
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePISA printing from CTA functionalized polymer scaffolds†
A. Priestera,
J. Yenga,
Y. Zhangb,
R. Wang b and
A. J. Convertine*a
b and
A. J. Convertine*a
aDepartment of Materials Science and Engineering, 1400 North Bishop Avenue, Rolla, MO 65409, USA. E-mail: convertinea@mst.edu
bDepartment of Chemistry, 400 West 11th Street, Rolla, MO 65409, USA
First published on 20th January 2024
Abstract
This study investigates DLP 3D printing using RAFT polymerization-induced self-assembly (RAFT PISA) printing, focusing on the impact of CTA (chain transfer agent) groups per scaffold on PISA printing times and mechanical properties of printed objects. We synthesized a solvophilic polymer scaffold from DMA (N,N-dimethylacrylamide) and HEAm (2-hydroxyethyl acrylamide) in a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 molar ratio, suitable for CTA functionalization and solubility in various solvents. Employing an aqueous, oxygen-tolerant PET RAFT process, we achieved a target degree of polymerization (DP) of 10000, introducing varying amounts of CTA groups by esterification (chain transfer agent functionalized scaffold or CFS), with the highest graft density resulting in a scaffold with an average of 74 CTA groups. This process was additionally repeated with a DP 500 scaffold by grafting with varying densities of CTAs. Photocurable DLP (digital light projection) resins based on the chain extension of core-forming DAAm (diacetone acrylamide) from the CFS macro-CTA were then prepared and used to print 3D objects. This study revealed that the increased number of CTA groups per scaffold decreased the normal exposure time for PISA printing when compared to our previous work which employed a difunctional macro-CTA. In mechanical property assessments, conducted across different DPs and CTA graft densities, we observed trends in modulus, strain-to-break, and toughness. The increasing modulus trend ceased beyond a DP of 500, suggesting a balance between the amounts of DAAm and CTA-functional scaffolds. Additionally, parts with higher graft densities demonstrated increased stiffness due to a higher density of physical crosslinks. The study also explored the dissolution behavior of these parts in DMF, with parts showing varying degrees of swelling and dissolution depending on their DP and CTA graft density. These findings indicate a significant advancement in the 3D PISA printing technique, offering new insights into the optimization of printing times and mechanical properties, potentially revolutionizing applications in areas like biomedical implants and tissue engineering scaffolds.
:10 molar ratio, suitable for CTA functionalization and solubility in various solvents. Employing an aqueous, oxygen-tolerant PET RAFT process, we achieved a target degree of polymerization (DP) of 10000, introducing varying amounts of CTA groups by esterification (chain transfer agent functionalized scaffold or CFS), with the highest graft density resulting in a scaffold with an average of 74 CTA groups. This process was additionally repeated with a DP 500 scaffold by grafting with varying densities of CTAs. Photocurable DLP (digital light projection) resins based on the chain extension of core-forming DAAm (diacetone acrylamide) from the CFS macro-CTA were then prepared and used to print 3D objects. This study revealed that the increased number of CTA groups per scaffold decreased the normal exposure time for PISA printing when compared to our previous work which employed a difunctional macro-CTA. In mechanical property assessments, conducted across different DPs and CTA graft densities, we observed trends in modulus, strain-to-break, and toughness. The increasing modulus trend ceased beyond a DP of 500, suggesting a balance between the amounts of DAAm and CTA-functional scaffolds. Additionally, parts with higher graft densities demonstrated increased stiffness due to a higher density of physical crosslinks. The study also explored the dissolution behavior of these parts in DMF, with parts showing varying degrees of swelling and dissolution depending on their DP and CTA graft density. These findings indicate a significant advancement in the 3D PISA printing technique, offering new insights into the optimization of printing times and mechanical properties, potentially revolutionizing applications in areas like biomedical implants and tissue engineering scaffolds.
Introduction
Additive manufacturing, particularly digital light projection (DLP), has revolutionized the production of complex parts across various industries, because of its high resolution and ability to print the entire XY plane simultaneously for each layer.1–3 Traditional DLP resins typically employ multifunctional alkenes and photoinitiators to form chemically crosslinked structures upon light exposure. Due to the crosslinked nature of these traditional DLP resins, they are not suitable for producing soluble materials. This inherent characteristic excludes them from various biomedical engineering applications that require biodegradable structures. Additionally, their use is limited in processes like 3D printing of microvasculature, where fugitive inks are employed to create negative structures, because these resins cannot be formulated into such inks.4–7Recent developments in photosensitive resins aim to introduce more diverse material properties, including flexibility, toughness, self-healing, and conductivity.8–10 A pivotal advancement in 3D printing has been the adoption of reversible deactivation radical polymerizations (RDRPs), particularly reversible addition–fragmentation chain transfer (RAFT) polymerization. This method significantly enhances control over polymer architecture.11–14 RAFT-based photopolymerization in DLP printing, exemplified by Bagheri et al., Boyer et al., Zhu et al. and others, showcases rapid polymerization and versatile build capabilities.15–23
Building upon these advancements, we recently reported the development of RAFT polymerization-induced self-assembly (RAFT PISA) printing. RAFT PISA printing enables the creation of 3D parts without requiring multifunctional crosslinking monomers. This method produces parts stabilized by physical, rather than chemical, crosslinks. A significant advantage of this approach is that the 3D printed parts can be dissolved in an appropriate solvent without needing degradable chemical linkages. Similar to conventional PISA, PISA printing utilizes a RAFT macro-chain transfer agent (macro-CTA) that undergoes chain extension with a monomer, leading to phase separation during block copolymer formation (Fig. 1A). However, our findings indicate that attaching at least two CTA residues to the macro-CTA is necessary to 3D print mechanically stable parts using the PISA printing process (Fig. 1B). This aligns with other studies that have shown enhancements in mechanical properties with the use of difunctional RAFT agents, which facilitate gelation or physical crosslinking in PISA nanostructures.24–28
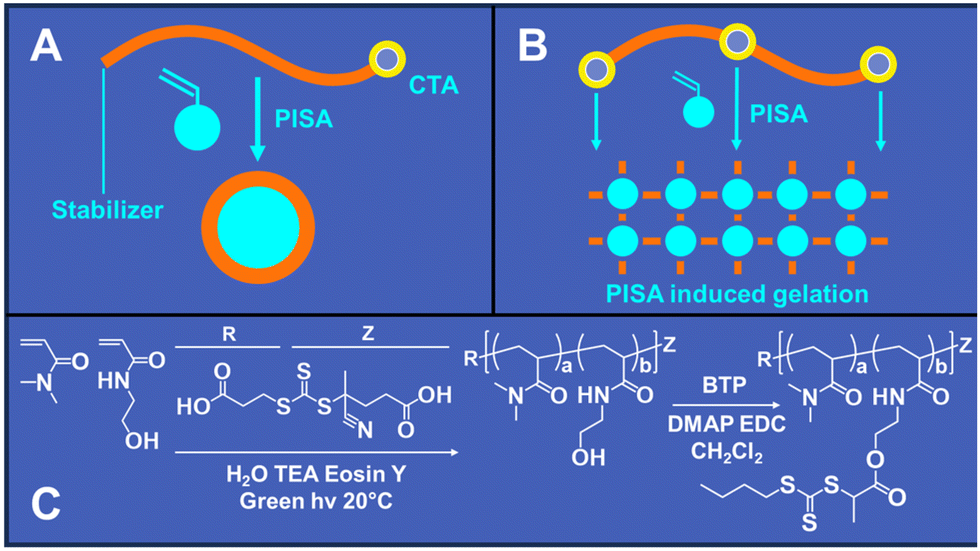 | ||
| Fig. 1 RAFT PISA involves extending a solvophilic RAFT macro-CTA with a monomer that turns solvophobic upon polymerization, leading to the formation of self-assembled nanoparticles. (A) Macro-CTAs with a single RAFT agent at the chain terminus typically yield soluble nanostructures or lightly gelled assemblies. (B) Increasing the number of CTA groups per scaffold induces gelation via corona entanglement and intraparticle bridging. (C) Synthetic scheme for preparation of CTA functionalized scaffolds (CFSs). | ||
Our initial work on RAFT PISA printing utilized a difunctional poly(ethylene glycol) (PEG) macro-CTA, chain extended with diacetone acrylamide (DAAm) in water, and 3D printed via DLP using lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) as a photo-initiator and phenol red as a photoabsorber.29 In this study, we observed the dissolution behavior of DAAm PISA frogs in N,N-dimethylformamide (DMF), noting that those without N′N-methylene bisacrylamide (MBAc) crosslinker dissolved completely, indicating the presence of physical crosslinks. Building on this foundation, our second PISA printing study aimed to develop formulations capable of dissolving in physiological fluids with controlled kinetics.30 Here, PISA printing was conducted from a highly branched multi-CTA scaffold in isopropanol, using acrylamide as the phase-separating monomer and varying ratios of the hydrophobic monomer N-(butoxymethyl)acrylamide (BAM) to modify the aqueous dissolution rates.
The current study further investigates PISA printing, aiming to understand how different macro-CTA architectures influence the mechanical properties, cure time, and dissolution characteristics of PISA printed parts. We aim to examine the interactions between various polymer scaffolds with increased CTA functionality and their impact on the final properties of the 3D printed materials. Furthermore, this research examines the potential of PISA printing in creating intricate, high-resolution structures for biomedical applications.
Materials and methods
Materials
N′N-Dimethylacrylamide (DMA, 99%) was obtained from Sigma-Aldrich. N-(2-Hydroxyethyl)acrylamide (HEAm, >98%), trimethylamine (TEA, >99%), tetrabromofluorescein (eosin Y, EY, >95%), diacetone acrylamide (DAAm, >98%), 2-butanone (MEK, >99%), and ethyl phenyl(2,4,6-trimethylbenzoyl)phosphinate (>95%) were obtained from TCI America. 2-(Butylthiocarbonothioylthio)propanoic acid (BTP, 95%), 4-((((2-carboxyethyl)thio)carbonothioyl)thio)-4-cyanopentanoic acid (CCC, 95%), and 4-cyano-4-(((dodecylthio)carbonothioyl)thio)pentanoic acid (DCT, 97%) were obtained from Boron Molecular. Lithium bromide (LiBr, >99%), dichloromethane (CH2Cl2, >99.5%), N′N-dimethylformamide (DMF, >99%), 4-dimethylaminopyridine (DMAP, >99%), and 3-(3-dimethylaminopropyl)-1-ethyl-carbodiimide hydrochloride (EDC, >98%) were obtained from Chem Impex Int.Synthesis of poly(DMA-co-HEAm) macro-CTA via PET RAFT
RAFT copolymerizations of DMA and HEAm (molar ratio 9:1, respectively) were conducted under aqueous conditions at an initial comonomer concentration of 2.5 M (∼20 wt%), targeting a degree of polymerization ([M]0/[CTA]0) 100, 250, 500, 1000, 5000 or 10000. A type II photoinitiating system based on eosin Y and TEA were employed in these polymerizations using a [TEA]0/[EY]0 ratio of 100. [CTA]0/[EY]0 ratios of 20, 15, 10, 5, 5 and 5 were used for DP targets 100, 250, 500, 1000, 5000 and 10000, respectively. A 0.1 molar equivalent DMF spike with respect to total comonomer concentration was used an internal NMR reference to determine polymer conversion. 1H NMR samples in D2O were measured before and after polymerization, and conversion was determined by integrating the reduction in the area of the vinyl peaks of DMA/HEAm (∼5.5–6.5 ppm) with respect to the DMF amide peak (∼8.02 ppm). Polymerizations were carried out in 500 mL beakers using a GLW 30 W LED green light (550 nm) at a distance of 6′′ from the light source (∼1450 lux as determined by the MT-912 light meter). Once placed under the light, polymerization was carried out for 48 hours followed by determining polymer conversion via NMR. Polymer solutions were then diluted with DI water (1:1 v/v) and placed in SpectrPor regenerated cellulose dialysis membranes (12–14 kDa) cutoff. Polymers were dialyzed first against methanol (3× over 3 days) to remove unreacted monomer and excess eosin Y followed by dialysis against water (3× over 2 days) to remove the methanol. Polymers were then frozen and lyophilized to obtain the final product. A representative procedure for macro-CTA synthesis for DP 10000 is as follows: to a 500 mL beaker, DMA (45.0 g, 454 mmol), HEAm (5.81 g, 50.4 mmol), 310 μL of a 50 mg mL−1 CCC stock in methanol (15.5 mg, 50.4 μmol), 262 μL of a 25 mg mL−1 eosin Y stock in methanol (6.55 mg, 10.1 μmol), 140 μL TEA (0.102 g, 1.01 mmol) and DMF (3.68 g, 50.4 mmol) were added and mixed homogeneously. The solution was then diluted with 202 mL of distilled water to a final comonomer concentration of ∼2.5 M (∼20 wt%).
Post-polymerization esterification of poly(DMA-co-HEAm) macro CTAs (CFS synthesis)
Following isolation, BTP RAFT agents were functionalized by grafting the polymers with varying densities of CTAs, in this case BTP. Two scaffolds were chosen for grafting and subsequent studies: DP 500 and DP 10000. These two DPs result in drastically different viscosities in water (the basis for the 3D PISA printing resin) which was hypothesized to affect ease of printing as well as print resolution. BTP CTA was grafted onto the hydroxyl residues of the HEAm units of the poly(DMA-co-HEAm) macro-CTA via Steiglich esterification. For the DP 10000, the grafting densities were 1, 2.5, 5 and 10% of HEAm units. For the DP 500 scaffold, graft densities were 4, 10, 20, 50 and 100% of HEAm units. For these esterifications, DMAP was employed as the catalyst, and EDC was employed as the carbodiimide coupling reagent. Since HEAm only makes up 10% of the DP 10000 CFS polymer, this means that no more than 100 BTP RAFT agents will be esterified to any one polymer chain. To begin, lyophilized CFS was dissolved into dichloromethane at a concentration of 10 wt% for ∼2 hours. Following complete dissolution, BTP and DMAP, respectively, were added into the solution. Lastly, EDC was slowly added to the solution in small increments. As the EDC reacted, the solution color changed in appearance from a bright yellow to a dark orange-red and back again. The solution was covered and left to react for 18 hours on a magnetic stir plate. For purification, the grafted solution were dialyzed against methanol (3× for 3 days) to remove any unreacted BTP in addition to DMAP and EDC. This was followed by dialysis against water, freezing and lyophilization to obtain the final large molecular weight, functionalized polymers (CFSs). A representative procedure for the 10% BTP grafted poly(DMA-co-HEAm) macro CTA is as follows: to a 100 mL beaker, 10 g of poly(DMA-co-HEAm) macro-CTA (1.14 g HEAm, 9.90 mmol R-OH) was dissolved in 68 mL of dichloromethane. The was followed by the addition of BTP (0.236 g, 0.991 mmol) and DMAP (0.181 g, 1.49 mmol). EDC (0.284 g, 1.49 mmol) was then slowly added into the solution and allowed to react overnight. An additional batch of poly(DMA-co-HEAm) macro CTA was grafted with 10% DCT instead of BTP so that experiments could be carried out with methacrylate monomers.
Synthesis of lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) photoinitiator
To a 250 mL beaker containing 100 mL of 2-butanone, ethyl phenyl(2,4,6-trimethylbenzoyl)phosphinate (7 g, 22.1 mmol) and lithium bromide (7.69 g, 88.4 mmol) were added and stirred into solution. Once dissolved, the mixture was heated in a 60 °C water bath for 24 hours. The precipitated product was vacuum-filtered, washed six times with 2-butanone, and vacuumed dry to obtain the pure white solid (5.53 g, ∼85% yield).Characterization of CFSs
CFSs were characterized by dynamic light scattering (DLS), gel permeation chromatography (GPC), 1H nuclear magnetic resonance (NMR) and UV-visible spectrophotometry (UV-Vis). 1 mg mL−1 samples of CFS in DI water were prepared, and 1 mL was pipetted into a 1.25 × 1.25 cm polystyrene cuvette. Polymer size was measured on the Malvern dynamic light scattering system. NMR of the purified DP 10000 poly(DMA-co-HEAm) and 10% grafted CFS were measured in CDCl3. 5 mg mL−1 samples of CFS in DMF were measured at a flow rate of 0.5 mL min−1 on the Agilent Infinity II series GPC. Number average molecular weight values (Mn) for the CFS was determined using a PMMA calibration curve. UV-Vis was used to determine moles of BTP per gram of polymer. This was done by preparing a 10 mg mL−1 in DI water and measuring the absorbance on the TECAN infinite M nano+ UV-Visible spectrophotometer and comparing to a BTP standard curve using Beer's law. The 10% DCT grafted polymer was quantified in the same manner, using a DCT standard curve instead. To account for the absorbance of any residual eosin Y within the CFS, the ungrafted and purified poly(DMA-co-HEAm) macro-CTA (concentration) was used as an absorbance blank that was subtracted out from the CFS sample absorbance. GPC traces, summarized molecular weight/molar mass dispersity values, DLS trace and NMR spectra can be found in ESI Fig. 1 and ESI Table I, ESI Fig. 2 and ESI Fig. 3,† respectively. UV-Vis spectra can be found in ESI Fig. 4.†
3D DLP printing of aqueous CFS PISA resins
For 3D DLP printing studies, the 10% BTP-grafted multifunctional CFS macro-CTA was utilized as the stabilizer block with DAAm serving at the solvophobic/PISA block in the aqueous-based PISA system. A target DP ([M]0/[CTA]0) of 500 was chosen to formulate the resin. The DP calculation was determined based on the CTA value of the CFS polymer as determined by UV-Vis. 0.5 wt% LAP photoinitiator and 0.005 wt% phenol red photoabsorber with respect solids were the other two components added to the system. Due to the high molecular weight of the CFS (∼1 MDa), curing trials were first conducted in 24 well plates to find a solids content that would result in a workable viscosity for DLP printing. From these trials, it was determined that 30 wt% would be chosen to formulate the final DLP resins. To make the resin, CFS was dissolved in distilled water and stirred into solution followed by the DAAm. The appropriate amounts of LAP and phenol red stocks in water were then added to the solution and mixed homogeneously. Resins were used to a print a treefrog model at three different normal exposure times: 5 s, 10 s, and 15 s per layer. Additionally, a “S&T” moniker was also printed to evaluate the resolution of these resins and whether they were subject to significant overcuring by comparing the DP 10000 to a 3D printed part using the DP 500 grafted CFS (10%, target DAAm DP of 500). Photoabsorber and photoinitiator concentrations were kept constant for the DP 500 CFS print. Lastly, a lattice cube was also printed with DP 500 CFS PISA system. All parts were printed on the Phrozen Sonic Mini 8K DLP printer (1.60 mW cm−2, 405 nm, resolution = 22 μm) using a bottom exposure time of 20 s and layer thickness of 50 μm. A representative procedure for CFS DAAm PISA resin is as follows: to a 100 mL beaker, 10% BTP grafted CFS (2.50 g, 0.183 mmol BTP) and DAAm (15.5 g, 91.6 mmol) were dissolved in 42.0 g of distilled water by hand-stirring. 1.79 g of a 50 mg g−1 LAP stock and 0.359 g of a 2.5 mg g−1 phenol red stock were then added to complete the resin. An additional PISA resin for a methacrylate-based system was formulated using HPMA and the 10% DCT grafted CFS to show that multiple monomer systems could be cured via this PISA mechanism.
Characterization of 3D DLP printed CFS PISA parts
Atomic force microscopy (AFM) and scanning electron microscopy (SEM) were used to evaluate the nanostructural and microstructural features of PISA-printed parts. For these studies, the 10% BTP-grafted CFS PISA resin with DAAm (target DP of 500) was utilized. For AFM, parts were prepared by pipetting a small amount of resin onto a metal substrate and curing under UV light for 2 minutes. For SEM, resins were cured under a 30 W UV light (405 nm) in a 12 well plate for 2 minutes. After removal, the cured parts were dialyzed against distilled water for one day to remove any residual monomer. They were then frozen and lyophilized. The resulting part was fractured and coated with Au on the Denton Sputter coater before being imaged on the PRISMA E SEM (25 kV).Dissolution studies for cured CFS PISA resins
Dissolution studies were performed for BTP and DCT grafted CFS resins to see how DP, graft density and monomer may affect the dissolution of parts following curing/printing. Due to the viscosity limitations associated with the 1% BTP grafted CFS resin formulations, all dissolution study resins were fixed at a solids content of 25 wt%. Five different DP targeted resins (100, 250, 500, 750, 1000) for each BTP graft density for both DP 10000 and DP 500 CFS grafted scaffolds were prepared in a manner similar to that described in the previous section. 1 g of reach resin was pipetted into a 24 well plate and exposed to UV light for 2 minutes on the Phrozen Sonic Mini 8K DLP printer. The cured parts were then placed in DMF, and dissolution (or lack thereof) was monitored over time. DMF should solubilize both the poly(DMA-co-HEAm) and DAAm blocks of the polymer. Similarly, a DP study of dissolution was also conducted for the 10% DCT grafted CFS with HPMA at a solids content of 25 wt%. In this case, DPs of 50, 100, 175, 250 and 500 were targeted, and the resin was cured for 5 minutes under UV light.
Additional dissolution studies were carried out on these resins with free RAFT agent. Free CCC RAFT agent was added at different concentrations to the 5% BTP grafted CFS DAAm resin to see if increasing free CTA may promote dissolution. CCC molar equivalents of 0.25, 0.5, 1, 2, 3.5, 5 and 10 with respect to grafted BTP concentration was added to the resins and cured in the same manner as previously described for dissolution studies. Curing times were 5 minutes in these studies. After curing, parts were placed in DMF to monitor dissolution.
Mechanical property studies for cured CFS PISA resins
Mechanical properties for the BTP-grafted CFS PISA resin with DAAm were measured and evaluated as a function of increasing target DP (degree of polymerization), increasing BTP graft density, and increasing solids concentration. In DP studies, the DP was varied (100, 250, 500, 750, 1000) while graft density (10%) and solids content (25 wt%) were kept fixed. In graft density studies, BTP grafts with respect to hydroxyls (1, 2.5, 5 and 10%) were varied while DP (1000) and solids content (25 wt%) were kept fixed. Lastly, for solids content studies, the solids were varied (12.5, 25 and 40 wt%) while the graft density (10%) and DP (500) were fixed. Additionally, mechanical properties for the 10% DCT grafted CFS PISA resin with HPMA were measured using a target DP of 250 and solids content of 25 wt%. To perform these studies, the various resins were pipetted into a silicone-coated ASTM D638 Type V dogbone mold (3 samples per set of conditions) and were cured for 2 minutes (DAAm) for 5 minutes (HPMA) under UV light (405 nm). Following removal, samples were tested on the Shimadzu EZ-LX tensile tester using a 5 kN load cell and a stroke of 10 mm min−1. Mechanical properties were not measured for the DP 500 scaffold.Results
The primary goals of this study were to investigate whether increasing the number of CTA groups per scaffold could reduce the time required for PISA printing and to determine if a greater number of CTAs per scaffold enhances the mechanical properties of the printed objects when compared to our previous PISA printing study that employed a difunctional PEG macro-CTA. To explore these objectives, we synthesized a solvophilic polymer scaffold based on a copolymer of DMA and HEAm in a 90:10 molar ratio (Fig. 1C). This specific ratio was chosen to provide hydroxyl residues appropriate for CTA functionalization and to ensure solubility across various aqueous and organic solvents. Our previous research indicated that this scaffold composition is effective in creating soluble polymer brushes with diverse target DPs.31,32
For the synthesis of the polymer scaffold, we use aqueous, oxygen-tolerant PET RAFT, targeting DPs of 500 and 10000. The copolymerization of DMA and HEAm was conducted under green light illumination, employing eosin Y and triethylamine ([CTA]0/[EY]0 = 1 (for 10000) or 10 (for 500), [TEA]0/[EY]0 = 100) as the photosensitizer and electron donor, respectively. To enable a comparative analysis of scaffolds with varying molecular weights, we optimized the oxygen-tolerant PET RAFT conditions for a range of target DPs (ESI Fig. 1†).
The DP 10000 polymerization reaction achieved complete conversion within 48 hours, resulting in a copolymer with an average molecular weight of 511000 Da and a molar mass dispersity of 1.68 (Table 1). The DP 500 CFS reached complete conversion within 8 hours with an average molecular weight of 46000 Da and a molar mass dispersity of 1.34. Post-synthesis of the polymer scaffold, CTA groups were incorporated through the esterification of BTP carboxylic acid groups, situated on the CTA R group, with 10% of the HEAm hydroxyl residues using EDC and DMAP (Fig. 1C). This process yielded a CTA functionalized scaffolds (CFS) with an average of 74 CTA groups per scaffold. Furthermore, we prepared a series of scaffolds with varied BTP graft densities, specifically at 1%, 2.5%, and 5%, to facilitate further investigations. The DP 500 target scaffold was also chosen to prepare a series of grafted scaffolds for further studies. DP 500 scaffolds were functionalized by grafting either 4, 10, 20, 50 or 100% of the hydroxyl residues on each linear chain with BTP. These results are also summarized in Table 1.
| CTS scaffold target DP | Conversion (%) | Mn, theo (Da) | Mn, GPC (Da) | Molar mass dispersity (Đ) | Targeted graft densities (%) | Experimental graft densities (%) |
|---|---|---|---|---|---|---|
| 500 | 99+ | 50000 |
46000 |
1.34 | 4% – BTP | ∼2% |
| 10% – BTP | ∼6% | |||||
| 20% – BTP | ∼10% | |||||
| 50% – BTP | ∼24% | |||||
| 100% – BTP | ∼44% | |||||
| 10000 |
95+ | 991000 |
511000 |
1.68 | 1% – BTP | ∼0.6% |
| 2.5% – BTP | ∼1.5% | |||||
| 5% – BTP | ∼3.2% | |||||
| 10% – BTP | ∼7.4% | |||||
| 10% – DCT | ∼6.5% |
PISA printing of CFS-based resins
To evaluate the impact of CTA graft density on the PISA printing process, diacetone acrylamide (DAAm) was polymerized in water in the presence of the CFSs. Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) and phenol red were used as the photoinitiator (0.5 wt% with respect to solids) and photoabsorber (0.006 wt%) with respect to solids), respectively, with a monomer-to-CTA ratio ([M]0/[CTA]0) of 500 and a total solids content of 30 wt%. In these experiments, the resins underwent normal exposure times of 5, 10, and 15 seconds. This approach contrasts with our earlier studies that required a longer exposure time of 45 seconds for polymer scaffolds with only two CTA residues. The current study showed that reducing the exposure time to 10 seconds was sufficient for successful 3D printing of detailed treefrog models without noticeable defects (Fig. 2A). The printed frogs displayed distinct features, such as individual digits and body ribbing. Notably, the frogs exhibited a yellow color due to the pH-sensitive phenol red photoabsorber, alongside a bluish-white hue near the legs and feet, attributed to light scattering from nanostructures formed during polymerization or UV curing, indicative of PISA activity. When put in the context of our previous study employing a linear difunctional PEG macro-CTA, the curing times were reduced from 45 s/layer in that study to just 10 s/layer in the present work. This significant improvement in reducing cure time was attributed to the higher density of anchor points or CTAs per chain (higher functionality), which may allow the material to gel at lower conversions when compared to the difunctional system. | ||
| Fig. 2 Photographs showing PISA printed parts from CFSs. (A) 10% BTP-grafted DP 10000 CFS DAAM PISA resin printed at a normal exposure time of 10 s per layer. (B) 10% BTP-grafted DP 500 CFS DAAm PISA resin printed lattice cube at a normal exposure time of 10 s per layer. (C) S&T moniker printed at a normal exposure times of 10 s per layer using 10% BTP-grafted DP 500 CFS DAAm PISA resin. | ||
Printing with a 15-second exposure time was successful, though it led to overcuring, especially noticeable on the frog's feet. Conversely, reducing the exposure time to 5 seconds resulted in part failure and defects, with the shorter exposure insufficiently curing the slices for the arms, causing the frog's front feet to detach. This printing displayed a more pronounced blue color near the arms and feet, indicating a lower monomer conversion of the solvophobic block during UV exposure. This likely stemmed from reduced polymerization, resulting in smaller nanostructures and increased light scattering. This observation aligns with Lowe et al.'s study on a poly(vinylpyridine)-based macro-CTA system combined with styrene under alcoholic RAFT PISA conditions,33 where higher conversion of the solvophobic styrene block led to complex particle morphologies and a color shift in the particle solution. The viscosity of this resin, particularly the DP 10000 CFS PISA-based resin, may also impact the resolution, degree of overcuring, and printing difficulty. Resolution could potentially be improved with increased photoabsorber concentration. However, this comes with the drawback of increased cure times.
To evaluate the potential detail of CFS resins, a standard 20-second exposure was used to print a lattice cube model and S&T logo with the less viscous DP 500 scaffold (Fig. 2B and C). Longer cure times were required as this was a slower curing resin at the same LAP and phenol red concentrations when compared to the DP 10000 scaffold. The resulting lattice cube model (2B) showed high detail but some overcuring, possibly due to resin getting trapped in the lattice holes and overcuring in subsequent layers. The 3D printed logo had clear lettering with minor overcuring, not significantly affecting print quality. This was contrasted with a logo printed with the more viscous DP 10000 CFS (ESI Fig. 5†), where more significant overcuring was noted. The letters were discernible under light, but overcuring obscured the gaps between them. Overall, PISA printing can effectively produce complex structures when using suitable scaffolds for resin preparation.
Dissolution studies of PISA-printed parts
The development of 3D printed materials with controllable dissolution rates is highly relevant for biomedical implants and tissue engineering scaffolds.34–40 Achieving this feature often involves integrating degradable linkages groups into the cured 3D network.41–44 In our initial PISA printing study we used a difunctional PEG macro CTA combined to 3D print DAAm. The resulting 3D printed parts were soluble in DMF, confirming the absence of chemical crosslinks. In a follow-up study, we PISA printing formulations that can be assembled in isopropanol to yield parts with controllable aqueous dissolution kinetics.In the current study, we anticipated a similar dissolution behavior with DMF as it effectively solubilizes both DAAm and CFS (grafted poly(DMA-co-HEAm)). However, contrary to expectations, complete dissolution was not observed in PISA printed parts polymerized in the presence of CFS. Instead, these parts underwent considerable swelling, reaching almost five times their original weight in DMF, but they did not fully dissolve. Over several weeks, the parts continued to expand in DMF, gradually weakening, swelling more, and eventually breaking down into gel-like fragments. This unexpected result prompted further exploration into the effects of target DP and graft density on dissolution. To this end, we conducted experiments on CFS with various DPs (100, 250, 500, 750, 1000) at four different graft densities (1, 2.5, 5, and 10%) for DP10000 CFS and at five different graft densities for DP500 CFS (4, 10, 20, 50, and 100%). For each condition, PISA printing resins were prepared and then subjected to UV light exposure (405 nm) for 2 minutes to initiate curing. Post-curing, the samples were immersed in DMF to observe their dissolution behavior.
In this study (Fig. 3A), parts grafted with a 1% BTP concentration successfully retained their shape at lower DPs (e.g. 100 and 250) although they exhibited a gel-like appearance. However, at higher DPs (e.g. 500, 750, 1000), these parts formed stable, well-defined shapes, despite their inherent fragility. For parts with 1% BTP grafting, dissolution in DMF occurred within hours, irrespective of their DP. Notably, parts with a higher DP of 1000 and 1% graft exhibited prolonged dissolution times, approximately 1–2 hours, indicating a degree of DP dependency in dissolution. In contrast, parts with lower target DPs dissolved more rapidly, typically within 15–30 minutes.
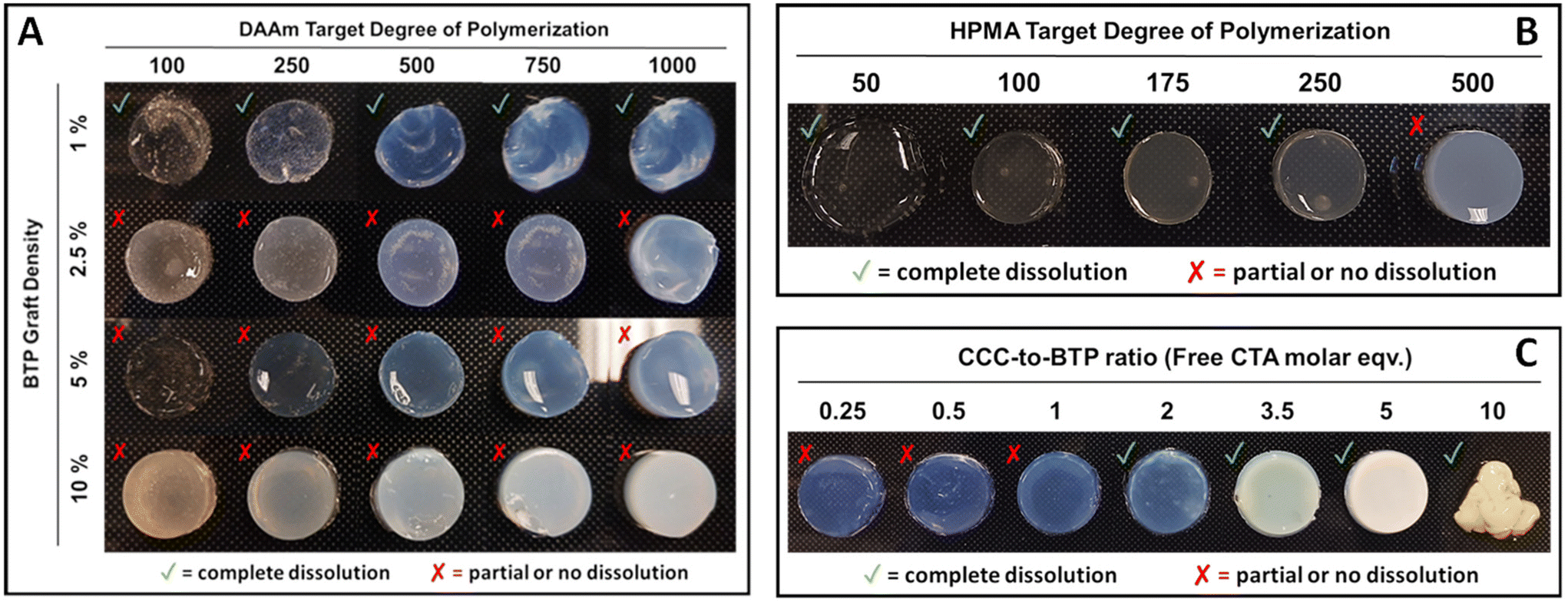 | ||
| Fig. 3 (A) Dissolution studies for CFS DAAm PISA resins at varying combinations of DP and CTA graft density of the CFS. All samples marked by a green checkmark indicate full dissolution of the parts following curing. Samples marked by a ‘x’ either did not dissolve at all or only partially dissolved after 24 hours in DMF. (B) Dissolution studies of 10% DCT-grafted CFS HPMA PISA resins. As shown, the target DPs of 250 or less for HPMA led to dissolution of the part while the DP 500 swelled but did not dissolve. (C) Dissolution studies of 5% BTP-grafted CFS DAAm PISA resins with free CCC. As shown, increasing free CCC concentration with respect to BTP promoted dissolution in DMF. | ||
In samples with a 2.5% graft density, the parts did not completely dissolve in DMF, but rather disintegrated into gel-like fragments over several hours. These fragments continued to break down over a week but did not achieve total dissolution. Parts with 5% and 10% graft densities did not dissolve or disintegrate but instead showed significant swelling during the same period. This pattern suggests that the target DP does not substantially influence the solubility of PISA printed parts across different CTA graft densities. The complete dissolution of all parts at 1% graft density, contrasted with the lack of dissolution at higher graft densities (2.5%, 5%, and 10%), implies the existence of a CTA graft threshold above which parts do not dissolve. The increase in the number of CTA groups per scaffold might lead to crosslinking via termination by coupling. Additionally, the brush architecture of the CTA-functionalized scaffolds used in these experiments likely affects the frequency of these chain coupling reactions. However, our preliminary work with hyperbranched scaffolds that have a high number of CTA groups/scaffold did yield soluble parts suggesting that the scaffold architecture does play an important role.
In order to determine if the solubility of the PISA printed part was affected by monomer class, hydroxypropyl methacrylate (HPMA) was polymerized from the 10% CFS. In these studies (Fig. 3B), a range of DPs were targeted including 50, 100, 175, 250, and 500. Because of the slower polymerization rates of HPMA relative to DAAm, the cure times per increased to 5 minutes per layer. Significantly, almost all parts from the HPMA system dissolved except for those with a DP of 500. It should be noted that these studies employed scaffolds grafted with 4-cyano-4-(((dodecylthio)carbonothioyl)thio)pentanoic acid (DCT) rather than BTP in order to provide a more appropriate R group for the methacrylate monomer. Additionally, the dissolution process in DMF was slow, taking one day for DP 50, a few days for DP 100, and up to a week for DPs 175 and 250, as shown in Fig. 3B.
Further experiments were conducted in order to probe the ability of free CTA to suppress crosslinking reactions leading to soluble 3D printed parts (Fig. 3C). In these studies, DAAm was polymerized in the presence of the 5% BTP-grafted CFS with the addition of free trithiocarbonate-based CTA (CCC). These experiments showed that PISA prints conducted with two or more equivalents of free CTA relative to CFS CTA groups were completely soluble in DMF. In contrast, PISA prints conduct at a free CTA to CFS CTA ratio of one and below resulted in substantial swelling (>5 times their original weight), but did not dissolve.
AFM and SEM imaging of PISA-printed parts
The microstructural and nanostructural features of the PISA printed parts were evaluated using a combination of SEM and AFM (Fig. 4). As shown in Fig. 4 (top), the SEM images show a highly porous and web-like nanostructure at the lowest magnification (top-left). At the highest magnification (top-right), there appears to be a bubble or popcorn-like structures on the surfaces of each web. The roughness on the surface also appears to consist of nanoporosity in between these surface structures. The structures themselves may be the result of self-assembled nanostructures that were produced via PISA in the 3D printing process. However, since the resolution was lacking at this high magnification, further investigation of the structure on the parts was studied via AFM. As shown in Fig. 4A–F, the morphology of the PISA printed nano-objects is predominately spherical with some irregular spheres. This result contrasts sharply with our previous PISA printing studies conducted from a difunctional PEG scaffold and a hyperbranched scaffold, in which a combination of well-defined spheres and worms were observed. It is possible that the high molecular weights of the CFSs disrupted the formation of highly uniform morphologies. | ||
| Fig. 4 (Top) Results for SEM imaging of cured, lyophilized part using 10% grafted CFS PISA resins with a DP 500 targeted DAAm block. As shown web-like microstructure was observed for the part with evidence of nanostructures composing the surfaces of each web. (Bottom) AFM imaging of 10% grafted CFS PISA resins with a DP 500 targeted DAAm. AFM images of DCT polymer: AFM topography image (A), corresponding peak force error image (B), and 2D roughness images (C)–(E). (F) 3D-image including the height information. Scale bar: 300 nm. | ||
Mechanical properties of cured PISA resins
The mechanical properties of the CFS PISA resins were examined as a function of target DP, graft density, and solids content for aqueous DAAm polymerizations conducted in the presence of the BTP-grafted scaffolds. The modulus was determined from the slope of the stress–strain curve within the linear elastic range. Strain-to-break was calculated by measuring the change in length relative to the initial gauge length. The toughness was estimated from the energy derived from the area under the stress–strain curve. The outcomes of these tests are graphically represented in Fig. 5. The mechanical characteristics were also assessed for the 10% DCT-grafted HPMA system, with the results summarized in ESI Table II.† For the DP experiments, the 10% scaffolds were used at a constant solids content of 25 wt% (Fig. 5A). The modulus was observed to increase until it reached a maximum value of 50.9 ± 3.30 kPa at DP 500, after which it began to decrease with rising DP. Strain-to-break diminished from approximately 1034 ± 117% at the lowest DP of 100 to merely 202 ± 21.2% at the highest DP of 1000. The combined influence of these two factors on the energy derived from the area under the stress–strain curve culminated in a peak toughness of 1853 ± 700 kJ m−3 at DP 250, followed by a sharp decline at higher DPs (<274 ± 23.2 kJ m−3). The large error bar for this measurement seems to be attributable to the larger error associated with the strain-to-break of these parts.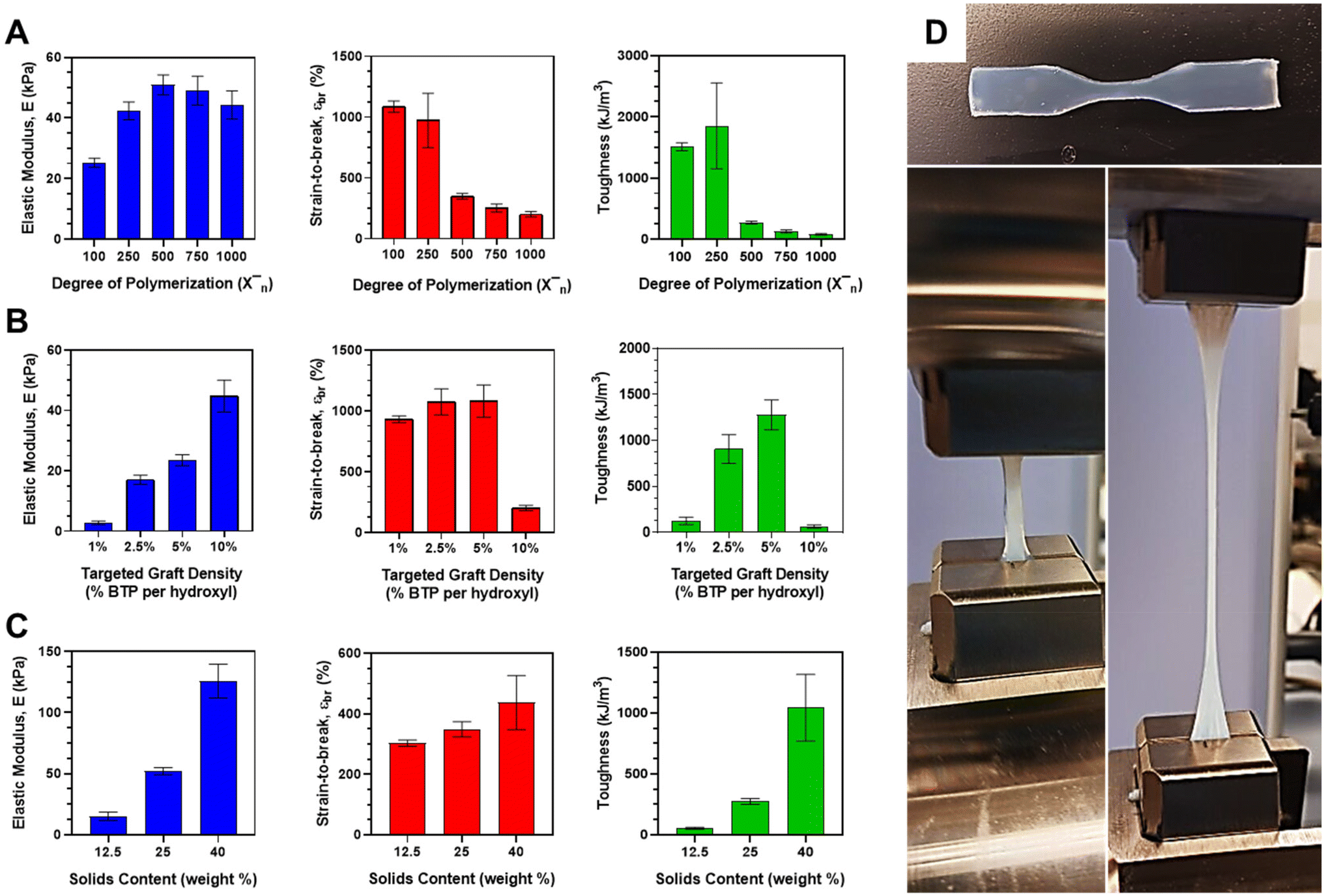 | ||
| Fig. 5 Mechanical properties for CFS DAAm PISA cured resins. (A) Modulus, strain-to-break and toughness properties as a function of DP using fixed solids content (25 wt%) and fixed BTP graft density (10%). (B) Modulus, strain-to-break, and toughness properties as a function of increasing BTP graft density at a fixed DP (1000) and fixed solids content (25 wt%). (C) Modulus, strain-to-break and toughness properties as function of solids content at a fixed DP 500 and fixed BTP graft density (10%). (D) DAAM CFS PISA resin cured dogbone shows high stretchability during mechanical property testing. Finally, the total solids content was varied for the 10% BTP graft density scaffolds using a DAAm target DP of 500. As shown, modulus increased with increasing solids content to a maximum of 126 ± 4.77 kPa at a solids content of 40 wt%. These same trends were observed with strain-to-break (maximum of 557 ± 164% at 40 wt%) and toughness (maximum of 1396 ± 596 kJ m−3 at 40 wt%). This seems to make intuitive sense. At just 12.5 wt% solids, parts were barely being held together and were made of almost 90 wt% water. In contrast, the 40 wt% part held its shape well and could be handled roughly without any issue. Since the part was only 60 wt% water, there was much more solids present per unit volume, resulting in tougher and stiffer parts. | ||
Given the high graft density of the 10% BTP-grafted CFS, the DAAm monomer composes most of the resin solids content (since target DP is based on moles of CTA). As DP increases, so does the proportion of monomer in the resin system. DAAm is a highly crystalline material that exhibits strong hydrogen bonding and a high Tg. Therefore, it was expected that increasing the DAAm should result in a higher modulus, which is what was observed up to DP 500. However, at higher target DPs, the modulus began to decrease. To explain this, we must look at the relative proportions of monomer and stabilizer in the system. For the 10% grafted system at high target DPs (>750), DAAm monomer constitutes >90% of the total solids with small amounts of the CFS stabilizing the large amounts of phase-separated domains. In other words, it may be behaving more like a bulk DAAm part since there is little stabilizer in the system. As its name suggest, the CFS or stabilizing segment is important for keeping the hydrophobic core-forming block and the overall part stable under aqueous conditions. Too little stabilizer and the hydrophobic chains will not be stabilized effectively. The higher target DP parts are thus mostly held together by self-assembled chains that are quite hydrophobic. In the absence of any chemical crosslinker, these high DP DAAm parts with little stabilizer should start to become weaker at high concentrations of core-forming block or DAAm monomer, which is what we observed with the decrease after DP 500. Resins targeting lower DPs with larger proportions of CFS yielded parts that were rubbery and compliant.
Similarly, this relationship between monomer concentration and stabilizer can be expanded to explain the trends of strain-to-break and toughness. The mass ratio of DAAm-to-stabilizer is 1.2 for DP 100 (1034%) at the experimental 10% graft density, which showed the highest strain-to-break. Compare that to a ratio of 3 for the DP 250 (973%) and ratio of 6 for DP 500 (349%), where the strain-to-break dropped off significantly. As the stabilizer in the system decreases, it reaches a point where the brittle DAAm blocks make up most of the system (>86% at DP 500). We would expect an increase in the brittle core-forming block to see a reduction in strain-to-break values, which is what was observed across all DPs. It appears that there is some critical concentration of DAAm with respect to stabilizer (>80%) in which the bulk properties begin to reflect those of poly(DAAm) in water without crosslinker, which would explain both the loss in strain-to-break and modulus values. Bulk poly(DAAm) is unstable in water and cannot hold its shape. The small addition of CFS allowed it to hold its shape via physical crosslinking formed by PISA printing, but the overall part was still exceptionally weak. Since toughness is contributed to by strain-to-break and modulus, it makes sense the energy under the curve would be significantly less in DPs 500–1000 given the low strain-to-break.
For graft density studies, the targeted DP for all cured resins was kept fixed at a value of 1000 with a fixed total solids content of 25 wt%. As shown in Fig. 2B, the modulus of the material increased nearly linearly from a minimum value of 2.80 ± 0.625 kPa to a maximum of 44.76 ± 5.27 kPa with increasing graft density. Strain-to-break remained nearly constant (or slightly increased) up to a maximum (1085 ± 133%) at a graft density of 5% before falling off considerably at a graft density of 10%. The contribution of both modulus and strain to energy and therefore toughness resulted in increasing toughness with graft density up to a peak toughness at 5% graft density (1280 ± 163 kJ m−3). Considering the sharp falloff in strain-to-break at the 10% graft density, it makes sense that the resulting toughness for this graft density was considerably lower than the graft densities at 2.5 and 5% and did not follow the increasing toughness with graft density trend that the others showed. In fact, this 10% graft density targeting a DP of 1000 was in good agreement with the previous results obtained in the DP studies of mechanical properties. Increasing modulus for these systems can be explained by the increasing density of physical crosslinks within the cured part with increasing CTA graft density. This means higher modulus or stiffness may be associated with the increased number of chain entanglements per unit volume. The sharp decrease in strain-to-break may be explained by the same phenomenon that was observed in the DP studies. While the relative amount of DAAm-to-scaffold in the 10% grafted PISA resin is high (meaning little stabilizer), there is much more scaffold present relative to DAAm in the 1, 2.5 and 5% graft densities at the same targeted DP. As mentioned previously, increasing amounts of CFS relative to DAAm leads to more compliant and rubbery parts, which explains the high strain-to-break values and toughness values associated with these two densities. On the opposite end, the 1% graft density has little monomer relative to the scaffold and few chain entanglements per unit volume because of the low graft density. The parts themselves were observed to be weak and barely hold their shape during dogbone mold removal, which may explain the low toughness associated with the 1% graft density.
Finally, the total solids content was varied for the 10% BTP graft density scaffolds using a DAAm target DP of 500. As shown, modulus increased with increasing solids content to a maximum of 126 ± 4.77 kPa at a solids content of 40 wt%. These same trends were observed with strain-to-break (maximum of 557 ± 164% at 40 wt%) and toughness (maximum of 1396 ± 596 kJ m−3 at 40 wt%). This seems to make intuitive sense. At just 12.5 wt% solids, parts were barely being held together and were made of almost 90 wt% water. In contrast, the 40 wt% part held its shape well and could be handled roughly without any issue. Since the part was only 60 wt% water, there was much more solids present per unit volume, resulting in tougher and stiffer parts. The higher solids parts, while mechanically stronger, will experience a less open or porous structure following drying which may be disadvantageous for applications such as tissue engineering and cell culture, if gelatin is incorporated. A combination of a mechanically-strong and porous materials could be selectively developed by targeting a solids content somewhere between 25 and 40 wt%, especially if these materials are targeted for cell culture and tissue engineering applications.
In comparison to our previous PISA printing studies, which employed a difunctional PEG macro-CTA stabilizer, the current CFS system displayed notable improvements in both curing rate and mechanical properties. CFS resins, targeting a DP of 500 at a solids content of 40 wt%, exhibited higher overall toughness (1396 kJ m−3, >50% higher) than the PEG system (910 kJ m−3). The stiffness of the CFS system (225 kPa) was lower than that of the PEG system (390 kPa), which can be attributed to the higher DAAm-to-grafted polymer ratios in the PEG system. The strain values for many of the CFS compositions were higher, indicating greater stretchability compared to the PEG system. Additionally, toughness values for several CFS compositions surpassed those of the PEG system, with some formulations exhibiting over double the toughness value. For instance, the DP 250-targeted 10% graft system had a toughness value of 1853 kJ m−3, more than twice that of the PEG system.
To put these results into context, they were compared to our previous linear difunctional PEG PISA system. In those studies, the DAAm PEG resins were cured using a target DP of 500 and a solids content of 40 wt%. Resulting cured parts had modest mechanical stiffness (390 kPa), strain (∼650%) and toughness (910 kJ m−3). These values were compared to mechanical property values in the 40 wt% system with a target DP of 500, conditions which mirror those used in the PEG resins. While the stiffness (225 kPa) was lower and the strain (557%) was comparable for the CFS system when compared to the PEG system, the energy calculated under the stress–strain curve resulted in a higher overall toughness (1396 kJ m−3, >50% higher) for these CFS materials/architectures. The higher stiffness in the PEG system could possibly be explained by our previous observation in this system. Higher DAAm-to-grafted polymer ratios can result in an increase in stiffness up to a point. Based on our observations in these studies, it could be possible that the stiffness may be higher in the PEG resin when compared to the CFS system. The larger contributor to the higher modulus may be a result of the architecture of the multifunctional macro-CTA, its conformation and the spacing between CTAs (growing DAAm chains). Interestingly, many of our compositions showed higher strain values than the PEG system (1, 2.5 and 5% graft densities and low DPs in the 10% graft density), meaning most materials cured using these architectures were much more stretchable. Toughness values for many of our compositions also surpassed that of the PEG system. For example, the DP 250-targted 10% graft system gave a toughness value of 1853 kJ m−3, which is over double that of the PEG system. Toughness values for the 2.5 and 5% graft densities were similar or higher than that of the PEG system even though they were cured at much lower solids content (25 wt% versus 40 wt%). As shown by our solids study, toughness increases with solids (over 5 fold increase from 25 to 40 wt%). If the 2.5 and 5% graft densities formulations were increased to similar solids contents as the PEG system, it seems possible their toughness could be measured to be 2–3× higher. While some conditions in this study produced lower modulus, toughness, and strain values, many conditions resulted in much more compliant and tougher materials when compared to the PEG system. This suggests that architecture of the macro-CTA does have a significant impact on mechanical properties for these 3D printable PISA resins. Further studies in the future with low graft densities and higher solids (40 wt%) may yield even tougher materials. Additionally, studies that study the effect of the macro-CTA scaffold DP on mechanical properties may be explored.
Lastly, these PISA printed parts with no crosslinker were compared to a set of PISA parts printed with 2.5 wt% MBAc (N′N-methylene(bisacrylamide)) crosslinker to observe any differences in mechanical properties. Results are summarized in ESI Table III.† Both sets of dogbones were fabricated at conditions using the DP 10000 10% grafted CFS with target DP 500 DAAm and 25 wt% solids. The only difference was the addition of the 2.5 wt% crosslinker with respect to solids. The parts with crosslinker showed a higher modulus or greater stiffness (174 kPa v. 50.9 kPa) and lower strain-to-break (96% v. 349%) when compared to the part without crosslinker. This is not surprising considering the addition of rigid chemical bonds should resist elongation and provide stiffness to the 3D printed network. The biggest consequence of adding the crosslinker was a reduction in toughness (59.8 kJ m−3 v. 274 kJ m−3), which can be attributed to the addition of the rigid chemical bonds reducing any rubbery or stretchy quality that parts without crosslinker would otherwise display.
Conclusions
In conclusion, this study looked at the application of CFSs to PISA print mechanically durable parts with lower cured times, specifically focusing on the impact of CTA graft density and DP on printing and mechanical properties. Incorporating CTA groups into polymer scaffolds facilitated efficient PISA printing and reduced exposure times for high-quality prints. Lower DP CFS scaffolds (500) appeared to produce more fluid resins, and consequently, high resolution prints. The printed parts displayed physical crosslinks, evidenced by light scattering from nanostructures formed during UV curing. Parts with higher CTA graft densities showed limited dissolution, suggesting a threshold of CTA graft density for solubility of the resulting part. SEM showed web-like, porous microstructures while AFM subtly displayed the presence of spheroidal nanostructures. Mechanical properties were closely linked to CTA graft density and DP, with increases in graft density enhancing modulus and toughness to a certain point before declining. The optimal mechanical performance was observed at a 5% graft density. This study highlights the potential of PISA printing for fabricating 3D parts with adjustable properties, useful for biomedical applications. The ability to modulate printing times and mechanical features through CTA graft density and DP offers avenues for material customization. However, the solubility challenges in parts with high CTA graft densities warrant further exploration into crosslinking mechanisms and scaffold architecture.Author contributions
A. J. C. conceived the idea. A. P. synthesized RAFT macro CTAs, designed resins formulations, PISA printed CFS resins, performed dissolution studies, and characterized the materials via GPC, NMR, DLS and UV-Vis. J. Y. performed mechanical testing of PISA cured dogbones. Y. Z. and R. W. imaged the materials by AFM.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank the center for biomedical research (CBR) at Missouri University of Science and Technology for providing seed funding to support this work.References
- A. Bagheri and J. Jin, Photopolymerization in 3D printing, ACS Appl. Polym. Mater., 2019, 1(4), 593–611 CrossRef.
- R. Chaudhary, P. Fabbri, E. Leoni, F. Mazzanti, R. Akbari and C. Antonini, Additional manufacturing by digital light processing: a review, Prog. Addit. Manuf., 2023, 8, 331–351 CrossRef.
- D. Srinivasan, M. Meignanmoorthy, M. Ravichandran, V. Mohanavel, S. V. Alagarsamy, C. Chanakyan, S. Sakthivelu, A. Karthick, T. R. Prabhu and S. Rajkumar, 3D Printing Manufacturing Techniques, Materials and Applications: An Overview, Adv. Mater. Sci. Eng., 2021, 5756563 Search PubMed.
- J. Borrello, P. Nasser, J. C. Iatridis and K. D. Costa, 3D printing a mechanically-tunable acrylate resin on a commercial DLP-SLA printer, Addit. Manuf., 2018, 23, 374–380 Search PubMed.
- V. S. D. Voet, T. Strating, G. H. M. Schnelting, P. Dijkstra, M. Tietema, J. Xu, A. J. J. Woortman, K. Loos, J. Jager and R. Folkersma, Biobased Acrylate Photocurable Resin Formulation for Stereolithography 3D Printing, ACS Omega, 2018, 3, 1403–1408 CrossRef PubMed.
- L. Shahzadi, F. Maya, M. C. Breadmore and S. C. Thickett, Functional Materials for DLP-SLA 3D Printing Using Thiol-Acrylate Chemistry: Resin Design and Postprint Applications, ACS Appl. Polym. Mater., 2022, 4, 3896–3907 CrossRef.
- L. Schittecatte, V. Geertsen, D. Bonamy, T. Ngyuen and P. Guenoun, From resin formulation and process parameters to the final mechanical properties of 3D printed acrylate materials, MRS Commun., 2023, 13, 357–377 CrossRef.
- V. Egorov, U. Gulzar, Y. Zhang, S. Breen and C. O'Dwyer, Evolution of 3D Printing Methods and Materials for Electrochemical Energy Storage, Adv. Mater., 2020, 32, 2000556 CrossRef PubMed.
- I. Bacskay, Z. Ujhelyi, P. Feher and P. Arany, The evolution of 3D-Printed Drug Delivery Systems: A Review, Pharmaceutics, 2022, 14, 1312 CrossRef PubMed.
- J. Zhang, Q. Hu, S. Wang, J. Tao and M. Gou, Digital Light Processing Based Three-Dimensional Printing for Medical Applications, Int. J. Bioprint., 2020, 6, 242 CrossRef.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, A comparison of RAFT and ATRP methods for controlled radical polymerization, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef PubMed.
- V. A. Bobrin, J. Zhang, N. Corrigan and C. Boyer, The Emergence of Reversible-Deactivation Radical Polymerization in 3D Printing, Adv. Mater. Technol., 2022, 8(5), 2201054 CrossRef.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Reversible-deactivation radical polymerization (controlled/living radical polymerization): From discovery to materials design and applications, Prog. Polym. Sci., 2020, 111, 101311 CrossRef.
- M. Ouchi and M. Sawamoto, Sequence-controlled polymers via reversible-deactivation radical polymerization, Polym. J., 2018, 50, 83–94 CrossRef.
- J. Li, C. Boyer and X. Zhang, 3D Printing based on Photopolymerization and Photocatalysts: Review and Prospect, Macromol. Mater. Eng., 2022, 307, 2200010 CrossRef.
- Z. Zhang, N. Corrigan, A. Bagheri, J. Jin and C. Boyer, A Verstaile 3D and 4D Printing System through Photocontrolled RAFT Polymerization, Angew. Chem., Int. Ed., 2019, 58, 17954–17963 CrossRef PubMed.
- A. Bagheri, K. E. Engel, C. W. A. Bainbridge, J. Zu, C. Boyer and J. Jin, 3D printing of polymeric materials based on photo-RAFT polymerization, Polym. Chem., 2020, 11, 641–647 RSC.
- B. Zhao, J. Li, Y. Xiu, X. Pan, Z. Zhang and J. Zhi, Xanthane-Based Photoiniferter RAFT Polymerization toward Oxygen-Tolerant and Rapid Living 3D Printing, Macromolecules, 2022, 55, 1620–1628 CrossRef.
- T. Maruyama, M. Mukai, R. Sato, M. Iijima, M. Sato, T. Furukawa and S. Maruo, Multifunctional 3D Printing of Heterogeneous Polymer Structures by Laser-Scanning Micro-Stereolithography Using Reversible Addition-Fragmentation Chain Transfer, ACS Appl. Polym. Mater., 2022, 4, 5515–5523 CrossRef.
- A. Bagheri, C. W. A. Bainbridge, K. E. Engel, G. G. Qiao, J. Xu, C. Boyer and J. Jin, Oxygen Tolerant PET-RAFT Facilitated 3D Printing of Polymeric Materials under Visible LEDs, ACS Appl. Polym. Mater., 2020, 2(2), 782–790 CrossRef.
- A. Bagheri, M. Asadi-Eydivand, A. A. Rosser, C. M. Fellows and T. C. Brown, 3D Printing of Customized Drug Delivery Systems with Controlled Architecture via Reversible Addition-Fragmentation Chain Transfer Polymerization, Adv. Eng. Mater., 2022, 25(10), 2201785 CrossRef.
- A. Bagheri, Application of RAFT in 3D Printing: Where are the Future Opportunities?, Macromolecules, 2023, 56(5), 1778–1797 CrossRef.
- B. Zhao, J. Li, Z. Li, X. Lin, X. Pan, Z. Zhang and J. Zhu, Photoinduced 3D Printing through a Combination of Cationic and Radical RAFT Polymerization, Macromolecules, 2022, 55(16), 7181–7192 CrossRef.
- R. Yamanaka, A. Sugawara-Narutaki and R. Takahashi, In Situ, Monitoring of Polymerization-Induced Self-Assembly and Gelation During the Synthesis of Triblock Copolymers via Time-Resolved Small-Angle X-Ray Scattering and Rheology, Macromolecules, 2023, 56, 4354–4361 CrossRef.
- P. Blais, O. Colombani, L. Bouteiller, F. Stoffelbach and J. Rieger, Unraveling the formation of BAB block copolymer assemblies during PISA in water, Polym. Chem., 2020, 11, 4568–4578 RSC.
- C. Gao, S. Li, Q. Li, P. Shi, S. A. Shah and W. Zhang, Dispersion RAFT polymerization: comparison between the monofunctional and bifunctional macromolecular RAFT agents, Polym. Chem., 2014, 5, 6957–6966 RSC.
- R. Zeng, Y. Chen, L. Zhang and J. Tan, R-RAFT or Z-RAFT? Well-Defined Star Block Copolymer Nano-Objects Prepared by RAFT-Mediated Polymerization-Induced Self-Assembly, Macromolecules, 2020, 53, 1557–1566 CrossRef.
- Z. Zeng, Z. Li, Q. Li, G. Song and G. Huo, Strong and Tough Nanostructure of Hydrogels and Organogels Prepared by Polymer-Induced Self Assembly, Small Methods, 2023, 7, 2201592 CrossRef.
- A. Priester, J. Yeng, Y. Zhang, R. Wang and A. J. Convertine, 3D printing soluble solids via PISA, Polym. Chem., 2023, 14, 2452–2456 RSC.
- A. Priester, J. Yeng, Y. Zhang, K. Hilmas, R. Wang and A. J. Convertine, PISA Printing Microneedles with Controllable Aqueous Dissolution Kinetics, ACS Appl. Polym. Mater., 2024 DOI:10.1021/acsapm.3c02796.
- D. D. Lane, D. Chiu, F. Y. Siu, S. Srinivasan, H. B. Kern, O. W. Press, P. S. Stayton and A. J. Convertine, Well-defined single polymer nanoparticles for the antibody-targeted delivery of chemotherapeutic agents, Polym. Chem., 2015, 6, 1286–1299 RSC.
- A. Kerr, M. Hartlieb, J. Sanchis, T. Smith and S. Perrier, Complex multiblock bottle-brush architectures by RAFT polymerization, Chem. Commun., 2017, 53, 11901–11904 RSC.
- A. B. Lowe, RAFT alcoholic dispersion polymerization with polymerization-induced self-assembly, Polymer, 2016, 106, 161–181 CrossRef.
- E. M. Maines, M. K. Porwal, C. J. Ellison and T. M. Reineke, Sustainable advances in SLA/DLP 3D printing materials and processes, Green Chem., 2021, 23, 6863–6897 RSC.
- V. S. D. Voet, J. Guit and K. Loos, Sustainable Photopolymers in 3D Printing: A Review on Biobased, Biodegradable, and Recyclable Alternatives, Macromol. Rapid Commun., 2020, 42, 2000475 CrossRef.
- L. E. Moore, S. Vucen and A. C. Moore, Trends in drug- and vaccine-based dissolvable microneedle materials and methods of fabrication, Eur. J. Pharm. Biopharm., 2022, 173, 54–72 CrossRef.
- S. N. Economidou and D. Douroumis, 3D printing as a transformative tool for microneedle systems: Recent advances, manufacturing considerations, and market potential, Adv. Drug Delivery Rev., 2021, 173, 60–69 CrossRef.
- R. Ali, P. Mehta, M. S. Arshad, I. Kucuk, M.-W. Chang and Z. Ahmad, Transdermal Microneedles-A Materials Perspective, AAPS PharmSciTech, 2020, 21, 12 CrossRef.
- M. Ali, S. Namjoshi, H. A. E. Benson, Y. Mohammed and T. Kumeria, Dissolvable polymer microneedles for drug delivery and diagnostics, J. Controlled Release, 2022, 347, 561–589 CrossRef.
- C. Heller, M. Schwentenwein, G. Russmueller, F. Varga, J. Stampfl and R. Liska, Vinyl esters: Low cytotoxicity monomers for the fabrication of biocompatible 3D scaffolds by lithography based additive manufacturing., J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6941–6954 CrossRef.
- J. H. Galarraga, A. P. Dhand, B. P. Enzmann III and J. A. Burdick, Synthesis, Characterization, and Digital Light Processing of Hydrolytically Degradable Hyaluronic Acid Hydrogel, Biomacromolecules, 2023, 24, 413–425 CrossRef.
- Y. Wu, M. C. Simpson and J. Jin, Fast Hydrolytically Degradable 3D Printed Object Based on Aliphatic Polycarbonate Thiol-yne Photoresins, Macromol. Chem. Phys., 2021, 222, 2000435 CrossRef.
- A. Oesterreicher, J. Wiener, M. Roth, A. Moser, R. Gmeiner, M. Edler, G. Pinter and T. Griesser, Tough and degradable photopolymers derived from alkyne monomers for 3D printing of biomedical devices and materials, Polym. Chem., 2016, 7, 5169–5180 RSC.
- A. S. Kuenstler, J. J. Hernandez, M. Trujillo-Lemon, A. Osterbaan and C. N. Bowman, Vat Photopolymerization Additive Manufacturing of Tough, Fully Recyclable Thermosets, ACS Appl. Mater. Interfaces, 2023, 15, 11111–11121 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00252g |
| This journal is © The Royal Society of Chemistry 2024 |