
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Niobium oxide coatings on nanostructured platinum electrocatalysts: benefits and limitations†
Annabelle M. K.
Hadley
 ,
Sakshi
Gautam
and
Byron D.
Gates
*
,
Sakshi
Gautam
and
Byron D.
Gates
*
Department of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, BC V5A 1S6, Canada. E-mail: bgates@sfu.ca
First published on 16th August 2024
Abstract
Development of durable nanoscale electrocatalysts is an important step towards improving the affordability and sustainability of fuel cell technology. Nanostructured platinum catalysts are used to facilitate the two half reactions for hydrogen fuel cells. The sluggish kinetics of the cathodic oxygen reduction reaction and the less than optimal stability of cathode catalysts provide motivation for additional efforts to improve the catalytic performance of platinum. Metal oxide coatings on electrocatalysts have been found to increase durability of nanostructured catalysts and to impart additional properties such as increased activity and resistance to poisoning by contaminants. Niobium oxides have been studied as supporting materials for platinum fuel cell catalysts and shown to have a relatively high stability. It has also been suggested that niobium oxides can impart an increased activity due to strong metal support interactions. However, the lack of electrical conductivity of niobium pentoxide limits its viability as a support. Herein, coatings of niobium oxide were applied to nanotextured platinum catalysts prepared by electrodeposition against self-assembled templates to explore the impact of the coatings on the durability and electrocatalytic activity of the catalyst both during and after an accelerated stress test. The catalysts were characterized via scanning electron microscopy, X-ray photoelectron spectroscopy, conductive atomic force microscopy, and electrochemical techniques. Increasing the thickness of the coating from ∼0.5 nm to ∼4.5 nm was found to preserve the initial nanostructured morphology of the electrodeposited platinum catalyst. The thicker coatings did, however, result in larger charge transfer resistances towards the oxygen reduction reaction. These studies provide further evidence of the utility of ultrathin coatings to improve the properties of nanostructured electrocatalysts.
Introduction
Nanoscale particles and nanostructures have long been a subject of interest in heterogeneous catalysis. This interest stems from the high surface areas and activities of these materials relative to bulk materials. Platinum nanoparticles are currently used commercially in hydrogen-based polymer electrolyte membrane (PEM) fuel cell technologies. The development and implementation of these low temperature hydrogen fuel cells is a vital part of shifting our dependence from fossil fuels to renewable energy sources.1,2 The high price of platinum is a motivating factor for optimizing the use of platinum and ensuring that the platinum within the PEM fuel cell retains as much of its initial activity for as long as possible. These motivations have led to many studies being performed to understand how these nanoparticle catalysts behave over the lifetime of a fuel cell and, specifically, what these behaviours mean in terms of catalytic performance. Over the course of fuel cell operation, platinum nanoparticles are known to lose activity by coalescing and forming larger particles, which results in an overall decrease in surface area. These catalysts can also lose activity through poisoning by contaminants, as well as through dissolution and a subsequent deposition onto other particles (Ostwald ripening) or migration into other components within the PEM fuel cell.3,4 These degradation mechanisms are most prevalent at the cathode of the PEM fuel cell where the oxygen reduction reaction (ORR) takes place. Platinum degradation is often a result of the formation of platinum oxide, which plays a central role in platinum dissolution.5 Minimization of platinum re-structuring is especially of interest for platinum catalysts prepared as nanostructured thin films (NSTFs) such as those developed by 3M, their collaborators, and others.6,7 Current explorations of methods to reduce the degree of platinum nanocatalyst degradation include improving nanoparticle-to-support interactions and modifying the compositions of nanoparticles, as well as optimizing PEM fuel cell operating procedures in search of conditions that minimize catalyst degradation.8–10Metal oxide supports can provide a means to stabilize nanoparticle catalysts but often have a limited electrical conductivity in comparison with more traditional carbon support materials.10 This work focuses instead on improving the stability of a platinum nanocatalyst by coating it with an ultrathin metal oxide. Catalysts coated with a stabilizing, non-catalytically active material can have the benefit of enhanced catalyst durability and protection against catalyst poisoning.11–14 Specifically, studies of Pt/C catalysts with coatings of TaOx or TiO2, both in half-cell and full cell configurations, have exhibited an increase in catalyst durability when compared to their uncoated counterparts.15,16 Furthermore, coatings provide a consistent, adjustable local environment at the catalyst surface and, depending on how they are applied, do not interfere with the electrical conductivity of the supporting material.17 Of primary interest for platinum fuel cell catalysts is the ability to utilize coatings as a means to stabilize catalytic nanoparticles against the multiple mechanisms of degradation. Naturally, given that this method involves covering the active surface of the catalyst, access to the catalyst needs to be carefully maintained by controlling the thickness, porosity, and composition of the encapsulating material.11 A series of studies by Takenaka et al. describe the optimization of porous SiO2 coated Pt/C fuel cell catalysts culminating in a method that resulted in the same electrochemically active surface area for both pristine and coated catalysts. The coated catalyst did, however, exhibit a superior durability to the pristine catalyst because of the formation of an encapsulating layer of porous SiO2. This set of studies with silica coatings illustrated the importance of the morphology of the coating material on the catalyst performance.18–21
Niobium oxide was chosen as the coating material for platinum catalysts in this study. This oxide has previously been explored as a platinum catalyst support in PEM fuel cells and was shown to stabilize platinum nanoparticles and increase their activity towards the ORR, which was attributed to strong metal support interactions.22,23 Niobium oxide also has a high stability and corrosion resistance under a variety of operating conditions including at elevated temperatures and relatively high acidities.24,25 This type of coating would be of interest for catalysts composed of either Pt nanoparticles supported on conductive carbon substrates, or a mesoporous Pt film (e.g., a NSTF) such that electron conduction does not need to occur through the electrically insulating Nb2O5 coating.6,7,26,27 An experimental design was, therefore, created to evaluate a series of Nb2O5 films of different thickness to assess their influence on charge transfer resistance during the ORR, as well as their ability to stabilize the Pt catalyst. Atomic layer deposition (ALD) was selected as the method to prepare these layers since this method enables a relatively high degree of control over the thickness of the deposited material. In addition, films deposited by ALD have also been shown to exhibit a highly conformal coating with a relatively uniform thickness across a variety of substrates.28
The Pt catalyst described herein was synthesized using electrodeposition of Pt against a self-assembled template of surfactants to form a catalyst layer that contained a nanostructured or mesoporous texture.26,27,29 Prior work by Eastcott et al. demonstrated that a ∼3 nm thick layer of Nb2O5 was able to minimize the degradation of a thin, planar Pt catalyst prepared by physical vapour deposition (PVD). Furthermore, this prior study demonstrated that a ∼3 nm thick layer of Nb2O5 did not affect the initial electrochemically active surface area (Aecsa) of the catalyst and that the Aecsa of the coated catalyst did not decrease as much as the Aecsa of the uncoated catalyst after being subjected to a stress test.30 The study reported herein builds upon this prior work by evaluating a series of Nb2O5 coatings of different thicknesses to explore their relative impact on charge transfer resistance during the ORR. Furthermore, these coatings are applied to nanostructured platinum catalysts designed to more closely mimic NSTFs, such as those designed by 3M and other types of mesoporous Pt catalysts, to further assess the ability of thin layers of Nb2O5 to mitigate Pt restructuring and dissolution.6,7,26 These degradation phenomena are prevalent under conditions where the nanostructured Pt is subjected to repetitive processes of oxidation and reduction.31,32 In nanoporous Pt catalysts, restructuring can also result from a collapse of the pores within these materials during oxidative and reductive processes.32 Minimizing these mechanisms for restructuring may be achieved by coating the catalyst material with niobium oxide as a means of maintaining their relatively high activity associated with their high surface area. An enhanced durability would prolong the useful lifetime of these catalysts, improving the economics and sustainability of PEM fuel cell technologies. Herein, thin films of Nb2O5 are found to be effective at minimizing changes to the morphology of nanostructured Pt catalysts when compared to the pristine, uncoated nanostructured Pt catalysts. The Nb2O5 coated Pt retains its morphology even when subjected to a rigorous durability test that promotes Pt dissolution and restructuring.
Results and discussion
Preparation and initial characterization of the pristine and coated catalysts
A high surface area, nanostructured Pt catalyst was prepared via electrodeposition onto a planar Pt film supported on a polished Si wafer. The electrodeposition of the nanostructured Pt was carried out over 150 s using a double-pulse or two-step process (Fig. 1a). Initially, a high overpotential was applied over a relatively short duration, in this case −0.6 VAg/AgCl for 30 s, followed by the application of a lower overpotential growth potential over a longer duration, −0.2 VAg/AgCl for 120 s.27,33 Higher overpotentials were not used for the nucleation stage of this process because it was found that more reducing potentials than −0.6 VAg/AgCl resulted in hydrogen gas evolution. This gas evolution resulted in the formation of a less uniform layer of porous Pt upon the substrate. The double-pulse, or two-stage electrodeposition process was used to prepare the mesoporous Pt with a nanoscale texture upon a layer of Pt prepared by PVD techniques. The resulting texture of the electrodeposited Pt layer was characterized by scanning electron microscopy (SEM) based techniques (Fig. 1c). The Pt catalyst was electrodeposited in the presence of Brij-78, a pore-forming agent, to further increase the active surface area of the catalyst from ∼6 cm2 to ∼9 cm2.27,29 The area measured by integration of the profiles recorded during electrodeposition is correlated to the charge passed during the formation of these textured Pt films. Assuming an efficient 4-electron process during electrodeposition (Pt4+ + 4e− → Pt), the average mass of Pt electrodeposited onto each 1 × 1 cm2 substrates was 42 ± 2 μg cm−2. This process was used to prepare a series of Pt catalysts with nanoscale textured morphologies. A series of these nanostructured Pt catalysts were coated with Nb2O5 films. Thickness of the niobia layer was progressively tuned to achieve a set of samples for comparing the relative influence of its thickness on Pt activity and durability.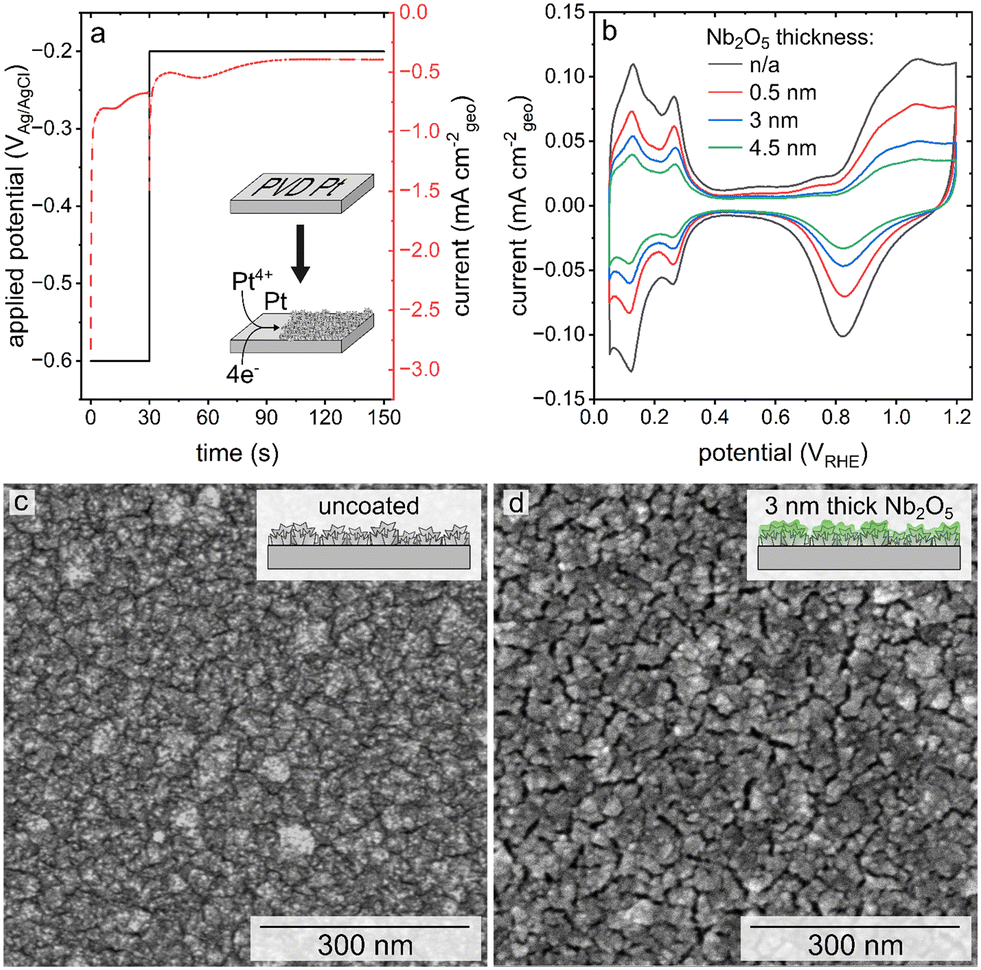 | ||
| Fig. 1 (a) Current density and potential recorded as a function of time during the sequential, double pulse electrodeposition of a textured Pt catalyst. (b) A series of cyclic voltammetry plots corresponding to the textured Pt catalysts (both without and with niobia or Nb2O5 coatings as indicated in the legend) before an accelerated stress test (AST). Scanning electron microscopy (SEM) images of pristine Pt with a nanoscale texture (c) and the same substrate after coating with a 3 nm thick layer of Nb2O5 (d). | ||
Atomic layer deposition was selected for creating the ultrathin coatings of Nb2O5 on the nanostructured Pt due to the relatively high degree of control of ALD over the thickness of the deposited layers, as well as its reported ability to prepare uniform and conformal films.28,34,35 The Nb2O5 coatings were targeted to have a nominal thickness of 0.5 nm, 3 nm, and 4.5 nm based on previously established methods.30 Representative SEM images of the electrodeposited Pt after applying the Nb2O5 coating (Fig. 1d, S1†) demonstrate the presence of visible recesses between the nanoscale features of these nanotextured films. These recesses follow a similar pattern to the grains of the PVD Pt that lies beneath (Fig. S2a†). Control experiments were performed to evaluate if the process of heating the substrates to 250 °C during the ALD process resulted in the formation of these recesses. In this control study, a sample of electrodeposited Pt was subjected to the same thermal cycles as performed during the ALD process. As shown in Fig. S2b,† similar recesses in the electrodeposited Pt can be seen after this thermal treatment demonstrating that they are a product of the ALD conditions and not specifically the deposition of the Nb2O5. The morphology of the coated samples also appears to have a relatively smooth topography in comparison to the features observed before applying the niobia coating, which could be a function of the oxide coating and/or the differences in electron conduction of the Pt and the Nb2O5 (e.g., relatively minor charging in the oxide coated regions of the substrates). It is notable that the ‘smoothness’ of the topography increases with thickness of the coating, indicating that the deposited niobia may be filling in some of the recessed nanotextured features of the electrodeposited Pt.
The texture and composition of the Pt sample coated with a 3 nm thick layer of Nb2O5 was further analyzed by transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDS) techniques (Fig. S3†). The samples were prepared by gently scraping the supported electrodes and suspending the obtained particulates in an ethanol solution to subsequently deposit them onto a TEM grid. The STEM and EDS images in Fig. S3† depict textured Pt particles with layers of niobium covering one-half of the particles, which were likely electrodeposited particles that had been a part of the mesoporous Pt substrate. These results were consistent with a partial coverage of Nb expected for these samples since the remainder of the Pt particle would have been either attached to the planar Pt support (prepared by PVD) or adjacent to other electrodeposited Pt particles. The layer of Nb visualized by EDS mapping was not perfectly uniform but had an average thickness of 2.5 ± 0.7 nm. It is possible that some of the non-uniformity of the niobia films arose from the harsh methods used during sample preparation (e.g., mechanical treatment of the substrate used to isolate particles from their surfaces), resulting in some Nb2O5 being dislodged from the surfaces as shown in Fig. S3.†
The contrast between the electrical conductivity of the amorphous Nb2O5 coating (reported to be from 10−12 to 10−11 S cm−1)36 and that of the Pt substrate (9.4 × 104 S cm−1) enabled another means of independently evaluating the coverage of the Nb2O5 films on the nanotextured Pt. Conductive atomic force microscopy (C-AFM) techniques were used to assess the uniformity of the niobia coatings through the use of this electrical contrast. By measuring the electrical conductivity of the sample while simultaneously mapping the surface morphology, it was possible to gain information with regards to the surface coverage of the Nb2O5 films upon the intact electrodeposited Pt substrates (i.e., without mechanical damage used to prepare the TEM samples). It was clear from conducting these measurements that the atomic layer deposited Nb2O5 coating was not as uniform or conformal as expected, which was more apparent for the thinner coatings of niobia (Fig. 2, 3c and d). As shown in Fig. 3, the measured electrical conductivity of the pristine Pt sample is an order of magnitude larger than that for the Nb2O5 coated samples as expected. This difference is apparent when comparing the peak current values measured at different locations on each type of sample. Some of the measured values on the 0.5 nm thick niobia coated substrates agree with the observed conductivity of the pristine, uncoated electrodeposited Pt. The scales of the peak currents extended to negative values, which included a few measurements at negative currents that were likely the result of some local surface charges. As noted in the inset histograms in Fig. 3, the majority of the currents were positive values. The imaging of the Pt coated with a 3 nm thick layer of Nb2O5 revealed much more consistent surface properties with only some small spikes in the maps of the electrical conductivity therein (Fig. 3e and f, S4†). The observed trends suggest that the Nb2O5 coating, although it does not form a film with a uniform thickness, was able to successfully form an oxide coating across the nanotextured Pt sample at film thicknesses >3 nm. Interestingly, height variations within the samples did not correlate with the observed non-uniformities in the conductivity across each sample. The non-uniformity of the niobia coatings may be a consequence of both the Pt morphology and changes therein during the ALD process as a result of heating to 250 °C.
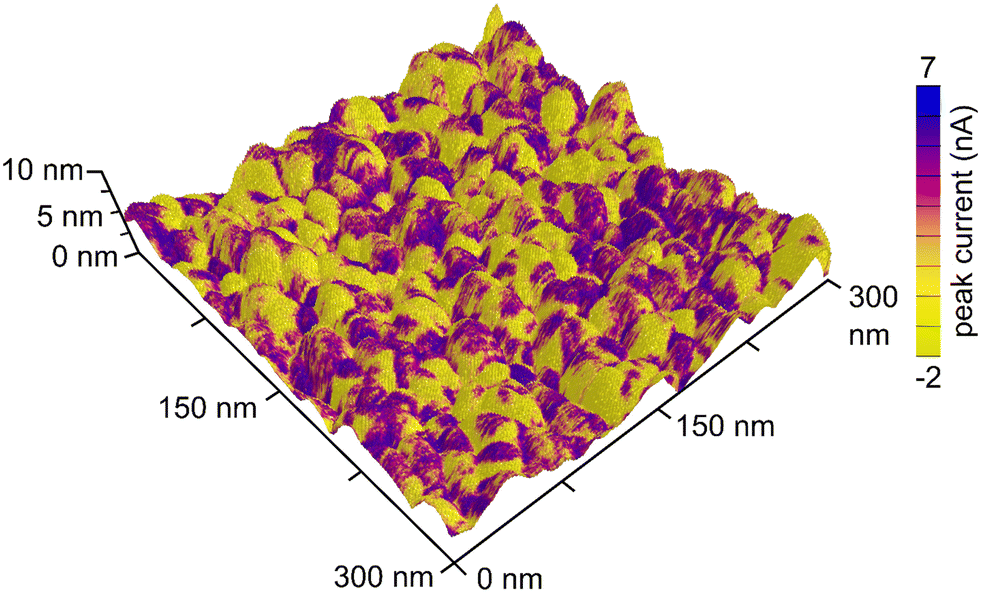 | ||
| Fig. 2 Three-dimensional image of the topography of electrodeposited Pt coated with a 0.5 nm thick layer of Nb2O5. The topography is overlaid with an electrical conductivity map (e.g., peak current values) as measured by conductive atomic force microscopy (C-AFM). | ||
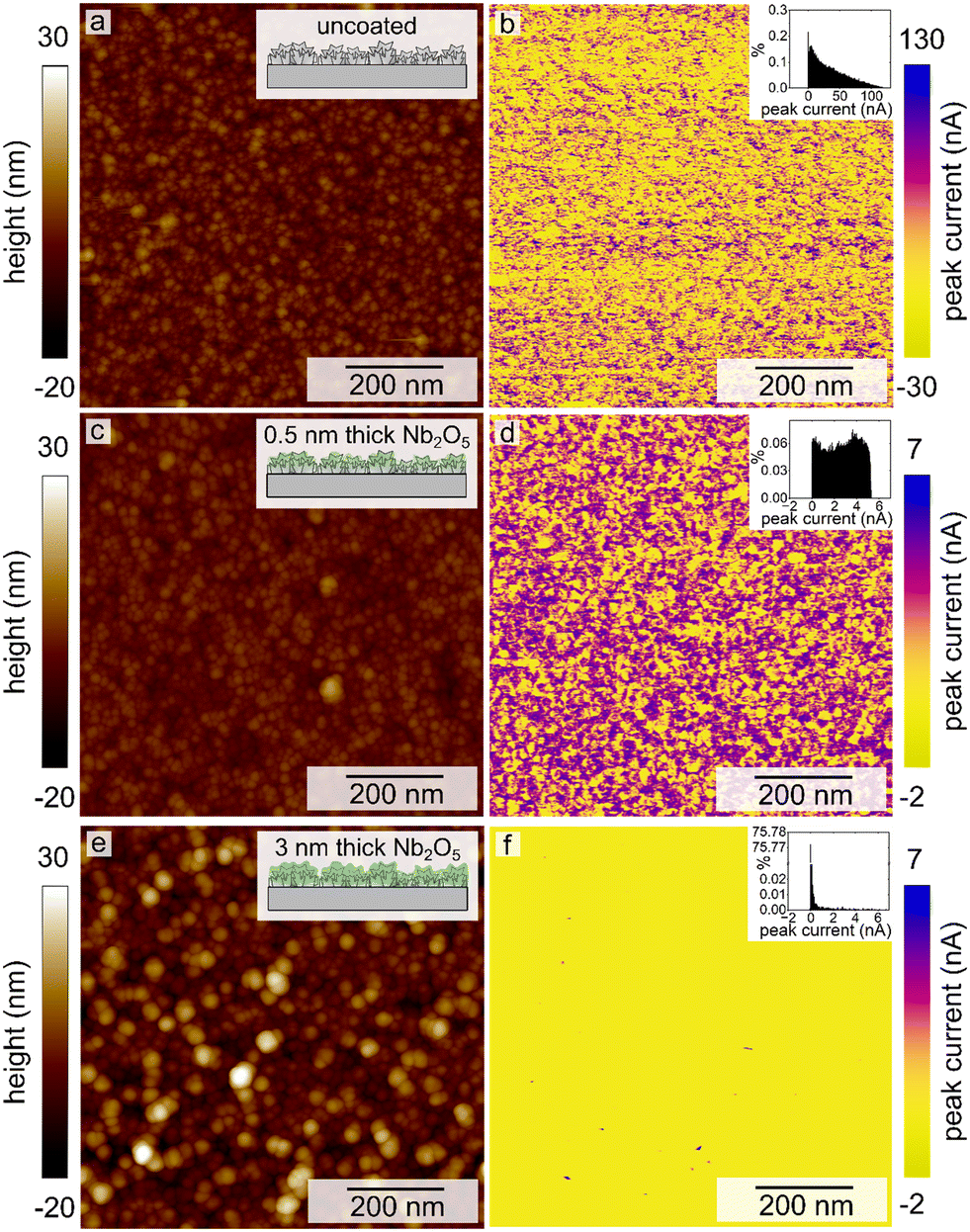 | ||
| Fig. 3 Conductive atomic force microscopy images displaying the measured sample height (a, c and e) and peak current (b, d and f) for a pristine, uncoated electrodeposited Pt catalyst (a and b), and these nanotextured Pt catalysts after coating with a 0.5 nm (c and d) and a 3 nm thick layer of Nb2O5 (e and f). Histograms of the measured peak currents are plotted as insets to each, corresponding CAFM map. | ||
The presence of Nb2O5 on the surfaces of the coated Pt electrodes was further confirmed using X-ray photoelectron spectroscopy (XPS) measurements. The binding energies of Nb 3d measured from high resolution XPS spectra match closely with those of the Nb5+ species found in Nb2O5 (Fig. 4a, Table S1†). Additionally, the binding energies of Pt 4f were also observed, which corresponded to Pt metal species (Fig. 4b, Table S1†). The ratios of Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nb for each type of sample were calculated from the atomic percentages obtained from the survey spectra. The ratio of Pt to Nb for each type of Nb2O5 coated, nanostructured Pt sample (i.e., each film thickness evaluated herein) is plotted in Fig. 4d. The Pt:Nb ratio decreased with an increasing niobia film thickness, changing from 1.8 to 1.4 to 0.50 for the 0.5 nm, 3 nm, and 4.5 nm thick Nb2O5 films, respectively. This trend supports the presence of Nb2O5 films of an increasing thickness across this series of samples. As the thickness of the Nb2O5 layer increases, fewer photoelectrons from Pt are able to pass through the film to reach the detector, leading to a decreased signal for Pt relative to that of the Nb species. The non-linear nature of the trend is, however, indicative of a non-uniform thickness of the Nb2O5 coating, which is consistent with the results of both the TEM and C-AFM imaging.
:Nb for each type of sample were calculated from the atomic percentages obtained from the survey spectra. The ratio of Pt to Nb for each type of Nb2O5 coated, nanostructured Pt sample (i.e., each film thickness evaluated herein) is plotted in Fig. 4d. The Pt:Nb ratio decreased with an increasing niobia film thickness, changing from 1.8 to 1.4 to 0.50 for the 0.5 nm, 3 nm, and 4.5 nm thick Nb2O5 films, respectively. This trend supports the presence of Nb2O5 films of an increasing thickness across this series of samples. As the thickness of the Nb2O5 layer increases, fewer photoelectrons from Pt are able to pass through the film to reach the detector, leading to a decreased signal for Pt relative to that of the Nb species. The non-linear nature of the trend is, however, indicative of a non-uniform thickness of the Nb2O5 coating, which is consistent with the results of both the TEM and C-AFM imaging.
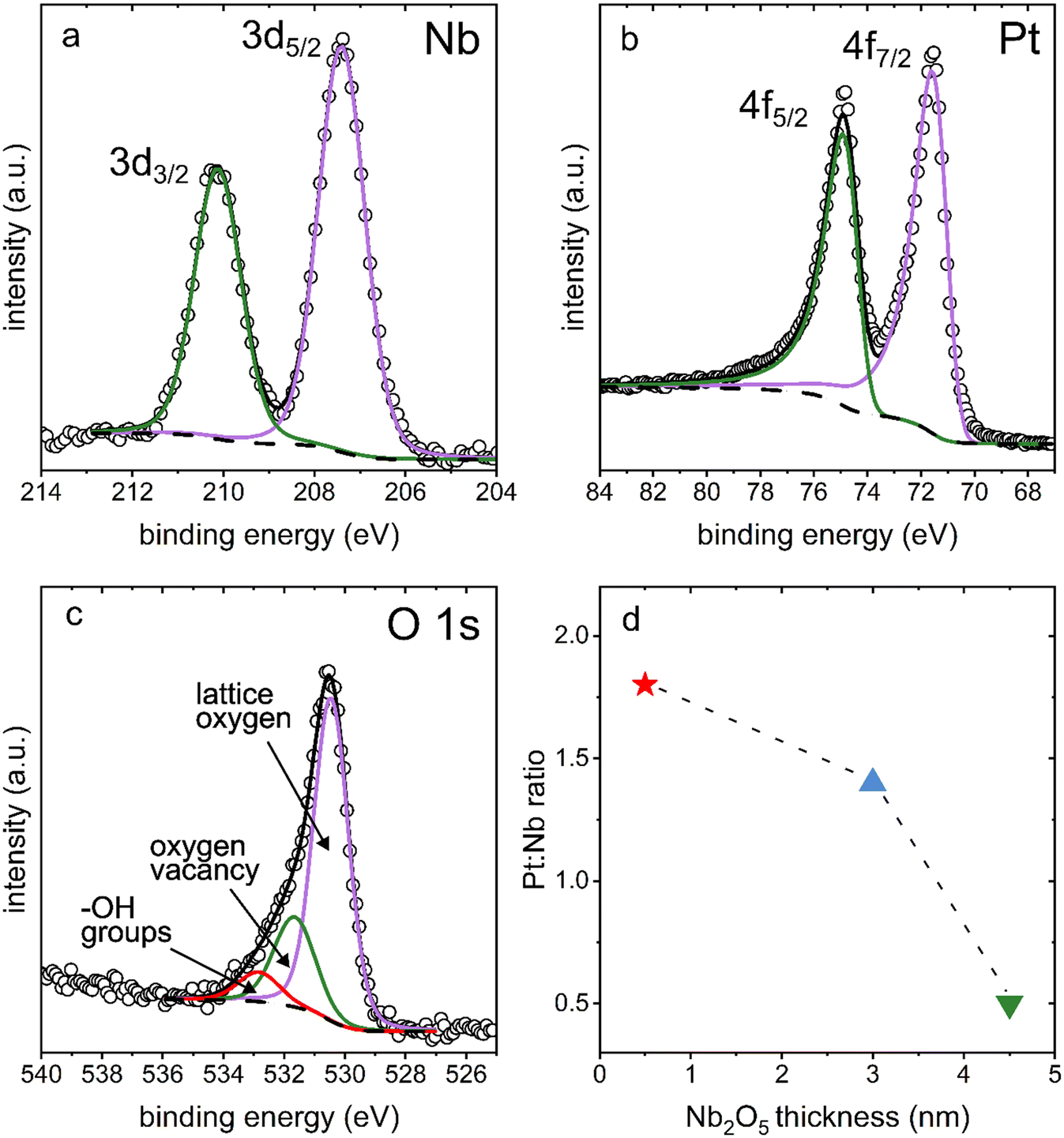 | ||
| Fig. 4 High-resolution X-ray photoelectron spectroscopy (XPS) analyses of the (a) Nb 3d, (b) Pt 4f, and (c) O 1s species present in the nanostructured Pt samples coated with a 3 nm thick film of Nb2O5. Raw data are plotted as hollow circles, and the overall fits are plotted as solid lines. (d) Plot of the ratio of Pt:Nb as calculated from survey spectra of the electrodeposited Pt after coating with (red star) 0.5 nm, (blue triangle) 3 nm, and (green triangle) 4.5 nm thick films of Nb2O5. The dashed line was added to draw notice to the non-linearity of the three plotted Pt:Nb ratios. | ||
Cyclic voltammetry-based analyses of both the pristine (uncoated) and niobia coated films of the nanotextured Pt exhibited characteristic Pt oxidation and reduction peaks from ∼0.8 to 1.2 VRHE and ∼1.1 to 0.6 VRHE, respectively (Fig. 1b). Both types of samples also exhibited hydrogen underpotential deposition (HUPD) from ∼0.05 to 0.4 VRHE. The Aecsa, which is proportional to the integrated area of the HUPD region, is negatively impacted by the presence of the Nb2O5 coating as seen in a relative decrease of the HUPD peak intensities with a proportional increase in the thickness of the Nb2O5 layer. The striking impact of the coating on the accessible Pt surface area was unexpected based on the previously observed negligible impact of a 3 nm thick Nb2O5 coating on the Aecsa of a thin film Pt catalyst.30 The reason for this is, however, attributed to the differences in morphology of each type of catalyst used in these separate studies. Herein, the catalyst was prepared specifically to have a high surface area (more akin to the mesoporous Pt demonstrated in prior studies) through the electrodeposition of Pt in the presence of Brij-78 to create Angstrom-to-nanoscale pores.6,7,26,27,29 In contrast, the thin Pt film prepared by Eastcott et al. had a morphology more akin to the initial, planar Pt substrates used herein that were prepared by PVD techniques (Fig. S2a†).30 These relatively smooth Pt substrates were used herein as supports to prepare the nanotextured films using Pt electrodeposition in the presence of a surfactant. The presence of pores in these nanotextured films likely had dimensions proportional to or smaller than the thickness of the niobium oxide coating, which could lead to a non-uniform deposition of the oxide. It is likely that the oxide coating could have sealed some or all of these relatively narrow pores, making the surfaces within them inaccessible to the electrolyte.37 The C-AFM studies demonstrated changes to the surface properties (i.e., electrical conductivity) of the nanotextured catalyst after coating with different thicknesses of the Nb2O5. The results suggest that the deposited Nb2O5 layers are fairly uniform but the mass transport (including proton transport when probing the HUPD region) could be hindered in some regions depending on the strength of the field lines within the pores versus at the outer edges of the coated pores.38 As a result, the surface areas as measured by the adsorption of species from the electrolyte would be hindered in the case of the coated samples.13 In addition, the observed changes in morphology of the Pt catalyst after the coating procedure (e.g., the heat treatment) could also contribute to the differences observed in the Aecsa between the pristine and coated catalysts.
Methods for measuring the Aecsa of Pt catalysts such as by HUPD and CuUPD have been thoroughly investigated in the literature.39,40 The addition of a metal oxide coating can change the properties of the catalyst surface, which is why two separate techniques to measure the Aecsa were employed herein. These two techniques can also be used to investigate the presence of cracks in a coating layer, as well as to probe the transport of species through a coating as a function of varying scan rate.13 The use of either technique to measure the Aecsa of the metal oxide coated catalysts can also reveal additional properties of the metal oxide overlayer including its ability to protect the catalyst against catalyst poisoning species.13 When evaluating the Aecsa as measured by HUPD at different scan rates, changes therein could arise from the influence of the metal oxide coating on the rates of adsorption and desorption, as well as diffusion of reactants through the coating. Prior studies have demonstrated a decreased Aecsa at higher scan rates when the H+ had to diffuse through relatively thick metal oxide coatings.13 Interestingly, the Aecsa measured by HUPD for the Nb2O5 coated catalysts barely changed (≤3%) with an increase in the scan rates even for the sample with the 4.5 nm thick niobia coatings (Table 1). This suggests that the coating is able to facilitate the transport of protons to the catalyst surfaces, which could be assisted through Å-level pores in the layer of Nb2O5. An increase in Nb2O5 coating thickness did, however, have an impact on the Aecsa as measured by CuUPD. The CuUPD method measured ∼85% of the Aecsa obtained by integrating the HUPD region for both the uncoated and 0.5 nm Nb2O5 coated sample. The measured CuUPD relative to the HUPD dropped to ∼55% for the samples coated with either a 3 nm or 4.5 nm thick layer of Nb2O5. The access of Cu2+ to the Pt surface was hindered by the presence of the thicker Nb2O5 coatings, which is indicative of the surface coverage of the thicker coatings and their ability to block potential contaminants from poisoning the catalyst surfaces.
The difference in activity between the pristine and niobia coated catalysts towards the oxygen reduction reaction was initially quite large. The difference in onset potential was initially ∼30 mV at −0.02 mA cmecsa−2. Following 100 cycles of the accelerated stress test (AST), the activity normalized against the Aecsa from HUPD for the coated catalysts overlapped with the normalized activity observed for the pristine Pt catalyst (Fig. S5a and c†). Electrochemical impedance spectroscopy (EIS) was used to assess changes in the capacitances and resistances of these systems throughout the AST. The EIS results exhibited a similar trend to the changes in Aecsa as a function of applying the AST. As the AST progressed, the charge transfer resistance of the niobia coated catalysts decreased and approached the resistance of the pristine Pt catalyst (Fig. S5b and d†). The increased activity and decreased charge transfer resistance of the Nb2O5 coated catalysts over 100 cycles of the AST was accompanied with an increase in its Aecsa (Fig. S6†The most significant changes were observed within the first 100 cycles of the AST for the coated samples, but the pristine samples were more consistent over this same duration of the AST. It is possible that the niobia coated samples may require an increased number of cycles to reach a stable state in comparison to the pristine Pt catalyst. This prolonged period of activation could be due to mass transport limitations of the Nb2O5 layer, which would need to be accounted for in any protocols for handling these coated Pt catalysts.
Comparing the longer term performance of the pristine and coated catalysts
The accelerated stress tests were selected to specifically target Pt degradation mechanisms that are triggered by repeating Pt oxidation and Pt oxide reduction. An example of a cyclic voltammetry plot depicting a single, representative cycle of the AST is provided in Fig. S7.† Changes to the morphology of the pristine Pt catalyst when comparing the sample before and after 500 cycles and 1000 cycles of the AST can be seen in the SEM images presented in Fig. S8.† The initial Pt texture contained faceted and dimpled surface features, which were transformed into porous, branched structures with smoother features. On the other hand, the catalyst coated with a 3 nm thick layer of Nb2O5 exhibited relatively little change in its morphology after 500 cycles of the AST (Fig. S8b and d†). After 1000 cycles of the AST, the SEM images of the niobia coated sample exhibited slightly smoother features (Fig. S8f†). It is at this stage that the coated Pt catalyst appears to exhibit a similar morphology to that of the pristine Pt catalyst after its AST (i.e., the initial stages of a transition to a smoother porous structure at the surface of the Pt). This trend is more clearly observable for the catalyst coated with the 0.5 nm thick layer of Nb2O5 (Fig. S9†). The initial morphology of this catalyst contains relatively jagged features with a relatively sharp contrast, but as the catalyst is electrochemically cycled the cracks or recesses between the features widen with progression of the testing along with the formation of smoother, porous features. A comparison of the morphology of the uncoated and coated samples after 1000 AST cycles is portrayed in Fig. 5 showing that an increase in the thickness of the Nb2O5 coating is associated with smaller changes in morphology and catalyst re-structuring. | ||
| Fig. 5 A series of scanning electron microscopy (SEM) images of (a) pristine Pt with a nanoscale texture and the same substrate after coating with a (b) 0.5 nm, (c) 3 nm, and (d) 4.5 nm thick layer of Nb2O5. These images were taken after 1000 cycles of an AST. | ||
The textures of the pristine Pt catalyst and the catalyst coated with 0.5 nm thick layer of Nb2O5 adopt morphologies after electrochemical cycling similar to those textures observed for dealloyed nanoporous materials.41,42 The morphologies of such dealloyed materials are understood to evolve from bulk alloys into nanoporous structures through a surface-diffusion based process. The proposed model for this process proceeds through a dissolution of one of the alloyed metals. The surface atoms of the remaining metal(s) are undercoordinated and coalesce with the other undercoordinated atoms to form islands through surface migration. As one metal continues to dissolve from the surfaces of the newly exposed alloy, the undercoordinated metal that remains in the substrate will continue to lead to further coalescence and the formation of branched, nanoporous networks.41,42 Although the work reported herein does not utilize a metal alloy, the AST is designed to induce a process of Pt dissolution that is akin to removing one metal from an alloy. As some Pt atoms dissolve into the electrolyte, the undercoordinated Pt atoms remaining at the surfaces of the catalyst appear to undergo a similar process of coalescence and formation of similarly branched, nanoporous structures. It is likely that the changes observed in the surface features of the pristine Pt and niobia coated Pt proceed through surface migration. The results further suggest that the Nb2O5 coating is hindering the process of reconstruction due to the relatively consistent morphology observed for the niobia coated Pt both before and after the AST (Fig. S8 and S9†).
The evolution of the samples coated with thicker layers of Nb2O5 (i.e., 3 nm and 4.5 nm) as prepared by atomic layer deposition is, however, distinct from that observed in the pristine nanostructured Pt and the Pt coated with a thinner coating of Nb2O5 (i.e., 0.5 nm thick niobia). After 5000 cycles of the AST, the Pt coated with the thicker layers of niobium oxide exhibit a series of relatively flat islands bounded by circular recesses (Fig. S10†). Analysis by C-AFM of the sample coated with 3 nm Nb2O5 after 5000 cycles of the AST showed no change in the surface electrical conductivity of the sample (Fig. S10c and d). Analysis by XPS demonstrated that the Pt:Nb ratio for the sample coated with a 3 nm thick layer of Nb2O5 increased from 1.4 prior to any AST cycles to 18 after the sample was subjected to 5000 AST cycles. The large decrease in the Nb signal relative to the Pt signal suggests that after 5000 AST cycles, a majority of the Nb2O5 film has been either dissolved or dislodged from the electrode surface despite the electrical conductivity remaining unchanged.
The layer of Nb2O5 successfully mitigated catalyst restructuring for up to at least 1000 cycles of the AST and the presence of the coating evidently affects the surface migration behaviour of the Pt atoms. Surface restructuring of the Pt can result from the processes of oxidation and subsequent reduction of the Pt oxide, such as during the transformation from chemically adsorbed oxygen to the incorporation of the oxygen into the Pt lattice.43–45 Additional surface reconstruction can result from the dissolution of Pt species into the electrolyte.46–48 The mechanistic role of the Nb2O5 coating is unclear from the observed trends in the changes in morphology (Fig. S8 and S9†) and the corresponding electrochemical response of each sample (Fig. 6 and S6†). To test the presence of dissolved platinum, aliquots of the electrolyte were analyzed by inductively coupled plasma-mass spectrometry (ICP-MS) following a set number of cycles in the AST (Fig. S11†). The ICP-MS results demonstrated that the pristine Pt electrode produced a slightly larger mass of dissolved Pt per geometric area (24.3 ± 0.2 μg Pt cmgeo−2) compared with the Pt electrode coated with 3 nm thick layer of Nb2O5 (19.9 ± 0.3 μg Pt cmgeo−2). In addition, the amount of Pt dissolution increased in a linear manner over the course of 5000 cycles of the AST. Since the Nb2O5 does not mitigate the majority of Pt dissolution, the differences between the coated and pristine catalyst morphologies after extensive AST cycling must be a result of the interaction between the undercoordinated Pt atoms and the Nb2O5. Calculations of the interaction between Pt and the oxygen in Nb2O5 in a bilayer catalyst show a strong adsorption energy compared with other niobium oxides.49 Further support for the mechanism behind the stabilization of the electrodeposited Pt by Nb2O5 lies in the many studies of Pt single atom catalysts showing that they can be stabilized on metal oxide supports by binding to excess oxygen species at the surface.50–52
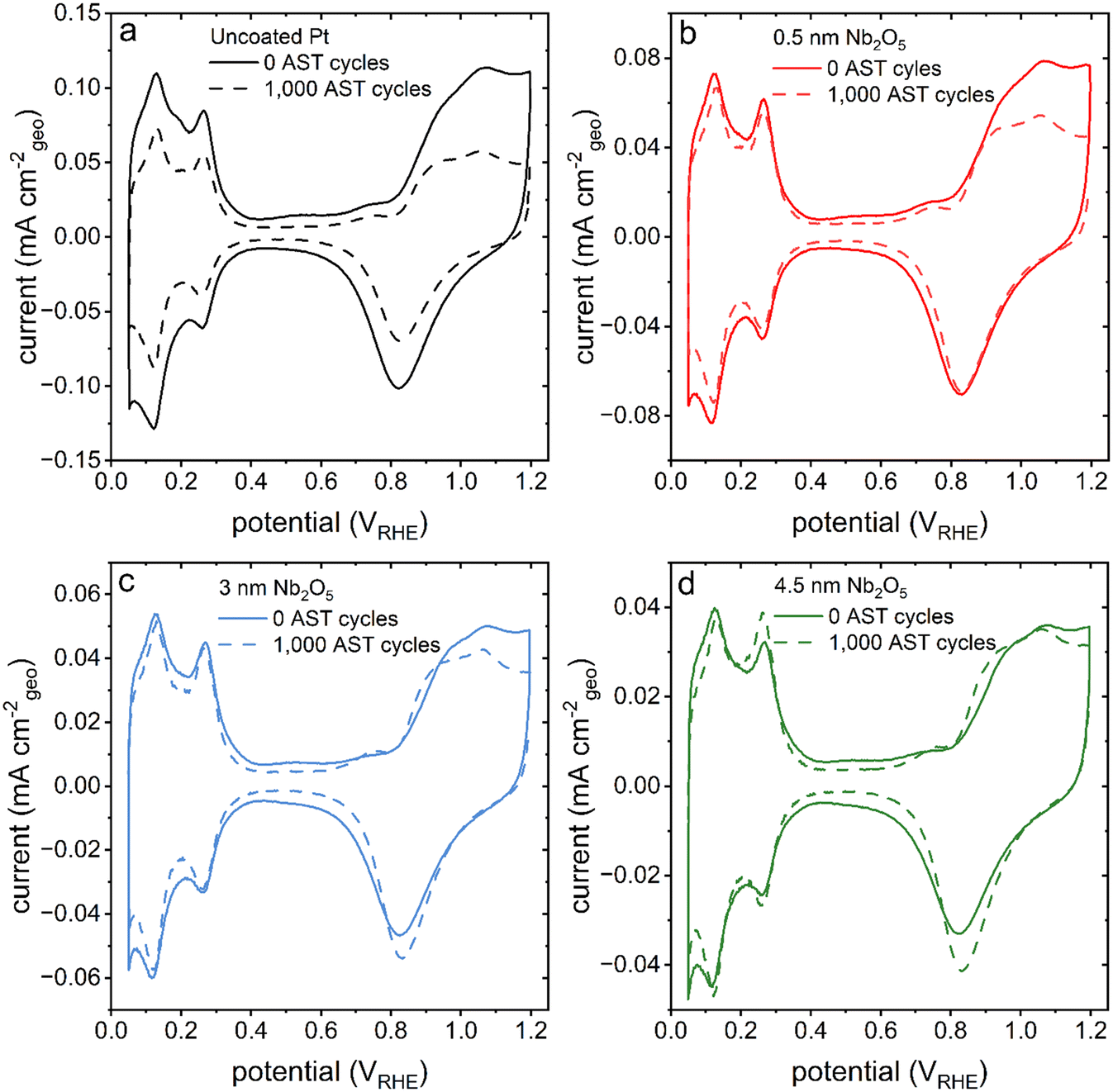 | ||
| Fig. 6 A series of cyclic voltammetry plots corresponding to the nanotextured Pt catalysts (solid line) before and (dashed line) after 1000 cycles of an accelerated stress test (AST). The colours of the traces correlate to differences in thickness of the Nb2O5 coatings, where (a) black represents uncoated or pristine nanotextured Pt, and (b) red is 0.5 nm, (c) blue is 3 nm, and (d) green is 4.5 nm thick niobia coated nanotextured Pt. | ||
After the AST, the activity of the catalyst towards the ORR was also probed by linear sweep voltammetry and electrochemical impedance spectroscopy techniques. The onset potential towards the ORR determined by linear sweep voltammetry was found to shift towards more negative potentials only for the pristine Pt catalyst (Fig. 7). The onset potential of the Nb2O5 coated catalysts remained relatively constant after 1000 cycles of the AST (Fig. 7 and S12†). The relatively high potentials used to perform these tests predominantly probed the charge transfer resistances of these systems.53 The charge transfer resistance of the pristine Pt catalyst increased by ∼30 Ω cmecsa−2 while the same resistance for the coated catalysts increased by ∼20 Ω cmecsa−2 over the course of the 1000 AST cycles (Fig. S6b†). These results collectively suggest that the Nb2O5 coating had a stabilizing effect on the activity of the Pt catalyst, in addition to its influence on the sample morphology.
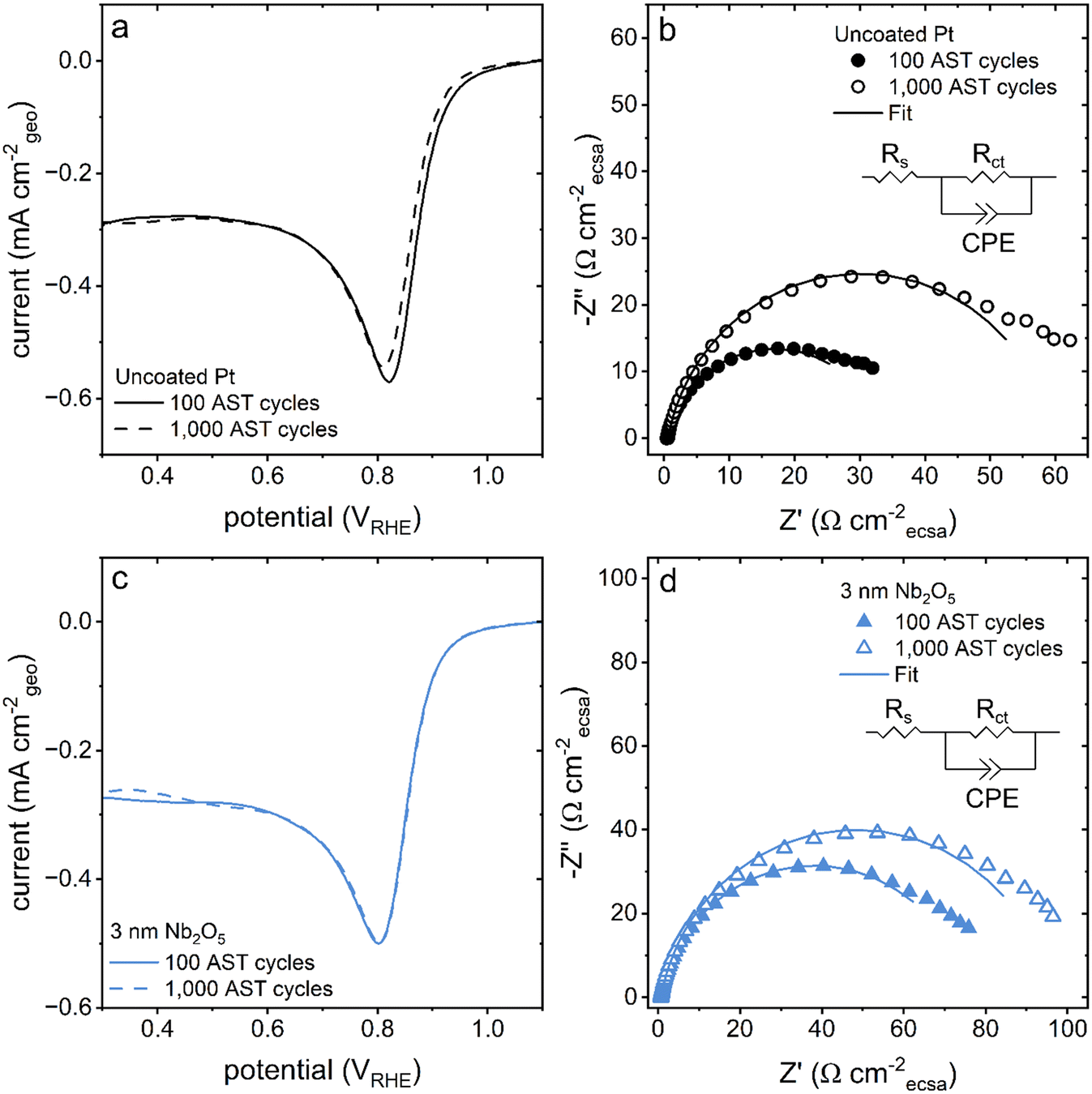 | ||
| Fig. 7 (a and c) Linear sweep voltammetry plots recorded at a scan rate of 10 mV s−1 in an O2 (g) saturated electrolyte for the pristine and coated Pt samples after performing (solid line) 100 cycles and (dashed line) 1000 cycles of the AST. (b and d) Nyquist plots recorded at 0.9 VRHE in an O2 saturated electrolyte with fits (solid lines) corresponding to the circuit displayed in the inset also obtained after performing (solid symbols) 100 cycles and (hollow symbols) 1000 cycles of the AST. The colours of the traces correspond to the thickness of the Nb2O5 films where black is uncoated, pristine Pt and blue is Pt with a 3 nm thick coating of Nb2O5. | ||
This study was conducted to build upon previous work that showed, via electrochemical techniques, an improved durability of PVD Pt coated with nanoscale, ∼3 nm thick films of Nb2O5 in comparison to bare PVD Pt. This work aimed to understand how these coatings performed when applied to more realistic Pt fuel cell catalysts with higher surface areas and nanoscale features.30 By working with the porous, electrodeposited Pt substrate used herein it became apparent that the design of niobia coated high surface area catalysts should be carefully considered to maintain the accessible surface area after applying the coating (e.g., ensure that pore and feature sizes are large enough that the coating is not able to fill in areas of the catalyst such that they are inaccessible to the electrolyte). This work saw similar stabilization of the catalyst activity and an improvement in the stabilization of the Aecsa. It was suggested that the cause of the improved durability of the ∼3 nm Nb2O5 layer was that the coating was mitigating Pt dissolution for both the PVD Pt catalysts and the electrodeposited Pt catalysts presented here. But a series of ICP-MS studies demonstrated that the mitigation of Pt dissolution was less than expected. Instead, it is hypothesized that the Nb2O5 coating increases the durability of the Pt by forming bonds between the undercoordinated surface Pt and O atoms in the Nb2O5. Additional work should be pursued with regards to the method(s) used to prepare the niobia coating to improve its impact on the nanostructured Pt, as well as to further correlate the nanoscale structure (e.g., porosity) and texture (e.g., including its atomic-scale uniformity) of the niobia film to the potential for Pt dissolution and the observed mechanisms for improved Pt durability.
Conclusions
A porous, high surface area Pt catalyst was synthesized by electrodeposition techniques in the presence of templates self-assembled from surfactants. The resulting nanostructured Pt catalyst was subsequently coated with different thicknesses of Nb2O5 using atomic layer deposition as confirmed by XPS and C-AFM, and this series of samples each exposed to an aggressive stress test designed to target Pt dissolution and re-structuring processes. In comparison to previous tests on planar Pt coated with Nb2O5,30 the three-dimensional structure of the Pt catalyst synthesized in this work enabled a more rigorous study of the stabilizing effect of the metal oxide coating. The durability of the catalyst was specifically evaluated as a function of the thickness of the applied niobium oxide coatings. Analysis of the samples did, however, reveal that the coating was not perfectly uniform across the surfaces of the textured Pt catalyst. This lack of uniformity is the likely reason that the dissolution of Pt was not as mitigated as expected, although the thicker (3 to 4.5 nm thick) non-uniform coatings were highly effective at stabilizing the catalyst morphology under aggressively oxidizing and reducing conditions. Furthermore, this work found that a coating with a thickness of 3 nm was able to both stabilize the catalyst structure over a range of applied potentials while minimizing the charge transfer resistance towards the ORR, which increased with the thickness of the niobia coating. In future studies, techniques such as in situ electrochemical TEM54,55 or single particle ICP-MS56 would be instrumental in probing the mechanism of re-structuring in the presence of metal oxide coatings. These techniques could show the distributions of the coating and platinum oxide in the catalyst during the processes of oxidation and reduction57 and could elucidate the mechanism(s) by which the catalyst is degrading (e.g., dissolution into Pt ions or by nanoparticles being dislodged from the surface).58 The knowledge gained over the course of this study can be used in the endeavor to stabilize nanomaterials to both decrease the financial investment and increase the lifetime of these materials for a variety of applications.Experimental section
Electrode fabrication
Material Characterization
:1 (v:v) solution of 2% HNO3:0.5 M H2SO4. The Pt standards were used within two weeks of their preparation. The subsequent dissolution of Pt from the electrodeposited films into the electrolyte during electrochemical testing was measured by taking 1 mL aliquots of the electrolyte at various intervals throughout a 5000 cycle AST. During the AST, the electrochemical cell was held at 25 °C and purged with N2 gas (99.999%, Linde Canada). The aliquot was then diluted 100× in 2% HNO3 and 1 mL of fresh electrolyte was added to the electrochemical cell to maintain the initial electrolyte volume. The samples were analyzed by ICP-MS on the same day that they were prepared.
Electrochemical characterization
All electrochemical characterization of the custom electrocatalysts were conducted while immersing the samples in 0.5 M H2SO4 at 25 °C in a three-electrode cell set up that was controlled using a Metrohm PGSTAT 302N potentiostat. A cylindrical graphite rod (Alfa Aesar, United States) was used for the counter electrode and a custom-built reversible hydrogen electrode (RHE) was used as the reference electrode. The upper, pristine half of the wafer supported electrocatalysts were held by a flat Cu clip soldered to a Cu wire that also provided an electrical connection during the characterization. Prior to the electrochemical analyses, the electrolyte was purged with N2 gas (99.999%, Linde Canada) to displace dissolved O2 gas. Each newly fabricated custom electrocatalyst was conditioned with via cyclic voltammetry (CV) techniques, scanning the potential from 0.05 to 1.2 VRHE at a scan rate of 50 mV s−1 for 40 scans. Electrochemical analysis was also performed in O2 (99.993%, Linde Canada) saturated electrolyte to test the activity of the catalyst towards the oxygen reduction reaction and to evaluate the effect of adding the Nb2O5 layer to the catalyst. Linear sweep voltammetry (LSV) scans were collected at a scan rate of 10 mV s−1 and electrochemical impedance spectroscopy (EIS) measurements were collected at an applied potential of 0.9 VRHE over the frequency range of 100 kHz to 0.1 Hz with an amplitude of 0.01 V. The accelerated stress test (AST) consisted of a series of CVs collected at a scan rate of 500 mV s−1 over the potential range from 0.6 to 1.5 VRHE for up to 5000 cycles.The Aecsa for each substrate was calculated from an average of the areas associated with the hydrogen underpotential adsorption and desorption regions recorded in the CV scans collected at a scan rate of 10 mV s−1 in N2 gas saturated 0.5 M H2SO4 electrolyte. The current arising from the double-layer capacitance was subtracted, applying the standard method for baseline correction.61 A conversion factor of 210 μC cm−2 for Pt was employed to convert the charge calculated from the region of hydrogen underpotential deposition into the Aecsa values for the Pt catalyst.62 The underpotential deposition of copper (Cu) was separately used to investigate the electrode surface areas. The desorption of underpotentially adsorbed Cu atoms was recorded by linearly scanning the potential from 0.36 VRHE to 1.0 VRHE at a scan rate of 10 mV s−1 or 20 mV s−1 after holding the electrode at 0.36 VRHE for 50 s in 0.5 M H2SO4 and 2 mM CuSO4 (ReagentPlus®, ≥98.0%, Sigma-Aldrich).13 A conversion factor of 420 μC cm−2 for Pt was employed to convert the charge calculated from the underpotential deposition of Cu into the Aecsa for the Pt catalysts.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors confirm no competing financial interest.Acknowledgements
This research was supported in part by the Natural Sciences and Engineering Research Council of Canada (NSERC; Grant No. RGPIN-2020-06522), NSERC CGS-D (Annabelle M. K. Hadley), by Simon Fraser University and Indian Oil R&D under the SFU-IOCL joint Ph.D. program in clean energy (Sakshi Gautam), Simon Fraser University Graduate Fellowships (Annabelle M. K. Hadley and Sakshi Gautam), and CMC Microsystems (MNT Grant No. 9858 and No. 10866). This work made use of the 4D LABS and the Center for Soft Materials shared facilities supported by the Canada Foundation for Innovation (CFI), British Columbia Knowledge Development Fund (BCKDF), Western Economic Diversification Canada, and Simon Fraser University. The authors thank Dr. Michael Wang of 4D LABS for assistance with the collection of the XPS data.References
- D. A. Cullen, K. C. Neyerlin, R. K. Ahluwalia, R. Mukundan, K. L. More, R. L. Borup, A. Z. Weber, D. J. Myers and A. Kusoglu, New Roads and Challenges for Fuel Cells in Heavy-Duty Transportation, Nat. Energy, 2021, 6(5), 462–474, DOI:10.1038/s41560-021-00775-z.
- S. Harichandan, S. K. Kar, R. Bansal and S. K. Mishra, Achieving Sustainable Development Goals through Adoption of Hydrogen Fuel Cell Vehicles in India: An Empirical Analysis, Int. J. Hydrogen Energy, 2023, 48(12), 4845–4859, DOI:10.1016/j.ijhydene.2022.11.024.
- I. Martens, A. Vamvakeros, N. Martinez, R. Chattot, J. Pusa, M. V. Blanco, E. A. Fisher, T. Asset, S. Escribano, F. Micoud, T. Starr, A. Coelho, V. Honkimäki, D. Bizzotto, D. P. Wilkinson, S. D. M. Jacques, F. Maillard, L. Dubau, S. Lyonnard, A. Morin and J. Drnec, Imaging Heterogeneous Electrocatalyst Stability and Decoupling Degradation Mechanisms in Operating Hydrogen Fuel Cells, ACS Energy Lett., 2021, 6(8), 2742–2749, DOI:10.1021/acsenergylett.1c00718.
- I. Martens, R. Chattot and J. Drnec, Decoupling Catalyst Aggregation, Ripening, and Coalescence Processes inside Operating Fuel Cells, J. Power Sources, 2022, 521, 230851, DOI:10.1016/j.jpowsour.2021.230851.
- K. Yasuda, A. Taniguchi, T. Akita, T. Ioroi and Z. Siroma, Platinum Dissolution and Deposition in the Polymer Electrolyte Membrane of a PEM Fuel Cell as Studied by Potential Cycling, Phys. Chem. Chem. Phys., 2006, 8(6), 746–752, 10.1039/B514342J.
- M. K. Debe, Tutorial on the Fundamental Characteristics and Practical Properties of Nanostructured Thin Film(NSTF) Catalysts, J. Electrochem. Soc., 2013, 160(6), F522, DOI:10.1149/2.049306jes.
- M. K. Debe, A. K. Schmoeckel, G. D. Vernstrom and R. Atanasoski, High Voltage Stability of Nanostructured Thin Film Catalysts for PEM Fuel Cells, J. Power Sources, 2006, 161(2), 1002–1011, DOI:10.1016/j.jpowsour.2006.05.033.
- A. A. Topalov, I. Katsounaros, M. Auinger, S. Cherevko, J. C. Meier, S. O. Klemm and K. J. J. Mayrhofer, Dissolution of Platinum: Limits for the Deployment of Electrochemical Energy Conversion?, Angew. Chem., Int. Ed., 2012, 51(50), 12613–12615, DOI:10.1002/anie.201207256.
- R. L. Borup, A. Kusoglu, K. C. Neyerlin, R. Mukundan, R. K. Ahluwalia, D. A. Cullen, K. L. More, A. Z. Weber and D. J. Myers, Recent Developments in Catalyst-Related PEM Fuel Cell Durability, Curr. Opin. Electrochem., 2020, 21, 192–200, DOI:10.1016/j.coelec.2020.02.007.
- S. Jayabal, G. Saranya, D. Geng, L.-Y. Lin and X. Meng, Insight into the Correlation of Pt–Support Interactions with Electrocatalytic Activity and Durability in Fuel Cells, J. Mater. Chem. A, 2020, 8(19), 9420–9446, 10.1039/D0TA01530J.
- D. V. Esposito, Membrane-Coated Electrocatalysts—An Alternative Approach To Achieving Stable and Tunable Electrocatalysis, ACS Catal., 2018, 8(1), 457–465, DOI:10.1021/acscatal.7b03374.
- C. Gao, F. Lyu and Y. Yin, Encapsulated Metal Nanoparticles for Catalysis, Chem. Rev., 2021, 121(2), 834–881, DOI:10.1021/acs.chemrev.0c00237.
- N. Y. Labrador, E. L. Songcuan, C. De Silva, H. Chen, S. J. Kurdziel, R. K. Ramachandran, C. Detavernier and D. V. Esposito, Hydrogen Evolution at the Buried Interface between Pt Thin Films and Silicon Oxide Nanomembranes, ACS Catal., 2018, 8(3), 1767–1778, DOI:10.1021/acscatal.7b02668.
- S. S. Hardisty, S. Frank, M. Zysler, R. Yemini, A. Muzikansky, M. Noked and D. Zitoun, Selective Catalyst Surface Access through Atomic Layer Deposition, ACS Appl. Mater. Interfaces, 2021, 13(49), 58827–58837, DOI:10.1021/acsami.1c20181.
- Z. Song, B. Wang, N. Cheng, L. Yang, D. Banham, R. Li, S. Ye and X. Sun, Atomic Layer Deposited Tantalum Oxide to Anchor Pt/C for a Highly Stable Catalyst in PEMFCs, J. Mater. Chem. A, 2017, 5(20), 9760–9767, 10.1039/C7TA01926B.
- W.-J. Lee, S. Bera, H. Woo, H. G. Kim, J.-H. Baek, W. Hong, J.-Y. Park, S.-J. Oh and S.-H. Kwon, In Situ Engineering of a Metal Oxide Protective Layer into Pt/Carbon Fuel-Cell Catalysts by Atomic Layer Deposition, Chem. Mater., 2022, 34(13), 5949–5959, DOI:10.1021/acs.chemmater.2c00928.
- Y. Liu, K. Y. Leung, S. E. Michaud, T. L. Soucy and C. C. L. McCrory, Controlled Substrate Transport to Electrocatalyst Active Sites for Enhanced Selectivity in the Carbon Dioxide Reduction Reaction, Comments Inorg. Chem., 2019, 39(5), 242–269, DOI:10.1080/02603594.2019.1628025.
- S. Takenaka, T. Miyazaki, H. Matsune and M. Kishida, Highly Active and Durable Silica-Coated Pt Cathode Catalysts for Polymer Electrolyte Fuel Cells: Control of Micropore Structures in Silica Layers, Catal. Sci. Technol., 2015, 5(2), 1133–1142, 10.1039/C4CY01301H.
- S. Takenaka, H. Matsumori, K. Nakagawa, H. Matsune, E. Tanabe and M. Kishida, Improvement in the Durability of Pt Electrocatalysts by Coverage with Silica Layers, J. Phys. Chem. C, 2007, 111(42), 15133–15136, DOI:10.1021/jp076120b.
- S. Takenaka, N. Susuki, H. Miyamoto, E. Tanabe, H. Matsune and M. Kishida, Highly Durable Carbon Nanotube-Supported Pd Catalysts Covered with Silica Layers for the Oxygen Reduction Reaction, J. Catal., 2011, 279(2), 381–388, DOI:10.1016/j.jcat.2011.02.008.
- S. Takenaka, H. Miyamoto, Y. Utsunomiya, H. Matsune and M. Kishida, Catalytic Activity of Highly Durable Pt/CNT Catalysts Covered with Hydrophobic Silica Layers for the Oxygen Reduction Reaction in PEFCs, J. Phys. Chem. C, 2014, 118(2), 774–783, DOI:10.1021/jp407928m.
- L. Zhang, L. Wang, C. M. B. Holt, B. Zahiri, Z. Li, K. Malek, T. Navessin, M. H. Eikerling and D. Mitlin, Highly Corrosion Resistant Platinum–Niobium Oxide–Carbon Nanotube Electrodes for the Oxygen Reduction in PEM Fuel Cells, Energy Environ. Sci., 2012, 5(3), 6156–6172, 10.1039/C2EE02689A.
- R. Alipour MoghadamEsfahani, S. K. Vankova, E. B. Easton, I. I. Ebralidze and S. Specchia, A Hybrid Pt/NbO/CNTs Catalyst with High Activity and Durability for Oxygen Reduction Reaction in PEMFC, Renewable Energy, 2020, 154, 913–924, DOI:10.1016/j.renene.2020.03.029.
- M. O. A. Ferreira, R. V. Gelamo, C. E. B. Marino, B. P. da Silva, I. V. Aoki, M. S. da Luz, N. D. Alexopoulos, N. B. Leite and J. A. Moreto, Effect of Niobium Oxide Thin Film on the Long-Term Immersion Corrosion of the 2198-T851 Aluminium Alloy, Materialia, 2022, 22, 101407, DOI:10.1016/j.mtla.2022.101407.
- M. S. Safavi, F. C. Walsh, L. Visai and J. Khalil-Allafi, Progress in Niobium Oxide-Containing Coatings for Biomedical Applications: A Critical Review, ACS Omega, 2022, 7(11), 9088–9107, DOI:10.1021/acsomega.2c00440.
- M. T. Y. Paul and B. D. Gates, Mesoporous Platinum Prepared by Electrodeposition for Ultralow Loading Proton Exchange Membrane Fuel Cells, Sci. Rep., 2019, 9(1), 4161, DOI:10.1038/s41598-019-38855-6.
- S. Gautam, A. M. K. Hadley and B. D. Gates, Controlled Growth of Platinum Nanoparticles during Electrodeposition Using Halide Ion Containing Additives, J. Electrochem. Soc., 2022, 169(11), 112508, DOI:10.1149/1945-7111/ac9e22.
- R. W. Johnson, A. Hultqvist and S. F. Bent, A Brief Review of Atomic Layer Deposition: From Fundamentals to Applications, Mater. Today, 2014, 17(5), 236–246, DOI:10.1016/j.mattod.2014.04.026.
- S. Gautam, S. Chugh and B. D. Gates, Evaluating the Effects of Surfactant Templates on the Electrocatalytic Activity and Durability of Multifaceted Platinum Nanostructures, ACS Appl. Energy Mater., 2023, 6(11), 5883–5898, DOI:10.1021/acsaem.3c00172.
- J. I. Eastcott, A. Parakh, M. T. Y. Paul, A. W. H. Lee, M. W. Bilton and B. D. Gates, Nanoscale Thin Films of Niobium Oxide on Platinum Surfaces: Creating a Platform for Optimizing Material Composition and Electrochemical Stability, Can. J. Chem., 2018, 96(2), 260–266, DOI:10.1139/cjc-2017-0595.
- Y. Chen, X. X. Wang, B. Li, X. X. Huang and J. N. Wang, Porous Pt Aggregates Deposited on a Carbon Nanotube Film with High Catalytic Activity and Durability, ChemElectroChem, 2016, 3(11), 1908–1914, DOI:10.1002/celc.201600349.
- B. Geboes, J. Ustarroz, K. Sentosun, H. Vanrompay, A. Hubin, S. Bals and T. Breugelmans, Electrochemical Behavior of Electrodeposited Nanoporous Pt Catalysts for the Oxygen Reduction Reaction, ACS Catal., 2016, 6(9), 5856–5864, DOI:10.1021/acscatal.6b00668.
- M. M. E. Duarte, A. S. Pilla, J. M. Sieben and C. E. Mayer, Platinum Particles Electrodeposition on Carbon Substrates, Electrochem. Commun., 2006, 8(1), 159–164, DOI:10.1016/j.elecom.2005.11.003.
- R. Zazpe, M. Knaut, H. Sopha, L. Hromadko, M. Albert, J. Prikryl, V. Gärtnerová, J. W. Bartha and J. M. Macak, Atomic Layer Deposition for Coating of High Aspect Ratio TiO2 Nanotube Layers, Langmuir, 2016, 32(41), 10551–10558, DOI:10.1021/acs.langmuir.6b03119.
- J. W. Elam, Coatings on High Aspect Ratio Structures, in Atomic Layer Deposition of Nanostructured Materials, John Wiley & Sons, Ltd, 2011, pp. 227–249, DOI:10.1002/9783527639915.ch10.
- M. Maček and B. Orel, Electrochromism of Sol–Gel Derived Niobium Oxide Films, Sol. Energy Mater. Sol. Cells, 1998, 54(1), 121–130, DOI:10.1016/S0927-0248(98)00062-2.
- J. S. Daubert, R. Wang, J. S. Ovental, H. F. Barton, R. Rajagopalan, V. Augustyn and G. N. Parsons, Intrinsic Limitations of Atomic Layer Deposition for Pseudocapacitive Metal Oxides in Porous Electrochemical Capacitor Electrodes, J. Mater. Chem. A, 2017, 5(25), 13086–13097, 10.1039/C7TA02719B.
- X. Wang, T. Liu, H. Li, C. Han, P. Su, N. Ta, S. P. Jiang, B. Kong, J. Liu and Z. Huang, Balancing Mass Transfer and Active Sites to Improve Electrocatalytic Oxygen Reduction by B,N Codoped C Nanoreactors, Nano Lett., 2023, 23(11), 4699–4707, DOI:10.1021/acs.nanolett.3c00202.
- S. Rudi, C. Cui, L. Gan and P. Strasser, Comparative Study of the Electrocatalytically Active Surface Areas(ECSAs) of Pt Alloy Nanoparticles Evaluated by Hupd and CO-Stripping Voltammetry, Electrocatalysis, 2014, 5(4), 408–418, DOI:10.1007/s12678-014-0205-2.
- M. Shao, J. H. Odell, S.-I. Choi and Y. Xia, Electrochemical Surface Area Measurements of Platinum- and Palladium-Based Nanoparticles, Electrochem. Commun., 2013, 31, 46–48, DOI:10.1016/j.elecom.2013.03.011.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Evolution of Nanoporosity in Dealloying, Nature, 2001, 410(6827), 450–453, DOI:10.1038/35068529.
- J. Weissmüller and K. Sieradzki, Dealloyed Nanoporous Materials with Interface-Controlled Behavior, MRS Bull., 2018, 43(1), 14–19, DOI:10.1557/mrs.2017.299.
- H. A. Baroody, G. Jerkiewicz and M. H. Eikerling, Modelling Oxide Formation and Growth on Platinum, J. Chem. Phys., 2017, 146(14), 144102, DOI:10.1063/1.4979121.
- A. A. Topalov, S. Cherevko, A. R. Zeradjanin, J. C. Meier, I. Katsounaros and K. J. J. Mayrhofer, Towards a Comprehensive Understanding of Platinum Dissolution in Acidic Media, Chem. Sci., 2013, 5(2), 631–638, 10.1039/C3SC52411F.
- T. Fuchs, V. Briega-Martos, J. Drnec, N. Stubb, I. Martens, F. Calle-Vallejo, D. A. Harrington, S. Cherevko and O. M. Magnussen, Anodic and Cathodic Platinum Dissolution Processes Involve Different Oxide Species, Angew. Chem., Int. Ed., 2023, 62(34), e202304293, DOI:10.1002/anie.202304293.
- X. Deng, F. Galli and M. T. M. Koper, In Situ Electrochemical AFM Imaging of a Pt Electrode in Sulfuric Acid under Potential Cycling Conditions, J. Am. Chem. Soc., 2018, 140(41), 13285–13291, DOI:10.1021/jacs.8b07452.
- N. Furuya, M. Ichinose and M. Shibata, Structural Changes at the Pt(100) Surface with a Great Number of Potential Cycles, J. Electroanal. Chem., 1999, 460(1), 251–253, DOI:10.1016/S0022-0728(98)00331-3.
- N. Arulmozhi, D. Esau, R. P. Lamsal, D. Beauchemin and G. Jerkiewicz, Structural Transformation of Monocrystalline Platinum Electrodes upon Electro-Oxidation and Electro-Dissolution, ACS Catal., 2018, 8(7), 6426–6439, DOI:10.1021/acscatal.8b00319.
- L. Zhang, L. Wang, C. M. B. Holt, T. Navessin, K. Malek, M. H. Eikerling and D. Mitlin, Oxygen Reduction Reaction Activity and Electrochemical Stability of Thin-Film Bilayer Systems of Platinum on Niobium Oxide, J. Phys. Chem. C, 2010, 114(39), 16463–16474, DOI:10.1021/jp104306j.
- S. Li, M. Dong, J. Yang, X. Cheng, X. Shen, S. Liu, Z.-Q. Wang, X.-Q. Gong, H. Liu and B. Han, Selective Hydrogenation of 5-(Hydroxymethyl)Furfural to 5-Methylfurfural over Single Atomic Metals Anchored on Nb2O5, Nat. Commun., 2021, 12(1), 584, DOI:10.1038/s41467-020-20878-7.
- H. V. Thang, G. Pacchioni, L. DeRita and P. Christopher, Nature of Stable Single Atom Pt Catalysts Dispersed on Anatase TiO2, J. Catal., 2018, 367, 104–114, DOI:10.1016/j.jcat.2018.08.025.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Single-Atom Catalysts Based on the Metal–Oxide Interaction, Chem. Rev., 2020, 120(21), 11986–12043, DOI:10.1021/acs.chemrev.0c00797.
- R. K. Singh, R. Devivaraprasad, T. Kar, A. Chakraborty and M. Neergat, Electrochemical Impedance Spectroscopy of Oxygen Reduction Reaction(ORR) in a Rotating Disk Electrode Configuration: Effect of Ionomer Content and Carbon-Support, J. Electrochem. Soc., 2015, 162(6), F489, DOI:10.1149/2.0141506jes.
- N. Hodnik, G. Dehm and K. J. J. Mayrhofer, Importance and Challenges of Electrochemical in Situ Liquid Cell Electron Microscopy for Energy Conversion Research, Acc. Chem. Res., 2016, 49(9), 2015–2022, DOI:10.1021/acs.accounts.6b00330.
- R. Yang, L. Mei, Y. Fan, Q. Zhang, H.-G. Liao, J. Yang, J. Li and Z. Zeng, Fabrication of Liquid Cell for in situ Transmission Electron Microscopy of Electrochemical Processes, Nat. Protoc., 2023, 18, 555–578, DOI:10.1038/s41596-022-00762-y.
- D. Mozhayeva and C. Engelhard, A Critical Review of Single Particle Inductively Coupled Plasma Mass Spectrometry – A Step Towards an Ideal Method for Nanomaterial Characterization, J. Anal. At. Spectrom., 2020, 35, 1740–1783, 10.1039/C9JA00206E.
- F. Shi, P. Tieu, H. Hu, J. Peng, W. Zhang, F. Li, P. Tao, C. Song, W. Shang, T. Deng, W. Gao, X. Pan and J. Wu, Direct in-situ Imaging of Electrochemical Corrosion of Pd-Pt Core-Shell Electrocatalysts, Nat. Commun., 2024, 15, 5084, DOI:10.1038/s41467-024-49434-3.
- M. Mansor, H. Alarcon, J. Xu, J. F. Ranville and M. D. Montaño, Simultaneous Insight into Dissolution and Aggregation of Metal Sulfide Nanoparticles through Single-Particle Inductively Coupled Plasma Mass Spectrometry, ACS Earth Space Chem., 2022, 6(3), 541–550, DOI:10.1021/acsearthspacechem.1c00368.
- W. Kern and D. Puotinen, RCA Rev., 1970, 31, 187 CAS.
- W. A. Syverson, M. J. Fleming and P. J. Schubring, ECS Symposium Proceedings, Second International Symposium on Cleaning Technology in Semiconductor Device Manufacturing, ed. J. Ruzyllo and R. E. Novak, The Electrochemical Society, Pennington, NJ, 1992, vol. 92–12, p. 10 Search PubMed.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stäb, P. M. Urban, D. M. Kolb and R. J. Behm, Characterization of High-Surface-Area Electrocatalysts Using a Rotating Disk Electrode Configuration, J. Electrochem. Soc., 1998, 145(7), 2354, DOI:10.1149/1.1838642.
- T. Biegler, D. A. J. Rand and R. Woods, Limiting Oxygen Coverage on Platinized Platinum; Relevance to Determination of Real Platinum Area by Hydrogen Adsorption, J. Electroanal. Chem. Interfacial Electrochem., 1971, 29(2), 269–277, DOI:10.1016/S0022-0728(71)80089-X.
Footnote |
| † Electronic supplementary information (ESI) available: Additional analyses of the niobium oxide coated electrocatalysts prepared herein such as their morphologies and composition as observed by scanning electron microscopy, transmission electron microscopy, conductive atomic force microscopy, and X-ray photoelectron spectroscopy techniques, their performance towards the oxygen reduction reaction and their analysis by electrochemical impedance spectroscopy (PDF). See DOI: https://doi.org/10.1039/d4lf00211c |
| This journal is © The Royal Society of Chemistry 2024 |