSC.
- C. Han, L. Zhong, Q. H. Sun, D. D. Che, T. T. Li, Y. Hu, J. J. Qian and S. M. Huang, J. Power Sources, 2021, 499, 229947 CrossRef CAS.
- X. Ren, T. Z. Wu, Y. M. Sun, Y. Li, G. Y. Xian, X. H. Liu, C. M. Shen, J. Gracia, H. J. Gao, H. T. Yang and Z. C. Xu, Nat. Commun., 2021, 12, 2608 CrossRef CAS PubMed.
- W. Mtangi, V. Kiran, C. Fontanesi and R. Naaman, J. Phys. Chem. Lett., 2015, 6, 4916–4922 CrossRef CAS PubMed.
- W. Y. Zhang, K. Banerjee-ghosh, F. Tassinari and R. Naaman, ACS Energy Lett., 2018, 3, 2308–2313 CrossRef CAS.
- G. Solomon, M. G. Kohan, M. Vagin, F. Rigoni, R. Mazzaro, M. M. Natile, S. J. You, V. Morandi, I. Concina and A. Vomiero, Nano Energy, 2021, 81, 105664 CrossRef CAS.
- W. Mtangi, F. Tassinari, K. Vankayala, A. V. Jentzsch, B. Adelizzi, A. R. A. Palmans, C. Fontanesi, E. W. Meijer and R. Naaman, J. Am. Chem. Soc., 2017, 139, 2794–2798 CrossRef CAS PubMed.
- F. Tassinari, K. Banerjee-ghosh, F. Parenti, V. Kiran, A. Mucci and N. R. Naama, J. Phys. Chem. C, 2017, 121, 15777–15783 CrossRef CAS.
- R. Naaman and D. H. Waldeck, J. Phys. Chem. Lett., 2013, 3, 2178–2187 CrossRef PubMed.
- W. Y. Zhang, W. Wang, Y. F. Hu, H. M. Guan, X. L. Yang and L. Y. Hao, Int. J. Hydrogen Energy, 2021, 46, 8922–8931 CrossRef CAS.
- S. Ghosh, B. P. Bloom, Y. Lu, D. Lamont and D. H. Waldeck, J. Phys. Chem. C, 2020, 124, 22610–22618 CrossRef CAS.
- K. B. Ghosh, W. Y. Zhang, F. Tassinari, Y. Mastai, O. Lidor-Shalev, R. Naaman, P. Möllers, D. Nürenberg, H. Zacharias, J. Wei, E. Wierzbinski and D. H. Waldeck, J. Phys. Chem. C, 2019, 123, 3024–3031 CrossRef CAS.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- L. L. Gao, C. Y. Tang, J. C. Liu, N. L. He, H. B. Wang, Z. J. Ke, W. Q. Li, C. Z. Jiang, D. He, L. Cheng and X. H. Xiao, Energy Environ. Sci., 2021, 4, 392–398 CAS.
- X. X. Li and J. L. Yang, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1314 Search PubMed.
- T. Bai, J. Ai, J. Ma, Y. Y. Duan, L. Han, J. G. Jiang and S. A. Che, Angew. Chem., Int. Ed., 2021, 60, 20036–20041 CrossRef CAS PubMed.
- W. E. Pickett and J. S. Moodera, Phys. Today, 2001, 54, 39 CrossRef CAS.
- J. H. Park, E. Vescovo, H. J. Kim, C. Kwon, R. Ramesh and T. Venkatesan, Nature, 1998, 392, 794–796 CrossRef CAS.
- X. Hu, Adv. Mater., 2012, 24, 294–298 CrossRef CAS PubMed.
- S. Jain and A. O. Adeyeyea, J. Appl. Phys., 2005, 97, 093713 CrossRef.
- S. Li, J. C. Tang, Q. L. Liu, X. M. Liu and B. Gao, Environ. Int., 2020, 138, 105639 CrossRef CAS PubMed.
- X. L. Wang, Y. G. Liu, H. Y. Han, Y. Y. Zhao, W. M. Ma and H. Y. Sun, Sustainable Energy Fuels, 2017, 1, 915–7922 RSC.
- B. Devi, M. Venkateswarulu, H. S. Kushwaha, A. Halder and R. R. Koner, Chem. – Eur. J., 2018, 24, 1–10 CrossRef PubMed.
- W. He, Z. Li, S. Lv, M. Niu, W. Zhou, J. Li, R. Lu, H. Gao, C. Pan and S. Zhang, Chem. Eng. J., 2021, 409, 128274 CrossRef CAS.
- E. W. Bohannan, H. M. Kothari, I. M. Nicic and J. A. Switzer, J. Am. Chem. Soc., 2004, 126, 488–489 CrossRef CAS PubMed.
- W. Y. Zhang, J. Li, G. X. Lu, H. M. Guan and L. Y. Hao, Chem. Commun., 2019, 55, 13390–13393 RSC.
- C. H. Liu, F. Wang, J. Zhang, K. Wang, Y. Y. Qiu, Q. Liang and Z. D. Chen, Nano-Micro Lett., 2018, 10, 1–13 CrossRef CAS PubMed.
- A. S. Bondarenko and G. A. Ragoisha, J. Solid State Electrochem., 2005, 9, 845–849 CrossRef CAS.
- B. A. Koiki, B. O. Orimolade, B. N. Zwane, D. Nkosi, N. Mabuba and O. A. Arotiba, Electrochim. Acta, 2020, 340, 135944 CrossRef CAS.
- J. W. Fang, H. Q. Fan, M. M. Li and C. B. Long, J. Mater. Chem. A, 2015, 3, 13819–13826 RSC.
- J. J. Tian, L. L. Lv, C. B. Fei, Y. J. Wang, X. G. Liu and G. Z. Cao, J. Mater. Chem. A, 2014, 2, 19653–19659 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00854a |
| This journal is © The Royal Society of Chemistry 2024 |