
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adenosine detection in serum using a surface plasmon resonance biosensor with molecularly imprinted polymers incorporating modified thymidine monomers†
Molly I.
Wild
,
Mark V.
Sullivan
 ,
Chester
Blackburn
and
Nicholas W.
Turner
*
,
Chester
Blackburn
and
Nicholas W.
Turner
*
Department of Chemistry, University of Sheffield, Dainton Building, 13 Brook Hill, Sheffield, S3 7HF, UK. E-mail: n.w.turner@sheffield.ac.uk
First published on 22nd May 2024
Abstract
Stress is a response to stimuli which disrupt the homeostasis of a cell or organism. Adenosine is a purine nucleoside which functions as an immunomodulator and signalling molecule, with elevated levels present in tissues exposed to stress. Current methods used to determine adenosine levels within the body involve chromatography coupled with mass spectrometry, which while sensitive is time consuming and costly, highlighting the need for a quicker and more cost-effective detection method. Six nanoMIPs were produced using solid-phase synthesis targeting adenosine: a plain nano-MIP, an acrylamide-dT nano-MIP (bearing an acrylamide-modified thymidine molecule), and a carboxy-dT nanoMIP (bearing a carboxy-modified thymidine molecule) were made using two different methods. The first involved glutaraldehyde as the linker molecule connecting the template to the solid phase, whilst the second used EDC/NHS coupling chemistry. This allowed us to alter the orientation of the template to present either the base or sugar outwards. SPR was used to test the nanoMIP binding affinities and selectivity against adenosine, thymidine, deoxyguanosine and deoxycytidine. It was found the binding affinities of the nanoMIPs increased with use of the modified thymidine monomers, with equilibrium dissociation constants (KD) values of the plain nanoMIP, acrylamide-dT nanoMIP and carboxy-dT nanoMIP being 221 nM, 9.35 nM, and 2.11 nM respectively for the glutaraldehyde method. The following KD values were obtained for the EDC/NHS method: 212 nM, 5430 nM, and 111 nM for the plain nanoMIP, acrylamide-dT nanoMIP and carboxy-dT nano-MIP respectively. This illustrated the glutaraldehyde method produced more effective nanoMIPs than using EDC/NHS. This is surprising as it is counter-intuitive to the imagined Watson–Crick pairing. When challenged with the other nucleosides, excellent selectivity was observed. Fetal bovine serum was used to test the capability of the nanoMIPs in complex matrixes with consistent results produced throughout.
Introduction
Adenosine is a purine nucleoside which functions as an immunomodulator within disease states.1,2 It has the potential to be a stress marker if a successful method for rapid and accurate detection can be developed. The structure of adenosine consists of an adenine base molecule coupled to a ribose molecule via glycosidic bonding (Fig. 1).2 There are various ways in which extracellular adenosine is produced within the body, one of which being the dephosphorylation of ATP (adenosine triphosphate) to adenosine.3 Under stressful conditions (such as trauma or disease) that result in the rupturing of cell membranes high concentrations of ATP are released into the extracellular space and is quickly catabolised into extracellular adenosine.4 These elevated levels of extracellular adenosine allow it to act as an immunomodulator through signalling via the G-protein coupled receptors on cell membranes. The A2B receptor has the lowest affinity for adenosine and so is usually activated under pathological conditions; whereas the A1, A2A and A3 receptors all have a much higher affinity and so are activated under normal conditions.1,5 | ||
| Fig. 1 Structure of adenosine with the ribose molecule (pink) and the adenine base molecule (blue). (Top) Attached via glutaraldehyde linker, presenting the sugar foremost. (Bottom) Attached via EDC/NHS linker presenting the base foremost. Green sphere representative of the silica solid phase. Dashed lines representative of linker chain. | ||
Stress is a response to stressors or stimuli which disrupt the homeostasis of a cell or organism.6,7 The stressors can be physical or environmental, with the stress itself either being acute or chronic. Acute stress is defined as short term or low intensity whereas chronic is long term or high intensity.8,9 Acute stress can typically be adapted to, however, long term stress can be detrimental to an individual's health and contribute towards certain disease states, such as heart disease, cancer and asthama.9,10 Stress can be measured within the body by stress biomarkers (stress-markers). A biomarker is a compound that is measured and the values characterised, with different values acting as indicators towards either normal physiology, pathogenic physiology or a response to medical treatment.11 Examples of these stress markers include cortisol, alpha amylase, serotonin and catecholamines.12,13
The stress response is activated upon exposure to stimuli by nervous, immune and endocrine mechanisms which initiate both the sympathetic-adrenomedullary (SAM) and the hypothalamus-pituitary-adrenal (HPA) axes and the immune system.14 On a cellular level the stress response is initiated by the release of norepinephrine and epinephrine from the adrenal medulla. These hormones interact with specific G-protein receptors on a cell's surface to induce the cAMP (cyclic adenosine monophosphate) intracellular signalling pathway.14 Monitoring stress markers is an effective way to determine the intensity of stress and the effects of it within the body after activation of the stress response. Often it is carried out through measurement of these markers within body fluid samples (saliva, sweat, blood, urine).13,15
Adenosine has the potential to be a stress-marker due to its immunomodulator effects and high concentrations of the compound present in various disease states. These include hypoxia, heart disease and inflammation.16 An example of the effect of elevated adenosine levels can be found in tumour microenvironments: the increased levels of adenosine activates a response pathway which suppresses the immune response to the tumour, allowing it to grow and spread.17,18 Normal functioning levels of extracellular adenosine are less than 1 μM, but elevated levels can reach up to 100 μM.19,20 The concentrations of adenosine fluctuate within and between individuals,21 and with roles in both normal and pathological processes it can make diagnostics more difficult which is one of the reasons it is not yet widely used as a stress marker.
Moreover, the half-life of adenosine is less than 10 seconds,22 which can make quantification of adenosine levels in real time difficult. One of the ways in which stress markers and adenosine are both currently measured is via HPLC-MS (high performance liquid chromatography-mass spectrometry) analysis. HPLC is considered to be the ‘gold standard’ of analysis due to its high accuracy and sensitivity23 however, these benefits come at a cost of being expensive and time consuming. A single run can take anywhere between 15–60 minutes to obtain a result which is not practical when real-time concentrations are required for point of care treatment and diagnosis,24 and portability is considered. In example, Marin et al. demonstrated a HPLC method that produced a LOD of 0.1 μM and a LOQ of 0.25 μM.25 However, more recently Löfgren et al. developed a method using UPLC (ultra performance liquid chromatography) with a LOQ of 2 nM,26 illustrating the improvement of the sensitivity of chromatography over the years. An alternative method to HPLC is the use of biosensors. They are less expensive and can also produce results more rapidly. A biosensor is a device which measures a reaction (biological or chemical) and produces a signal proportional to the concentration of the target analyte.27 An example being the glucose biosensor which is used to determine blood glucose concentrations in real time by utilising glucose enzymes to oxidise the glucose in the blood sample, producing an electrical signal.28 Biosensors also commonly use antibodies as the biological agent to bind to the target. Whilst very effective with high sensitivity and selectivity, they are unsuitable for long term storage due to their fragility with fluctuations in temperature and pH.29 An alternative to using biological molecules in a biosensor is to replace them with nanoMIPs (nanoparticle-sized molecularly imprinted polymers). These synthetic counterparts to antibodies have greater resistance to changes in their environment, making them more suitable for long term storage but with similar sensitivities and selectivity.30
A molecularly imprinted polymer is a synthetic polymer that contains binding sites within its polymeric matrix that are complimentary to a specific target molecule in shape, size and functional groups.31 These binding sites are formed through a series of steps: first a pre-polymerisation complex is formed between a template molecule and functional monomers through non-covalent interactions,32,33 the monomers are then polymerised around the template, and the template molecule is then removed leaving the complimentary binding cavities.31 This paper uses nanoMIPs (nanoparticle sized molecularly imprinted polymers) synthesised through a solid phase approach. These nanoMIPs have a greater range of options for targets and monomers to consider, and is still a simple procedure compared to other MIP synthesis methods.33 Moreover, the addition of modified thymidine monomers provides a novel approach to increasing the specificity towards the target molecule. After researching the success of incorporating acrylamide-modified thymidine into DNA strands,34,35 we elected to explore the potential of using a polymerizable thymidine (modified with the addition of a carboxyl or acrylamide group onto the C5 position) to allow for it to be incorporated into the polymer matrix during the formation of the nanoMIP. This would hopefully add improved recognition based on Watson–Crick pairing.
Surface plasmon resonance is a spectroscopic method which examines real time interactions of an analyte and ligand together,36 providing information on the binding affinities and selectivity of the nanoMIPs against target and non-target molecules.37 SPR has been shown to be very beneficial in the analysis of antibody–antigen interactions, and it has also been adapted for the effective analysis of MIPs. The work carried out in this paper has focused on the development of an optical biosensor using nanoMIPs and SPR for the detection of adenosine in complex matrices.
Experimental
Materials
Acrylic acid (AA), 3-aminopropyltrimethyloxy-silane (APTMS), ammonium persulfate (APS), 1-ethyl-3-(3-dimethylamino-propyl) carbodiimide (EDC), glutaraldehyde (GA), glycine, N-(3-aminopropyl) methacrylamide hydrochloride (NAPA), N,N′-methylenebisacrylamide (BIS), N-hydroxy succinimide (NHS), N-isopropylacrylamide (NIPAm), N-tert-butylacrylamide (TBAm), and tetramethylethyldiamide (TEMED), were all obtained from Sigma-Aldrich (Poole, Dorset, UK).Acetone, acetonitrile (dry), Celite®, chloroform, dimethylformamide (DMF), dimethyl sulfoxide (DMSO), ethanol, ethanolamine (EA), ethylenediaminetetraacetic acid (EDTA), methanol, methyl acrylate (MA), palladium acetate, potassium chloride, potassium dihydrogen phosphate, sodium chloride, sodium dihydrogen phosphate, sodium hydroxide, toluene (anhydrous), tributylamine and Tween 20 were all obtained from Fisher Scientific UK (Loughborough, Leicestershire, UK).
Adenosine, deoxycytidine hydrochloride (deoxycytidine), deoxyguanosine, thymidine and 5-iodo-2′-deoxyuridine were obtained from LGC LINK Technologies Ltd (Bellshill, Scotland, UK).
Glass beads (75 μm diameter) were obtained from Microbeads AG, (Brugg, Switzerland).
Carboxymethyl Dextran Hydrogel Surface Sensor chips were obtained from Reichert Technologies Life Sciences, Buffalo, New York, USA.
Phosphate buffered saline (PBS) was made using a PBS tablet (Fisher Scientific UK) and 0.01% Tween20 in 200 mL distilled water at 10 mM and pH 7.4 for use in the nanoMIP synthesis. PBS running buffer for the SPR was made fresh daily in the laboratory with 0.01% Tween20, made up to 10 mM and pH 7.4.
Fetal bovine serum was obtained from Gibco, (Fisher Scientific, UK) and was used as is without dilution.
All chemicals and solvents used in this work were used as received without further filtration.
Methods
This was then irradiated in a microwave (Discover 2.0, CEM, Oxford, UK) using a dynamic control setting whereby a maximum power of 300 W is used to ramp the temperature to 100 °C, and minimal power (approximately 2–5 W) is then used to hold the reaction at 100 °C for 10 minutes. The solution was then allowed to cool (under air stream within the reactor). The resultant solution was then filtered through approximately 5 g of Celite® in a glass sintered funnel. Cold chloroform (approx. 50–80 mL) was added to the solid in a round bottomed flask and stored in the freezer overnight. The solution was filtered through a Buchner funnel with filter paper. Upon filtering, the solid was a pale red colour so to improve the purity it was redissolved in warm chloroform (20 mL at 30 °C) then filtered again through a Buchner funnel with filter paper and left to dry in a desiccator overnight. This yielded a pale pink solid.
The sample was studied using NMR (Jeol ECZ 600 MHz) with deuterated dimethyl sulfoxide (DMSO) as the solvent. 1H NMR (600 MHz, DMSO-D6) δ 11.50 (s, 1H), 8.63 (t, J = 3.4 Hz, 2H), 8.28 (s, 1H), 7.13 (d, 0H), 6.92 (d, J = 1.3 Hz, 1H), 6.25–6.15 (m, 1H), 6.12–6.04 (m, 2H), 5.59–5.53 (m, 1H), 5.21 (d, J = 4.3 Hz, 1H), 5.10 (t, J = 2.1 Hz, 1H), 4.52 (t, J = 5.8 Hz, 2H), 4.25–4.19 (m, 1H), 3.78–3.73 (m, 1H), 3.65–3.52 (m, 2H), 2.16–2.07 (m, 2H) (Fig. S1†).
This was then irradiated in a microwave (Discover 2.0, CEM, Oxford, UK) using a dynamic control setting whereby a maximum power of 300 W is used to ramp the temperature to 100 °C, and minimal power (approximately 2–5 W) is then used to hold the reaction at 100 °C for 10 minutes. The solution was then allowed to cool (under air stream within the reactor), then refrigerated overnight. The solution was then filtered through approximately 5 g of Celite® in a glass sintered funnel. Cold chloroform (approx. 50–80 mL) was added to the solid in a round bottomed flask and stored in the freezer overnight. The solution was then filtered through a Buchner funnel with filter paper and dried in a desiccator, yielding very pale pink powder.
The sample was studied using NMR (Jeol ECZ 600 MHz) with deuterated dimethyl sulfoxide (DMSO) as the solvent. 1H NMR (600 MHz, DMSO-D6) δ 11.60 (s, 1H), 8.38 (d, J = 2.1 Hz, 1H), 7.34–7.30 (m, 1H), 6.81 (ddd, J = 15.6, 4.1, 1.8 Hz, 1H), 6.09 (t, J = 1.9 Hz, 1H), 5.21 (dd, J = 4.5, 1.4 Hz, 1H), 5.13 (t, J = 2.6 Hz, 1H), 4.24–4.18 (m, 1H), 3.76 (q, J = 3.6 Hz, 1H), 3.65–3.63 (m, 3H), 3.62–3.52 (m, 1H), 2.19–2.07 (m, 2H) (Fig. S2†).
Polymer synthesis
In a 50 mL glass beaker 30 g of beads (75 μm diameter) were boiled in 24 mL of 4 M NaOH for 15 minutes to activate them. The beads were washed with approximately 8 × 100 mL (per 30 g of beads) distilled water until they reached a pH of approximately 7. They were rinsed with 2 × 100 mL acetone and dried at 60 °C for 2 hours. Once dried, the beads were put into a solution of 3% v/v APTMS in 12 mL anhydrous toluene under nitrogen and then incubated at 60 °C for 24 hours. After this, they were washed with approximately 8 × 100 mL acetone, then 2 × 100 mL methanol and oven-dried at 150 °C for 30 minutes. Two following steps were then completed depending on the intended orientation of the template.
| Method | NanoMIP (mg) | NIPAm (mg) | BIS (mg) | AA (μL) | NAPA (mg) | TBAm (mg) (in 250 μL ethanol) | Acrylamide-dT (mg) (in 250 μL DMF) | Carboxy-dT (mg) (in 250 μL DMF) | APS (mg)/TEMED (μL) |
|---|---|---|---|---|---|---|---|---|---|
| GA | Plain | 20 | 1 | 2.2 | 7 | 10 | N/A | N/A | 15/12.5 |
| Acrylamide-dT | 20 | 1 | 2.2 | 7 | 7 | 12.5 | N/A | 15/12.5 | |
| Carboxy-dT | 20 | 1 | 2.2 | 7 | 7 | N/A | 8.5 | 15/12.5 | |
| EDC/NHS | Plain | 6.6 | 0.3 | 0.7 | 2.3 | 3.3 | N/A | N/A | 5/4.1 |
| Acrylamide-dT | 6.6 | 0.3 | 0.7 | 2.3 | 2.3 | 4 | N/A | 5/4.1 | |
| Carboxy-dT | 6.6 | 0.3 | 0.7 | 2.3 | 2.3 | N/A | 2.8 | 5/4.1 |
The nanoMIP solutions were stored at 4 °C. The above methods were used for all nanoMIPs produced.
Using SPR, the nanoMIPs were each first immobilised onto a carboxymethyl dextran functionalised gold chip by running PBS over the chip for 10 minutes at 10 μL min−1. A 1 mL EDC/NHS (40 mg & 10 mg respectively) solution in PBS was then passed over the chip for 6 minutes at 10 μL min−1 to activate the carboxy groups on the chip. To activate the –NH functional groups on the nanoMIP, 0.01 M sodium acetate was added to the resuspended nanoMIP, and this was passed over the left channel (working channel) for 1 minute. After which, the reaction with EDC/NHS was stopped by passing over 1 M ethanolamine for 8 minutes, “capping” any unreacted carboxy functional groups on the chip's surface. PBS was finally passed over the chip also at 10 μL min−1 as before. Injections were taken when the baseline was stable.
The rebinding method used was taken from Sullivan et al.39 to measure the kinetics of the rebinding by measuring the binding affinity and selectivity for the nanoMIPs against the target molecule adenosine and three other nucleosides. It comprised of a 2-minute injection window (association), followed by a 5-minute wash of PBS (dissociation) and a 1-minute regeneration using regeneration buffer of 0.01 M Glycine-HCl at pH 2 (to remove target), finished by a 1-minute wash of PBS for each run. PBS with Tween20 was used for all analysis, and an analyte concentration range of 8, 16, 32, 64, 128 nM with a blank concentration of 0 initially. Each analysis was repeated at least three times, with the calibration curves produced from an average of three runs.
For analysis in a complex matrix, fetal bovine serum (FBS) was spiked with the above concentration range of the analyte (8–128 nM). The samples were then ran as above with an initial blank concentration of 0.
Reichert TraceDrawer software was used to fit the SPR sensorgrams with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Langmuir binding model. Equilibrium dissociation constants (KD) for each concentration were calculated using the equation:
:1 Langmuir binding model. Equilibrium dissociation constants (KD) for each concentration were calculated using the equation:
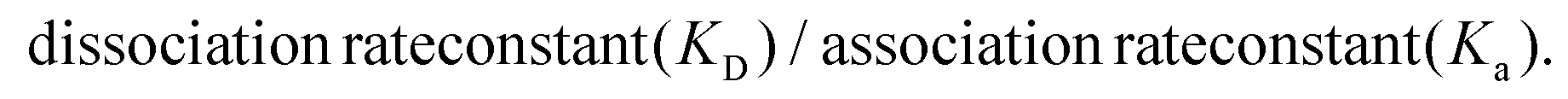
Results and discussion
Firstly, two different polymerisable thymidine monomers were synthesised via a Heck reaction before incorporation into the nanoMIPs. Three different nanoMIPs were produced for two different orientations for each of the nanoMIPs produced, providing six MIPs in total. A plain nanoMIP, acrylamide-dT nanoMIP and carboxy-dT nanoMIP were all synthesised first using the GA method, and then using the EDC/NHS method to give a total of six nanoMIPs. The first method involved the use of glutaraldehyde as the cross-linker, which resulted in the nanoMIPs with the thymidine monomers to have the ribose sugar exposed. The second method used EDC/NHS coupling chemistry for the cross-linking which resulted in the nanoMIPs with the thymidine monomers having the thymidine base exposed.Both methods produced: a plain nanoMIP without any thymidine monomers (the control nanoMIP), an acrylamide-dT nanoMIP containing an acrylamide-modified thymidine monomer, and a carboxy-dT nanoMIP containing a carboxy-modified thymidine monomer. The target for these nanoMIPs was adenosine, with selectivity being tested against deoxyguanosine, deoxycytidine and thymidine due to their similar weights and structures. The methodology for the monomer synthesis was adapted from Allabush et al.,34 whilst the methodology used within the nanoMIP syntheses was adapted from our previous work.38
The monomer synthesis produced yields of 64 mg (6%) and 569 mg (65%) of the acrylamide-dT and carboxy-dT respectively. 1H NMR analysis was conducted to confirm the monomer synthesis had been successful before incorporation into the nanoMIPs (Fig. S1 and S2† for the acrylamide-dT and carboxy-dT respectively).
For the GA nanoMIP synthesis approximately 150 mL of solution was produced for each nanoMIP, with the concentrations being 40 ± 18 μg mL−1, 128 ± 2 μg mL−1, and 245 ± 2 μg mL−1 for the plain nanoMIP, acrylamide-dT nanoMIP and carboxy-dT nanoMIP respectively.
During the EDC/NHS synthesis of the nanoMIPs the reactions were scaled down threefold to reduce waste. Approximately 50 mL of solution was obtained for each nanoMIP with the concentrations for each being 110 μg mL−1, 95 μg mL−1 and 96 μg mL−1 for the plain nanoMIP, acrylamide-dT nanoMIP and carboxy-dT nanoMIP respectively.
DLS was used to approximate the size of the nanoMIPs produced. Those made using the GA method produced nanoMIPs which averaged 100 nm in diameter. The peaks produced were narrow and singular, illustrating particle size homogeneity (Fig. S3(A–C)†). This is consistent for all three nanoMIPs.
The EDC/NHS method produced nanoMIPs which averaged 130 nm in diameter. The peaks produced for the plain nanoMIP were narrow and singular (Fig. S4(A)†), illustrating particle size homogeneity. There was some variation and aggregation occurring when the measurements for the Acrylamide-dT (Fig. S4(B)†) and Carboxy-dT (Fig. S4(C)†) nanoMIPs were taken, however, the peak illustrating particle size was clear enough for analysis. Three runs were obtained for each nanoMIP for an average n = 3.
Immobilisation of the nanoMIPs onto the gold chip occurred through coupling via the well understood Steglich-type EDC/NHS chemistry40 exploiting the presence of –COOH on the SPR chip. Ethanolamine was then used to ‘cap’ any unreacted carboxyl groups and wash away any of the nanoMIPs that had not bound to the surface. It was expected that there would be some unbound nanoMIPs because they were added in excess to ensure maximum coverage.
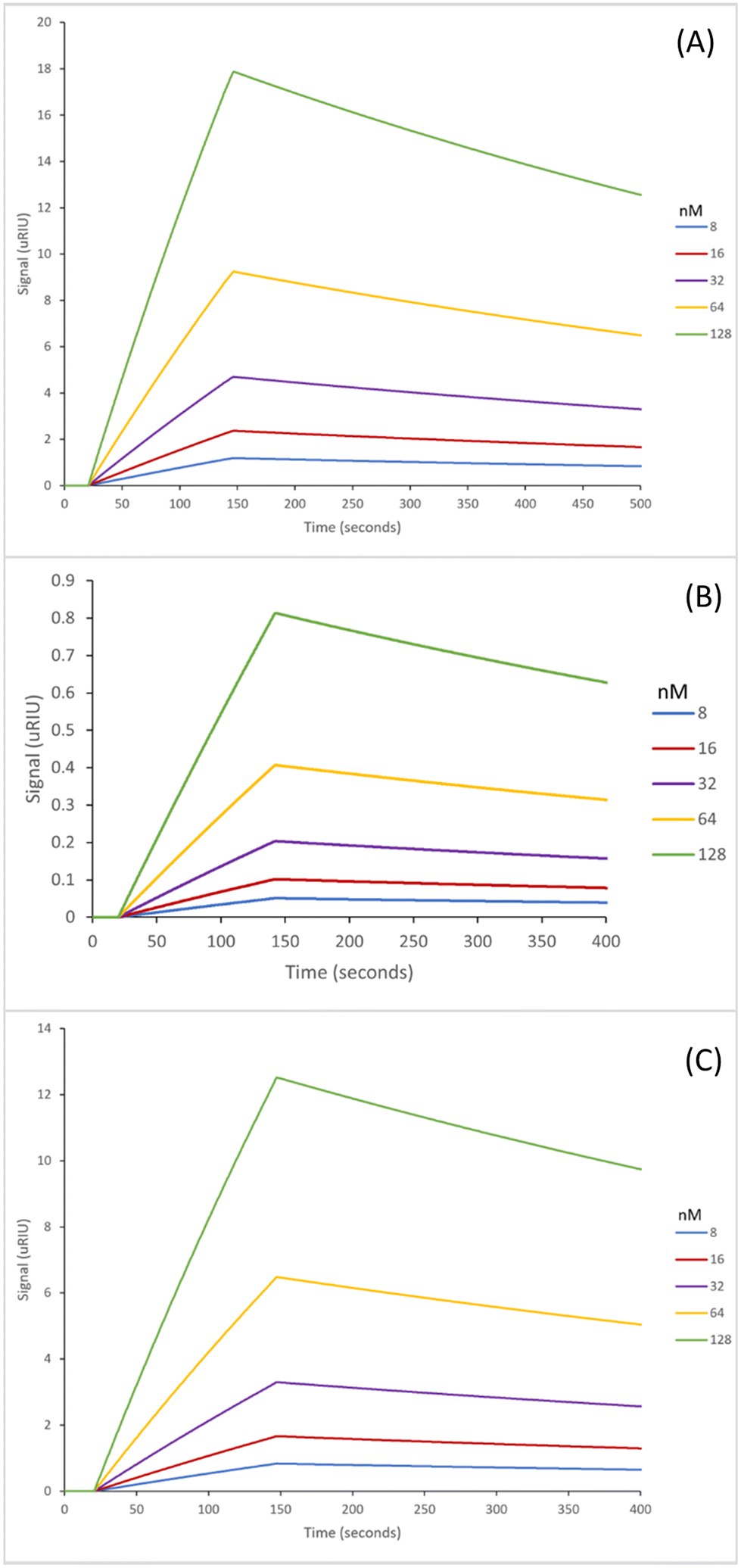
The SPR sensorgrams shown in Fig. 2 illustrate the interaction of the three GA nanoMIPs with five different concentrations of the target molecule adenosine. Each graph shows the concentration range of 8–128 nM. SPR sensorgrams illustrating the selectivity of the nanoMIPs against deoxycytidine, deoxyguanosine and deoxythymidine can be found in Fig. S5.† A 1:1 Langmuir binding model was used to calculate the equilibrium dissociation constant (KD) for each nanoMIP interaction with the target. Table 2 illustrates the binding affinities and selectivity for the plain nanoMIP, acrylamide-dT nanoMIP and carboxy-dT nanoMIP synthesized using GA against adenosine and the other nucleosides.
 | ||
| Fig. 2 SPR curves illustrating the binding affinities of the nanoMIPs synthesized using the glutaraldehyde method to adenosine; plain nanoMIP (A), acrylamide-dT nanoMIP (B) and carboxy-dT nanoMIP (C) in PBS. | ||
| Method | NanoMIP | Concentration (μg mL−1) | Diameter (nm) |
|---|---|---|---|
| GA | Plain | 40 ± 18 | 98.6 ± 4.4 |
| Acrylamide-dT | 128 ± 2 | 101.1 ± 7.9 | |
| Carboxy-dT | 245 ± 2 | 99.5 ± 2.2 | |
| EDC/NHS | Plain | 110 | 122.9 ± 1.6 |
| Acrylamide-dT | 95 | 134.2 ± 12.2 | |
| Carboxy-dT | 96 | 136.8 ± 13.4 |
The KD values for the GA nanoMIPs targeting adenosine were 221 (±200) nM, 9.35 (±1.8) nM, and 2.11 (±1.1) nM for the plain nanoMIP, acrylamide-dT nanoMIP and carboxy-dT nanoMIP respectively. The acrylamide-dT nanoMIP performed 30× better than the plain nanoMIP, whilst the carboxy-dT performed 130× better than the plain nanoMIP (Fig. 2). There was a 4× increase in performance between the acrylamide-dT nanoMIP and the carboxy-dT nanoMIP, which is supported by the work from Sullivan et al. who developed a hybrid aptaMIP using an acrylamide and carboxy-dT also.39 They achieved an increase in performance of approximately 1.5× when using the carboxy-dT compared to the acrylamide-dT, which is consistent with the results found in this paper. The carboxy-dT nanoMIPs have better performance due to the reduced flexibility thanks to the shorter carboxy chains compared to those of the acrylamide. The difference is lesser in the Sullivan et al. paper due to the use of the aptamers which already reduced flexibility with the acrylamide-dT.
The KD values produced for this work are similar to other recent work on nanosensors developed for the detection of adenosine. Kurt et al. have produced an optical-based thin-film nanosensor for the detection of adenosine, with a binding affinity of 57.8 nM (KD calculated by Scatchard),41 which is greater than the value achieved for the plain nanoMIP (221 nM (Table 3)) however, it is not better than the values achieved for the modified deoxythymidine nanoMIPs at 9.35 nM and 2.11 nM for the acrylamide-dT and carboxy-dT nanoMIPs respectively (Table 3).
| K D (nM) | |||
|---|---|---|---|
| NanoMIP | Acrylamide-dT nanoMIP | Carboxy-dT nanoMIP | |
| Adenosine | 221 (±200) | 9.35 (±1.8) | 2.11 (±1.1) |
| Deoxycytidine | 4170 (±940) | 2010 (±900) | 3460 (±75) |
| Deoxyguanosine | 27800 (±4100) |
8860 (±980) | 16800 (±6900) |
| Thymidine | 2120 (±640) | 19400 (±8900) |
3360 (±2000) |
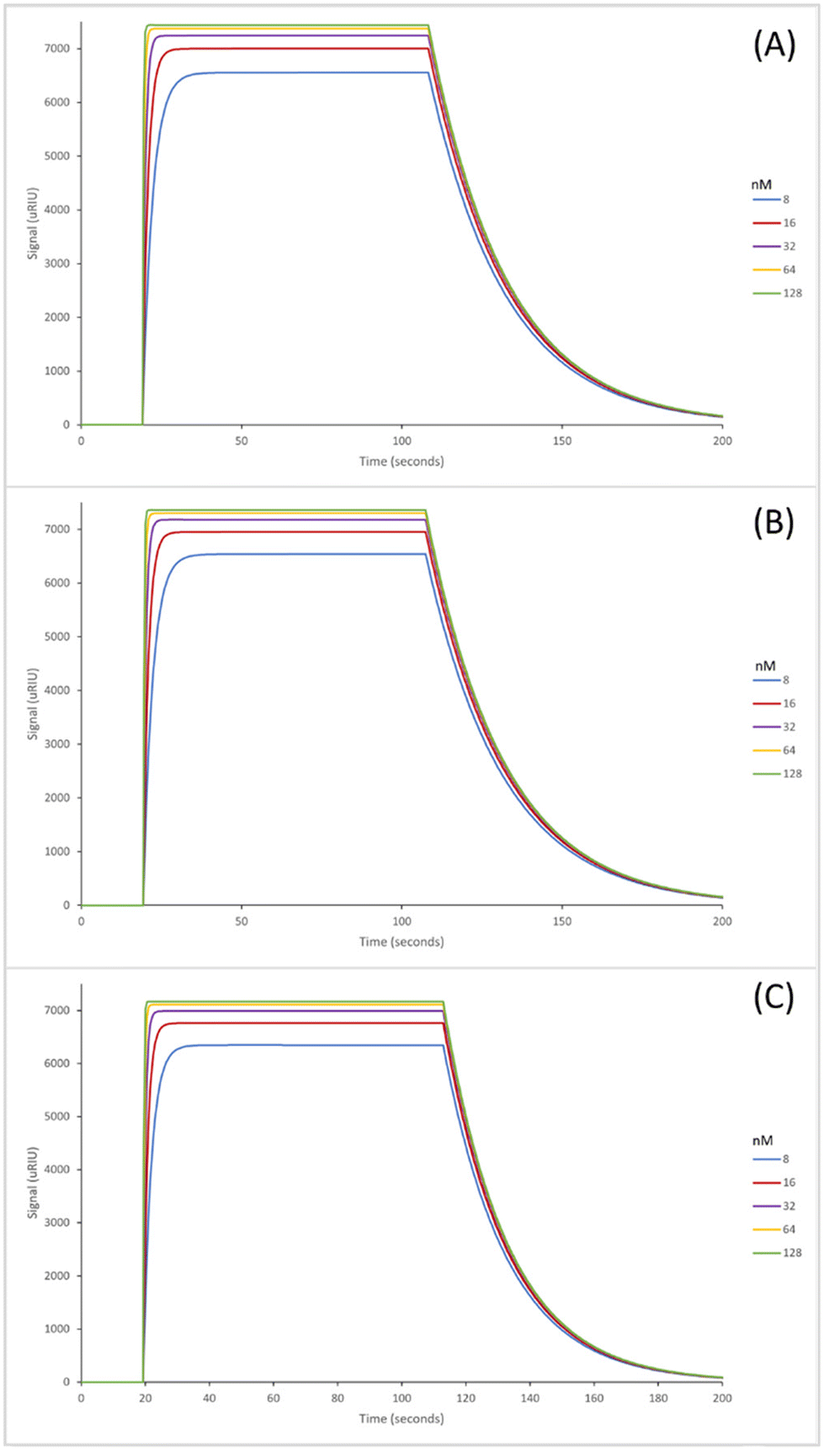
The SPR sensorgrams displayed in Fig. 3 illustrate the interaction of the three EDC/NHS nanoMIPs with five different concentrations of the target molecule adenosine. Each graph shows the concentration range of 8–128 nM. SPR sensorgrams illustrating the selectivity of the nanoMIPs against deoxycytidine, deoxyguanosine and deoxythymidine can be found in Fig. S6.† Again, a 1:1 Langmuir binding model was used to calculate the equilibrium dissociation constant (KD) for each nanoMIP interaction with the target. Table 3 highlights the binding affinities and selectivity for the plain nanoMIP, acrylamide-dT nanoMIP and carboxy-dT nanoMIP synthesized using EDC/NHS against adenosine and the other nucleosides. The KD values for the EDC/NHS nanoMIPs targeting adenosine were 212 (±170) nM, 96.4 (±2.7) nM, and 111 (±17) nM for the plain nanoMIP, acrylamide-dT nanoMIP and carboxy-dT nanoMIP respectively. The carboxy-dT nanoMIP performed approximately 2× better than the plain nanoMIP, whilst the acrylamide-dT nanoMIP performed slightly better than the carboxy-dT nanoMIP. When comparing the results for the GA nanoMIPs (Table 3) to the EDC/NHS nanoMIPs (Table 4) overall, the GA nanoMIPs performed more effectively in PBS.
 | ||
| Fig. 3 SPR curves illustrating the binding affinities of the nanoMIPs synthesized using the EDC/NHS method to adenosine; plain nanoMIP (A), acrylamide-dT nanoMIP (B) and carboxy-dT nanoMIP (C) in PBS. | ||
| K D (nM) | |||
|---|---|---|---|
| NanoMIP | Acrylamide-dT nanoMIP | Carboxy-dT nanoMIP | |
| Adenosine | 212 (±170) | 96.4 (±2.7) | 111 (±17) |
| Deoxycytidine | 2750 (±55) | 2630 (±440) | 780 (±21) |
| Deoxyguanosine | 475 (±7.7) | 7800 (±1100) | 3660 (±1300) |
| Thymidine | 3420 (±780) | 320 (±77) | 1440 (±70) |
The fact that the plain nanoMIP controls for both methods are very similar –the GA method had a KD of 221 nM (Table 3) compared to the EDC/NHS method which had a KD of 212 nM (Table 4) – illustrates a strong methodology for the polymer synthesis and can confirm the synthesis between the two methods is consistent.
It was expected that the EDC/NHS method which left the thymidine base exposed would result in improved binding affinities for adenosine (Table 4) compared to the GA method (Table 3) which left the ribose sugars exposed. This is due to the expectation of Watson–Crick base pairing between the adenosine and thymidine bases to occur. However, this may have been prevented by steric hindrance which then reduced the effectiveness of the nanoMIPs. This is supported by the similar pattern of results for the acrylamide and carboxy-dT nanoMIPs across both methods – as both were more effective when synthesised via the GA method. A possible reason for this is the occurrence of interactions between the adenosine sugar hydroxyl groups and thymidine monomers which strengthened the binding affinities.
Overall, the most successful nanoMIP was the GA carboxy-dT nanoMIP; not only was it most effective in binding to adenosine, but it also had excellent selectivity against the other nucleosides as illustrated in Table S1,† followed by the acrylamide-dT. The selectivity factors for the EDC/NHS methodology (Table S2†) also support the statement that the GA method is superior as they are lower compared to the GA nanoMIPs (by at least an order of magnitude as observed in both dT nanoMIPs).
The next step in the experiment was to then test the binding affinities of the six nanoMIPs in fetal bovine serum (FBS) to determine the effectiveness of the nanoMIPs in a complex biological matrix as such the capability of our sensor. The adenosine concentrations were spiked in undiluted FBS for analysis. The SPR sensorgrams for which are exhibited in Fig. 4 (GA method) and Fig. 5 (EDC/NHS method).
 | ||
| Fig. 4 Average SPR curves illustrating the binding affinities of the nanoMIPs synthesized using the GA method to adenosine; plain nanoMIP (A), acrylamide-dT nanoMIP (B) and carboxy-dT nanoMIP (C) in FBS. | ||
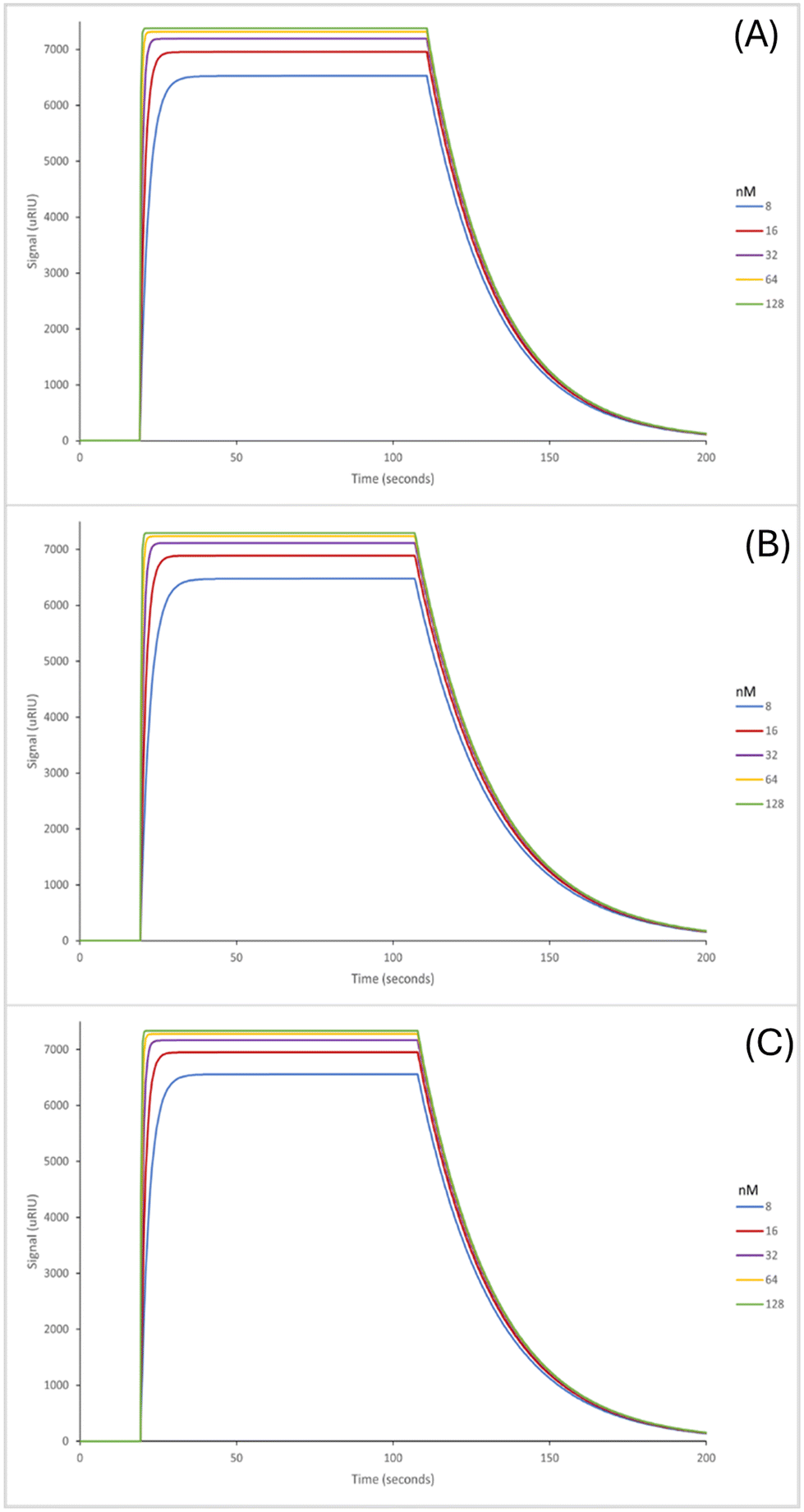 | ||
| Fig. 5 Average SPR curves illustrating the binding affinities of the nanoMIPs synthesized using the EDC/NHS chemistry. Method to adenosine; plain nanoMIP (A), acrylamide-dT nanoMIP (B) and carboxy-dT nanoMIP (C) in FBS. | ||
The GA method carboxy-dT nanoMIP has the best binding affinity out of the six, at 1.15 nM (Table 5), allowing for low nanomolar detection while the rest can reach sub micromolar detection levels. During stress levels of adenosine can range from <1 μM up to potentially 100 μM so the nanoMIPs would still be capable of detecting the target within serum in a stressed environment. Interestingly, similar results were obtained for both the GA and EDC/NHS methodologies. As illustrated in Fig. 4 and 5, the SPR curves for all six nanoMIPs are comparable, this is supported by the KD values obtained in Table 5. It can be seen that there is comparable affinity and performance between the PBS and FBS results. This illustrated the capability of the nanoMIPs performing in both buffer and a biological matrix. However, there is a difference in signal intensity between the SPR curves in PBS (Fig. 2 and 3) compared to the curves in FBS (Fig. 4 and 5).
| Sugar exposed orientation (using GA) | Base exposed orientation (using EDC/NHS) | |||||
|---|---|---|---|---|---|---|
| NanoMIP | Acrylamide-dT nanoMIP | Carboxy-dT nanoMIP | NanoMIP | Acrylamide-dT nanoMIP | Carboxy-dT nanoMIP | |
| Adenosine in FBS (KD (nM)) | 413 (±5.3) | 856 (±51) | 1.15 (±0.021) | 109 (±8.5) | 775 (±44) | 105 (±3.0) |
| Theoretical LOD of adenosine in FBS (nM) | 1.26 | 0.65 | 2.97 | 0.80 | 0.15 | 1.62 |
Those in FBS have a higher signal due to the matrix effect occurring from the FBS. This was expected due to the higher density and colour difference of the FBS compared to the PBS – as SPR relies on monitoring changes in refractive index there is more interference. This is a common occurrence with SPR analysis, with it also seen in our previous work utilising surine and FBS.38,42 Any changes were compared to a blank sample (FBS with zero adenosine present), therefore the changes in signal generated can be attributed to adenosine binding rather than matrix effect. The concentration calibration curves for the nanoMIPs’ performance in FBS (Fig. S6 and S7†) were able to be plotted using the SPR sensorgrams obtained in Fig. 4 and 5. The calibration curves were then used to calculate and estimated theoretical limit of detection (LOD) for each of the nanoMIPs (Table 5). The results obtained for the FBS experiment indicates the occurrence of matrix effect, as the other molecules within the complex matrix are causing absorbance in the dextran layer of the SPR chip and that SPR is a refractive index technique that will be affected by the nature of the serum. This is observed by the difference signal scale (y-axes in Fig. 2vs.Fig. 4 and 5). This raises the baseline causing saturation of the signal as observed in the sensorgrams in Fig. 4 and 5. Given that normal functioning levels of extracellular adenosine are less than 1 μM, but elevated levels can reach up to 100 μM,19,20 our sensor is currently operating in the expected concentration range. The advancement factor (AF) for the nanoMIPs was calculated by dividing the LOD for the UPLC and HPLC methods by the calculated theoretical LOD for each nanoMIP as shown in Table 6. Therefore, the AF is a measure of our work benchmarked against existing LC methods. As illustrated, the nanoMIPs produced in this work have a much greater sensitivity than the HPLC method by Marin et al.,25 with the lowest AF being 84 for the carboxy-dT GA nanoMIP and the highest AF reaching 1666 for the acrylamide-dT EDC/NHS nanoMIP. When compared to the UPLC method by Löfgren et al.,26 all have an AF of at least 1, aside from the carboxy-dT GA nanoMIP which has a comparable LOD of 2.97 nM compared to 2 nM; indicating a potential for it to still be competitive to the UPLC method.
Conclusions
This paper has focused on the development of nanoMIPs with incorporated modified thymidine monomers for the detection and recognition of adenosine. The methodology used was solid phase synthesis using either glutaraldehyde or EDC/NHS coupling chemistry as the cross-linker. Six nanoMIPs were produced, a plain nanoMIP, acrylamide-dT nanoMIP and a carboxy-dT nanoMIP all made using both methodologies. NanoMIPs produced using the GA method achieved high binding affinity to adenosine and excellent selectivity against the other nucleosides tested. It was determined that the addition of the modified thymidine monomers into the nanoMIP structure produced more selective rebinding to adenosine and were more effective compared to the plain nanoMIPs. Of interest the expected Watson–Crick pairing of the polymerisable thymidine, and the adenosine did not provide the greatest affinity materials.To confirm the nanoMIPs were successful in recognising adenosine in a biological matrix as well as a buffer solution fetal bovine serum was spiked with adenosine, the nanoMIPs exhibited a high affinity for the target. This identifies a potential for the nanoMIPs to be used in the detection of adenosine in biological samples, providing a solid foundation for further work to be built upon. We have produced effective nanoMIPs which are able to detect adenosine within its normal and pathological function concentration range as illustrated by the theoretical LODs calculated in Table 5. The synthesis of the nano-MIPs is straight forward and low cost which is beneficial when considering future work in further developing the biosensor. Nonetheless, there have been some weaknesses identified which must be brought to attention. Although the method is simple the nanoMIPs have not yet been synthesized on a large scale, so we have been unable to identify issues with reproducibility or batch variation on such a scale. Saturation effects have been observed in the PBS and FBS which would require new calibration curves to be made up each time, which adds additional time onto the experimental and data analysis aspects. Moreover, to aid with reducing the saturation effects samples may require dilution before analysis. Further exploration is needed on the detection of the target within complex matrixes to improve detection levels, alongside exploring the detection of adenosine within a mixture of other nucleosides. We are working on these issues, primarily by taking our MIPs and moving towards electrochemical detection and will present our work in due course.
Author contributions
MIW: investigation, formal analysis, writing – original draft; CB: formal analysis, methodology; MVS: formal analysis, methodology; NWT: conceptualization, funding acquisition, supervision, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the University of Sheffield Faculty of Science PhD scholarship program. We also wish to acknowledge EPSRC grant number EP/V056085/2.References
- H. Liu and Y. Xia, J. Appl. Physiol., 2015, 119, 1173–1182 CrossRef CAS PubMed.
- J. C. Shryock and L. Belardinelli, Am. J. Cardiol., 1997, 79, 2–10 CrossRef CAS PubMed.
- T. V. Dunwiddie and S. A. Masino, Annu. Rev. Neurosci., 2001, 24, 31–55 CrossRef CAS PubMed.
- B. B. Fredholm, Cell Death Differ., 2007, 14, 1315–1323 CrossRef CAS PubMed.
- B. N. Cronstein, J. Appl. Physiol., 1994, 76, 5–13 CrossRef CAS PubMed.
- K. Kagias, C. Nehammer and R. Pocock, Front. Genet., 2012, 3, 222 Search PubMed.
- I. Kyrou and C. Tsigos, Curr. Opin. Pharmacol., 2009, 9, 787–793 CrossRef CAS PubMed.
- Y. M. Ulrich-Lai and J. P. Herman, Nat. Rev. Neurosci., 2009, 10, 397–409 CrossRef CAS PubMed.
- H. Yaribeygi, Y. Panahi, H. Sahraei, T. P. Johnston and A. Sahebkar, EXCLI J., 2017, 16, 1057–1072 Search PubMed.
- B. S. McEwen, Ann. N. Y. Acad. Sci., 1998, 840, 33–44 CrossRef CAS PubMed.
- Biomarkers Definitions Working Group, Clin. Pharmacol. Ther., 2001, 69, 89–95 CrossRef PubMed.
- K. Dhama, S. K. Latheef, M. Dadar, H. A. Samad, A. Munjal, R. Khandia, K. Karthik, R. Tiwari, M. I. Yatoo, P. Bhatt, S. Chakraborty, K. P. Singh, H. M. N. Iqbal, W. Chaicumpa and S. K. Joshi, Front. Mol. Biosci., 2019, 6, 91 CrossRef CAS PubMed.
- A. J. Steckl and P. Ray, ACS Sens., 2018, 3, 2025–2044 CrossRef CAS PubMed.
- B. Chu, K. Marwaha, T. Sanvictores and D. Ayers, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2023 Search PubMed.
- R. S. Vasan, Circulation, 2006, 113, 2335–2362 CrossRef PubMed.
- R. Guieu, J.-C. Deharo, B. Maille, L. Crotti, E. Torresani, M. Brignole and G. Parati, J. Clin. Med., 2020, 9, 1366 CrossRef CAS PubMed.
- K. Sek, C. Mølck, G. D. Stewart, L. Kats, P. K. Darcy and P. A. Beavis, Int. J. Mol. Sci., 2018, 19, 3837 CrossRef PubMed.
- S. Gessi, S. Merighi, V. Sacchetto, C. Simioni and P. A. Borea, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1400–1412 CrossRef CAS PubMed.
- Y. Liu, J. Chen, X. Li, X. Zhou, Y. Hu, S. Chu, Y. Peng and N. Chen, CNS Neurosci. Ther., 2019, 25, 899–910 CrossRef PubMed.
- G. Haskó and B. N. Cronstein, Trends Immunol., 2004, 25, 33–39 CrossRef PubMed.
- T. Simard, R. G. Jung, P. Di Santo, F. D. Ramirez, A. Labinaz, C. Gaudet, P. Motazedian, S. Parlow, J. Joseph, R. Moreland, J. Marbach, P. Boland, S. Promislow, J. J. Russo, A.-Y. Chong, D. So, M. Froeschl, M. Le May and B. Hibbert, Clin. Transl. Sci., 2021, 14, 354–361 CrossRef CAS PubMed.
- D. Faulds, P. Chrisp and M. M.-T. Buckley, Drugs, 1991, 41, 596–624 CrossRef CAS PubMed.
- M. W. Dong, LCGC North Am., 2013, 31, 472–479 CAS.
- G. Haink and A. Deussen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 784, 189–193 CrossRef CAS PubMed.
- R. M. Marin, K. G. Franchini and S. A. Rocco, J. Sep. Sci., 2007, 30, 2473–2479 CrossRef CAS PubMed.
- L. Löfgren, S. Pehrsson, G. Hägglund, H. Tjellström and S. Nylander, PLoS One, 2018, 13, e0205707 CrossRef PubMed.
- N. Bhalla, P. Jolly, N. Formisano and P. Estrela, Essays Biochem., 2016, 60, 1–8 CrossRef PubMed.
- J. P. Chambers, B. P. Arulanandam, L. L. Matta, A. Weis and J. J. Valdes, Curr. Issues Mol. Biol., 2008, 10, 1–12 CAS.
- J. E. Dover, G. M. Hwang, E. H. Mullen, B. C. Prorok and S.-J. Suh, J. Microbiol. Methods, 2009, 78, 10–19 CrossRef CAS PubMed.
- A. N. Hasanah, N. Safitri, A. Zulfa, N. Neli and D. Rahayu, Molecules, 2021, 26, 5612 CrossRef CAS PubMed.
- G. Ertürk and B. Mattiasson, Sensors, 2017, 17, 288 CrossRef PubMed.
- M. V. Sullivan, A. Henderson, R. A. Hand and N. W. Turner, Anal. Bioanal. Chem., 2022, 414, 3687–3696 CrossRef CAS PubMed.
- C. Alvarez-Lorenzo and A. Concheiro, Handbook of Molecularly Imprinted Polymers, Smithers Rapra, Shrewsbury, 2013 Search PubMed.
- F. Allabush, P. M. Mendes and J. H. R. Tucker, RSC Adv., 2019, 9, 31511–31516 RSC.
- A. Poma, H. Brahmbhatt, H. M. Pendergraff, J. K. Watts and N. W. Turner, Adv. Mater., 2015, 27, 750–758 CrossRef CAS PubMed.
- D. Capelli, V. Scognamiglio and R. Montanari, TrAC, Trends Anal. Chem., 2023, 163, 117079 CrossRef CAS.
- N. J. de Mol and M. J. E. Fischer, in Surface Plasmon Resonance: Methods and Protocols, ed. N. J. Mol and M. J. E. Fischer, Humana Press, Totowa, NJ, 2010, pp. 1–14 Search PubMed.
- C. Blackburn, M. V. Sullivan, M. I. Wild, A. J. O′ Connor and N. W. Turner, Anal. Chim. Acta, 2024, 1285, 342004 CrossRef CAS PubMed.
- M. V. Sullivan, O. Clay, M. P. Moazami, J. K. Watts and N. W. Turner, Macromol. Biosci., 2021, 21, 2100002 CrossRef CAS PubMed.
- A. Jordan, K. D. Whymark, J. Sydenham and H. F. Sneddon, Green Chem., 2021, 23, 6405–6413 RSC.
- Z. T. Kurt, D. Çimen, A. Denizli and N. Bereli, ACS Omega, 2023, 8, 18839–18850 CrossRef CAS PubMed.
- A. Henderson, M. V. Sullivan, R. A. Hand and N. W. Turner, J. Mater. Chem. B, 2022, 10, 6792–6799 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1HNMR spectra for acrylamide-dT and carboxy-dT, DLS particle size intensity graphs, SPR sensorgrams for selectivity studies and selectivity factors for all nanoMIPs. See DOI: https://doi.org/10.1039/d4lp00059e |
| This journal is © The Royal Society of Chemistry 2024 |