
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc adeninate metal–organic framework-coated optical fibers for enhanced luminescence-based detection of rare earth elements†
Scott E.
Crawford
 *a,
Ward A.
Burgess
ab,
Ki-Joong
Kim
ab,
John P.
Baltrus
a and
Nathan A.
Diemler
ab
*a,
Ward A.
Burgess
ab,
Ki-Joong
Kim
ab,
John P.
Baltrus
a and
Nathan A.
Diemler
ab
aNational Energy Technology Laboratory, 626 Cochran Mill Road, Pittsburgh, Pennsylvania 15236, USA. E-mail: Scott.Crawford@netl.doe.gov
bNETL Support Contractor, 626 Cochran Mill Road, Pittsburgh, Pennsylvania 15236, USA
First published on 7th March 2024
Abstract
Luminescent sensing platforms for metal ions show significant promise due to their low cost, portability, and ease-of-use. Here, we demonstrate a portable system for monitoring visible light-emitting rare earth elements by immobilizing a zinc adeninate metal–organic framework using a sol–gel method onto a fiber optic tip, which is then integrated with a compact lab-built spectrometer. The sensor is capable of sensitizing emission from terbium, europium, dysprosium and samarium down to part-per-billion concentrations. Unlike conventional solution-based luminescent approaches, immobilization of the sensing material provides a straightforward method for removing solvent molecules after exposure to rare earth element-containing solutions, thus reducing vibrational quenching and enhancing signal while also facilitating recovery, regeneration, and reuse of the sensing material over at least 10 sensing cycles. When the sensor system is applied to a simulated rare earth element process stream, good qualitative agreement with results obtained by inductively-coupled plasma mass spectrometry is exhibited. The sensor system is also capable of detecting sub-part-per-million concentrations of Tb and Eu in a spiked acid mine drainage sample. This work therefore demonstrates immobilization of luminescent sensing material films on fiber optics as a promising strategy for improving metal ion detection in complex environments.
Introduction
Rare earth elements (REEs) are critical to emerging clean energy and defense technologies, but their supply chain is threatened by environmental and geopolitical factors.1,2 Alternative REE-rich resources such as coal utilization byproducts are a promising source for sustainably meeting increasing REE demand while providing a more robust domestic supply chain.3–6 However, slow and expensive metals characterization techniques such as inductively-coupled plasma mass spectrometry (ICP-MS), which are required for prospecting and process monitoring during REE production, present a significant barrier to economic viability.7,8 Luminescence-based sensors offer an attractive alternative due to their low instrumentation costs, ease-of-use, high sensitivity, and portability, facilitating transport into the field and between labs.7–11 Moreover, REEs are a particularly suitable sensing target because, unlike other metals, several REEs are inherently luminescent: REE emission bands are narrow, element distinct and well-resolved from one another, enabling multiple metals to be detected and distinguished simultaneously.2,12 However, the optical transitions that give rise to these emission bands are parity forbidden, rendering direct excitation inefficient.12 Thus, a sensing material must be used that is capable of transferring excited state energy to the REE, which in turn induces REE emission.13–15 A range of high-performance sensing materials have recently been developed for the luminescence-based detection of REEs.2Anionic zinc adeninate metal–organic frameworks, a type of biological metal–organic framework (BioMOF), are capable of detecting and distinguishing part-per-billion levels of terbium, europium, dysprosium, samarium, ytterbium, and neodymium, and this response is observed even in complex matrices such as spiked acid mine drainage samples.14,15 Multiple portable instruments have been developed specifically for rare earth element detection, and the BioMOF sensing material has successfully been integrated with several of these platforms.16,17 However, in these cases the sensing material is dispersed in solution during sensing measurements and is not recovered for reuse. Moreover, the sensing signal in solution is weakened by solvent molecules, which are known to significantly quench REE emission.18 Solution-based luminescent sensing also makes real-time monitoring a challenge because new sensing material must constantly be introduced. Alternatively, immobilization of the sensing material onto an optical fiber presents opportunities for real-time monitoring while facilitating material recovery and solvent removal;19 this approach has been recently applied using metal–organic frameworks for the optical detection of metal ions such as copper(II)20 chromium(VI).21
Here, a sol–gel method22 is used to functionalize an optical fiber tip with a BioMOF powder, and this tip is integrated with a patented portable spectroscopic system23 for REE detection in aqueous solutions. Part-per-billion concentrations of visible-light emitting REEs (Tb, Eu, Dy, and Sm) are detected in water, with significant signal enhancement upon drying the MOF-coated tip following REE exposure. Moreover, immobilization of the sensing material facilitates regeneration and reuse; the sensor may be exposed to 0.3 M hydrochloric acid to remove the absorbed REEs and their associated characteristic luminescent signal, and then the sensor can be reused in another REE-containing sample. The sensor is successfully applied to solutions from a simulated Tb and Eu extraction process as well as a spiked acid mine drainage sample, showing the potential to deploy the sensing system under environmentally or industrially relevant conditions. Critically, the method presented here should be compatible with a range of solid-state REE sensitizers.
Experimental
Materials
Reagent grades of zinc nitrate hexahydrate, europium(III) chloride hexahydrate, terbium(III) nitrate hexahydrate, dysprosium(III) chloride hexahydrate, samarium(III) nitrate hexahydrate, ethylenediaminetetraacetic acid (EDTA) disodium salt, strontium acetate, calcium chloride, adenine, nitric acid, hydrochloric acid, 1,3,5′-benzenetricarboxylate (BTC), and dimethylformamide (DMF) were purchased from Millipore Sigma (St. Louis, MO). Tetraethyl orthosilicate (TEOS, 98%), aluminum(III) nitrate nonahydrate (99+%), and iron(III) nitrate nonahydrate were purchased from Acros Organics (Geel, Belgium). Methanol (certified ACS grade), sodium sulfate (anhydrous) and magnesium chloride hexahydrate were purchased from Fisher Scientific (Waltham, MA). De-ionized water (purity of 18.2 MΩ cm, Barnstead EASYpure LF system) was used for all syntheses and measurements (typical pH: ∼5.8).Synthesis of the BTC-1 BioMOF sensor
The BioMOF sensor (BTC-1) was synthesized according to an existing literature protocol.24 Briefly, 5 mL of a 0.05 M adenine solution in DMF was combined with 5 mL of a 0.05 M zinc acetate dihydrate solution in DMF (dissolved by heating and stirring at 130 °C followed by cooling). This was followed by the addition of 2.5 mL of a 0.2 M BTC solution in DMF, and 1 mL of 1 M nitric acid in DMF. The solution was mixed briefly by sonication, placed in a sealed, Teflon-lined autoclave and heated at 130 °C for 24 hours. After the synthesis the autoclave was cooled for 1 hour on a metal plate. The MOF powders were then washed three times with DMF and dried using nitrogen gas and gentle heating at 45 °C.Functionalization of the BioMOF on a fiber optic tip
A ∼5 inch piece of UV-compatible fiber optic cable (FG910UEC, Thorlabs) was carefully cleaved. A few cm of the polymer coating was removed on both sides. One side was first treated for 10 minutes by placing the tip into a vial of 1 M hydrochloric acid. Meanwhile, in a 2 mL vial, 220 μL of TEOS and 180 μL of methanol were combined. After 10 minutes, 40 μL of 1 M hydrochloric acid was added, and this mixture sat for at least an additional ten minutes and was used or discarded within a four day time period or when solid formation was observed. The optical fiber tip was then placed in the TEOS solution for 15 minutes, was then quickly and directly pressed into BioMOF powder to form a film on the tip, and was held 2 inches away from a heating gun set to 700 °C where it was annealed for 30 seconds. The tip was rinsed gently with water to remove excess and poorly attached BioMOF, followed by drying under nitrogen.X-ray diffraction
XRD of the BTC-1 powder was collected using a Malvern Panalytical Empyrean X-ray diffractometer using Cu kα radiation (0.1542 nm) operating at 45 kV and 40 mA. The sample was scanned from a 2 theta range of 10° to 40°.Scanning Electron microscopy
SEM characterization was performed using a FEI Quanta 600 F field emission scanning electron microscope operated at 20 kV at a 10 mm working distance.Design of a Portable Fiber Optic Probe
The patented portable fiber optic probe has been described elsewhere.9 The custom-built system uses a mounted 275 nm light-emitting diode (LED, Thorlabs, M275L4, 45 mW) as the light source, and the UV light is focused using a Thorlabs LA4025 lens through a 240–395 nm bandpass filter (Thorlabs FGUV5M). This light then enters into the 9-fiber arm of a bifurcated solarization-resistant optical fiber cable (Thorlabs BF19Y2HS02, solarization-resistant 19–200 μm 0.22NA fiber bifurcated cable bundle). The 19-fiber end of the bifurcated cable connects to a terminal clamp containing the BTC-1-coated fiber tip, which is exposed to an REE-containing sample. Emission signal from the REEs@BioMOF moves back through the 10-fiber arm of the bifurcated cable, then passes through an aspheric lens (Thorlabs, C220TME-A) to a 400 nm longpass filter (Thorlabs, FELH0400), then passes again through another aspheric lens (Thorlabs, C220TME-A) and into the OceanOptics QE Pro spectrometer. An integration time of 5 seconds was used with 5 scans being averaged during each period of data collection. The LED was controlled using a Thorlabs DC2100 LED driver. Background signal, taken with the excitation source turned off, was collected and subtracted from all spectra. All measurements were taken in a dark room. Moreover, the system was placed into a covered cardboard box and the fiber tip was placed into an amber vial inside of another box to block any ambient light. The system is sufficiently compact to fit into a drawer, box, or backpack for easy transport and storage.Luminescence measurements
During luminescent measurements, at least 5 mL of liquid sample was added to an amber vial. The optical fiber tip was placed into the REE-containing solution and monitored until the emission signal stabilized (typically within 3 minutes), using an excitation wavelength of 275 nm. After the measurement was recorded, the fiber tip was removed and dried for 1 minute using a heat gun set to 400 °C. The tip was then placed into an empty amber vial and the signal was re-recorded. For limit of detection studies, emission was first recorded in water and then after heat gun drying. Then, various concentrations of different REEs were added to the liquid, and the emission properties at each concentration level were recorded under wet and dry conditions. At least 7 data points were collected at each concentration level, and at least 7 different concentrations were measured. For each measurement, the ratio of the most intense REE peak (for example, 543 nm for Tb) versus the ratio of the MOF peak at 453 nm was taken, and the data points for each concentration level were averaged. The standard deviation of the intensity ratio at the lowest REE concentration tested (s) was used for noise estimation, and sensitivity was estimated by recording the slope (m) of a calibration curve of average signal (e.g., average intensity ratio at each REE concentration level minus the average intensity ratio of the 0 ppm REE measurement) versus the REE concentration, with the slope forced through 0. Limits of detection (LOD) were then estimated using eqn (1):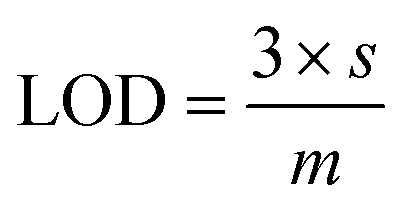 | (1) |
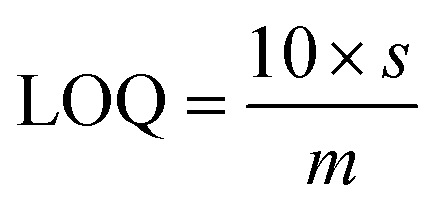 | (2) |
Analysis of a rare earth purification process stream
A simulated process source solution containing low concentrations of REEs (∼2.7 ppm La, Nd, Y, Eu, and Tb) along with high concentrations of impurities (∼4000 ppm aluminum and calcium) was prepared using metal chloride salts and was subjected to a proprietary purification process using a dilute HCl extraction solution. Over time, the REEs are selectively transferred from the process source solution into the extraction solution. Unfortunately, undesired transfer of Ca and Al impurities continues even after REE recovery is nearly complete. Sensors can be used to monitor the REE concentration in both the process source and extraction solution to determine the time for optimal recovery of REEs. Here, 1.5 mL samples of the process source and extraction solutions were collected at times varying from 0.25 to 46.6 h. A fresh MOF-coated tip was exposed to each sample for 5 minutes, followed by drying with a heat gun and fluorescence measurement. Following the measurement of each sample, the fiber tip was cleaved and fresh MOF was attached to the fiber tip prior to analysis of the next sample.Evaluation of sensor stability under stirring
The MOF-coated fiber optic tip was immersed approximately half-way into a 20 mL vial filled with 20 mL total volume of water and different REEs. The vial was equipped with a stir bar and was placed on the center of a stir plate set to 1150 rpm. The strip chart function on the Ocean Optics spectrometer was set to measure the intensity ratio of the rare earth element and the BTC-1 MOF continuously over time.Acid mine drainage studies
A sample of acid mine drainage was characterized by inductively-coupled plasma mass spectrometry, and a pH meter was used to record pH. 2.5 mL aliquots of sample were measured for each experiment, with microliter additions of 10 mM stock solutions of terbium nitrate, europium nitrate, and dysprosium nitrate added. The BioMOF-coated fiber optic tip was immersed in the sample for 5 minutes prior to measuring emission spectra.Interference studies
20 mL samples containing 50 μL of 10 mM Tb(NO3)3 and 50 μL of 1 M metal salts (Fe(NO3)3, Al(NO3)3, Zn(NO3)2, Sr(CH3COO)2, CaCl2, MgCl2, and Na2SO4) were prepared, as well as a 20 mL solution containing 50 μL of 10 mM Tb(NO3)3 and 5 μL of 100 mM EDTA and a control solution containing only terbium in water with no other metals added. For each sample, the BTC-1 coated tip was immersed for 10 minutes, and the emission was then measured in water and after drying. Additional tests were conducted on samples containing 20 mL of water with 50 μL of 1 M solutions of all 7 metals added, along with 50 μL of 10 mM Tb(NO3)3, Eu(NO3)3, or Dy(NO3)3, and once again a 10 minute exposure time was used before each measurement. At least three independent trials were conducted for each sample.Results and discussion
The detection of trace concentrations of economically critical REEs in complex streams using low-cost, portable instrumentation poses a significant analytical challenge,2 requiring innovations in the sensing material25,26 and platform.17 The narrow, element-specific emission bands of sensitized luminescent REEs makes photoluminescent-based approaches attractive, since compact and sensitive platforms may be developed for analysis, and multiple REEs may be simultaneously detected and distinguished.2 However, REE sensitization in solution is a significant challenge due to solvent-induced vibrational quenching of REE emission.27 In addition, while the ideal sensing system should facilitate regeneration and reuse of the sensing material to lower costs and minimize waste,28 recovery of the sensing material in solution can be challenging and resource-intensive. Immobilization of the sensing material onto a sensor probe provides a straightforward method for removing solvent post-REE chelation while also facilitating recovery and reuse of the sensing material.Here, a BioMOF material24 known to sensitize visible-emitting REEs in fairly complex environments with high sensitivity15 is immobilized onto a fiber optic tip using a simple sol–gel coating method with TEOS (Fig. 1). Importantly, the MOF-coated fiber tip is solarization-resistant, enabling operation with deep UV-light, and also has a high diameter (910 μm core diameter) to maximize signal. The MOF-coated tip was incorporated into a lab-built, compact fiber optic probe (Fig. 2).8,9 Briefly, the MOF-coated tip was affixed to the base of a solarization-resistant bifurcated fiber optic cable using a fiber terminator clamp; one branch of the cable ran from the fiber to a deep UV light-emitting diode (LED, 275 nm), and the other branch was connected to an Ocean Optics QE Pro spectrometer. The MOF structure was confirmed by X-ray diffraction (Fig. S1†). The lab-built fiber optic system9,10 was initially tested on aqueous solutions of four visible-emitting REEs: terbium, europium, samarium, and dysprosium in solution and after drying the fiber-optic tip following REE exposure (Scheme 1). Characteristic emission peaks were observed from each of the REEs; this emission resulted from energy transfer from the immobilized BTC-1 MOF to the encapsulated REE cations, which are absorbed by the anionic MOF. This energy transfer process, known as “sensitization,” induces element-specific, narrow REE-centered emission bands that enable REEs to be both detected and distinguished from one another.12 Importantly, drying the MOF-coated sample enhanced the REE emission intensity due to the removal of solvent molecules that typically act as vibrational quenchers of REE emission.29
 | ||
| Fig. 1 Side view of a section of an optical fiber in a fiber optic terminator functionalized on one end by a BioMOF (A) (inset: MOF-coated tip with the 275 nm LED turned on), front view of the MOF-coated fiber optic tip (B), comparison of the fiber optic tip before (C) and after (D) MOF functionalization with the 275 nm LED turned on, optical microscope image of the MOF-functionalized fiber optic tip (E), and SEM image of the MOF-coated fiber optic tip (F). | ||
 | ||
| Fig. 2 Photograph of the portable fiber optic probe. The system is sufficiently compact to fit within a drawer or box and includes a bifurcated cable connecting the BioMOF-coated tip to both the UV LED excitation source and the ocean optics QEPro spectrometer. | ||
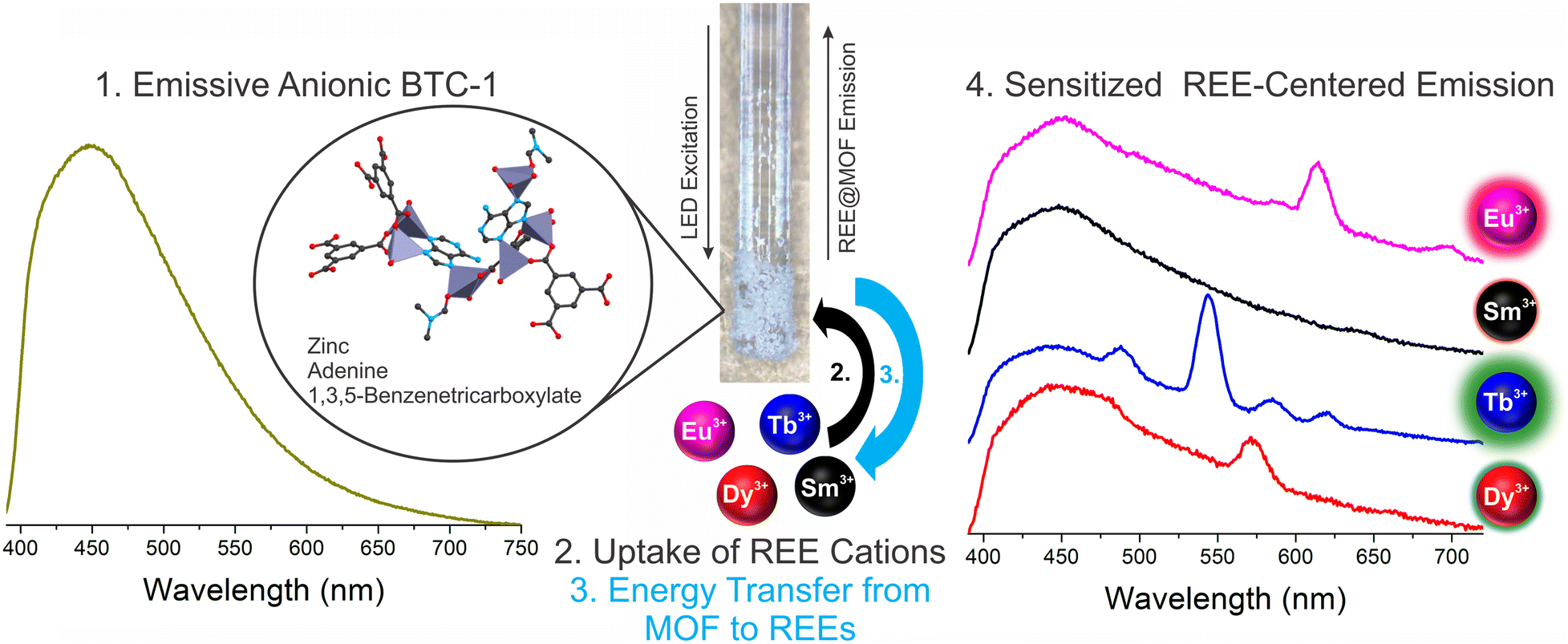 | ||
| Scheme 1 Mechanism of REE detection. A blue-emitting anionic metal–organic framework, BTC-1, is immobilized on a fiber optic cable and subjected to 275 nm excitation (1). REE cations in solution are then absorbed by BTC-1 MOF (2), and excited state energy is transferred from the MOF to the encapsulated REEs (3). This sensitization process produces narrow REE-centered emission bands that are unique to each REE. | ||
In order to better estimate the sensitivity of the sensor, limit of detection studies were conducted in which the emission was measured at different REE concentration levels both in solution and after drying the fiber optic tip; detection limits within the part-per-billion range were achieved in water for all four visible-emitting REEs (Table 1). To account for variations in emission intensity due to inconsistencies in the concentration of MOF material on the fiber optic tip from batch-to-batch, as well as potential signal reduction due to gradual leaching of the sensing material from the optical fiber tip into solution, a ratiometric approach was used,30–32 in which the ratio of emission from the primary REE emission band was divided by the intensity of the MOF peak at 453 nm (Fig. S2–S5†). A marked improvement in the detection limits were observed upon drying (Fig. 3): for example, the LOD for Tb decreased from 140 to 60 ppb, while the LOD for Eu decreased from 110 to 90 ppb (Table 1).
| REE | Solution | Dry | |
|---|---|---|---|
| Tb | LOD (ppb) | 140 ± 20 | 60 ± 10 |
| LOQ (ppb) | 450 ± 80 | 210 ± 40 | |
| Dy | LOD (ppb) | 600 ± 80 | 440 ± 20 |
| LOQ (ppb) | 2000 ± 200 | 1460 ± 60 | |
| Sm | LOD (ppb) | 780 ± 80 | 620 ± 80 |
| LOQ (ppb) | 2600 ± 300 | 2100 ± 300 | |
| Eu | LOD (ppb) | 110 ± 20 | 90 ± 10 |
| LOQ (ppb) | 380 ± 60 | 290 ± 30 | |
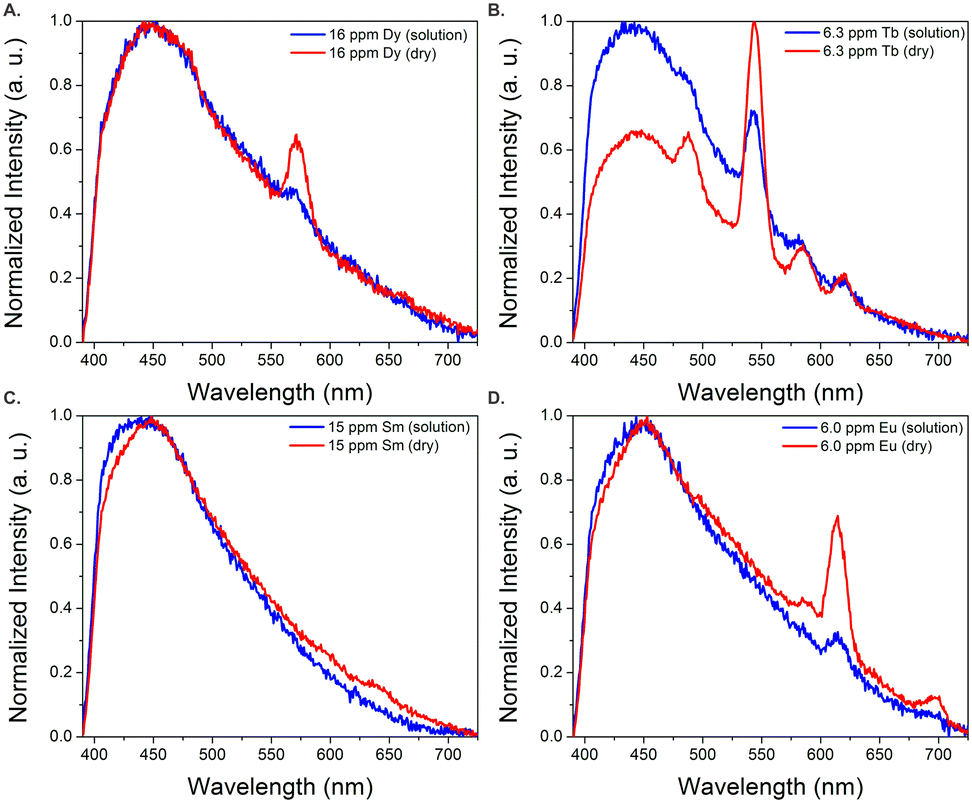 | ||
| Fig. 3 Normalized emission spectra of BTC-1 with Dy (A), Tb (B), Sm (C), and Eu (D) in solution and after drying. Drying significantly enhances the emission signal from the REEs. | ||
Increased sensitivity as a result of drying is one advantage of having the sensing material immobilized onto the optical fiber; in a solution-phase measurement, it would be much more tedious to isolate, concentrate, and dry the sensing material after exposure to rare earth elements. Importantly, the sensor is capable of detecting all four visible-emitting REEs (Tb, Eu, Dy, and Sm) when they are present simultaneously (Fig. S6†), with signal again improved for each REE upon drying. It should be noted that although characteristic Sm(III) emission is observed at low concentrations, the signal is quite weak, thus Sm(III) detection with this method would likely only be practical in simple matrices with high Sm(III) content and purity. Tb(III) and Eu(III), which produce the strongest emission, are likely the most logical sensing targets using a sensitization-based approach compared to the weaker-emitting Dy(III) and Sm(III).
Regeneration of the sensing material
A number of strategies have been developed to regenerate REE sensing materials, including the introduction of fresh solvent,33 the application of a chelation agent,34 and/or the introduction of an acid.35 Regeneration is desirable because it increases the lifetime of the sensing material, reduces chemical waste, and saves time that would be spent synthesizing additional material.2,36 Here, exposing the BioMOF-coated tip to dilute (0.3 M) hydrochloric acid for ∼1 minute resulted in the removal of the REEs and elimination of their associated emission signal, a phenomenon that has been observed for other MOF-based REE sorbents.35 However, the REE signal returned upon exposure to the REE solution. Qualitative regeneration of the REE sensing material could thus be achieved across multiple cycles by repeatedly alternating between dilute HCl and REE-containing solutions, up to at least 10 cycles (Fig. 4). It should be noted that as an alternative, the portion of the optical fiber coated by the MOF can simply be either cleaned or cleaved, followed by dipcoating in fresh BioMOF. Given the miniscule amount of BioMOF used per measurement, the cost of each measurement is roughly estimated to be a few cents based on the price of MOF precursors (Table S1†) if the fiber is cleaned, or tens of cents if 1 cm of fiber is cleaved.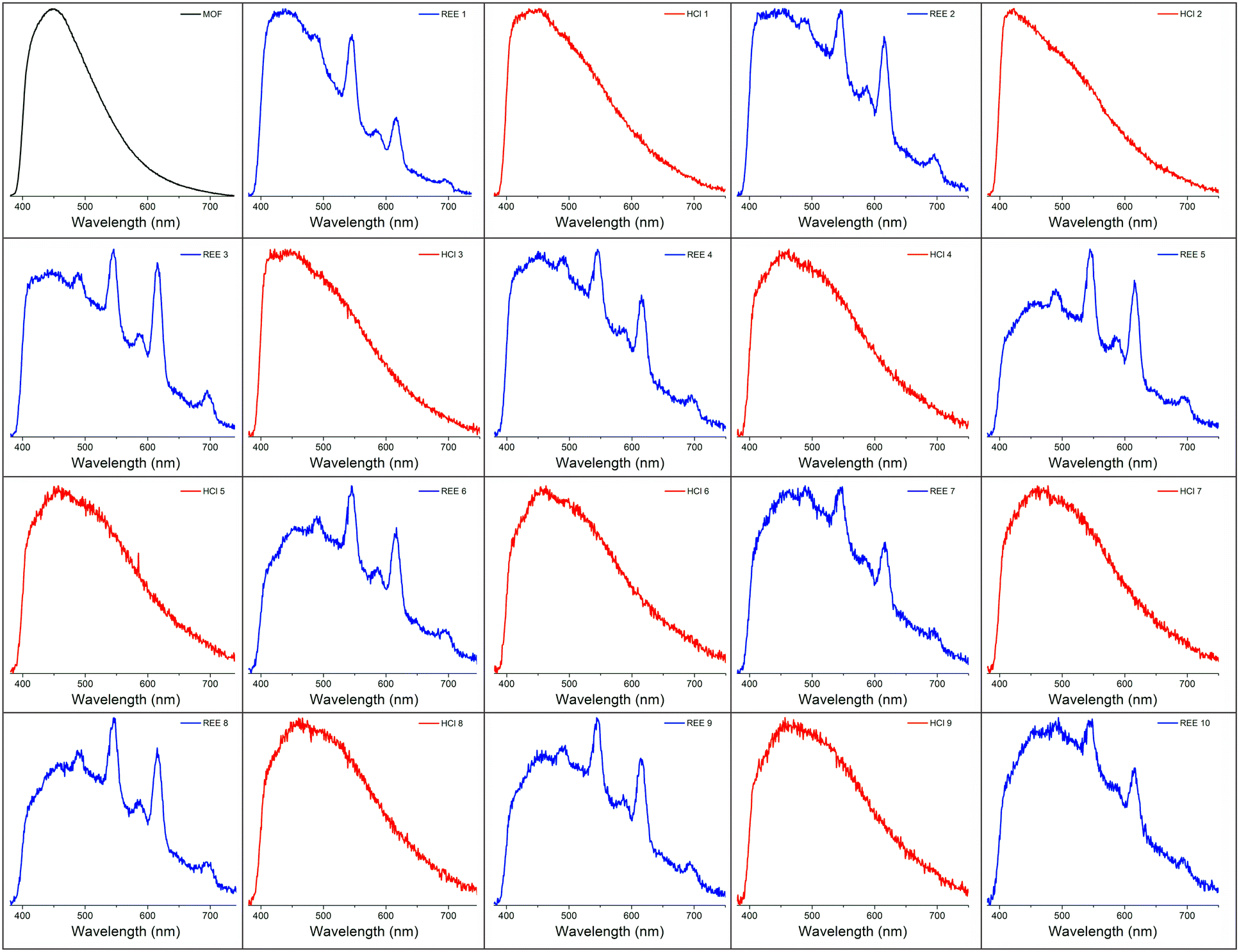 | ||
| Fig. 4 Demonstration of qualitative reversible sensing of ∼6 ppm terbium and europium across 10 sensing cycles; 0.3 M hydrochloric acid is used in between each measurement to restore the original emission signal of the BTC-1 MOF. | ||
Deployment in a simulated process stream
A crucial application area for REE sensor technologies is in process monitoring, where the efficiency and effectiveness of REE extraction steps can be analyzed in real time. A simulated process source solution containing ∼3 part-per-million concentrations of terbium and europium with significantly higher (∼4000 ppm) aluminum and calcium concentrations was subjected to a purification process in which REEs are selectively transferred over time into an HCl-based extraction solution. Optimizing the time for transfer into the extraction solution is critical; after all of the REEs have been transferred to the extraction solution, impurity metals such as calcium and aluminum still continue to be transferred, decreasing the purity of the REE product. Monitoring REE signal at different time points can therefore provide valuable information for optimizing the processing time used to maximize the purity of the extracted REEs. Representative spectra of the dried BioMOF-coated tip taken after exposure to extraction and process source solutions at different time points are included in the supporting information along with corresponding ICP-MS trends for Tb(III) and Eu(III) content in both the process source and extraction solution over time (Fig. S7–S8†).Maximum REE signal was achieved after ∼20 hours in the extraction solution, and, for clarity, Fig. 5 shows representative spectra of the process source and extraction solutions during the initial stages of the experiment and after reaching the optimum REE signal for the extraction solution at ∼27 hours. These experiments follow the expected trend: weak signal from Tb(III) and Eu(III) are observed in the process source solution at reaction times of less than 5 hours, with little to no signal observed in the extraction solution. By the 20 hour mark, signal from Tb(III) and Eu(III) are evident in the extraction solution but not in the initial process source solution. Critically, the observed trends over time in both the process source and extraction solutions (Fig. S9†) qualitatively match results obtained using ICP-MS, where Tb and Eu are fully recovered in the extraction solution after ∼20 hours. ICP-MS data taken at different time points indicates that low (∼3 ppm) concentrations of Tb and Eu are detected even in the presence of significantly higher concentrations of potentially interfering ions such as aluminum and calcium, along with equivalent concentrations of non-emissive or near-infrared emitting Y, La, and Nd (Table S2†). Moreover, both the process source and extraction solution are highly acidic, with pH values of 0.59 and −0.12, respectively. The ability of the sensor to at least qualitatively detect Tb(III) and Eu(III) in streams with low pH and/or high concentrations of competing ions undergoing a real extraction process is an encouraging step towards deployment in real-world applications. There are, of course, still limitations to be overcome with this sensing platform for process monitoring: while the qualitative trends generally follow ICP-MS results, weak REE signal and batch-to-batch variations in signal pose challenges, and these difficulties are evident in the standard errors of Fig. S7–S9.† Real samples, containing other potentially interfering ions, will likely be far more difficult to analyze than a simulated stream, and these barriers must be overcome before practical deployment.
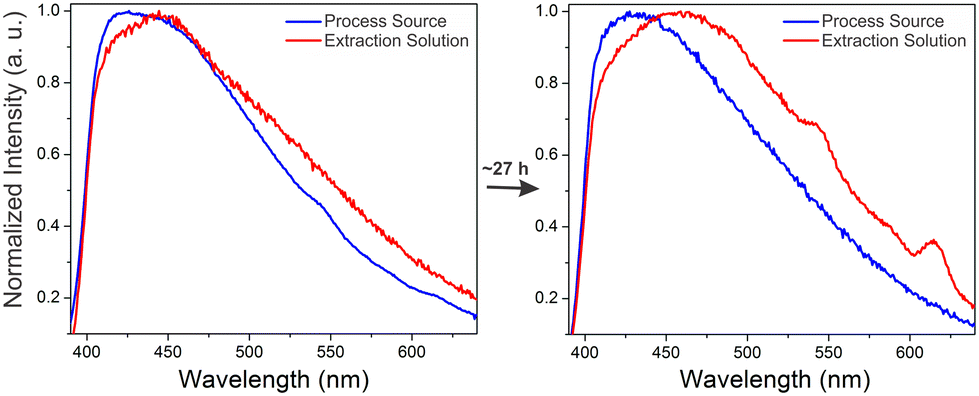 | ||
| Fig. 5 Normalized emission spectra of the dry immobilized BTC-1 MOF after incubation in the process source and extraction solution at the beginning of the experiment and after ∼1610 minutes. Initially, weak signal from terbium (545 nm) and europium (617 nm) are observed only in the process source, with no signal in the extraction solution. After ∼27 hours, stronger Tb and Eu signals are observed in the extraction solution and no signal is observed in the process source, indicating that the REEs have been transferred from the process source to the extraction solution. Additional time points are included in Fig. S7–S9, and Table S2† includes ICP-MS characterization at different time points during the experiment. | ||
Analysis of spiked acid mine drainage samples
To further probe the ability of the sensor to function in complex streams, additional tests were conducted in an acid mine drainage sample. The acid mine drainage sample had a pH of 3.3, with 59 ppm Ca, 10 ppm Al, 15 ppm Na, 29 ppm Mg, and 1.3 ppm Zn, among other metals (Table S3†). While the natural Tb(III), Eu(III), and Dy(III) concentrations (∼1 ppb for each) were too low to be detected, sub-ppm spikes of Tb(III) (Fig. S10†), Dy(III) (Fig. S11†), and Eu(III) (Fig. S12†) could be discerned (Fig. 6), particularly after drying. Such experiments indicate that the sensing system is sufficiently selective to withstand acidic conditions as well as moderately high concentrations of potentially competing metal ions, such as aluminum, calcium, iron, sodium, and others.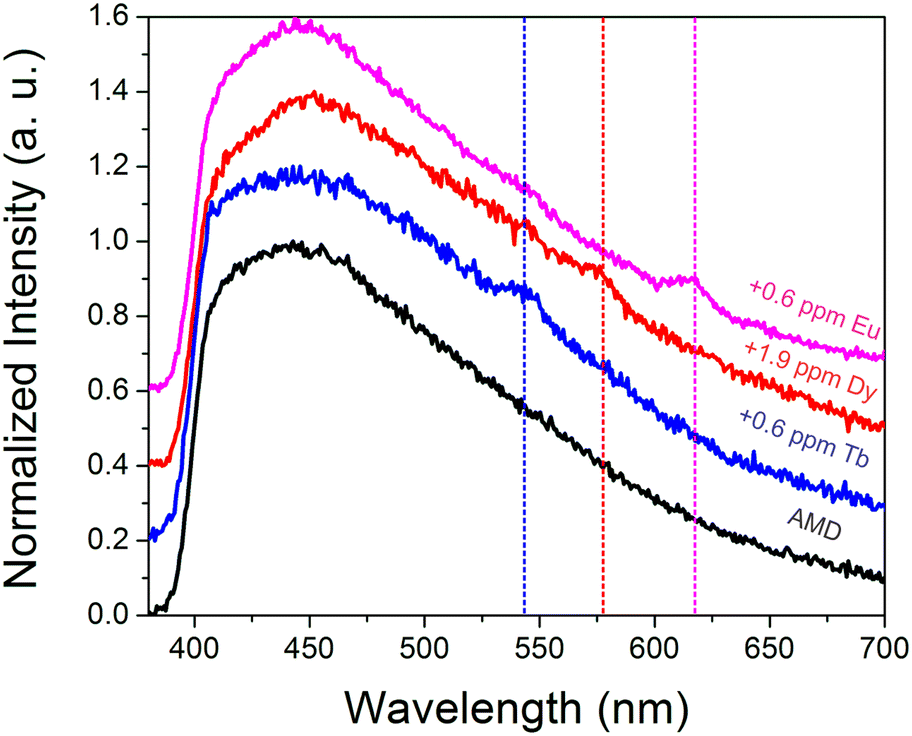 | ||
| Fig. 6 Normalized emission spectra (offset for clarity) of the dried BioMOF sensing material after exposure to acid mine drainage (black), acid mine drainage with a 0.6 ppm spike of terbium (blue) and europium (pink), and with a 1.9 ppm spike of dysprosium (red). Characteristic emission peaks, guided by dotted lines, are easily discerned for each spiked sample. | ||
To gain additional insights into the impact that common components of acid mine drainage may have on the emission signal from REEs, additional experiments were conducted in which Tb(III) signal was measured in 100× higher concentrations of some of the most abundant potentially competing species in coal streams (e.g. Fe(III), Al(III), Ca(II), Sr(II), Na(I), Zn(II),and Mg(II)), both in solution and after drying (Fig. S13†). It should be noted that the metal salts tested contained different anions, including sulfate, acetate, nitrate, and chlorides, which may also be encountered in environmental streams. A widely-used chelator, ethylenediaminetetraacetate (EDTA) was also evaluated to determine how chelating agents added during processing may impact REE sensing. Some quenching (relative to a control in which no interfering metals or chelators were added) was generally observed in solution with the introduction of interferants; these effects were far less pronounced upon drying, however. The most drastic quenching was observed when iron was added, which we have previously reported for this system;15 both iron and EDTA completely quenched REE signal in solution. However, characteristic Tb emission could still be easily distinguished upon drying. Interestingly, the introduction of Al(III) enhanced Tb(III) signal upon drying relative to the control; similar enhancements have been reported for Eu(III)@MOF systems in which silver ions were introduced,37 and this metal-enhanced emission mechanism could potentially enable the design of REE sensors with improved sensitivity. Given the complex interactions between REEs and the added metals, additional experiments were conducted in which 100:1 ratios of all 7 interfering metals were added simultaneously to Tb(III), Eu(III), and Dy(III) solutions (Fig. S14†). Very weak Dy(III) signal could only be detected upon drying, while characteristic Eu(III) and Tb(III) signal could be discerned in solution, with significant enhancement upon solvent removal. These experiments reveal that REE detection is still feasible even in complex, high ionic strength environments. However, the sensor performance will vary significantly depending upon the metal composition of the stream being analyzed and will be in particular less effective in iron-rich streams as well as in systems in which chelating molecules have been introduced.
While the process stream and sensitivity experiments presented here were obtained under stagnant conditions, the sensing material coating on the optical fiber is sufficiently robust to withstand stirring at 1150 rpm for times exceeding 1 hours (Fig. S15†) without any decrease in signal, an important consideration when monitoring processes with flowing liquids. The sensor is also capable of detecting spiked REE in real-time; here, the sensor provided a readout of the intensity ratio of Tb to that of the MOF (543 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :453 nm) as a function of time, and varying concentrations of Tb(III) were added at different time points. Sharp increases in intensity corresponding to these additions could be observed following REE addition, which plateaued after several minutes (Fig. S16†). Similar results were also obtained for Eu(III) (Fig. S17†). It should be noted that, while these results are promising indicators that the sensing material may be deployed for at least hour long intervals, the original study on BTC-1 reported that the MOF lost crystallinity upon soaking in water for 24 hours, suggesting that more water-stable sensitizers are needed for longer-term monitoring.24
:453 nm) as a function of time, and varying concentrations of Tb(III) were added at different time points. Sharp increases in intensity corresponding to these additions could be observed following REE addition, which plateaued after several minutes (Fig. S16†). Similar results were also obtained for Eu(III) (Fig. S17†). It should be noted that, while these results are promising indicators that the sensing material may be deployed for at least hour long intervals, the original study on BTC-1 reported that the MOF lost crystallinity upon soaking in water for 24 hours, suggesting that more water-stable sensitizers are needed for longer-term monitoring.24
Comparison to other sensing methods for REEs
ICP-MS and other atomic emission techniques offer extraordinary sensitivity (down to part-per-trillion concentration levels) as well as the ability to simultaneously characterize multiple metals.38 Nonetheless, ICP-MS and related techniques have several drawbacks, including high equipment and operating costs, interference from spectral overlap, and a lack of portability that prevents field deployment.2,39 Handheld x-ray fluorescence spectrometers (XRF)40 and laser-induced breakdown spectrometers (LIBS)41 offer portability and lower costs, however these instruments have typically been limited to solid state samples with sensitivities in the tens or hundreds of part-per-million range.42–44 For REE extraction and production, detection limits in the part-per-billion range or better are typically required; thus, electrochemical and optical platforms are promising alternatives that offer both high sensitivity and potential for portability.45 A notable disadvantage of optical and electrochemical methods is that they typically use materials that can only sensitize and distinguish individual or a limited number of REEs, whereas ICP-MS can detect and distinguish all REEs.A direct comparison of the sensing performance (sensitivity, selectivity, response time, range, etc.) of the fiber optic sensing platform presented here versus other optical and electrochemical REE sensors is challenging due to the large number of sensing materials that have been reported.2,45 Specific advantages that the BioMOF sensing material may offer relative to other reported REE-responsive materials is its ease-of-synthesis (using only commercially available materials and a simple 24 hour solvothermal reaction), its ability to detect and distinguish multiple REEs simultaneously with high sensitivity, and capacity to function in low pH (∼0) aqueous environments.14,15
The BioMOF-coated fiber optic sensor offers capabilities that may complement other high-performance REE sensors that have been reported; for example, Cotruvo's group has developed an extraordinarily selective and sensitive sensor capable of directly detecting Tb(III) in acid mine drainage.25 However, this sensor has not been extended to other REEs such as Eu, and has not been used in more acidic conditions. Thus, the sensing platform reported here may be less suitable for acid mine drainage, but better suited for harsher conditions such as coal fly ash leachate. Pan and co-workers developed a highly sensitive MOF-based luminescent sensor for Tb(III) and Dy(III) (with sub-ppb detection levels for Tb(III)), however this material was evaluated in DMF and therefore may be ideal for characterizing REEs extracted into organic solvents but less suitable for deployment in aqueous leachates.46 In theory, any of these sensing materials could be integrated with the fiber optic sensing platform presented in this work, which would facilitate solvent removal to improve signal as well as regeneration of the sensing material for use across multiple cycles. One can also envision incorporating different ion-selective sensing materials onto individual detachable optical fiber tips, which could allow multiple elements to be quickly detected by swapping out the different ion-responsive tips. Thus, there is potential for optical sensors such as the system presented in this work to compete with ICP-MS as a less expensive and portable alternative when information on only a few metals are needed, and the system may also complement ICP-MS as a rapid screening tool for specific metals prior to full characterization of all metals.
Conclusions
Simple, sensitive, and compact sensors for REEs are critical for REE prospecting and process monitoring. Fiber optic sensing platforms for high value metals offer many attractive advantages, including low cost, portability, and ease-of-use. Here, we demonstrate the sensitive detection of part-per-billion concentrations of rare earth elements in complex environments by coating the sensing material onto a detachable fiber optic tip that can be cut to any desired length to suit specific application requirements. The BioMOF-coated tip is attached to a compact, inexpensive optical setup comprised of a deep UV LED excitation source, bifurcated cable, and spectrometer. This set-up facilitates transport of the system between processing facilities or to the field for metals prospecting. Immobilization of the sensing material (as opposed to dispersing the material into solution) eases solvent removal, which significantly improves emission signal during REE sensitization, enables regeneration of the sensing material for multiple uses, and is suitable for real-time monitoring of REE concentrations. As a proof-of-concept, the sensor is applied for qualitatively monitoring the extraction of ∼3 ppm terbium and europium from a complex process source solution into an acidic extraction solution, an important process for REE purification. Moreover, the sensor can detect sub-ppm spikes of Tb, Eu, and Dy in a real acid mine drainage matrix. Beyond the proof-of-concept demonstrations in this work, the optical set-up described here should be amenable to other solid-state REE sensing materials as well; future innovations in the development of REE sensors will further improve the sensitivity and selectivity of the probe. Notably, other synthetic innovations, such as decreases in the size of the MOF sensing material,47 should also improve performance by providing a higher quality film coating on the optical fiber. Alternative optical configurations may be envisioned that could further reduce the size of the sensing platform; for example, a shorter bifurcated fiber optic cable could be used, or the bifurcated cable could be replaced entirely using a beam splitter.48 Finally, the development of low-cost, portable near-infrared fluorescence detectors would further expand the practicality of this technology by enabling the detection of Yb and Nd, two near-infrared elements that can be sensitively detected by zinc adeninate MOFs.14 Taken together, this work presents a simple, effective method for REE detection using immobilized sensing materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the United States Department of Energy, National Energy Technology Laboratory, in part, through a site support contract. Neither the United States Government nor any agency thereof, nor any of their employees, nor the support contractor, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.Notes and references
- M. De Boer and K. Lammertsma, ChemSusChem, 2013, 6, 2045–2055 CrossRef CAS PubMed.
- S. E. Crawford, P. R. Ohodnicki and J. P. Baltrus, J. Mater. Chem. C, 2020, 8, 7975–8006 RSC.
- S. Dai and R. B. Finkelman, Int. J. Coal Geol., 2018, 186, 155–164 CrossRef CAS.
- W. Franus, M. M. Wiatros-Motyka and M. Wdowin, Environ. Sci. Pollut. Res., 2015, 22, 9464–9474 CrossRef CAS PubMed.
- B. C. Hedin, R. S. Hedin, R. C. Capo and B. W. Stewart, Int. J. Coal Geol., 2020, 231, 103610 CrossRef CAS.
- B. C. Hedin, R. C. Capo, B. W. Stewart, R. S. Hedin, C. L. Lopano and M. Y. Stuckman, Int. J. Coal Geol., 2019, 208, 54–64 CrossRef CAS.
- J. A. Mattocks, J. V. Ho and J. A. Cotruvo Jr, J. Am. Chem. Soc., 2019, 141, 2857–2861 CrossRef CAS PubMed.
- J. C. Ahern, Z. L. Poole, J. Baltrus and P. R. Ohodnicki, IEEE Sens. J., 2017, 17, 2644–2648 CAS.
- S. E. Crawford, K.-J. Kim, N. Diemler and J. P. Baltrus, ACS Appl. Opt. Mater., 2023, 1, 587–597 CrossRef CAS.
- S. E. Crawford, K. J. Kim and J. P. Baltrus, J. Mater. Chem. C, 2022, 10, 16506–16516 RSC.
- F. Sainz-Gonzalo, C. Elosua, J. Fernández-Sánchez, C. Popovici, I. Fernández, F. Ortiz, F. Arregui, I. Matias and A. Fernández-Gutiérrez, Sens. Actuators, B, 2012, 173, 254–261 CrossRef CAS.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- S. E. Crawford, C. M. Andolina, D. C. Kaseman, B. H. Ryoo, A. M. Smith, K. A. Johnston and J. E. Millstone, J. Am. Chem. Soc., 2017, 139, 17767–17770 CrossRef CAS PubMed.
- S. E. Crawford, X. Y. Gan, P. C. Lemaire, J. E. Millstone, J. P. Baltrus and P. R. Ohodnicki Jr, ACS Sens., 2019, 4, 1986–1991 CrossRef CAS PubMed.
- S. E. Crawford, J. E. Ellis, P. R. Ohodnicki and J. P. Baltrus, ACS Appl. Mater. Interfaces, 2021, 13, 7268–7277 CrossRef CAS PubMed.
- H. Lan, S. Crawford, Z. Splain, T. Boyer, P. Ohodnicki, J. Baltrus, R. Zou, M. Wang and K. P. Chen, in Conference on Lasers and Electro-Optics, Optical Society of America, 2019 JW2A. 14 Search PubMed.
- Z. Wu, S. E. Crawford, M. Buric, Z. Splain and K. P. Chen, IEEE Sens. J., 2023, 23, 11574–11581 CAS.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- Y. Cai, M. Li, M. Wang, J. Li, Y.-n. Zhang and Y. Zhao, Front. Phys., 2020, 8, 598209 CrossRef.
- S. Menon, S. Dutta, N. Madaboosi and V. Sai, ACS Appl. Nano Mater., 2023 DOI:10.1021/acsanm.3c04104.
- S. Menon, S. P. Usha, H. Manoharan, P. V. N. Kishore and V. Sai, ACS Sens., 2023, 8, 684–693 CrossRef CAS PubMed.
- S. A. Shahamirifard, M. Ghaedi and S. Hajati, Sens. Actuators, B, 2018, 259, 20–29 CrossRef CAS.
- J. C. Ahern, P. R. Ohodnicki Jr, J. P. Baltrus and J. L. Poole, Luminescence based fiber optic probe for the detection of rare earth elements. US Pat., 11170986, 2021 Search PubMed.
- J. An, S. J. Geib, M.-G. Kim, S. Y. Choi and W. T. Lim, J. Porous Mater., 2015, 22, 867–875 CrossRef CAS.
- E. R. Featherston, E. J. Issertell and J. A. Cotruvo Jr, J. Am. Chem. Soc., 2021, 143, 14287–14299 CrossRef CAS PubMed.
- D. Bhowmik and U. Maitra, Chem. Sci., 2023, 14, 4901–4904 RSC.
- W.-S. Lo, W.-T. Wong and G.-L. Law, RSC Adv., 2016, 6, 74100–74109 RSC.
- R. Goswami, S. C. Mandal, B. Pathak and S. Neogi, ACS Appl. Mater. Interfaces, 2019, 11, 9042–9053 CrossRef CAS PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. Gareth Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- B. Fu, J. Chen, Y. Cao, H. Li, F. Gao, D.-Y. Guo, F. Wang and Q. Pan, Sens. Actuators, B, 2022, 369, 132261 CrossRef CAS.
- J.-F. Feng, T.-F. Liu, J. Shi, S.-Y. Gao and R. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 20854–20861 CrossRef CAS PubMed.
- X. Zheng, R. Fan, Y. Song, A. Wang, K. Xing, X. Du, P. Wang and Y. Yang, J. Mater. Chem. C, 2017, 5, 9943–9951 RSC.
- L. Chen, K. Tan, Y.-Q. Lan, S.-L. Li, K.-Z. Shao and Z.-M. Su, Chem. Commun., 2012, 48, 5919–5921 RSC.
- M. Shamsipur, M. Mohammadi, A. A. Taherpour, V. Lippolis and R. Montis, Sens. Actuators, B, 2014, 192, 378–385 CrossRef CAS.
- W. Hua, T. Zhang, M. Wang, Y. Zhu and X. Wang, Chem. Eng. J., 2019, 370, 729–741 CrossRef CAS.
- J. Ellis, S. Crawford and K.-J. Kim, Mater. Adv., 2021, 2, 6169–6196 RSC.
- J.-N. Hao and B. Yan, J. Mater. Chem. A, 2014, 2, 18018–18025 RSC.
- K. Benkhedda, H. G. Infante, E. Ivanova and F. C. Adams, J. Anal. At. Spectrom., 2001, 16, 995–1001 RSC.
- T.-S. Lum and K. S.-Y. Leung, J. Anal. At. Spectrom., 2016, 31, 1078–1088 RSC.
- R. Schramm, Phys. Sci. Rev., 2016, 1, 20160061 Search PubMed.
- T. F. Akhmetzhanov, T. A. Labutin, D. M. Korshunov, A. A. Samsonov and A. M. Popov, J. Anal. At. Spectrom., 2023, 38, 2134–2143 RSC.
- C. R. Bhatt, D. Hartzler, J. C. Jain and D. L. McIntyre, Opt. Laser Technol., 2020, 126, 106110 CrossRef CAS.
- M. Martin, R. C. Martin, H. B. Andrews, S. Allman, D. Brice, S. Martin and N. Andre, Appl. Spectrosc., 2022, 76, 937–945 CrossRef CAS PubMed.
- G. Simandl, R. Stone, S. Paradis, R. Fajber, H. Reid and K. Grattan, Miner. Deposita, 2014, 49, 999–1012 CrossRef CAS.
- D. Paderni, L. Giorgi, V. Fusi, M. Formica, G. Ambrosi and M. Micheloni, Coord. Chem. Rev., 2021, 429, 213639 CrossRef CAS.
- M. Li, G. Ren, F. Wang, Z. Li, W. Yang, D. Gu, Y. Wang, G. Zhu and Q. Pan, Inorg. Chem. Front., 2019, 6, 1129–1134 RSC.
- L. H. T. Nguyen, Y. Thi Dang, T. T. T. Nguyen, B. Q. G. Le, N. X. D. Mai, H. V. Nguyen, M.-T. Le, T. B. Phan and T. L. H. Doan, New J. Chem., 2022, 46, 6630–6635 RSC.
- B. Zhou, K. Fan, J. Guo, J. Feng, C. Yang, Y. Li, S. Shi and L. Kong, Sci. Adv., 2023, 9, eadg0218 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00001c |
| This journal is © The Royal Society of Chemistry 2024 |