
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of ceria/precious metal interfaces in catalysis
Michele
Melchionna
 *a and
Paolo
Fornasiero
*ab
*a and
Paolo
Fornasiero
*ab
aDepartment of Chemical and Pharmaceutical Sciences, Center for Energy, Environment and Transport “Giacomo Ciamician”, INSTM UdR Trieste, University of Trieste, Via Licio Giorgieri 1, 34127 Trieste, Italy. E-mail: melchionnam@units.it; pfornasiero@units.it
bICCOM-CNR, Unit of Trieste, Italy
First published on 29th November 2023
Abstract
The popularity of ceria (CeO2) supports has been increasing over the last three decades on account of the rich redox chemistry of such an oxide. The ability to act as an oxygen buffer and switch the oxidation state of Ce depending on conditions implies that the role of this oxide goes beyond the conventional function of stabilizing nanoparticles. In fact, ceria can actively participate in catalytic reactions by interacting with the supported metal, in particular precious metals, promoting various types of dynamic processes that are beneficial for catalysis. This perspective put into light such interfacial synergy, and the effects in several catalytic processes, encompassing the most traditional applications up to the most modern reaction schemes.
Introduction
Precious metals (PMs) and cerium dioxide (also known as “ceria”, CeO2) have been sharing a long relationship in the history of heterogeneous catalysis. This is explained by the successful combination of their properties that made many catalytic conversions with high efficiency feasible. Much of the initial interest, which dates back to at least six decades ago, was derived from the knowledge that some metal oxides (e.g. zirconia, thoria etc.) are excellent oxygen carriers. Such a property could be exploited to promote the catalytic activity of noble metals in important reactions, such as hydrogenation of olefins.1 Continuing evidence of some metal oxide effects led to the terminology “active supports” to define metal oxides such as CeO2, alternatively referred to as “promoters” or “non-innocent supports”. In the 1980s, the literature on the specific PM–CeO2 interaction started to appear, focusing in particular on Pt as the PM, mainly reporting the oxygen (or other gases) adsorption behaviour of Pt nanoparticles deposited on ceria.2 Other probe molecules such as H2 or CO were lately investigated in-depth, as they represent typical reactants for the most studied catalytic processes. Defining a paradigm of the adsorption behaviour in the framework of PM/CeO2 composites has been highly challenging, and not yet fully achieved, on account of the high adsorption capacity of CeO2 towards H2 and CO, which makes it difficult to discriminate between metal and metal support contributions. The initial strongest driving force for research on Pt/CeO2 was related to the needs of the automotive exhaust industry, where emission control in three-way catalysts heavily depended on the use and improvement of catalysts integrating Pt/CeO2.3 Our research group has been contributing to this topic for several years and with growing enthusiasm,4 and it became evident that the effects of ceria as a promoter for PM nanoparticles went beyond the mere stabilization and dispersion of the NP, but it involved an improved functionality of the PM/CeO2 system. Of utmost importance, the reducibility of CeO2 harnesses the release of active oxygen so that the support actively participates in the catalytic cycle.5 It has been demonstrated that for many reactions the catalytic site is located at the interface of PM NPs and CeO2,6 where synergy is maximum, and this led to synthetic efforts aiming at the maximization of the interfacial area. This perspective article illustrates some of the most meaningful work that over the years contributed to shedding light on the role of interfaces in PM NPs supported on CeO2 for various catalytic processes.Seminal works on thermal catalysis by PM/CeO2
One of our seminal studies showed that the supramolecular-inspired assembly of Pd/CeO2 nanoparticles with a precise core–shell configuration allowed almost complete methane oxidation conversions to be reached at a lower temperature (400 °C) (Fig. 1), surpassing the performance of what at that time were the state-of-the-art catalysts.7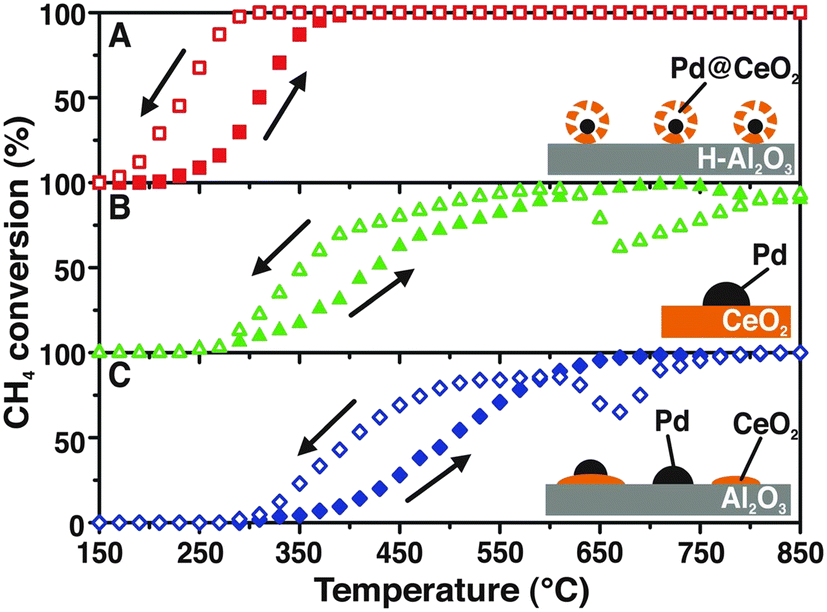 | ||
| Fig. 1 Heating and cooling during CH4 conversion for: A) Pd@CeO2/hydrophobic Al2O3 (H-Al2O3) core–shell catalyst; B) Pd/CeO2 prepared by incipient wetness impregnation, and C) Pd/CeO2/Al2O3 prepared by coimpregnation of Pd and Ce precursors. Reproduced from ref. 7 with permission from AAAS, copyright 2012. | ||
This work gives a clear demonstration of how assembling structures with a controlled hierarchy impacts to a large extent the properties of the catalytic system. In the above specific case, the core–shell hierarchy provides extra stability for the Pd NPs, avoiding their sintering at high temperature, and the activity benefits from the extended Pd–CeO2 interaction. Optimisation of the oxide–metal interaction is a key factor for maximising catalytic performance, and a number of strategies have been proposed.8
As hinted earlier, the oxygen buffer functionality of CeO2 is dependent on its defectivity, and within this framework the role of oxygen vacancies is known to be the most critical. However, in contrast with the majority of metal oxides such as TiO2, where vacancy formation is instrumental to create binding sites within the structure, in the case of ceria the results of oxygen removal are accompanied by significant electronic changes. In particular, the presence of empty f orbitals on Ce results in a localization of the excess electron that caused change in the valence state of Ce4+ to Ce3+ of the two Ce centres adjacent to the vacancy. It turns out that thorough knowledge of the oxygen vacancy formation, type and distribution is of high relevance to predict CeO2 catalytic behaviour and appropriately design PM/CeO2 catalysts. To this end, scanning tunnelling microscopy (STM) has proven to be a powerful tool for understanding the details of vacancy formation, and the evolution of the CeO2 surface and subsurface in terms of electron localization, valence states and the coordinative surrounding of the Ce cationic species.9 Based on the observed synergy between PM and CeO2 for several catalytic processes, intensive research on the exploitation of such an interaction has been proliferating over the years. The so-called “strong metal–support interaction” (SMSI) coined by Tauster et al. to describe the significant detrimental effects of the TiO2 support on the H2 and CO chemisorption properties of Pt NPs10 was reanalysed in the case of Pt/CeO2 to explain how this interaction differs and resulted in enhanced catalytic performance. Sanchez and Gazquez modelled the SMSI for fluorite-type supports such as CeO2 and compared it to that observed in rutile supports, observing a remarkably different behaviour of the supported PM NP in terms of stability and catalytic activity. In fact, they noticed that the CeO2 type of structure presents a cationic sub-lattice structure able to create a diffusion barrier for the metal, preventing its penetration into the bulk, in contrast with rutile-type structures. This feature is one of the descriptors for explaining the catalytically beneficial SMSI effect of PM/CeO2 catalysts.11 Moreover, experiments for the characterization of PM/CeO2 conducted under reducing conditions with H2 flows could be set at temperatures higher than 773 K, an upper limit typically dictated by the TiO2 support. This possibility allowed a better understanding of the nature of the metal/support interaction to be obtained, and Bernal et al. reported several detailed investigations by means of high-resolution electron microscopy (HREM) of the structural evolutions of PM/CeO2 under both high temperature reducing and oxidizing conditions. A remarkable difference is that, in contrast to PM/TiO2 systems, the high temperature H2 treatment does not cause covering of the metal microcrystal by the reduced support (Fig. 2).12
 | ||
| Fig. 2 HREM images of various CeO2-supported Pt and Rh catalysts reduced with H2 at different temperatures. Profile view images corresponding to: (A1–A2) Pt(7%)/CeO2-MS; (B1–B2) Pt(4%)/CeO2-LS; (C1–C2) Rh(2.4%)/CeO2-LS (MS and LS denotes two different ceria samples). Reproduced from ref. 12c with permission from Elsevier, copyright 1999. | ||
Inspired by Frost's junction effect theory,13 Acerbi et al. first put into relation the reducibility of ceria with the work function of various precious metals such as Au, Ag, Pd, Pt, Rh, Ru to rationalize the different SMSIs.14 However, the same authors later claimed that relationship with the d-band centres of the precious metals was a better descriptor of the SMSI as it provides a more satisfactory correlation of the ceria H2 chemisorption with the PM electronic structure.15 The SMSI has been fruitfully exploited in applications such as thermal hydrogenation reactions: in a seminal work, Trovarelli et al. described the use of transient and steady-state thermal hydrogenation of CO and CO2 by Rh/CeO2, which could reveal the effect of the metal–metal oxide support,16 while in another article the importance of the generated bulk vacancies on ceria by means of reduction at high temperatures was discussed.17 More recently, the selective hydrogenation of CO2 to CO, avoiding methanation, was achieved by tuning the chemical state of Ir species supported on CeO2, with the SMSI effect being critical for controlling efficiently the Ir chemical state.18 Hydrogenation of phenol to cyclohexanone at room temperature was achieved by using Pd/CeO2, thanks to the ability of CeO2 to chemisorb phenoxy species and facilitate attack by Pd-activated hydrogen.19
Campbell et al. introduced the term “electronic metal–support interaction” (EMSI) to better include all the complex and synergistic charge redistributions that were at the origin of improved catalytic performance of PMs and CeO2.20 Bruix et al. proposed a new type of metal–support interaction for Pt/CeO2 on the basis of photoemission measurements and DFT calculations while studying the water-gas shift reaction (WGSR). The observed catalytic activity was then related to a dramatic charge distribution occurring for the Pt adatoms on ceria, with favorable Pt → CeO2 charge transfer and CeO2 → Pt oxygen transfer. Following this electronic interaction, the adsorption of H2O on Pt becomes more favourable, and the dissociation of the O–H of water becomes a very exothermic process, enhancing H2 evolution (Fig. 3).21
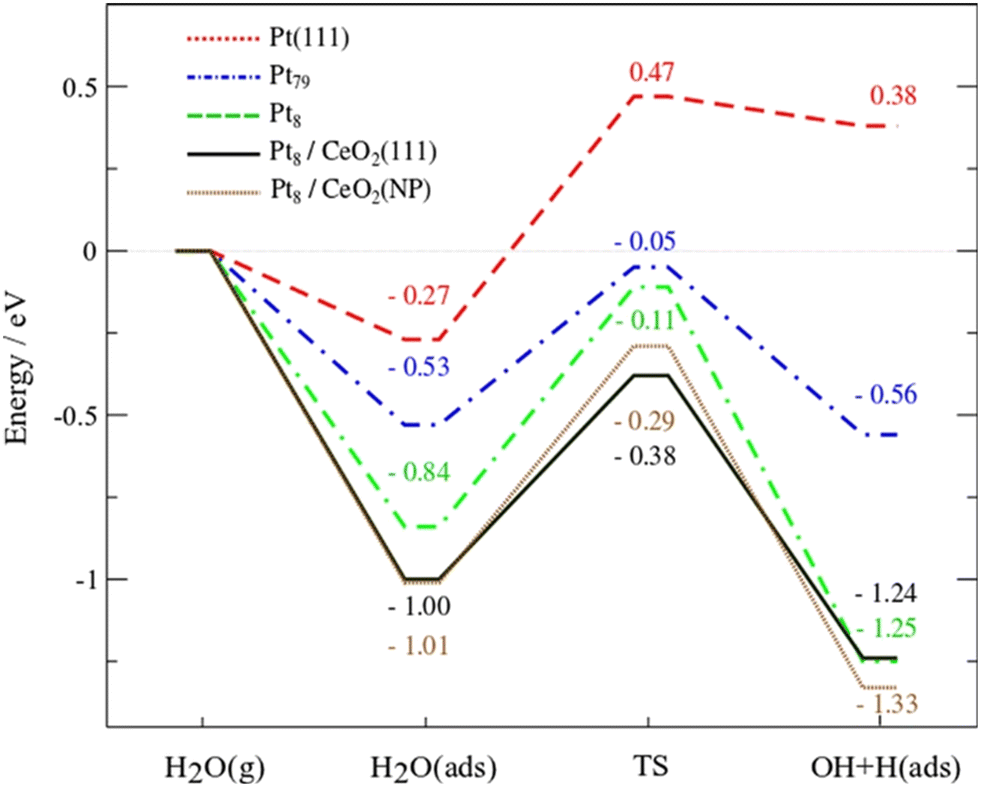 | ||
| Fig. 3 DFT-calculated energy profiles for the dissociation of water on Pt(111), Pt79 and Pt8 clusters, and Pt8 on CeO2(111) or Ce40O80 NPs. Reproduced from ref. 17 with permission from American Chemical Society, copyright 2012. | ||
Nevertheless, this type of electronic metal–support interaction is also linked to the size of the metal NP. A quantitative analysis of the charge transfer occurring between Pt and CeO2 in well-defined composites was performed by a combination of STM, XPS and DFT, unveiling that the EMSI is closely linked to the size of the Pt NPs. The maximum charge transfer occurs for Pt NPs composed of 30 to 70 atoms, while for smaller or larger nanoparticles the charge transfer is hindered either by nucleation at defect sites (smaller NPs) or directly by the support (larger NPs). The result is of relevance for the preparation of optimum PM NP sizes, but it also highlights the dependence of the synergistic effects from subtle structural differences.
Model catalysts
Given the complexity of the metal–support fundamental phenomena, it became evident that model systems were an inevitable choice for studying ceria–PM interfaces. A large variety of well-defined metal–ceria systems were prepared and investigated in relation to a few specific catalytic processes, such as CO oxidation, CO2 hydrogenation, WGSR and methane or alcohol reforming. In previous studies, the assembly of such model catalysts was based on synthetic routes (deposition of metallic Ce followed by oxidation, or oxidation of metal–Ce alloys) that could not establish a perfect control of the structural order.22 More recently, controlled growth of CeO2 nanoparticles could be achieved via epitaxy and the ceria phase structure and composition can be accurately controlled by means of post-growth thermal treatments.23 Rodriguez et al. provided a comprehensive review on the most important aspects that are involved in the design, synthesis and characterization of metal–ceria model catalysts, emphasizing the strong dependence of the catalytic activity from a large number of variables, which have to be controlled ad hoc for a specific process.24 It is interesting to note the resorting to inverse model catalysts, where the two components (PM and CeO2) switch roles for a more complete understanding of the functionality of the system and identifying optimum activity conditions. An important advantage of inverse model catalysts lies in the possibility to unravel novel interfacial effects by means of highly resolved electron-based techniques such as low energy electron microscopy (LEEM) and X-ray photoemission electron microscopy (XPEEM).25 The spatial resolution of such techniques is compromised on conventional model catalysts because of charging effects occurring on insulating metal oxides during probing, a problem that is alleviated in inverse catalysts where the conducting area is much larger as it is made of metals. This variant has mostly involved the synthesis of CeO2 nanoparticles grown on (111) facets of metal surfaces, distinguishing the cases of oxidisable, for instance Cu(111) and non-oxidisable metals, such as Au(111) (Fig. 4A).26 This distinction has been used to determine both the mode of CeO2 growth and catalytic reaction-relevant phenomena, such as oxygen spillover.27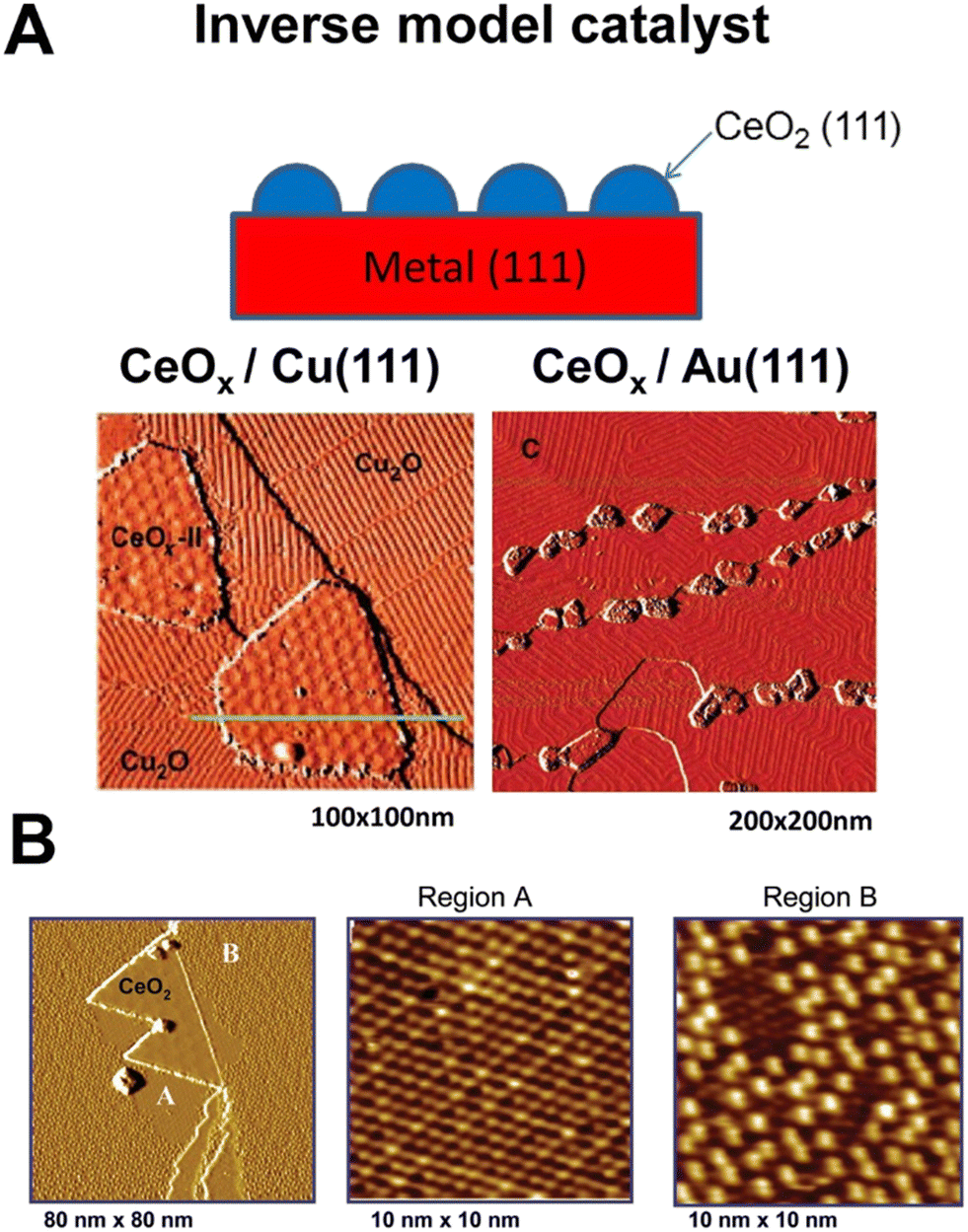 | ||
| Fig. 4 A) Top: Schematic of an inverse model catalyst; bottom: STM images for inverse CeOx/Cu2O/Cu(111) and CeOx/Au(111) model catalysts; reprinted with permission from ref. 22, copyright 2013 American Chemical Society. B) STM images of ceria promoted surface oxidation of Cu(111); the first panel shows the CeO2/Cu(111) surface exposed to 5 × 10−7 Torr O2 at 550 K for 5 min. Reproduced from ref. 24 with permission from American Chemical Society, copyright 2011. | ||
For example, in the oxidation reaction of carbon monoxide (2CO + O2 → 2CO2), the poor interaction and dissociation of O2 with respect to the Cu(111) surface is significantly improved by the presence of the CeO2 NP, which spills oxygen to Cu and allows the formation of an oxygen-deficient Cu2O1+x phase, more active for the oxidation of CO. The details of CeO2 growth and formation of the new Cu2O1+x phase could be uncovered thanks to the precise crystalline order of the inverse catalyst and the use of scanning tunnelling microscopy (STM), confirming that the ceria-promoted oxidation of Cu proceeds with the formation of both ordered Cu2O(111) and Cu2O1+x (Fig. 4B).28 Recent studies evaluated the redox phenomena occurring at the Pt/CeO2 interface in inverse model catalysts consisting of epitaxially grown CeO2 on single crystal Pt(111). Interestingly, the authors could distinguish the different sites where reduced ceria is stabilized by the metal as opposed to sites far away from the Pt.29
Model catalysts vs. bulk catalysts
Despite the atomic scale understanding that model systems offer, such knowledge cannot be straightforwardly transferred to bulk composites prepared by more conventional protocols. In those situations, a combination of a variety of characterization techniques is required to hypothesize and confirm the synergistic properties of metal–ceria. A first consideration relates to the fact that many studies incorporate a third phase in addition to ceria and metals, and in several cases such an extra component may considerably affect the overall properties of ceria and in turn its interfacial dynamics with PM NPs. As our group demonstrated, this situation occurs for example with the incorporation of carbon nanostructures (CNSs) within the nanocomposite. CNSs can often influence the reducibility of CeO2 and the Ce4+/Ce3+ ratio, implying a differential interaction with the metal nanoparticles.30 Moreover, CeO2 can be a useful stabilizing phase in Pt/CNS catalysts, as demonstrated during selective glycerol-to-glyceric acid (GLYA) catalytic oxidation by Pt/CNT/CeO2 (CNT = carbon nanotubes), where ceria improves the anti-poisoning of the Pt nanoparticle by GLYA by modulation of the Pt electronic structure (Fig. 5).31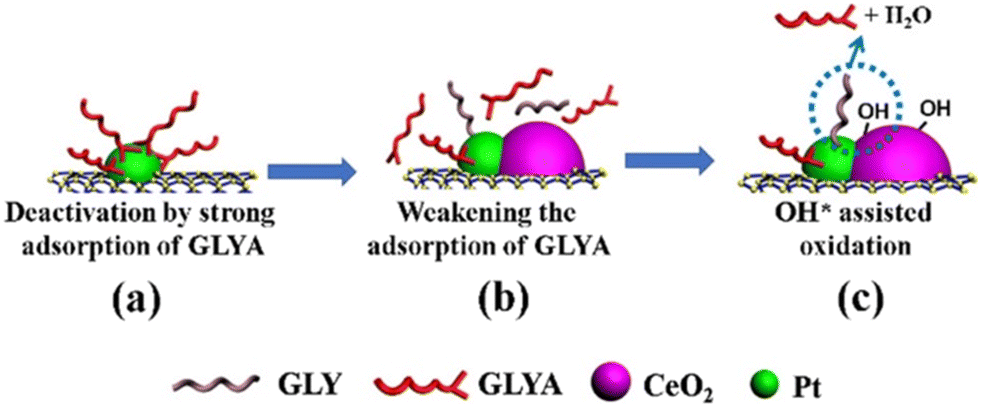 | ||
| Fig. 5 Proposed mechanisms for overcoming the deactivation of Pt/CNTs by introducing CeO2. Reproduced from ref. 27 with permission from American Chemical Society, copyright 2020. | ||
Besides, preparative routes based on conventional methods lead to bulk materials with important sample inhomogeneity and at times the presence of unexpected impurities that play a role. Unfortunately, not always all these possibilities are taken into account, which has resulted in the proliferation of articles on Pt/CeO2 catalysts where support effects and synergy are somehow taken for granted, causing misleading interpretation of results. A seminal article by Fu et al. posed critical questions on the contentious identification of the catalytically active sites in PM/CeO2 used in WGSR, CO oxidation and other oxidation; specifically whether these are invariably located at the metal–ceria interface. The investigation focused on Au/CeO2 and Pt/CeO2 catalysts for the WGSR, whereby careful analysis on the kinetics of leached and unleached composite catalysts evidenced that the WGSR catalytic activity was not due to interfacial effects. Metallic Au and Pt NPs were ineffective for the catalytic activity, which instead originated from the isolated Au or Pt ions stabilized on CeO2 through lattice substitution.32 As a matter of fact, the work represents the first example of single-atom catalysis in the WGSR, which the authors further corroborated in the following studies, highlighting the essential presence of Pt-alkali-Ox(OH)y species,33 as well as the fact that the activity is not an exclusiveness of the CeO2 support.34 An important contribution into the field of Pt-SAC/CeO2 was provided by Nie et al. who explained how ceria is able to activate atomically dispersed Pt2+ metal ions by means of active surface lattice oxygen formed during aging of the catalyst at 750 °C. Hence, the catalytic activity towards CO oxidation was greatly improved without sacrificing the ionic nature of isolated Pt2+, which is generally compromised by the very strong binding of CO molecules to the metal.35 High activities for the water-promoted CO oxidation by Pt-SAC/CeO2 were also observed by Wang et al., who found that the lattice hydroxyl species at the interface between Pt and CeO2 played a critical role in the reaction, yielding the carboxyl reaction intermediate.36
Electrocatalytic applications
The examples provided above and the discussion of the growing understanding on PM and CeO2 interfaces were rightly focused on the most deeply explored thermal catalysis. Nevertheless, researchers soon realized that the synergistic effects had potential for exploitation in other types of catalytic applications. Electrocatalysis gained attention towards the beginning of the 2000's and after that Marina et al. fabricated electrodes based on Gd-doped CeO2/Au NPs for the electrochemical oxidation of methane.37 While the work did not directly studied the interfacial phenomena occurring at the Au–ceria junction, it triggered successive investigations focused more on the understanding on the promoting role of ceria. Obvious motivations were dictated by the oxygen buffer ability of ceria, which was for instance an advantage for the electrocatalytic oxygen reduction reaction (ORR). In this framework, the facile oxygen adsorption and desorption on the ceria component was recognized to be instrumental in supplying oxygen to the metal NP.38 However, in most of the examples, PM/CeO2 is employed as fuel cell-relevant anodic electrocatalysts for the oxidation of fuels (H2, methanol, etc.). The hydrogen oxidation reaction (HOR) is one of the explored reactions where Pd/CeO2 proved to be very competitive, taking advantage of the facile Ce4+/Ce3+ redox process. The quest for synthetic strategies to maximize the interfacial contact between Pd and CeO2, which results in higher electrocatalytic activity, is an immediate demonstration of the criticality of the two-component cooperative mechanism.39 The saturation of CeO2 with OH− in alkaline media and their transfer to the metal nanoparticles to form the active PdIOHads species is one of the most important catalytic steps for obtaining high HOR activity.40 However, because of the insulating properties of ceria, the catalyst design must inevitably incorporate conductive phases, typically in the form of carbon black or carbon nanostructures.41 The resulting interfacial effects in such ternary composites are more complex to analyze, but it is particularly meaningful that the nanostructuring of the composite catalyst allows for improved dispersion and stabilization of the Pd NP on CeO2. Recently, Ogada et al. carried out calculations on the electronic band structures and the projected density of states (PDOS) for nanocomposites where Pd NPs were supported on onion-like carbon (OLC) with and without CeO2. It turned out that the available states in the CeO2-free composites were fewer than those in the case of Pd–CeO2/OLC (Fig. 6). This electronic modulation of Pd by CeO2 was responsible for the significantly improved performance.42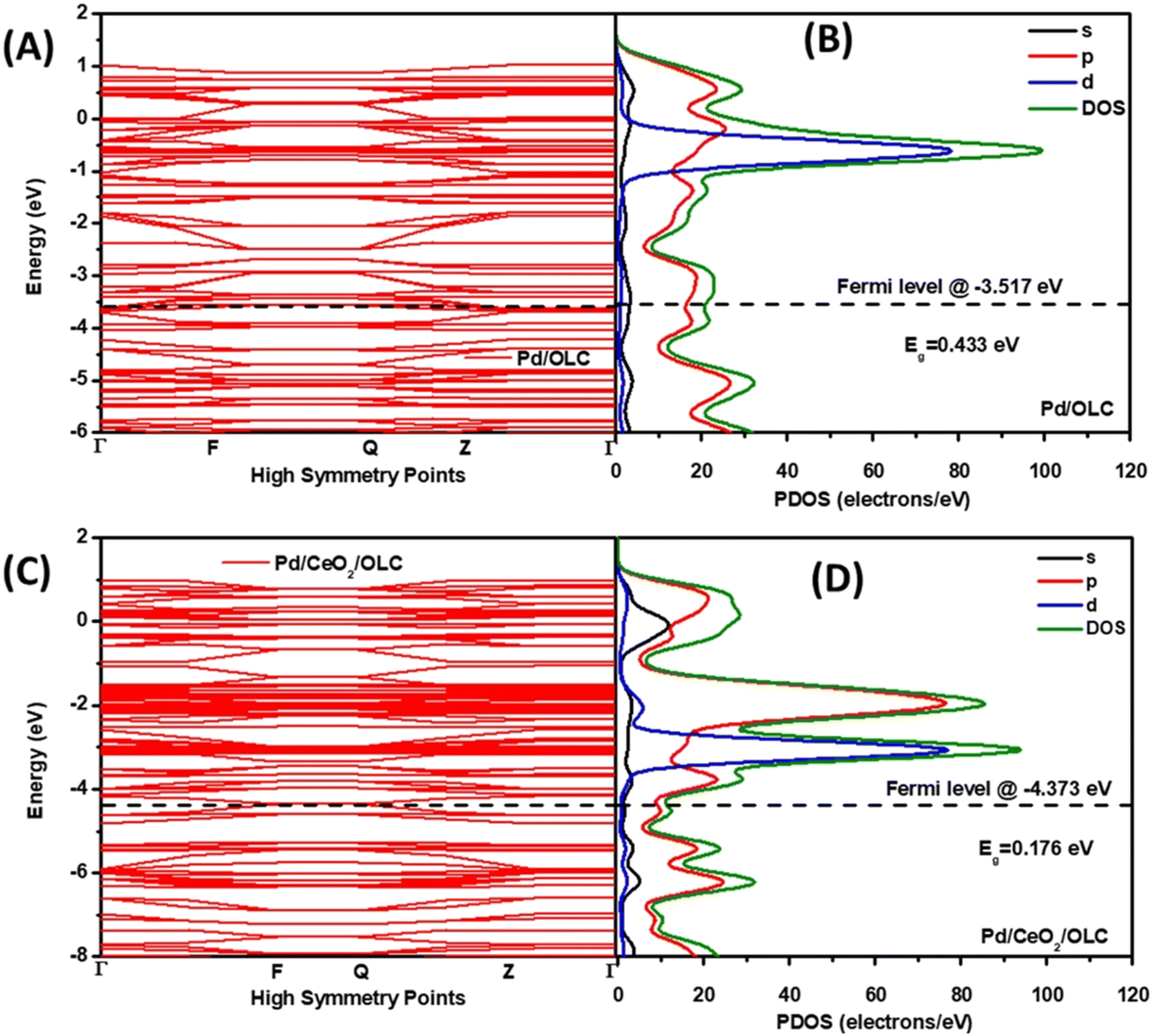 | ||
| Fig. 6 (A and C) Band structure and (B and D) PDOS of (A and B) Pd/OLC and (C and D) Pd–CeO2/OLC. Reproduced from ref. 36 with permission from American Chemical Society, copyright 2022. | ||
New exploits on PM/CeO2 catalysts
After decades of research on PM/CeO2 interaction and synergy, on the catalytic possibilities of such system, above all thermal catalysis but in more recent years also electrocatalysis, it may appear that this particular field has reached complete maturity. Hence, is there still anything to learn? It seems that some work is still devoted to computationally refining some structural details that has been of less interest, but that can still project some effects on catalytic reactions. Monte Carlo simulations have been performed to evaluate the role of the perimeter sites at the Pt/CeO2 interface during CO oxidation,43 while DFT combined with first-principles molecular dynamics confirmed the existence of a new class of SMSIs in Pt-SAC/CeO2 systems, identifying several well-defined and dynamically interconnected charge states originating from the phonon-assisted adjustment of the Ce(4f) level.44 Other endeavours tackled unexplored synthetic approaches to improve the regulation of the PM–CeO2 interaction. Plasma irradiation was applied to engineer the surface of CeO2 nanorods, enhancing the surface roughness and oxygen vacancy content. As a consequence, they obtain the double advantage of a better dispersion of Pt NPs and an increased amount of extra electrons on the ceria surface, and proposed that the latter effect could reverse the electron transfer between Pt and ceria, with the former exhibiting lower CO chemisorption on account of the higher electron density. The better CO tolerance resulted in higher methanol electro-oxidation activity and stability.45A few years ago, a nanofence structure was assembled by atomic layer deposition (ALD), with CeOx selectively deposited on Pt(111), which was thus stabilized, leaving Pt(100) exposed and available to catalyse CO oxidation.46 Despite the large interest in establishing metal–support interfaces in connection with typically observed better catalytic performances, it has been recognised that these systems also suffer from their specific drawbacks. One of these lies in the fact that during long-time operation catalysis, the interface is readily subject to the action of the reactive environment or to an interface reconstruction which alters the properties of the interface and causes loss of catalytic performance, either in terms of activity or selectivity. A recent intriguing opportunity has been offered by the assembly of the so-called “dual-active site” as an alternative configuration of PM/CeO2 catalysts. In these types of systems, rather than the interface, it is the two different sites (on the metal and on the support) which individually adsorb the two different molecules taking part in the reaction, and activate the overall catalytic process. For example, hydrogenation of nitroarenes was shown to proceed with a cooperative effect between Pd NPs and CeO2 porous nanorods, whereby the strong adsorption affinity of Pd towards H2 was flanked by the ability of oxygen vacancy-rich CeO2 to selectively adsorb the nitroarene. Crucial for the high activity was the sub-nanometric size of the Pd NPs, which disfavoured nitroarene coordination and promoted selective H2 adsorption.47 The dual-active site concept was reprised very recently for the reverse water-gas shift reaction (RWGS) by Pt/CeO2. Here, rather than depending on the metal–support interface, a strong coordination of the CO2 nanorods was enabled by the presence of frustrated Lewis pairs constructed on the surface of porous CeO2 nanorods. In parallel, the Pt clusters could readily adsorb and dissociate H2, finally undergoing H spillover on ceria, where CO2 was hydrogenated and released CO thanks to its weak interaction with the catalyst.48
Very recently, an interesting piece of work addressed the possibility to design smart catalysts based on Pd/CeO2 where sub-nanometric Pd clusters are able to undergo reversible speciation by means of reactive environment modulation.49 This is particularly useful for automotive exhaust applications, where differential temperature stages are in place.
In situ and operando characterization of the interface
From the latest publications, it is evident that one aspect of growing interest in the realm of PM/CeO2 catalysts is the necessity to elucidate the role of interfaces under real catalytic conditions. Traditional characterization was usually carried out on bulk materials, fresh or spent catalysts, but the properties so far unravelled have not necessarily been those emerging under real catalytic conditions, where the environment is completely different. Triple (or more) interfaces of varying nature can be formed between the PM, the CeO2 and the solid, liquid or gaseous reactive mixtures (or a combination of them), requiring specificity of the analytic technique. For example, simulations were used to demonstrate that ceria surfaces behave as proton conductors upon wetting with water, as the water can undergo dissociation into H+ and OH− and alter the local pH. The increased local acidity gives rise to a Grotthus-like diffusion mechanism of the ionic species that occurs at the Pt/CeO2 junction region, and that from there can be channelled along the interface, which has essential repercussions in various applications.50 In order to probe the dynamic evolution of the multicomponent catalyst in particular at the interfacial sites, and/or understand the adsorption kinetics and thermodynamics of intermediates over the course of the reaction, in the last decade the scene has been dominated by operando or in situ investigations. In reality, such techniques have been available for more than 20 years,51 although only scarcely employed, while recent years have witness a proliferation of articles using them. In situ Raman spectroscopy shed some light on the still controversial mechanism of CO oxidation at metal–oxide interfaces. In particular, in the case of CeO2-based catalysts, the participation of the ceria lattice oxygen is often invoked, as an alternative to the conventional description that the oxidation proceeds merely through superoxide or peroxide species as the active sites. The fabrication of Au@Pt/CeO2 composites permitted the application of surface-enhanced Raman spectroscopy (SERS), which considerably increased the sensitivity of the technique. Thanks to isotopic substitution experiments it was revealed that much of the activity during CO oxidation originated from chemisorbed Pt–O species at the interfacial Ce3+ defect sites, rather than lattice Ce–O species.52Operando Raman was also used to study toluene oxidation by Pt nanoclusters introduced on MOF-based 1D CeO2, identifying key intermediates and the role of oxygen vacancies at the Pt/CeO2 interface (Fig. 7).53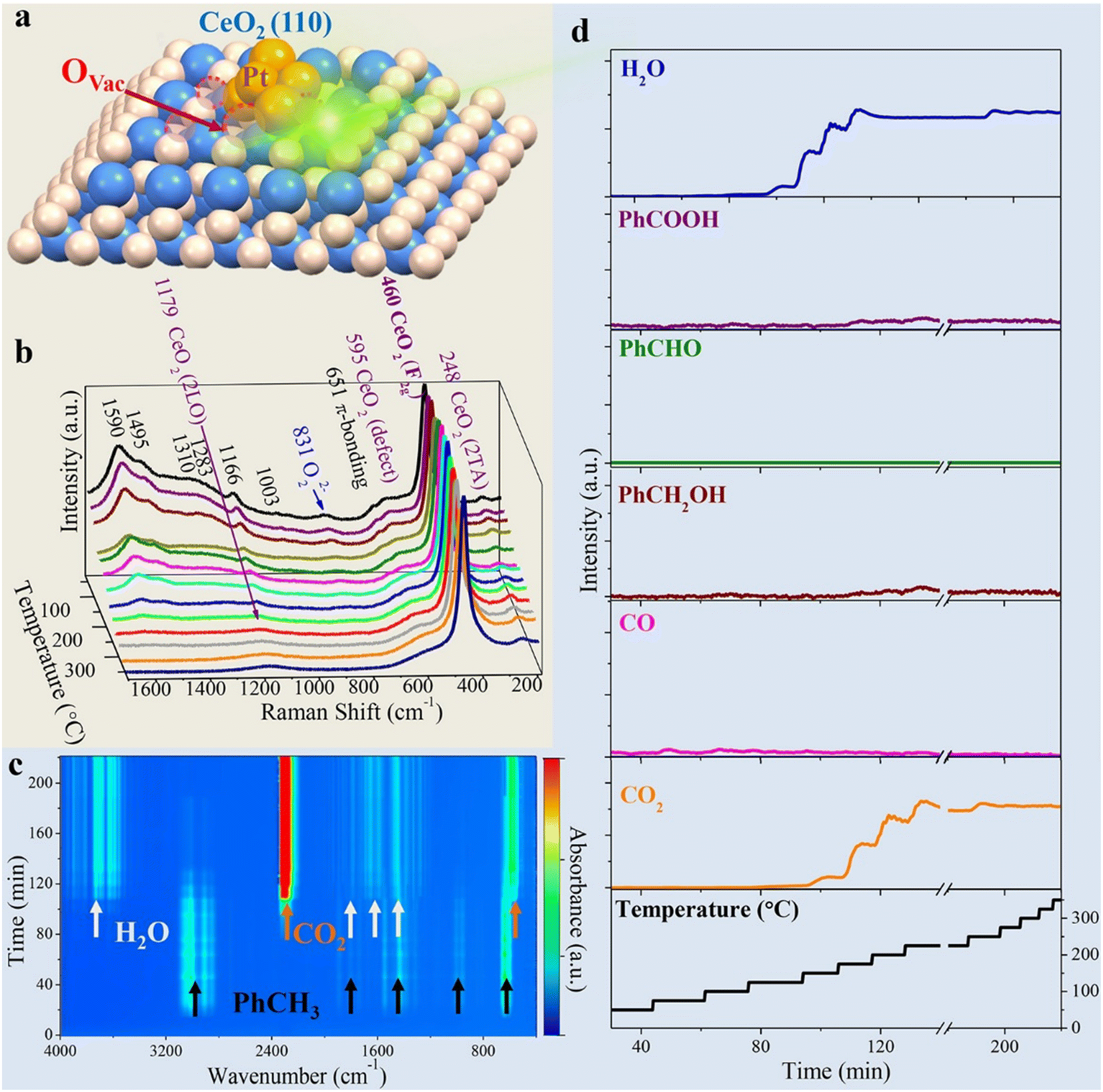 | ||
| Fig. 7 a) Graphical illustration of the Pt@CeO2–BDC catalyst with oxygen vacancies (OVac); b) 3D plot of the operando Raman spectra; c) 3D contour plot of the online FTIR spectra; d) plots of reactor temperature program and the corresponding concentration of products. Reproduced from ref. 47 with permission from American Chemical Society, copyright 2021. | ||
A combination of in situ Raman and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was critical to understand the activity of Au supported on CeO2 nanorods in the WGSR. Specifically, by comparing Au NPs with smaller size Au nanoclusters, the authors found evidence that the dominating catalytically active species involved a bridged surface –OH group formed at the gold–ceria interface, which explained the considerably higher activity of the nanoclusters.54 Time-resolved operando DRIFTS was used to study the structural evolution of the Au/CO2 interface in the case of CO oxidation, once again tracing the development of critical carbonate, bicarbonate and formate intermediates at the perimeter of the Au–CeO2 junction, where Auδ+ was presumed to be formed.55 The conversion of CH4 to olefins, aromatic compounds and H2 catalysed by Pt/CeO2 has been recently investigated using operando X-ray absorption spectroscopy (XAS) in order to unravel the importance of atomically dispersed Pt. It was found that while Pt SACs are active catalysts, aggregated Pt clusters exhibit a comparable activity, thanks to an increased Pt–Ce interaction, which was proposed on the basis of operando X-ray absorption near-edge spectroscopy (XANES) during the reaction with CH4 at 975 °C. The experiments also confirmed a sintering phenomenon of the single site Pt upon heating (Fig. 8).56
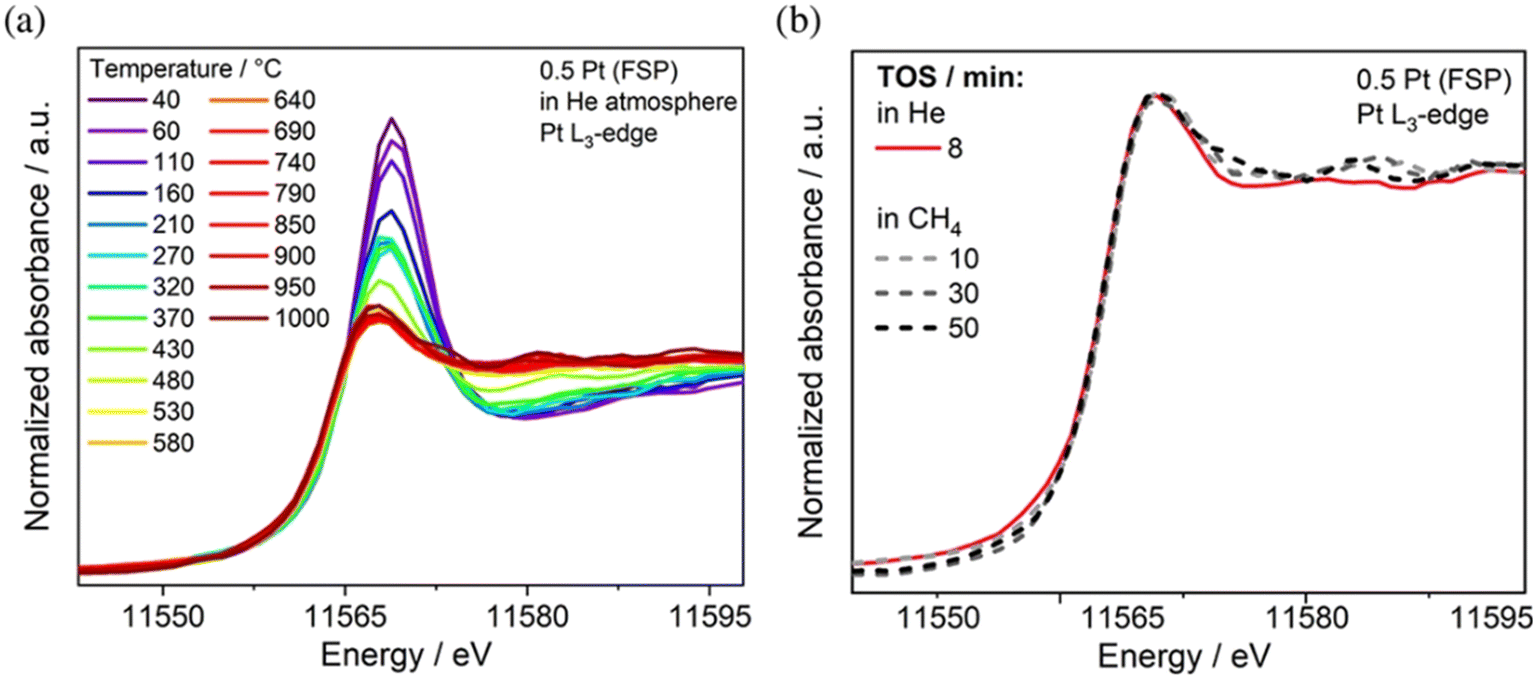 | ||
| Fig. 8 a) XANES spectra recorded at the Pt L3-edge of 0.5 Pt (FSP) during heating in He; b) XANES spectra under the reaction at 975 °C in 90% CH4/He. Reproduced from ref. 50 with permission from American Chemical Society, copyright 2022. | ||
Recently, Pollastri et al. addressed the stability of Pt/CeO2 catalysts in the context of proton-exchange membrane fuel cells (PEMFC) by means of operando XAS,57 while Wang et al. studied the methanation of CO2 by Ru/CeO2 by means of different in situ and operando techniques including XANES, Raman and steady-state isotope transient kinetic analysis (SSITKA)-type in situ DRIFTS, confirming that the methanation proceeds via the formate route with this catalyst.58In situ electron microscopy has also been used to understand the structural evolution of Pd NPs in a core–shell configuration with CeO2 during air calcination at different temperatures.59
Conclusions and perspectives
The exploitation of PM/CeO2 interfaces for catalytic applications has enjoyed a great success in the last 30 years, and it seems that it still represents a fertile soil for new discoveries. The primary objective was the stabilization of the PM nanoparticles by the CeO2 support, which as expected is at the centre of industry's attention for economic reasons, and in general sense for improving the sustainability of catalytic processes. Moreover, stabilization effects take the lead in the case of sub-nanometric PM particles and clusters, and even more in SAC systems, where strong anchoring is critical. However, the role of the interphase between ceria and PMs has revealed its complexity through a large number of research articles, going well beyond the pure structural stabilization of the NP by the support. The dynamics of the PM–ceria “communication” are at times complex, involving electron and/or atom (H spillover), as well as structural alteration of the support structure, predominantly described in terms of oxygen removal. In perspective, such dynamics will play an important role in those applications where the selectivity of the process is the real goal, such as tandem catalysis or switchable catalysts. Therefore, it clearly emerges that the complexity of the PM/CeO2 interaction is on the basis of the synergistic PM/CeO2 catalytic behaviour, and in most cases the synergy originates from the interface region, which coincides with the most active catalytic site. With the advancement of characterization techniques, researchers have been able to ignite ever renewed interest due to new fascinating discoveries and acquired skills to master the synthesis at a notably precise level. Applications have gradually moved from the traditional thermal catalysis to embrace more modern processes of more sustainable nature such as electrocatalysis and, more recently, photocatalysis. As the most abundant rare earth metal, cerium is hardly to be replaced in the design of catalytic nanocomposites, given the exclusive redox behaviour of rare earth metal oxides. On the other hand, efforts for more sustainable materials may be directed in the replacement of PMs with more abundant transition metals. A large number of notable reports are already available where some catalytic reactions can transcend PM active metals and resort instead to other readily available metals exhibiting interfacial effects, obtaining an overall high efficiency.60 New paradigms are continuously established, and much of the merits are derived from ingenious strategies to alter catalyst structures with innovative methodologies, although the presence of the metal–ceria interface always remains the cornerstone. After many years of studying ceria-based catalysts, our feeling is that much is yet to come.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Trieste, INSTM, the European Commission (H2020 – RIA-CE-NMBP-25 Program, Grant No. 862030). We acknowledge the COST Action 21101. M. M. acknowledges the project P2022WANKS funded by the Ministero dell'Università e della Ricerca (MUR) and the project FRA2022 funded by the University of Trieste.References
- E. B. Maxted and S. Akhtar, J. Chem. Soc., 1960, 1995–1998 RSC.
- (a) L. Mendelovich, H. Tzehoval and M. Steinberg, Appl. Surf. Sci., 1983, 17, 175–188 CrossRef; (b) D. W. Daniel, J. Phys. Chem., 1988, 92, 3891–3899 CrossRef CAS; (c) J. Z. Shyu and K. Otto, J. Catal., 1989, 115, 16–23 CrossRef CAS.
- (a) B. Engler, E. Koberstein and P. Schubert, Appl. Catal., 1989, 48, 71–92 CrossRef CAS; (b) G. Kim, Ind. Eng. Chem. Prod. Res. Dev., 1982, 21, 267–274 CrossRef CAS.
- (a) J. Kašpar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285–298 CrossRef; (b) J. Kašpar, P. Fornasiero and N. Hickey, Catal. Today, 2003, 77, 419–449 CrossRef.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- (a) T. Bunluesin, R. J. Gorte and G. W. Graham, Appl. Catal., B, 1998, 15, 107–114 CrossRef CAS; (b) A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439–520 CrossRef CAS.
- M. Cargnello, J. J. D. Jaén, J. C. H. Garrido, K. Bakhmutsky, T. Montini, J. J. C. Gámez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- M. Cargnello, P. Fornasiero and R. J. Gorte, Catal. Lett., 2012, 142, 1043–1048 CrossRef CAS.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS PubMed.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- M. G. Sanchez and J. L. Gazquez, J. Catal., 1987, 104, 120–135 CrossRef CAS.
- (a) S. Bernal, J. J. Calvino, M. A. Cauqui, G. A. Cifredo, A. Jobacho and J. M. Rodríguez-Izquierdo, Appl. Catal., A, 1993, 99, 1–8 CrossRef CAS; (b) S. Bernal, J. J. Calvino, M. A. Cauqui, J. A. Pérez Omil, J. M. Pintado and J. M. Rodríguez-Izquierdo, Appl. Catal., B, 1998, 16, 127–138 CrossRef CAS; (c) S. Bernal, J. J. Calvino, M. A. Cauqui, J. M. Gatica, C. Larese, J. A. Pérez Omil and J. M. Pintado, Catal. Today, 1999, 50, 175–206 CrossRef CAS.
- J. C. Frost, Nature, 1988, 334, 577–580 CrossRef CAS.
- N. Acerbi, S. C. Tsang, S. Golunski and P. Collier, Chem. Commun., 2008, 1578–1580 RSC.
- N. Acerbi, S. C. E. Tsang, G. Jones, S. Golunski and P. Collier, Angew. Chem., Int. Ed., 2013, 52, 7737–7741 CrossRef CAS PubMed.
- (a) A. Trovarelli, C. de Leitenburg and G. Dolcetti, J. Chem. Soc., Chem. Commun., 1991, 472–473 RSC; (b) C. Deleitenburg and A. Trovarelli, J. Catal., 1995, 156, 171–174 CrossRef CAS.
- A. Trovarelli, C. Deleitenburg, G. Dolcetti and J. L. Lorca, J. Catal., 1995, 151, 111–124 CrossRef CAS.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef CAS PubMed.
- N. C. Nelson, J. S. Manzano, A. D. Sadow, S. H. Overbury and I. I. Slowing, ACS Catal., 2015, 5, 2051–2061 CrossRef CAS.
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed.
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed.
- (a) C. Hardacre, G. M. Roe and R. M. Lambert, Surf. Sci., 1995, 326, 1–10 CrossRef CAS; (b) U. Berner and K.-D. Schierbaum, Phys. Rev. B, 2002, 65, 235404 CrossRef.
- (a) P. Luches and S. Valeri, Materials, 2015, 8, 5818–5833 CrossRef CAS PubMed; (b) P. Luches, F. Pagliuca and S. Valeri, J. Phys. Chem. C, 2011, 115, 10718–10726 CrossRef CAS; (c) P. Luches, F. Pagliuca and S. Valeri, Phys. Chem. Chem. Phys., 2014, 16, 18848–18857 RSC.
- J. A. Rodriguez, D. C. Grinter, Z. Liu, R. M. Palomino and S. D. Senanayake, Chem. Soc. Rev., 2017, 46, 1824–1841 RSC.
- (a) D. C. Grinter, C. L. Pang, C. A. Muryn, F. Maccherozzi, S. S. Dhesi and G. Thornton, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 13–17 CrossRef CAS; (b) B. Kaemena, S. D. Senanayake, A. Meyer, J. T. Sadowski, J. Falta and J. I. Flege, J. Phys. Chem. C, 2013, 117, 221–232 CrossRef CAS; (c) J. I. Flege and D. C. Grinter, Prog. Surf. Sci., 2018, 93, 21–45 CrossRef CAS.
- S. D. Senanayake, D. Stacchiola and J. A. Rodriguez, Acc. Chem. Res., 2013, 46, 1702–1711 CrossRef CAS PubMed.
- W. C. Conner, Jr. and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef.
- F. Yang, J. Graciani, J. Evans, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, J. Am. Chem. Soc., 2011, 133, 3444–3451 CrossRef CAS PubMed.
- P. Luches, G. Gasperi, M. Sauerbrey, S. Valeri, J. Falta and J. I. Flege, Front. Chem., 2019, 7 Search PubMed.
- (a) A. Beltram, M. Melchionna, T. Montini, L. Nasi, R. J. Gorte, M. Prato and P. Fornasiero, Catal. Today, 2015, 253, 142–148 CrossRef CAS; (b) M. Melchionna, A. Beltram, A. Stopin, T. Montini, R. W. Lodge, A. N. Khlobystov, D. Bonifazi, M. Prato and P. Fornasiero, Appl. Catal., B, 2018, 227, 356–365 CrossRef CAS; (c) G. Valenti, M. Melchionna, T. Montini, A. Boni, L. Nasi, E. Fonda, A. Criado, A. Zitolo, S. Voci, G. Bertoni, M. Bonchio, P. Fornasiero, F. Paolucci and M. Prato, ACS Appl. Energy Mater., 2020, 3, 8509–8518 CrossRef CAS.
- X. Zhang, D. Zhou, X. Wang, J. Zhou, J. Li, M. Zhang, Y. Shen, H. Chu and Y. Qu, ACS Catal., 2020, 10, 3832–3837 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2010, 329, 1633–1636 CrossRef CAS PubMed.
- (a) M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed; (b) M. Yang, J. Liu, S. Lee, B. Zugic, J. Huang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2015, 137, 3470–3473 CrossRef CAS PubMed.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- C. Wang, X.-K. Gu, H. Yan, Y. Lin, J. Li, D. Liu, W.-X. Li and J. Lu, ACS Catal., 2017, 7, 887–891 CrossRef CAS.
- O. A. Marina and M. Mogensen, Appl. Catal., A, 1999, 189, 117–126 CrossRef CAS.
- (a) K. Kwon, K. H. Lee, S.-A. Jin, D. J. You and C. Pak, Electrochem. Commun., 2011, 13, 1067–1069 CrossRef CAS; (b) D.-H. Lim, W.-D. Lee, D.-H. Choi and H.-I. Lee, Appl. Catal., B, 2010, 94, 85–96 CrossRef CAS.
- H. A. Miller, M. Bellini, D. R. Dekel and F. Vizza, Electrochem. Commun., 2022, 135, 107219 CrossRef CAS.
- H. A. Miller, A. Lavacchi, F. Vizza, M. Marelli, F. Di Benedetto, F. D'Acapito, Y. Paska, M. Page and D. R. Dekel, Angew. Chem., Int. Ed., 2016, 55, 6004–6007 CrossRef CAS PubMed.
- (a) A. Lenarda, M. Bellini, A. Marchionni, H. A. Miller, T. Montini, M. Melchionna, F. Vizza, M. Prato and P. Fornasiero, Inorg. Chim. Acta, 2018, 470, 213–220 CrossRef CAS; (b) R. K. Singh, E. S. Davydova, J. Douglin, A. O. Godoy, H. Tan, M. Bellini, B. J. Allen, J. Jankovic, H. A. Miller, A. C. Alba-Rubio and D. R. Dekel, Adv. Funct. Mater., 2020, 30, 2002087 CrossRef CAS.
- J. J. Ogada, A. K. Ipadeola, P. V. Mwonga, A. B. Haruna, F. Nichols, S. Chen, H. A. Miller, M. V. Pagliaro, F. Vizza, J. R. Varcoe, D. M. Meira, D. M. Wamwangi and K. I. Ozoemena, ACS Catal., 2022, 12, 7014–7029 CrossRef CAS.
- N. Bosio, M. Di, M. Skoglundh, P.-A. Carlsson and H. Grönbeck, J. Phys. Chem. C, 2022, 126, 16164–16171 CrossRef CAS.
- N. Daelman, M. Capdevila-Cortada and N. López, Nat. Mater., 2019, 18, 1215–1221 CrossRef CAS PubMed.
- L. Tao, Y. Shi, Y.-C. Huang, R. Chen, Y. Zhang, J. Huo, Y. Zou, G. Yu, J. Luo, C.-L. Dong and S. Wang, Nano Energy, 2018, 53, 604–612 CrossRef CAS.
- K. Cao, L. Shi, M. Gong, J. Cai, X. Liu, S. Chu, Y. Lang, B. Shan and R. Chen, Small, 2017, 13, 1700648 CrossRef PubMed.
- S. Zhang, C.-R. Chang, Z.-Q. Huang, J. Li, Z. Wu, Y. Ma, Z. Zhang, Y. Wang and Y. Qu, J. Am. Chem. Soc., 2016, 138, 2629–2637 CrossRef CAS PubMed.
- W. Li, J. Gan, Y. Liu, Y. Zou, S. Zhang and Y. Qu, Angew. Chem., Int. Ed., 2023, 62, e202305661 CrossRef CAS PubMed.
- D. Jiang, G. Wan, J. Halldin Stenlid, C. E. García-Vargas, J. Zhang, C. Sun, J. Li, F. Abild-Pedersen, C. J. Tassone and Y. Wang, Nat. Catal., 2023, 6, 618–627 CrossRef CAS.
- M. Farnesi Camellone, F. Negreiros Ribeiro, L. Szabová, Y. Tateyama and S. Fabris, J. Am. Chem. Soc., 2016, 138, 11560–11567 CrossRef CAS PubMed.
- G. Jacobs, P. M. Patterson, L. Williams, E. Chenu, D. Sparks, G. Thomas and B. H. Davis, Appl. Catal., A, 2004, 262, 177–187 CrossRef CAS.
- D.-Y. Wei, M.-F. Yue, S.-N. Qin, S. Zhang, Y.-F. Wu, G.-Y. Xu, H. Zhang, Z.-Q. Tian and J.-F. Li, J. Am. Chem. Soc., 2021, 143, 15635–15643 CrossRef CAS PubMed.
- Q. Wang, Y. Li, A. Serrano-Lotina, W. Han, R. Portela, R. Wang, M. A. Bañares and K. L. Yeung, J. Am. Chem. Soc., 2021, 143, 196–205 CrossRef CAS PubMed.
- X.-P. Fu, L.-W. Guo, W.-W. Wang, C. Ma, C.-J. Jia, K. Wu, R. Si, L.-D. Sun and C.-H. Yan, J. Am. Chem. Soc., 2019, 141, 4613–4623 CrossRef CAS PubMed.
- S. Chen, L. Luo, Z. Jiang and W. Huang, ACS Catal., 2015, 5, 1653–1662 CrossRef CAS.
- D. Eggart, X. Huang, A. Zimina, J. Yang, Y. Pan, X. Pan and J.-D. Grunwaldt, ACS Catal., 2022, 12, 3897–3908 CrossRef CAS.
- S. Pollastri, M. Bogar, R. Fiala, H. Amenitsch, Y. Yakovlev, A. Lavacchi, G. Aquilanti and V. Matolin, Int. J. Hydrogen Energy, 2022, 47, 8799–8810 CrossRef CAS.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- S. Zhang, C. Chen, M. Cargnello, P. Fornasiero, R. J. Gorte, G. W. Graham and X. Pan, Nat. Commun., 2015, 6, 7778 CrossRef PubMed.
- (a) A. Parastaev, V. Muravev, E. Huertas Osta, A. J. F. van Hoof, T. F. Kimpel, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2020, 3, 526–533 CrossRef CAS; (b) A. Chen, X. Yu, Y. Zhou, S. Miao, Y. Li, S. Kuld, J. Sehested, J. Liu, T. Aoki, S. Hong, M. F. Camellone, S. Fabris, J. Ning, C. Jin, C. Yang, A. Nefedov, C. Wöll, Y. Wang and W. Shen, Nat. Catal., 2019, 2, 334–341 CrossRef CAS; (c) Y. Wang, Q. Jiang, L. Xu, Z.-K. Han, S. Guo, G. Li and A. Baiker, ACS Appl. Mater. Interfaces, 2021, 13, 61078–61087 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |