
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTiO2 core–shell and core-dual-shell nanoparticles with tunable heterojunctions and visible to near-infrared extinctions†
Riddhiman
Medhi
 a,
Sarawut
Plengjaroensirichai
b,
Nhat
Ngo
b,
Maria D.
Marquez
b,
Pannaree
Srinoi
c,
Hung-Vu
Tran
d,
Allan J.
Jacobson
b,
Tai-Chou
Lee
e and
T. Randall
Lee
*b
a,
Sarawut
Plengjaroensirichai
b,
Nhat
Ngo
b,
Maria D.
Marquez
b,
Pannaree
Srinoi
c,
Hung-Vu
Tran
d,
Allan J.
Jacobson
b,
Tai-Chou
Lee
e and
T. Randall
Lee
*b
aDepartment of Chemistry, University of Scranton, 800 Linden Street, Scranton, Pennsylvania 18510, USA
b5004 SERC, Department of Chemistry and the Texas Center for Superconductivity, University of Houston, 4800 Calhoun Road, Houston, Texas 77204-5003, USA. E-mail: trlee@uh.edu; Fax: +1 (281) 754-4445; Tel: +1 (713) 743-2724
cDepartment of Chemistry, Faculty of Science, Kasetsart University, Bangkok 10900, Thailand
dNTT Hi-Tech Institute, Nguyen Tat Thanh University, 298-300A Nguyen Tat Thanh Street, District 4, Ho Chi Minh City 7280, Viet Nam
eDepartment of Chemical and Materials Engineering, National Central University, 300 Jhongda Road, Jhongli City 32001, Taiwan
First published on 12th January 2024
Abstract
The popular use of titanium dioxide (TiO2) among metal oxides in a variety of optoelectronic applications can be attributed to its low cost and strong extinction in the UV region. However, TiO2 suffers from an inability to utilize a major part of the solar spectrum, and from rapid electron–hole recombination of photo-generated charge carriers. These challenges can be resolved by incorporating gold–silver nanoshells (GS–NS) with localized surface plasmon resonances (LSPR) into TiO2 nanostructures, with the overall goal of understanding near-field LSPR activation as well as electron-transfer and charge-trapping characteristics in core–shell and core-dual-shell systems. A method for the synthesis of core–shell nanoparticles consisting of a hollow gold–silver nanoshell core with a TiO2 shell (GS–NS@TiO2) is presented. Synthesis of core-dual-shell nanoparticles with either a semiconducting or insulating interlayer between the GS–NS core and TiO2 shell (GS–NS@SnO2@TiO2 and GS–NS@SiO2@TiO2) is also presented. In addition to a strong tunable surface plasmon resonance in the visible to near-IR region that allows better utilization of the solar spectrum by the TiO2 shell, incorporation of a GS–NS core leads to significant suppression of electron–hole recombination processes in TiO2, further demonstrating the advantages of the plasmonic core and metal oxide shell architecture. The near-field LSPR properties and charge transfer characteristics were further modified by the incorporation of SnO2 or SiO2 interlayers, with the SnO2 interlayer providing the most effective suppression of charge recombination. Reliable methods to fabricate composite TiO2-based nanoparticles with finely tunable optical and electrical properties are reported.
1. Introduction
Wide band gap (2–4 eV) metal oxide semiconductors are important materials for the fabrication of transparent electronics, thin-film transistors (TFTs), acoustic devices, solar cells, light-emitting diodes (LEDs), and sensors.1–11 With the demand for these materials expanding rapidly, especially in the field of photocatalysis, the cost-effective fabrication of materials in the form of nanoscale colloidal particles, is an important task.7 Recently, metal oxide nanoparticles have been shown to be very effective materials for photocatalytic applications like water splitting, hydrogen generation, CO2 reduction, degradation of dyes, organic and industrial pollutants, as well as for antibacterial treatment.5,7 For photocatalysis, which is primarily a surface process, a higher surface area per mass of material becomes a critical factor for commercial utilization. In addition to their deposition as finely structured thin films, colloidal nanoparticles also offer flexibility for use in suspensions, colloidal solution-phase reactions, and incorporation within polymeric structures.12–16 Titanium dioxide (TiO2) is the earliest discovered and most widely used metal oxide photocatalyst by virtue of its low cost, low toxicity, high photoactivity, and good chemical and thermal stability.17 Like many other metal oxides, however, TiO2 faces two major challenges to widespread practical use in solar light-induced photocatalytic and photovoltaic applications. The first limitation is its inability to utilize the majority of the solar spectrum that reaches the earth's surface; due to its wide band gap, extinction is limited to the UV region only.17 The second limitation for TiO2 is the poor quantum yield caused by rapid recombination of photogenerated electrons and holes.16–18Noble metal nanoparticles are a class of materials known for strong extinction of light in visible to near-IR regions. Localized surface plasmon resonance (LSPR) leads to strong extinction of light with a frequency matching that of the oscillating electrons in the nanoparticle. In noble metal nanoparticles, these resonances correspond to strong extinctions in the visible and near-IR wavelengths.19–21 Gold nanoparticles (Au NPs) are the most commonly used type of materials for this purpose, with their extinction in the range between 520–560 nm.19,22 This LSPR extinction can be extended towards the longer wavelengths by changing the morphology to nanorods, nanocubes, nanostars, nanoshells, or nanospheres of larger sizes.23–27 Various types of gold nanoparticles have been studied in core–shell systems with TiO2 and have effectively enhanced their visible-light photoactivity.27–32 Recently, hollow nanoshells of gold alloyed with silver (GS–NS) were demonstrated to be a much more stable alternative to gold nanoparticles, affording a wider LSPR signal using lesser and cheaper material.33 The extinction of GS–NS is tunable across the entire range between 500–1000 nm.16 In a previous paper, our groups showed that incorporating a GS–NS core inside a SnO2 shell can activate intense extinction of light in visible and near-IR regions.34 Additionally, the GS–NS core led to a remarkable near-complete suppression in the rate of electron–hole recombination in the SnO2 shell; demonstrating the distinct advantages of the metal core- metal oxide shell architecture.
Encouraging results observed with SnO2 suggests that GS–NSs could have a similar potential to overcome the limitations of TiO2 as well. SnO2 and TiO2 are similar in terms of their crystal structure (rutile and anatase) and bond lengths (∼1.9 Å), giving rise to comparable band gaps (∼3.2–3.6 eV).35,36 However, the optical and electronic properties can be quite different. Theoretical studies of their electronic structures (both rutile and anatase) show that the conduction band (CB) of TiO2 has several dense energetically flat levels, while the CB of SnO2 has a single highly dispersive level.35 As a consequence, TiO2 absorbs UV wavelengths much more strongly compared to SnO2. To add to the strong UV extinction, this work attempts to utilize the properties of a GS–NS core with metal oxide shell architecture for enhancing visible and near-IR utilization and charge separation of TiO2. Most recipes for uniform TiO2 nanoparticle synthesis involve controlled hydrolysis of titanium alkoxides in non-aqueous solvents.37–41 This route is typically preferred because titanium alkoxides spontaneously hydrolyze to form TiO2 when exposed to water, hence controlled growth of TiO2 shells cannot be performed in an aqueous environment.37–41 However, most popular methods for plasmonic noble metal nanoparticle synthesis involve aqueous systems.16,42–45 This report describes an effective synthetic strategy to circumvent this problem to achieve the controlled growth of TiO2 shells with various thickness around plasmonic GS–NS cores.
In addition to noble metal nanoparticle coupling, other strategies explored to increase electron–hole pair separation efficiency in TiO2 are nanoscale heterostructures with other semiconductors, which have received attention due to their excellent charge separation and catalytic activities.46,47 Among the semiconductor/TiO2 heterojunction systems, the SnO2/TiO2 heterostructure attracts interest in particular due to their structural analogy. Both oxides have the same crystal symmetry (tetragonal) and have two molecular units per unit cell (Z = 2).46 SnO2 and TiO2 form a type-II heterojunction. The valence band (VB) of TiO2 is energetically positioned between the VB and CB of SnO2, while the CB of TiO2 is positioned above the VB and CB of SnO2.46 When activated simultaneously, this staggered conformation of energy levels means that the CB of SnO2 can act as a sink for photogenerated electrons.46 The photogenerated holes move in the opposite direction and accumulate in the VB of TiO2. This formation of an excitonic charge separation state leads to an effective separation of electrons and holes, reducing recombination.48 This feature is beneficial to applications such as photovoltaic devices and photocatalysts. The SnO2/TiO2 core–shell heterojunction characteristics have been extremely effective for dye-sensitized solar cells due to an effective inhibition of back-electron transfer between the oxidized dye and reduced semiconductor.47,49,50 So far, such metal oxide heterojunctions have been mostly fabricated and studied as thin films, and nanowire arrays.46,51 In this current study, a strategy to synthesize this heterojunction in a colloidal nanoparticle is described. Previously, our group has synthesized the TiO2/SnO2 heterojunction with TiO2 as the core and SnO2 as the shell, making a reduction photocatalyst material, with electrons collecting at the outer SnO2 shell.36 The nanoparticles reported in this paper feature the reverse heterojunction, employing a TiO2 shell outside a SnO2-based core which would concentrate holes on the TiO2 shell outside, making them more effective as an oxidation photocatalyst. Photogenerated holes oxidize hydroxide ions to hydroxide radicals which act as the major oxidizing species in oxidation photocatalysis.51 Importantly, in this report, a three-layer composite with TiO2 shells around a plasmonic GS–NS with a SnO2 interlayer is synthesized to generate the plasmonically-activated SnO2/TiO2 heterojunction core-dual-shell nanoparticle - GS–NS@SnO2@TiO2.
According to a previous report by Li et al., photocatalytic activation in ZnIn2S4 was greatly enhanced by incorporating a GS–NS core, which was further improved by the presence of an insulating SiO2 interlayer.16 As a different kind of dielectric to SnO2, an insulating SiO2 interlayer is expected to block electron movement between the TiO2 shell and GS–NS core. However, there is no direct evidence in the literature to confirm this hypothesis. To study this effect, GS–NS@SiO2@TiO2 nanoparticles were also synthesized. A facile and versatile method is reported herein for the controlled growth of TiO2 shells directly on a metal core such as GS–NSs, as well as on GS–NSs with an insulating (SiO2) and semiconducting (SnO2) interlayers. The effects of the different interlayers on the near-field LSPR properties and charge transfer between the GS–NS core and TiO2 shell has been investigated. A better understanding of these different types of interlayers in such a system adds an extra degree of control in tuning the optical and electrical properties on the nanoscale. All of the nanoparticles were characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), energy dispersive X-ray spectroscopy (EDX), X-ray photoelectron spectroscopy (XPS), and X-ray diffraction (XRD). Separately, the optical properties were evaluated by UV-vis spectroscopy and the rates of electron–hole recombination in these various TiO2-based systems were compared using photoluminescence spectroscopy (PL).
2. Materials and methods
2.1. Synthesis of hollow gold–silver nanoshells (GS–NS)
Hollow gold–silver nanoshells (GS–NS) were prepared as outlined in Scheme S1 (ESI†), using the recipe previously reported by our group.34 After removing the supernatant, the residue was redispersed in 10 mL of Milli-Q water.2.2. Synthesis of GS–NS@TiO2 core–shell nanoparticles
The TiO2 coating recipe, outlined in Scheme 1, is a modification of the recipe reported by Lee et al.52 The first step is phase-transfer of the GS–NSs to an ethanol solvent. Specifically, 0.02 g of hydroxypropyl cellulose (HPC) was added to a 10 mL solution of GS–NSs in Milli-Q-water from the previous step and sonicated for 90 min for functionalization. After functionalization with HPC, 15 mL of EtOH was added to this dispersion, sonicated again for 10–15 min, and then centrifuged at 8000 rpm for 15 min. The supernatant was discarded, 15 mL of EtOH was added, and the solution sonicated again for 15 min. To reduce or eliminate the formation of unwanted TiO2 from hydrolysis of TBOT in free solution, anhydrous EtOH from a freshly opened bottle was used at each step. This solution was then centrifuged at 8000 rpm for 15 min, the supernatant was discarded to remove any remaining water, and then finally redispersed in 1 mL of EtOH. | ||
| Scheme 1 Synthesis route for GS–NS@TiO2, GS–NS@SnO2@TiO2, and GS–NS@SiO2@TiO2 nanoparticles. | ||
Subsequently, this concentrated solution of GS–NSs in EtOH was transferred to a glass vial, and 0.02 g of HPC was added. To activate the controlled hydrolysis of TBOT, 300 μL of Milli-Q-water was also added to this vial, and the contents were sonicated for 90 min. Subsequently, 180 μL (0.528 mmol) of TBOT was dissolved in 4 mL of EtOH from a freshly opened bottle. Using a syringe pump, 3 mL of this TBOT solution was then slowly added to the HPC-functionalized GS–NSs over 30 min under stirring. The reaction vial was then stirred overnight at rt. The contents of the reaction vial were then transferred to a 75 mL round-bottom flask and refluxed at 85 °C for 90 min. The contents were then cooled to rt and centrifuged, typically at 8000 rpm for 15 min, washed with EtOH, centrifuged again, and finally redispersed in 1 mL of EtOH. The nanoparticles thus obtained after the first TiO2 coating are denoted as GS–NS@TiO2(×1). To increase the thickness of the TiO2 shell, a second coating was performed on the GS–NS@TiO2(×1) nanoparticles. Specifically, 0.02 g of HPC was added to 1 mL of GS–NS@TiO2(×1) and sonicated for 90 min. Subsequently, 180 μL (0.528 mmol) of TBOT was dissolved in 4 mL of EtOH from a freshly opened bottle. Using a syringe pump, 3 mL of this TBOT solution was then slowly added to the HPC-functionalized GS–NSs over 30 min under stirring. The reaction was stirred overnight at rt, then the contents were transferred to a 75 mL round-bottom flask and refluxed at 85 °C for 90 min. The contents were then centrifuged, typically at 8000 rpm for 15 min, washed twice with EtOH, centrifuged again, and finally redispersed in 1 mL of EtOH. The nanoparticles thus obtained after the second TiO2 coating are denoted as GS–NS@TiO2(×2).
2.3. Synthesis of GS–NS@SnO2 core–shell nanoparticles
As outlined in Scheme 1, the recipe reported previously by our group was used to synthesize the SnO2-coated GS–NS.34 The particles were washed once after synthesis and finally redispersed in 10 mL of Milli-Q water.2.4. Synthesis of GS–NS@SiO2 core–shell nanoparticles
The SiO2 coated GS–NS were adapted from a previously reported method, as outlined in Scheme 1.16 Briefly, 10 mL of the GS–NS solution was diluted to 20 mL and added to a 150 mL round bottom flask. Separately, 2 mL of ammonium hydroxide was mixed with 45 mL of fresh ethanol and this mixture was added to the flask containing the diluted GS–NS solution. After 10 min of stirring, 35 μL of TEOS was added to this mixture and stirred overnight at rt. The contents were then centrifuged at 8000 rpm for 15 min, the supernatant was discarded, and the residue was redispersed in 10 mL of ethanol.2.5. Synthesis of GS–NS@SnO2@TiO2 and GS–NS@SiO2@TiO2 core-dual-shell nanoparticles
The recipe for phase transfer of GS–NS@SnO2 core is similar to that for the GS–NS core after HPC functionalization, as outlined in Scheme 1. Specifically, 10 mL of the GS–NS@SnO2 in water from the previous step was taken, 15 mL of EtOH was added to this dispersion, sonicated for 10–15 min, and then centrifuged at 8000 rpm for 15 min. The supernatant was discarded, 15 mL of EtOH was added, and the solution sonicated again for 15 min. This solution was then centrifuged again at 8000 rpm for 15 min, the supernatant was discarded to remove any remaining water and then finally redispersed in 1 mL of EtOH. The phase transfer of the GS–NS@SiO2 core is simpler since the nanoparticles are already dispersed in EtOH. Specifically, 10 mL of the GS–NS@SiO2 in EtOH from the previous step was concentrated by centrifuging and directly redispersing into 1 mL of EtOH. After generating a concentrated solution of GS–NS@SnO2 or GS–NS@SiO2 in EtOH, the TiO2 coating recipe is exactly the same as described for the GS–NS core. The nanoparticles thus obtained after the first TiO2 coating are denoted as GS–NS@SnO2@TiO2(×1) and GS–NS@SiO2@TiO2(×1), while those obtained after the second TiO2 coating are denoted as GS–NS@SnO2@TiO2(×2) and GS–NS@SiO2@TiO2(×2), respectively.2.6. Synthesis of GS–NS@SiO2@TiO2-seed nanoparticles
The recipe for GS–NS@SiO2@TiO2 nanoparticles is modified to synthesize GS–NS@SiO2 nanoparticles decorated with TiO2-seeds. Specifically, 10 mL of the GS–NS@SiO2 in EtOH from the previous step was concentrated by centrifuging and redispersed into 1 mL of EtOH. This concentrated solution of GS–NS@SiO2 in EtOH was transferred to a glass vial and 0.02 g of HPC was added. To activate the controlled hydrolysis of TBOT, 50 μL of Milli-Q-water was also added to this vial, and the contents were sonicated for 90 min. Subsequently, 60 μL (0.176 mmol) of TBOT was dissolved in 4 mL of EtOH from a freshly opened bottle. Using a syringe pump, 3 mL of this TBOT solution was then slowly added to the HPC-functionalized GS–NS@SiO2 over 30 min under stirring. The reaction vial was then stirred for 90 min at rt. The contents of the reaction vial were transferred to a 75 mL round-bottom flask and refluxed at 85 °C for 90 min. The reaction mixture was allowed to cool down to rt and then sonicated for 90 min. The contents were then cooled to rt and centrifuged, typically at 8000 rpm for 15 min, and finally redispersed in 1 mL of EtOH. The nanoparticles thus obtained after the TiO2 seeding are denoted as GS–NS@SiO2@TiO2-seed nanoparticles. This seeding recipe can be repeated again to increase the number of TiO2 seeds on the surface.3. Results and discussion
3.1. Synthesis and morphological study of the nanoparticles
In this report, hydroxypropyl cellulose (HPC) was chosen as the surfactant. In addition to successful phase transfer, hydroxypropyl cellulose can also stabilize and disperse the final TiO2 coated core–shell composites synthesized by hydrolysis of TBOT in ethanol.52 Lee et al. compared the growth of TiO2 shells on SiO2 cores from TBOT with and without HPC and found that the addition of HPC significantly improves the dispersion of the final nanoparticles.53 Although, eccentric TiO2-coated Au nanostructures were obtained by Seh et al. using HPC,54 our goal is to synthesize uniform shells of TiO2 onto gold–silver alloy systems and such uniform shells require the use of TBOT as the TiO2 precursor.52 Thus, in our method we attempted to exploit the dual-ability of HPC as a phase-transfer agent as well as a stabilizer and combine it with the attributes of TBOT hydrolysis to generate uniform TiO2 shells around aqueously synthesized metal nanoparticles. The GS–NS solution functionalized with HPC, was washed with anhydrous ethanol multiple times to eliminate water. Secondly, the as-prepared HPC-functionalized GS–NS solutions were concentrated 10-fold due to the need for a high NP concentration to reduce free solvent volume further, diminishing the chances for formation of free TiO2. A minimal amount of Milli-Q water was then added to the concentrated core nanoparticle solution along with hydroxypropyl cellulose and sonicated to selectively disperse the HPC and water molecules on the surface of the GS–NSs. The slow addition of TBOT precursor and overnight stirring followed by reflux leads to the formation of TiO2 shells via controlled hydrolysis.
The SEM and TEM images demonstrate the successful formation of TiO2 shells around the GS–NSs. As evident from the SEM image in Fig. 1, TiO2 is exclusively hydrolyzed only on the surface of the GS–NS, and the formation of free TiO2 particulates is dramatically reduced or completely eliminated. Obtaining a thicker TiO2 shell in a single step by increasing water or TBOT concentration is complicated by the formation of free TiO2. Instead, to obtain thicker shells with good uniformity, the coating process is repeated on the already coated nanoparticles. The TEM images in Fig. 1 show that the TiO2 shell becomes thicker after the second coating. Using statistical image analysis of the SEM images (Table 1), after one coating, the average diameter of the GS–NS@TiO2(×1) nanoparticles was determined to be 70 ± 10 nm. According to these measurements, the TiO2 shells have an average thickness of 7 ± 1 nm. After a second coating, the average diameter increased to 112 ± 31 nm, indicating an increase in TiO2 shell thickness to 28 ± 11 nm for the GS–NS@TiO2(×2) nanoparticles.
 | ||
| Fig. 1 (a)–(c) SEM images, (d)–(f) TEM images, and (g)–(i) Size distribution histograms for GS–NSs, GS–NS@TiO2(×1), and GS–NS@TiO2(×2) nanoparticles, respectively. The fuzzy appearance of the particles in many of the SEM and TEM images can be attributed to polycrystalline nature of the TiO2 coatings and the resolution of our microscopy instruments. | ||
| Particle | GS–NS core diameter (nm) | SnO2 shell thickness (nm) | SiO2 shell thickness (nm) | TiO2 shell thickness (nm) | Total diameter (nm) |
|---|---|---|---|---|---|
| GS–NS | 56 ± 9 | — | — | — | 56 ± 9 |
| GS–NS@TiO2(×1) | 56 ± 9 | — | — | 7 ± 1 | 70 ± 10 |
| GS–NS@TiO2(×2) | 56 ± 9 | — | — | 28 ± 11 | 112 ± 31 |
| GS–NS@SnO2 | 56 ± 9 | 11 ± 3 | — | — | 77 ± 15 |
| GS–NS@SnO2@TiO2(×1) | 56 ± 9 | 11 ± 3 | — | 4 ± 1 | 85 ± 15 |
| GS–NS@SnO2@TiO2(×2) | 56 ± 9 | 11 ± 3 | — | 17 ± 4 | 110 ± 23 |
| GS–NS@SiO2 | 56 ± 9 | — | 23 ± 7 | — | 102 ± 22 |
| GS–NS@SiO2@TiO2(×1) | 56 ± 9 | — | 23 ± 7 | 5 ± 4 | 112 ± 30 |
| GS–NS@SiO2@TiO2(×2) | 56 ± 9 | — | 23 ± 7 | 10 ± 3 | 122 ± 28 |
The synthesis procedure for SiO2 coated GS–NS was adopted from a previously reported method, as outlined in Scheme 1.16 TEOS was used as the SiO2 precursor. The thickness of the SiO2 shell can be varied by changing the amount of TEOS. In our experiments, the amount of TEOS was fixed at 35 μL. The SEM image in Fig. 2g shows the morphology of the GS–NS@SiO2 nanoparticles. Using statistical image analysis of the SEM images, after one coating, the average diameter of the GS–NS@SiO2 nanoparticles was determined to be 102 ± 22 nm. According to these measurements, the SiO2 shells have an average thickness of 23 ± 7 nm.
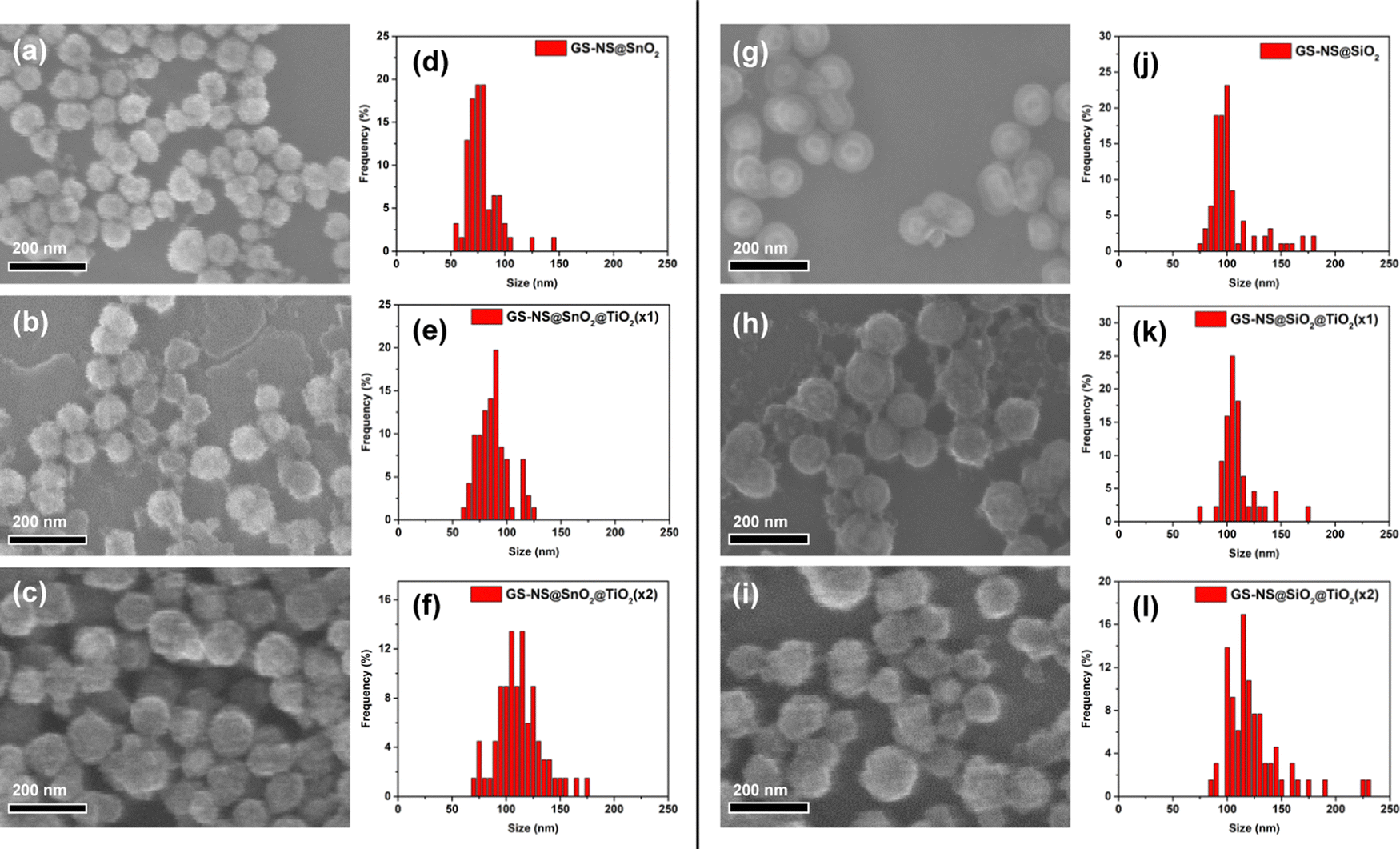 | ||
| Fig. 2 (a)–(c) SEM images and (d)–(f) Size distribution histograms for stepwise coating of TiO2 shells on GS–NS cores with a SnO2 interlayer. (g)–(i) SEM images and (j)–(l) Size distribution histograms for stepwise coating of TiO2 shells on GS–NS cores with a SiO2 interlayer. The fuzzy appearance of the particles in many of the SEM and TEM images can be attributed to polycrystalline nature of the TiO2 coatings and the resolution of our microscopy instruments. | ||
 | ||
| Fig. 3 TEM images of (a) GS–NS@TiO2(×2), (b) GS–NS@SnO2@TiO2(×2), and (c) GS–NS@SiO2@TiO2(×2) nanoparticles, respectively. | ||
The TiO2 shell formation process is similar on both the GS–NS@SnO2 and GS–NS@SiO2 nanoparticles, with similar shell thicknesses obtained for both sets of nanoparticles. These results suggest that the SnO2-coated nanoparticles can easily adapt to recipes utilized for SiO2-coated systems. The TiO2 shell-growth process on GS–NS@SnO2 cores is also extremely well controlled under identical reaction parameters. SiO2 has so far been the go-to shell material for nanoparticle stabilization because of its ability to be further encapsulated by other coating materials. SnO2, here, demonstrates similar abilities. Both materials are largely transparent to visible light and form amorphous or polycrystalline shells.55–57 Thus both materials do not block incident visible/near-IR-light and small chemical species from accessing the inner core.55–57 As observed in our previous paper,34 the SnO2 coating recipe is also facile and well-controlled, similar to the recipes for the formation of SiO2 shells.58 The SnO2 shell is also more stable than a SiO2 shell across a wider range of pH values, which makes them more robust shell materials.58 This feature allows them to be available for further functionalization that may require basic conditions. Unlike SiO2, which is an insulator, SnO2 is a wide band gap semiconductor. Furthermore, the band gap, light extinction, and electrical conductivity of SnO2 can be carefully tuned via doping,34,56 which would not be possible for SiO2. Thus, the SnO2 shell offers a very viable and tunable semiconductor alternative to traditional SiO2 coatings for nanoparticle stabilization and functionalization.
 | ||
| Fig. 4 (a) and (c) SEM images and (b) and (d) TEM images of GS–NS@SiO2 and GS–NS@SiO2@TiO2-seed nanoparticles, respectively. | ||
3.2. Compositional analysis of the nanoparticles
 | ||
| Fig. 5 STEM-EDX mapping images of (a) GS–NS@TiO2(×2), (b) GS–NS@SnO2@TiO2(×2), and (c) GS–NS@SiO2@TiO2(×2) nanoparticles, showing the distribution of the constituent elements in the respective nanoparticles. | ||
Fig. S4 (ESI†) shows that the EDX spectra for the final core–shell nanoparticles after two rounds of TiO2 coatings contains the characteristic peaks for Ag, Au, Ti, and Sn/Si in their respective nanoparticles. The relative atomic concentrations of Ag, Au, Si, Ti, and Sn obtained from EDX measurements listed in Table S1 (ESI†), roughly match the relative volume occupied by their corresponding phases in the respective nanoparticle architectures. For example, the relative concentration of Ti is highest in the GS–NS@TiO2(×2) since this nanoparticle has the thickest TiO2 shell. The relative concentrations in GS–NS@SiO2@TiO2(×2) nanoparticles are dominated by the element Si due to the very thick SiO2 shell. The relative concentration of Ag and Au are also the lowest in this particle as it has the thickest overall shell thickness (SiO2 + TiO2). The relative concentration for Ag and Au in GS–NS@SnO2@TiO2(×2) is lower than the particle with no interlayer but higher than the particle with a thicker SiO2 interlayer, in accordance with size measurements. The concentration of Sn is also lower than Ti in this particle since the thickness of the SnO2 interlayer is less than the total thickness of the TiO2 shell.
 | ||
| Fig. 6 XPS spectra for (a) Ti 2p region, (b) O 1s region, (c) Ag 3d region, and (d) Au 4f region for the GS–NSs, GS–NS@TiO2(×1), and GS–NS@TiO2(×2) nanoparticles. | ||
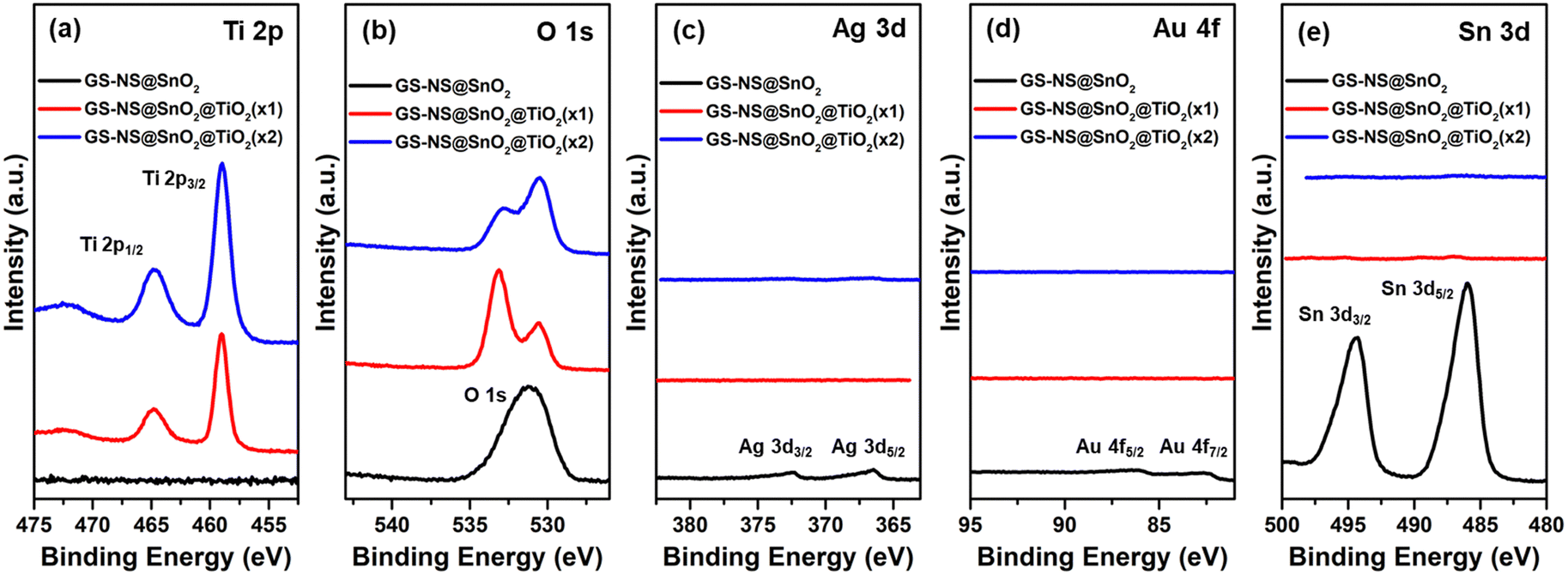 | ||
| Fig. 7 XPS spectra for the (a) Ti 2p region, (b) O 1s region, (c) Ag 3d region, (d) Au 4f region, and (e) Sn 3d region for the GS–NS@SnO2, GS–NS@SnO2@TiO2(×1), and GS–NS@SnO2@TiO2(×2) nanoparticles. | ||
 | ||
| Fig. 8 XPS spectra for the (a) Ti 2p region, (b) O 1s region, (c) Ag 3d region, (d) Au 4f region, and (e) Si 2p region for the GS–NS@SiO2, GS–NS@SiO2@TiO2(×1), and GS–NS@SiO2@TiO2(×2) nanoparticles. | ||
| Sample | Relative atomic concentration (%) | |||||
|---|---|---|---|---|---|---|
| Ti (2p) | O (1s) | Ag (3d) | Au (4f) | Si (2p) | Sn (3d5/2) | |
| GS–NS | 0 | 43 | 34 | 19 | 4 | 0 |
| GS–NS@TiO2(×1) | 19 | 75 | 3 | 2 | 1 | 0 |
| GS–NS@TiO2(×2) | 20 | 79 | 0 | 0 | 1 | 0 |
| GS–NS@SnO2 | 0 | 60 | 2 | 1 | 0 | 38 |
| GS–NS@SnO2@TiO2(×1) | 12 | 87 | 0 | 0 | 0 | 1 |
| GS–NS@SnO2@TiO2(×2) | 19 | 78 | 1 | 0 | 1 | 1 |
| GS–NS@SiO2 | 0 | 67 | 0 | 0 | 31 | 0 |
| GS–NS@SiO2@TiO2(×1) | 5 | 58 | 0 | 0 | 36 | 0 |
| GS–NS@SiO2@TiO2(×2) | 19 | 79 | 0 | 0 | 2 | 0 |
Fig. 7 shows the XPS spectra of the particles with a SnO2 interlayer before and after coating with the TiO2 shells. After the first round of TiO2 coating, Ti 2p3/2 and 2p1/2 peaks appear in the spectra, and the intensity and relative concentration of Ti 2p increases gradually with each coating step. The peak intensities for Ag 3d and Au 4f are faint because of the SnO2 shell (11 ± 3 nm). The peaks for Ag and Au, eventually disappear after coating with TiO2. The peaks for Sn 3d5/2 and 3d3/2 at 486.0 eV and 494.5 eV, respectively, can be clearly observed for GS–NS@SnO2 nanoparticles,56 becomes negligible after the first round of TiO2 coating and eventually disappears after the second round of TiO2 coating. All these observations are in good agreement with thicknesses of the SnO2 = 11 ± 3 nm, TiO2(×1) = 4 ± 1 nm, and TiO2(×2) = 17 ± 4 nm shells in this system. The O 1s spectrum shows a broad peak at around 532 eV for the GS–NS@SnO2 nanoparticles which becomes broader after one round TiO2 coating and eventually splits up into two peaks after a second TiO2 coating. These observations can be attributed to the presence of two different oxides in these nanoparticles.52 As reported in prior literature, in SnO2@TiO2 composites structures, the peak at 532.9 eV corresponds to SnO2, while the peak at 530.2 eV corresponds to TiO2.61,65,66 The intensity of the peak at 532.9 eV, arising from SnO2, gradually becomes weaker, while the peak at 530.2 eV, arising from TiO2, becomes stronger with each round of TiO2 coating. This observation is in good agreement with the above assignment for the two peaks.
In the case of a TiO2 shell grown on a SiO2 interlayer, the XPS spectra in Fig. 8 shows that the intensity and relative concentration of the Ti 2p peaks increases gradually with each coating step. The peak intensities for Ag 3d and Au 4f are negligible compared to the GS–NS@SnO2 nanoparticles because the SiO2 shell (23 ± 7 nm) is thicker than the SnO2 shell (11 ± 3 nm). The Si 2p peak remains evident after the first round of TiO2 coating but eventually disappears after the second round of TiO2 coating. All these observations further confirm the thicknesses of the SiO2 = 23 ± 7 nm, TiO2 (×1) = 5 ± 4 nm, and TiO2 (×2) = 10 ± 3 nm shells in this system. As observed with SnO2, the O 1s peak splits up in the presence of both SiO2 and TiO2. As reported in prior literature, in structures containing both SiO2 and TiO2, the peak at 532.6 eV corresponds to O–Si in SiO2, while the peak at 530.2 eV corresponds to O–Ti in TiO2.52,62,65 The intensity of the peak at 533 eV, corresponding to O–Si gradually becomes weaker, while the peak at 530 eV, corresponding to O–Ti, becomes stronger with each step of the TiO2 coating, in good agreement with the assignment for the two peaks. As calculated in Table 2, the relative atomic concentrations of Au and Ag are much lower in the XPS measurements, compared to EDX results (Table S1, ESI†), since XPS mainly probes the surface composition, which further confirms that the Au NP and GS–NS cores are consistently covered by TiO2/SnO2/SiO2 shells. Additionally, all the relative atomic concentrations of the TiO2 coated nanoparticles after the second coating step, are quite similar for all the nanoparticles. Thus, the outer surface of the GS–NS@TiO2(×2), GS–NS@SiO2@TiO2(×2), and GS–NS@SnO2@TiO2(×2), comprise almost exclusively of titanium and oxygen confirming complete encapsulation of the GS–NS, GS–NS@SiO2 and GS–NS@SnO2 cores with a TiO2 shell.
3.3. Structural analysis of the nanoparticles
After coating the nanoparticles with a TiO2 shell, XRD analysis was conducted to determine the crystallinity of the shell. The as-synthesized TiO2-coated nanoparticles do not show any peaks for TiO2 (as illustrated in Fig. 9). This result indicates that the TiO2 shell is not crystalline at this stage, consistent with previous studies.52 The nanoparticles are then annealed at 450 °C for 4 h. The SEM image of GS–NS@SiO2@TiO2(×2) in Fig. S3 (ESI†) shows that the discrete morphology is largely retained after the annealing process. XRD analysis of the annealed samples shows additional peaks corresponding to the anatase phase of TiO2 (JCPDS 21-1272).69 The peaks at 2θ = 25.38°, 48.05°, 53.95°, 55.04°, 62.85°, and 75.12° correspond to the (101), (200), (105), (211), (204), and (215) planes of anatase TiO2.70 The peak at 2θ = 48.05° and the twin peaks at 2θ = 53.95° and 55.04° can be clearly observed; all of which are fingerprint peaks for the anatase phase of TiO2. Thus, all three types of architectures are amorphous as-synthesized and adopt the anatase phase after annealing. Annealing at 450 °C for 3–4 hours is an established protocol to obtain anatase phase TiO2.52 The anatase phase is desirable since the position of the conduction band is ideal to produce hydrogen gas from splitting water.71 The XRD peak intensities for the anatase phase, albeit weak, are consistent with previous reports for similar morphology and recorded shell thicknesses of the TiO2 shell synthesized from TBOT.52 Interestingly, the TiO2 shell grown on top of a rutile SnO2 layer adopts the anatase phase as well. Rutile SnO2 synthesized using the sodium stannate method yields polycrystalline SnO2 with very small crystallites.34,56 Hence, the peaks for rutile SnO2 are not distinctly visible in comparison to peaks arising from the much more crystalline Au–Ag alloy and anatase TiO2 phases. However, the broad (211) peak characteristic of SnO2 nanoparticles prepared using this sodium stannate method,34,56 can be weakly observed at 2θ = 51.51° for the GS–NS@SnO2@TiO2(×2) samples. Since SiO2 is amorphous, no peaks are observed for SiO2 in the corresponding XRD.52
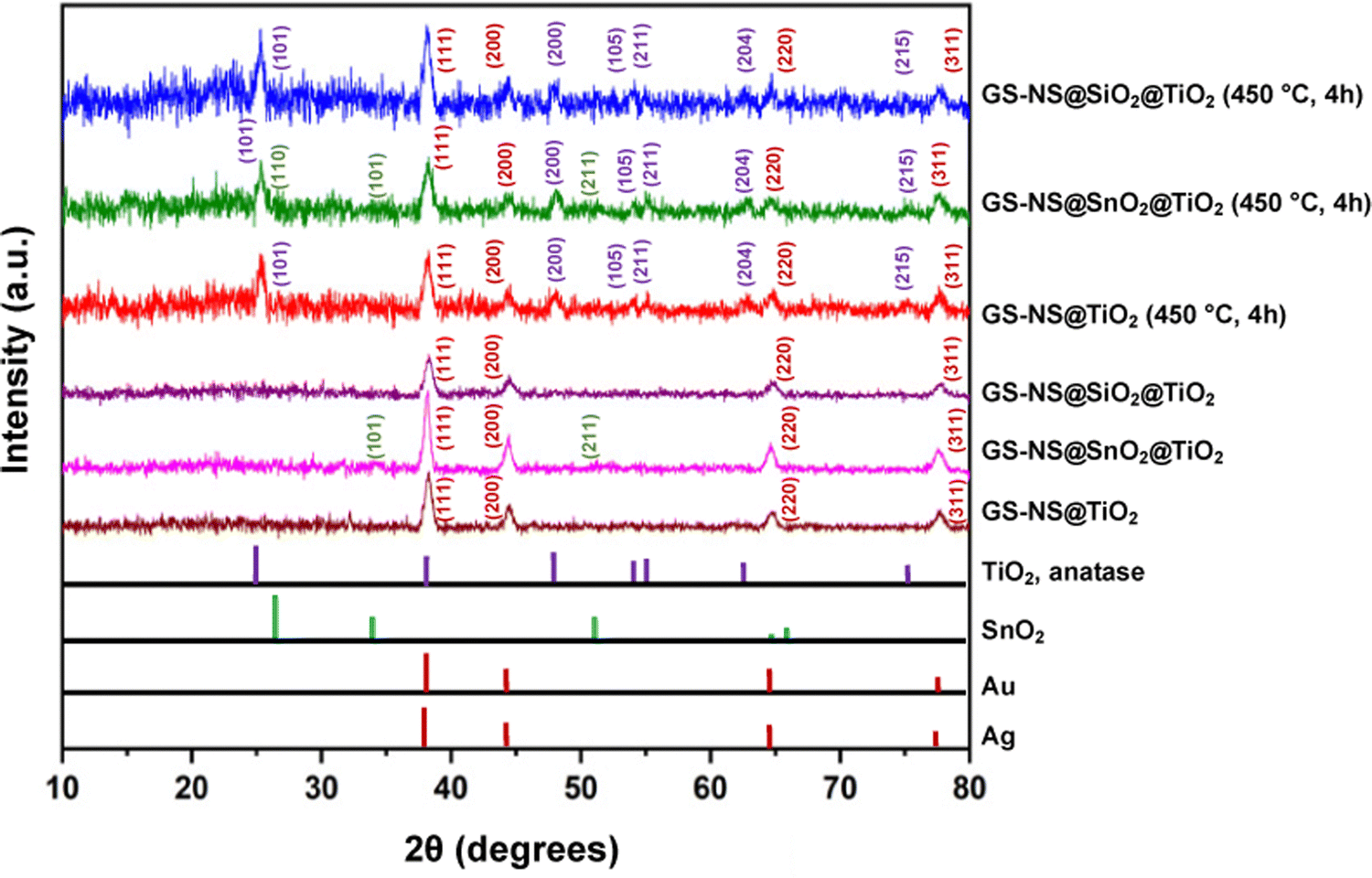 | ||
| Fig. 9 Powder XRD patterns for GS–NS@TiO2(×2), GS–NS@SiO2@TiO2(×2), and GS–NS@SnO2@TiO2(×2) nanoparticles before and after annealing along with their respective reference line patterns. | ||
3.4. Optical and electrical properties
 | ||
| Fig. 10 (a) Extinction spectra of GS–NSs demonstrating tunable LSPR maximum (λmax). Spectra normalized with GS–NS = 1. (b) Extinction spectra of GS–NS@SiO2 nanoparticles showing relative extinction in visible and UV regions after coating with TiO2 seeds and TiO2 shells. Spectra normalized with TiO2 = 1. | ||
 | ||
| Fig. 11 Extinction spectra in the range of 400–1000 nm for the (a)–(c) GS–NSs, GS–NS@SiO2, and GS–NS@SnO2 nanoparticles before and after each round of coating with TiO2 shells. | ||
The LSPR peak from the GS–NS core largely retains its plasmonic characteristics after the various stages of coating. The LSPR peak position, however, shows a red-shift after coating with SiO2/SnO2 and TiO2 shells (see Fig. 11a–c). This red-shift is consistent with previous observations of coating plasmonic nanoparticles by materials with higher refractive index (R.I.); notably, the R.I. of SnO2 (∼2.01), SiO2 (∼1.46), and TiO2 (∼2.41) are greater than that of water (∼1.33).75,76 In the case of the GS–NS@TiO2 nanoparticles, shell thickness after the first and second round of coating are 7 ± 1 nm and 28 ± 11 nm, respectively. Fig. 11a shows a red-shift in the LSPR peak of the GS–NS by about ∼40 nm after the first round of coating. After the second round of coating, the position of the LSPR peak is unchanged. Any further red-shift of the peak is masked by broadening as contributions from the extinction tail of the very thick TiO2 shell increase significantly. In the case of GS–NS@SiO2@TiO2 nanoparticles, the thickness of the SiO2 shell is 23 ± 7 nm, while the thicknesses of the TiO2 shells after the first and second round of coating are 5 ± 4 nm and 10 ± 3 nm, respectively. Since, the R.I. of SiO2 is not too different from water, there is a small red-shift of about ∼20 nm in the LSPR peak for GS–NS after coating with SiO2 (see Fig. 11c). After the TiO2 coating, the position of the LSPR peak remains constant, and no significant red-shift is observed. This phenomenon can perhaps be attributed to the thickness of the SiO2 shell, which is >20 nm. Hence, further coating with TiO2 fails to exert a strong influence on the LSPR properties of the inner GS–NS core.
This hypothesis is further proved in the case of the GS–NS@SnO2@TiO2 nanoparticles. The thickness of the SnO2 shell is 11 ± 3 nm, while the thickness of the TiO2 shell after the first and second round of coating are 4 ± 1 nm and 17 ± 4 nm, respectively. There is an initial red-shift of about ∼60 nm in the LSPR peak position for the GS–NS after coating with a SnO2 shell. Notably, this red-shift is higher than that for just a single shell of SiO2 since R.I.(SnO2) > R.I.(SiO2). Interestingly, this red-shift is also higher than just a single shell of TiO2 in the GS–NS@TiO2 nanoparticles, although the R.I.(TiO2) > R.I.(SnO2). This phenomenon arises because the thickness of the SnO2 shell (11 ± 3 nm) is more than the thickness of a single TiO2 shell (7 ± 1 nm), increasing overall red-shift. In the case of the GS–NS@SnO2@TiO2 nanoparticles, Fig. 11b shows a consistent red-shift with every step of TiO2 coating. Since the thickness of the SnO2 shell is only 11 ± 3 nm, another red-shift of ∼15 nm is observed after the first round of TiO2 coating. Also, since the first round of TiO2 coating produces a TiO2 shell with a thickness of only 4 ± 1 nm, the total combined shell thickness (SnO2 + TiO2) is about 15 ± 4 nm, which is still <20 nm; therefore, another red-shift is observed in the LSPR peak position by ∼15 nm after the second round of TiO2 coating. Thus, the LSPR field of the GS–NS is clearly influenced by the TiO2 coating in each step as long as the combined shell thickness is within ∼20 nm. Additionally, as evident from Fig. 11b, since the total TiO2 shell thickness after both rounds of TiO2 coatings in GS–NS@SnO2@TiO2(×2) is only 17 ± 4 nm thick, hence there is not a strong contribution to the LSPR peak from the extinction tail of TiO2. Thus, the total red-shift of the LSPR peak alone in the case of GS–NS@SnO2@TiO2(×2) nanoparticles is about ∼ 90 nm. From these observations, it can be concluded that the LSPR field of hollow GS–NS extends to a range of about ∼20 nm. Beyond this range, the LSPR induced field of the GS–NS is not affected as strongly. Thus, the LSPR peak can be red-shifted most effectively by coating them with multiple thin shells (<20 nm) of materials with a high refractive index (≥2.0) rather than a single thick shell.
These observations also indicate that the LSPR induced field of the GS–NS only interacts up to a range of ∼20 nm. There have been similar indications of this phenomenon in previous literature as well. For example, Li et al. observed that the photocatalytic efficiency of a GS–NS@ZnIn2S4 photocatalyst was improved when a 17 nm thick SiO2 interlayer was introduced.16 However, the sample with an even thicker SiO2 interlayer of 42 nm thickness performed the worst, even worse than the sample with no interlayer. The range of LSPR field of GS–NS was thus thought to extend up to a maximum of ∼40 nm. However, results in this present study provide clear evidence that the LSPR properties of the GS–NS actually extend up to a maximum range of ∼20 nm.
Looking at the UV-Vis spectra for the GS–NS@SiO2@TiO2 (λmax ∼ 700 nm) system as an example (Fig. 10b), one can also evaluate the relative extinctions in visible and UV regions for the various TiO2 coated systems. As observed, coating TiO2 introduces a characteristic peak in the UV region corresponding to the excitation of electrons across its band gap. As expected from theoretical predictions, UV extinction by TiO2 is significantly more intense than the SnO2-coated nanoparticles.35 The relative UV extinction due to the TiO2 band-gap excitation increases rapidly with increasing thickness for all samples. When the amounts of TBOT precursor and water for hydrolysis are increased, thicker shells are obtained, as illustrated by the SEM imaging and size measurements (see Fig. 1–3). With a second round of coating on the TiO2 coated particles, the thickness increases further, and the UV extinction also increases, as expected. As demonstrated earlier in Fig. 4, the TiO2 coating recipe can also be modified to synthesize TiO2-seed decorated nanoparticles. Fig. 10b shows the UV-Vis spectra of TiO2-seed decorated nanoparticles compared to the ones with complete TiO2 shells after one and two rounds of coatings. Apparent from the data, relative UV and visible-light extinction intensities in this nanoparticle system can be tuned simply by changing between TiO2-seeds and TiO2 shells with various thicknesses. The UV light extinction in the TiO2-seed decorated nanoparticles is nearly equal to the LSPR extinction in the visible region. These results also hold true for TiO2-seeding and TiO2 shells on just GS–NSs and on GS–NS@SnO2 cores as well. Thus, one can add equivalent levels of UV extinction to plasmonic nanoparticles simply by seeding TiO2 nanoparticles on its surface, without necessarily coating an entire shell. Overall, using this TiO2 coating recipe, the relative extinctions in the visible and UV regions can be easily tuned. This feature adds further tunability and versatility to the optical properties of this nanoparticle system.
In a previous paper,34 our groups studied a different design with the plasmonic Au NP as the core and the metal oxide as the outer shell. An extremely effective suppression in the rate of electron–hole recombination was observed per particle. When the PL intensities were normalized with respect to the volume of tin oxide material per particle, a nearly complete suppression of the recombination is observed. This result outlined a significant advantage of having the noble metal nanoparticle as the core and the metal oxide as the outer shell. Although Au NPs have been commonly employed for decreasing electron–hole recombination, the use of gold–silver nanoshells was also examined for this purpose. The suppression of electron–hole recombination per volume of tin oxide appears to be more effective with a GS–NS core compared to the ones with an Au NP core, affording a near-complete suppression.
As discussed earlier, TiO2 absorbs light much more strongly than SnO2, leading to the creation of a much greater number of electron–hole pairs.35 This study examines whether the effect of GS–NS core on the electron–hole recombination of TiO2 also shows similar quenching as observed for SnO2.34 To examine the effect of the GS–NS core in quenching the electron–hole recombination in TiO2, photoluminescence studies were conducted. Photoluminescence intensity in metal oxide semiconductors arises from the energy emitted by the electrons that fall back from the conduction band to recombine with the holes in the valence band. Hence, a drop in the photoluminescence intensity is indicative of a decrease in the rate of electron–hole recombination.22 Photoluminescence measurements were carried out for all the samples with excitation wavelength at 300 nm to ensure complete band gap excitation in the TiO2 component. Photoluminescence spectra of the core–shell and core-dual-shell nanoparticles are compared to commercial TiO2 nanoparticles. For the purpose of meaningful comparison, photoluminescence intensities were normalized with respect to particle concentration and volume of TiO2 per particle, as shown in Fig. 12. As illustrated in Fig. 12, the GS–NS core effects significant suppression in the rate of electron–hole recombination in TiO2 as well. This extremely effective quenching of electron–hole recombination in TiO2 further reiterates the importance of this design where the metal oxide material is employed as the shell with the noble metal core inside. These results prove that the GS–NS core design is effective for the suppression of electron–hole recombination in metal oxides with much higher number of excitons than SnO2 as well. When the PL intensities are normalized with respect to volume of TiO2 per particle (see Fig. 12b), the effect of the interlayers becomes more evident in the three different types of TiO2 core–shell materials synthesized. Among the three types of particles, the recombination is quenched most effectively in the presence of a SnO2 interlayer, at ∼68%, even better than the particle with no interlayer. The quenching due to the GS–NS core in the GS–NS@TiO2 was found to be ∼56%, while for the GS–NS@SiO2@TiO2 nanoparticles, the quenching was the lowest at ∼49%. However, these results suggest that even in the presence of an insulating SiO2 interlayer between the GS–NS and TiO2, the GS–NS can still accept electrons from the TiO2 shell and increase the lifetime of charge carriers. This phenomenon can plausibly be attributed to tunneling due to the mesoporous nature of the SiO2 shell, or to the presence of small TiO2 particulates within the SiO2 interlayer, facilitating electron transfer to the GS–NS core.
 | ||
| Fig. 12 Photoluminescence spectra of TiO2, GS–NS@TiO2(×2), GS–NS@SiO2@TiO2(×2), and GS–NS@SnO2@TiO2(×2) nanoparticles normalized with respect to (a) particle concentration, (b) particle concentration + volume of TiO2 per particle, and (c) TiO2 extinction intensity. | ||
To verify these trends, the photoluminescence spectra were normalized with respect to the intensity of UV extinction due to TiO2 in the corresponding samples (see Fig. 12c). As expected, the trends in Fig. 12c closely correlate those in Fig. 12b, confirming the effect of the interlayers on the charge transfer and recombination characteristics. The quenching in GS–NS@TiO2, GS–NS@SnO2@TiO2, and GS–NS@SiO2@TiO2 nanoparticles was calculated to be ∼58%, ∼68%, and 50%, respectively, when normalized with TiO2 extinction intensity. These results together confirm that electron–hole recombination of TiO2 is most effectively suppressed in the presence of a SnO2 interlayer, which can likely be attributed to the type-II heterojunction characteristics between the SnO2 interlayer and the TiO2 shell. In addition to charge-carrier trapping by the GS–NS core, additional electrons can be trapped in the conduction band of SnO2, increasing their lifetime. As reported in previous studies, electrons move from the CB of TiO2 to the CB of SnO2, while the holes from VB of SnO2 move in the opposite direction towards VB of TiO2 in such a system.46,48 This orchestration allows for highly effective separation of charge carriers.48 Given the polycrystalline porous nature of both the TiO2 and SnO2 shells,34,36,52,58 all parts of the nanoparticle are accessible; chemical species in solution can easily access any part of the nanoparticle to react with either holes or electrons.
In a previous paper, our groups had also demonstrated the ability to modify the GS–NS@SnO2 recipe to generate doped tin oxide shells with antimony and zinc,34 which was achieved simply by adding sodium antimonate and sodium zincate to sodium stannate in the tin oxide coating step. When combined with the ability to alter the band gap and conductivity of the tin oxide interlayer material by doping with antimony and zinc, this recipe allows us to design finely tailored heterojunctions with TiO2 while keeping the chemistry consistent, which will greatly enhance our ability to modulate electronic structures on the nanoscale.
4. Conclusions
We successfully demonstrated a strategy to compensate for the drawbacks and enhance the utility of TiO2. A single facile and versatile solution was developed for the controlled growth of TiO2 shells directly on a noble metal core consisting of GS–NSs, as well as on GS–NSs with a semiconducting (SnO2) and insulating (SiO2) interlayer. The versatility of both this method and optimization approach allows us to utilize a single synthesis procedure to coat uniform TiO2 shells on multiple diverse cores, such as a crystalline metal, polycrystalline metal oxide, and amorphous silica. Measurements of the optical properties showed that the plasmonic core–shell and core-dual-shell nanoparticles exhibit strong extinctions in the visible and near-IR regions, respectively, and that the position of extinction maximum (λmax) can be successfully tuned within this region. This strong LSPR extinction can extend the utility of TiO2 well beyond the UV region. Further, the relative extinctions in the UV, visible, and near-IR regions can be carefully tailored by changing the ratio of Au/Ag in the GS–NS alloy and/or by controlled TiO2 coating/seeding methods reported herein. The range of the LSPR field for the GS–NSs was also thoroughly investigated and found to extend up to ∼20 nm. Incorporation of the GS–NS core also demonstrated a remarkable suppression in the rate of electron–hole recombination in TiO2. This observation further establishes the utility of plasmonic gold–silver nanoshells and paves the way for their use in enhancing the properties of other traditional semiconductor materials as well. Importantly, this exceptional reduction of the electron–hole recombination further demonstrates the effectiveness of designing the plasmonic nanoparticle as the core and the metal oxide as the shell.This synthesis method is easily transferable to generate TiO2 shells on GS–NS with a semiconducting SnO2 interlayer or an insulating SiO2 interlayer. Moreover, by virtue of the facile and consistent synthetic method described herein, SnO2 shells now offer a viable semiconductor alternative to traditional silica shells. Like SiO2, the SnO2 shell can also be easily functionalized using recipes used for SiO2, while possessing multiple advantages such as having a tunable semiconducting band gap34,56 and stability across a wider pH range.58 The results demonstrate that the presence of different interlayers between the GS–NS and TiO2 shell can change the localized plasmonic effects, energy-transfer, charge-transfer, and recombination properties in these systems. The extinction spectra of the three different types of nanoparticles demonstrate that the LSPR field of GS–NS extend to a range of 20 nm, and can be red-shifted most effectively with successive addition of thin interlayers (<5 nm each) with a high refractive index (>2.0) instead of a single thick layer. The nanoparticles with an SnO2 interlayer also afforded the greatest suppression of TiO2 recombination. The successful synthesis of visible/near-IR-active SnO2/TiO2 composite nanoparticles with proposed type-II heterojunction characteristics opens the door for novel optoelectronic possibilities. Combined with the ability to vary the SiO2 shell thickness81 and modulate the band gap of SnO2via doping,34,56 this versatile TiO2 coating method affords us multiple handles to tune the electrical properties in nanoscale colloids. This work provides a good foundation for the synthesis of core-dual-shell tunable-heterojunction nanoparticles, which can be further developed in terms of the shell smoothness by employing templated methods such as using block copolymers as nanoreactors, which has been reported previously.82–86 On the whole, a combination of these optical and electrical properties should render these unique hybrid particles effective materials for photovoltaics, photocatalysis, optoelectronic devices, and sensors.
Author contributions
Riddhiman Medhi: conceptualization, investigation, methodology, data curation, formal analysis, visualization, writing – original draft. Sarawut Plengjaroensirichai: investigation, methodology. Nhat Ngo: investigation, methodology. Maria D. Marquez: supervision, validation, project administration, writing – review & editing. Pannaree Srinoi: investigation, methodology. Hung-Vu Tran: project administration. Allan J. Jacobson: conceptualization, supervision. Tai-Chou Lee: conceptualization, funding acquisition. T. Randall Lee: funding acquisition, project administration, conceptualization, supervision, validation, formal analysis, resources, writing – review and editing.Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank the Air Force Office of Scientific Research (FA9550-20-1-0349; 20RT0302 and FA9550-23-1-0581; 23RT0567) and the Robert A. Welch Foundation (Grant No. E-1320 and E-0024) for generously supporting this research. The authors would also like to thank Zhenglin Zhang for aiding with the PL measurements.References
- A. Dey, Semiconductor Metal Oxide Gas Sensors: A Review, Mater. Sci. Eng., B, 2018, 229, 206–217 CrossRef CAS.
- C. Ray and T. Pal, Recent Advances of Metal–Metal Oxide Nanocomposites and their Tailored Nanostructures in Numerous Catalytic Applications, J. Mater. Chem. A, 2017, 5, 9465–9487 RSC.
- X. Yu, T. J. Marks and A. Facchetti, Metal Oxides for Optoelectronic Applications, Nat. Mater., 2016, 15, 383 CrossRef CAS PubMed.
- Y. Liu, W. Wang, X. Xu, J.-P. Marcel Veder and Z. Shao, Recent Advances in Anion-Doped Metal Oxides for Catalytic Applications, J. Mater. Chem. A, 2019, 7, 7280–7300 RSC.
- A. Raghunath and E. Perumal, Metal Oxide Nanoparticles as Antimicrobial Agents: A Promise for the Future, Int. J. Antimicrob. Agents, 2017, 49, 137–152 CrossRef CAS PubMed.
- R. Daghrir, P. Drogui and D. Robert, Modified TiO2 For Environmental Photocatalytic Applications: A Review, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CrossRef CAS.
- R. Medhi, M. D. Marquez and T. R. Lee, Visible-Light-Active Doped Metal Oxide Nanoparticles: Review of their Synthesis, Properties, and Applications, ACS Appl. Nano Mater., 2020, 3, 6156–6185 CrossRef CAS.
- Y.-T. Chen, R. Medhi, I. Nekrashevich, D. Litvinov, S. Xu and T. R. Lee, Specific Detection of Proteins Using Exceptionally Responsive Magnetic Particles, Anal. Chem., 2018, 90, 6749–6756 CrossRef CAS PubMed.
- R. Medhi, P. Srinoi, N. Ngo, H.-V. Tran and T. R. Lee, Nanoparticle-Based Strategies to Combat COVID-19, ACS Appl. Nano Mater., 2020, 3, 8557–8580 CrossRef CAS PubMed.
- B. Zeng, S. Wang, Y. Xiao, G. Zeng, X. Zhang, R. Li and C. Li, Surface Phosphate Functionalization for Boosting Plasmon-Induced Water Oxidation on Au/TiO2, J. Phys. Chem. C, 2022, 126, 5167–5174 CrossRef CAS.
- D. J. Da Silva, A. G. Souza, G. D. S. Ferreira, A. Duran, A. D. Cabral, F. L. A. Fonseca, R. F. Bueno and D. S. Rosa, Cotton Fabrics Decorated with Antimicrobial Ag-Coated TiO2 Nanoparticles Are Unable to Fully and Rapidly Eradicate SARS-CoV-2, ACS Appl. Nano Mater., 2021, 4, 12949–12956 CrossRef CAS PubMed.
- A. Jaroenworaluck, W. Sunsaneeyametha, N. Kosachan and R. Stevens, Characteristics of Silica-Coated TiO2 and its UV Absorption for Sunscreen Cosmetic Applications, Surf. Interface Anal., 2006, 38, 473–477 CrossRef CAS.
- C. M. Staller, Z. L. Robinson, A. Agrawal, S. L. Gibbs, B. L. Greenberg, S. D. Lounis, U. R. Kortshagen and D. J. Milliron, Tuning Nanocrystal Surface Depletion by Controlling Dopant Distribution as a Route Toward Enhanced Film Conductivity, Nano Lett., 2018, 18, 2870–2878 CrossRef CAS PubMed.
- K. M. Noone and D. S. Ginger, Doping for Speed: Colloidal Nanoparticles for Thin-Film Optoelectronics, ACS Nano, 2009, 3, 261–265 CrossRef CAS PubMed.
- S. E. Lohse and C. J. Murphy, Applications of Colloidal Inorganic Nanoparticles: From Medicine to Energy, J. Am. Chem. Soc., 2012, 134, 15607–15620 CrossRef CAS PubMed.
- C.-H. Li, M.-C. Li, S.-P. Liu, A. C. Jamison, D. Lee, T. R. Lee and T.-C. Lee, Plasmonically Enhanced Photocatalytic Hydrogen Production from Water: The Critical Role of Tunable Surface Plasmon Resonance from Gold–Silver Nanoshells, ACS Appl. Mater. Interfaces, 2016, 8, 9152–9161 CrossRef CAS PubMed.
- P. Mazierski, A. Mikolajczyk, B. Bajorowicz, A. Malankowska, A. Zaleska-Medynska and J. Nadolna, The Role of Lanthanides in TiO2-Based Photocatalysis: A Review, Appl. Catal., B, 2018, 233, 301–317 CrossRef CAS.
- B. Ohtani, Titania Photocatalysis beyond Recombination: A Critical Review, Catalysts, 2013, 3, 942–953 CrossRef CAS.
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Plasmonic Photocatalysts: Harvesting Visible Light with Noble Metal Nanoparticles, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC.
- E. Petryayeva and U. J. Krull, Localized Surface Plasmon Resonance: Nanostructures, Bioassays and Biosensing—A Review, Anal. Chim. Acta, 2011, 706, 8–24 CrossRef CAS PubMed.
- W. W. Brian, R. Medhi, M. D. Marquez, S. Rittikulsittichai, M. Tran and T. R. Lee, Porous Silver-Coated pNIPAM-co-AAc Hydrogel Nanocapsules, Beilstein J. Nanotechnol., 2019, 10, 1973–1982 CrossRef PubMed.
- M. M. Khan, S. A. Ansari, M. E. Khan, M. O. Ansari, B.-K. Min and M. H. Cho, Visible Light-Induced Enhanced Photoelectrochemical and Photocatalytic Studies of Gold Decorated SnO2 Nanostructures, New J. Chem., 2015, 39, 2758–2766 RSC.
- V. Vongsavat, B. M. Vittur, W. W. Bryan, J.-H. Kim and T. R. Lee, Ultrasmall Hollow Gold–Silver Nanoshells with Extinctions Strongly Red-Shifted to the Near-Infrared, ACS Appl. Mater. Interfaces, 2011, 3, 3616–3624 CrossRef CAS PubMed.
- C. Zhang, Y. Zhou, A. Merg, C. Song, G. C. Schatz and N. L. Rosi, Hollow Spherical Gold Nanoparticle Superstructures with Tunable Diameters and Visible to Near-Infrared Extinction, Nanoscale, 2014, 6, 12328–12332 RSC.
- L. Zhang, K. Xia, Z. Lu, G. Li, J. Chen, Y. Deng, S. Li, F. Zhou and N. He, Efficient and Facile Synthesis of Gold Nanorods with Finely Tunable Plasmonic Peaks from Visible to Near-IR Range, Chem. Mater., 2014, 26, 1794–1798 CrossRef CAS.
- I. G. Theodorou, Z. A. R. Jawad, Q. Jiang, E. O. Aboagye, A. E. Porter, M. P. Ryan and F. Xie, Gold Nanostar Substrates for Metal-Enhanced Fluorescence through the First and Second Near-Infrared Windows, Chem. Mater., 2017, 29, 6916–6926 CrossRef CAS.
- T.-M. Chen, G.-Y. Xu, H. Ren, H. Zhang, Z.-Q. Tian and J.-F. Li, Synthesis of Au@TiO2 Core–Shell Nanoparticles with Tunable Structures for Plasmon-Enhanced Photocatalysis, Nanoscale Adv., 2019, 1, 4522–4528 RSC.
- J. Goebl, J. B. Joo, M. Dahl and Y. Yin, Synthesis of Tailored Au@TiO2 Core–Shell Nanoparticles for Photocatalytic Reforming of Ethanol, Catal. Today, 2014, 225, 90–95 CrossRef CAS.
- X.-F. Wu, H.-Y. Song, J.-M. Yoon, Y.-T. Yu and Y.-F. Chen, Synthesis of Core−Shell Au@TiO2 Nanoparticles with Truncated Wedge-Shaped Morphology and Their Photocatalytic Properties, Langmuir, 2009, 25, 6438–6447 CrossRef CAS PubMed.
- W.-L. Liu, F.-C. Lin, Y.-C. Yang, C.-H. Huang, S. Gwo, M. H. Huang and J.-S. Huang, The Influence of Shell Thickness of Au@TiO2 Core–Shell Nanoparticles on the Plasmonic Enhancement Effect in Dye-Sensitized Solar Cells, Nanoscale, 2013, 5, 7953–7962 RSC.
- R. Wang, J. Zhang, Y. Zhu, Z. Chai, Z. An, X. Shu, H. Song, X. Xiang and J. He, Selective Photocatalytic Activation of Ethanol C–H and O–H Bonds over Multi-Au@SiO2/TiO2: Role of Catalyst Surface Structure and Reaction Kinetics, ACS Appl. Mater. Interfaces, 2022, 14, 2848–2859 CrossRef CAS PubMed.
- J. Xue, L. Wu, C. Deng, D. Tang, S. Wang, H. Ji, C. Chen, Y. Zhang and J. Zhao, Plasmon-Mediated Electrochemical Activation of Au/TiO2 Nanostructure-Based Photoanodes for Enhancing Water Oxidation and Antibiotic Degradation, ACS Appl. Nano Mater., 2022, 5, 11342–11351 CrossRef CAS.
- H. Wu, P. Wang, H. He and Y. Jin, Controlled Synthesis of Porous Ag/Au Bimetallic Hollow Nanoshells with Tunable Pasmonic and Catalytic Properties, Nano Res., 2012, 5, 135–144 CrossRef CAS.
- R. Medhi, C.-H. Li, S. H. Lee, P. Srinoi, M. D. Marquez, F. Robles-Hernandez, A. J. Jacobson, T.-C. Lee and T. R. Lee, Antimony- and Zinc-Doped Tin Oxide Shells Coated on Gold Nanoparticles and Gold–Silver Nanoshells Having Tunable Extinctions for Sensing and Photonic Applications, ACS Appl. Nano Mater., 2020, 3, 8958–8971 CrossRef CAS.
- M. Dou and C. Persson, Comparative Study of Rutile and Anatase SnO2 and TiO2: Band-Edge Structures, Dielectric Functions, and Polaron Effects, J. Appl. Phys., 2013, 113, 083703 CrossRef.
- R. Medhi, N. Ngo, T.-C. Lee and T. R. Lee, Type-II Heterojunctions in SnO2-Coated TiO2 Core–Shell Nanoparticles, Curr. Res. Mater. Sci., 2021, 3, 112 Search PubMed.
- G. Yang, Z. Jiang, H. Shi, T. Xiao and Z. Yan, Preparation of Highly Visible-Light Active N-Doped TiO2 Photocatalyst, J. Mater. Chem., 2010, 20, 5301–5309 RSC.
- J. Shao, W. Sheng, M. Wang, S. Li, J. Chen, Y. Zhang and S. Cao, In Situ Synthesis of Carbon-Doped TiO2 Single-Crystal Nanorods with a Remarkably Photocatalytic Efficiency, Appl. Catal., B, 2017, 209, 311–319 CrossRef CAS.
- R. Jaiswal, J. Bharambe, N. Patel, A. Dashora, D. C. Kothari and A. Miotello, Copper and Nitrogen Co-Doped TiO2 Photocatalyst with Enhanced Optical Absorption and Catalytic Activity, Appl. Catal., B, 2015, 168–169, 333–341 CrossRef CAS.
- A. Eshaghi and H. Moradi, Optical and Photocatalytic Properties of the Fe-Doped TiO2 Nanoparticles Loaded on the Activated Carbon. Adv, Powder Technol., 2018, 29, 1879–1885 CrossRef CAS.
- S. P. Takle, S. D. Naik, S. K. Khore, S. A. Ohwal, N. M. Bhujbal, S. L. Landge, B. B. Kale and R. S. Sonawane, Photodegradation of Spent Wash, A Sugar Industry Waste, using Vanadium-Doped TiO2 Nanoparticles, RSC Adv., 2018, 8, 20394–20405 RSC.
- S. H. Lee, D. M. Hoffman, A. J. Jacobson and T. R. Lee, Transparent, Homogeneous Tin Oxide (SnO2) Thin Films Containing SnO2-Coated Gold Nanoparticles, Chem. Mater., 2013, 25, 4697–4702 CrossRef CAS.
- N. G. Bastús, F. Merkoçi, J. Piella and V. Puntes, Synthesis of Highly Monodisperse Citrate-Stabilized Silver Nanoparticles of up to 200 nm: Kinetic Control and Catalytic Properties, Chem. Mater., 2014, 26, 2836–2846 CrossRef.
- P. Yuan, R. Ma, N. Gao, M. Garai and Q.-H. Xu, Plasmon Coupling-Enhanced Two-Photon Photoluminescence of Au@Ag Core–Shell Nanoparticles and Applications in the Nuclease Assay, Nanoscale, 2015, 7, 10233–10239 RSC.
- A. M. Brito-Silva, R. G. Sobral-Filho, R. Barbosa-Silva, C. B. de Araújo, A. Galembeck and A. G. Brolo, Improved Synthesis of Gold and Silver Nanoshells, Langmuir, 2013, 29, 4366–4372 CrossRef CAS PubMed.
- C. Wang, C. Shao, X. Zhang and Y. Liu, SnO2 Nanostructures-TiO2 Nanofibers Heterostructures: Controlled Fabrication and High Photocatalytic Properties, Inorg. Chem., 2009, 48, 7261–7268 CrossRef CAS PubMed.
- M. K. Gish, A. M. Lapides, M. K. Brennaman, J. L. Templeton, T. J. Meyer and J. M. Papanikolas, Ultrafast Recombination Dynamics in Dye-Sensitized SnO2/TiO2 Core/Shell Films, J. Phys. Chem. Lett., 2016, 7, 5297–5301 CrossRef CAS PubMed.
- X. Huang, L. Shang, S. Chen, J. Xia, X. Qi, X. Wang, T. Zhang and X.-M. Meng, Type-II ZnO Nanorod–SnO2 Nanoparticle Heterostructures: Characterization of Structural, Optical and Photocatalytic Properties, Nanoscale, 2013, 5, 3828–3833 RSC.
- N. S. McCool, J. R. Swierk, C. T. Nemes, C. A. Schmuttenmaer and T. E. Mallouk, Dynamics of Electron Injection in SnO2/TiO2 Core/Shell Electrodes for Water-Splitting Dye-Sensitized Photoelectrochemical Cells, J. Phys. Chem. Lett., 2016, 7, 2930–2934 CrossRef CAS PubMed.
- L. Troian-Gautier, R. N. Sampaio, E. J. Piechota, M. D. Brady and G. J. Meyer, Barriers for Interfacial Back-Electron Transfer: A Comparison between TiO2 and SnO2/TiO2 Core/Shell Structures, J. Chem. Phys., 2018, 150, 041719 CrossRef PubMed.
- K. Vinodgopal, I. Bedja and P. V. Kamat, Nanostructured Semiconductor Films for Photocatalysis. Photoelectrochemical Behavior of SnO2/TiO2 Composite Systems and Its Role in Photocatalytic Degradation of a Textile Azo Dye, Chem. Mater., 1996, 8, 2180–2187 CrossRef CAS.
- J.-W. Lee, S. Kong, W.-S. Kim and J. Kim, Preparation and Characterization of SiO2/TiO2 Core–Shell Particles with Controlled Shell Thickness, Mater. Chem. Phys., 2007, 106, 39–44 CrossRef CAS.
- J. W. Lee, K. Hong, W. S. Kim and J. Kim, Effect of HPC Concentration and Ultrasonic Dispersion on the Morphology of Titania-Coated Silica Particles, J. Ind. Eng. Chem., 2005, 11, 609–614 CAS.
- Z. W. Seh, S. Liu, S.-Y. Zhang, K. W. Shah and M.-Y. Han, Synthesis and Multiple Reuse of Eccentric Au@TiO2 Nanostructures as Catalysts, Chem. Commun., 2011, 47, 6689–6691 RSC.
- H.-M. Song, B.-S. Chon, S.-H. Jeon, P. Rai, Y.-T. Yu and P. K. Dutta, Synthesis of Au@SnO2 Core–Shell Nanoparticles with Controllable Shell Thickness and their CO Sensing Properties, Mater. Chem. Phys., 2015, 166, 87–94 CrossRef CAS.
- R. Medhi, C.-H. Li, S. H. Lee, M. D. Marquez, A. J. Jacobson, T.-C. Lee and T. R. Lee, Uniformly Spherical and Monodisperse Antimony- and Zinc-Doped Tin Oxide Nanoparticles for Optical and Electronic Applications, ACS Appl. Nano Mater., 2019, 2, 6554–6564 CrossRef CAS.
- Q. Chen, Y. Ge, H. Granbohm and S.-P. Hannula, Effect of Ethanol on Ag@Mesoporous Silica Formation by In Situ Modified Stöber Method, Nanomaterials, 2018, 8, 362 CrossRef PubMed.
- S. H. Lee, I. Rusakova, D. M. Hoffman, A. J. Jacobson and T. R. Lee, Monodisperse SnO2-Coated Gold Nanoparticles Are Markedly More Stable than Analogous SiO2-Coated Gold Nanoparticles, ACS Appl. Mater. Interfaces, 2013, 5, 2479–2484 CrossRef CAS PubMed.
- J. Sheng, H. Tong, H. Xu and C. Tang, Preparation and Photocatalytic Activity of SnO2@TiO2 Core–Shell Composites Modified by Ag, Catal. Surv. Asia, 2016, 20, 167–172 CrossRef.
- D. P. Anderson, R. H. Adnan, J. F. Alvino, O. Shipper, B. Donoeva, J.-Y. Ruzicka, H. Al Qahtani, H. H. Harris, B. Cowie, J. B. Aitken, V. B. Golovko, G. F. Metha and G. G. Andersson, Chemically Synthesised Atomically Precise Gold Clusters Deposited and Activated on Titania. Part II, Phys. Chem. Chem. Phys., 2013, 15, 14806–14813 RSC.
- A. Kumar, L. Rout, L. S. K. Achary, A. Mohanty, R. S. Dhaka and P. Dash, An Investigation into the Solar Light-Driven Enhanced Photocatalytic Properties of a Graphene Oxide–SnO2–TiO2 Ternary Nanocomposite, RSC Adv., 2016, 6, 32074–32088 RSC.
- M. Andrulevičius, S. Tamulevičius, Y. Gnatyuk, N. Vityuk, N. Smirnova and A. Eremenko, XPS Investigation of TiO2/ZrO2/SiO2 Films Modified with Ag/Au Nanoparticles, Mater. Sci. Medzg, 2008, 14, 8–14 Search PubMed.
- Z. Yang, X. Lu, W. Tan, J. Zhao, D. Yang, Y. Yang, Y. He and K. Zhou, XPS Studies of Nitrogen Doping Niobium used for Accelerator Applications, Appl. Surf. Sci., 2018, 439, 1119–1126 CrossRef CAS.
- J. B. Gilbert, M. F. Rubner and R. E. Cohen, Depth-Profiling X-Ray Photoelectron Spectroscopy (XPS) Analysis of Interlayer Diffusion in Polyelectrolyte Multilayers, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6651 CrossRef CAS PubMed.
- G. M. Ingo, S. Dirè and F. Babonneau, XPS Studies of SiO2–TiO2 Powders Prepared by Sol-Gel Process, Appl. Surf. Sci., 1993, 70–71, 230–234 CrossRef CAS.
- Z. Zhang, Y. Ma, X. Bu, Q. Wu, Z. Hang, Z. Dong and X. Wu, Facile One-Step Synthesis of TiO2/Ag/SnO2 Ternary Heterostructures with Enhanced Visible Light Photocatalytic Activity, Sci. Rep., 2018, 8, 10532 CrossRef PubMed.
- A. Shahzad, W.-S. Kim and T. Yu, A Facile Synthesis of Ag/AgCl Hybrid Nanostructures with Tunable Morphologies and Compositions as Advanced Visible Light Plasmonic Photocatalysts, Dalton Trans., 2016, 45, 9158–9165 RSC.
- J. Helmlinger, M. Heise, M. Heggen, M. Ruck and M. Epple, A Rapid, High-Yield and Large-Scale Synthesis of Uniform Spherical Silver Nanoparticles by a Microwave-Assisted Polyol Process, RSC Adv., 2015, 5, 92144–92150 RSC.
- D. Zhang, F. Jing, F. Gao, L. Shen, D. Sun, J. Zhou, Y. Chen and S. Ruan, Enhanced Performance of a TiO2 Ultraviolet Detector Modified with Graphene Oxide, RSC Adv., 2015, 5, 83795–83800 RSC.
- H. Zhu, J. Tao and X. Dong, Preparation and Photoelectrochemical Activity of Cr-Doped TiO2 Nanorods with Nanocavities, J. Phys. Chem. C, 2010, 114, 2873–2879 CrossRef CAS.
- A. Ganguly, O. Anjaneyulu, K. Ojha and A. K. Ganguli, Oxide-Based Nanostructures for Photocatalytic and Electrocatalytic Applications, CrystEngComm, 2015, 17, 8978–9001 RSC.
- C.-H. Li, A. C. Jamison, S. Rittikulsittichai, T.-C. Lee and T. R. Lee, In Situ Growth of Hollow Gold–Silver Nanoshells within Porous Silica Offers Tunable Plasmonic Extinctions and Enhanced Colloidal Stability, ACS Appl. Mater. Interfaces, 2014, 6, 19943–19950 CrossRef CAS PubMed.
- S. W. Verbruggen, M. Keulemans, J. A. Martens and S. Lenaerts, Predicting the Surface Plasmon Resonance Wavelength of Gold–Silver Alloy Nanoparticles, J. Phys. Chem. C, 2013, 117, 19142–19145 CrossRef CAS.
- R. G. Navarro Julien and F. Lerouge, From Gold Nanoparticles to Luminescent Nano-Objects: Experimental Aspects for Better Gold-Chromophore Interactions, Nanophotonics, 2017, 6, 71–92 CrossRef.
- G. Oldfield, T. Ung and P. Mulvaney, Au@SnO2 Core–Shell Nanocapacitors, Adv. Mater., 2000, 12, 1519–1522 CrossRef CAS.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Synthesis of Nanosized Gold−Silica Core−Shell Particles, Langmuir, 1996, 12, 4329–4335 CrossRef.
- P. Riente and T. Noël, Application of Metal Oxide Semiconductors in Light-Driven Organic Transformations, Catal. Sci. Technol., 2019, 9, 5186–5232 RSC.
- E. Fudo, A. Tanaka and H. Kominami, AuOx Surface Oxide Layer as a Hole-Transferring Cocatalyst for Water Oxidation over Au Nanoparticle-Decorated TiO2 Photocatalysts, ACS Appl. Nano Mater., 2022, 5, 8982–8990 CrossRef CAS.
- O. Khantamat, C.-H. Li, S.-P. Liu, T. Liu, H. J. Lee, O. Zenasni, T.-C. Lee, C. Cai and T. R. Lee, Broadening the Photoresponsive Activity of Anatase Titanium Dioxide Particles via Decoration with Partial Gold Shells, J. Colloid Interface Sci., 2018, 513, 715–725 CrossRef CAS PubMed.
- A. Stevanovic, S. Ma and J. T. Yates, Effect of Gold Nanoparticles on Photoexcited Charge Carriers in Powdered TiO2−Long Range Quenching of Photoluminescence, J. Phys. Chem. C, 2014, 118, 21275–21280 CrossRef CAS.
- P. Srinoi, M. D. Marquez, T.-C. Lee and T. R. Lee, Hollow Gold–Silver Nanoshells Coated with Ultrathin SiO2 Shells for Plasmon-Enhanced Photocatalytic Applications, Materials, 2020, 13, 4967 CrossRef CAS PubMed.
- X. Pang, L. Zhao, W. Han, X. Xin and Z. Lin, A General and Robust Strategy for the Synthesis of Nearly Monodisperse Colloidal Nanocrystals, Nat. Nanotechnol., 2013, 8, 426–431 CrossRef CAS PubMed.
- X. Pang, Y. He, J. Jung and Z. Lin, 1D Nanocrystals with Precisely Controlled Dimensions, Compositions, and Architectures, Science, 2016, 353, 1268–1272 CrossRef CAS PubMed.
- Y. Liu, J. Wang, M. Zhang, H. Li and Z. Lin, Polymer-Ligated Nanocrystals Enabled by Nonlinear Block Copolymer Nanoreactors: Synthesis, Properties, and Applications, ACS Nano, 2020, 14, 12491–12521 CrossRef CAS PubMed.
- Y. He, Y. J. Yoon, Y. W. Harn, G. V. Biesold-McGee, S. Liang, C. H. Lin, V. V. Tsukruk, N. Thadhani, Z. Kang and Z. Lin, Unconventional Route to Dual-Shelled Organolead Halide Oerovskite Nanocrystals with Controlled Dimensions, Surface Chemistry, and Stabilities, Sci. Adv., 2019, 5, eaax4424 CrossRef CAS PubMed.
- X. Li, J. Iocozzia, Y. Chen, S. Zhao, X. Cui, W. Wang, H. Yu, S. Lin and Z. Lin, From Precision Synthesis of Block Copolymers to Properties and Applications of Nanoparticles, Angew. Chem., Int. Ed., 2018, 57, 2046 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, materials used and characterization methods; synthesis scheme and SEM images for Ag NPs and GS-NSs; additional TEM images, STEM-EDX spectra and STEM-EDX line spectra for GS-NS@TiO2(×2), GS-NS@SnO2@TiO2(×2), and GS-NS@SiO2@TiO2(×2) NPs; are included in the Supplementary Data. See DOI: https://doi.org/10.1039/d3ma00756a |
| This journal is © The Royal Society of Chemistry 2024 |