
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymerizable BODIPY probe crosslinker for the molecularly imprinted polymer-based detection of organic carboxylates via fluorescence†
Yijuan
Sun
 ,
Kornelia
Gawlitza
,
Virginia
Valderrey
,
Jérémy
Bell
and
Knut
Rurack
*
,
Kornelia
Gawlitza
,
Virginia
Valderrey
,
Jérémy
Bell
and
Knut
Rurack
*
Chemical and Optical Sensing Division, Bundesanstalt für Materialforschung und-prüfung (BAM), Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: knut.rurack@bam.de
First published on 21st March 2024
Abstract
This contribution reports the development of a polymerizable BODIPY-type fluorescent probe targeting small-molecule carboxylates for incorporation into molecularly imprinted polymers (MIPs). The design of the probe crosslinker includes a urea recognition site π-conjugated to the 3-position of the BODIPY core and two methacrylate moieties. Titration experiments with a carboxylate-expressing antibiotic, levofloxacin (LEVO), showed a blue shift of the absorption band as well as a broadening and decrease in emission, attributed to hydrogen bonding between the probe's urea group and the carboxylate group of the antibiotic. Using this probe crosslinker, core–shell particles with a silica core and a thin MIP shell were prepared for the detection of LEVO. The MIP exhibited highly selective recognition of LEVO, with an imprinting factor of 18.1 compared to the non-imprinted polymer. Transmission electron microscopy confirmed the core–shell structure and spectroscopic studies revealed that the receptor's positioning leads to a unique perturbation of the polymethinic character of the BODIPY chromophore, entailing the favourable responses. These features are fully preserved in the MIP, whereas no such response was observed for competitors such as ampicillin. The sensory particles allowed to detect LEVO down to sub-micromolar concentrations in dioxane. We have developed here for the first time a BODIPY probe for organic carboxylates and incorporated it into polymers using the imprinting technique, paving the way for BODIPY-type fluorescent MIP sensors.
Introduction
Boron-dipyrromethenes (BODIPYs) have emerged as a powerful class of fluorescent dyes due to their exceptional properties. These include their high fluorescence quantum yield, large molar absorption coefficient, absorption and emission bands in the visible-to-near infrared wavelength range, and excellent photostability. Additionally, BODIPY dyes are characterized by a chemically versatile core, which allows facile functionalization in a multitude of ways. Due to these properties, BODIPYs are widely employed in a range of applications, especially in bioimaging and (bio)chemical sensing.1 Since the first BODIPY-based pH and metal ion probes have been reported,2,3 numerous examples have followed,4,5 and the ranges of analytes have been expanded to include reactive oxygen, nitrogen and sulphur species,6,7 and biomolecules and biochemical structures that can be conjugated to or stained with BODIPY derivatives.8,9 However, probes for anions that recognize these species and do not react with them10 are rare11–13 and, to our knowledge, have not yet been reported for the recognition of organic carboxylates.Opto-chemical sensors, specifically utilising fluorescent molecularly imprinted polymers (MIPs), offer a highly sensitive and miniaturisable solution for (bio)chemical sensing outside of laboratories.14,15 These MIPs, often immobilized on silica substrates, enable selective detection of a wide range of small-molecule analytes.16–18 They possess advantageous properties such as format adaptability, reversibility, and chemical and thermal stability.19 By incorporating fluorescent probe monomers or crosslinkers into a polymer matrix, MIPs can undergo spectroscopic changes upon analyte binding, allowing for direct analyte detection.20 Conceptually, the approach is generic in that such MIPs are prepared by copolymerizing a complex between probe monomer or crosslinker and the analyte of interest as the so-called template with structural comonomers and crosslinkers, thereby imprinting the chemical structure as well as the electronic, electrostatic, hydrogen bonding and/or π-stacking requirements of the template into a rigid but porous polymer network. However, the selection of suitable dye scaffolds as fluorescent probe monomers or crosslinkers is limited. This limitation arises from specific chemical, supramolecular and application-oriented criteria that such a probe must fulfil. These criteria include the ability to tolerate polymerization conditions, exhibit spectroscopic responses to an analyte even when immobilized within a confined matrix, and generate a distinctive fluorescence signal.20 Consequently, chromophores with double bonds or photophysical processes relying on larger molecular motions are mostly unsuitable. In addition, analyte-induced spectral band shifts, as opposed to intensity-only changes, are favoured. So far, this narrowed down the available options to benzoxadiazoles,21–24 naphthalimides,25–27 phenazines28,29 and phenoxazinones.30 Although BODIPY units have been incorporated into various types of polymers used in optoelectronic materials, sensors, biotherapy and imaging,31–33 MIPs containing BODIPY-type probes have not yet been reported, despite their popularity, versatility and performance. Therefore, and drawing from our experience with BODIPY probes in detecting small-molecule analytes,34–36 we were motivated to explore the potential of the BODIPY chromophore as a polymerizable fluorescent probe.
As actions to support the UN's Sustainable Development Goals become increasingly important and new legislation to limit harmful residues in water and food is continually being enacted,37–39 there is a growing demand for analytical techniques that can be applied directly at a point of need.40 Early and preventive detection is particularly important for compounds such as antibiotics, which play a key role in modern human and veterinary medicine but often have to be dosed quite high to compensate for the partly poor absorption by the body and are therefore frequently eliminated from the body,41 leading to residues and accumulation in animal tissues as well as the environment, and ultimately to resistance that endangers human health.42,43 Levofloxacin (LEVO) is the most commonly used third-generation fluoroquinolone antibiotic, with greater activity against Gram-positive bacteria and atypical intracellular pathogens, generally used to treat bacterial infections of the urinary and respiratory tract and for intensive poultry farming in most developing countries.44 However, due to its high stability, it is not fully metabolized and the toxicity of LEVO slows down its biodegradation in water and soil.45 Traditional techniques for its analysis include electrochemical46 and HPLC-based approaches,47,48 which are laboratory-bound, time-consuming and costly, while there is a lack of strategies that can be developed for field use for better (waste)water management. MIP-based fluorescence approaches offer a promising alternative and have been used for drug residue detection.49–53 However, current methods mostly rely on non-fluorescent MIPs combined with competition assays using fluorescently labelled derivatives49 or luminescent quantum dots (QDs) sterically or covalently incorporated into MIPs.50–52 In this study, we introduce a polymerizable BODIPY probe crosslinker (1, Scheme 1) for organic carboxylates, specifically focussing on the relevant model analyte LEVO. This probe serves as both the recognition and signalling unit within fluorescent sensory MIPs.
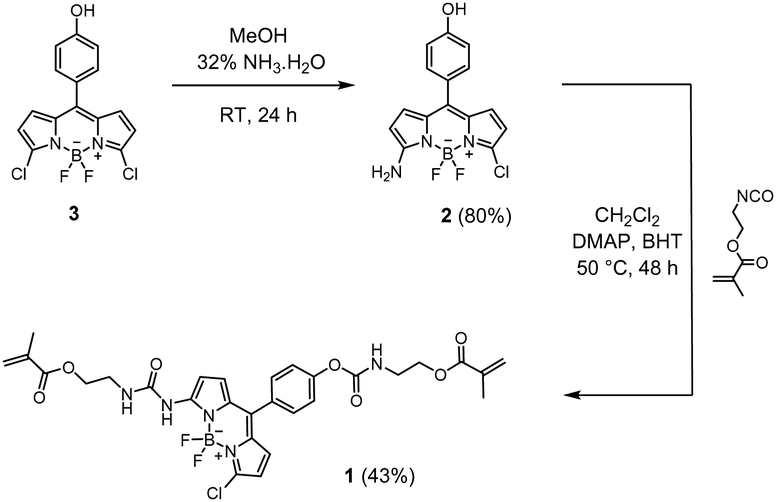 | ||
| Scheme 1 Synthetic route to fluorescent BODIPY probe crosslinker 1 (yields in brackets). | ||
Experimental
Materials and instruments
All reagents from commercial suppliers were used without further purification unless otherwise stated. All solvents were obtained from Chemsolute, except for chloroform, spectroscopic methanol, toluene (Merck), spectroscopic 1,4-dioxane, tetrahydrofuran, anhydrous toluene (Acros Organics), and anhydrous dichloromethane (Alfa Aesar). Ammonia solution (32%) and tetraethyl orthosilicate were purchased from Merck, 2,6-di-tert-butyl-4-methylphenol (BHT) and ethyl chloroformate from Fluka, 4-dimethylaminopyridine (DMAP), 2-isocyanatoethyl methacrylate, 3-aminopropyltriethoxysilane (APTES), benzyl methacrylate (BMA), ethylene glycol dimethacrylate (EGDMA), tetrabutylammonium (TBA) hydroxide, amoxicillin (AMOX) and ampicillin (AMPI) from Sigma-Aldrich, 4-cyano-4-(thiobenzoylthio)pentanoic acid (CPDB) from abcr, 2,2′-azobis(2,4-dimethylvaleronitrile) (ABDV) from Wako Chemicals and levofloxacin (LEVO) from Glentham Life Sciences. Milli-Q water was obtained via a Milli-Q water purification system (Millipore). The preparation of the TBA of the templates/analytes is described in the ESI.†1H and 13C NMR spectra were obtained with a Mercury 400 NMR spectrometer and referenced to the residual proton signals of the deuterated solvent. Ultrahigh-performance liquid chromatography electrospray ionization mass spectrometry (UPLC-ESI-MS) was performed on an Acquity UPLC with an LCT Premier XE time-of-flight mass detector (Waters). Absorption spectra were recorded on a Specord 210 Plus spectrometer (Analytik Jena). The binding constants were assessed with BindFit software.54 Fluorescence spectra were recorded with a FluoroMax-4P spectrofluorometer (HORIBA Scientific). Fluorescence lifetimes were determined with a customized laser impulse fluorometer with picosecond time resolution55 equipped with a streak camera (Hamamatsu) as detection unit. The fluorescence lifetime profiles were analysed with the High-Performance Digital Temporal Analyzer (HPD-TA) software package including the TA-fit with the global deconvolution fitting module (Hamamatsu). Transmission electron microscopy (TEM) images were taken using a ThermoFisher Scientific Talos F200S scanning/transmission electron microscope. Zeta potential measurements were performed with a Zetasizer Nano-ZS from Malvern, thermogravimetric analyses (TGA) on a STA7200 thermobalance (Hitachi High-Tech Analytical Science).
Single crystal X-ray diffraction data were collected on a Bruker D8 Venture diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Data reduction was performed using the Bruker AXS SAINT and SADABS software packages.56,57 Direct method of SHELXT 2018 was used to solve the structure of the crystal,58 followed by successive Fourier and difference Fourier syntheses. All H atoms bonded directly to C atoms were fixed at their ideal positions.
Synthesis of amino-BODIPY 2
Compound 3 (66 mg, 0.19 mmol) was prepared as in ref. 59 and dissolved in methanol (10 mL). After addition of ammonia solution (32%, 0.12 mL), the solution was stirred at room temperature for 24 h. The reaction mixture was then poured into water (10 mL), and the resulting crude product was extracted with ethyl acetate (3 × 30 mL). The combined extracts were washed with water and brine, dried over Na2SO4, and evaporated to dryness under reduced pressure. The residue was purified by silica gel chromatography with ethyl acetate/cyclohexane 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) as eluent to give 50 mg (80% yield) orange solid of compound 2. 1H NMR (400 MHz, CD3OD) δ: 7.26 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 4.1 Hz, 2H), 6.86 (d, J = 2.0 Hz, 1H), 6.24 (d, J = 4.8 Hz, 2H), 6.10 (d, J = 3.8 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ: 164.01, 159.72, 136.55, 134.95, 133.26, 132.77, 131.24, 128.40, 126.30, 119.07, 116.15, 112.44. MS: m/z cal. for [C15H11BClF2N3O–H]−: 332.0574, found: 332.0579.
:1 (v/v) as eluent to give 50 mg (80% yield) orange solid of compound 2. 1H NMR (400 MHz, CD3OD) δ: 7.26 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 4.1 Hz, 2H), 6.86 (d, J = 2.0 Hz, 1H), 6.24 (d, J = 4.8 Hz, 2H), 6.10 (d, J = 3.8 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ: 164.01, 159.72, 136.55, 134.95, 133.26, 132.77, 131.24, 128.40, 126.30, 119.07, 116.15, 112.44. MS: m/z cal. for [C15H11BClF2N3O–H]−: 332.0574, found: 332.0579.
Synthesis of fluorescent probe crosslinker 1
Compound 2 (70 mg, 0.21 mmol), 4-dimethylaminopyridine (5 mg, 0.04 mmol) and butylated hydroxytoluene (5 mg, 0.02 mmol) were dissolved in anhydrous CH2Cl2 (20 mL) under argon. After addition of 2-isocyanatoethyl methacrylate (65 μL, 0.46 mmol), the mixture was refluxed for 48 h and quenched with water. The resulting solution was extracted with CH2Cl2, washed with brine, and dried over Na2SO4. The solvent was evaporated in vacuo, the residue was purified using a chromatography column (MeOH/CH2Cl2 v/v 1:25) to yield pure 1 (58 mg, 43%) as a red solid. 1H NMR (400 MHz, CDCl3) δ: 8.26 (s, 1H), 7.47 (d, J = 8.5 Hz, 2H), 7.27 (d, J = 5.0 Hz, 2H), 7.25 (s, 1H), 6.96 (d, J = 4.9 Hz, 1H), 6.55 (d, J = 4.0 Hz, 1H), 6.27 (d, J = 4.0 Hz, 1H), 6.18 (s, 1H), 6.15 (s, 1H), 5.80 (s, 1H), 5.65 (s, 1H), 5.62 (s, 1H), 5.43 (s, 1H), 4.37 (t, J = 5.2 Hz, 2H), 4.31 (t, J = 5.2 Hz, 2H), 3.66 (m, 4H), 1.99 (s, 3H), 1.97 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 167.73, 167.54, 154.37, 152.50, 152.02, 136.21, 136.08, 136.02, 135.20, 132.17, 131.53, 130.39, 126.47, 126.39, 126.08, 121.73, 114.70, 63.65, 63.59, 40.81, 39.86, 18.45, 18.40. MS: m/z cal. for [C29H29BClF2N5O7−H]−: 642.1738, found: 642.1794.
Synthesis of fluorescent core–shell MIPs
Monodisperse SiO2 cores were prepared via a Stöber method and modified with 3-aminopropyltriethoxysilane (APTES), followed by functionalization with a RAFT reagent (4-cyano-4-(thiobenzoylthio)pentanoic acid, CPDB) as we have previously reported, see Section I, (ESI†).29 For MIP1 particles, probe 1 (0.96 mg, 1.49 μmol), the tetrabutylammonium salt of levofloxacin (LEVO.TBA, 0.90 mg, 1.49 μmol, see Section II, ESI† for details on preparation), BMA (4.0 μL, 22.4 μmol), EGDMA (21.0 μL, 110 μmol) and RAFT-functionalized SiO2 particles (10 mg) were mixed in dioxane (1 mL) and sonicated for 10 min to disperse the particles, before ABDV (0.7 mg) was added and the solution was degassed with a stream of Argon for 5 min at 0 °C. Subsequently, the mixture was stirred at 50 °C for 18 h and further aged for 2 h at 70 °C. After the reaction was completed, the particles were washed twice with dioxane and acetonitrile, respectively, with intervening centrifugation at 9140 × g for 8 min to remove the solvent. A solution of methanol and acetic acid (90:1 v/v) was then mixed into the particles, and the suspension was left on a rotator at 40 rpm for 30 min to remove the template, followed by centrifugation at 6931 × g for 5 min. The washing process, consisting of washing with acetonitrile and centrifugation at 6931 × g for 5 min, was repeated 3 times, after which the particles were dried in a vacuum. The non-imprinted polymer (NIP1) particles were prepared according to the same procedure and recipe as described before only in the absence of LEVO.TBA.
Fluorescence titration analysis
To evaluate the recognition characteristics of 1, the MIP and NIP toward LEVO.TBA, fluorescence titration experiments were performed in a quartz cuvette (10 mm path length) at 22 ± 3 °C. Either a stock solution of 1 was used or a dispersion of the core–shell particles was prepared in dioxane (0.05 mg mL−1 of MIP1, 0.03 mg mL−1 of NIP1) and aliquots of 1 mM stock solutions of the analytes were added stepwise until saturation of the fluorescence signal change was reached. After each addition, the solution/suspension was equilibrated by stirring for 2 min prior to fluorescence measurement.The ability for specific binding of the MIP was assessed via the imprinting factor (IF), which is calculated as the relative fluorescence intensity change of MIP vs. NIP according to
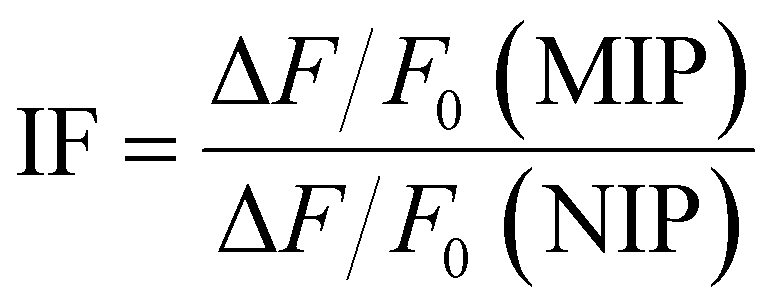
The selectivity of the MIPs for LEVO against other potential competitors was evaluated via the discrimination factor (DF) according to
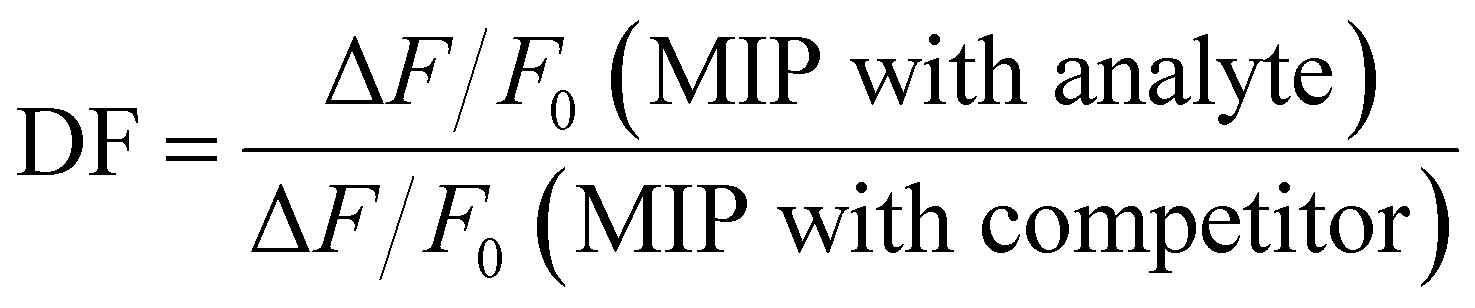
Results and discussion
Considering BODIPY dyes, we found it most promising to integrate the analyte-responsive group at the 3- (aka 5-) position, as this promises to yield more pronounced spectral shifts based on molecular orbital considerations60,61 than introduction via the 1,2,6,7-positions of the dipyrrin moiety or the meso-position at the central methine group.62 Asymmetric substitution at the 3- or 5-position distorts the cross-conjugated polymethine-like BODIPY chromophore,63 which usually leads to hypsochromic absorption shifts and fluorescence changes,64 commonly including a broadening of the emission spectra.62,65 Given the versatility of the BODIPY core and the fact that we have recently found that probe crosslinkers show better performance than probe monomers,30 the aim of this study was to develop fluorescent probe crosslinker 1 (Scheme 1).Synthesis and characterization of probe crosslinker
The BODIPY-based probe 1 was synthesized in a first step by reacting the precursor dichloro-BODIPY 3, which was prepared in 18% overall yield according to a described procedure,59 with ammonia in methanol to afford mono-substituted amino-BODIPY 2 in 80% yield. The probe 1 was then prepared in good yield via the reaction of 2 with 2-isocyanatoethylmethacrylate in the presence of DMAP as catalyst and BHT as polymerization inhibitor in anhydrous CH2Cl2, using two equivalents of methacrylate and DMAP for the doubly linker-substituted crosslinker 1. All obtained products were characterized by 1H NMR, 13C NMR and high-resolution mass spectrometry (HRMS), see ESI† for more details. Additionally, the molecular structure of crosslinker 1 in the solid state was confirmed via X-ray diffraction of a single crystal grown by slow evaporation of a hexane/CH2Cl2 solution (Fig. 1). The selected crystallographic data and refined details are given in Table S1 (ESI†).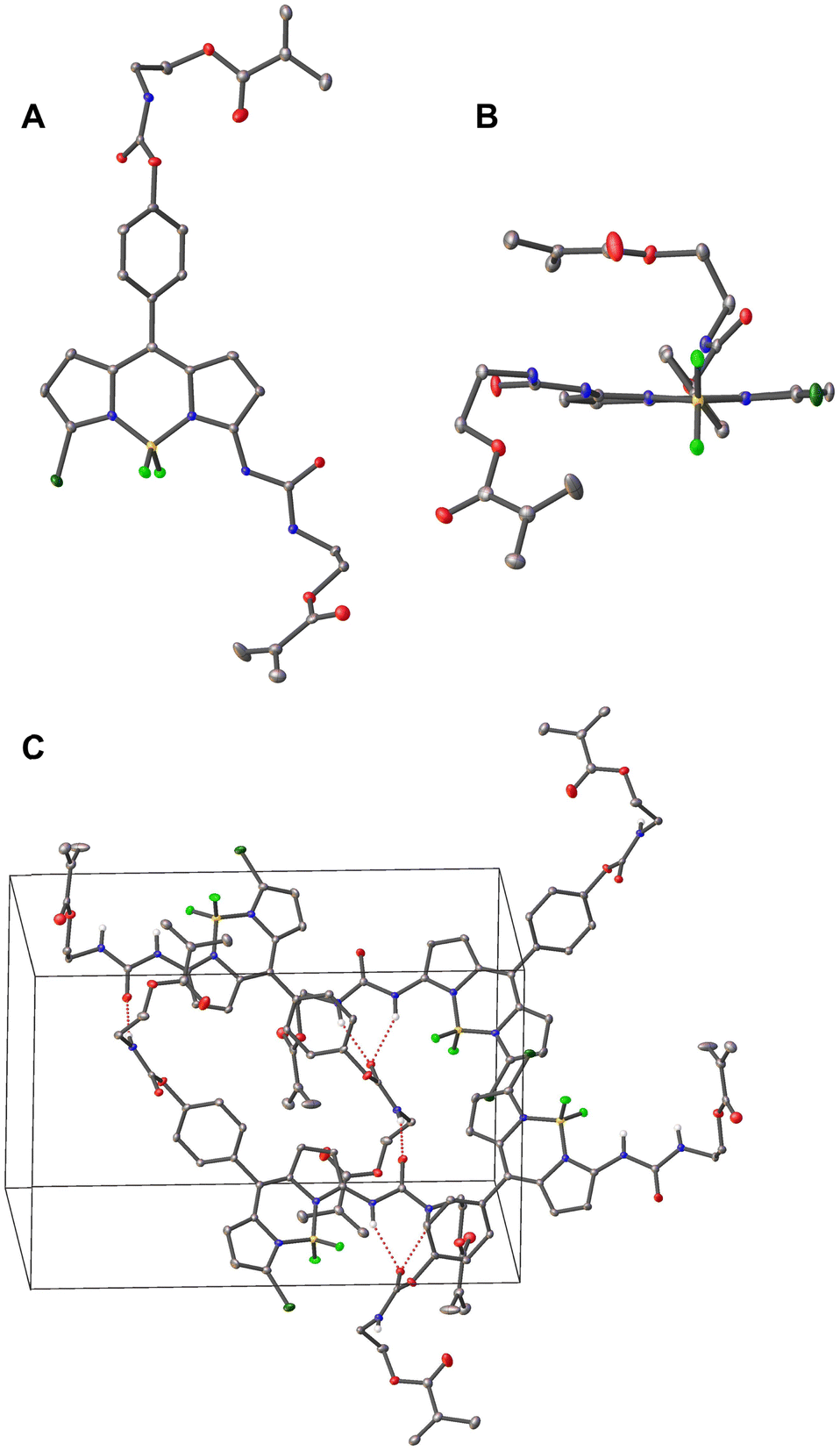 | ||
| Fig. 1 Crystal structure of probe 1 with the thermal ellipsoids plotted at the 30% probability, (A) top view, (B) front view. (C) Molecular packing pattern in the crystal structure of 1, red dashed lines showing H-bonding interactions. Red for O, blue for N, bright green for F, dark green for Cl, yellow for B, and grey for C atoms. For clarity, hydrogen atoms, except for the N–H in (C), and solvent molecules (CH2Cl2) are omitted. | ||
Compound 1 crystallizes in the monoclinic P21/n space group with one molecule of 1 and two CH2Cl2 guest solvent molecules. As shown in Fig. 1, two methacrylate moieties were coupled to the BODIPY scaffold via an SN2 reaction. The boron atom is coordinated by two nitrogen and two fluorine atoms in a tetrahedral geometry. The BODIPY core displays a nearly planar conformation, and the dihedral angle between the meso-phenyl group and the dipyrrin plane amounts to 54.9°, which is in good agreement with other meso-phenyl BODIPYs that carry hydrogen atoms at the BODIPY's 1,7-positions and at the meso-phenyl's 2,6-positions.62,65 The molecules are packed in a head-to-tail orientation in the unit cell and adjacent BODIPY molecules are connected by intermolecular hydrogen bonds between the carbonyl-O of the carbamate motif and the urea's N–H atoms as well as the amido-H of the carbamate motif and the urea's carbonyl-O atom (Fig. 1(c)).
Spectroscopic characteristics of probe crosslinker
To gain insight into its photophysical properties, the fluorescent probe was investigated by absorption and fluorescence spectroscopy in a variety of solvents of increasing polarity from toluene to methanol, and the data are presented in Table 1. As shown in Fig. 2 and Fig. S1, ESI,†1 exhibits the characteristic spectroscopic features of BODIPY derivatives, i.e., narrow absorption bands and an emission band that is a mirror image of the lowest-energy absorption band. The lowest-energy absorption maximum at ca. 520 nm can be assigned to the typical S1 ← S0 transition, while the second maximum or shoulder on the high-energy side is attributed to a 0–1 vibrational transition.62,66 The weaker and broader absorption band at ca. 345 nm, respectively, is assigned to the S2 ← S0 transition.55 Quantum chemical calculations support these assignments, see Section VI, Fig. S2–S4 and Table S3, ESI.†| Solventa | λ abs [nm] | λ em [nm] |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) abs-em
[cm−1]
abs-em
[cm−1] |
Φ f | τ f [ns] |
k
effr
[108 s−1] |
k
effnr
[108 s−1] |
|---|---|---|---|---|---|---|---|
a For relevant solvent parameters, see Table S2 (ESI).
b Absorption maximum.
c Emission maximum.
d Stokes shift.
e Fluorescence quantum yield relative to rhodamine 6G in ethanol (Φf = 0.91).69
f Average fluorescence lifetime:  with relative amplitudes ai and lifetimes τi of the single decay species, see Table S4 (ESI).
g Effective radiative and nonradiative rate constants of the main emissive species: keffr = Φ2/τ2 and keffnr = (1 − Φ2/τ2); for τ2 and Φ2, see Table S4 (ESI). with relative amplitudes ai and lifetimes τi of the single decay species, see Table S4 (ESI).
g Effective radiative and nonradiative rate constants of the main emissive species: keffr = Φ2/τ2 and keffnr = (1 − Φ2/τ2); for τ2 and Φ2, see Table S4 (ESI).
|
|||||||
| Toluene | 521 | 539 | 641 | 0.25 | 0.80 | 2.2 | 6.9 |
| Dioxane | 516 | 536 | 723 | 0.19 | 0.74 | 1.8 | 7.9 |
| CHCl3 | 520 | 538 | 678 | 0.34 | 0.90 | 2.1 | 4.4 |
| THF | 515 | 535 | 796 | 0.16 | 0.46 | 2.2 | 12.4 |
| MeOH | 514 | 534 | 764 | 0.12 | 0.35 | 2.4 | 19.1 |
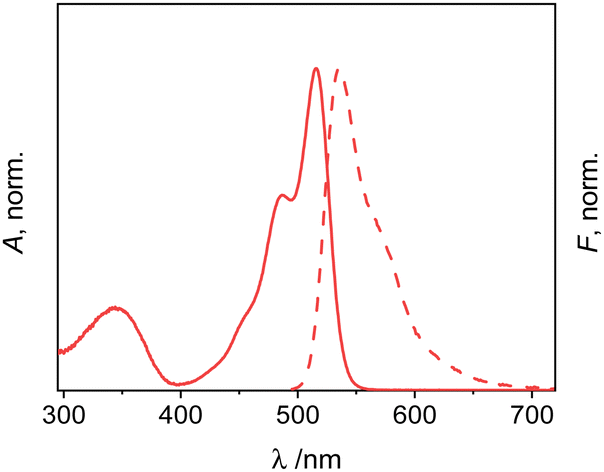 | ||
| Fig. 2 Normalized absorption (solid) and emission (dashed) spectra of 1 in dioxane; c1 = 2 μM; λexc = 478 nm. | ||
The absorption and emission maxima of 1 are not significantly influenced by solvent polarity, but only by dispersive interactions.66 Accordingly, the Stokes shifts also do not show pronounced trends. The fluorescence excitation spectra match the absorption spectra within measurement uncertainty (for a representative example, see Fig. S1, ESI†). The fluorescence quantum yields of bis-methacrylate-substituted 1 are moderate, in agreement with previous reports on 3-amido-substituted BODIPY derivatives.67,68 The fluorescence lifetime data support this interpretation as they generally follow the same trend (Table 1). As is described in detail in Section VII, ESI,† two decay components were found for 1 in all the solvents studied, with one of them being mainly responsible for the dye's emission (Table S4, ESI†). Accordingly, when analysing the respective data, effective radiative rate constants keffr were found that agree well with kr commonly found for green BODIPY dyes.3 Furthermore, neglecting solvent-related peculiarities, the effective non-radiative rate constants keffnr increase with solvent polarity and proticity, see Table 1.
Binding studies with levofloxacin at dilute concentrations
The interaction of 1 with LEVO was studied by absorption and fluorescence spectroscopy. LEVO carries a carboxylic acid group that can be deprotonated by tetrabutylammonium (TBA) hydroxide to give the tetrabutylammonium salt of levofloxacin (LEVO.TBA). The carboxylate group of LEVO.TBA can then form two directional hydrogen bonds with the urea moiety of 1 (Scheme 2), resulting in spectroscopic changes.70 When using 1 for anion recognition, it is important to use a solvent in which the spectroscopic properties of the probe are favourable (e.g., high Φf) and the spectroscopic changes upon analyte binding are as pronounced as possible, while the solvent itself does not interfere with host–guest interactions or the desired formation of the sensory polymer network. Based on the findings in Table 1, spectroscopic investigations were thus conducted in chloroform and dioxane. However, as will be discussed in detail in the part on MIPs below, the latter was identified as the better candidate for our study, which is why we will focus on dioxane here. As shown in Fig. 3, stepwise addition of LEVO.TBA to a dioxane solution of 1 resulted in the formation of the complex 1-LEVO. | ||
| Scheme 2 Possible complex conformers including hydrogen bonding motifs of 1 with LEVO. TBA+ counterions, omitted here for clarity, remain in the vicinity, but do not participate in complexation. | ||
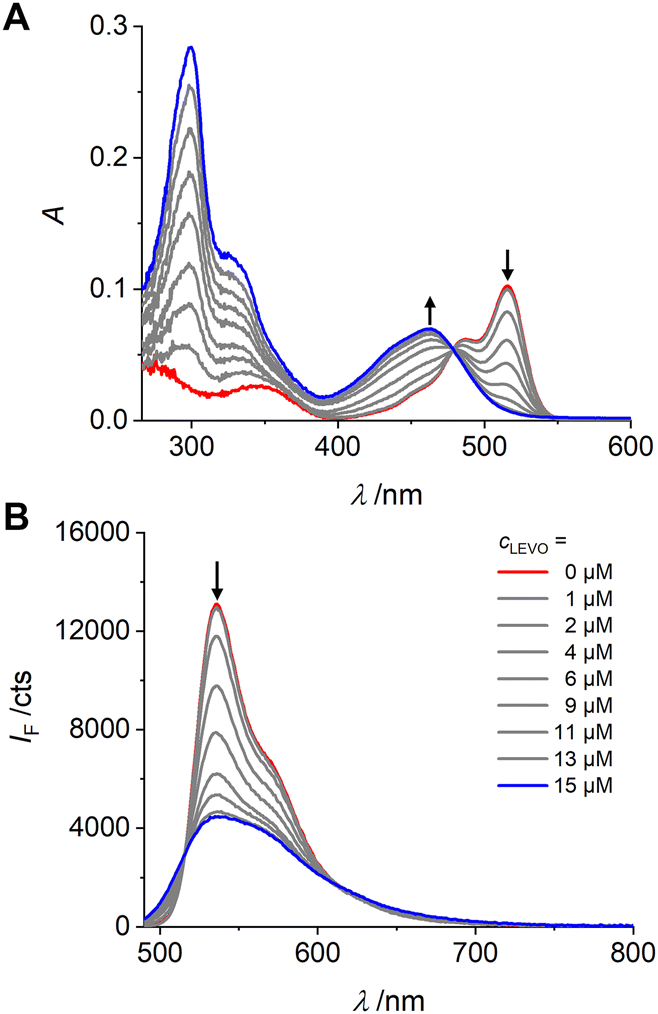 | ||
| Fig. 3 Absorption (A) and emission (B) spectra of 1 (c1 = 2 μM, red spectra) upon addition of LEVO.TBA (concentrations indicated in lower graph) in dioxane; λexc = 478 nm. | ||
The typical BODIPY absorption band at ca. 515 nm gradually decreased, while a new band formed at ca. 460 nm, and a clear isosbestic point was observed at 478 nm. The binding constant for 1 with LEVO.TBA was determined to K1-LEVO.TBA = 2.73 (± 0.06) × 105 M−1, respectively, from the absorption data using a 1:1 binding model of BindFit software (Fig. S5, ESI†). By plotting the change in the A462/A516 absorbance ratio vs. concentration, a typical titration curve was obtained for binding to the LEVO anion (Fig. S6A, ESI†). The fluorescence intensity of 1 at 536 nm decreased progressively with the addition of LEVO.TBA when excited at the isosbestic point in absorption, indicating that the complex emits less strongly than the probe. Fluorescence lifetime measurements of the complex in dioxane confirmed the results (Tables S5 and S6, ESI†).
A detailed analysis of the fluorescence decays also seems to support our interpretation of the two emissive species of 1 (see Section X, ESI†). Whereas the short lifetime component is increased, the long lifetime component is decreased, finally resulting in two species with a reduced keffr = 0.9 × 108 s−1, both being present in almost equimolar amounts (Tables S5 and S6, ESI†). Assuming that binding to the bulky LEVO anion facilitates the outward orientation of the urea group compared to the uncomplexed 1, and considering that the angles between the urea group and dipyrrin core were similar for the inward and outward orientation of 1–LEVO (Section VI, ESI†), but significantly larger than for 1 (approximately 27° vs. 4°), it is reasonable to assume that complexation causes a redistribution of species and influences the planarity of the polymethine auxochrome (dipyrrin) and the attached substituent (urea). This results in an overall quenching effect and broadening of the spectrum. The latter, a broadening of the emission band, has also already been observed when a weaker electron-donating group in the 3-position is exchanged for a stronger electron-donating group in such a cross-conjugated polymethinic system,64,65,71 which essentially happens when a negatively charged carboxylate group binds to a neutral urea group. The plots of the relative changes in emission intensity are consistent with these results and allow analytical evaluation (Fig. S6B, ESI†). Again, for the complex, the fluorescence excitation spectra match the absorption spectra (for a representative example, see Fig. S6C, ESI†) and the theoretical investigations described in Section VI, ESI† support the experimentally observed changes and enable a mechanistic explanation.
Binding studies with levofloxacin at polymerization concentrations
The fluorescent probe crosslinker 1 was designed for covalent integration into responsive polymer networks by co-polymerization. Before synthesizing the materials, it is therefore necessary to ensure that the response observed in dilute solution is maintained at the higher concentrations required for polymer formation. In addition, the complex between 1 and LEVO.TBA must remain intact under polymerization conditions, i.e., in the presence of other species required for polymerization such as structural crosslinkers and comonomers, to achieve imprinting. Based on the considerations regarding a suitable solvent, see above, our experience with fluorescent MIPs for small-molecule carboxylates and after a six-step optimization process involving MIP preparation, see below, benzyl methacrylate (BMA) and ethylene glycol dimethyl methacrylate (EGDMA) were chosen as structural building blocks. BMA is an aromatic monofunctional methacrylate monomer that provides a phenyl group for π–π interaction with the aromatic moiety of LEVO.TBA and has been shown to be an effective functional monomer for imprinting of analytes bearing aromatic moieties.72,73 EGDMA is a widely used hydrophilic cross-linking agent that allows high affinity to polar analytes due to its polar character. In addition, EGDMA provides a certain flexibility to the cross-linked MIP-shells, which facilitates the access of the analyte molecules to the imprinted cavities.74 As shown in Fig. 4, the spectroscopic changes at 0.25 and 1 equiv. of LEVO.TBA agree well with the pattern observed in dilute solution. Furthermore, the addition of 15 equiv. of BMA and 74 equiv. of EGDMA did not result in any pronounced additional changes, indicating that the complexes remained stable under these conditions. (Note that the other five recipes realised during the optimisation process also resulted in favourable responses under pre-polymerisation conditions.) To verify these results, 1H NMR titrations were performed with 1 and LEVO.TBA (Fig. S7, ESI†). In the presence of 1 equiv. of LEVO.TBA, the NH proton of the urea attached to the pyrrole shifted downfield by 0.26 ppm (Hb) and the other NH proton of the urea shifted downfield by 0.74 ppm (Ha), confirming the formation of hydrogen bonds between 1 and LEVO.TBA.75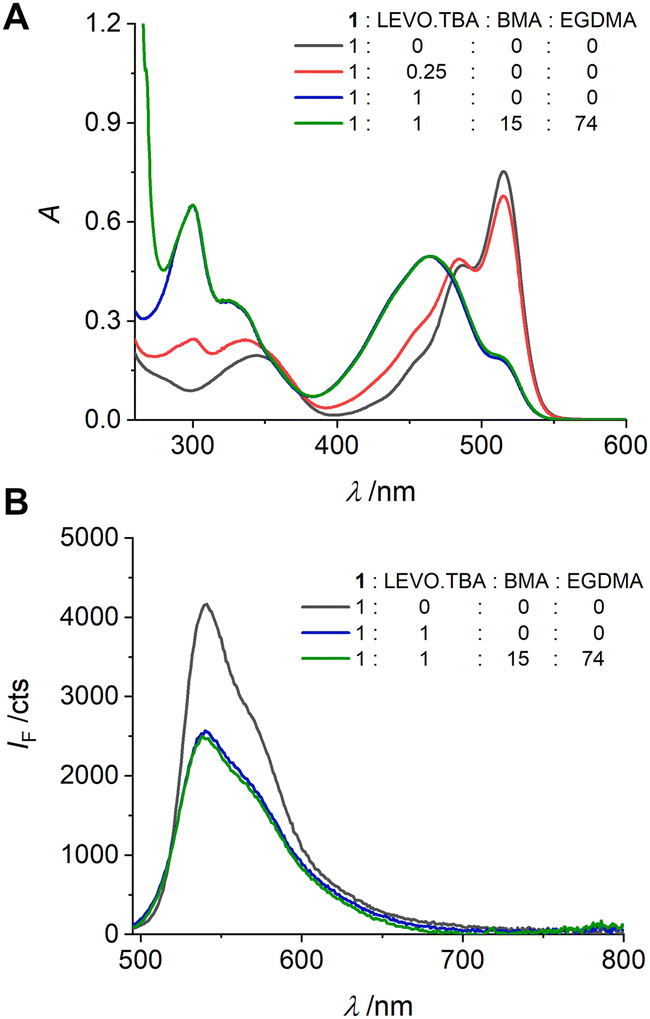 | ||
| Fig. 4 Absorption (A) and emission (B) spectra of the pre-polymerization solutions of 1 (c1 = 1.49 mM) upon addition of LEVO.TBA and/or BMA and EGDMA (ratios as indicated) in dioxane; λexc = 478 nm. | ||
Preparation and characterization of MIPs incorporating probe 1
Having established that the complex between the polymerizable fluorescent BODIPY probe 1 and the anionic LEVO remains stable under pre-polymerization conditions, we proceeded to molecular imprinting and MIP synthesis. Since conventional bulk MIPs can lead to uneven distribution of recognition sites, incomplete template removal and slow template diffusion,76 synthesis of a thin MIP layer on a solid core particle was chosen to avoid these limitations. If a thin MIP layer is grafted from a supporting particle core of micro- or nanometric dimension, not only faster diffusion of the template to the binding sites is ensured, but also a more efficient response due to the possibility of complete template removal and uniform distribution of the imprinted cavities.77 To this end, sub-micron-sized silica particles are commonly the vehicle of choice for the preparation of core–shell MIPs due to their thermal stability and favourable monodispersity.76 In addition, reversible addition–fragmentation chain transfer (RAFT) polymerization from the surface of such supports is an effective method to obtain a thin polymer shell and has been intensively explored for the preparation of core–shell MIPs.78 Based on these considerations, nanometrically thin MIP layers incorporating BODIPY-type crosslinker 1 against the antibiotic LEVO have been prepared (Scheme 3).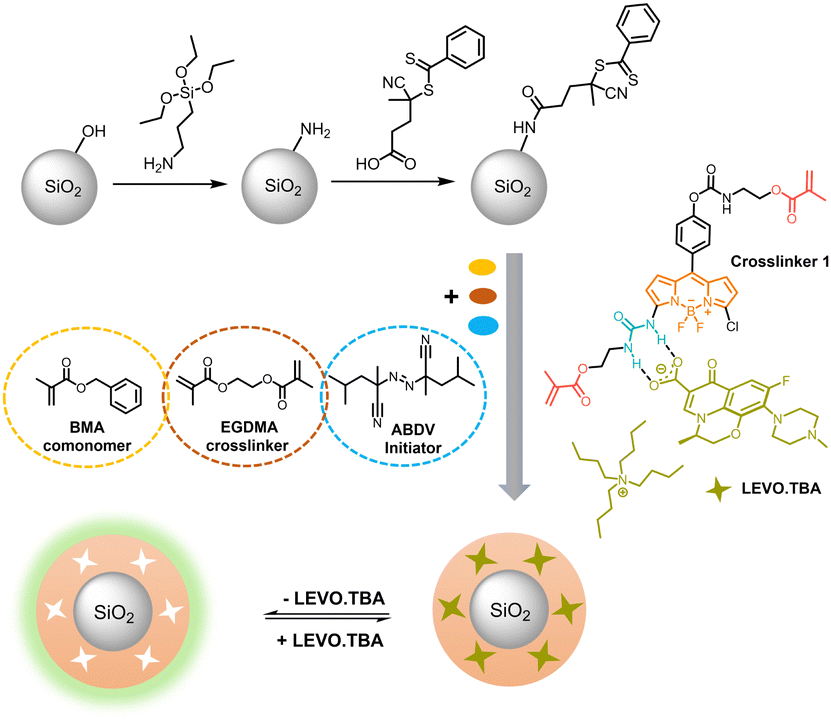 | ||
| Scheme 3 Schematic for the preparation of fluorescent core–shell MIPs. | ||
Briefly, monodisperse SiO2 nanoparticles were obtained following a modified Stöber approach,29 followed by the functionalization of the silica surface with 3-aminopropyltriethoxysilane (APTES) as anchor points for condensation of the RAFT agent (4-cyano-4-(thiobenzoylthio)pentanoic acid). The chemical modifications of each step were investigated by zeta potential measurements of the respective particles in water at pH 5 (Fig. S8, ESI†). While the net surface charge of the neat SiO2 particles was negative, −48 ± 1 mV, indicating the presence of surface silanolate groups, APTES modification resulted in an increase of +61 mV and a charge reversal to +13 ± 1 mV, indicating an excess of protonated amino groups on the particle surface. A second charge reversal back to −28 ± 1 mV indicates efficient condensation of APTES with the RAFT reagent. The density of RAFT groups was calculated to be 1.1 nm−2 based on the sulphur content (0.19 ± 0.01%) from elemental analysis and the BET surface area (17.1 ± 0.1 m2 g−1) from porosimetry. Thermogravimetric (TGA) analysis of the particles revealed that up to ca. 160 °C, mass loss is due to evaporation of adsorbed water and organic solvents (Fig. S8, ESI†). Between 160 and 550 °C, the mass loss of 8.4% and 8.6% of APTES-SiO2 and RAFT-SiO2 particles, respectively, is due to the decomposition of the organic layer coated onto the particles. The total mass loss of RAFT-SiO2 is 13.8%, i.e., 0.8% more than that of APTES-SiO2; the difference is thus attributed to the amount of RAFT groups in the system.
After RAFT-mediated growth of the fluorescent MIP1 layer using BODIPY probe 1, LEVO.TBA, BMA, EGDMA and 2,2′-azobis(2,4-dimethylvaleronitrile) as initiator in dioxane, the template was removed by washing with an acidic solution, disrupting the hydrogen bonded complex and leaving cavities which are complementary in shape and electronic demand to the template, ready to selectively rebind deprotonated LEVO as analyte. For comparison, the non-imprinted polymer control particles NIP1 were prepared in an identical manner to MIP1 without the addition of LEVO.TBA. In addition to the considerations on a suitable solvent for MIP preparation as mentioned above, Section XIII, ESI,† including Fig. S9 and Table S7, collects a more detailed account on system optimization.
The size and morphology of the SiO2 core particles and the core–shell MIPs and NIPs were investigated by transmission electron microscopy (TEM). The SiO2 particles appeared monodisperse and spherical with an average diameter of 275 ± 10 nm, calculated from the particle size distribution (Fig. S10, ESI†). Similar particle morphologies were observed for the MIPs and NIPs. The shell thicknesses were determined from TEM images to be 13 ± 6 nm and 15 ± 7 nm for MIP1 and NIP1, respectively (Fig. 5). As mentioned above, such thin imprinted layers are ideal for an accelerated response and at the same time allow for good sensitivity.
 | ||
| Fig. 5 TEM images of MIP1 (A) and NIP1 (B). | ||
The spectroscopic properties of the 1-containing polymers in dioxane are virtually identical to those of 1 in the same solvent, i.e., the absorption and emission maxima are centred at 519 and 537 nm for MIP1 and 519 and 539 nm for NIP1 (Fig. 6 and Fig. S11, ESI†). As would be expected upon covalent incorporation into a polymer matrix, the fluorescence of 1 is somewhat stronger than in solution, exemplified by an increase in the average fluorescence lifetime τf from 0.74 ns for 1 in dioxane (Table 1) to 1.31 ns in MIP1 and 1.65 ns in NIP1. This further increase in the NIP reflects well the fact that a denser network can be formed around 1 upon polymerization in the absence of the template. The fluorescence decays are nonexponential, consistent with a different microenvironment around each single emitter.79,80
 | ||
| Fig. 6 Fluorescence responses of MIP1 (A) and NIP1 (B) upon addition of LEVO.TBA (concentrations indicated in upper graph) in dioxane; λexc = 478 nm. | ||
To estimate the sensitivity of the fluorescent MIPs, MIP1 and NIP1 were first titrated with increasing amounts of LEVO.TBA (from 0 to 78 μM) in dioxane. As can be seen in Fig. 6(A), the fluorescence intensity of MIP1 decreased significantly with the addition of LEVO.TBA, and the decreased fluorescence was accompanied by a broadening of the band and a 34 nm bathochromic shift of the emission maximum; the absorption maximum was shifted by ca. 60 nm to shorter wavelengths (Fig. S11, ESI†). The fluorometric analytical figures of merit, limit of blank (LOB) and limit of detection (LOD), were determined to be 0.10 μM and 0.34 μM, respectively (see Section XVII and Fig. S12, ESI† for details), with a dynamic working range of 0.9–23 μM. In case of NIP1, addition of the same amount of LEVO.TBA resulted in less efficient quenching, while the emission maximum remained unchanged (Fig. 6(B)). This suggests successful imprinting for MIP1.
Important for the generation of responsive polymers is the fact that the different spectroscopic responses of MIP1 and NIP1 can be exploited either by evaluating the fluorescence changes at, e.g., 650 nm, that is unique to the MIP, or by evaluating the quotient of the signals at 540 and 650 nm. Using the simpler, single-wavelength approach, a promising imprinting factor (IF) of 18.1 was determined for MIP1, which hints at a specific recognition. The imprinting factor is the quotient of the signal obtained with the MIP divided by the signal obtained with the NIP at a given analyte concentration, see Experimental section.
Such pronounced differences in the accommodation of the analyte molecule in the MIP and the NIP suggest that the system may also have good selectivity for LEVO over other potential competitors. Accordingly, two other antibiotics carrying a carboxylic acid, an amino and a phenyl group, amoxicillin (AMOX) and ampicillin (AMPI) as their TBA salts (Fig. 7(A)), were measured as potential competitors. Fluorescence titration experiments showed that MIP1 undergoes significantly weaker fluorescence intensity changes and no spectral emission shifts when AMOX.TBA and AMPI.TBA were added (Fig. 6(A), (B), 7(B) and Fig. S13, ESI†). The discrimination factor (DF), defined as the ratio of the change in fluorescence intensity of MIP1 with LEVO.TBA to MIP1 with the competitors, was determined to be 5.0 and 5.1 against AMOX.TBA and AMPI.TBA, respectively. Overall, these results indicate that fluorescent probe 1 containing two polymerizable units was efficiently immobilized in the polymer matrix, resulting in selective recognition of LEVO.TBA.
 | ||
Fig. 7 (A) Chemical structures of competitors, AMOX.TBA and AMPI.TBA. (B) Fluorescence changes of MIP1 (■) and NIP1 ( ) particles towards LEVO.TBA and its competitors AMOX.TBA ( ) particles towards LEVO.TBA and its competitors AMOX.TBA ( ) and AMPI.TBA ( ) and AMPI.TBA ( ) at 650 nm. ) at 650 nm. | ||
In contrast to other fluorescent probe monomers and crosslinkers, BODIPY 1 exhibits pronounced spectroscopic changes upon guest binding, which are also observed in a MIP matrix. However, distinct differences are obvious. The characteristic BODIPY emission band at ca. 535 nm decreases in all combinations of MIP1 and NIP1 with LEVO.TBA, AMOX.TBA and AMPI.TBA, with the most pronounced effect observed for MIP1 and LEVO.TBA. Additionally, the emission band broadens significantly, with a full width at half maximum (FWHM) of 53 nm for MIP1 increasing to 137 nm for MIP1/LEVO.TBA. In contrast, NIP1/LEVO.TBA shows a narrower broadening (FWHM = 77 nm) and MIP1 in the presence of other competitors exhibits even smaller broadening (FWHM < 60 nm). Considering that the binding of LEVO by 1 leads to a broadening of the emission band from FWHM = 49 nm to 88 nm (Fig. 3(B)), these findings suggest that the responses of NIP1 and MIP1 to the competitors are due to nonspecific binding, possibly by probe crosslinkers near the MIP–solution interface or in regions of the polymer shell with lower crosslinking density. These probe crosslinkers are not located within fully formed cavities and are thus susceptible to complexation by a broader range of organic carboxylates. However, analysis of the responses in the longer wavelength range (Fig. 7(B)) indicates that these arise from probe crosslinkers situated well within the polymer network and within well-formed cavities. The relative abundance of probe crosslinkers near the interface compared to those in the polymer layer depends on the thickness of the latter, offering potential for sensitivity enhancement through careful tuning. Importantly, previous findings have demonstrated that this aspect can be addressed by measuring both the MIP and NIP in an assay to correct for nonspecific binding.30 Furthermore, our present results suggest that BODIPY 1 is not only a powerful probe for carboxylate detection but also an excellent candidate for reporting successful MIP formation.
Interestingly, compared with the diaryl urea-type BODIPY probes reported in ref. 65, probe 1 with an alkyl-aryl-substituted urea shows a ca. 6-fold higher fluorescence quantum yield while the spectral band positions are virtually identical. Furthermore, in that case spectroscopic responses were only observed in the presence of fluoride as the target anion, but not in the presence of oxoanions such as H2PO4− or HSO4−, showing a clear advantage of our approach when it comes to addressing this much broader class of analytes.
Conclusions
In summary, a novel urea-functionalized BODIPY probe with two additional polymerizable units was synthesized and incorporated into thin MIP shells on submicron-sized silica particles to selectively target levofloxacin, a carboxylate containing antibiotic. Compared to other chromophores commonly used in sensory polymer matrices, the probe crosslinker does not only exhibit favourable spectroscopic properties such as absorption and emission in the blue/green region of the spectrum, but also considerable fluorescence quantum yields of 15–20%, which is an order of magnitude higher than those of most benzoxadiazole-, naphthalimide-, phenazine- or phenoxazinone-type probe monomers and crosslinkers. 1 displayed high affinity (2.73 × 105 M−1) for LEVO.TBA and demonstrated a favourable fluorescence response—red-shift, broadening, decrease—which resulted from the modulation of the cross-conjugated polymethinic BODIPY fluorophore by binding of the anion to the asymmetrically attached urea receptor. Molecular imprinting facilitated the probe's selectivity toward LEVO.TBA, showing favourable discrimination factors of 5 against other carboxylate, amino and phenyl group-containing antibiotics. The findings highlight the potential of BODIPY chromophores as versatile platforms for developing anion-responsive indicators that operate through direct, hydrogen bond-mediated binding and fluorescence modulation instead of irreversible probe degradation,81 analyte-induced inner filter effects82 or other indirect processes as for many QD-based systems. The dyes’ ability to absorb well within the visible or near-infrared spectral region without relying on unbridged double bonds makes them particularly suitable for polymer-type sensing membranes. Furthermore, it is anticipated that such fluorescent crosslinkers can function not only as thin layers on carrier particles but also as thin films on layered macroscopic supports or optical fibres.Author contributions
YS: methodology, investigation, formal analysis, funding acquisition, writing – original draft. KG: investigation, supervision, writing – review & editing. VV: methodology, supervision, writing – review & editing. JB: investigation, formal analysis, writing – review & editing. KR: conceptualization, methodology, supervision, funding acquisition, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 21 March 2024. The footnotes for Table 1 were updated for readability.Acknowledgements
We thank the China Scholarship Council for a fellowship to Y. S. (no. 201908330307), the European Commission for funding under the Horizon 2020 research program through a Research and Innovation Action (RIA), grant agreement no 848098 (REVERT), B. Bhattacharya (BAM, Structural Analysis Div.) for X-ray analysis, M. Grüneberg (BAM, Chemical and Optical Sensing Div.) for TGA, A. Meckelburg (BAM, Inorganic Reference Materials Div.) for elemental analysis, A. Zimathies (BAM, Structure Analysis Div.) for porosimetry and J. Lisec (BAM, Organic Trace and Food Analysis Div.) for MS measurements.References
- P. Kaur and K. Singh, J. Mater. Chem. C, 2019, 7, 11361–11405, 10.1039/c9tc03719e.
- T. Werner, C. Huber, S. Heinl, M. Kollmannsberger, J. Daub and O. S. Wolfbeis, Fresenius’ J. Anal. Chem., 1997, 359, 150–154, DOI:10.1007/s002160050552.
- M. Kollmannsberger, K. Rurack, U. Resch-Genger and J. Daub, J. Phys. Chem. A, 1998, 102, 10211–10220, DOI:10.1021/jp982701c.
- E. V. Antina, N. A. Bumagina, A. I. V’Yugin and A. V. Solomonov, Dyes Pigm., 2017, 136, 368–381, DOI:10.1016/j.dyepig.2016.08.070.
- P. E. Hande, Y. G. Shelke, A. Datta and S. J. Gharpure, ChemBioChem, 2022, 23, e202100448, DOI:10.1002/cbic.202100448.
- S. Kolemen and E. U. Akkaya, Coord. Chem. Rev., 2018, 354, 121–134, DOI:10.1016/j.ccr.2017.06.021.
- V. N. Nguyen, J. Ha, M. Cho, H. D. Li, K. M. K. Swamy and J. Yoon, Coord. Chem. Rev., 2021, 439, 213936, DOI:10.1016/j.ccr.2021.213936.
- E. Antina, N. Bumagina, Y. Marfin, G. Guseva, L. Nikitina, D. Sbytov and F. Telegin, Molecules, 2022, 27, 1396, DOI:10.3390/molecules27041396.
- A. Barattucci, S. Campagna, T. Papalia, M. Galletta, A. Santoro, F. Puntoriero and P. Bonaccorsi, ChemPhotoChem, 2020, 4, 647–658, DOI:10.1002/cptc.202000073.
- L. Y. Wang, H. Ding, X. G. Ran, H. Tang and D. R. Cao, Dyes Pigm., 2020, 172, 107857, DOI:10.1016/j.dyepig.2019.107857.
- Y. J. Iv, J. Xu, Y. Guo and S. J. Shao, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 95–101, DOI:10.1007/s10847-011-9946-1.
- Y. J. Lv, J. Xu, Y. Guo and S. J. Shao, Chem. Pap., 2011, 65, 553–558, DOI:10.2478/s11696-011-0033-2.
- I. S. Shin, S. W. Bae, H. Kim and J. I. Hong, Anal. Chem., 2010, 82, 8259–8265, DOI:10.1021/ac1017293.
- O. Y. F. Henry, D. C. Cullen and S. A. Piletsky, Anal. Bioanal. Chem., 2005, 382, 947–956, DOI:10.1007/s00216-005-3255-8.
- X. Z. Huang, L. Xia and G. K. Li, Chemosensors, 2023, 11, 168, DOI:10.3390/chemosensors11030168.
- M. C. Moreno-Bondi, F. Navarro-Villoslada, E. Benito-Pena and J. L. Urraca, Curr. Anal. Chem., 2008, 4, 316–340, DOI:10.2174/157341108785914925.
- J. Wackerlig and P. A. Lieberzeit, Sens. Actuators, B, 2015, 207, 144–157, DOI:10.1016/j.snb.2014.09.094.
- W. Y. X. Yang, Y. X. Ma, H. Sun, C. X. Huang and X. T. Shen, TrAC, Trends Anal. Chem., 2022, 152, 116608, DOI:10.1016/j.trac.2022.116608.
- K. Gawlitza, W. Wan, S. Wagner and K. Rurack, in Advanced Molecularly Imprinting Materials, ed. A. Tiwari and L. Uzun, Scrivener, Beverly, MA, 2016, pp. 89–128 DOI:10.1002/9781119336181.ch3.
- W. Wan, S. Wagner and K. Rurack, Anal. Bioanal. Chem., 2016, 408, 1753–1771, DOI:10.1007/s00216-015-9174-4.
- Q. J. Li, S. Shinde, G. Grasso, A. Caroli, R. Abouhany, M. Lanzillotta, G. Q. Pan, W. Wan, K. Rurack and B. Sellergren, Sci. Rep., 2020, 10, 9924, DOI:10.1038/s41598-020-66802-3.
- S. J. Xu, Y. W. Zou and H. Q. Zhang, Talanta, 2020, 211, 120711, DOI:10.1016/j.talanta.2020.120711.
- V. Valderrey, K. Gawlitza and K. Rurack, Chem. – Eur. J., 2022, 28, e202104525, DOI:10.1002/chem.202104525.
- W. L. Zhang, Q. Li and H. Q. Zhang, Molecules, 2023, 28, 1077, DOI:10.3390/molecules28031077.
- R. Wagner, W. Wan, M. Biyikal, E. Benito-Pena, M. C. Moreno-Bondi, I. Lazraq, K. Rurack and B. Sellergren, J. Org. Chem., 2013, 78, 1377–1389, DOI:10.1021/jo3019522.
- X. A. Ton, V. Acha, P. Bonomi, B. T. S. Bui and K. Haupt, Biosens. Bioelectron., 2015, 64, 359–366, DOI:10.1016/j.bios.2014.09.017.
- N. Y. Limaee, S. Rouhani, M. E. Olya and F. Najafi, Polymer, 2019, 177, 73–83, DOI:10.1016/j.polymer.2019.05.067.
- M. Kimani, E. Kislenko, K. Gawlitza and K. Rurack, Sci. Rep., 2022, 12, 14151, DOI:10.1038/s41598-022-16825-9.
- W. Wan, A. B. Descalzo, S. Shinde, H. Weisshoff, G. Orellana, B. Sellergren and K. Rurack, Chem. – Eur. J., 2017, 23, 15974–15983, DOI:10.1002/chem.201703041.
- S. Wagner, J. Bell, M. Biyikal, K. Gawlitza and K. Rurack, Biosens. Bioelectron., 2018, 99, 244–250, DOI:10.1016/j.bios.2017.07.053.
- Y. Q. Fan, J. J. Zhang, Z. Y. Hong, H. Y. Qiu, Y. Li and S. C. Yin, Polymers, 2021, 13, 75, DOI:10.3390/polym13010075.
- U. Haldar and H. I. Lee, Polym. Chem., 2018, 9, 4882–4890, 10.1039/c8py01232f.
- P. T. Lu, K. Y. Chung, A. Stafford, M. Kiker, K. Kafle and Z. A. Page, Polym. Chem., 2021, 12, 327–348, 10.1039/d0py01513j.
- P. Ashokkumar, H. Weisshoff, W. Kraus and K. Rurack, Angew. Chem., Int. Ed., 2014, 53, 2225–2229, DOI:10.1002/anie.201307848.
- E. Climent, M. Biyikal, K. Gawlitza, T. Dropa, M. Urban, A. M. Costero, R. Martinez-Manez and K. Rurack, Chem. – Eur. J., 2016, 22, 11138–11142, DOI:10.1002/chem.201601269.
- P. Ashokkumar, J. Bell, M. Buurman and K. Rurack, Sens. Actuators, B, 2018, 256, 609–615, DOI:10.1016/j.snb.2017.09.201.
- US Environmental Protection Agency, Clean Water Act, https://www.epa.gov/laws-regulations/summary-clean-water-act, (accessed 04/2023).
- European Parliament and Council, Directive 2000/60/EC establishing a framework for Community action in the field of water policy, https://environment.ec.europa.eu/topics/water/water-framework-directive_en, (accessed 04/2023).
- International Water Management Institute (IWMI), https://www.iwmi.cgiar.org/, (accessed 04/2023).
- J. Han, M. Ishigaki, Y. Takahashi, H. Watanabe and Y. Umebayashi, Anal. Sci., 2023, 39, 133–137, DOI:10.1007/s44211-023-00271-2.
- M. E. Levison and J. H. Levison, Infect. Dis. Clin. North Am., 2009, 23, 791–815, DOI:10.1016/j.idc.2009.06.008.
- T. P. Van Boeckel, J. Pires, R. Silvester, C. Zhao, J. Song, N. G. Criscuolo, M. Gilbert, S. Bonhoeffer and R. Laxminarayan, Science, 2019, 365, eaaw1944, DOI:10.1126/science.aaw1944.
- C. Manyi-Loh, S. Mamphweli, E. Meyer and A. Okoh, Molecules, 2018, 23, 795, DOI:10.3390/molecules23040795.
- S. A. Kraemer, A. Ramachandran and G. G. Perron, Microorganisms, 2019, 7, 180, DOI:10.3390/microorganisms7060180.
- A. Sitovs, I. Sartini and M. Giorgi, Res. Vet. Sci., 2021, 137, 111–126, DOI:10.1016/j.rvsc.2021.04.031.
- T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, MedChemComm, 2019, 10, 1719–1739, 10.1039/c9md00120d.
- S. Siewert, J. Pharm. Biomed. Anal., 2006, 41, 1360–1362, DOI:10.1016/j.jpba.2006.02.010.
- S. Schulte, T. Ackermann, N. Bertram, T. Sauerbruch and W. D. Paar, J. Chromatogr. Sci., 2006, 44, 205–208, DOI:10.1093/chromsci/44.4.205.
- S. Carrasco, E. Benito-Pena, D. R. Walt and M. C. Moreno-Bondi, Chem. Sci., 2015, 6, 3139–3147, 10.1039/c5sc00115c.
- L. Zhang and L. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 16248–16256, DOI:10.1021/acsami.6b04381.
- K. C. Chen, R. He, X. Y. Luo, P. Z. Qin, L. Tan, Y. W. Tang and Z. C. Yang, Biosens. Bioelectron., 2017, 94, 609–615, DOI:10.1016/j.bios.2017.03.059.
- Y. Y. Geng, M. L. Guo, J. A. Tan, S. Y. Huang, Y. W. Tang, L. Tan and Y. Liang, Sens. Actuators, B, 2018, 268, 47–54, DOI:10.1016/j.snb.2018.04.065.
- A. D. Hudson, O. Jamieson, R. D. Crapnell, K. Rurack, T. C. C. Soares, F. Mecozzi, A. Laude, J. Gruber, K. Novakovic and M. Peeters, Mater. Adv., 2021, 2, 5105–5115, 10.1039/d1ma00192b.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323, 10.1039/c0cs00062k.
- Z. Shen, H. Rohr, K. Rurack, H. Uno, M. Spieles, B. Schulz, G. Reck and N. Ono, Chem. – Eur. J., 2004, 10, 4853–4871, DOI:10.1002/chem.200400173.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112–122, DOI:10.1107/S0108767307043930.
- G. M. Sheldrick, SADABS Version 2.10, University of Göttingen, 2002 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
- M. Baruah, W. W. Qin, N. Basaric, W. M. De Borggraeve and N. Boens, J. Org. Chem., 2005, 70, 4152–4157, DOI:10.1021/jo0503714.
- J. Bañuelos Prieto, F. López Arbeloa, V. Martínez Martínez and I. López Arbeloa, Chem. Phys., 2004, 296, 13–22, DOI:10.1016/j.chemphys.2003.09.010.
- J. Bañuelos, F. López Arbeloa, T. Arbeloa, S. Salleres, J. L. Vilas, F. Amat-Guerri, M. Liras and I. López Arbeloa, J. Fluoresc., 2008, 18, 899–907, DOI:10.1007/s10895-008-0320-7.
- M. Hecht, T. Fischer, P. Dietrich, W. Kraus, A. B. Descalzo, W. E. S. Unger and K. Rurack, ChemistryOpen, 2013, 2, 25–38, DOI:10.1002/open.201200039.
- S. Dähne and F. Moldenhauer, in Progress in Physical Organic Chemistry, ed. R. W. Taft, John Wiley & Sons, New York, 1985, pp. 1–130 DOI:10.1002/9780470171943.ch1.
- O. Przhonska, Y. Slominsky, A. Kachkovsky, U. Stahl, M. Senoner and S. Daehne, AIP Conf. Proc., 1996, 364, 197–203, DOI:10.1063/1.50194.
- E. Ganapathi, S. Madhu, T. Chatterjee, R. Gonnade and M. Ravikanth, Dyes Pigm., 2014, 102, 218–227, DOI:10.1016/j.dyepig.2013.10.038.
- W. Qin, M. Baruah, M. Van der Auweraer, F. C. De Schryver and N. Boens, J. Phys. Chem. A, 2005, 109, 7371–7384, DOI:10.1021/jp052626n.
- J. G. Knight, R. B. Alnoman and P. G. Waddell, Org. Biomol. Chem., 2015, 13, 3819–3829, 10.1039/c4ob02626h.
- M. Deng, D. Gong, S.-C. Han, X. Zhu, A. Iqbal, W. Liu, W. Qin and H. Guo, Sens. Actuators, B, 2017, 243, 195–202, DOI:10.1016/j.snb.2016.11.139.
- C. Wurth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550, DOI:10.1038/nprot.2013.087.
- T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117, DOI:10.1016/j.ccr.2006.08.017.
- C. S. Gutsche, B. F. Hohlfeld, K. J. Flanagan, M. O. Senge, N. Kulak and A. Wiehe, Eur. J. Org. Chem., 2017, 3187–3196, DOI:10.1002/ejoc.201700264.
- W. Wan, M. Biyikal, R. Wagner, B. Sellergren and K. Rurack, Angew. Chem., Int. Ed., 2013, 52, 7023–7027, DOI:10.1002/anie.201300322.
- S. Jiang, K. Gawlitza and K. Rurack, in Molecularly Imprinted Polymers: Methods and Protocols, ed. A. Martín-Esteban, Humana, New York, NY, 2021, pp. 195–208 DOI:10.1007/978-1-0716-1629-1_17.
- N. S. Shaipulizan, S. N. A. Jamil, S. Kamaruzaman, N. N. S. Subri, A. A. Adeyi, A. H. Abdullah and L. C. Abdullah, Polymers, 2020, 12, 423, DOI:10.3390/polym12020423.
- M. Boiocchi, L. Del Boca, D. Esteban-Gomez, L. Fabbrizzi, M. Licchelli and E. Monzani, Chem. – Eur. J., 2005, 11, 3097–3104, DOI:10.1002/chem.200401049.
- B. Fresco-Cala, A. D. Batista and S. Cardenas, Molecules, 2020, 25, 4740, DOI:10.3390/molecules25204740.
- L. X. Chen, S. F. Xu and J. H. Li, Chem. Soc. Rev., 2011, 40, 2922–2942, 10.1039/c0cs00084a.
- E. Abdollahi, M. Abdouss, M. Salami-Kalajahi and A. Mohammadi, Polym. Rev., 2016, 56, 557–583, DOI:10.1080/15583724.2015.1119162.
- O. Przhonska, M. Bondar, J. Gallay, M. Vincent, Y. Slominsky, A. Kachkovski and A. P. Demchenko, J. Photochem. Photobiol., B, 1999, 52, 19–29, DOI:10.1016/S1011-1344(99)00096-2.
- A. Y. Jee, S. Park, H. Kwon and M. Lee, Chem. Phys. Lett., 2009, 477, 112–115, DOI:10.1016/j.cplett.2009.06.088.
- W. Y. Mao, L. Y. Xia and H. X. Xie, Angew. Chem., Int. Ed., 2017, 56, 4468–4472, DOI:10.1002/anie.201612495.
- Z. Q. Xu, X. F. Yi, Q. Wu, Y. C. Zhu, M. R. Ou and X. P. Xu, RSC Adv., 2016, 6, 89288–89297, 10.1039/c6ra19459a.
Footnote |
| † Electronic supplementary information (ESI) available: Materials preparation and characterization, X-ray crystal data and NMR spectra of 1, additional spectroscopic data of 1 and materials, determination of binding constants, complex formation by NMR, MIP optimization, determination of limit of detection (PDF). CCDC 2259775. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ma00476g |
| This journal is © The Royal Society of Chemistry 2024 |